
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCompartmentalized drug localization studies in extracellular vesicles for anticancer therapy†
Arunkumar
Pitchaimani‡
 *a,
Miguel
Ferreira
a,
Annalisa
Palange
a,
Martina
Pannuzzo
a,
Claudia
De Mei
a,
Raffaele
Spano
a,
Roberto
Marotta
b,
Beatriz
Pelacho
c,
Felipe
Prosper
cd and
Paolo
Decuzzi
*a
*a,
Miguel
Ferreira
a,
Annalisa
Palange
a,
Martina
Pannuzzo
a,
Claudia
De Mei
a,
Raffaele
Spano
a,
Roberto
Marotta
b,
Beatriz
Pelacho
c,
Felipe
Prosper
cd and
Paolo
Decuzzi
*a
aNanotechnology for Precision Medicine, Fondazione Istituto Italiano di Tecnologia (IIT), Genova, GE, Italy. E-mail: arunkumar.pitchaimani@vit.ac.in; paolo.decuzzi@iit.it
bElectron Microscopy Facility, Fondazione Istituto Italiano di Tecnologia (IIT), Genova, GE, Italy
cCentre for Applied Medical Research (CIMA), University of Navarra, Navarra, Spain
dClinica Universidad de Navarra, CCUN, IDISNA and CIBERONC, Navarra, Spain
First published on 8th November 2023
Abstract
In the development of therapeutic extracellular vesicles (EVs), drug encapsulation efficiencies are significantly lower when compared with synthetic nanomedicines. This is due to the hierarchical structure of the EV membrane and the physicochemical properties of the candidate drug (molecular weight, hydrophilicity, lipophilicity, and so on). As a proof of concept, here we demonstrated the importance of drug compartmentalization in EVs as an additional parameter affecting the therapeutic potential of drug-loaded EVs. In human adipose mesenchymal stem cell (hADSC) derived EVs, we performed a comparative drug loading analysis using two formulations of the same chemotherapeutic molecule – free doxorubicin (DOX) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE) lipid-conjugated doxorubicin (L-DOX) – to enhance the intracellular uptake and therapeutic efficacy. By nano surface energy transfer (NSET) and molecular simulation techniques, along with cryo-TEM analysis, we confirmed the differential compartmentalization of these two molecules in hADSC EVs. L-DOX was preferentially adsorbed onto the surface of the EV, due to its higher lipophilicity, whereas free DOX was mostly encapsulated within the EV core. Also, the L-DOX loaded EV (LDOX@EV) returned an almost three-fold higher DOX content as compared to the free DOX loaded EV (DOX@EV), for a given input mass of drug. Based on the cellular investigations, L-DOX@EV showed higher cell internalization than DOX@EV. Also, in comparison with free L-DOX, the magnitude of therapeutic potential enhancement displayed by the surface compartmentalized L-DOX@EV is highly promising and can be exploited to overcome the sensitivity of many potential drugs, which are impermeable in nature. Overall, this study illustrates the significance of drug compartmentalization in EVs and how this could affect intracellular delivery, loading efficiency, and therapeutic effect. This will further lay the foundation for the future systematic investigation of EV-based biotherapeutic delivery platforms for personalized medicine.
Understanding the role of extracellular vesicles (EVs) in inter-cellular communications is gaining enormous interest in the fields of cell biology and disease pathogenesis.1–3 Based on their cellular origin, size, and biomarker distribution, EVs are characterized as microvesicles, exosomes, and apoptotic bodies.3 Among these, exosomes and ectosomes (smaller membrane vesicles) are identified as nanosized vesicles confined by a lipid bilayer and containing cell cytosolic and membrane constituents, including proteins, lipids, metabolites, DNA, and microRNA.4–7 Extracellular vesicles are endogenous membrane-bound vesicles, which are believed to play a huge role in communications between cells via the exchange of specific biomolecular information. Due to their inherent biocompatibility, non-immunogenic nature, and selective cell-targeting, EVs could be employed as natural drug delivery platforms in the treatment of a variety of diseases, including cancer. However, these endogenous vehicles, including EVs, are self-limited with their natural membrane architecture, to restrict biomolecular cargo and small molecule passivation and thus, limit their clinical exploitation as drug delivery systems. Several bioengineering strategies, including the use of viral peptides, fusogenic liposomes, and bio-conjugate linkers, allow one to modify the surface and core of the EV to carry small molecules, peptides, and lipids of choice.8–10 Especially in cancer, such a reprogramming strategy is advantageous in modulating the immune resistance, angiogenesis, drug resistance, and metastasis formation in more personalized approaches for better therapy and bioimaging applications.4,9,11,12 However, maintaining the integrity and nature of biomolecular load after surface modification is highly challenging.
Despite significant advancements in EV bioengineering, their clinical drug encapsulation efficiency continues to be much lower, when compared with the synthetic nanoplatforms/nanomedicines.13 The drug loading efficacy in EVs is dependent on various physico-chemical properties including molecular weight, hydrophilic/hydrophobicity, lipophilicity, chemical nature, mechanical stiffness, etc. As a pilot study, here we have incorporated free doxorubicin molecules (DOX) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE) lipid-conjugated doxorubicin (L-DOX) into human adipose-derived mesenchymal stem cell (hADSC) EVs for personalized drug delivery applications. In this study, the term ‘EV’ refers to endogenous exosomes and ectosomes, which are in the size range of less than 200 nm. It is reported that mesenchymal stem cell (MSC) EVs have functions similar to those of their parent cells in repairing tissue damage, modulating the immune system, and so on.14,15 Here we demonstrated the importance of drug compartmentalization in EVs for modulating the effectiveness of the therapeutic potential of the drug. For this, we designed a comparative drug loading strategy of hADSC EVs using two forms of the same drug – free DOX and lipid-conjugated DOX (L-DOX), for maximizing the intracellular uptake (ESI, Fig. S1†) and to enhance the therapeutic efficacy of the encapsulated drug in the glioblastoma model using the U87-MG cell line, for a personalized drug delivery approach. Though the DOX molecule has very limited brain penetrance, recent advances in EV-mediated DOX delivery have shown higher blood–brain barrier (BBB) permeation, through systemic and tri-geminal pathways (nose-brain delivery). For this reason, we have chosen EV-associated DOX for our study.
