
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePivotal role of the protein corona in the cell uptake of fluorinated nanoparticles with increased sensitivity for 19F-MR imaging†
Lodovico
Gatti‡§
 ab,
Cristina
Chirizzi‡
a,
Giulia
Rotta¶
c,
Pietro
Milesi
a,
María
Sancho-Albero||
def,
Victor
Sebastián
def,
Anna
Mondino
c,
Jesús
Santamaría
def,
Pierangelo
Metrangolo
a,
Linda
Chaabane**
*b and
Francesca Baldelli
Bombelli
*a
ab,
Cristina
Chirizzi‡
a,
Giulia
Rotta¶
c,
Pietro
Milesi
a,
María
Sancho-Albero||
def,
Victor
Sebastián
def,
Anna
Mondino
c,
Jesús
Santamaría
def,
Pierangelo
Metrangolo
a,
Linda
Chaabane**
*b and
Francesca Baldelli
Bombelli
*a
aDepartment of Chemistry, Materials and Chemical Engineering “Giulio Natta”, Politecnico di Milano, 32, Milano, 20131, Italy. E-mail: francesca.baldelli@polimi.it
bInstitute of Experimental Neurology (INSpe) and Experimental Imaging Center (CIS), IRCCS San Raffaele Scientific Institute, Via Olgettina, 58, Milano, 20132, Italy. E-mail: linda.chaabane@cnr.it
cLymphocyte Activation Unit, Division of Immunology, Transplantation and Infectious Diseases, IRCCS San Raffaele Scientific Institute, Via Olgettina, 58, Milan, 20132, Italy
dInstituto de Nanociencia y Materiales de Aragón (INMA), CSIC-Universidad de Zaragoza, Calle Pedro Cerbuna, 12, Zaragoza, 50009, Spain
eDepartment of Chemical Engineering and Environmental Technologies, University of Zaragoza, Calle Pedro Cerbuna, 12, Zaragoza, 50009, Spain
fNetworking Research Center on Bioengineering Biomaterials and Nanomedicine (CIBER-BBN), Calle Monforte de Lemos, 3-5, Madrid, 28029, Spain
First published on 8th June 2023
Abstract
In vivo cell tracking by non-invasive imaging technologies is needed to accelerate the clinical translation of innovative cell-based therapies. In this regard, 19F-MRI has recently gained increased attention for unbiased localization of labeled cells over time. To push forward the use of 19F-MRI for cell tracking, the development of highly performant 19F-probes is required. PLGA-based NPs containing PERFECTA, a multibranched superfluorinated molecule with an optimal MRI profile thanks to its 36 magnetically equivalent fluorine atoms, are promising 19F-MRI probes. In this work we demonstrate the importance of the surface functionalization of these NPs in relation to their interaction with the biological environment, stressing the pivotal role of the formation of the protein corona (PC) in their cellular labelling efficacy. In particular, our studies showed that the formation of PC NPs strongly promotes the cellular internalization of these NPs in microglia cells. We advocate that the formation of PC NPs in the culture medium can be a key element to be used for the optimization of cell labelling with a considerable increase of the detection sensitivity by 19F-MRI.
Introduction
Cell therapy is becoming a potent treatment approach for multiple human diseases. To support the development and accelerate the clinical translation of such ‘living’ systems, longitudinal cell imaging tools would be needed and they allow the monitoring of cell biodistribution, therapy outcomes and prevent off-target toxicity. Accordingly, the Food and Drug Administration (FDA) recommended the development and, subsequently, the use of non-invasive imaging techniques to track cells. Magnetic resonance imaging (MRI), when compared to other approaches, has the advantage of being safe, exploitable in longitudinal studies, and able to provide whole-body data also useful for clinical use.Nanoparticles (NPs) containing conventional MRI contrast agents (i.e., gadolinium or iron based paramagnetic agents) can be used to label cells and render them detectable by MRI. A large number of preclinical studies with superparamagnetic iron oxide (SPIO) NPs have shown promising results.1 However, a rather poor signal specificity, a low cellular uptake and the fact that SPIO NP concentration is not linearly proportional to MRI signal are considered limiting factors, preventing a quantitative and accurate analysis.2,3 The detection of SPIO NPs is unspecific since it is determined as signal loss in the tissue and could lead to misinterpretation and to rise of artefacts because these NPs strongly interfere with the proton signal (1H-MRI). Beyond the incompatibility of SPIO NPs detection with standard MRI analysis, these contrast agents could be confused with vessels, haemorrhage, necrosis or endogenous iron traces normally present in healthy or diseased tissues.
A number of studies recently highlighted the potential of fluorinated NPs as labelling agents for cell tracking by 19F-MRI.3–8 Indeed, as endogenous fluorine in tissues is not detected by MRI, the signal originating from fluorinated NP-loaded cells specifically refers to the infused product and is directly quantifiable by 19F-MRI.3–7,9 Of note, the 19F-MRI signal is directly proportional to the amount of 19F atoms, it is stable and does not interfere with the standard signal from 1H. Indeed, 19F-MRI can be combined with any 1H-MRI protocols on the same scanner. Therefore, 19F-MRI is a suitable strategy for unbiased localization and tracking of labelled cells over time. The low sensitivity of 19F-MRI currently limits its potential applicability and translation to the clinic. While the use of high field MRI scanner and cryogenic radiofrequency probes could improve the sensitivity,10,11 the customized design of better performing fluorinated tracers bears the potential to maximize imaging performance. In this respect, we previously reported that a superfluorinated molecule, named PERFECTA, showed ideal chemical and structural properties for 19F-MRI for improving imaging sensitivity, thanks to its large number of 19F atoms per molecule.12–15 Importantly, the 36 19F atoms are symmetrically distributed in this molecule resulting in intense MRI signals at a specific and unique chemical shift.12 In addition to a high 19F payload, the physicochemical properties of the formulation, necessary to disperse these fluorinated molecules in physiological solutions as 19F-NPs, could be tuned to obtain optimal internalization by cells without affecting their functionality.
Considering that NP internalization mainly occurs through endocytosis mechanisms,16–18 their uptake is strongly dependent on NP size, shape, charge and composition.19–22 Moreover, proteins can be adsorbed onto the NP surface, and form a layer called protein corona (PC) impacting on the surface and colloidal properties of NPs with consequences on the cellular uptake mechanism. This biomolecular layer is characterized by two components: a “hard” (HC) and a “soft” corona, formed by layers more strongly and more loosely bound to the NP layers, respectively. Since the inner HC layer persists on the NP surface for times long-enough to ensure cellular interaction, it is considered more biologically relevant.23 Thus, the PC adsorbed on NPs creates a new bio–nano interface with direct consequences on cellular uptake mechanisms.24–26 For example, adsorption of opsonin on the NP surface can preferentially activate the phagocytic pathway.27
In particular, we have developed polylactic-co-glycolic acid (PLGA) NPs loaded with PERFECTA at high yields with different surface functionalizations. Specifically, PERFECTA@PLGA NPs (19F–PLGA NPs) were stabilized by three different biocompatible coating agents, namely poly-vinyl alcohol (PVA), sodium cholate (NaC) and chitosan (CS), forming NPs with a neutral, negative and slightly positive surface electrokinetic potential, respectively. As expected, these NPs were characterized by a specific bio–nano interface in the biological environment23–28 with the formation, in some cases, of PC NPs with larger size and different surface properties (composition and electrokinetic potential) compared to the original NPs.28,29 The PC formation can be essential to enhance cell labelling performance as it was demonstrated with ferumoxytol NPs for stem cell labelling.30 In other cases, it has been shown that PC can strongly affect the relaxation times of different SPIO NPs depending on their surface properties and PC composition.31,32 We tested these 19F–PLGA NPs as labelling agents on microglial cells (BV2), which are characterized by a high phagocytic capacity33 similar to the resident immune cells of the central nervous system that play a fundamental role in immune defence.34 The in vivo tracking of myeloid cells with a natural capacity to phagocyte NPs such as macrophages, microglia, and dendritic cells (DCs) by 19F-MRI helped monitor the progression of different inflammations,35,36 including the dynamics of cancer-associated immune response.37 In a first clinical trial it was used to trace immunotherapeutic DCs in colorectal adenocarcinoma patients.3 More recent studies exploited human peripheral blood mononuclear cells to validate clinical scanners and evaluate safety compliance.38 In spite of technology improvements, sensitivity remains an issue when tracking small number of cells. Various NPs have been formulated to maximize the 19F loading, to increase the interaction with cells and to facilitate their internalization to overcome the sensitivity limits.38,39 In the present work, we performed an optimization of superfluorinated PLGA-based nanoformulations modulating their surface properties to further improve cell labelling efficiency and imaging sensitivity. We compared the cell labelling efficiency of 19F–PLGA NPs in relation to their surface features by a systematic 19F-MRI study. The NPs with a higher tendency to form a PC in the culture medium resulted to be the most efficient labelling agents enabling the best 19F-MRI response (Fig. 1). Moreover, studies on NP cellular uptake mechanisms, using pharmacological inhibitors for specific endocytosis pathways, showed that the highest NP uptake was associated with an enhancement of the phagocytic pathway indicating a significant role of the PC formation in this process.
