
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot green bio-assisted synthesis of highly active catalytic palladium nanoparticles in porcine gastric mucin for environmental applications†
Roman
Nudelman
a,
Shir
Zuarets
a,
Meiron
Lev
a,
Shira
Gavriely
ab,
Louisa
Meshi
c,
Ines
Zucker
 b and
Shachar
Richter
*a
b and
Shachar
Richter
*a
aDepartment of Materials Science and Engineering, University Center for Nanoscience and Nanotechnology, The Iby and Aladar Fleischman Faculty of Engineering, Tel Aviv University, 69978, Tel-Aviv, Israel. E-mail: srichter@tauex.tau.ac.il
bSchool of Mechanical Engineering, The Porter School of Environmental and Earth Sciences, The Iby and Aladar Fleischman Faculty of Engineering, Tel Aviv University, 69978, Tel-Aviv, Israel
cDepartment of Materials Engineering, Ben-Gurion University of the Negev, PO Box 653, Beer-Sheva 84105, Israel
First published on 7th September 2023
Abstract
In this work, palladium nanoparticles were synthesized using one-pot synthesis utilizing porcine gastric mucin glycoproteins as reducing and capping agents. It is shown that the particles exhibited noticeable catalytic activity through both nitrophenol reduction and Suzuki–Miyaura coupling reactions. The catalytic performance was demonstrated with exceptionally high product yield, a fast reaction rate, and low catalyst use. The palladium–mucin composites obtained could be used in particle solution and as hydrogel catalysts to increase their reusability for at least ten reaction cycles with minimum loss in their catalytic effectiveness.
Introduction
Transition metal nanoparticles (NPs) are of substantial interest in fundamental and practical science due to their unique properties, such as the quantum size effect and optical, electrical, magnetic, and catalytic activity.1 NPs made from platinum-group metals are the most studied and industrially used in numerous applications, including electronics, optics, biomedical, and catalysis2–5 Among platinum-group metals, palladium nanoparticles (PdNPs) are one of the most studied and application-oriented nanomaterials.6–8 These particles have shown excellent catalytic properties compared to other metal NPs and could also be used as an important component in bimetallic NP structures.9,10 PdNPs are mainly used as chemical catalysts in organic chemistry (in wet processes) and the automobile industry (in the solid–gas phase). In the latter, PdNPs are utilized in exhaust systems of billions of petrol-fuelled vehicles to oxidize hydrocarbons and carbon monoxide into less polluting carbon dioxide.11 Not surprisingly, this excessive use of PdNPs results in a considerable economic footprint. As of 2019, about 300 metric tons of palladium are used as catalysts which takes a toll on its price and availability due to the finite amount of palladium ore. Thus, enhancing palladium catalytic effectiveness and recyclability is necessary to minimize its consumption.8,12Common approaches for synthesizing PdNPs include chemical reduction13 and encapsulation,14 electrochemical deposition,15 microwave-assisted synthesis,16 and UV-assisted synthesis.17 Among all the methods, chemical reduction and encapsulation of PdNPs are considered relatively simple and reproducible, resulting in the homogeneous formation of PdNPs in terms of shape and size.18–21 This methodology utilizes reducing and capping agents to reduce Pd cations and stabilize the formed nanoparticles. The resulting PdNP structure and size can be manipulated by adjusting the reagents' pH, temperature, and concentration. However, this method is still limited from an applicative point of view due to the chemical stability of the particles.19–21 In addition, chemical capping and reduction agents often involve toxic and environmentally unfriendly materials, resulting in considerable amounts of toxic waste.
In this respect, green biosynthesis of PdNPs might help overcome these obstacles. The approach uses natural biomimetic synthetic strategies to produce various types of NPs in a green and easy fashion.22–25 In some cases, green biosynthesis allows biomolecules to function as reducing and capping agents, where the host biomolecule reduces that metal cation and naturally stabilizes the formed PdNPs. Thus, green biosynthesis usually adopts a one-pot strategy in which the metal precursor, biomolecule, and reaction medium are mixed in a single, straightforward synthesis step. For example, Anand et al.26 demonstrated the synthesis of spherical PdNPs from Moringa oleifera peel extract, and Sathishkumar and co-workers27 studied the formation of crystalline PdNPs from the bark of the Cinnamomum zeylanicum tree. Mikheenko et al.28 reported PdNP synthesis by sulfidogenic bacteria for organic catalytic reactions. Overall, PdNPs formed via green synthesis show excellent promise as a catalyst material29,30 and are considered less polluting as the reaction reagents and possible by-products are non-toxic and produce minimal waste.
Recently, proteins and biopolymers have also been recognized as beneficial biomatrix platforms for metal nanoparticle synthesis. Bachar et al.31 showed the synthesis of PdNPs in a stable protein 1 biomatrix. Scott and coworkers produced stable PdNPs using liposome enzymes extracted from bacteria, further showcasing their applicative use as catalysts for Suzuki and Heck reactions. However, current bio-derived green biosynthesis of PdNPs from protein and polymer substances often involves complicated and unsolved molecular structures and compositions, thus making it challenging to predict and control reaction products and optimize them. Therefore, the use of an appropriate bio-polymer with a fully characterized structure, high commercial availability, and good reducing properties is needed.
Recently, we identified mucin glycoproteins as a highly effective protein platform for metal nanoparticle synthesis, showing high reproducibility and control over produced nanoparticles. Examples include Au, Ag and even complex metal–insulator structures.32–38 Mucin glycoproteins belong to a large family of mucus proteins,39 a complex viscous adherent secretion synthesized by specialized goblet cells in the epithelium for shielding functions. These cell lines are present in organs exposed to the external environment, including the respiratory, gastrointestinal, and reproductive tracts.40 Mucins are of high molecular weight—ranging between 0.5 and 20 million Da with heavily glycosylated oligosaccharide chains.41 These chains are arranged in a “bottle brush” configuration around the protein core, which is arranged into distinct regions of tandem repeats that are rich in serine, threonine, and proline (STP repeats).42 Interspersed between the STP repeats are hydrophobic regions with an amino acid composition with a high proportion of cysteine (>10%). Cysteines are known to be strong nucleophiles and metal binders.35 Cysteine can, therefore, act as a reducing agent, while the hydrophobic pockets can act as natural capping agents.43–45
In this study, we utilize the extraordinary reducing properties of porcine gastric mucin (PGM), a commercially available mucin, to synthesize reproducible and highly effective PdNPs. We further show that these stable PdNPs can be used in a liquid environment or incorporated into a stable solid biodegradable hydrogel, allowing their reusability.46 To assess their catalytic activity, we performed two Pd-catalyzed reactions: reduction of nitrophenol47 for kinetic rate calculations and the Suzuki–Miyaura carbon bond coupling reaction48 to explore the reaction yield. Compared to other PdNP catalysts synthesized by other biological methods, our PdNPs exhibit higher catalytic efficiency in terms of reaction rate per weight of the used catalyst in a common nitrophenol reduction reaction. Moreover, the extraordinary efficiency demonstrated in the Suzuki–Miyaura carbon coupling suggests that this synthesis is appealing for organic chemistry uses.