Human adipose-derived mesenchymal stem cell exosomes (EVs) were isolated via an ultracentrifugation method, from conditional media upon starving hADSC under serum-free conditions for 48 h. Then, the purified hADSC EVs were stabilized in trehalose PBS.16 The extracted EV presented a regular spherical shape with a size diameter of 120 ± 77 nm, as documented by the cryo-EM image in Fig. 1A. This was also confirmed by the dynamic light scattering (DLS) analyses that showed a hydrodynamic diameter of 154 ± 46 nm and a surface ζ-potential of −22 ± 3 mV (ESI, Fig. S2†) respectively. The western blot analysis on EVs in the presence and the absence of the proteinase-K (PK) treatment showed the presence of the characteristic EV biomarkers CD-9 and CD-81 (ESI, Fig. S3†). By treating the EV with PK, it can cleave only the surface-accessible protein present on the surface of the EV (ESI, Fig. S3†). In our case, CD-81 is a membrane-accessible sensitive protein, which gets cleaved by the PK treatment, as reported earlier.17
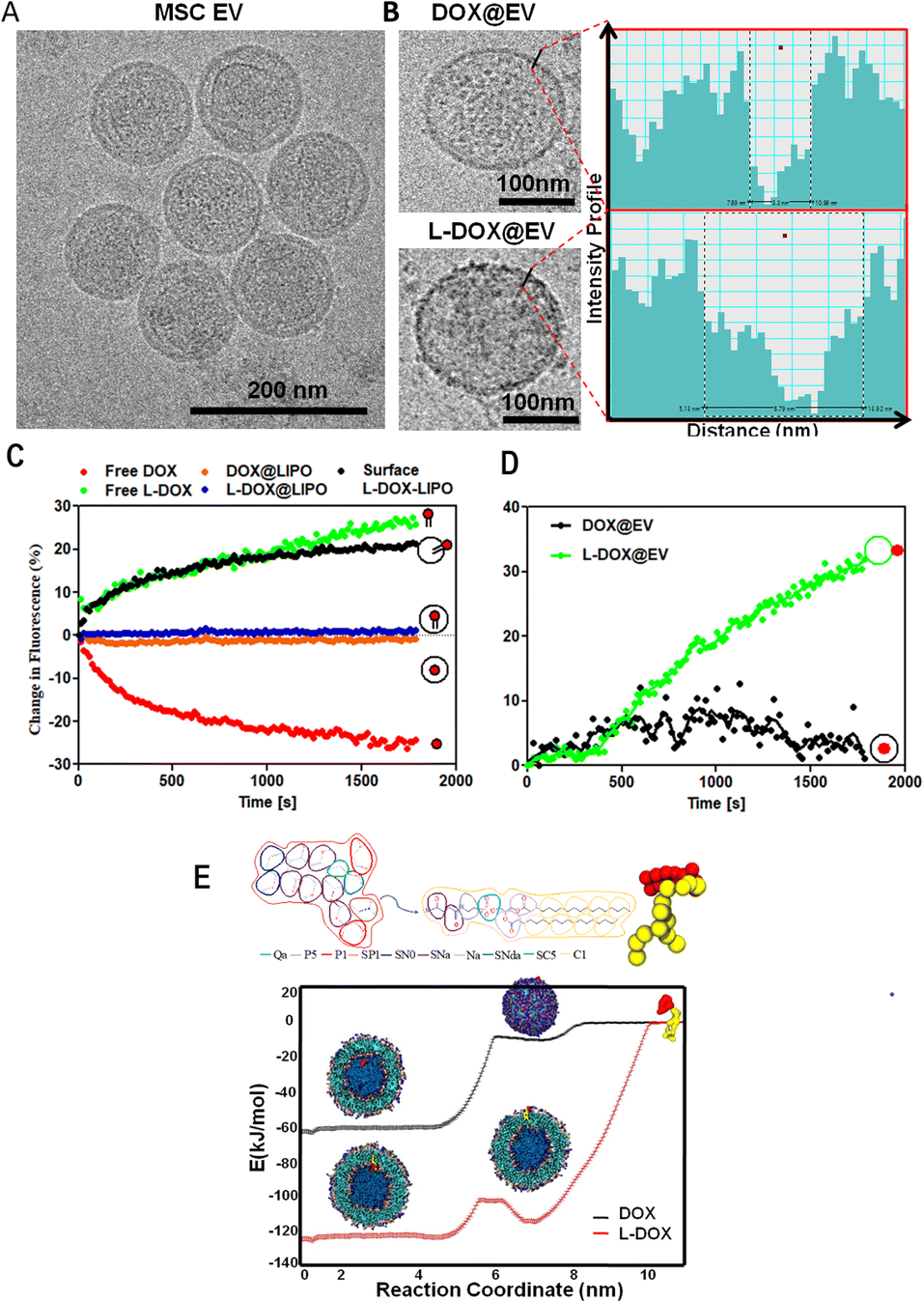 | ||
| Fig. 1 Physico-chemical characterization of doxorubicin-associated extracellular vesicles. (A) Cryo-EM analysis of extracellular vesicles (EV) isolated via ultracentrifugation from human adipose-derived mesenchymal stem cells (hADSC). (B) Cryo-EM images of individual EVs associated with free DOX (top) and L-DOX (bottom). On the right, line intensity profile spectrum analysis of the corresponding EV membrane topology. (C) NSET analysis quantifying the quenching or enhancement of the fluorescent signal emitted by doxorubicin for free DOX, L-DOX, DOX@liposomes, L-DOX@liposome, and surface-L-DOX@liposome, when reacting with negatively charged AuNPs. (D) Nanoparticle surface energy transfer (NSET) fluorescent enhancement analysis for DOX and L-DOX associated EVs. (E) Coarse-Grained Molecular Dynamics simulation of the interaction between lipid vesicles and free DOX or L-DOX. | ||
With the focus of developing therapeutic EVs with anti-cancer agents, EVs were incubated with free DOX (DOX@EV) and lipid-conjugated DOX (L-DOX@EV), at tested concentrations ranging from 25 to 150 μg ml−1 of DOX equivalent dose. The 1,2-distearoyl-3-sn-glycerophosphoethanolamine molecule (DSPE) was used as a lipid for the lipid-conjugated DOX (L-DOX) complex, as per our previous report.18 The association of the therapeutic molecules with EVs was assessed using multiple and complementary techniques, including dynamic light scattering, zeta sizer, cryo-electron microscopy, and nanoparticle surface energy transfer (NSET) analysis.
The incubation of EVs with free-DOX and DSPE-DOX (L-DOX) led to a progressive increase in their hydrodynamic size from the native ∼150 nm up to ∼300 nm, at the highest incubation dose of 150 μg ml−1. Specifically, the hydrodynamic diameter changed from 165 ± 12 to 277 ± 11 nm for the DOX@EV, and from 165 ± 12 to 321 ± 69 nm for L-DOX@EV (ESI, Fig. S4A and B†). Interestingly, the zeta potential value of the DOX@EV (−23 ± 1 mV) shows no significant change even at higher drug concentrations tested, whereas L-DOX@EV shows a steady decrease in its zeta potential from −22 ± 3 to −16 ± 0.8 mV, at the higher concentration tested (ESI, Fig. S4†). This decrease in characteristic zeta potential would suggest that the L-DOX@EV was adsorbed on the surface rather than encapsulated in the core of the EV. These observations stimulated additional characterization of the DOX-associated EV. Specifically, the cryo-EM analysis of the DOX@EV and