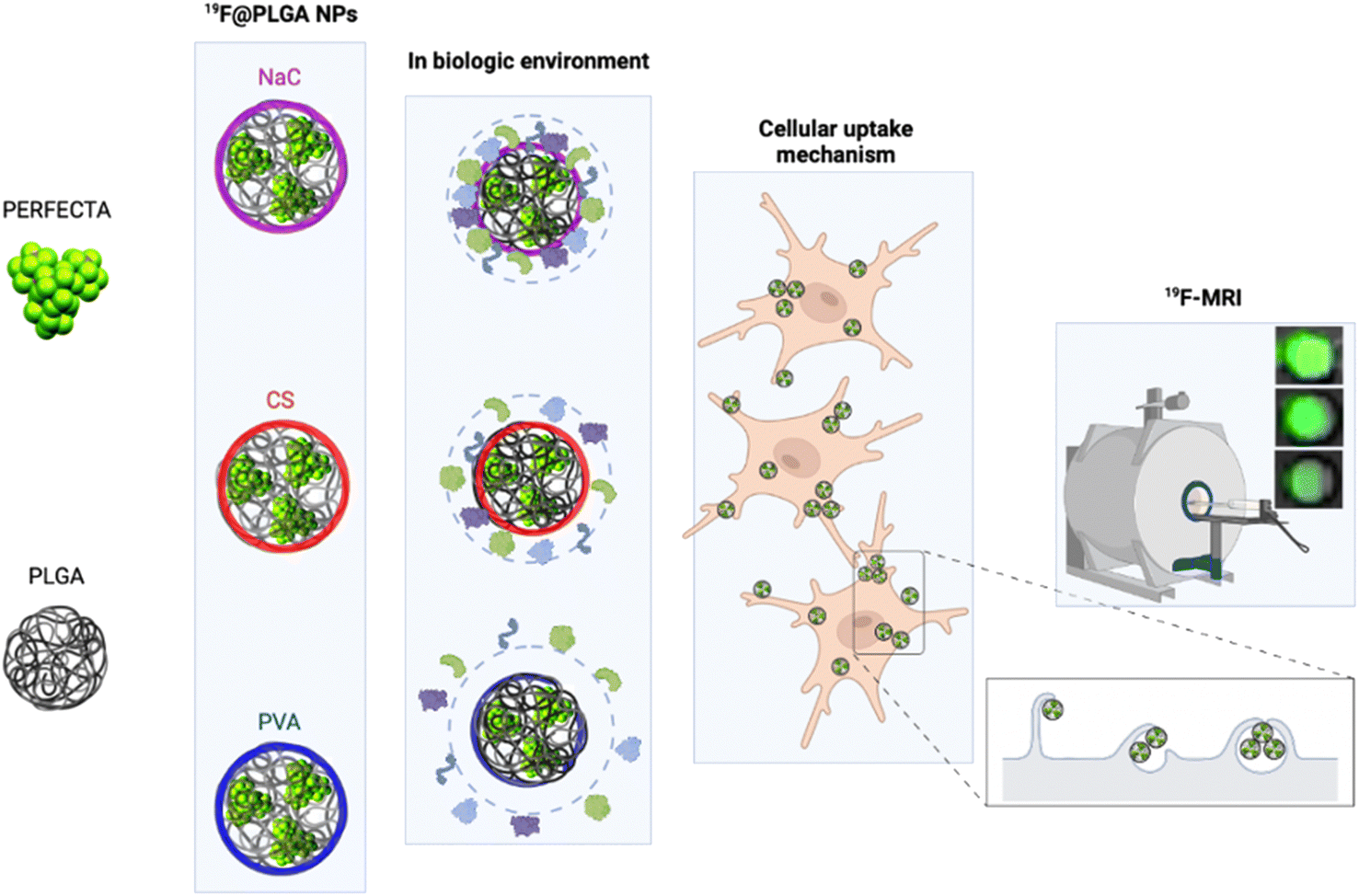 | ||
| Fig. 1 Schematic representation of the components used to produce 19F–PLGA NPs containing the superfluorinated probe (PERFECTA) and coated by different stabilizers. The obtained 19F–PLGA NPs show a different behaviour in the biological environment with a diverse tendency to form the protein corona (PC). The PC modulates cell labelling efficiency through different internalization pathways and has a significant impact on 19F-MRI sensitivity. | ||
Results and discussion
Physico-chemical characterization of 19F–PLGA NPs
19F–PLGA NPs were stabilized by sodium cholate (NaC), polyvinyl alcohol (PVA) or chitosan (CS), to obtain NPs with different surface properties.40 While 19F–PLGA–NaC and 19F–PLGA–PVA NPs were formulated following an established procedure,15 an initial study was dedicated to optimize the preparation protocol of positively charged NPs, which should exhibit a higher electrostatic interaction with the negatively charged cell membrane.20,41,42 The optimization was carried out to obtain small and positively charged 19F–PLGA–PVA–CS NPs with a homogeneous size distribution, a fine colloidal stability and high PERFECTA loading (ESI, Section 1.2 and Fig. S1†). Overall, using a CS/PLGA (w/w) ratio of 0.8 and PVA content of 2% (w/v) we obtained NPs with a positive electrokinetic potential (19 ± 8 mV) and the best colloidal properties among all the tested conditions to produce 19F–PLGA–PVA–CS NPs (Fig. S1E and F†).On the other hand, 19F–PLGA NPs coated with either NaC or PVA were characterized as expected by negative and neutral electrokinetic potentials, respectively (Table 1). Dynamic Light Scattering (DLS) characterization in aqueous solution showed stable monodisperse NP size distributions for all the NPs ranging from 100 to 200 nm depending on the stabilizer (Fig. S2A–C†). Of note, 19F–PLGA NPs stabilized with NaC showed a particularly smaller hydrodynamic size, around 100 nm compared to the others. Interestingly, comparable results have been reported on similar NaC coated PLGA NPs loaded with a hydrophilic drug.43,44 A different level of fluorine encapsulation was found through 19F-NMR experiments with an external standard for the NPs, ranging from 2 to 4 × 1020 atoms per ml (Table 1).
| Surfactants | NP sizeb | Particle chargec | Fluorine contentd | NPs per mle (×1011) | ||
|---|---|---|---|---|---|---|
| D H (nm) | PDI | Zeta potential (mV) | Encapsulation (%) | Atoms per ml (×1020) | ||
| a All reported values are the mean of six independent measurements performed on different NP batches. b Hydrodynamic diameter (DH) and polydispersity index (PDI) obtained by cumulant fitting of DLS measurements. c Particle charge determined by ζ-potential measurement. d Fluorine content determined by 19F-NMR. e NP concentration determined by Nanoparticle Tracking Analysis (NTA). | ||||||
| NaC | 104 ± 10 | 0.20 ± 0.03 | −49 ± 5 | 48 ± 10 | 2.1 ± 0.4 | 8.1 ± 1.2 |
| PVA–CS | 215 ± 51 | 0.16 ± 0.06 | +19 ± 8 | 47 ± 9 | 3.0 ± 0.6 | 11.2 ± 6.7 |
| PVA | 206 ± 9 | 0.12 ± 0.03 | −13 ± 1 | 61 ± 12 | 4.0 ± 0.8 | 69.0 ± 27.0 |
Considering the concentrations of NPs measured by NTA (Table 1), the average amount of fluorine (PERFECTA) per single NP can be assumed to be considerably higher in PVA–CS and NaC–19F–PLGA NPs (almost 27 × 107 atoms per NP) rather than in PVA formulation (around 5.8 × 107 atoms per NP).
Further analysis by HAADF-STEM confirmed the presence of spherical and colloidally stable NPs (Fig. S3A–C and G–I†) while semi-quantitative PERFECTA loading was verified by EDS chemical analysis (Fig. S3D–F†). Furthermore, the relaxation times of the three 19F–PLGA NPs were in the same range and suitable for 19F-MRI, with relatively short T1 (260–350 ms, Table S1†) and long T2 (198–220 ms, Table S1†).
The colloidal stability of the NPs was also investigated under physiological conditions and in the presence of proteins. Specifically, size variations of the 19F–PLGA NPs dispersed in cell culture medium (DMEM), with and without FBS, were evaluated by DLS over time up to 48 h (Fig. 2 and S2†). 19F–PLGA–NaC NPs, when incubated with FBS, immediately showed after 30 min of incubation (Fig. 2A) an increase in size, while NP size remained stable in pure DMEM (Fig. 2A and D). This behavior was likely due to the NaC coating, which provides a good electrostatic stabilization in buffer solution, but it is known to promote strong interactions with serum proteins leading to the formation of aggregates.45 Importantly, 19F–PLGA–NaC NPs did not show sedimentation or precipitation effects during the overall experimental window. On the other hand, 19F–PLGA–PVA NPs, stabilized by steric hindrance, showed the highest colloidal stability for both conditions, without effective interactions with serum proteins (Fig. 2C and F). For 19F–PLGA–PVA–CS NPs, a slight increase of the hydrodynamic size was observed only in DMEM, indicating a stabilization effect of serum proteins (Fig. 2B and E).
