
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHollow @CuMgAl double layered hydrotalcites and mixed oxides with tunable textural and structural properties, and thus enhanced NH3-NOx-SCR activity
Tomasz
Kondratowicz
 *ab,
Ondřej
Horký
a,
Stanislav
Slang
c,
Lada
Dubnová
a,
Marta
Gajewska
d,
Lucjan
Chmielarz
e and
Libor
Čapek
*a
*ab,
Ondřej
Horký
a,
Stanislav
Slang
c,
Lada
Dubnová
a,
Marta
Gajewska
d,
Lucjan
Chmielarz
e and
Libor
Čapek
*a
aUniversity of Pardubice, Faculty of Chemical Technology, Department of Physical Chemistry, Studentská 573, 532 10 Pardubice, Czech Republic. E-mail: tomasz.kondratowicz@chem.ox.ac.uk; libor.capek@upce.cz
bChemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, Oxford, OX1 3TA, UK
cUniversity of Pardubice, Faculty of Chemical Technology, Center of Materials and Nanotechnologies, Studentská 95, 532 10 Pardubice, Czech Republic
dAcademic Centre for Materials and Nanotechnology, AGH University of Science and Technology, Mickiewicza 30, 30-059 Kraków, Poland
eJagiellonian University, Faculty of Chemistry, Gronostajowa 2, 30 387 Kraków, Poland
First published on 18th May 2023
Abstract
Well-organized, spherical, mesoporous hollow @CuMgAl-LDHs (layered double hydroxides) are prepared by the controlled removal of the SiO2 from SiO2@CuMgAl-LDH core–shell hybrids that in turn are synthesized via a bottom-up strategy. The materials are prepared with various Cu/Mg molar ratios (Cu/Mg = 0.05–0.50) while keeping the ratio of Cu and Mg constant, (Cu + Mg)/Al = 2. The effect of Cu doping and the silica core removal process (conducted for 4 h at 30 °C using 1 M NaOH) on the chemical composition, morphology, structure, texture and reducibility of the resulting materials are described. @CuMgAl-MOs (mixed oxides) obtained by thermal treatment of the @CuMgAl-LDHs are active and selective catalysts for the selective catalytic reduction of NOx using ammonia, and effectively operate at low temperatures. The N2 yield increases with increased Cu content in the CuMgAl shell, which is associated with the easier reducibility of the Cu species incorporated into the MgAl matrix. @CuMgAl-MOs show better catalytic performance than bulk CuMgAl MOs.
Introduction
NOx is primarily made up of nitric oxide (NO) and nitrogen dioxide (NO2) and belongs to a significant group of air pollutants which have a number of negative impacts on human health and the environment, including e.g., photochemical smog, ozone depletion and acid rain.1 The anthropogenic emission of NOx originates mainly from the stationary combustion of fossil fuels, such as that found in steel plants, power plants, cement plants and waste incineration. NOx emissions also originate from mobile sources, with a major contribution from the use of diesel engines. Considering the above, NOx emissions should be reduced by 42% for all EU member countries over the years 2020–2029.2Up to now, the most recognized and effective technology for NOx abatement is the selective catalytic reduction of NOx using ammonia (NH3-SCR), urea, or hydrocarbons (HC-SCR).3 Commercial NH3-SCR technology has been implemented in thermal power plants using V2O5-WO3(MoO3)/TiO2 catalysts with relatively high efficiency in the temperature range of 300–400 °C.4 However, exhaust gas temperatures in non-energy industries are often much lower, rendering vanadium-based SCR catalysts less efficient. Therefore, the development of catalysts with a satisfactory low-medium temperature catalytic activity window (<250 °C) and good resistance stability (e.g., in the presence of SO2 or HCl) is a permanent challenge for chemists and materials scientists for both economic and environmental reasons.4–6 Among the many proposed systems, metal oxides, such as Cu,7,8 Fe9,10 and Mn,11,12 are widely recognized and considered to be effective active components for low-medium temperature SCR catalysts, mainly due to their high catalytic activity, long lifetimes, wide availability and relatively low production costs. Besides the qualitative composition of the catalysts, the transition metal content also greatly influences the efficiency in the NH3-SCR process.13–15 However, synthesis of multicomponent oxide catalysts by traditional preparation methods, e.g., coprecipitation, hydrothermal synthesis and sol–gel methods, often leads to a heterogeneous distribution of the active components, as well as agglomeration and sintering of particles, which may significantly limit the potential of these catalysts for practical applications.1
Therefore, mixed oxides (MOs) originating from layered double hydroxides (LDHs) with different combinations of metal ions have been successfully tested as SCR catalysts since the late 1990s.1 Such great interest in this class of materials is related to their unique properties, such as having excellent and uniform metal oxide dispersions, high thermal stabilities and memory effects. Moreover, their properties and surface compositions can be easily tuned during the synthesis, their preparation methods are well known, and they are environmentally friendly materials that are relatively cheap to produce. However, frequently LDH materials prepared using conventional methods have poor morphologies, such as aggregated powders composed of irregular particles of various sizes (stone-like morphologies).16 This is the result of severe platelet aggregation caused by ab-face stacking during the drying process and it may negatively affect the porosity of the LDH materials and significantly limits their utilization.
Given the unique and important properties of pristine LDHs and MOs, various functionalization strategies have been applied to bring even more exciting performance outcomes. One such strategy is the dispersion of LDHs via aqueous miscible organic solvent treatment (AMOST) into thin nanosheets or via exfoliation into single layers.17,18 Another class of LDHs modification is the generation of additional porosity, and thus a significant expansion of the specific surface area, in pristine LDHs, which enables better dispersions of the LDHs, exposes more active sites and facilitates the mass transport of reagents towards and from the surface. This goal can be achieved through alkaline etching that utilizes the amphoteric features of the Al3+ ion in the LDHs crystal19,20 or by the introduction of a foreign substance during the synthesis of the LDH materials. The synthesis can be performed in the presence of carbon-containing precursors, such as activated carbon,21 date palm ash,22 and all types of ionic and non-ionic surfactant molecules,23–26 which can then be removed by thermal treatment, helping to fine-tune the surface properties of the resulting mixed oxides.
In addition to the above-mentioned methods to tailor LDHs, approaches based on the immobilization of LDH on supports is a relatively new field of materials chemistry, proposed for the first time by Zhang et al. in 2009.27 In particular, when hard templates with well-defined shapes and dimensions are employed, three-dimensional (3D) hierarchical LDH-based materials can be constructed to further improve the diffusion limitations, accessibility of the active sites, texture properties as well as the irregular shapes and sizes of the LDHs.28,29 Up to now, various types of LDH-based core–shell nanostructures with a variety of cores have been reported, e.g., nonporous SiO2,30 mesoporous silicas (e.g., MCM-41, MCM-48, SBA-15),31 FexOy,32 Cu2O,33 TiO2,34 zeolites (e.g., ZSM-5, Y, TS-1),35 MOFs36 and carbon.37
However, the current state of the literature clearly shows that SiO2@MgAl-LDH hybrids16,30,38–44 are the most explored and well-described in the literature. This is probably related to (i) the relatively fast and uncomplicated synthesis of the spherical SiO2 core with tunable control of particle size using the Stöber process,45 (ii) the high stability of the SiO2@LDH composite due to the Si–O–M covalent bonds,46 as well as (iii) the MgAl phase being among the most popular variety of LDHs. A few papers have described other SiO2@LDH core–shell systems in which the LDH consists of NiAl,39,47,48 NiFe, CoFe, CoNi47 and MgGa.39 However, core–shell materials containing silica as a core and more than two elements in the LDH shell have not been thoroughly examined. To date, only three structures, SiO2@CeMgAl-LDH,49 SiO2@MgAlFe-LDH and SiO2@MgAlNi-LDH,50 have been described in detail.
The study focusses on hollow @CuMgAl-LDHs and hollow @CuMgAl-MOs and is a natural continuation and extension of our previous work,38 in which we presented a controlled removal of the SiO2 core from a SiO2@MgAl-LDH system to form hollow @MgAl-LDHs. Additionally, this work perfectly fills the space describing core–shell systems containing a SiO2 core and ternary LDHs as a shell. SiO2@CuMgAl-LDHs materials with various Cu/Mg molar ratios (0.05–0.50) are synthesized while keeping the (Cu + Mg)/Al molar ratio at a constant value of 2. Additionally, the conditions for the SiO2 core removal which facilitate the formation of hollow @CuMgAl-LDH are described. The effect of doping MgAl-LDH with various amounts of Cu, the implementation of the silica core removal process and the effect of thermal treatment (to form mixed oxides) on the morphology, structural, textural and redox properties of the core–shell and hollow sphere materials are described here. Moreover, the potential of such materials in environmental catalysis (selective catalytic reduction of NO with ammonia (NH3-SCR)) has also been demonstrated.
Experimental
Synthesis
The spherical core–shell SiO2@CuMgAl-LDH materials (with various Cu/Mg molar ratios with a constant Cu + Mg/Al ratio of 2) were prepared according to our previous work.38 The method involves the in situ coprecipitation of Cu, Mg and Al precursors in the presence of non-porous spherical SiO2 particles of uniform size with ca. 400 nm diameters. Briefly, 100 mg of spherical SiO2 particles were dispersed in 20.0 mL of deionized water using an ultrasound bath. After 1 h, 0.96 mmol of Na2CO3 (Lach-Ner, 100.1%) was added to the milky solution and dispersed for another 10 minutes. Subsequently, to the as-prepared suspension was added dropwise (1 mL min−1) a 19.2 mL water solution containing various amounts of Cu(NO3)2·3H2O (Lach-Ner, 101.9%), Mg(NO3)2·6H2O (Lach-Ner, 100.8%), and Al(NO3)3·9H2O (Lach-Ner, 98.6%) (total metal concentration = 1.44 mmol). The pH was maintained at 10.00 ± 0.03 using an aqueous NaOH solution (1 M) during titration. After 2 h of stirring (400 rpm), the obtained core–shell particles were filtered, washed with 2 L of deionized water and dried at 60 °C overnight. Dried samples are denoted as SiO2@CuMgAl_x-LDH, where x expresses the nominal Cu/Mg molar ratio (0.05, 0.1, 0.2, 0.3, 0.4 and 0.5).In the next step, the silica core was etched under static conditions by treatment with a NaOH solution (1 M) at 30 °C for 4 h. To remove remaining NaOH, the obtained particles were thoroughly washed with distilled water (2 L) and then dried at 60 °C. The obtained materials are denoted as @CuMgAl_x-LDH.
CuMgAl-based mixed oxides were obtained by thermal treatment of the LDH-like precursor in air at 450 °C for 4 h (heating rate of 5 °C min−1) in a muffle oven. Calcined materials are denoted as @CuMgAl_x-MO.