Experimental
Materials
Porcine gastric mucin, PdCl2, phosphate buffered saline, p-nitrophenol, sodium boron hydride, iodobenzene, chloroform, phenylboronic acid, potassium carbonate, sodium hydroxide, Pluronic F-127, and 2-hydroxy-4′-(2-hydroxyethyl)-2-methylpropiophenone (Irgacure 2959) were used. All materials were purchased from Sigma-Aldrich.PdNP synthesis
Briefly, 50 mg of commercially available porcine gastric mucin was dissolved in 5 ml of deionized water for 60 min until an entirely homogeneous solution was obtained. Next, 2 ml of phosphate-buffered saline (PBS 1 M) was added to maintain the pH of the solution at pH 7–8. Finally, PdCl2 salt was added to the protein solution at concentrations of 0.0075 M, 0.011 M, 0.015 M, and 0.019 M and stirred until a homogeneous dispersion was formed. The resulting solutions were heated to 70 °C and stirred in the dark for 24 h until a dark brownish solution was observed. The solution was then divided into three fractions: (i) the supernatant (SUP) fraction, which contained PdNPs and remained in the solution after centrifugation; (ii) the whole (WHO) fraction that contains PdNP solution as is without any centrifugation. (iii) and a PdNP-containing precipitate (PREP), collected from centrifuged PdNP solution (5 min, 200 rpm). All the fractions were tested for catalytic activity assays and the preparation of hydrogel catalysts (Fig. 1).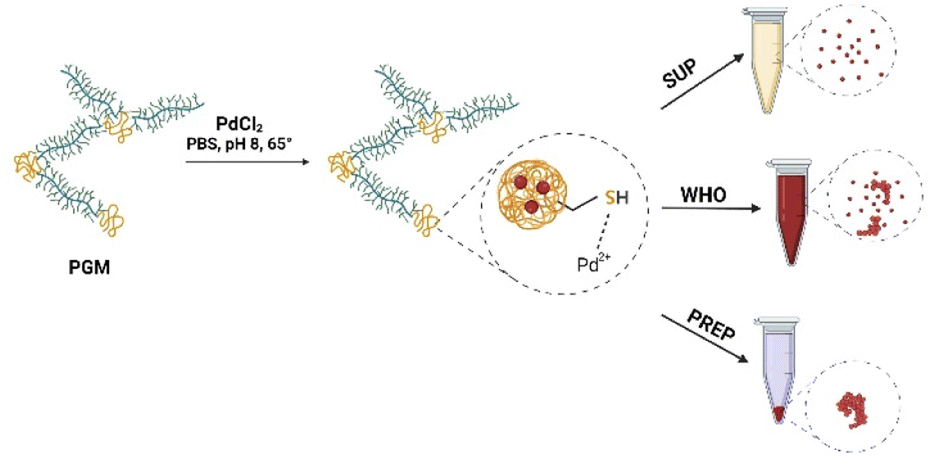 | ||
| Fig. 1 Synthesis process of PdNPs (see the text for details). | ||
Preparation of PGM–PdNP hydrogels
PGM–PdNP hydrogels were prepared by adding PGM–PdNP solution to a synthetic polymer solution of methacrylate Pluronic F-127 (Sigma-Aldrich) followed by UV-crosslinking.46 Shortly, Pluronic F-127 solution was prepared by dissolving 500 mg of Pluronic in 2 ml of 75/25 v%/v% deionized water/PBS buffer. Due to its temperature sensitivity, the polymer was dissolved at 0 °C. After full dissolution of the Pluronic compound, 200 μl of PdNP solution from WHO and SUP fractions and 10 μl of the PREP fraction were added separately to form three hydrogel solutions. To crosslink the formed hydrogel, 10 mg of Irgacure UV curing agent was added per 1 ml of hydrogel solution. Circular hydrogels of 100 mg weight and 5 mm in diameter were formed and crosslinked for 5 min under UV light (365 nm, 2 W) irradiation for further use in the nitrophenol reduction reaction.Characterization of PdNPs
Transmission electron microscopy (TEM, JEOL JEM-2010, 200 kV) and an environmental scanning electron microscope (ESEM, Thermo Fisher, Quanta 200 FEG ESEM 20 kV) were used to image and analyze the formed particles' crystalline structure and atomic composition. For TEM analysis, PdNP samples were drop-cast on a 200 mesh copper grid for 30 min, then the drop was removed, and the grid was dried under vacuum. The ESEM imaging samples were dried on carbon tape and transferred to the imaging stage. Additional elemental analysis was performed using an energy dispersive spectroscopy EDS detector (Thermo Fisher).To determine the Pd concentration in the formed particle solutions prepared from the initial concentration of 0.0075–0.019 M of PdCl2 precursor. The resulting solution was centrifuged for 5 min at 500 rpm. The supernatant and the mixed solution were diluted 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times with HNO3 3%. The resolved solution was then diluted 200 times in HCl 37%, followed by another dilution (×100) with HNO3 3%. Pd2+ concentrations were determined by inductively coupled plasma mass spectroscopy (ICP-MS, 7800, Agilent, US).
000 times with HNO3 3%. The resolved solution was then diluted 200 times in HCl 37%, followed by another dilution (×100) with HNO3 3%. Pd2+ concentrations were determined by inductively coupled plasma mass spectroscopy (ICP-MS, 7800, Agilent, US).
Assessment of catalytic activity of PdNPs
Calculations of nitrophenol reduction reaction rates of different fractions of the PdNP catalyst were based on a pseudo-first-order kinetic reaction model.28 In this case, the use of this model is acceptable due to the significant concentration of one of the reactants (sodium borohydride). The plot of ln(Ct/C0), where Ct is the concentration at time t and C0 is the initial concentration of p-NLP versus reaction time, allowed us to extract the constant reaction rate, k.