L-DOX@EV showed distinct surface morphologies, with a notable roughness of the L-DOX associated EV (Fig. 1B). By analysing the DOX@EV and L-DOX@EV in cryo-TEM, the surface of the L-DOX@EV shows more pronounced electron-dense regions, when compared to the other. Using a line intensity profile spectrum (Gatan digital micrograph Inc), the outer membranes of the DOX and L-DOX-associated EV were further characterized. The line intensity phase gap identified a thickness of the DOX@EV membrane of 3 ± 1 nm as opposed to the 8 ± 3 nm, measured for the L-DOX@EV (Fig. 1B inset and ESI, Fig. S5†). The higher line intensity phase gap spectrum of L-DOX@EV would be possible due to the surface deposition of L-DOX molecules (>2 kDa).
To further understand the association of DOX and L-DOX with EVs, a nanoparticle surface energy transfer (NSET) analysis was conducted. This is a dipole-surface energy transfer process that can be used to measure biomolecular interactions at the nanoscale (ESI, Fig. S6†). Specifically, depending on the relative distance between gold nanoparticles (AuNP) and fluorescent biomolecules, the optical signal resulting from the AuNP-biomolecule interaction can be either quenched (short distance) or enhanced (long distance).19,20 In the current assay, DOX and L-DOX are the fluorescent biomolecules to be localized with respect to the EV membrane. For calibrating the NSET analysis, we first characterized the interaction between liposomes, as a model of EVs, and doxorubicin. Three different configurations were considered, namely liposomes carrying free DOX dispersed in the aqueous core (DOX@LIPO), liposomes carrying DSPE-DOX dispersed within the aqueous core (L-DOX@LIPO), and liposomes carrying DSPE-DOX adsorbed on the membrane (surface L-DOX@LIPO), following previous reports by the authors21,22 (ESI, Table S1†). The direct interaction of citrate-capped 40 nm gold nanoparticles with free DOX in solution led to rapid quenching of the DOX fluorescent signal due to its surface conjugation with the negatively charged metallic nanoparticles (short distance) (Fig. 1C – red curve). Differently, the signal associated with the interaction between the AuNP and free L-DOX was enhanced (Fig. 1C – green curve), possibly due to the presence of the DSPE spacer (conjugation of the DSPE lipid protects the amine in DOX). Then, the NSET interaction for the doxorubicin-carrying liposomes was assessed, showing no NSET enhancement or quenching of the DOX signal in the case of DOX@LIPO and L-DOX@LIPO, whereas the signal for the surface L-DOX@LIPO was enhanced (Fig. 1C – orange, blue, and black curves, respectively). The lack of quenching or enhancement in the fluorescent signal would suggest that the AuNPs were too far to interact with doxorubicin, thus confirming that the fluorescent biomolecules are mostly confined within the aqueous core of the DOX@LIPO and L-DOX@LIPO, while the AuNP is external to the liposomes. Conversely, the enhancement observed for the surface L-DOX@LIPO, similar to that observed for the free L-DOX in solution, would suggest that L-DOX chains in this liposomal membrane configuration are sufficiently close to the surface of the AuNP. Having completed this series of calibration tests, we further characterized DOX@EV and L-DOX@EV with the NEST technique. While DOX@EV showed no change, L-DOX@EV returned a strong enhancement in DOX fluorescence, confirming the exposure of L-DOX on the surface of the EV and the core confinement of free DOX in the DOX@EV configuration (Fig. 1D).