Characterization
Quantitative energy-dispersive X-ray spectroscopy (EDX) measurements were performed on an electron microscope (LYRA3, Tescan) equipped with an EDX analyzer AZtec X-Max 20 (Oxford Instruments) at an acceleration voltage of 20 kV (an accelerating voltage possessing a signal from a depth of micrometres, which is sufficient for the studied core–shell particles with dimensions of approximately 800 nm). The samples were covered with 20 nm of carbon using a Leica EM ACE200 coater and subsequently 5 different spots with a size of 200 × 200 μm were measured and the obtained compositions were averaged. A scanning electron microscope (LYRA3, Tescan) was used for the surface morphology study using a 10 kV acceleration voltage. The samples were coated with a thin 18 nm layer of gold (Leica EM ACE200) prior to the scanning electron microscopy (SEM) analysis. Transmission electron microscopy (TEM) measurements were carried out on a FEI Tecnai TF20 X-TWIN (FEG) microscope equipped with an energy-dispersive X-ray spectrometer (EDAX), working at an accelerating voltage of 200 kV. Samples for the TEM observations were prepared by drop-casting on carbon-coated copper grids. The crystallographic structure of samples was determined using a Rigaku MiniFlex 600 diffractometer with a PDF-2 database and D/teX Ultra detector using Cu Kα radiation operated at 40 kV and 15 mA. The X-ray diffraction (XRD) patterns were recorded in the range of 2θ = 5–80° with a step of 0.02°. Nitrogen adsorption–desorption isotherms were recorded at −196 °C using a Micromeritics TriStar II instrument. Before the measurements, samples were degassed for 6 h under vacuum at 130 °C or 150 °C for the LDH and MO phases, respectively. Specific surface areas (SBET) were determined using the Brunauer–Emmett–Teller (BET) method. Total pore volumes (Vtotal) were obtained from the amounts of nitrogen adsorbed at a relative pressure of 0.99, whereas the micropore (Vmicro) and mesopore (Vmeso) volumes were calculated using a t-plot and Barrett–Joyner–Halenda (BJH) models, respectively. In turn, the volume of macropores (Vmacro) was calculated as the difference between Vtotal and the sum of Vmicro and Vmeso. Temperature-programmed reduction with hydrogen (H2-TPR) was carried out in a flow system using a Micromeritics AutoChem II 2920 equipped with a TCD detector. Before a TPR experiment, 100 mg of a sample was outgassed in a flow of helium (25 mL min−1) at 450 °C and then for 30 min in an oxygen atmosphere at 450 °C, followed by cooling to 150 °C. At 150 °C, the oxygen atmosphere was swapped to a helium atmosphere to flush the reactor with an inert gas and the reactor was cooled down to 25 °C. After a freezer was added to the system, the hydrogen uptake from a stream of 5 vol% of H2 in Ar (a flow rate of 25 mL min−1) was analyzed over a temperature range of 25–900 °C at a heating rate of 10 °C min−1. The number of redox sites was calculated from the integrals of the signal after their integration with external calibration.Catalytic tests
The catalysts were tested in the selective catalytic reduction of NO with ammonia (NH3-SCR). Catalytic studies were performed on an experimental system consisting of a fixed-bed quartz microreactor with the outlet connected directly to the detector – a quadrupole mass spectrometer (QMS, PREVAC). The used gas mixture contained 0.25 vol% NO, 0.25 vol% NH3 and 2.5 vol% O2 diluted in pure helium (total flow rate of 40 mL min−1). Prior to the catalytic run, 100 mg of the sample (particle size in the range of 250–315 μm) was placed in the quartz microreactor and outgassed in the flow of pure helium at 450 °C for 30 min. After the microreactor was cooled down to 100 °C, the catalytic test was initiated with a linear heating rate of 10 °C min−1, over a temperature range of 100–400 °C. Catalytic tests were performed under atmospheric pressure.Moreover, for the most active catalyst, an isothermal long-term stability NH3-SCR test at 275 °C for 24 h was done with the same composition of the reaction mixture and mass of the catalyst sample. A catalytic study of the ammonia oxidation reaction was carried out using the same experimental system used in the case of the NH3-SCR tests. Prior to the catalytic tests, the samples (100 mg, particle sizes of 250–315 μm) were outgassed in a flow of pure helium (20 mL min−1) at 450 °C for 30 min. Catalytic runs were conducted in the temperature range of 100–450 °C with a gas mixture containing: [NH3] = 0.5 vol% and [O2] = 2.5 vol%, diluted in pure helium used as a balance gas (total flow rate 40 mL min−1). All catalytic experiments were performed at a constant gas hourly space velocity (GHSV) and a volumetric hourly space velocity (VHSV) of approx. 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 and 24000 cm3 h−1 g−1, respectively.
000 h−1 and 24000 cm3 h−1 g−1, respectively.
Results and discussion
Features of the spherical SiO2@CuMgAl-LDH and @CuMgAl-LDHs
Table 1 gives the Cu/Mg, (Cu + Mg)/Al and the Si/(Si + Cu + Mg + Al) molar ratios of the pristine core–shell SiO2@CuMgAl-LDHs and the hollow @CuMgAl-LDHs obtained after the leaching of the SiO2 core for 4 h. To keep a constant (Cu + Mg)/Al molar ratio, magnesium was isomorphically substituted by copper during the synthesis. However, the experimental Cu/Mg and (Cu + Mg)/Al molar ratios for the SiO2@CuMgAl-LDHs were slightly higher than the theoretical values. Similar discrepancies between the theoretical and real Cu/Mg molar ratios in LDHs have been reported previously in the literature.51–54 The observed differences may result from the preferential incorporation of Cu2+ ions into the crystal lattice during the growth of the LDH phase over Mg2+ ions. It could be connected with the pH value, as the coprecipitation of various ions requires different pH values.55 Thus, the pH value of 10.0, used in the synthesis of SiO2@CuMgAl-LDHs, could slightly favour the incorporation of Cu2+ ions. It can also be explained by the preferential dissolution of the Mg2+ ions over Cu2+ during the washing of the solid materials with water. This hypothesis may be supported by the fact that Mg(OH)2 is about 3 orders of magnitude more water-soluble than Cu(OH)2 (the solubility equilibrium at 25 °C is 5.61 × 10−12vs. 1.99 × 10−20, respectively). Additionally, the partial dissolution of Al3+ and Cu2+ in NaOH solution, due to their amphoteric properties, cannot be excluded.19,20,56 However, considering the relatively low NaOH concentration used during the modification, the impact may by negligible. In our previous study,38 it was reported that the silica core cannot be completely removed from the SiO2@MgAl-LDHs even at long leaching times (up to 48 h) or at much stronger leaching conditions (2 M, 50 °C, 20 h). For the synthesis of @MgAl-LDHs, the optimal conditions chosen for the leaching of the SiO2 core with respect to its catalytic behaviour were a 1 M NaOH solution for 4 h. Therefore, based on that, the same conditions for leaching the SiO2 core of SiO2@CuMgAl-LDHs were implemented. It is a compromise between the degree of silica core removal from the core–shell system on one side, and the resulting morphology, textural, basic and catalytic properties on the other. For the pristine SiO2@CuMgAl-LDHs, the Si/(Si + Cu + Mg + Al) molar ratio was in the range of 0.47–0.52 (Table 1). After 4 h of SiO2 template leaching, the Si/(Si + Cu + Mg + Al) molar ratio decreased, and it was found to range from 0.14 to 0.28. It should be emphasized that despite application of the same conditions (4 h, 1 M NaOH), it was not possible to achieve the same level of the SiO2 core removal for all the @CuMgAl-LDHs (Table 1).| Sample | Cu/Mg | (Cu + Mg)/Al | Si/(Si + Cu + Mg + Al) |
|---|---|---|---|
| SiO2@CuMgAl_0.05-LDH | 0.07 | 2.04 | 0.52 |
| SiO2@CuMgAl_0.10-LDH | 0.12 | 2.04 | 0.50 |
| SiO2@CuMgAl_0.20-LDH | 0.25 | 2.12 | 0.48 |
| SiO2@CuMgAl_0.30-LDH | 0.39 | 2.16 | 0.47 |
| SiO2@CuMgAl_0.40-LDH | 0.50 | 2.18 | 0.47 |
| SiO2@CuMgAl_0.50-LDH | 0.60 | 2.15 | 0.48 |
| @CuMgAl_0.05-LDH | 0.06 | 2.06 | 0.14 |
| @CuMgAl_0.10-LDH | 0.12 | 2.07 | 0.21 |
| @CuMgAl_0.20-LDH | 0.24 | 2.06 | 0.26 |
| @CuMgAl_0.30-LDH | 0.35 | 2.11 | 0.16 |
| @CuMgAl_0.40-LDH | 0.45 | 2.11 | 0.17 |
| @CuMgAl_0.50-LDH | 0.56 | 2.11 | 0.28 |
Fig. 1A and B show XRD patterns of the SiO2@CuMgAl-LDHs and @CuMgAl-LDHs, respectively. Also shown is the diffractogram of the reference material CuMgAl_ref-LDH. The XRD patterns of all materials contained diffraction lines at 11.6°, 23.4°, 34.8°, 39.2°, 46.8°, 60.6° and 62.0° that could be assigned to the (0 0 3), (0 0 6), (0 1 2), (0 1 5), (0 1 8), (1 1 0) and (1 1 3) planes, respectively, typical for classical double-layered structures with the trigonal R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group.57,58 The shape of the diffraction lines does not vary significantly with increasing copper content (Cu/Mg molar ratio), neither in intensity nor in peak position shift. Moreover, for the pristine SiO2@CuMgAl-LDHs, a broad hump at 2θ ≈ 22° is distinctly visible and is related to the presence of the amorphous SiO2 core in the core–shell particles.38,59 After 4 h of alkali treatment, none of the positions of the LDH diffraction lines change, while their intensities slightly increased, and the broad hump almost disappears (the slight bend in the baseline between the (003) and (012) planes is still visible). This confirms a decrease in the silica content of the @CuMgAl-LDHs relative to their corresponding pristine structures. Table 2 contains the lattice parameters a (a = 2d110), c (c = 3d003), LDH basal spacing (also known as the d value or interlayer distance along the c axis, calculated from d(003)60) as well as the LDH crystallite size in the stacking direction D(003) and plane direction D(110). The lattice parameters and LDH basal spacings are almost identical for all SiO2@CuMgAl-LDHs regardless of the LDH composition. Additionally, these parameters were maintained around the same values in the @CuMgAl-LDHs (after leaching of the SiO2 core for 4 hours). The determined unit cell in the crystal lattice and the d value for the SiO2@CuMgAl-LDHs and hollow @CuMgAl-LDHs correspond to the previously reported SiO2@MgAl-LDHs and hollow@MgAl-LDHs38 as well as other classic bulk double-layered structures with the similar M2+/Al3+ molar ratios.60,61 This may be explained by the almost identical sizes of the octahedral ionic radii of Cu2+ (0.73 Å) and Mg2+ (0.72 Å).62 On the other hand, the calculated unit cell dimensions are slightly lower than those of various SiO2@LDH core–shell systems (LDH = MgAl, NiAl, MgGa, M2+/M3+ = 3).39,40 This difference may be explained by the different ionic radii in the other systems as well as higher molar ratio between M2+ and M3+ in the brucite-like layers. In the case of the LDH crystallite size in the SiO2@CuMgAl-LDHs, the calculated dimension in the stacking direction D(003) is slighter shorter than in the plane direction D(110) and are in the range of 6.9–7.8 and 12.3.1–13.2 nm, respectively. After treatment in an alkaline environment, these values remained almost unchanged in the @CuMgAl-LDHs (6.6–7.2 and 13.4–14.7 nm, respectively). Crystallite sizes that are higher in the plane direction than in the stacking direction have also been reported for other core–shell materials constructed with SiO2 cores and NiAl-, MgGa-,39 and MgAl-LDH38–40 shells. Previously reported CuMgAl-LDHs with a similar composition (Cu/Mg = 0.08 and (Cu + Mg)/Al = 2.03) and synthesized via a co-precipitation method, but without the SiO2 particles, resulted in much higher crystallite sizes (75 nm).53 The rapid nucleation of LDH compactly packed on the surface of the core, with simultaneous faster growth of the LDH crystal on the (110) plane than on the (003) plane, is known to ensure vertical orientation of the LDH plates onto the surface of the support.46,63 Consequently, it is suggested that the CuMgAl-LDH platelets are anchored perpendicularly onto the SiO2 surface in the SiO2@CuMgAl-LDH core–shell materials.