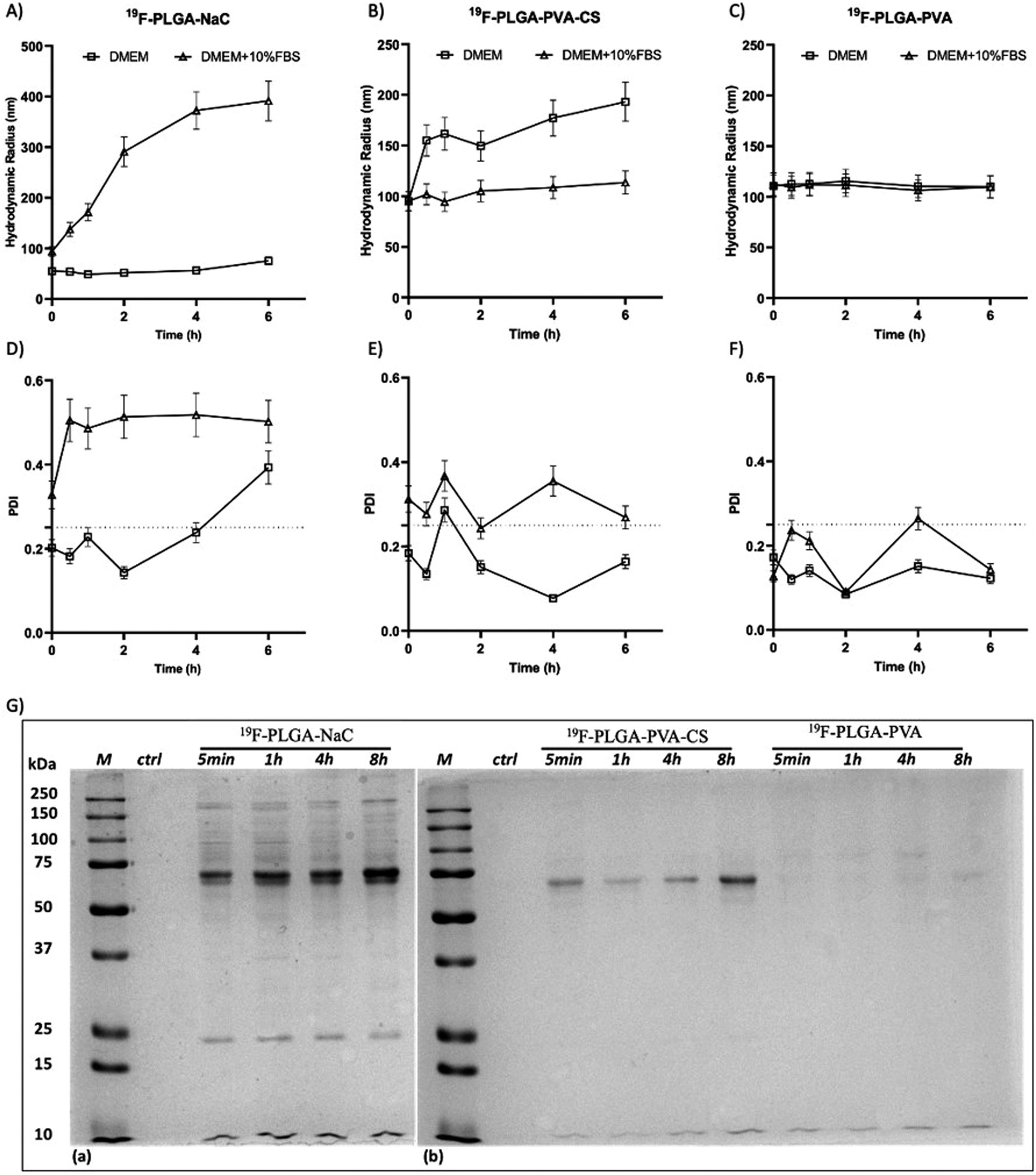 | ||
| Fig. 2 Hydrodynamic radius and PDI of all the NPs incubated in DMEM (square) or DMEM supplemented with 10% FBS (triangle) at 37 °C for time points from 5 min to 6 hours. Hydrodynamic radius and PDI values of PLGA–NaC (A–D), PLGA–PVA–CS (B–E) and PLGA–PVA (C–F). 1D-SDS–PAGE gel (G) of isolated HC PC from 19F–PLGA–NaC NPs, 19F–PLGA–PVA–CS and 19F–PLGA–PVA NPs. “M” indicates the protein ladder reference, while “ctrl” is the control of the cell culture medium treated with the same procedure but without NPs. | ||
Overall, we developed three PERFECTA loaded NPs with high yield of encapsulation, good colloidal stability and different patterns of interaction with the serum proteins as a function of their surface functionalization.
Protein corona characterization
In cell culture medium, 19F–PLGA–NaC (Fig. 2A) showed a rapid increase of NP size, indicative of the formation of a protein corona (PC) due to the binding of medium-derived components. 19F–PLGA–PVA–CS and 19F–PLGA–PVA did not show such a relevant variation of size, while the increase of PDI values was related to the presence of the protein background.46,47 The hard corona (HC) composition was then investigated upon NP incubation in cell culture medium supplemented with 10% FBS in the experimental window – from 5 min to 8 h – in accordance with cellular labelling conditions. SDS–PAGE gel analysis indicated the formation of an extensive (HC) PC already within the first 5 min of incubation with 19F–PLGA–NaC NPs (Fig. 2G). In the case of 19F–PLGA–PVA–CS, few bands at 75 kDa becoming more intense with the incubation time were observed (Fig. 2G). In detail, PLGA–NaC NPs showed a rich PC profile with two more intense bands at 75 and 25 kDa. Of note, no PC formation was observed for PVA NPs at all incubation times, indicating the ability of PVA to prevent NP–protein interactions (Fig. 2G).As expected, the PC composition was strongly dependent on the surface chemical composition of the NPs, increasing their complexity at the nano–bio interface.48–50 Here, we demonstrate that 19F–PLGA NPs coated with ionic stabilizers are more prone to form PC NPs than non-ionic analogues stabilized with PVA.
19F–PLGA NP cellular uptake studies
To test the cellular uptake and toxicity effects, microglial cells (BV2) were incubated with the 19F-NP formulations for 4, 6 and 8 h, keeping constant the dose of fluorine content (i.e., 1.54 mM PERFECTA corresponding to 1 × 1014 19F per cell). Under all tested conditions, cell viability was similar to that of untreated cells (Fig. 3A), while 19F labelling efficiency increased over time for all formulations. As expected, with the neutrally charged PLGA–PVA NPs, 19F uptakes by cells were the lowest and limited over time (Fig. 3B), while an increasing trend and higher 19F cellular uptake was observed for PLGA–NaC and PLGA–PVA–CS compared to PLGA–PVA NPs. Although the physical characteristics are almost similar, the main differences between 19F–PLGA–PVA NPs and 19F–PLGA–NaC or 19F–PLGA–PVA–CS NPs are related both to the 19F loading in single NP (lowest for 19F–PLGA–PVA) and to the interaction with serum proteins, with formation of a rich PC for 19F–PLGA–NaC and 19F–PLGA–PVA–CS NPs.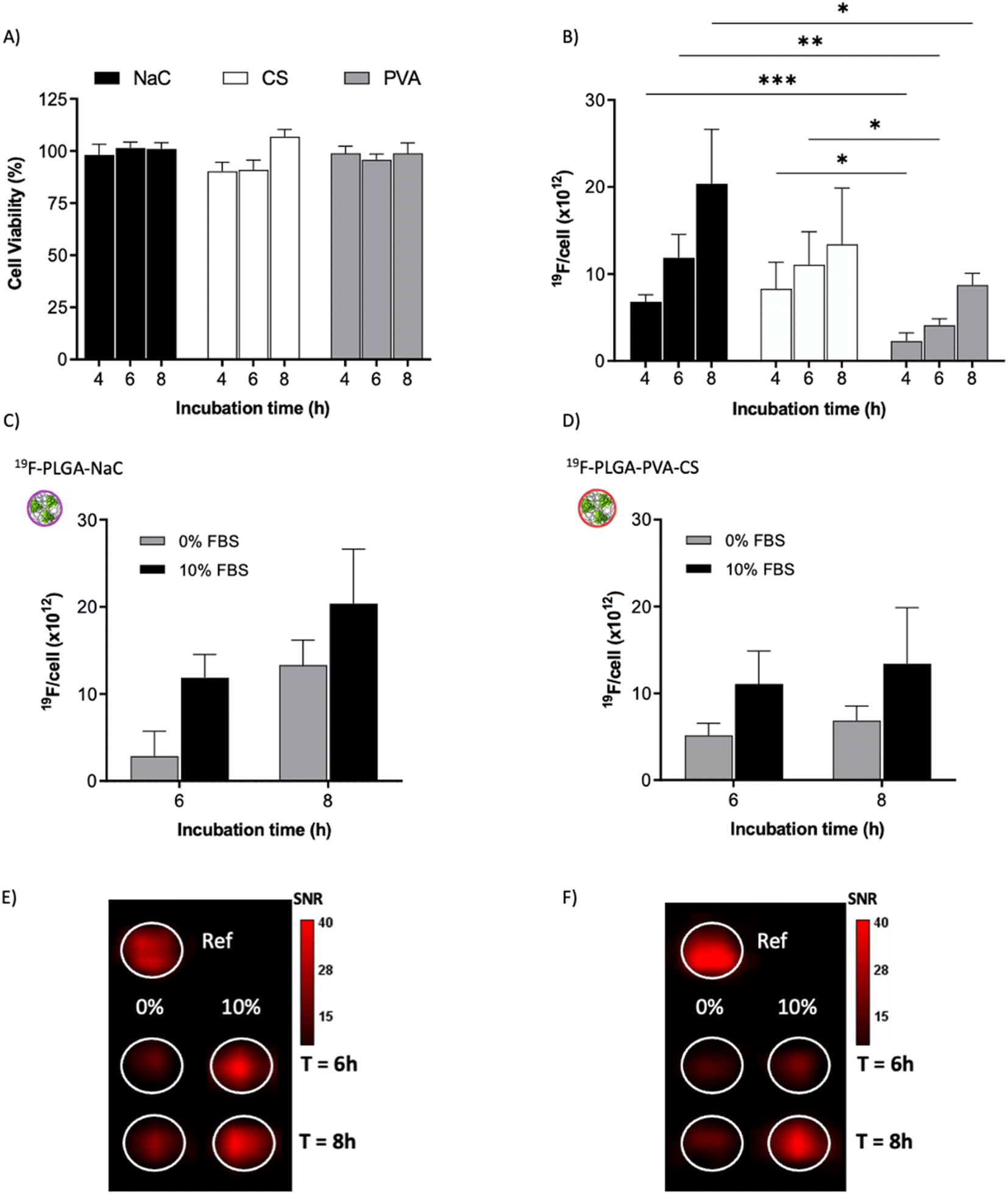 | ||
Fig. 3 Labelling efficiency. (A) Cell viability of labeled BV2 cells (normalized to the cell survival of unlabeled cells, ctrl) evaluated by standard trypan blue staining after 4, 6 and 8 h of incubation with the different 19F–PLGA-NPs. (B) 19F cellular uptake as a function of incubation time measured by 19F-MRI. (C and D) Labelling efficiency analyzed by 19F-MRI in DMEM with or without FBS (0%; 10% FBS) for 19F–PLGA–NaC (C) and 19F–PLGA–PVA–CS (D) NPs. (E and F) 19F-MRI phantoms of cells labeled with 19F–PLGA–NaC (E) or 19F–PLGA–PVA–CS (F) with or without FBS (0% or 10%, 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per sample, ref. = 5 × 1019 fluorine atoms). All reported data are mean and standard error from 6 to 10 samples related to 3 to 5 independent experiments (n = 3 for 0% FBS). 000 cells per sample, ref. = 5 × 1019 fluorine atoms). All reported data are mean and standard error from 6 to 10 samples related to 3 to 5 independent experiments (n = 3 for 0% FBS). | ||
Interestingly, the PC seems to enhance NP uptake by cells that consequently increase the 19F-MRI signal and thus improve the detection sensitivity (Fig. 3C–F). Such a role of the PC in the cellular internalization process was supported by the lower uptake of NPs in the absence of serum, which resulted in a strong reduction of the 19F-MRI signal. Thus, the improved efficiency to label cells shown by NaC and PVA–CS coated NPs seems not only due to higher 19F concentrations in single NPs, but in particular due to the bio–nano interface formed once NPs were in contact with the biological environment. Importantly, NaC coated NPs also showed a significant increase in size upon incubation with serum proteins, likely affecting the cellular NP internalization pathways.