Suzuki–Miyaura coupling
The typical reaction of Suzuki–Miyaura coupling is reported elsewhere.48 In short, 60 mg of phenylboronic acid was mixed with 160 mg of potassium carbonate in a 5 ml 1:1 solution of deionized water and ethanol. Next, 100 μl of iodobenzene (Sigma-Aldrich) was added dropwise and vigorously stirred. Next, the PdNP catalyst was added, and the reaction solution was immediately heated to 65 °C. The reaction was stopped at different time intervals of 15, 20, 30, 45, and 60 min and cooled to room temperature to allow crystallization of the produced biphenyl.
Biphenyl product yield was calculated by comparing the resulting product from our reaction to the theoretical calculation of the 100% product from the molecular masses of the reactants. H-Nuclear magnetic resonance H-NMR (Bruker 400 MHz, a 5 mm probe) and infrared IR analysis (Tensor 27, Platinum ATR) of biphenyl samples were conducted. For IR analysis, samples were used as is without the addition of solvent. Samples for H-NMR were dissolved in chloroform.
Results and discussion
Particle formation and characterization
The prepared PdNPs were obtained by one-pot green synthesis, where PGM was utilized as a reducing and capping agent, while PBS buffer served as a neutral pH mediator. The reduction process took 24 hours, resulting in a brownish solution with a visible precipitate (Fig. 1 and S3†). Notably, the pH of the reaction plays a crucial role in determining the reaction products. It was previously shown that at neutral to slightly alkaline pH, PGM proteins tend to be in their dispersed form in which the disulfide bonds in the hydrophobic pockets on the protein core are stabilized. Thus, the reduction of Pd ions occurs mainly in the cysteine-rich hydrophobic moieties in which the latter serve as an electron transfer relay via a process mediated by surrounding water.32 In this process, cysteine binds palladium ions through its thiol group and then oxidizes due to the transfer of an electron to the positively charged palladium ion. The nearby water molecules then deprotonate to form an intermediate thiol side group, which tends to form a more stabilized di-sulphide bond ready to bind another palladium ion. The buffer solution further enhances the reaction as it stabilizes the water protonation and deprotonation processes during the reaction.To analyze the size distribution of the synthesized PdNPs, we performed an HR-TEM analysis as a function of the initial concentrations of PdCl2 and the obtained PdNP fractions. Fig. 2A–C show selected size distribution histograms obtained from 10 mg PdCl2. An average particle size of 2–4 nm was obtained for all fractions measured. This homogeneity is not surprising as the main factors controlling the particle size are the PGM protein concentration and solution pH, which remained the same in all fractions (7.15 mg ml−1 of PGM and pH = 8). The PdNP population in the PREP fraction showed a higher tendency to form large agglomerates (150–500 nm in diameter), which can be attributed to the precipitation of particles (Fig. S4†). The diffraction pattern of PdNP PREP 10 mg PdCl2 clearly shows the FCC crystalline structure exhibiting (1,1,1), (2,0,0), and (2,2,0) planes (Fig. 2D).
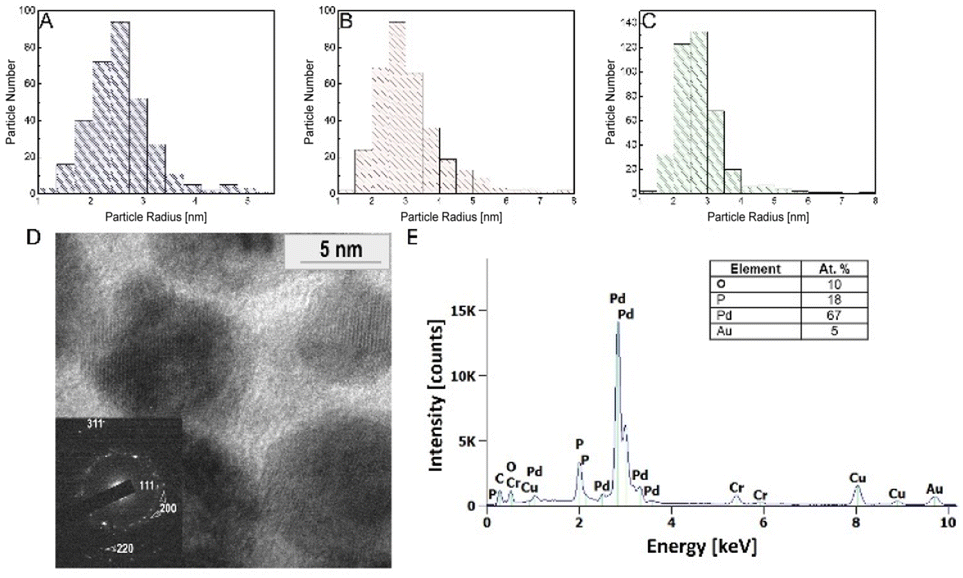 | ||
| Fig. 2 Characteristics of the PdNPs. (A–C) Particle size histograms of the PdNPs collected from 3 different fractions: SUP (A), PREP (B), and WHO (C). (D) TEM image of the PdNPs and corresponding selective area diffraction pattern (inset). (E) EDS spectrum of PdNPs. | ||
Energy dispersive X-ray spectroscopy (EDS) further confirms that the particles are composed of palladium without chlorine contamination (Fig. 2E). Table 1 shows the ICP-MS analysis of the Pd content in the different samples.
| PdCl2 (M) | PREP (mol) | SUP (mol) | WHO (mol) |
|---|---|---|---|
| 0.0075 | (1.81 ± 0.1) × 10−7 | (4.21 ± 0.2) × 10−8 | (7.61 ± 1.01) × 10−8 |
| 0.011 | (2.70 ± 0.1) × 10−7 | (1.02 ± 0.03) × 10−7 | (1.46 ± 0.09) × 10−7 |
| 0.015 | (4.95 ± 0.2) × 10−7 | (1.24 ± 0.03) × 10−7 | (1.76 ± 0.1) × 10−7 |
| 0.019 | (8.72 ± 0.5) × 10−7 | (1.29 ± 0.04) × 10−7 | (2.41 ± 0.1) × 10−7 |
The most considerable amount of Pd was in the PREP fraction, where the gross palladium weight was between 0.02 and 0.09 mg per 5 mg of sample, depending on the initial concentration of the PdCl2 salt. On the other hand, WHO and SUP fractions showed two orders of magnitude lower concentrations. The high content of Pd in PREP suggests that most of the Pd ions were reduced and aggregated into a large cluster of PdNPs. The increased initial PdCl2 ionic salt concentration resulted in an increase in the Pd0 molar concentration for all fractions.