m space group.57,58 The shape of the diffraction lines does not vary significantly with increasing copper content (Cu/Mg molar ratio), neither in intensity nor in peak position shift. Moreover, for the pristine SiO2@CuMgAl-LDHs, a broad hump at 2θ ≈ 22° is distinctly visible and is related to the presence of the amorphous SiO2 core in the core–shell particles.38,59 After 4 h of alkali treatment, none of the positions of the LDH diffraction lines change, while their intensities slightly increased, and the broad hump almost disappears (the slight bend in the baseline between the (003) and (012) planes is still visible). This confirms a decrease in the silica content of the @CuMgAl-LDHs relative to their corresponding pristine structures. Table 2 contains the lattice parameters a (a = 2d110), c (c = 3d003), LDH basal spacing (also known as the d value or interlayer distance along the c axis, calculated from d(003)60) as well as the LDH crystallite size in the stacking direction D(003) and plane direction D(110). The lattice parameters and LDH basal spacings are almost identical for all SiO2@CuMgAl-LDHs regardless of the LDH composition. Additionally, these parameters were maintained around the same values in the @CuMgAl-LDHs (after leaching of the SiO2 core for 4 hours). The determined unit cell in the crystal lattice and the d value for the SiO2@CuMgAl-LDHs and hollow @CuMgAl-LDHs correspond to the previously reported SiO2@MgAl-LDHs and hollow@MgAl-LDHs38 as well as other classic bulk double-layered structures with the similar M2+/Al3+ molar ratios.60,61 This may be explained by the almost identical sizes of the octahedral ionic radii of Cu2+ (0.73 Å) and Mg2+ (0.72 Å).62 On the other hand, the calculated unit cell dimensions are slightly lower than those of various SiO2@LDH core–shell systems (LDH = MgAl, NiAl, MgGa, M2+/M3+ = 3).39,40 This difference may be explained by the different ionic radii in the other systems as well as higher molar ratio between M2+ and M3+ in the brucite-like layers. In the case of the LDH crystallite size in the SiO2@CuMgAl-LDHs, the calculated dimension in the stacking direction D(003) is slighter shorter than in the plane direction D(110) and are in the range of 6.9–7.8 and 12.3.1–13.2 nm, respectively. After treatment in an alkaline environment, these values remained almost unchanged in the @CuMgAl-LDHs (6.6–7.2 and 13.4–14.7 nm, respectively). Crystallite sizes that are higher in the plane direction than in the stacking direction have also been reported for other core–shell materials constructed with SiO2 cores and NiAl-, MgGa-,39 and MgAl-LDH38–40 shells. Previously reported CuMgAl-LDHs with a similar composition (Cu/Mg = 0.08 and (Cu + Mg)/Al = 2.03) and synthesized via a co-precipitation method, but without the SiO2 particles, resulted in much higher crystallite sizes (75 nm).53 The rapid nucleation of LDH compactly packed on the surface of the core, with simultaneous faster growth of the LDH crystal on the (110) plane than on the (003) plane, is known to ensure vertical orientation of the LDH plates onto the surface of the support.46,63 Consequently, it is suggested that the CuMgAl-LDH platelets are anchored perpendicularly onto the SiO2 surface in the SiO2@CuMgAl-LDH core–shell materials.
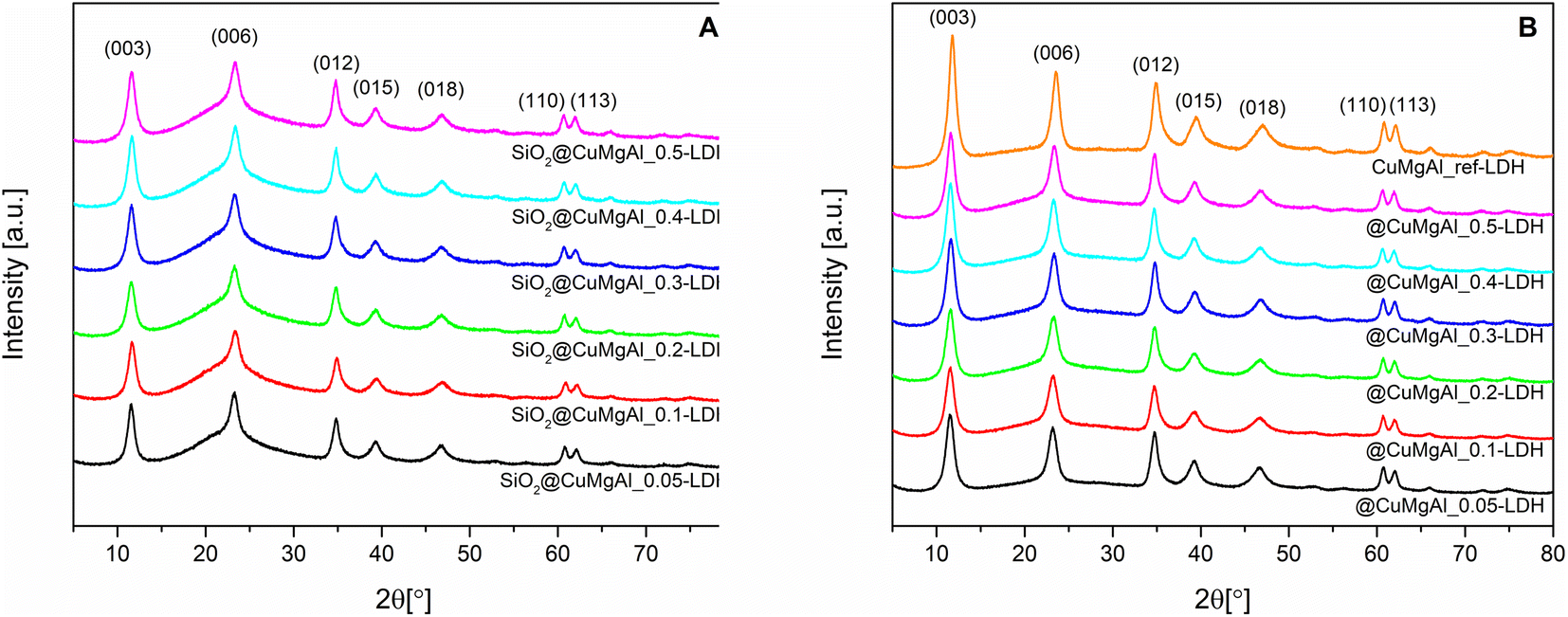 | ||
| Fig. 1 XRD patterns of the SiO2@CuMgAl-LDH composites (A) before and (B) after SiO2 core etching with 1 M NaOH solution. A pattern for a CuMgAl-LDH reference sample is also shown. | ||
| Sample | Lattice parameters [nm] | LDH basal spacing [nm] | Crystallite size [nm] | ||
|---|---|---|---|---|---|
| a | c | d | D(003) | D(110) | |
| SiO2@CuMgAl_0.05-LDH | 0.305 | 2.291 | 0.764 | 7.8 | 12.7 |
| SiO2@CuMgAl_0.1-LDH | 0.304 | 2.297 | 0.766 | 7.7 | 13.2 |
| SiO2@CuMgAl_0.2-LDH | 0.305 | 2.299 | 0.766 | 7.0 | 13.1 |
| SiO2@CuMgAl_0.3-LDH | 0.305 | 2.293 | 0.764 | 7.5 | 13.0 |
| SiO2@CuMgAl_0.4-LDH | 0.305 | 2.295 | 0.765 | 7.1 | 12.3 |
| SiO2@CuMgAl_0.5-LDH | 0.305 | 2.295 | 0.765 | 6.9 | 12.6 |
| @CuMgAl_0.05-LDH | 0.305 | 2.297 | 0.766 | 7.2 | 13.9 |
| @CuMgAl_0.1-LDH | 0.305 | 2.300 | 0.767 | 6.9 | 14.0 |
| @CuMgAl_0.2-LDH | 0.305 | 2.293 | 0.764 | 6.6 | 14.6 |
| @CuMgAl_0.3-LDH | 0.305 | 2.290 | 0.763 | 7.2 | 14.7 |
| @CuMgAl_0.4-LDH | 0.306 | 2.293 | 0.764 | 7.0 | 14.4 |
| @CuMgAl_0.5-LDH | 0.305 | 2.285 | 0.762 | 7.1 | 13.4 |
Regardless of the composition of the CuMgAl-LDH in the SiO2@CuMgAl-LDHs core–shell particles, no considerable changes in their shape and size were observed using microscopic techniques. Therefore, SEM and TEM images were obtained for the material with the highest Cu content, i.e., SiO2@CuMgAl_0.5-LDH (Fig. 2A and B, respectively). The prepared core–shell structures are evenly covered by the LDH phase with the CuMgAl-LDH platelets oriented mainly perpendicularly on the SiO2 template surface. The smooth surface of the SiO2 is not visible in any case. Moreover, the material shows uniform size distribution with an average diameter of the particles of ca. 800 nm and a thickness of the CuMgAl-LDH shell of ≈150 nm (estimated from more than 70 particles). After partial leaching of the SiO2 core from the SiO2@CuMgAl-LDHs for 4 h, a significant change of morphology is observed in the resulting @CuMgAl-LDH materials due to a decrease of the Si/(Si + Cu + Mg + Al) molar ratio (Fig. 2C and D). The formed hollow @CuMgAl-LDH spheres do not collapse, retaining their original spherical shape derived from the core–shell system, although a certain part of spheres is cracked with an obviously visible void inside the grains. The possible causes of these cracks were considered previously.38 The most likely explanation comes from the fact that the shells could have been formed on two or more silica spheres that were close to each other (in aggregated forms), thus blocking LDH growth in the space between the balls (contact surface). In such cases, a more complex system exists resembling, e.g., a raspberry-like shape, and after SiO2 leaching and/or pulverizing in a mortar, may burst, resulting in visibly broken, empty shells. Additionally, a change in the contrast between the core and the shell is visible (Fig. 2B and D) as a result of the partial removal of the SiO2 from the materials (Si/(Si + Cu + Mg + Al) molar ratio decreased from 0.48 to 0.28). Moreover, the above-mentioned diversity (between the core and shell of the core–shell and hollowed particles) is slightly less visible than was the case in our last study,38 where TEM images of an analogous system (@MgAl-LDH with a Si/(Si + Mg + Al) molar ratio of 0.11) were studied. Additionally, a similar effect was also reported by Chen et al.,16 who investigated the influence of the SiO2@MgAl-LDH synthesis conditions (temperature, pH) on the formation of core–shell, yolk–shell and hollow particles. However, the authors did not provide the chemical composition of the different types of spherical particles formed but suggested that the interface between the SiO2 and LDH phases is more stable than the silica in the core.