Finally, we also carried out Coarse-Grained Molecular Dynamics simulations to estimate the affinity, partitioning, and relative orientation of DOX and L-DOX with respect to a DPPC (Distearoylphosphatidylcholine) liposomal vesicle. The translocation of DOX molecules and L-DOX chains across a lipid bilayer, from the outer aqueous environment to the aqueous core of a liposome, was modelled using the MARTINI forcefield.23 The results in terms of free energy profiles are shown in Fig. 1E for DOX molecules (black curve) and L-DOX chains (red curve). Individual DOX molecules preferred to dwell within the core of the liposomes and an ∼60 kJ mol−1 energy barrier was estimated to cross the lipid bilayer. The free energy profile reduces steadily from the outer environment to the inner core suggesting that, given the appropriate time and mixing conditions, DOX molecules could spontaneously accumulate within the core of the liposomes. This is indeed in agreement with our results and current literature.24 Differently, for the L-DOX chains, two wells of similar depth in the free energy profile were observed right at the liposomal inner and outer layers with respect to the surrounding aqueous environment of ∼120 kJ mol−1, indicating a preference for L-DOX chains to avidly adsorb at the lipid membrane exploiting the higher lipophilicity of the pro-drug, as imparted by the DSPE appendix. The approach used for the estimation of drug partitioning in this work does not account for the free energy contribution associated with the flip-flop of L-DOX (switch to an antiparallel orientation) translocating from the outer to the inner lipid layer. In the case of DSPC molecules, a strong flip-flop energy penalty deriving from moving the hydrophilic headgroup across the hydrophobic core of the membrane has been experimentally determined to be ∼105 ± 2 kJ mol−1 at 40 °C.25 In the case of the L-DOX conjugate, the hydrophilic headgroup of DSPE is substituted by the large amphipathic DOX moiety, and the flip-flop barrier may be even larger due to steric hindrance. Therefore, L-DOX likely redistributes mainly in the outer layer of the vesicle where it remains kinetically trapped. Finally, L-DOX exhibits an overall stronger affinity for the liposome system (∼120 kJ mol−1) than free DOX (60 kJ mol−1), supporting the experimental evidence for its higher encapsulation efficiency. Based on these extensive and complementary characterization studies, one can conclude that upon direct incubation of doxorubicin with hADSC-derived EVs, free DOX molecules would mostly accumulate in the aqueous core whilst L-DOX chains would be adsorbed on the EV membrane.
Pharmacological features and cytotoxic potential of doxorubicin-associated EVs
After localizing the therapeutic agents within the EV compartments (core vs. membrane), the loading of doxorubicin was assessed using high performance liquid chromatographic techniques. For the free DOX molecules, loading was about 9% at 25 μg ml−1 of drug concentration but it reduced and stabilized around 6 ± 1% for all other cases up to 150 μg ml−1 (Fig. 2A). Indeed, the absolute drug content increased from an ∼2.5 μg to ∼10 μg per EV batch. Differently, for the L-DOX chains, loading increased proportionally with the input concentration reaching 17 ± 4%, corresponding to ∼30 μg of doxorubicin, at 150 μg ml−1 (Fig. 2B). Further, the release kinetics of doxorubicin from DOX@EV and L-DOX@EV was investigated under physiological (pH = 7.4) and acidic (pH = 5.8) conditions (Fig. 2C and D). For both formulations, a sustained release profile was documented under physiological conditions with about 80% of the associated drug being released within the first 48 hours. Differently, as expected, about 70% of the encapsulated drug was released in the first 6 h in an acidic environment. In general, the release of doxorubicin from the L-DOX@EV appeared to be slightly faster than from the DOX@EV, possibly because of the different drug compartmentalization, as discussed above. Further, the DLS of the DOX@EV and the L-DOX@EV stability in 25 mM trehalose-PBS buffer solution (by measuring their hydrodynamic size) were also tested for 30 day-storage at −20 °C. The results showed that the drug-loaded EVs (DOX@EV and the L-DOX@EV) depict excellent stability by maintaining their structure intactness. Based on the stability and maximum drug loading efficiency, the formulation with the input ratio of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) μg of the drug in 200μg of EVs was chosen for both DOX and L-DOX batches, and the same formulation was chosen for further biological studies (ESI, Fig. S7†). The biological stability of the drug-loaded EV was investigated using a serum stability assay (ESI, Fig. S8†). The results illustrated that both DOX and L-DOX-loaded EVs show higher biological stability in 90% FBS at 37 °C (ESI, Fig. S8†).