Catalytic activity of PdNPs
To assess the catalytic activity and calculate the rate constant of PdNPs present in different concentrations and solution fractions, we used the reference reaction of conversion of nitrophenol to aminophenol. The decrease in the concentration of nitrophenol was measured by UV-vis spectroscopy (Fig. 3A and B). In a reference reaction, i.e. in the absence of PdNPs, no decrease in the reactant concentration was observed. Since sodium borohydride was used in excess (0.2 M) compared to the concentration of nitrophenol (2–0.5 mM), one can assume a first-order reaction rate model.22,27 The reaction rate constants (k) of the different fractions and salt contents are summarized in Table S1.†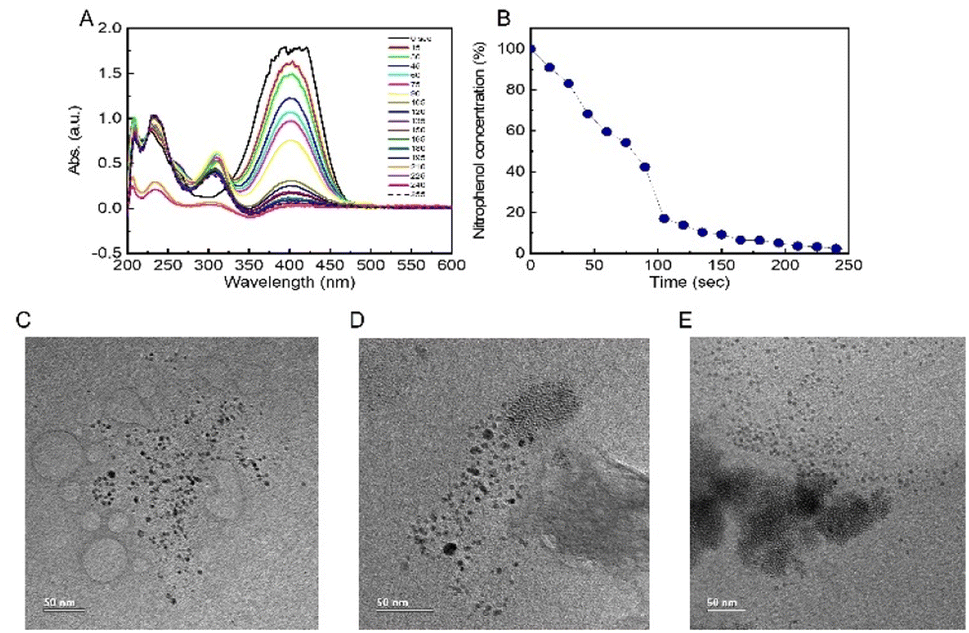 | ||
| Fig. 3 (A) UV-vis spectra of nitrophenol reduction reaction progression (decrease of the absorbance peak at 404 nm), (B) nitrophenol concentration vs. reaction time. HRTEM images of PdNPs isolated from 3 different fractions, SUP (C), WHO (D), PREP (E), showcasing the difference in particle aggregation in different fractions. | ||
The SUP fraction exhibited the highest constant rate with k = 0.0027 to k = 0.0187 sec−1 corresponding to concentrations of 0.0075 M to 0.019 M of PdCl2, respectively. The PREP fraction showed similar results but lagged in concentrations of 0.015 M and 0.019 M (k = 0.0102 and 0.0144 sec−1, respectively), and the WHO fraction showed the slowest reaction constants. Overall, the slowest reaction time until >90% reactant transition was 11 min in the WHO fraction (0.0075 M PdCl2) and the fastest in the SUP fraction (0.019 M PdCl2) with less than 4 min. Interestingly, the obtained reaction rates cannot be explained by the large PdNP content of the PREP samples (Table 1). However, this observation might be explained given the HRTEM results (Fig. 3C), which clearly show that PdNPs in the SUP fractions are smaller and well dispersed compared to the significantly larger and agglomerated PdNPs in PREP and WHO (Fig. 3D and E) fractions. We can thus conclude that the agglomeration of PdNPs decreases their reactivity due to the smaller active surface available for absorption of large aggregates' reactants compared to well-dispersed PdNPs formed in the SUP fraction. In addition, the WHO fraction also exhibits large particle aggregates, which also hinder their reactivity.
PdNPs embedded in hydrogels and their catalytic efficiency
To fabricate a reusable catalytic material from the PdNPs, we encapsulated them in a biocompatible, biodegradable, and water-insoluble stable hydrogel matrix in the form of a methacrylated Pluronic polymer. The encapsulation process allowed us to reuse the PdNP catalyst in several cycles and store it until further use. To assess the catalytic efficiency of the hydrogel for more extensive and reusable uses, we conducted ten cycles of the nitrophenol reduction reaction with a 0.015 M PdNP concentration as it showed more stable and higher reaction rate constants (see also Table S1† for rate constants). The results clearly indicate that even after ten reaction cycles (Fig. 4A), the PdNP hydrogel is still efficient and shows relatively high rate constants while giving a conversion efficiency of 85% at 20–80 min. Notably, we observed some fluctuations in rate constants during the catalytic cycles. This can be attributed to the partial decomposition of the hydrogels during the reactions in which the surface and the inner parts of the hydrogel pellets containing PdNPs were gradually exposed to the reactants (Fig. 4B–D) and degraded while the hydrogel that didn't undergo any catalytic reaction remains largely intact (Fig. S5†).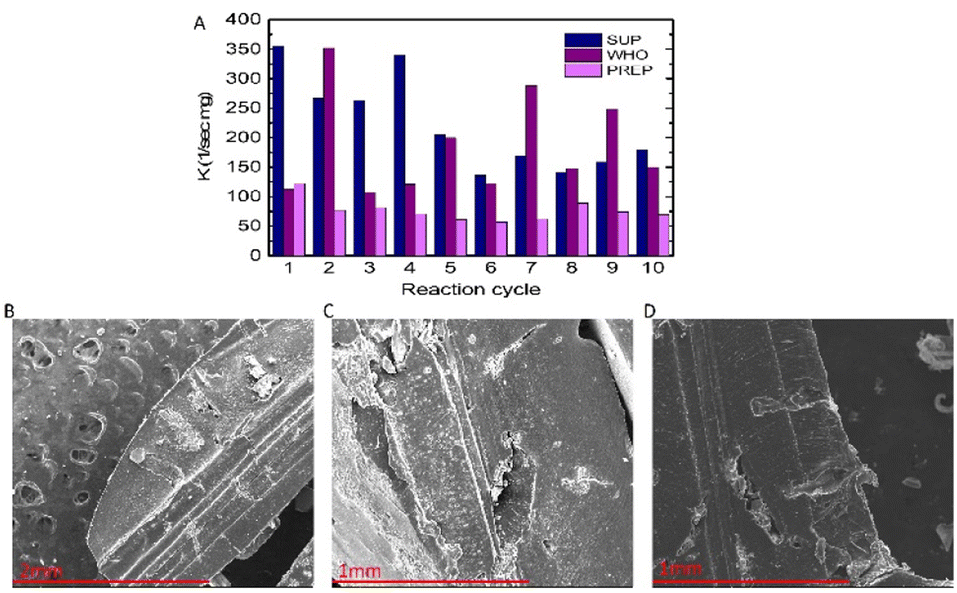 | ||