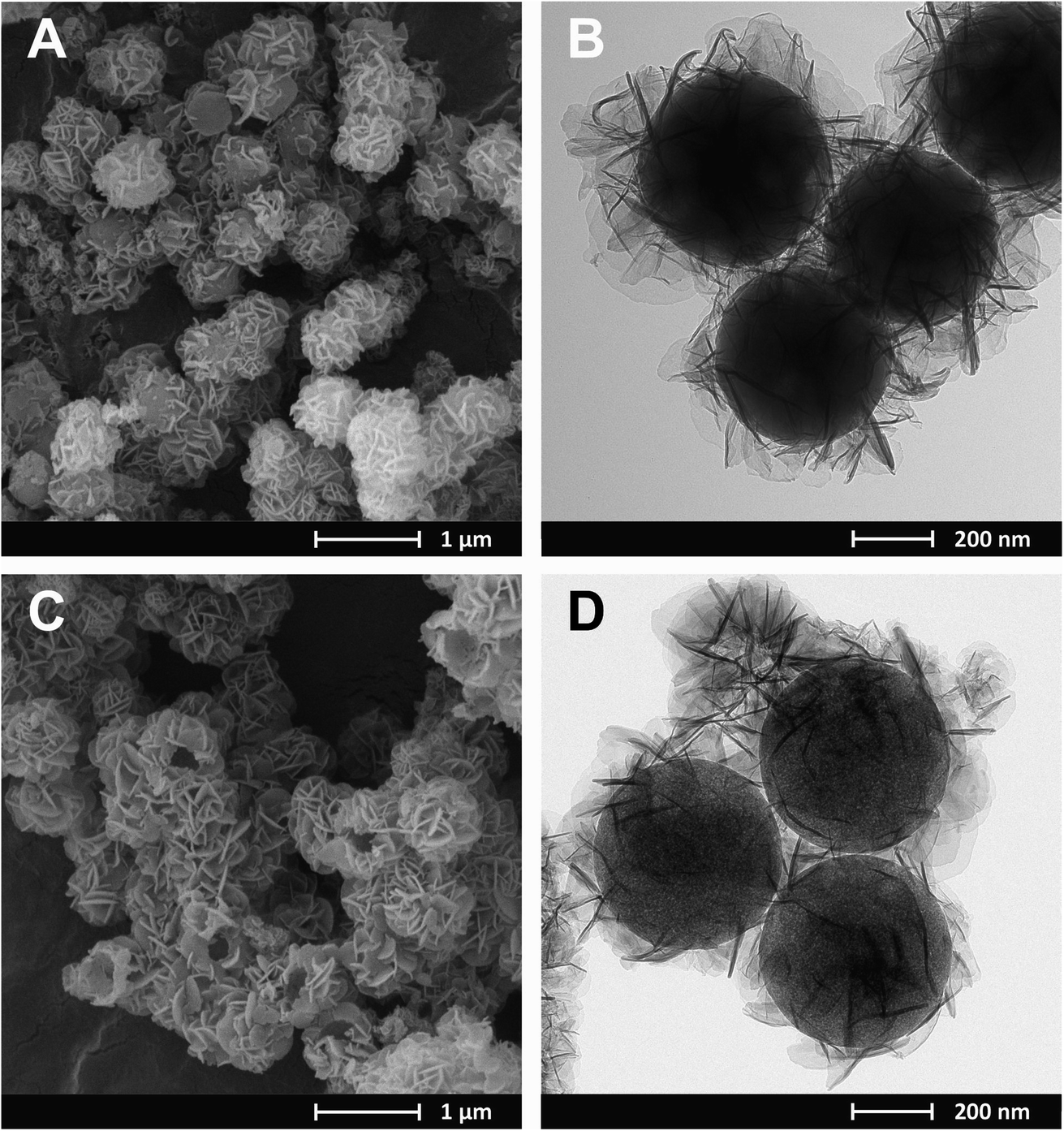 | ||
| Fig. 2 SEM and TEM images of SiO2@CuMgAl-LDH (A, B) and @CuMgAl-LDH (C, D). | ||
The isotherms for the SiO2@CuMgAl-LDHs and @CuMgAl-LDHs are shown in Fig. 3A and B, respectively, and the determined textural parameters are presented in Table 3. All SiO2@CuMgAl-LDH core–shell particles show IVa type N2 adsorption isotherms, according to the IUPAC classification, with H3 hysteresis loops characteristic for mesoporous materials with slit-shape pores.64 Regardless of the amount of copper introduced, the volume of the mesopores was similar (0.18–0.19 cm3 g−1), and the total pore volume ranged from 0.32 to 0.39 cm3 g−1. The specific surface area of the core–shell systems slightly increases with an increase in the Cu/Mg molar ratio (from 76 to 84 m2 g−1). This correlates with the lowest specific surface area of SiO2@MgAl-LDH (without Cu, 65 m2 g−1).38 Considering that (i) the silica core is a non-porous solid (SBET ≈ 10 m2 g−1)38 and (ii) that the influence it has on the textural properties of all the SiO2@CuMgAl-LDH core–shell materials is similar in all cases, its impact on the above trend can be ignored. Thus, the growth of the specific surface area of the core–shell systems as the Cu/Mg molar ratio increases can only be attributed to the LDH phase. The partial elimination of silica, reached after 4 hours of etching, results in an opening of the pore structure. This effect was observed in the measured isotherms as the significant increase in the volume of N2 adsorbed (especially noticeable at relatively low p/p0) and should be related to the appearance of new pores in the @CuMgAl-LDH materials. The values of Vtotal = 0.66–0.71 cm3 g−1, Vmeso = 0.39–0.47 cm3 g−1 and Vmacro = 0.21–0.28 cm3 g−1 demonstrated in Table 3 confirm this hypothesis. For the sake of completeness, it should be mentioned that the micropore volume for @CuMgAl-LDHs also increased by 2 to 3 times compared to the pristine core–shell materials, but the determined volume (0.01–0.02 cm3 g−1) has a negligible effect on the total porosity of the studied materials. The formation of new pores affected the specific surface area, which increased to 210–270 m2 g−1. However, an interesting correlation between SBET and the chemical composition of the @CuMgAl-LDH systems was found (Fig. 4). Materials with the nominal Cu/Mg molar ratios of 0.05, 0.30 and 0.40 (group A) exhibit lower SBET values, whereas the @CuMgAl-LDHs with 0.10, 0.20 and 0.50 (group B) Cu/Mg molar ratios have higher SBET values. This can be explained by the effectiveness of the removal of the silica core from the SiO2@CuMgAl-LDHs. It is clearly visible that in group A, the process of leaching the silica core was more effective than in group B (despite applying the same conditions). The lower level of silica core leaching in group B probably leads to the creation of new textured surfaces (more porous) inside the core–shell particles, which leads to an increase in SBET. In group A, it can be taken that greater removal of the SiO2 core is achieved, which leads to a less porous SiO2 species.
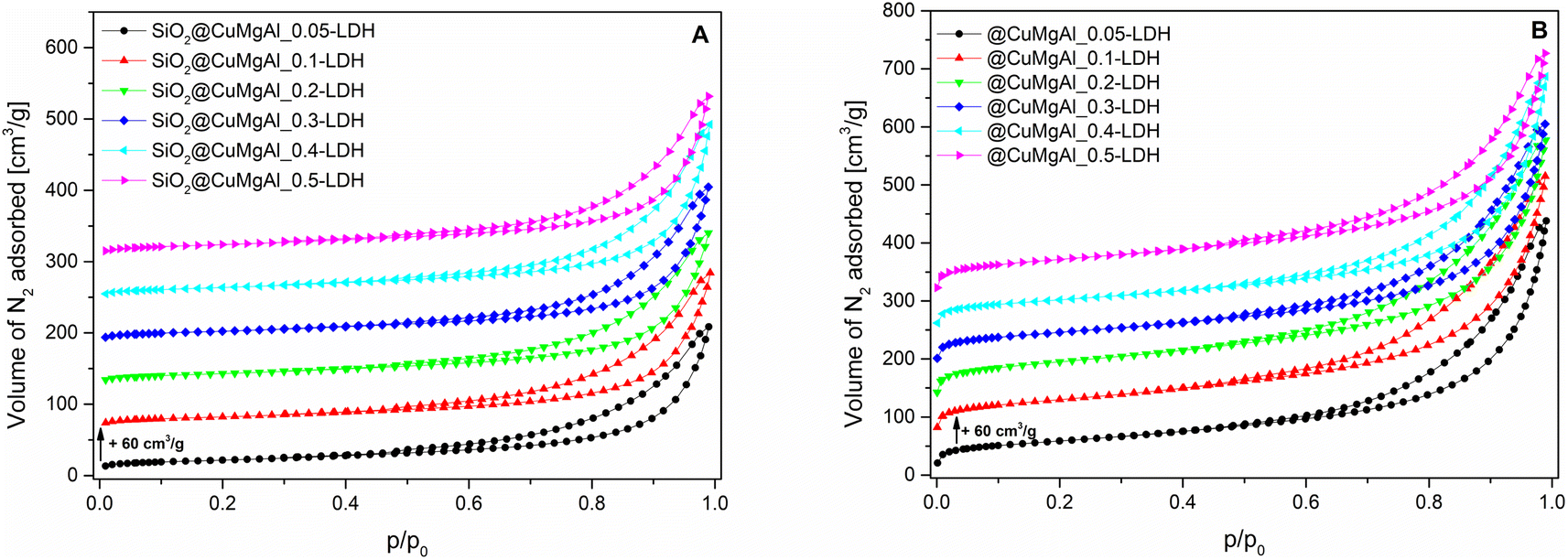 | ||
| Fig. 3 N2 adsorption isotherms for the SiO2@CuMgAl-LDH composites (A) before and (B) after SiO2 core etching. | ||
| Sample | S BET [m2 g−1] | V micro [cm3 g−1] | V meso [cm3 g−1] | V macro [cm3 g−1] | V total [cm3 g−1] |
|---|---|---|---|---|---|
| SiO2@CuMgAl_0.05-LDH | 76 | 0.007 | 0.180 | 0.135 | 0.322 |
| SiO2@CuMgAl_0.1-LDH | 78 | 0.006 | 0.188 | 0.152 | 0.347 |
| SiO2@CuMgAl_0.2-LDH | 80 | 0.007 | 0.188 | 0.143 | 0.340 |
| SiO2@CuMgAl_0.3-LDH | 79 | 0.007 | 0.181 | 0.159 | 0.347 |
| SiO2@CuMgAl_0.4-LDH | 83 | 0.007 | 0.190 | 0.193 | 0.390 |
| SiO2@CuMgAl_0.5-LDH | 84 | 0.007 | 0.189 | 0.162 | 0.358 |
| @CuMgAl_0.05-LDH | 210 | 0.013 | 0.389 | 0.276 | 0.677 |
| @CuMgAl_0.1-LDH | 251 | 0.015 | 0.474 | 0.214 | 0.703 |
| @CuMgAl_0.2-LDH | 270 | 0.017 | 0.443 | 0.247 | 0.707 |
| @CuMgAl_0.3-LDH | 236 | 0.019 | 0.387 | 0.251 | 0.657 |
| @CuMgAl_0.4-LDH | 222 | 0.018 | 0.392 | 0.280 | 0.691 |
| @CuMgAl_0.5-LDH | 258 | 0.021 | 0.384 | 0.255 | 0.660 |
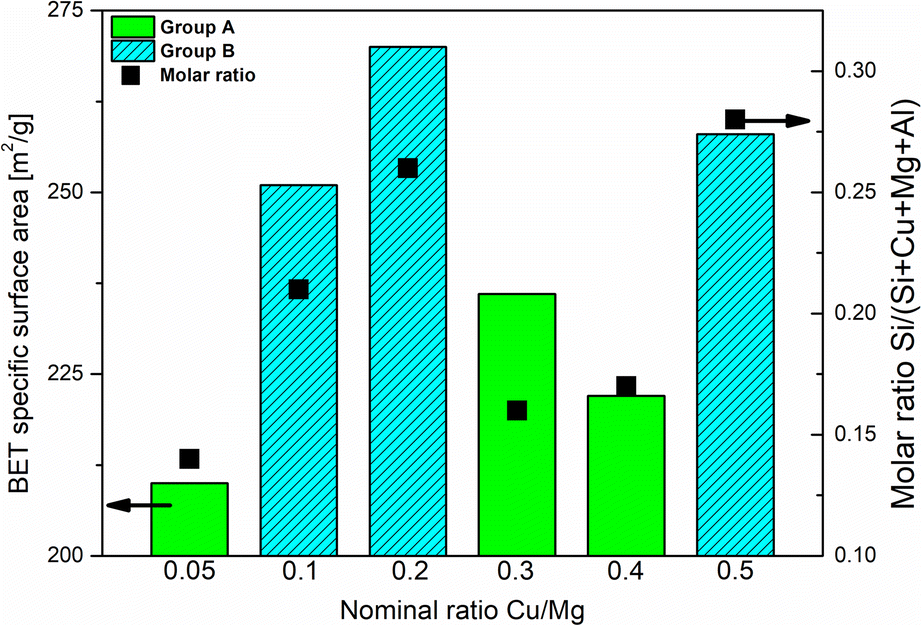 | ||
| Fig. 4 Correlation between the degree of SiO2 core etching and the textural properties of the @CuMgAl-LDHs. | ||
Spherical @CuMgAl-MO catalysts
After calcination of the LDHs at 450 °C, all characteristic peaks of the double layered structure disappeared and XRD patterns (Fig. 5) show the presence of a poorly crystalline MgO periclase phase with three characteristic diffraction lines at 2θ of approximately 35.1, 43.2 and 62.5° (PDF card no. 00-004-0829). For all the MOs, a slight shift of the reflections with respect to pure MgO is evident. However, irrespective of the Cu content, no highly crystalline Cu-oxide phases (e.g., Cu2O, CuO) were noted. Additionally, neither was a well-defined CuAl2O4 spinel phase detected since the calcination temperature is too low.65–67 Therefore, these observations indicate that the Cu and Al species should be well dispersed in the MgO matrix, forming a solid solution, or that well-dispersed Cu-contained phases under XRD detection limit are formed.54,62,68 With an increase in the Cu/Mg molar ratio, the diffraction lines become less pronounced, indicating a decrease in the crystallinity of the MOs.69 However, a large dependence between the intensity of the above-mentioned reflections and the calculated Si/(Si + Cu + Mg + Al) molar ratio was not found.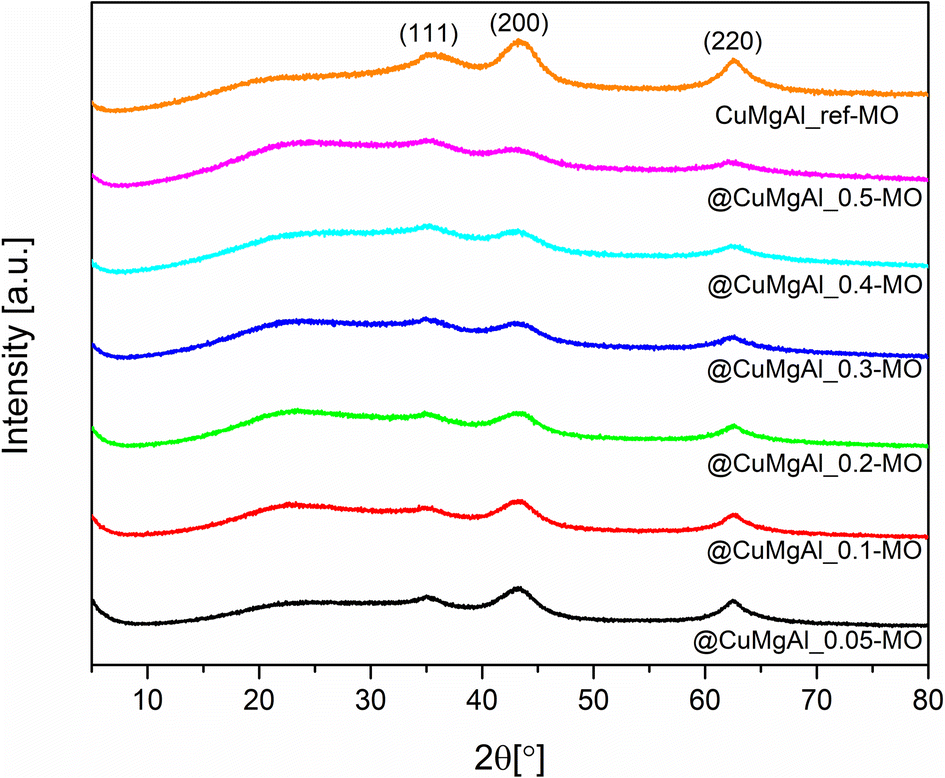 | ||