Of note, 19F-uptake was more variable at longer incubation times probably in relation to the active proliferation of BV2 cells. To further investigate this aspect, signal stability of BV2 cells was also monitored by 19F-MRI from 24 to 72 h post labelling with 19F–PLGA–NaC NPs. While cells rapidly proliferated as expected maintaining a good viability (Fig. 4A and B), 19F signal remained stable over time although the collected cells were washed before imaging (Fig. 4C). Internalized NPs should be redistributed among dividing cells, as shown by the dilution of 19F signal per cell (Fig. 4D). Cell culture supernatants failed to reveal significant 19F signal (<1% of the total uptaken 19F content, Fig. S4†) supporting redistribution of the internalized PERFECTA among dividing cells rather than release.
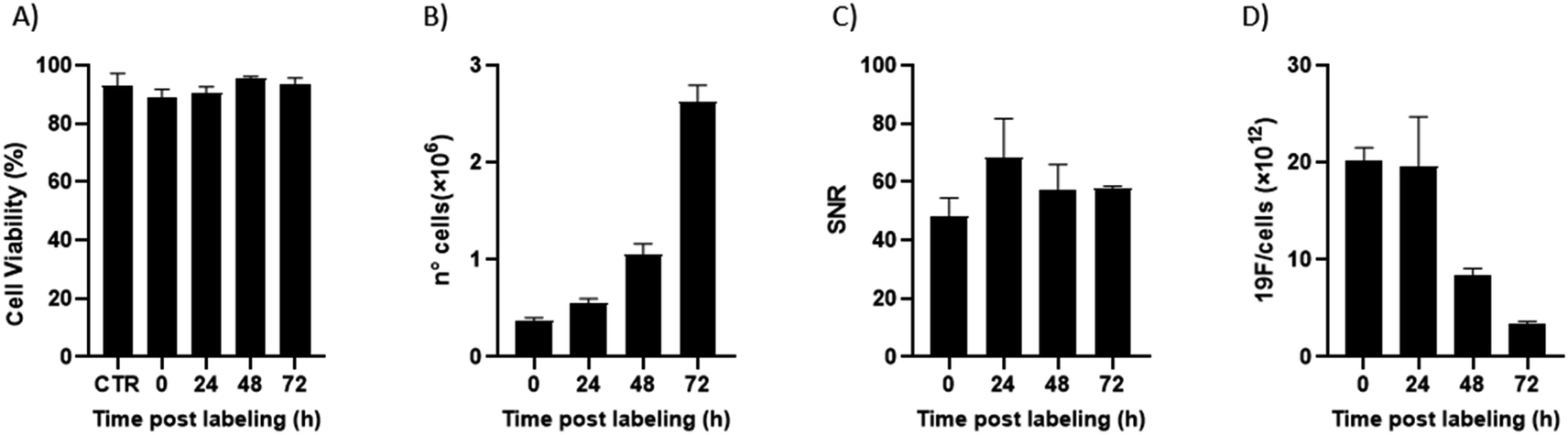 | ||
| Fig. 4 Labelling stability during BV2 cell proliferation. Cell viability (A) and proliferation (B) of BV2 cells over time post labelling with 19F–PLGA–NaC NPs. (C) Signal to noise ratio (SNR) related to the 19F-signal from cell samples collected at each time post labelling. (D) 19F content per cell decreased over time due to cell proliferation. All reported data are related to two independent experiments. | ||
Cellular internalization mechanism studies
It is known that the active endocytic mechanism is a common process for NP uptake by cells,51 but different pathways could be involved as a function of NP size and surface properties. Considering the size range of the 19F–PLGA-NPs (100–200 nm), phagocytosis and micropinocytosis pathways should be predominant.51,52 Using specific pharmacological inhibitors, we selectively evaluated the four main internalization pathways of phagocytic cells:51–54 phagocytosis, macropinocytosis, clathrin-mediated micropinocytosis and caveolin-mediated micropinocytosis (details are reported in Table S2†). Optimal doses were chosen based on significant inhibition with a limited effect on cell viability (Fig. S5†). Inhibition assays were carried out on the most efficient formulations for cell labelling, namely 19F–PLGA NPs formed with NaC or PVA–CS (Fig. 3B), and at short incubation time (4 h) to limit cell stress due to inhibitors which might alter the cell uptake mechanism. We used flow cytometry analysis to simultaneously assess cell viability (Fig. 5A and C) and NP uptake (Fig. 5B and D, reported singularly in Fig. S6†). To this end, fluorescent 19F–PLGA NPs were prepared by including a small amount of poly(lactide-co-glycolide)–rhodamine B in the formulation. While no effects were detected with the macropinocytosis inhibitor, those inhibiting phagocytosis and clathrin/caveolin-mediated uptakes significantly limited NP internalization (Fig. 5B and D). In the case of 19F–PLGA–PVA–CS NPs, clathrin/caveolin mediated processes were equally involved with an inhibition of about 32% compared to untreated cells, and almost 25% for the phagocytic pathway. For 19F–PLGA–NaC, phagocytosis was the predominant activated pathway, with a decrease of 61% in NP uptake followed by clathrin- and caveolin-mediated with 42 and 32% reduction, respectively. These differences in the uptake mechanisms could be expected considering that the size and composition of the protein corona were dependent on the pristine electrokinetic potential of the NPs.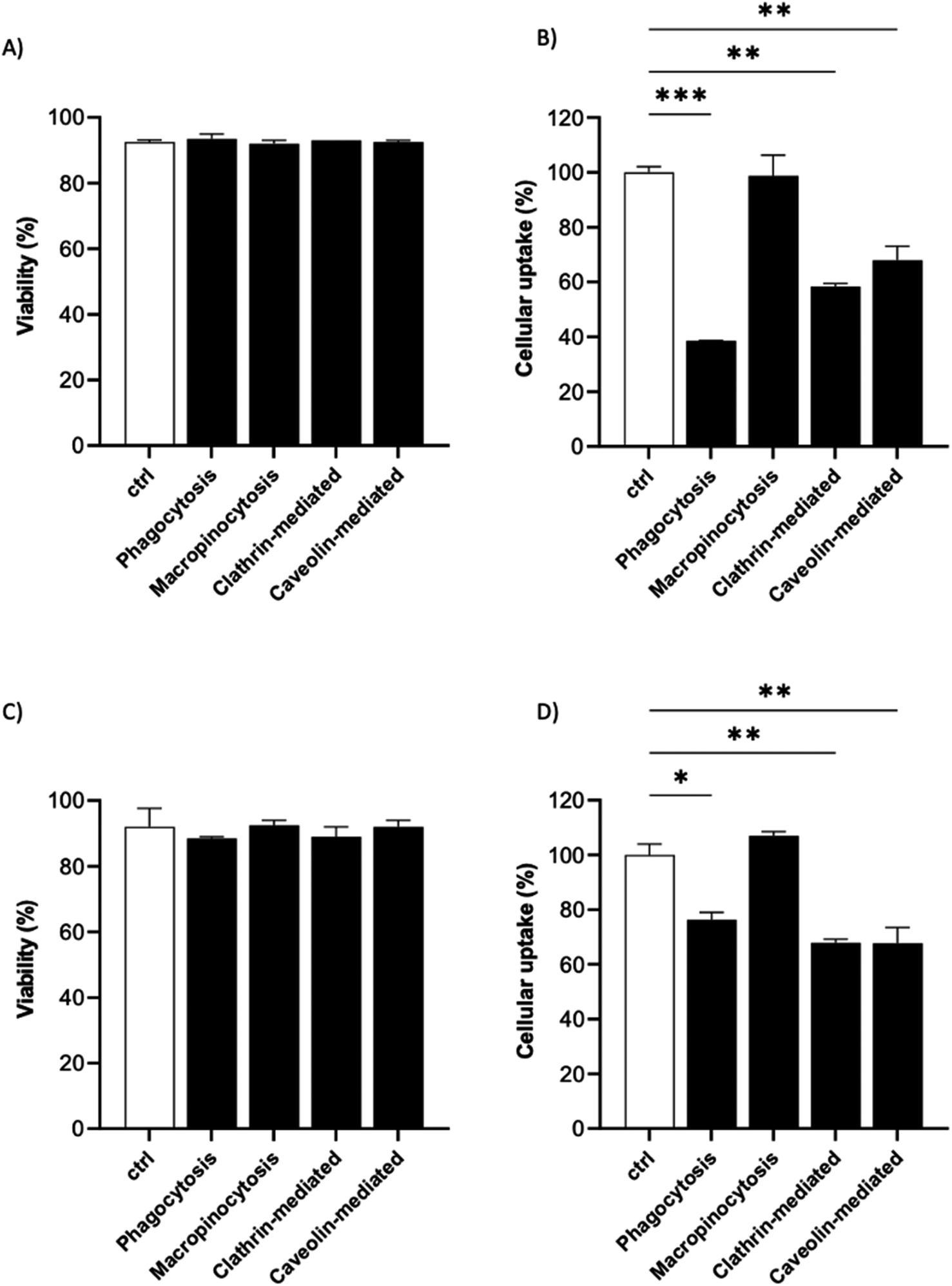 | ||
| Fig. 5 Preferential endocytic pathways of 19F–PLGA-NPs. The cell viability and uptake of 19F–PLGA–NaC (A and B) and 19F–PLGA–PVA–CS (C and D) NPs in BV2 cells were evaluated in the presence of pharmacological inhibitors of specific endocytic route and compared to untreated labeled cells (ctrl). Data were obtained by flow cytometry in independent experiments and expressed as follows: *p < 0.05; **p < 0.01; ***p < 0.001 (one way ANOVA test compared to the ctrl). | ||
In fact, with increased size and rich PC, it was expected that phagocytosis would have been the main mechanism in the case of 19F–PLGA–NaC NPs.
Our results are in line with previous reports highlighting that perfluorocarbon loaded polymeric NPs with more hydrophilic domains are mainly internalized by clathrin-mediated endocytosis, while more hydrophobic NPs rely on different mechanisms.53 Considering the overall results obtained, we suggest that these differences are mainly related to the interactions of the NPs with the biological media (formation of PC-NPs).
Conclusions
Overall, our data indicate that a careful design of 19F-nanoprobes together with a comprehensive analysis of their properties within biological fluids is needed to exploit 19F-MRI as a valuable imaging method for cell tracking. Here we reported the development of biocompatible 19F–PLGA-NPs containing PERFECTA and stabilized with different coating agents in order to modulate their surface properties and interaction with the biological environment and promote their cellular uptake. All the NPs were characterized by high 19F loading and excellent 19F-imaging properties, but they showed distinctive affinities to serum proteins with formation of PC NPs of diverse size and composition as a function of their surface properties. In this sense, we demonstrated that the PC composition was pivotal to promote NP uptake by phagocytic cells as microglia and maximize 19F-MRI response. Surface charged NPs, NaC and CS stabilized, showed a more significant interaction with serum proteins with consequently higher cell internalization and 19F-MRI signals. The bio–nano interface should then be considered