| Fig. 4 (A) Kinetic effectiveness constant K (sec−1 mg−1) of the PdNP hydrogel catalyst obtained for 10 cycles of the nitrophenol reduction reaction. (B–D) SEM images of hydrogel pellets containing PdNPs decomposed after 2 (B), 5 (C) and 8 (D) cycles of the nitrophenol reduction reaction. | ||
Suzuki–Miyaura reaction
The Suzuki–Miyaura reaction is a widely used carbon–carbon coupling reaction in organic chemistry research and industrial use. Pd is commonly used as the catalyst material for Suzuki–Miyaura reactions and shows excellent yields and high purity of the reaction products.The amounts of PdNPs used were like those utilized for catalytic rate calculations. Fig. 5 and S1† show the reaction product yields from the selected SUP fraction in 4 different amounts of initial PdCl2 ionic salt. Importantly, due to the particles' high reactivity, we could obtain more than 90% of the product in a much shorter reaction time (Fig. 5A, 15–60 min) compared to results reported in the literature (∼4–24 h).
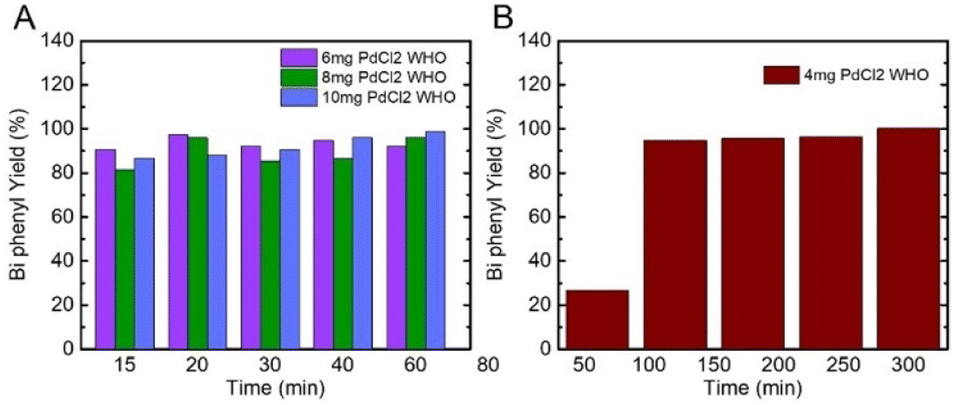 | ||
| Fig. 5 (A) and (B) Biphenyl yield% collected after different reaction times from the SUP fraction prepared from 4, 6, 8, and 10 mg of initial PdCl2 ionic salt. | ||
In all fractions, PdNPs synthesized from 4 mg of PdCl2 proved the least reactive, resulting in 20–50% yield after 60 min (Fig. 5B). Nevertheless, in 4 mg of PREP and WHO fractions after 120 min, we obtained more than 90% of product yield, while the SUP fraction, which contains fewer particles, resulted in a yield of 58% (Table S2†). After 5 h, all the fractions of PdNPs in 4 mg resulted in a yield of 100%, which is comparable to other published results.22,27,47
Chemical analyses show the formation of the bi-phenyl product: H-NMR measurement (Fig. S2†) shows a typical doublet at 7.63–7.61 ppm, a double triplet at 7.48–7.45 and 7.39–7.36, and a singlet at 7.26 which can be attributed to chloroform residues present in the solvent. No signals that can be attributed to the reaction reactants were present in the spectrum.
FTIR measurements show a typical spectrum with C–H bond stretch at 3000 (cm−1), carbon–carbon coupled bonds at 1750 (cm−1), and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C ring stretch at 1500 (cm−1), all of which are comparable to those in the commercial biphenyl FTIR database (see Fig. S2†).
C ring stretch at 1500 (cm−1), all of which are comparable to those in the commercial biphenyl FTIR database (see Fig. S2†).
Catalytic effectiveness comparison study
Fig. 6 depicts a comparison study of catalytic effectiveness (K [sec−1 mg−1]) between our PdNPs and other PdNPs synthesized with the help of biomolecules for the nitrophenol reduction reaction. The comparison indicates that our PdNP catalyst was at the same level of effectiveness as other reported results.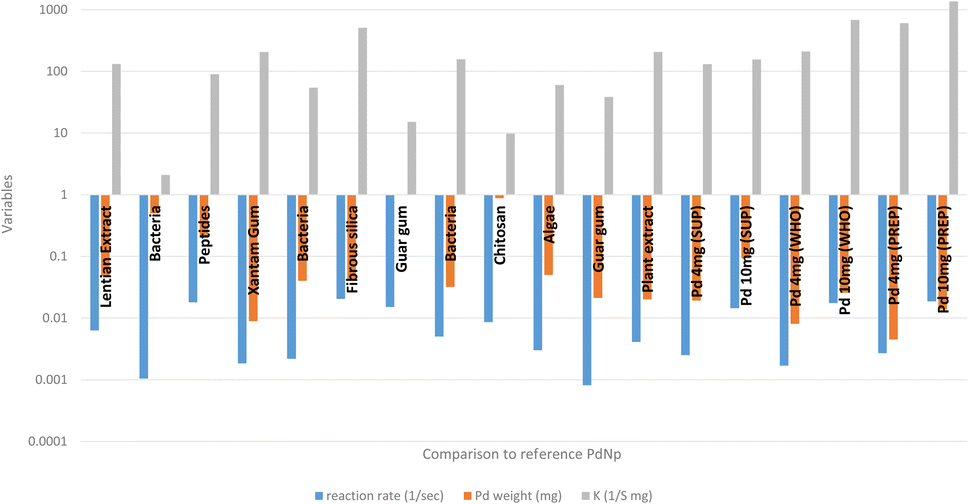 | ||
| Fig. 6 The nitrophenol reduction – comparison study.49–59 | ||
On the other hand, comparing the ratio between the product concentration and PdNP concentration used for the catalysis of the Suzuki–Miyaura reaction (K*) indicates the excellent characteristics of our PdNPs. It is evident that the K* value of our PdNPs was superior to the results of other biologically synthesized PdNP catalysts (Fig. 7).