| Fig. 5 XRD patterns of the @CuMgAl-MO systems and CuMgAl-MO reference catalyst. | ||
The shape of the @CuMgAl-MOs N2 adsorption isotherms did not change notably in comparison to the pristine LDH-based materials (Fig. 6), implying that the mesoporous character of the hollow spheres with slit-like pores was preserved after heat treatment. In turn, all designated textural parameters (we did not consider Vmicro, which has a negligible impact on the total porosity of studied materials, Table 4) increased after transformation of the LDHs to the MOs, which is in line with the literature.70–72 We noted a growth in the specific surface area by 14–24% (SBET = 261–308 m2 g−1), while maintaining the trend related to the presence of the silica core in the @CuMgAl materials (lower degree of SiO2 removal means higher SBET). The reported SBET values are approximately two times higher than for similar CuMgAl systems with comparable compositions made at similar annealing temperatures but without spherical ordering.54,70–74 Therefore, it can be concluded that the formation of mixed oxides from core–shell systems is beneficial to their resulting properties. In turn, the volume of meso- and macropores, as well as total porosity, increased after calcination by an average of 19, 31 and 23%, respectively. Considering that Vtotal was in the range of 0.81–0.87 cm3 g−1, it can be unequivocally stated that the mesopores (Vmeso = 0.45–0.54 cm3 g−1) play a dominant role in the pore systems of the @CuMgAl-MOs, which are supplemented by macroporous voids (Vmacro = 0.29–0.37 cm3 g−1).
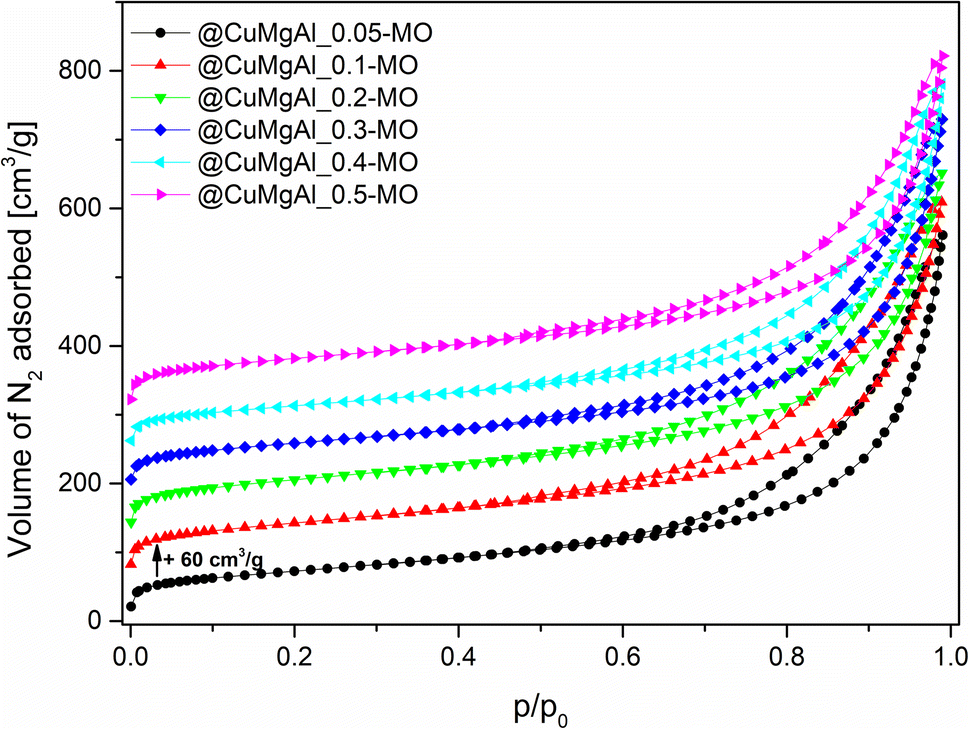 | ||
| Fig. 6 N2 adsorption isotherms of the @CuMgAl-MO catalysts. | ||
| Sample | S BET [m2 g−1] | V micro [cm3 g−1] | V meso [cm3 g−1] | V macro [cm3 g−1] | V total [cm3 g−1] |
|---|---|---|---|---|---|
| @CuMgAl_0.05-MO | 261 | 0.013 | 0.485 | 0.370 | 0.867 |
| @CuMgAl_0.1-MO | 294 | 0.015 | 0.536 | 0.297 | 0.849 |
| @CuMgAl_0.2-MO | 308 | 0.018 | 0.511 | 0.293 | 0.822 |
| @CuMgAl_0.3-MO | 283 | 0.019 | 0.473 | 0.357 | 0.850 |
| @CuMgAl_0.4-MO | 262 | 0.016 | 0.479 | 0.341 | 0.836 |
| @CuMgAl_0.5-MO | 296 | 0.021 | 0.449 | 0.335 | 0.806 |
The reducibility of the @CuMgAl-MO samples was studied using H2-TPR. The collected TPR profiles show two fundamental peaks of hydrogen consumption (Fig. 7). The first maximum is in the range of 230–350 °C (low-thermal peak), whereas the second one is at approximately 700 °C (high-thermal peak). It can be easily seen that the intensity and position of the observed low-thermal peak depends on the content of Cu in the CuMgAl mixed oxides. An increase in the Cu loading (Cu/Mg molar ratio) resulted in a higher amount of H2 consumed as well as a shift of the reduction process towards lower temperatures. However, different peak shapes are observed. Above a threshold of Cu/Mg = 0.2, a clear splitting or shouldering of signals is visible. Typically, CuO reduction occurs stepwise, the reduction of Cu2+ to Cu+ is closely followed by the reduction of Cu+ to Cu0.75 Therefore, in our case the low-thermal peak may be attributed to the complete reduction of the Cu2+ species incorporated in the MgAl phase to metallic Cu.76,77 Additionally, the segregated CuO phase present on the surface of the MgAl oxide is rather excluded, since reduction of CuO takes place at lower temperatures.71,78 The reduction temperature is connected to the strength of interaction between the Cu species and the MgAl matrix. Thus, it seems that Cu2+ ions present in the @CuMgAl_MO systems are strongly stabilised against reduction in a Mg-reach MgAl oxide matrix. In our study, reduction begins at temperatures as high as 260 °C (for @CuMgAl_0.05-MO) and gradually drops to 140 °C (for @CuMgAl_0.5-MO). A similar stabilisation of the Cu species deposited on pure MgO and on MgAl mixed oxides was recently reported by Basąg et al.77 and Luggren et al.79 Other possible explanations could be the formation of more aggregated Cu-species (still under XRD detection level) dispersed in the samples that have higher Cu content. Possibly, these more aggregated species are more easily reduced than smaller ones. In turn, the high-thermal H2-TPR peak observed between 600 and 900 °C, with a maximum at approximately 700 °C, is associated with the reduction of CuAl2O4 or the more complex Cu1−xMgxAl2O4 spinel phase.54,71 However, it should be stressed that the studied materials were prepared by thermal treatment