an essential element in the design of imaging nanoprobes for cell labelling, especially in the case of non-phagocytic cells. For example, in the challenging cases of labelling T-cells and stem cells, the modulation of the PC can be of major importance since NP internalization is limited due to the restricted intracellular space and poor phagocytic activity. In fact, while the formation of a PC is often avoided for in vivo administration of drug delivery agents (to avoid sequestration of the NPs by the RES and immune response activation), we strongly advocate its exploitation for ex vivo cell labelling, as it might be key for increasing 19F sensitivity and consequently in vivo cell tracing by 19F-MRI tracking. In this sense, the labelling procedure of therapeutic cells for clinical applications should be performed with human serum, with the possibility to use patient-derived serum. This approach would allow the formation of a “personalized protein corona”, as it has been recently proposed for the use of different nanomedicines with diagnostic and therapeutic purposes.55–57Experimental
Materials
PERFECTA was synthesized as previously described.58 Poly(lactic-co-glycolic acid) (PLGA; Resomer 502H, 50:50, 7–17 kDa), polyvinyl alcohol (PVA; 30–70 kDa, 87–90% hydrolyzed), sodium cholate (NaC), and ethyl acetate (EtOAc, purity ≥ 99.5%) were supplied by Sigma-Aldrich (Germany). Water ultrapure Type-I Milli-Q water (mQw) (18.2 mΩ cm−1) was obtained using a Simplicity® Water Purification System (MERCK, Germany). Chitosan (CS, 100–300 kDa; 85% deacetylated) was purchased from Fisher Scientific.
For cellular labelling, the immortalized murine microglial cell line (BV2) was kindly provided by Dr R. Furlan (Clinical Neuroimmunology Unit, IRCCS Ospedale San Raffaele). Cell culture medium DMEM (Lonza Bioscience, Italy) was supplemented with 10% fetal bovine serum (FBS, Lonza Bioscience, Italy), 100 mg ml−1 of streptomycin, 100 U ml−1 of penicillin and 2 mM of L-glutamine (Gibco-Invitrogen). Trypsin (Invitrogen) allowed the dissociation of adherent cells from culture dishes and their collection. Trypan blue exclusion assay (0.4% solution, Sigma-Aldrich, Germany) was used to evaluate cell viability. Paraformaldehyde (PFA, 4% v/v in water, Sigma-Aldrich, Germany) was used to fix cells before MRI analysis. For the inhibition of cellular uptake, phenylarsine oxide (PAO), filipin III, amiloride hydrochloride, chlorpromazine hydrochloride, Bodipy–LacCER and transferrin-Alexa Fluor 488 were supplied by Invitrogen and Thermo Fisher, while dextran–rhodamine B (44 kDa or 155 kDa) was provided by Sigma-Aldrich.
For flow cytometry (FCM) analysis a fluorescent version of the polymer was used. In detail, PLGA–rhodamine B (50:50, 10–30 kDa; Sigma-Aldrich) was used in the NP formulation at 2% w/w with respect to unlabeled PLGA. Finally, the impact of NPs on cell viability was evaluated by 7-aminoactinomycin D dye as a live–dead marker (7-AAD; Invitrogen).
Preparation of 19F–PLGA NPs
19F–PLGA NPs were formulated using an optimized protocol previously reported.15 Briefly, PERFECTA was firstly dissolved in 1.5 ml of EtOAc and then heated at 65 °C. PLGA was dissolved in the same organic solvent (1.5 ml) and heated at 65 °C. The two solutions were combined and the resulting organic solution was rapidly mixed and added dropwise to the aqueous solution, containing either 0.6% w/v NaC or 2% w/v PVA, depending on the formulation type. For the NPs coated with CS, PVA (2% w/v) was added to an aqueous solution where CS was previously dissolved (CS/PLGA w/w = 0.8, see the ESI, Section 1.2†). The solution was then sonicated using a tip sonicator at 60 W for 25 s. Samples were left stirring for 3 h and the excess of organic solvent was then removed by rotary evaporation. NPs were collected and purified by centrifugation (10733g × 14 min for 19F–PLGA–PVA–CS NPs and 10733g × 40 min for both 19F–PLGA–PVA and 19F–PLGA–NaC NPs). The final product was then resuspended in mQw.
Characterization of 19F–PLGA nanoparticles
NP colloidal stability and morphology were evaluated by multi-angle Dynamic Light Scattering (DLS, ALV/CGS-3 system, ALV-Laser Vertriebsgesellschaft.m.b.H., Germany). Hydrodynamic diameter, size distribution and polydispersity (PDI) were determined by a cumulant fitting of the auto-correlation function obtained through DLS measurements. For these analyses, the stock solution was diluted 1:100 (v/v) in water and measured at 25 °C. The colloidal stability of the NPs in a biological environment was assessed diluting each formulation 1:6 (v/v) in water, DMEM and DMEM + 10% FBS. Samples were incubated at 37 °C and measured at t0, 30 min, 1, 2, 4, 6, 24 and 48 h at a dilution 1:25 (v/v) in water through DLS measurements. The concentration of NPs was estimated using the Nanoparticle Tracking Analysis (NTA, Nanosight NS3000, Malvern Panalytical, Malvern, England), the measurements were done at a dilution of 1:5000 for 19F–PLGA–NaC and 1:10000 for 19F–PLGA–PVA and 19F–PLGA–PVA–CS (v/v) in water. The electrokinetic potential was obtained through ζ-potential measurements (Zetasizer Nano ZS, Malvern Panalytical, Malvern, England) at a dilution of 1:5 (v/v) in water. Encapsulation efficiency of PERFECTA was determined from the quantification of 19F atoms through 19F-NMR experiments (Bruker AV400, Biospec; Bruker Biospin, Germany) with an external reference (TFA, trifluoroacetic acid). 19F-MRI experiments were also performed to characterize and compare the MRI signal properties of each 19F–PLGA-NP.