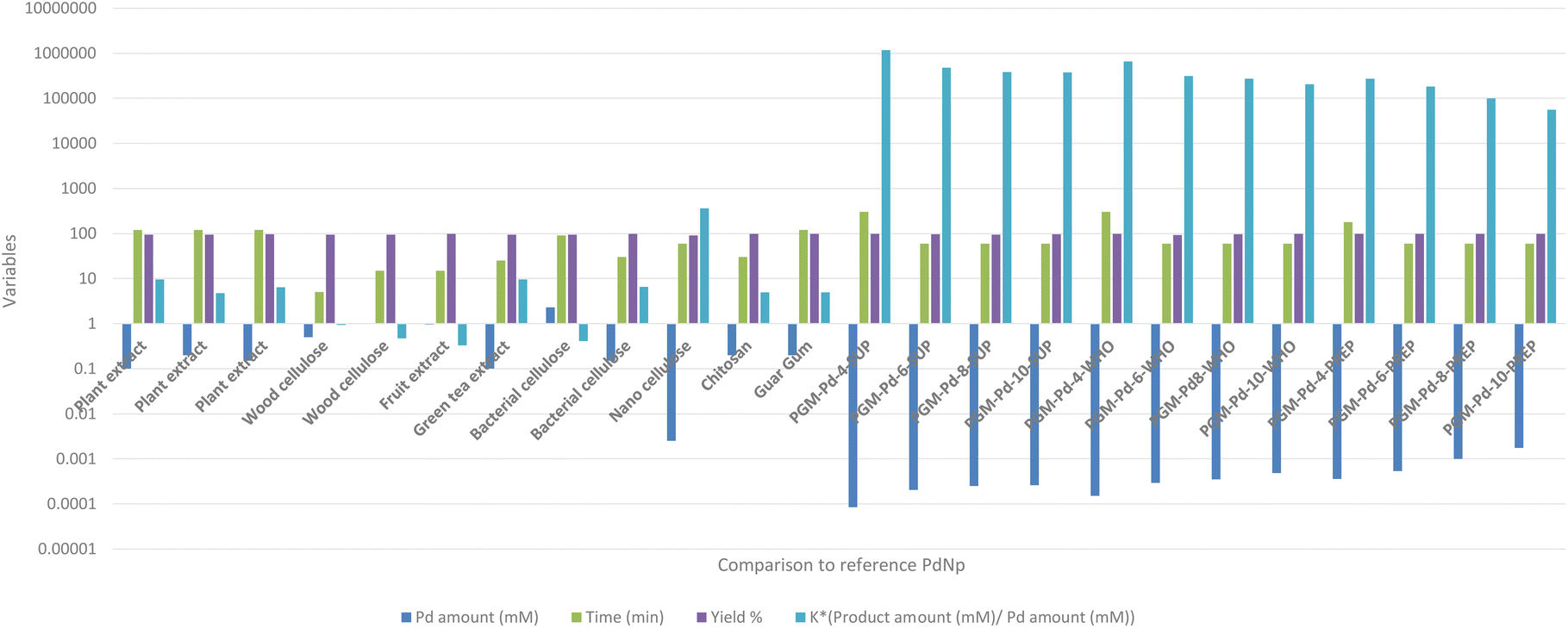 | ||
| Fig. 7 Suzuki–Miyaura reaction – comparison study.60–71 | ||
For example, the highest K* value reported by Zhang et al.70 was K* = 360, while our best K* value was K* = 1174928, four orders of magnitude better than the previously reported results. This dramatic difference can be explained by the small concentration of the PdNP catalyst, 8.42 × 10−5 mM used in our reaction compared to the best reported 2.5 × 10−3 mM concentration. Also, the amount of the bi-phenyl product produced in our reaction was 100 times greater than that in all the previously reported studies. This huge difference in catalytic activity can be explained by several factors: (1) the large surface-to-volume ratio of our PdNsP and (2) the accessibility of these particles towards the reaction reagents. These factors are clearly noticeable when comparing our data to those of Zhang et al.70 in which the synthesized PdNP average size was between 18 and 30 nm, about 9 times larger than our PdNPs. Additionally, in previously reported studies, the produced PdNPs were located both on the surface of and inside the nanocellulose matrix which limited their activity, in contrast to our study. This means that the active surface available for catalysis was further decreased. Additional referenced studies by Anuradha et al.71 and Kandathil et al.69 which synthesized 5–7 nm PdNPs on chitosan and cellulose, respectively, which are comparable to PdNPs synthesized in PGM, indicate that their active surface was rather limited due to encapsulation of PdNPs in a cellulose matrix and chitosan therefore limiting their overall catalytic activity.
Conclusions
In summary, we report a simple and green method to synthesize PdNPs with exceptionally high catalytic activity. In this one-pot step method, we utilize a single biomolecule to act as a reducing and capping agent, simplifying both the synthesis and optimization of the resulting PdNPs. It was shown that this methodology could be used in various organic syntheses where PdNPS are utilized as catalysts. Encapsulation of the PdNPs in hydrogels allowed their reusability for at least ten cycles, which makes them appealing for practical uses. Due to the small amount of Pd used and their large surface-to-volume ratio, these products show extraordinary efficiency in catalysis reactions, which makes them appealing for future practical uses.Author contributions
Roman Nudelman, Shir Zuarets, Meiron Lev and Shira Gavrieli performed the experiments. Louisa Meshi performed the TEM analysis, and Innes Zucker and Shachar Richter supervised the students. Shachar Richter and Roman Nudelman wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SR would like to thank The Israel Ministry of Environmental Protection fund: “circular economy grant” (# 221-5-6) for financial support. SG would like to thank the Adams Fellowships Program of the Israel Academy of Sciences and Humanities for their support.Notes and references
- A. A. Tedstone, D. J. Lewis and P. O'Brien, Chem. Mater., 2016, 28, 1965–1974 CrossRef CAS.