at 450 °C. According to Kovanda et al.,65 a well-defined CuAl2O4 spinel phase formed from CuMgAl LDHs only appears after calcination above 800 °C. However, it might be that the above-mentioned phase can be formed in situ under the conditions of the TPR experiment. On the other hand, the high-temperature peak may also reflect the reaction of H2 with highly stable carbonates and/or arise from residual template species that do not decompose during thermal treatment at 450 °C. Based on the TPR curves, the total amount of redox sites was calculated from all peaks in the range of 25–900 °C (Table 5). The total number of redox sites increases with the Cu/Mg molar ratio and ranges from 0.28 to 2.66 [mmol gcatalyst−1]. However, the two samples with the highest nominal Cu/Mg molar ratio (0.4 and 0.5) possess almost identical H2 consumption values. This can be explained by the comparable Cu content in both @CuMgAl_0.4-MO and @CuMgAl_0.5-MO (17.1 and 16.9 wt% of Cu, respectively), due to the fact that the latter possess more silica in the spherical system (Si/(Si + Cu + Mg + Al) is 0.22 and 0.32, respectively). The calculated values of H2 consumed/H2 theoretical were below 1 at all Cu/Mg molar ratios. However, an increase in this value is observed in line with the increase in Cu content (from 0.42 to 0.95). Similar findings were also reported previously for bulky CuMgAl mixed oxides with various Cu loadings54 and indicate that the copper ions are only partially reduced under the applied experimental conditions. Additionally, the formation of small amounts of Cu2O during the calcination step cannot be excluded.80
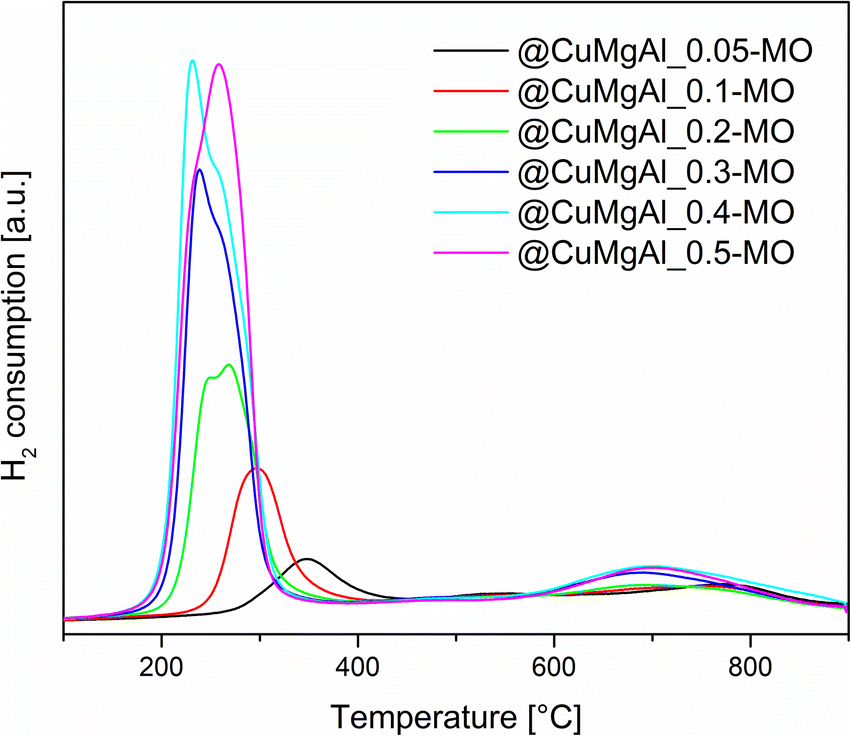 | ||
| Fig. 7 H2-TPR profiles of the @CuMgAl-MO systems pre-treated at 450 °C. | ||
| Sample | H2 consumption [mmol gcatalyst−1] | H2 consumed/H2 theoretical |
|---|---|---|
| @CuMgAl_0.05-MO | 0.28 | 0.42 |
| @CuMgAl_0.1-MO | 0.60 | 0.58 |
| @CuMgAl_0.2-MO | 1.13 | 0.70 |
| @CuMgAl_0.3-MO | 1.90 | 0.83 |
| @CuMgAl_0.4-MO | 2.59 | 0.96 |
| @CuMgAl_0.5-MO | 2.66 | 0.95 |
Catalytic performance of @CuMgAl-MO
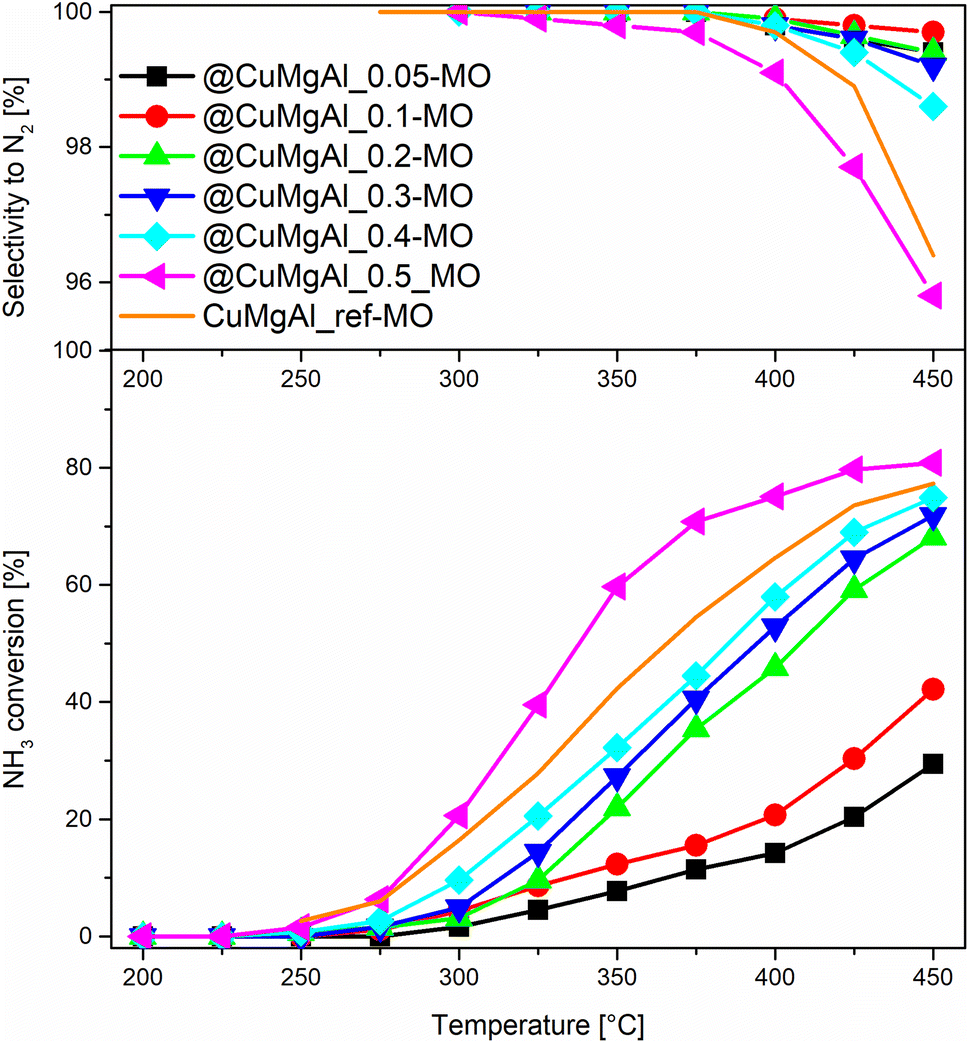
The catalytic performance of the @CuMgAl-MOs as well as a CuMgAl-MO reference material was tested in the NH3-SCR reaction. The relationship between the NO conversion and N2 selectivity, N2 being the desired product, vs. temperature is presented in Fig. 8. The influence of the Cu content of the @CuMgAl-MOs on the NO conversion is very visible. The reduction of NO by ammonia in the presence of @CuMgAl-MOs started at around 150 °C and it gradually increased with increasing reaction temperature. Increasing the Cu loading in the @CuMgAl-MOs causes a significant shift of the NO conversion profiles toward lower temperatures. The maximum NO conversion level of 89% was obtained for the @CuMgAl_0.5-MO catalyst at a temperature of 275–300 °C. The increased NO conversion of the @CuMgAl-MOs catalysts at higher Cu/Mg molar ratios can be associated with the results of the H2-TPR measurements, where the copper species were more easily reduced at higher Cu loadings (Fig. 7). However, in the high temperature region, a decrease in the NO conversion, assigned to the side process of direct NH3 oxidation by O2, was observed. Additionally, this effect is more pronounced for the MOs that demonstrate higher NO conversions. It should be noted that the N2 selectivity was studied over the entire temperature range and, regardless of the composition of the @CuMgAl-MO hollow spheres, was found to be very high (over 99.7%), which indicates a high selectivity for NO to N2 conversion.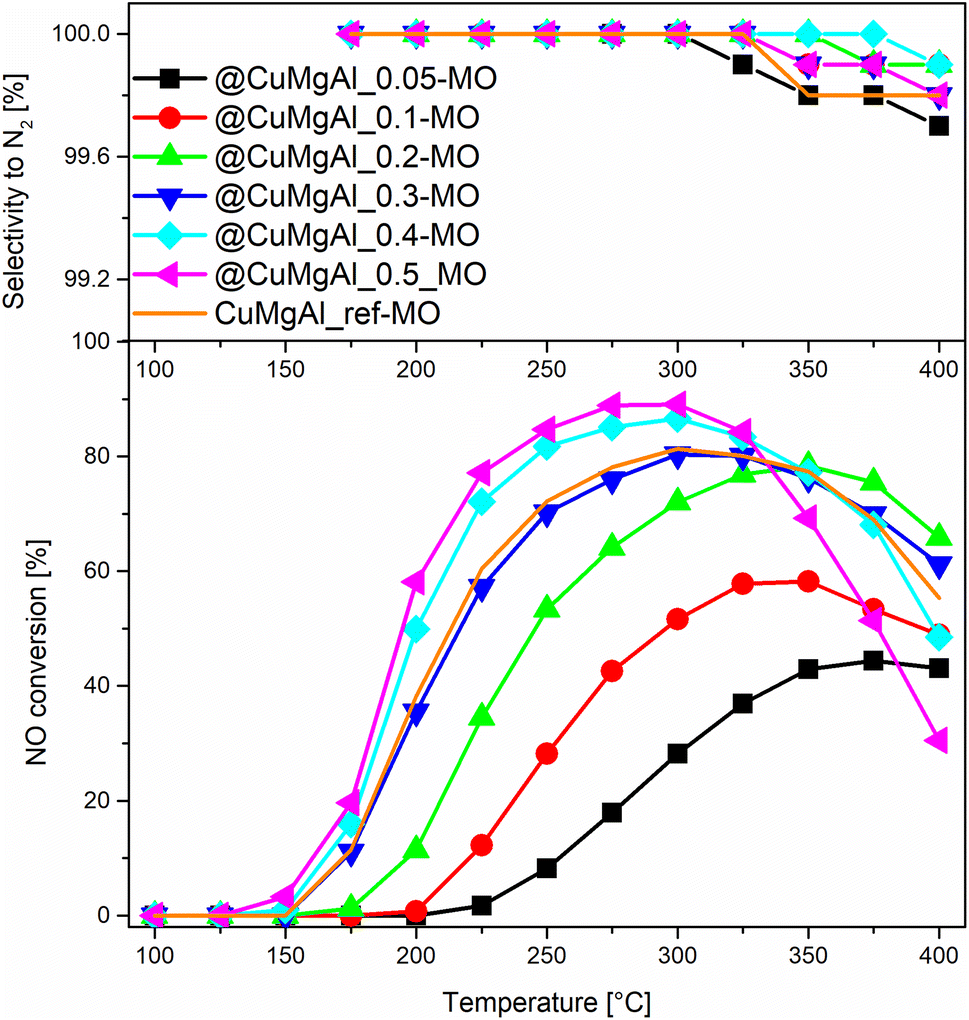 | ||
| Fig. 8 Results from the NH3-SCR catalytic tests for the @CuMgAl-MOs and CuMgAl-MO reference catalyst. | ||
To compare the catalytic behaviour of the @CuMgAl-MO hollow sphere catalysts with bulk CuMgAl-MO catalysts, a classical bulk CuMgAl-MO catalyst was prepared by coprecipitation,81 and both types of catalysts were studied under the same reaction conditions. Although the CuMgAl_ref-MO material possesses a little more Cu content than the @CuMgAl_0.4-MO and @CuMgAl_0.5-MO core–shell catalysts (7.0 vs. 5.8 and 5.7 at%, respectively), the former shows a slightly weaker catalytic performance. The NO conversion profile for CuMgAl_ref-MO is most similar to that found for @CuMgAl_0.3-MO. Thus, it can be concluded that the @CuMgAl-MO hollow sphere catalyst demonstrated higher NO conversion than the bulk CuMgAl-MO reference material. The lower NO conversion of bulk CuMgAl-MO may be due to the fact that the Cu-species present in this sample are harder to reduce, as concluded from the H2-TPR measurements. Moreover, the impact of its poor stone-like morphology, relatively low surface area (SBET = 86 m2 g−1), poor surface acidity (37.5 μmol g−1, ca. five times lower than for the hollow systems) and dissimilar textural properties, which may determine the accessibility of active sites for the reactants, cannot be excluded either.