Cell labelling assays
Cellular uptake of 19F–PLGA NPs were investigated using the immortalized murine microglial cell line (BV2). Cells were seeded at a density of 3.3 × 105 cells per ml in a 6-well plate and incubated with 1.54 mM of PERFECTA (corresponding to 1 × 1014 19F per cell) at 37 °C and 5% CO2 for 4, 6 and 8 h. Cells were then washed 3–5 times with supplemented culture medium in the well until complete removal of free NPs as assessed by light field microscopy. Labeled cells were then collected and washed in abundant culture medium (2 × 300g for 10 min). In addition, for determining the potential contribution due to the presence of free NPs that persist after repeated washing steps, cells incubated with NPs for few seconds (t0) were also included. As blank control, cells were treated with an equal volume of mQw and underwent the same experimental procedures. After labelling, cell viability was evaluated through trypan blue exclusion test. BV2 cells were washed in PBS and subsequently fixed with 4% PFA for 20 min and stored at 4 °C until MRI acquisitions.For FBS-free labelling tests, BV2 were treated as previously described and the culture medium was replaced with FBS-free DMEM before incubation with NPs.
Labelling stability
To test the labelling stability upon incubation with 19F–PLGA NPs (6 h with 1 × 1014 19F per cell), BV2 cells were washed to remove the not internalized NPs and then were kept in culture. Cells were then collected at t0, 24, 48 and 72 h. For each time point, cells were washed in abundant culture medium (2 × 300g for 10 min), and PBS (300g × 10 min), counted and fixed with 4% PFA. The collected cells were analyzed by 19F-MRI. The cell culture supernatant was recovered and the 19F content was evaluated by 19F NMR, except for t0.Cell labelling inhibition assays
To study the endocytic pathways, phenylarsine oxide (PAO), amiloride chloride, chlorpromazine hydrochloride (CPZ) and filipin III were used to assess phagocytosis, macropinocytosis, and caveolin-mediated and clathrin-mediated pathways, respectively (see Table S2†). Each inhibitor was initially tested to determine the optimal dose using selective commonly used substrates, namely dextran–rhodamine B (44 kDa or 155 kDa) for phagocytosis and macropinocytosis, Bodipy–LacCER for caveolin-mediated and transferrin-Alexa Fluor 488 for clathrin-mediated pathways. The inhibitory effect was assessed by flow cytometry as a decreasing fold in the mean fluorescence intensity (MFI) value compared to the uninhibited condition.BV2 cells were seeded at a density of 3.3 × 105 cells per ml in a 6-well plate and pre-incubated for 30 minutes with 0.0075, 400, 7.5 and 14 μM of PAO, amiloride hydrochloride, filipin III and chlorpromazine hydrochloride, respectively. Fluorescent 19F–PLGA NPs were added and incubated using a PERFECTA concentration equal to 0.84 mM for 4 h. Cells were then washed until the complete removal of free NPs, counted and analyzed by flow cytometry.
19F-magnetic resonance imaging
The relaxometric properties (T1 and T2) of 19F–PLGA NPs were assessed by MRI. In detail, 19F-MRI analysis was performed on all formulations at four different concentrations ranging from 6 to 12 × 1018 19F atoms in 100 μl of mQw. Each sample was loaded in 0.2 ml Eppendorf and placed into a phantom containing 2% agar gel.The quantification of 19F content in labeled cells was performed on pellets of cells, collected by centrifugation and placed in 0.2 ml tubes. The total amount of 19F atoms was estimated using standards of PERFECTA formulations as reference (4–10 ×1018 19F).
All experiments were performed on a 7T scanner (Biospec; Bruker-Biospin) using a dual-transmit receive 19F/1H volume coil (35 × 59 mm) equipped with a horizontal bore magnet (ultra-shielded). 19F-MR images were acquired at PERFECTA specific resonance frequency (−73.4 ppm). For all MRI acquisitions, a 3D turbo-spin echo sequence was used with the same field of view (45 × 30 × 24 mm) for 1H-MRI (TR/TE = 250/15 ms, matrix = 128 × 64 × 8, 6 averages) and for 19F-MRI (TR/TE = 1500/40 ms, 64 × 32 × 8, 32 averages). The relaxation times of 19F–PLGA NPs were assessed using a RARE sequence with multiple echo times (10TE from 16 to 304 ms) and multiple repetition times (6TR from 350 to 1500 ms). For each sample, T1 and T2 were extrapolated from the fitting of the signal intensity as a function of TE or TR, respectively, using the scanner software tools (ISA tools Bruker Paravision 6). For each sample, the 19F signal to noise ratio (SNR) was calculated by measuring the signal in each sample using the image analysis tool of the scanner (Paravision 6.0, Bruker-Biospin) and divided by the standard deviation of the noise (image area outside of the sample).
Protein corona characterization
The protein binding on the NP surface was investigated through the incubation of NPs in DMEM supplemented by 10% FBS (Biowest; South America) for different times from 5 minutes to 8 hours at 37 °C and 95% of humidity. NPs were diluted 1:6 v/v in order to mimic the experimental conditions of a cell labelling experiment. After incubation, NP–protein complexes were isolated by sedimentation through a sucrose cushion (CARLO ERBA Reagents; Italy) adapting a protocol that was already investigated.59 In detail, NP–protein samples were loaded over the sucrose cushion (0.7 M). The mixture was then centrifuged through the cushion in order to isolate NP–protein complexes from unbound proteins (10733 × g, 4 °C for 30 minutes). Isolation was followed by three washes in mQw water and re-suspensions in 50 μl of mQw. The last supernatants and protein–NP pellets were analyzed by DLS (at 90°) to check the successful isolation and purification.
SDS–PAGE electrophoresis
A qualitative determination of hard-corona (HC) patterns was investigated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) analysis. All reagents were purchased from Bio-Rad, Sigma-Aldrich. In detail, 5 μl of SDS–PAGE loading buffer (Bio-Rad, 65.8 mM Tris–HCl, pH 6.8, 2.1% SDS, 26.3% (w/v) glycerol, 0.01% bromophenol blue), supplemented with β-mercaptoethanol (1:20) was added to 15 μl of each sample and kept at 99 °C for 5 minutes for digestion. Digested HC samples and control (ctrl) were loaded in the wells of SDS–PAGE gels. Ctrl consisted of a sample of FBS that underwent the same purification protocol. The gel was composed of a 4% polyacrylamide stacking gel (125 mM Tris–HCl, pH 6.8, 0.1%, m/v, SDS) and a 12% resolving polyacrylamide gel (in 375 mM Tris–HCl, pH 8.8, 0.1%, m/v, SDS buffer). A Tris–glycine buffer at pH 8.3 (with 0.1% SDS, m/v) was used as the cathode, whereas a Tris buffer at pH 8.8 represented the anode. Electrophoresis was performed in three steps with increasing voltage: the first step was set at 50 V for 20 min, second step at 100 V for 40 min and third step at 150 V until the dye front reached the bottom of the gel. Staining and distaining were performed with Colloidal Coomassie Blue (Serva) and 7% (v/v) acetic acid in water, respectively. Finally, the SDS–PAGE gels were scanned with a VersaDoc imaging system (Bio-Rad).
High-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) supplemented by energy-dispersive X-ray (EDS) analysis
Scanning transmission imaging was performed using a STEM-HAADF and EDS in an FEI aberration corrected scanning transmission electron microscopy (Cs-corrected STEM) TITAN electron microscope operating at 300 kV. Samples for electron microscopy-analysis were prepared by drop casting 5 μl concentrated particle solution onto a carbon coated TEM-grid and allowed to dry at room temperature overnight. Nanoparticle size distribution was determined with ImageJ software.NMR experiments
For each formulation, 19F-loading was quantified through 19F-NMR experiments with an external standard, more precisely a sealed capillary containing 100 μl of 0.35 M trifluoroacetic acid (TFA) in D2O. All NMR spectra acquisitions were performed on a Bruker Avance III HD 400 spectrometer (Bruker BioSpin, Rheinstetten, Germany) using a Bruker Automatic Sample Changer (SampleXpress). 19F-NMR spectra were recorded in the range of −65 to −85 ppm compared to TFA (−75 ppm). The NMR probe was maintained at 300 K for the entire duration of the experiment. A delay time between repetitions of 14.5 s was adopted to ensure full relaxation and 256 scans were collected. All 19F-NMR spectra were automatically phased and baseline corrected for accurate quantitative analysis through MestReNova software. Each set of NPs was analyzed by 19F-NMR for fluorine quantification. In detail, 19F-NMR spectra were performed on NP stocks using TFA reference as previously described. From the ratio of integrals of the peaks, the fluorine content was calculated.Flow cytometry analysis
19F–PLGA NPs were formulated with 2% of PLGA–rhodamine B to make NPs detectable by flow cytometry. For inhibition tests, cells were stained with the 7-AAD dye to assess their viability. The inhibition of NP uptake was evaluated as decreasing fold of the mean fluorescence intensity (MFI) compared to the uninhibited conditions.All flow cytometry analyses were performed using a FACS Canto II (BD Biosciences) with the following laser configuration: 488 nm laser and 585/42 filter (detecting rhodamine B), 488 nm laser and 530/30 filter (detecting FITC and Alexa Fluor 488) and 488 nm laser and LP670 filter (detecting 7-AAD). Post-processing analyses were performed using the FlowJo software (BD Biosciences).