- S.-W. Hsu, K. On and A. R. Tao, J. Am. Chem. Soc., 2011, 133, 19072–19075 CrossRef CAS PubMed.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- G. A. Rudakov, K. B. Tsiberkin, R. S. Ponomarev, V. K. Henner, D. A. Ziolkowska, J. B. Jasinski and G. Sumanasekera, J. Magn. Magn. Mater., 2019, 472, 34–39 CrossRef CAS.
- R. Narayanan and M. A. El-Sayed, J. Phys. Chem. B, 2005, 109, 12663–12676 CrossRef CAS PubMed.
- D. Wu, K. Kusada, T. Yamamoto, T. Toriyama, S. Matsumura, S. Kawaguchi, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2020, 142, 13833–13838 CrossRef CAS PubMed.
- M. Azharuddin, G. H. Zhu, D. Das, E. Ozgur, L. Uzun, A. P. F. Turner and H. K. Patra, Chem. Commun., 2019, 55, 6964–6996 RSC.
- Y. Zhang, H. chang, C. Saliba and A. Hasnaoui, Resour. Policy, 2022, 78, 102924 CrossRef.
- M. Zahmakıran and S. Özkar, Nanoscale, 2011, 3, 3462–3481 RSC.
- Y.-H. Li, J.-Y. Li and Y.-J. Xu, EnergyChem, 2021, 3, 100047 CrossRef CAS.
- M. Omrani, M. Goriaux, Y. Liu, S. Martinet, L. Jean-Soro and V. Ruban, Environ. Pollut., 2020, 257, 113477 CrossRef CAS PubMed.
- S. McCarthy, A. Lee Wei Jie, D. C. Braddock, A. Serpe and J. D. E. T. Wilton-Ely, Molecules, 2021, 26, 5217 CrossRef CAS PubMed.
- S. McCarthy, D. C. Braddock and J. D. Wilton-Ely, Coord. Chem. Rev., 2021, 442, 213925 CrossRef CAS.
- R. W. J. Scott, H. Ye, R. R. Henriquez and R. M. Crooks, Chem. Mater., 2003, 15, 3873–3878 CrossRef CAS.
- I. E. Espino-López, M. Romero-Romo, M. G. M. de Oca-Yemha, P. Morales-Gil, M. T. Ramírez-Silva, J. Mostany and M. Palomar-Pardavé, J. Electrochem. Soc., 2018, 166, D3205 CrossRef.
- A. Ameri, M. Shakibaie, H.-R. Rahimi, M. Adeli-Sardou, M. Raeisi, A. Najafi and H. Forootanfar, Biol. Trace Elem. Res., 2020, 197, 132–140 CrossRef CAS PubMed.
- S. Navaladian, B. Viswanathan, T. K. Varadarajan and R. P. Viswanath, Nanoscale Res. Lett., 2009, 4, 181–186 CrossRef CAS PubMed.
- I. Saldan, Y. Semenyuk, I. Marchuk and O. Reshetnyak, J. Mater. Sci., 2015, 50, 2337–2354 CrossRef CAS.
- N. Joudeh, A. Saragliadis, G. Koster, P. Mikheenko and D. Linke, Front. Nanotechnol., 2022, 4, 1–24 Search PubMed.
- R. Narayanan and M. A. El-Sayed, J. Am. Chem. Soc., 2003, 125, 8340–8347 CrossRef CAS PubMed.
- V. Leso and I. Iavicoli, Int. J. Mol. Sci., 2018, 19, 503 CrossRef PubMed.
- K. S. Siddiqi and A. Husen, Nanoscale Res. Lett., 2016, 11, 482 CrossRef PubMed.
- R. R. Palem, G. Shimoga, S.-Y. Kim, C. Bathula, G. S. Ghodake and S.-H. Lee, J. Ind. Eng. Chem., 2022, 106, 52–68 CrossRef CAS.
- S. Ying, Z. Guan, P. C. Ofoegbu, P. Clubb, C. Rico, F. He and J. Hong, Environ. Technol. Innovation, 2022, 26, 102336 CrossRef CAS.
- Y. Yu, E. Jung, H. J. Kim, A. Cho, J. Kim, T. Yu and J. Lee, ACS Appl. Nano Mater., 2020, 3, 10487–10496 CrossRef CAS.
- K. Anand, C. Tiloke, A. Phulukdaree, B. Ranjan, A. Chuturgoon, S. Singh and R. M. Gengan, J. Photochem. Photobiol., B, 2016, 165, 87–95 CrossRef CAS PubMed.
- M. Sathishkumar, K. Sneha, I. S. Kwak, J. Mao, S. J. Tripathy and Y.-S. Yun, J. Hazard. Mater., 2009, 171, 400–404 CrossRef CAS PubMed.
- I. P. Mikheenko, J. A. Bennett, J. B. Omajali, M. Walker, D. B. Johnson, B. M. Grail, D. Wong-Pascua, J. D. Moseley and L. E. Macaskie, Appl. Catal., B, 2022, 306, 121059 CrossRef CAS.
- G. Park, J. Park, Y. Cho and C. Lee, Int. J. Electrochem. Sci., 2016, 11, 4539–4549 CrossRef CAS.
- H. Jiang, Q. Yan, Y. Du and R. Chen, React. Kinet., Mech. Catal., 2016, 117, 307–317 CrossRef CAS.
- O. Bachar, M. M. Meirovich, R. Kurzion and O. Yehezkeli, Chem. Commun., 2020, 56, 11211–11214 RSC.
- R. Nudelman, H. Alhmoud, B. Delalat, I. Kaur, A. Vitkin, L. Bourgeois, I. Goldfarb, A. Cifuentes-Rius, N. H. Voelcker and S. Richter, J. Nanobiotechnol., 2022, 20, 1–13 CrossRef PubMed.
- S. Gavriely, W. Hadibrata, R. Nudelman, K. Aydin and S. Richter, Adv. Sustainable Syst., 2021, 5, 2100099 CrossRef CAS.
- R. Nudelman, H. Alhmoud, B. Delalat, S. Fleicher, E. Fine, T. Guliakhmedova, R. Elnathan, A. Nyska, N. H. Voelcker, M. Gozin and S. Richter, Adv. Funct. Mater., 2019, 29, 1902783 CrossRef.
- N. Hendler, L. Fadeev, E. D. Mentovich, B. Belgorodsky, M. Gozin and S. Richter, Chem. Commun., 2011, 47, 7419 RSC.
- R. Nudelman, S. Gavriely, D. Bychenko, M. Barzilay, T. Gulakhmedova, E. Gazit and S. Richter, J. Nanobiotechnol., 2021, 19, 1–10 CrossRef PubMed.
- S. Gavriely, T. Gulakhmedova, Y. Yecheskel, A. E. Rubin, B. Xing, S. Richter and I. Zucker, NanoImpact, 2022, 27, 100417 CrossRef CAS PubMed.
- L. R. Steinberger, T. Gulakhmedova, Z. Barkay, M. Gozin and S. Richter, Adv. Sustainable Syst., 2019, 1900016 CrossRef.
- R. Bansil and B. S. Turner, Curr. Opin. Colloid Interface Sci., 2006, 11, 164–170 CrossRef CAS.
- J. P. Celli, B. S. Turner, N. H. Afdhal, R. H. Ewoldt, G. H. McKinley, R. Bansil and S. Erramilli, Biomacromolecules, 2007, 8, 1580–1586 CrossRef CAS PubMed.
- D. Ince, T. M. Lucas and S. A. Malaker, Curr. Opin. Chem. Biol., 2022, 69, 102174 CrossRef CAS PubMed.
- J. Celli, B. Gregor, B. Turner, N. H. Afdhal, R. Bansil and S. Erramilli, Biomacromolecules, 2005, 6, 1329–1333 CrossRef CAS PubMed.
- R. Nudelman, E. Gloukhikh, A. Rekun and S. Richter, Protein Sci., 2016, 25, 1918–1923 CrossRef CAS PubMed.
- J. Gotta, T. B. Shalom, S. Aslanoglou, A. Cifuentes-Rius, N. H. Voelcker, R. Elnathan, O. Shoseyov and S. Richter, Adv. Funct. Mater., 2018, 28, 1870167 CrossRef.
- N. Hendler, B. Belgorodsky, E. D. Mentovich, M. Gozin and S. Richter, Adv. Mater., 2011, 23, 4261–4264 CrossRef CAS PubMed.
- E. Gioffredi, M. Boffito, S. Calzone, S. M. Giannitelli, A. Rainer, M. Trombetta, P. Mozetic and V. Chiono, Procedia CIRP, 2016, 49, 125–132 CrossRef.
- A. Iben Ayad, D. Luart, A. Ould Dris and E. Guénin, Nanomater, 2020, 10, 1169 CrossRef PubMed.
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685–4696 CrossRef CAS PubMed.
- Z. Han, L. Dong, J. Zhang, T. Cui, S. Chen, G. Ma, X. Guo and L. Wang, RSC Adv., 2019, 9, 38265–38270 RSC.
- K. Mallikarjuna, L. V. Reddy, S. Al-Rasheed, A. Mohammed, S. Gedi and W. K. Kim, Crystals, 2021, 11, 134 CrossRef CAS.
- H. Veisi, M. Pirhayati, A. Kakanejadifard, P. Mohammadi, M. R. Abdi, J. Gholami and S. Hemmati, ChemistrySelect, 2018, 3, 1820–1826 CrossRef CAS.
- A. Santoshi kumari, M. Venkatesham, D. Ayodhya and G. Veerabhadram, Appl. Nanosci., 2015, 5, 315–320 CrossRef CAS.
- M. P. Desai, R. V. Patil and K. D. Pawar, Process Biochem., 2020, 98, 172–182 CrossRef CAS.
- Z. Dong, X. Le, C. Dong, W. Zhang, X. Li and J. Ma, Appl. Catal., B, 2015, 162, 372–380 CrossRef CAS.
- M. Liu, F. Cui, Q. Ma, L. Xu, J. Zhang, R. Zhang and T. Cui, New J. Chem., 2020, 44, 4042–4048 RSC.
- L. Xiong, X. Zhang, Y.-X. Huang, W.-J. Liu, Y.-L. Chen, S.-S. Yu, X. Hu, L. Cheng, D.-F. Liu and H.-Q. Yu, ACS Appl. Nano Mater., 2018, 1, 1467–1475 CrossRef CAS.
- S. Chatterjee and S. K. Bhattacharya, ACS Omega, 2021, 6, 20746–20757 CrossRef CAS PubMed.
- Y. Rodriguez Mejía and N. K. R. Bogireddy, RSC Adv., 2022, 12, 18661–18675 RSC.
- F. Anjum, S. Gul, M. I. Khan and M. A. Khan, Green Process. Synth., 2020, 9, 63–76 Search PubMed.
- C.-H. Liu, J. Liu, Y.-Y. Zhou, X.-L. Cai, Y. Lu, X. Gao and S.-D. Wang, Carbon, 2015, 94, 295–300 CrossRef CAS.
- R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- F. Ahmadi, M. Hekmati, M. Yousefi and H. Veisi, Asian J. Nanosci. Mater., 2018, 1, 104–114 Search PubMed.
- D. Baruah, R. N. Das, S. Hazarika and D. Konwar, Catal. Commun., 2015, 72, 73–80 CrossRef CAS.
- J. R. Anasdass, P. Kannaiyan, R. Raghavachary, S. C. B. Gopinath and Y. Chen, PLoS One, 2018, 13, e0193281 CrossRef PubMed.
- H. Veisi, M. Ghorbani and S. Hemmati, Mater. Sci. Eng., C, 2019, 98, 584–593 CrossRef CAS PubMed.
- Z. Xiang, Y. Chen, Q. Liu and F. Lu, Green Chem., 2018, 20, 1085–1094 RSC.
- A. Palliyarayil, K. K. Jayakumar, S. Sil and N. S. Kumar, Johnson Matthey Technol. Rev., 2018, 62, 60–73 CrossRef CAS.
- H. Gholami Derami, P. Gupta, R. Gupta, P. Rathi, J. J. Morrissey and S. Singamaneni, ACS Appl. Nano Mater., 2020, 3, 5437–5448 CrossRef CAS.
- V. Kandathil, M. Kempasiddaiah, B. S. Sasidhar and S. A. Patil, Carbohydr. Polym., 2019, 223, 115060 CrossRef CAS PubMed.
- K. Zhang, M. Shen, H. Liu, S. Shang, D. Wang and H. Liimatainen, Carbohydr. Polym., 2018, 186, 132–139 CrossRef CAS PubMed.
- Anuradha, S. Kumari, S. Layek and D. D. Pathak, New J. Chem., 2017, 41, 5595–5604 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00385j |
| This journal is © The Royal Society of Chemistry 2023 |