μg of the drug in 200μg of EVs was chosen for both DOX and L-DOX batches, and the same formulation was chosen for further biological studies (ESI, Fig. S7†). The biological stability of the drug-loaded EV was investigated using a serum stability assay (ESI, Fig. S8†). The results illustrated that both DOX and L-DOX-loaded EVs show higher biological stability in 90% FBS at 37 °C (ESI, Fig. S8†).
 | ||
| Fig. 2 Drug loading efficiency and release kinetics of the hADSC extracellular vesicles. (A and B) Drug loading efficiency and the quantitative drug content in the DOX and L-DOX loaded hADSC EVs were analysed using HPLC. (C and D) Drug release profiles of DOX and L-DOX loaded hADSC EVs under physiological conditions (pH = 7.4) and under acidic conditions (pH = 5.8) respectively. | ||
Then, the internalization and cytotoxic potential of these doxorubicin-associated EVs were tested on a human glioblastoma cell line – U87-MG cells. For cellular studies, EVs were labelled with PKH67 dye and they show a high stability under physiological conditions (ESI, Fig. S9†). The naïve hADSC-derived EVs were labelled in green with PKH67, while the nucleus of the U87-MG cells was stained in blue. After 6 h of incubation, fluorescent microscopy images show a prominent accumulation of the EVs in a perinuclear area (Fig. 3A). A time-dependent uptake of EVs in the U87-MG cells is observed (Fig. 3B). Furthermore, qualitative and quantitative analyses were conducted on naïve hADSC-derived EVs and a significant difference in cellular uptake was observed upon proteinase-K treatment26 (Fig. 3C and D). This molecule is reported to cause the proteolysis of the surface receptors in extracellular vesicles, thus inhibiting receptor-mediated endocytosis, which is the main intracellular uptake mechanism for EVs. By treating the EV with PK, it can cleave only the surface-accessible protein present on the surface of the EV. In our case, CD-81 is a membrane-accessible sensitive protein, which gets cleaved by the PK treatment. Similarly, the untreated control EV shows the presence of CD-81. Similar results have also been reported before.17 Since the EV uptake in U87-MG is higher at 12 h incubation time, two representative common times points were considered, 24 h – long incubation time and 6 h – short incubation time, to confirm that receptor depletion leads to a significant decrease in uptake. This is clearly shown in Fig. 3D for both arbitrarily selected time points. Notably, in the case of cell receptor depletion, the EV internalization efficiency appears to be independent of the incubation time, suggesting that the smaller percentage of internalized EVs is rapidly taken up within the first few hours of incubation. After 6 and 24 h of incubation, the PK-treated EV showed a 50% reduction in U87-MG cell uptake as opposed to the untreated EV (Fig. 3D). Further, the cellular uptake of therapeutic EVs was determined using fluorescent microscopy and flow cytometry assays. In cell culture, at 6 h post incubation, fluorescent microscopy imaging confirmed the avid uptake of the therapeutic EV by the U87-MG cells (Fig. 3E and ESI, Fig. S10†). The green signal of the EV membranes and the red signal for doxorubicin, both in the free DOX and L-DOX configurations, were observed to co-localize in a perinuclear position. Notably, the colocalization of the two signals would also suggest