Statistical analysis
All measurements reported in the present work are given as the average (±standard deviation) of at least n = 3 replicates for each condition. Cellular experiments were run at least twice, and each assay contained two replicates (n = 4 measures/condition). Data were plotted in Prism Software (GraphPad Software 9) and statistical significance was calculated by one-way ANOVA followed by Dunnett post hoc test. Statistical significance was considered for p < 0.05.Author contributions
L. C. and F. B. B. designed and supervised the research work. L. G., C. C., L. C. and F. B. B. designed the experimental work. L. G., C. C., M. S. A., V. S. and P. M. performed the experimental work. G. R. supervised flow cytometry acquisitions and helped to analyse the obtained results. L. G., C. C., L. C. and F. B. B. wrote the manuscript with the support of all other authors. All authors discussed the results and contributed to the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. B. B. and C. C. acknowledge financial support from the P2RY12 project, ID: GR-2016-02361325 (funded by the Italian Ministry of Health). F. B. B. and P. M. are thankful to the project NiFTy funded by MUR (PRIN2017, no. 2017MYBTXC) and to the NEWMED project, ID: 1175999 (funded by Regione Lombardia POR FESR 2014 2020). The authors acknowledge the Experimental Imaging Center at IRCCS Ospedale San Raffaele, part of the Italian Molecular Imaging node of Euro-BioImaging (https://www.eurobioimaging.eu), for providing access to advanced imaging technologies and services. L. C. and C. C. acknowledge the support of FISM (Fondazione Italiana Sclerosi Multipla onlus) grant no. 2016/R/8. A. M. and G. R. acknowledge the support of Associazione Italiana sulla Ricerca sul Cancro (AIRC: IG 2018, ID. 21763). M. S.-A. thanks the Spanish Government for an FPU PhD Research fellowship and AIRC Foundation for cancer research for a postdoctoral fellowship in Italy. CIBER-BBN is an initiative funded by the VI National R&D&i Plan 2008–2011 financed by the Instituto de Salud Carlos III with the assistance of the European Regional Development Fund.References
- I. J. M. D. Vries, W. J. Lesterhuis and J. O. Barentsz, et al., Magnetic resonance tracking of dendritic cells in melanoma patients for monitoring of cellular therapy, Nat. Biotechnol., 2005, 23(11), 1407–1413, DOI:10.1038/nbt1154.
- J. W. M. Bulte, In Vivo MRI Cell Tracking: Clinical Studies, Am. J. Roentgenol., 2009, 193(August), 314–325, DOI:10.2214/AJR.09.3107.
- E. T. Ahrens and J. W. M. Bulte, Tracking immune cells in vivo using magnetic resonance imaging, Nat. Rev. Immunol., 2013, 13(10), 755–763, DOI:10.1038/nri3531.
- M. Srinivas, A. Heerschap, E. T. Ahrens, C. G. Figdor and I. J. M. de Vries, 19F MRI for quantitative in vivo cell tracking, Trends Biotechnol., 2010, 28(7), 363–370, DOI:10.1016/j.tibtech.2010.04.002.
- P. Boehm-Sturm, L. Mengler, S. Wecker, M. Hoehn and T. Kallur, In Vivo tracking of human neural stem cells with 19F magnetic resonance imaging, PLoS One, 2011, 6(12), e29040, DOI:10.1371/journal.pone.0029040.
- J. M. Gaudet, E. J. Ribot, C. Yuhua, K. M. Gilbert and P. J. Foster, Tracking the Fate of Stem Cell Implants with Fluorine-19 MRI, PLoS One, 2015, 1–11, DOI:10.1371/journal.pone.0118544 . Published online..
- S. S. Moonshi, C. Zhang and H. Peng, et al., A unique 19F MRI agent for the tracking of non phagocytic cells: in vivo, Nanoscale, 2018, 10(17), 8226–8239, 10.1039/c8nr00703a.
- X. Tang, L. Ao and C. Zu, et al., Water-Soluble Chemically Precise Fluorinated Molecular Clusters for Interference-Free Multiplex 19F MRI in Living Mice, ACS Nano, 2023, 17(5), 5014–5024 CrossRef CAS PubMed.
- J. Ruiz-Cabello, B. P. Barnett, P. A. Bottomley and J. W. M. Bulte, Fluorine (19F) MRS and MRI in biomedicine, NMR Biomed., 2010, 24(2), 114–129 CrossRef PubMed.
- M. Srinivas, P. Boehm-Sturm, C. G. Figdor, I. J. de Vries and M. Hoehn, Labeling cells for in vivo tracking using 19F MRI, Biomaterials, 2012, 33(34), 8830–8840, DOI:10.1016/j.biomaterials.2012.08.048.
- S. Waiczies, J. M. Mil and L. Starke, et al., Enhanced Fluorine-19 MRI Sensitivity using a Cryogenic Radiofrequency Probe: Technical Developments and Ex Vivo Demonstration in a Mouse Model of Neuroinflammation, Sci. Rep., 2017, 7, 9808, DOI:10.1038/s41598-017-09622-2.
- I. Tirotta, V. Dichiarante and C. Pigliacelli, et al., 19 F Magnetic Resonance Imaging (MRI): From Design of Materials to Clinical Applications, Chem. Rev., 2015, 115(2), 1106–1129, DOI:10.1021/cr500286d.
- C. Chirizzi, D. De Battista and I. Tirotta, et al., Multispectral MRI with dual fluorinated probes to track mononuclear cell activity in mice, Radiology, 2019, 291(2), 351–357, DOI:10.1148/radiol.2019181073.
- C. Chirizzi, C. Morasso and A. A. Caldarone, et al., A Bioorthogonal Probe for Multiscale Imaging by 19F-MRI and Raman Microscopy: From Whole Body to Single Cells, J. Am. Chem. Soc., 2021, 143(31), 12253–12260, DOI:10.1021/jacs.1c05250.
- C. Chirizzi, L. Gatti and M. Sancho-albero, et al., Colloids and Surfaces B: Biointerfaces Optimization of superfluorinated PLGA nanoparticles for enhanced cell labelling and detection by 19 F-MRI, Colloids Surf., B, 2022, 220(July), 112932, DOI:10.1016/j.colsurfb.2022.112932.
- D. Dutta and J. G. Donaldson, Search for inhibitors of endocytosis: Intended specificity and unintended consequences, Cellular Logistics, 2012, 2(4), 203–208, DOI:10.4161/cl.23967.
- I. Canton and G. Battaglia, Endocytosis at the nanoscale, Chem. Soc. Rev., 2012, 41(7), 2718–2739, 10.1039/C2CS15309B.
- M. de Almeida, E. Susnik, B. Drasler, P. Taladriz-Blanco, A. Petri-Fink and B. Rothen-Rutishauser, Understanding nanoparticle endocytosis to improve targeting strategies in nanomedicine, Chem. Soc. Rev., 2021, 50(9), 5397–5434, 10.1039/D0CS01127D.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Determining the Size and Shape Dependence of Gold Nanoparticle Uptake into Mammalian Cells, Nano Lett., 2006, 6(4), 662–668, DOI:10.1021/nl052396o.
- R. R. Arvizo, O. R. Miranda and M. A. Thompson, et al., Effect of Nanoparticle Surface Charge at the Plasma Membrane and Beyond, Nano Lett., 2010, 10(7), 2543–2548, DOI:10.1021/nl101140t.
- C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles, Biomaterials, 2010, 31(13), 3657–3666, DOI:10.1016/j.biomaterials.2010.01.065.
- D. Montizaan, K. Yang, C. Reker-smit and A. Salvati, Comparison of the uptake mechanisms of zwitterionic and negatively charged liposomes by HeLa cells, Nanomedicine, 2020, 30, 102300, DOI:10.1016/j.nano.2020.102300.
- D. Nierenberg, A. R. Khaled and O. Flores, Formation of a protein corona influences the biological identity of nanomaterials, Rep. Pract. Oncol. Radiother., 2018, 23(4), 300–308, DOI:10.1016/j.rpor.2018.05.005.
- M. P. Monopoli, C. Åberg, A. Salvati and K. A. Dawson, Biomolecular coronas provide the biological identity of nanosized materials, Nat. Nanotechnol., 2012, 7(December), 779–786, DOI:10.1038/nnano.2012.207.
- K. Yang, B. Mesquita, P. Horvatovich and A. Salvati, Tuning liposome composition to modulate corona formation in human serum and cellular uptake, Acta Biomater., 2020, 106, 314–327, DOI:10.1016/j.actbio.2020.02.018.
- A. Aliyandi, C. Reker-smit, R. Bron, I. S. Zuhorn and A. Salvati, Correlating Corona Composition and Cell Uptake to Identify Proteins Affecting Nanoparticle Entry into Endothelial Cells, ACS Biomater. Sci. Eng., 2021, 7(12), 5573–5584, DOI:10.1021/acsbiomaterials.1c00804.
- S. M. Pustulka, K. Ling, S. L. Pish and J. A. Champion, Protein Nanoparticle Charge and Hydrophobicity Govern Protein Corona and Macrophage Uptake, ACS Appl. Mater. Interfaces, 2020, 12(43), 48284–48295, DOI:10.1021/acsami.0c12341.
- M. P. Monopoli, F. B. Bombelli and K. A. Dawson, Nanoparticle coronas take shape, Nat. Nanotechnol., 2011, 6, 11–12, DOI:10.1038/nnano.2011.267.
- D. Walczyk, F. B. Bombelli, M. P. Monopoli, I. Lynch and K. A. Dawson, What the Cell “Sees” in Bionanoscience, J. Am. Chem. Soc., 2010, 132(16), 5761–5768, DOI:10.1021/ja910675v.
- H. Nejadnik, S.-M. Taghavi-Garmestani and S. J. Madsen, et al., The Protein Corona around Nanoparticles Facilitates Stem Cell Labeling for Clinical MR Imaging, Radiology, 2018, 286(3), 938–947, DOI:10.1148/radiol.2017170130.
- C. Zhang, K. Yan, C. Fu, H. Peng, C. J. Hawker and A. K. Whittaker, Biological Utility of Fluorinated Compounds: from Materials Design to Molecular Imaging, Therapeutics and Environmental Remediation, Chem. Rev., 2022, 167–208, DOI:10.1021/acs.chemrev.1c00632.
- A. Houshang, B. Lorenzo and L. Alessandro, et al., Protein corona affects the relaxivity and MRI contrast efficiency of magnetic nanoparticles, Nanoscale, 2013, 5, 8656–8665, 10.1039/c3nr00345k.
- B. Stansley, J. Post and K. Hensley, A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease, J. Neuroinflammation, 2012, 9, 115, DOI:10.1186/1742-2094-9-115.
- M. B. Graeber, W. Li and M. L. Rodriguez, Role of microglia in CNS inflammation, FEBS Lett., 2011, 585(23), 3798–3805, DOI:10.1016/j.febslet.2011.08.033.
- S. Saini, A.-S. Vanherwegen and S. Liang, et al., Fluorine MR Imaging Probes Dynamic Migratory Profiles of Perfluorocarbon-Loaded Dendritic Cells After Streptozotocin-Induced Inflammation, Mol. Imaging Biol., 2022, 24(2), 321–332, DOI:10.1007/s11307-021-01701-1.
- G. Neri, G. Mion and A. Pizzi, et al., Fluorinated PLGA Nanoparticles for Enhanced Drug Encapsulation and 19F NMR Detection, Chem.–Eur. J., 2020, 26(44), 10057–10063 CrossRef CAS PubMed.
- D. Croci, R. S. Méndez and S. Temme, et al., Multispectral fluorine-19 MRI enables longitudinal and noninvasive monitoring of tumor-associated macrophages, Sci. Transl. Med., 2022, 14, eabo2952, DOI:10.1126/scitranslmed.abo2952.
- C. Fink, J. M. Gaudet and M. S. Fox, et al., 19F-perfluorocarbon-labeled human peripheral blood mononuclear cells can be detected in vivo using clinical MRI parameters in a therapeutic cell setting, Sci. Rep., 2018,(September 2017), 1–13, DOI:10.1038/s41598-017-19031-0.
- E. Mahon, A. Salvati, F. Baldelli Bombelli, I. Lynch and K. A. Dawson, Designing the nanoparticle–biomolecule interface for “targeting and therapeutic delivery, J. Controlled Release, 2012, 161(2), 164–174, DOI:10.1016/j.jconrel.2012.04.009.
- S. M. A. Sadat, S. T. Jahan and A. Haddadi, Effects of Size and Surface Charge of Polymeric Nanoparticles on in Vitro and in Vivo Applications, J. Biomater. Nanobiotechnol., 2016, 7, 91–108, DOI:10.4236/jbnb.2016.72011.
- C. Foged, B. Brodin, S. Frokjaer and A. Sundblad, Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model, Int. J. Pharm., 2005, 298(2), 315–322, DOI:10.1016/J.IJPHARM.2005.03.035.
- Y. Phanse, B. R. Carrillo-Conde and A. E. Ramer-Tait, et al., Functionalization of polyanhydride microparticles with di-mannose influences uptake by and intracellular fate within dendritic cells, Acta Biomater., 2013, 9(11), 8902–8909, DOI:10.1016/J.ACTBIO.2013.06.024.
- E. Luque-michel, A. Larrea and C. Lahuerta, et al., A simple approach to obtain hybrid Au-loaded polymeric nanoparticles with a tunable metal load, Nanoscale, 2016, 8, 6495–6506, 10.1039/c5nr06850a.
- O. Esim, N. K. Bakirhan, M. Sarper, A. Savaser and S. A. Ozkan, Influence of emulsifiers on the formation and in vitro anticancer activity of epirubicin loaded PLGA nanoparticles, J. Drug Delivery Sci. Technol., 2020, 60(August), 102027, DOI:10.1016/j.jddst.2020.102027.
- N. Ghosh, R. Mondal and S. Mukherjee, Hydrophobicity Is the Governing Factor in the Interaction of Human Serum Albumin with Bile Salts, Langmuir, 2015, 31(3), 1095–1104, DOI:10.1021/la504270a.
- T. Cedervall, I. Lynch and M. Foy, et al., Detailed Identification of Plasma Proteins Adsorbed on Copolymer Nanoparticles, Angew. Chem., Int. Ed., 2007, 46(30), 5754–5756, DOI:10.1002/anie.200700465.
- W. Richtering, I. Alberg and R. Zentel, Nanoparticles in the Biological Context: Surface Morphology and Protein Corona Formation, Small, 2020, 16(39), 2002162, DOI:10.1002/smll.202002162.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(38), 14265–14270, DOI:10.1073/pnas.0805135105.
- D. Chen, S. Ganesh, W. Wang and M. Amiji, Plasma protein adsorption and biological identity of systemically administered nanoparticles, Nanomedicine, 2017, 12(17), 2113–2135, DOI:10.2217/nnm-2017-0178.
- L. Digiacomo, F. Giulimondi, M. Mahmoudi and G. Caracciolo, Effect of molecular crowding on the biological identity of liposomes: an overlooked factor at the bio-nano interface, Nanoscale Adv., 2019, 1(7), 2518–2522, 10.1039/C9NA00195F.
- J. D. Torres-Vanegas, J. C. Cruz and L. H. Reyes, Delivery systems for nucleic acids and proteins: barriers, cell capture pathways and nanocarriers, Pharmaceutics, 2021, 13(3), 1–38, DOI:10.3390/pharmaceutics13030428.
- I. L. Hsiao, Y. K. Hsieh, C. Y. Chuang, C. F. Wang and Y. J. Huang, Effects of silver nanoparticles on the interactions of neuron- and glia-like cells: toxicity, uptake mechanisms, and lysosomal tracking, Environ. Toxicol., 2017, 32(6), 1742–1753, DOI:10.1002/tox.22397.
- R. Day, D. Estabrook, C. Wu, J. Chapman, A. Togle and E. Sletten, Systematic study of perfluorocarbon nanoemulsions stabilized by polymer amphiphiles, ACS Appl. Mater. Interfaces, 2020, 12(35), 38887–38898, DOI:10.1021/acsami.0c07206.
- D. Fitzner, M. Schnaars and D. Van Rossum, et al., Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis, J. Cell Sci., 2011, 124(3), 447–458, DOI:10.1242/jcs.074088.
- J. Ren, R. Cai and J. Wang, et al., Precision Nanomedicine Development Based on Specific Opsonization of Human Cancer Patient-Personalized Protein Coronas, Nano Lett., 2019, 19(7), 4692–4701 CrossRef CAS PubMed.
- M. Mahmoudi, M. P. Landry, A. Moore and R. Coreas, The protein corona from nanomedicine to environmental science, Nat. Rev. Mater., 2023, 8, 422–438, DOI:10.1038/s41578-023-00552-2.
- Y. Ju, H. G. Kelly and L. F. Dagley, et al., Person-Specific Biomolecular Coronas Modulate Nanoparticle Interactions with Immune Cells in Human Blood, ACS Nano, 2020, 14(11), 15723–15737, DOI:10.1021/acsnano.0c06679.
- I. Tirotta, A. Mastropietro and C. Cordiglieri, et al., A superfluorinated molecular probe for highly sensitive in vivo 19F-MRI, J. Am. Chem. Soc., 2014, 136(24), 8524–8527, DOI:10.1021/ja503270n.
- D. Docter, U. Distler and W. Storck, et al., Quantitative profiling of the protein coronas that form around nanoparticles, Nat. Protoc., 2014, 9(9), 2030–2044, DOI:10.1038/nprot.2014.139.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00229b |
| ‡ These authors have equally contributed. |
| § Department of Experimental Oncology, IEO, European Institute of Oncology IRCCS, Via Adamello 16, Milano, 20139, Italy. |
| ¶ Philochem AG, Libernstrasse 3, Otelfingen, 8112, Switzerland. |
| || Department of Biochemistry and Molecular Pharmacology, Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Via Mario Negri 2, Milano, 20156, Italy. |
| ** Euro-BioImaging ERIC, Institute of Biostructures and Bioimaging (IBB), Italian National Research Council (CNR), Via Nizza 52, Torino, 10126, Italy. |
| This journal is © The Royal Society of Chemistry 2023 |