The comparison of NO conversion rates achieved using the @CuMgAl-MO hollow spheres (a maximum NO conversion level of 89% was obtained using the @CuMgAl_0.5-MO catalyst at a temperature of 275–300 °C) with the values reported in literature is dependent on the different reaction conditions. For example, Wu et al.82 reported about 80% conversion of NO and 75% selectivity to N2 at a temperature of 250 °C using a CuMgAl catalyst with a molar ratio of Cu:Mg:Al = 2:1:1. Zhang et al.83 studied a similar system with a molar ratio of Cu:Mg:Al = 1:1:1 and reported a maximum NO conversion level of 58% at a temperature of 240 °C. In turn, Basąg et al.53 achieved 92% conversion of NO and 96% selectivity to N2 at a temperature of 325 °C, despite a much lower copper content (Cu:Mg:Al molar ratio equal to ca. 0.2:1.9:1.0). Chmielarz et al.15 studied LDH-derived catalysts with Cu:Mg:Al molar ratios of 0.2:2.3:1.0, 0.35:2.0:1.0 and 0.7:1.8:1.0, which presented NO conversions at 250 °C of 85, 94 and 95%, respectively. The selectivity to nitrogen was above 90% for all these catalysts. However, a drop in the NO conversion at above 250 °C, especially for the catalysts with the highest copper loading, was observed. In the case of binary CuAl catalysts, ca. a 70–95% conversion of NO is achieved over temperature ranges of 200–250 °C, depending on the composition and method of catalyst preparation.84–87
The catalytic behaviour of the studied catalysts in the side process of direct ammonia oxidation by oxygen was verified by conducting additional catalytic tests. As can be seen in Fig. 9, the catalytic activity of the studied samples increased with an increase in the copper loading. However, what is very important, nearly all ammonia is oxidised to dinitrogen. Thus, residual ammonia, not converted in the NH3-SCR reaction, could be selectively oxidised to N2, which is important in the case of the possible ammonia slip.
 | ||
| Fig. 9 Results of NH3 oxidation in the presence of the @CuMgAl-MOs and CuMgAl-MO reference catalysts. | ||
The stability of the most active NH3-SCR catalyst (@CuMgAl_0.5-MO) was also examined at 275 °C (Fig. 10). The collected results clearly confirm that no noticeable changes in catalytic efficiency are observed during the 24 hour catalytic run. Over the entire duration of the test, the NO conversion was stable, reaching 87.2–88.9% while the N2 selectivity was 100%.
 | ||
| Fig. 10 Results of long-term isothermal NH3-SCR catalytic test for the @CuMgAl_0.5-MO catalyst at 275 °C. | ||
Conclusions
The studied materials were found to be active and selective catalysts for a low-temperature selective catalytic reduction of NOx using ammonia (NH3-SCR). The activity of the catalysts increases with increasing copper content. Moreover, the temperature window for effective NO conversion is shifted to lower temperatures for the catalysts with the highest copper loadings. Thus, the copper content is very important for the formulation of effective catalysts for the low-temperature NH3-SCR process. It was shown that the temperature for copper reduction decreases with increasing copper content in the materials. The reducibility of the catalysts is, in general, correlated with their activity in the NH3-SCR process. Thus, the reducibility of the copper species seems to be very important for the activity of the studied catalysts in the low-temperature range. Comparison of the catalytic performance of the hollow @CuMgAl_0.5_MO sample and a reference catalyst originating from a hydrotalcite mixed metal oxide with a very similar copper loading showed significantly better activity for the hollow sample. These catalysts differ very significantly with respect to their specific surface areas (296 m2 g−1 for the hollow @CuMgAl_0.5_MO sample vs. 86 m2 g−1 for refence classical hydrotalcite sample). Thus, the surface accessibility of copper is possibly another very important parameter that should be considered when designing effective catalysts for the low-temperature NH3-SCR process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully thank the Czech Science Foundation of the Czech Republic (Project No. 19-22978S). T. Kondratowicz also thanks European Regional Development Fund-Project “International mobility of employees of the University of Pardubice II” (No. CZ.02.2.69/0.0/0.0/18_053/0016969)”. S. Slang thanks used infrastructure (project LM2023037).References
- A. Mytareva, D. Bokarev and A. Y. Stakheev, Kinet. Catal., 2021, 62, 1–32 CrossRef CAS.
- European Parliament, Council of the European Union, Directive (EU) 2016/2284 of the European Parilament and of the Council of 14 December 2016 on the reduction of national emissions of certain atmospheric pollutants, 2016, https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=uriserv%3AOJ.L_.2016.344.01.0001.01.ENG Search PubMed.
- M. Jablonska and R. Palkovits, Catal. Sci. Technol., 2016, 6, 49–72 RSC.
- L. Han, M. Gao, C. Feng, L. Shi and D. Zhang, Environ. Sci. Technol., 2019, 53, 5946–5956 CrossRef CAS.
- P. Wang, L. Yan, Y. Gu, S. Kuboon, H. Li, T. Yan, L. Shi and D. Zhang, Environ. Sci. Technol., 2020, 54, 6396–6405 CrossRef CAS PubMed.
- L. Kang, L. Han, J. He, H. Li, T. Yan, G. Chen, J. Zhang, L. Shi and D. Zhang, Environ. Sci. Technol., 2018, 53, 938–945 CrossRef PubMed.
- J.-W. Shi, Y. Wang, R. Duan, C. Gao, B. Wang, C. He and C. Niu, Catal. Sci. Technol., 2019, 9, 718–730 RSC.
- S. Ali, L. Chen, F. Yuan, R. Li, T. Zhang, X. Leng, X. Niu and Y. Zhu, Appl. Catal., B, 2017, 210, 223–234 CrossRef CAS.
- F. Liu, W. Shan, Z. Lian, J. Liu and H. He, Appl. Catal., B, 2018, 230, 165–176 CrossRef CAS.
- H. Chang, T. Zhang, H. Dang, X. Chen, Y. You, J. W. Schwank and J. Li, Catal. Sci. Technol., 2018, 8, 3313–3320 RSC.
- Y. Zhu, Y. Zhang, R. Xiao, T. Huang and K. Shen, Catal. Commun., 2017, 88, 64–67 CrossRef CAS.
- K. Song, C. Gao, P. Lu, D. Ma, Y. Cheng and J.-W. Shi, Fuel, 2023, 331, 125861 CrossRef CAS.
- Q. Yan, S. Chen, C. Zhang, Q. Wang and B. Louis, Appl. Catal., B, 2018, 238, 236–247 CrossRef CAS.
- Y. Nie, Q. Yan, S. Chen, D. O'Hare and W. Wang, Catal. Commun., 2017, 97, 47–50 CrossRef CAS.
- L. Chmielarz, P. Kuśtrowski, A. Rafalska-Łasocha, D. Majda and R. Dziembaj, Appl. Catal., B, 2002, 35, 195–210 CrossRef CAS.
- C. Chen, R. Felton, J.-C. Buffet and D. O'Hare, Chem. Commun., 2015, 51, 3462–3465 RSC.
- Q. Wang and D. O'Hare, Chem. Commun., 2013, 49, 6301–6303 RSC.
- C. Chen, M. Greenwood, J.-C. Buffet and D. O'Hare, Green Chem., 2020, 22, 3117–3121 RSC.
- X. Wang, F. Wu, J. Fan, A. Tian, Y. Cheng and S. Yang, J. Alloys Compd., 2021, 888, 161502 CrossRef CAS.
- L. Chen, X. Yang, Y. Tian, Y. Wang, X. Zhao, X. Lei and F. Zhang, Front. Energy Res., 2022, 9, 810568 CrossRef.
- M. N. Pahalagedara, L. R. Pahalagedara, D. Kriz, S.-Y. Chen, F. Beaulieu, W. Thalgaspitiya and S. L. Suib, Appl. Catal., B, 2016, 188, 227–234 CrossRef CAS.
- N. I. Blaisi, M. Zubair, S. Ali, T. S. Kazeem, M. S. Manzar, W. Al-Kutti and M. A. Al Harthi, Environ. Sci. Pollut. Res., 2018, 25, 34319–34331 CrossRef CAS PubMed.
- L. Deng, H. Zeng, Z. Shi, W. Zhang and J. Luo, J. Colloid Interface Sci., 2018, 521, 172–182 CrossRef CAS PubMed.