that the DOX@EV and L-DOX@EV are still intact within the U87-MG endosomes (Fig. 3E, top row) at 6 h incubation. Such a co-localization was however lost after 12 h suggesting the progressive release of doxorubicin in the cytosol (ESI, Fig. S11†). When directly comparing the internalization capacity of free DOX against L-DOX complexes and DOX@EV against L-DOX@EV via flow cytometry at 6 h post incubation, no significant difference was observed. However, the EV-based delivery of doxorubicin resulted in a significantly higher cellular uptake than the free drug (Fig. 3F).
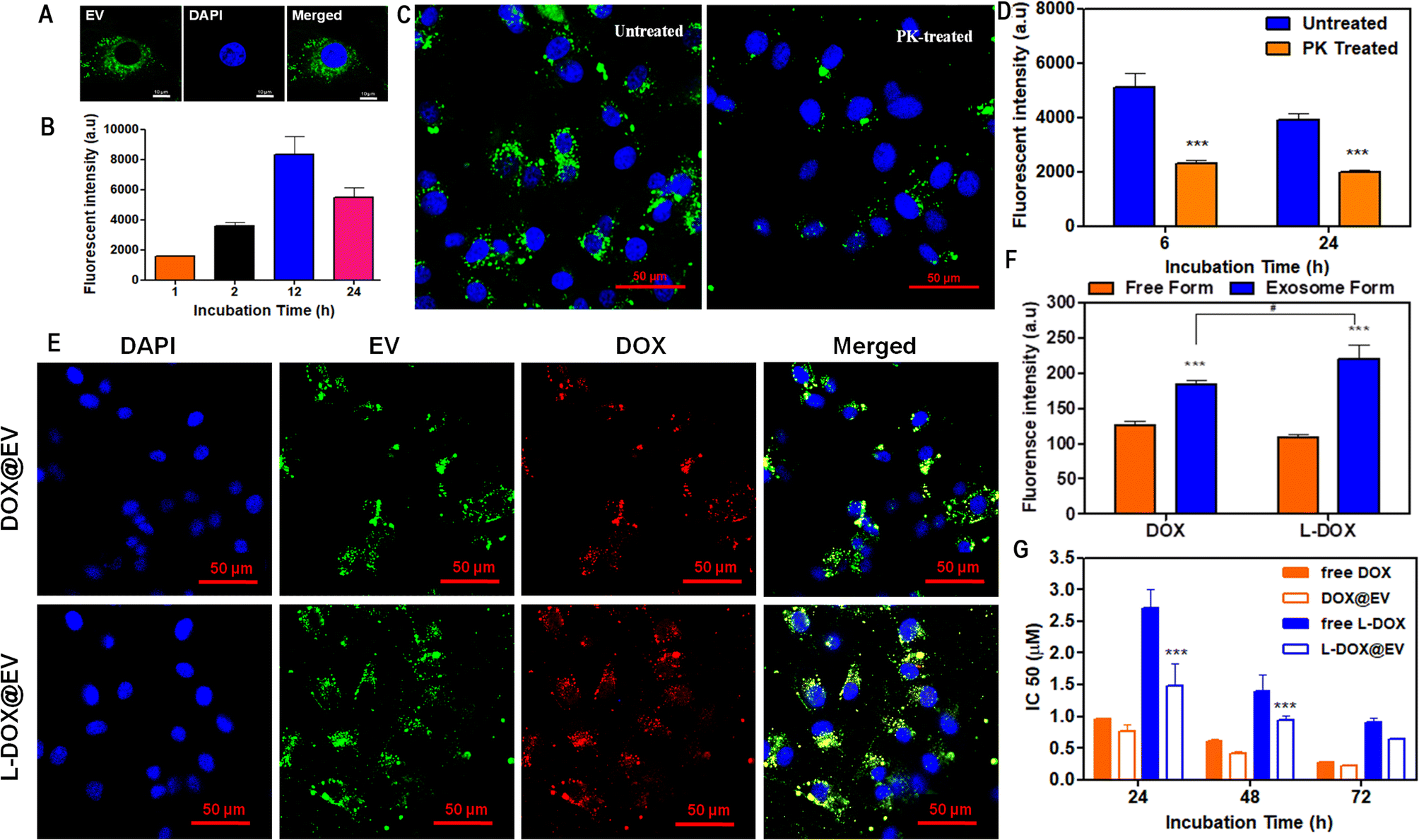 | ||
| Fig. 3 Cellular interaction and biological characterization of doxorubicin-associated extracellular vesicles. (A) Distribution of PKH67-labelled EVs within human glioblastoma cells (U87-MG) at 6 h post incubation. (B) Quantitative time-dependent EV uptake by U87-MG cells by FACS analysis. (C) Confocal analysis on differential uptake of U87-MG cells treated with PKH67-labelled EVs in the presence and the absence of proteinase-K treatment. (D) Quantitative flow cytometry-based analysis of EV uptake in U87-MG cells, at 6 and 24 h post-incubation, with and without proteinase-K treatment. (E) Confocal laser scanning micrographs of DOX and L-DOX associated EV uptake in U87-MG cells at 6 h post incubation. (F) Flow cytometry analysis of DOX and L-DOX uptake into U87-MG cells for EV formulations versus free drug treatment after 6 h incubation. (G) IC50 of free DOX and L-DOX associated EVs in U87-MG cells at 24, 48, and 72 h incubation, respectively. Data were presented as mean ± standard deviation. Values of p < 0.001 (***), p < 0.01 (**), and p < 0.05 (*) were considered significant. | ||
Finally, the cytotoxic effect of the free DOX, free L-DOX, DOX@EV, and L-DOX@EV was tested against U87-MG cells using an MTT assay. As expected, the therapeutic potential of the free drug (free DOX) is stronger than that of L-DOX at all tested time points, namely 24, 48, and 72 h (Fig. 3G). This should be associated with the reduced cytotoxicity of L-DOX and possibly a modest change in doxorubicin's configuration (DSPE lipid masking effect). Interestingly, the EV delivery of doxorubicin returned enhanced cytotoxic potentials comparable to or even higher than that of the free drug form. Specifically, the IC50 values of free DOX and DOX@EV were found to be 0.28 ± 0.01 and 0.22 ± 0.02 μM at 72 h post incubation, indicating a slightly stronger effect of the EV over the free drug. In L-DOX-treated cells, a significant change was observed between free L-DOX and L-DOX@EV at 24 and 48 h respectively. At 72 h a similar trend of cytotoxicity was observed between free L-DOX and L-DOX@EV, which returned IC50 values of 0.9 ± 0.08 and 0.65 ± 0.01 μM, respectively. The observed differential therapeutic response should be ascribed to the higher internalization propensity of the EV over the free DOX or free L-DOX, as observed above in Fig. 3F. At lower incubation times (24 and 48 h), the EV-associated L-DOX shows a significant therapeutic effect and at a higher incubation time (72 h) the difference diminishes as the free L-DOX can eventually enter the cell and exploit in full its killing potential of U87-MG cells. This trend is preserved across both DOX and L-DOX-associated EVs in U87-MG, showing the therapeutic advantage of the EV-mediated drug delivery system.
Conclusions
Extracellular vesicles (EVs) represent a promising tool for the delivery of a wide spectrum of therapeutic agents, such as small molecules, peptides, proteins, and nucleic acids, in the treatment of various biomedical conditions, including cancer.27–32 In this study, a model anti-cancer drug doxorubicin association with human adipose-derived mesenchymal stem cell-derived EVs was explored. Doxorubicin was used both in its free molecular form (DOX) as well as in the form of a pro-drug (L-DOX), resulting from the direct conjugation of DSPE lipid chains with the drug itself.18 DOX and L-DOX were associated with EVs through a passive loading and incubation method. The rationale behind the use of the L-DOX pro-drug was that of loading the chemotherapeutic molecules in different EV compartments (aqueous core vs. lipid membrane). Indeed, using extensive and complementary characterization assays, this work showed that while free doxorubicin was preferentially encapsulated in the aqueous core of the EV, the L-DOX prodrug adsorbed over the lipid bilayer. This is indeed consistent with the notion that DSPE lipid chains tend to fuse with other natural lipids, such as those forming the EV biomembrane.33,34 The naïve hADSC EVs isolated via ultracentrifugation were intact with a narrow size distribution and a diameter of about 150 nm. The DOX and L-DOX-associated EVs presented a larger size ranging from ∼150 to ∼300 nm, depending on drug loading. A cryo-TEM analysis highlighted significant differences in the EV membrane, depending on the payload, where L-DOX-associated EVs presented membrane pixel differences as opposed to the DOX-associated EV.It is well accepted that drug loading into EVs is lower than in synthesized nanocarriers, and this is due to the hierarchical structure of the EV membrane which is much more complex than the lipid bilayer of liposomes.8,35 For enhancing drug loading in EVs, several biochemical strategies have been already proposed, including electroporation, saponin treatment, sequential freeze–thaw cycles, and so on.10,13,36,37 In this work, a simpler strategy was adopted which was that of conjugating the therapeutic agent with a lipid chain. Indeed, L-DOX almost triplicated the total doxorubicin content in the EV, for a given input drug mass, when compared with free DOX, returning loading close to 20%.
Overall based on the observations, EV-mediated delivery of DOX and L-DOX shows superior advantages over the free drugs in their intracellular delivery and cytotoxic potential. However, in comparison with free L-DOX, the magnitude of therapeutic potential enhancement displayed by the surface compartmentalized L-DOX@EV is highly promising and can be exploited to overcome the sensitivity of many potential drugs, which are impermeable in nature. Though L-DOX@EV is not as potent as free DOX and DOX@EV, the compartmentalization effect of L-DOX in EVs enhances the therapeutic response of L-DOX. Thus the compartmentalization strategy is highly advantageous in delivering lipid-based therapeutics using extracellular vesicles to target cells in a personalized approach. Overall, this study illustrates the significance of the compartmentalization of drugs within extracellular vesicles, which might play a huge role in their therapeutic efficacy, regardless of their encapsulation efficiency. This will further lay the foundation for the future systematic investigation of EV-based biotherapeutic delivery platforms for personalized medicine.
Author contributions
AK and PD conceived the idea and wrote the manuscript. AK performed the experiments. MM, AP, CD, and RS helped in the cell culture experiments and assisted with manuscript writing. Dr RM helped in the cryo-TEM imaging experiments. BPS and FPC helped in experimental data analysis and revised the manuscript. PD supervised the research activities. All authors provided critical feedback, helped shape the research, and contributed to the manuscript.Conflicts of interest
All authors declared no conflict of interest.Acknowledgements
The authors acknowledge the support from the European Union’s Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie grant agreement no. 754490 (COFUND 2018 “MINDED”) and the Fondazione Istituto Italiano di Tecnologia, Italy. AK acknowledges the support from the Department of Biotechnology, Government of India for the DBT-Ramalingaswami Re-Entry Fellowship, Government of India. The authors thank Dr Peter James Gawne for his valuable comments and discussions.Notes and references
- A. A. Patil and W. J. Rhee, Biotechnol. Bioprocess Eng., 2019, 24, 689–701 CrossRef CAS.
- B. Yue, H. Yang, J. Wang, W. Ru, J. Wu, Y. Huang, X. Lan, C. Lei and H. Chen, Cell Proliferation, 2020, 53, e12857 CrossRef CAS PubMed.
- Y. Zhang, Y. Liu, H. Liu and W. H. Tang, Cell Biosci., 2019, 9, 19 CrossRef PubMed.
- B. Adem, P. F. Vieira and S. A. Melo, Trends Cancer, 2020, 6, 20–30 CrossRef CAS PubMed.
- R. Bandu, J. W. Oh and K. P. Kim, Exp. Mol. Med., 2019, 51, 1–10 CrossRef CAS PubMed.
- J. S. Brzozowski, H. Jankowski, D. R. Bond, S. B. McCague, B. R. Munro, M. J. Predebon, C. J. Scarlett, K. A. Skelding and J. Weidenhofer, Lipids Health Dis., 2018, 17, 211 CrossRef PubMed.
- A. Yokoi, A. Villar-Prados, P. A. Oliphint, J. Zhang, X. Song, P. D. Hoff, R. Morey, J. Liu, J. Roszik, K. Clise-Dwyer, J. K. Burks, T. J. O'Halloran, L. C. Laurent and A. K. Sood, Sci. Adv., 2019, 5, eaax8849 CrossRef CAS PubMed.
- I. K. Herrmann, M. J. A. Wood and G. Fuhrmann, Nat. Nanotechnol., 2021, 16, 748–759 CrossRef CAS PubMed.
- S. C. Jang, O. Y. Kim, C. M. Yoon, D.-S. Choi, T.-Y. Roh, J. Park, J. Nilsson, J. Lötvall, Y.-K. Kim and Y. S. Gho, ACS Nano, 2013, 7, 7698–7710 CrossRef CAS PubMed.
- H. Peng, W. Ji, R. Zhao, J. Yang, Z. Lu, Y. Li and X. Zhang, J. Mater. Chem. B, 2020, 8, 7591–7608 RSC.
- R. Ren, H. Sun, C. Ma, J. Liu and H. Wang, Cell Biosci., 2019, 9, 62 CrossRef PubMed.
- Z. Zhang, J. A. Dombroski and M. R. King, Cell. Mol. Bioeng., 2020, 13, 1–16 CrossRef CAS PubMed.
- C. Schindler, A. Collinson, C. Matthews, A. Pointon, L. Jenkinson, R. R. Minter, T. J. Vaughan and N. J. Tigue, PLoS One, 2019, 14(3), 1–19 CrossRef PubMed.
- F. M. Barros, F. Carneiro, J. C. Machado and S. A. Melo, Front. Immunol., 2018, 9, 730 CrossRef PubMed.
- H. Y. Kim, H. Kumar, M.-J. Jo, J. Kim, J.-K. Yoon, J.-R. Lee, M. Kang, Y. W. Choo, S. Y. Song, S. P. Kwon, T. Hyeon, I.-B. Han and B.-S. Kim, Nano Lett., 2018, 18, 4965–4975 CrossRef CAS PubMed.
- S. Bosch, L. de Beaurepaire, M. Allard, M. Mosser, C. Heichette, D. Chrétien, D. Jegou and J.-M. Bach, Sci. Rep., 2016, 6, 1–11 CrossRef PubMed.
- A. Cvjetkovic, S. C. Jang, B. Konečná, J. L. Höög, C. Sihlbom, C. Lässer and J. Lötvall, Sci. Rep., 2016, 6, 36338 CrossRef CAS PubMed.
- M. Ferreira, I. F. Rizzuti, A. L. Palange, M. G. Barbato, V. Di Francesco, M. Di Francesco and P. Decuzzi, Front. Bioeng. Biotechnol., 2020, 8, 5 CrossRef PubMed.
- Y. Chen, M. B. O'Donoghue, Y.-F. Huang, H. Kang, J. A. Phillips, X. Chen, M.-C. Estevez and W. Tan, J. Am. Chem. Soc., 2010, 132, 16559–16570 CrossRef CAS PubMed.
- C. Chen, C. Midelet, S. Bhuckory, N. Hildebrandt and M. H. V. Werts, J. Phys. Chem. C, 2018, 122, 17566–17574 CrossRef CAS.
- A. Pitchaimani, T. D. T. Nguyen, H. Wang, S. H. Bossmann and S. Aryal, RSC Adv., 2016, 6, 36898–36905 RSC.
- A. Pitchaimani, T. D. T. Nguyen and S. Aryal, Biomaterials, 2018, 160, 124–137 CrossRef CAS.
- M. Pannuzzo, A. Felici and P. Decuzzi, Biomacromolecules, 2022, 23, 4678–4686 CrossRef CAS PubMed.
- A. C. Alves, A. Magarkar, M. Horta, J. L. F. C. Lima, A. Bunker, C. Nunes and S. Reis, Sci. Rep., 2017, 7, 1–11 CrossRef CAS.
- T. C. Anglin, M. P. Cooper, H. Li, K. Chandler and J. C. Conboy, J. Phys. Chem. B, 2010, 114, 1903–1914 CrossRef CAS PubMed.
- O. Betzer, N. Perets, A. Angel, M. Motiei, T. Sadan, G. Yadid, D. Offen and R. Popovtzer, ACS Nano, 2017, 11, 10883–10893 CrossRef CAS PubMed.
- T. Martins-Marques, M. J. Pinho, M. Zuzarte, C. Oliveira, P. Pereira, J. P. G. Sluijter, C. Gomes and H. Girao, J. Extracell. Vesicles, 2016, 5, 32538 CrossRef.
- S. Luo, L. Du and Y. Cui, Mol. Pharm., 2020, 17, 1447–1457 CrossRef CAS PubMed.
- G. Zhang, X. Huang, H. Xiu, Y. Sun, J. Chen, G. Cheng, Z. Song, Y. Peng, Y. Shen, J. Wang and Z. Cai, J. Extracell. Vesicles, 2020, 10, e12030 CrossRef CAS.
- C. Chen, M. Sun, J. Wang, L. Su, J. Lin and X. Yan, J. Extracell. Vesicles, 2021, 10, e12163 CrossRef CAS PubMed.
- N. Salmond and K. C. Williams, Nanoscale Adv., 2021, 3, 1830–1852 RSC.
- J. Osborn, J. E. Pullan, J. Froberg, J. Shreffler, K. N. Gange, T. Molden, Y. Choi, A. Brooks, S. Mallik and K. Sarkar, Nanoscale Adv., 2020, 2, 3411–3422 RSC.
- D. Paolino, M. L. Accolla, F. Cilurzo, M. C. Cristiano, D. Cosco, F. Castelli, M. G. Sarpietro, M. Fresta and C. Celia, Colloids Surf., B, 2017, 155, 266–275 CrossRef CAS PubMed.
- L. Viitala, S. Pajari, L. Gentile, J. Määttä, M. Gubitosi, J. Deska, M. Sammalkorpi, U. Olsson and L. Murtomäki, Langmuir, 2019, 35, 3999–4010 CrossRef CAS.
- Z. Wang, J. Rich, N. Hao, Y. Gu, C. Chen, S. Yang, P. Zhang and T. J. Huang, Microsyst. Nanoeng., 2022, 8, 1–11 CrossRef PubMed.
- M. Piffoux, A. K. A. Silva, C. Wilhelm, F. Gazeau and D. Tareste, ACS Nano, 2018, 12, 6830–6842 CrossRef CAS PubMed.
- S. Rayamajhi, T. D. T. Nguyen, R. Marasini and S. Aryal, Acta Biomater., 2019, 94, 482–494 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed methodology and supplementary results. See DOI: https://doi.org/10.1039/d3na00207a |
| ‡ Present address: Centre for Biomaterials, Cellular and Molecular Theranostics (CBCMT), Vellore Institute of Technology, Vellore-632014, TN, India. |
| This journal is © The Royal Society of Chemistry 2023 |