- J. Xie, T. Yamaguchi and J.-M. Oh, J. Solid State Chem., 2021, 293, 121758 CrossRef CAS.
- M. N. Pahalagedara, L. R. Pahalagedara, C.-H. Kuo, S. Dharmarathna and S. L. Suib, Langmuir, 2014, 30, 8228–8237 CrossRef CAS PubMed.
- G. Varga, Z. Somosi, Z. Kónya, Á. Kukovecz, I. Pálinkó and I. Szilagyi, J. Colloid Interface Sci., 2021, 581, 928–938 CrossRef CAS PubMed.
- H. Zhang, D. Pan, K. Zou, J. He and X. Duan, J. Mater. Chem., 2009, 19, 3069–3077 RSC.
- C. Chen, L. K. Yee, H. Gong, Y. Zhang and R. Xu, Nanoscale, 2013, 5, 4314–4320 RSC.
- M. Shao, F. Ning, J. Zhao, M. Wei, D. G. Evans and X. Duan, J. Am. Chem. Soc., 2012, 134, 1071–1077 CrossRef CAS PubMed.
- S. Zhao, W.-C. Tsen, F. Hu, F. Zhong, H. Liu, S. Wen, G. Zheng, C. Qin and C. Gong, J. Mater. Sci., 2020, 55, 2967–2983 CrossRef CAS.
- H. Suo, H. Duan, C. Chen, J.-C. Buffet and D. O'Hare, RSC Adv., 2019, 9, 3749–3754 RSC.
- F. Mi, X. Chen, Y. Ma, S. Yin, F. Yuan and H. Zhang, Chem. Commun., 2011, 47, 12804–12806 RSC.
- C. Wang, B. Ma, S. Xu, D. Li, S. He, Y. Zhao, J. Han, M. Wei, D. G. Evans and X. Duan, Nano Energy, 2017, 32, 463–469 CrossRef CAS.
- Y. Dou, S. Zhang, T. Pan, S. Xu, A. Zhou, M. Pu, H. Yan, J. Han, M. Wei and D. G. Evans, Adv. Funct. Mater., 2015, 25, 2243–2249 CrossRef CAS.
- R. Li, T. Xue, R. Bingre, Y. Gao, B. Louis and Q. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 34834–34839 CrossRef CAS PubMed.
- M. Lyu, C. Chen, J.-C. Buffet and D. O'Hare, New J. Chem., 2020, 44, 10095–10101 RSC.
- Y. Ni, L. Yao, Y. Wang, B. Liu, M. Cao and C. Hu, Nanoscale, 2017, 9, 11596–11604 RSC.
- T. Kondratowicz, S. Slang, L. Dubnová, O. Kikhtyanin, P. Bělina and L. Čapek, Appl. Clay Sci., 2022, 216, 106365 CrossRef CAS.
- M. Shirotori, S. Nishimura and K. Ebitani, J. Mater. Chem. A, 2017, 5, 6947–6957 RSC.
- M. Shirotori, S. Nishimura and K. Ebitani, RSC Adv., 2018, 8, 28024–28031 RSC.
- C. Chen, P. Wang, T.-T. Lim, L. Liu, S. Liu and R. Xu, J. Mater. Chem. A, 2013, 1, 3877–3880 RSC.
- W. L. Kwok, D.-G. Crivoi, C. Chen, J.-C. Buffet and D. O'Hare, Dalton Trans., 2018, 47, 143–149 RSC.
- D. Cosano, D. Esquivel, A. J. Puertas, F. J. Romero-Salguero, C. Jiménez-Sanchidrián and J. R. Ruiz, Microporous Mesoporous Mater., 2021, 323, 111247 CrossRef CAS.
- K. Wang, X. Huang, Y. Liu, W. Fei and Z. Gu, J. Nanopart. Res., 2020, 22, 1–14 CrossRef.
- W. Stober, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 1973 CrossRef.
- X. Chen, F. Mi, H. Zhang and H. Zhang, Mater. Lett., 2012, 69, 48–51 CrossRef CAS.
- C. Zhang, M. Shao, L. Zhou, Z. Li, K. Xiao and M. Wei, ACS Appl. Mater. Interfaces, 2016, 8, 33697–33703 CrossRef CAS PubMed.
- M. Shao, F. Ning, Y. Zhao, J. Zhao, M. Wei, D. G. Evans and X. Duan, Chem. Mater., 2012, 24, 1192–1197 CrossRef CAS.
- K. Wang, Q. Mao, W. Fei, L. Kong, X. Cao and Z. Gu, RSC Adv., 2021, 11, 8375–8383 RSC.
- H. Suo, C. Chen, J.-C. Buffet and D. O'Hare, Dalton Trans., 2018, 47, 16413–16417 RSC.
- M. Liu, H. Wang, P. Zhu, J. Wang, H. Tan and Z. Zheng, Catal. Commun., 2019, 129, 105752 CrossRef CAS.
- Y. Shao, K. Sun, Q. Li, Q. Liu, S. Zhang, Q. Liu, G. Hu and X. Hu, Green Chem., 2019, 21, 4499–4511 RSC.
- S. Basąg, K. Kocoł, Z. Piwowarska, M. Rutkowska, R. Baran and L. Chmielarz, React. Kinet. Mech. Catal., 2017, 121, 225–240 CrossRef.
- S. Tanasoi, N. Tanchoux, A. Urdă, D. Tichit, I. Săndulescu, F. Fajula and I.-C. Marcu, Appl. Catal., A, 2009, 363, 135–142 CrossRef CAS.
- C. Johnson and F. Glasser, Clay Miner., 2003, 51, 1–8 CrossRef CAS.
- N. Abushrenta, X. Wu, J. Wang, J. Liu and X. Sun, Sci. Rep., 2015, 5, 1–9 Search PubMed.
- G. Mitran, T. Cacciaguerra, S. Loridant, D. Tichit and I.-C. Marcu, Appl. Catal., A, 2012, 417, 153–162 CrossRef.
- G. Cui, F. Wang, S. He and M. Wei, RSC Adv., 2016, 6, 105406–105411 RSC.
- T. Kondratowicz, M. Drozdek, A. Rokicińska, P. Natkański, M. Michalik and P. Kuśtrowski, Microporous Mesoporous Mater., 2019, 279, 446–455 CrossRef CAS.
- J. J. Creasey, A. Chieregato, J. C. Manayil, C. M. Parlett, K. Wilson and A. F. Lee, Catal. Sci. Technol., 2014, 4, 861–870 RSC.
- L. Smoláková, K. Frolich, J. Kocík, O. Kikhtyanin and L. Čapek, Ind. Eng. Chem. Res., 2017, 56, 4638–4648 CrossRef.
- S. Kannan, A. Dubey and H. Knozinger, J. Catal., 2005, 231, 381–392 CrossRef CAS.
- Z. Gu, J. J. Atherton and Z. P. Xu, Chem. Commun., 2015, 51, 3024–3036 RSC.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- F. Kovanda, K. Jirátová, J. Rymeš and D. Koloušek, Appl. Clay Sci., 2001, 18, 71–80 CrossRef.
- M. Salavati-Niasari, F. Davar and M. Farhadi, J. Sol-Gel Sci. Technol., 2009, 51, 48–52 CrossRef CAS.
- J. Yanyan, L. Jinggang, S. Xiaotao, N. Guiling, W. Chengyu and G. Xiumei, J. Sol-Gel Sci. Technol., 2007, 42, 41–45 CrossRef.
- C. A. Antonyraj, M. Gandhi and S. Kannan, Ind. Eng. Chem. Res., 2010, 49, 6020–6026 CrossRef CAS.
- N. Blanch-Raga, A. E. Palomares, J. n. Martínez-Triguero, G. Fetter and P. Bosch, Ind. Eng. Chem. Res., 2013, 52, 15772–15779 CrossRef CAS.
- K. A. Ferreira, N. F. Ribeiro, M. M. Souza and M. Schmal, Catal. Lett., 2009, 132, 58–63 CrossRef CAS.
- P. Benito, A. Vaccari, C. Antonetti, D. Licursi, N. Schiarioli, E. Rodriguez-Castellón and A. M. R. Galletti, J. Clean. Prod., 2019, 209, 1614–1623 CrossRef CAS.
- H. Zhang, Q. Jia, F. Yan and Q. Wang, Green Energy Environ., 2022, 7, 105–115 CrossRef CAS.
- J. Shi, Y. He, K. Ma, S. Tang, C. Liu, H. Yue and B. Liang, Catal. Today, 2021, 365, 318–326 CrossRef CAS.
- O. M. Perrone, F. Lobefaro, M. Aresta, F. Nocito, M. Boscolo and A. Dibenedetto, Fuel Process. Technol., 2018, 177, 353–357 CrossRef CAS.
- Z. Liu, M. D. Amiridis and Y. Chen, J. Phys. Chem. B, 2005, 109, 1251–1255 CrossRef CAS PubMed.
- J. Han, H.-Y. Zeng, S. Xu, C.-R. Chen and X.-J. Liu, Appl. Catal., A, 2016, 527, 72–80 CrossRef CAS.
- S. Basąg, Z. Piwowarska, A. Kowalczyk, A. Węgrzyn, R. Baran, B. Gil, M. Michalik and L. Chmielarz, Appl. Clay Sci., 2016, 129, 122–130 CrossRef.
- X.-Y. Xi, Z.-H. Sun, H.-T. Cao, Y.-T. Pei, G. H. ten Brink, P. J. Deuss, K. Barta and H. J. Heeres, Catalysts, 2020, 10, 996 CrossRef CAS.
- P. J. Luggren, C. R. Apesteguia and J. I. Di Cosimo, Appl. Catal., A, 2015, 504, 256–265 CrossRef CAS.
- B. Montanari, A. Vaccari, M. Gazzano, P. Käßner, H. Papp, J. Pasel, R. Dziembaj, W. Makowski and T. Lojewski, Appl. Catal., B, 1997, 13, 205–217 CrossRef CAS.
- F. Kovanda, E. Jindova, B. Dousova, D. Kolousek, J. Plestil and Z. Sedlakova, Acta Geodyn. Geomater., 2009, 6, 111–119 CAS.
- X. Wu, J. Liu, X. Liu, X. Wu and Y. Du, J. Catal., 2022, 407, 265–280 CrossRef CAS.
- Y.-S. Zhang, C. Li, C. Yu, T. Tran, F. Guo, Y. Yang, J. Yu and G. Xu, Chem. Eng. J., 2017, 330, 1082–1090 CrossRef CAS.
- Q. Yan, Y. Nie, R. Yang, Y. Cui, D. O'Hare and Q. Wang, Appl. Catal., A, 2017, 538, 37–50 CrossRef CAS.
- Q. Yan, Y. Gao, Y. Li, M. A. Vasiliades, S. Chen, C. Zhang, R. Gui, Q. Wang, T. Zhu and A. M. Efstathiou, Appl. Catal., B, 2019, 255, 117749 CrossRef CAS.
- H. Meng, J. Liu, Y. Du, B. Hou, X. Wu and X. Xie, Catal. Commun., 2019, 119, 101–105 CrossRef CAS.
- W. Wang, L. Wang, Y. Rao, Y. Huang, R. Li, F. Wei, H. Mei and J. Cao, Appl. Surf. Sci., 2023, 618, 156638 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |