
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA high activity mesoporous Pt@KIT-6 nanocomposite for selective hydrogenation of halogenated nitroarenes in a continuous-flow microreactor†
Kejie
Chai
a,
Xilin
Yang
a,
Runqiu
Shen
a,
Jianli
Chen
ab,
Weike
Su
 *a and
An
Su
*c
*a and
An
Su
*c
aKey Laboratory of Pharmaceutical Engineering of Zhejiang Province, National Engineering Research Center for Process Development of Active Pharmaceutical Ingredients, Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals, Zhejiang University of Technology, Hangzhou, 310014, P. R. China. E-mail: pharmlab@zjut.edu.cn
bCollege of New Materials Engineering, Jiaxing Nanhu University, Jiaxing, 314000, P. R. China
cCollege of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, P. R. China. E-mail: ansu@zjut.edu.cn
First published on 14th September 2023
Abstract
In this study, we designed a Pt@KIT-6 nanocomposite prepared by impregnating platinum nanoparticles on the nanopores of the KIT-6 mesoporous material. This Pt@KIT-6 nanocomposite was used as a catalyst in a micro fixed bed reactor (MFBR) for the continuous-flow hydrogenation of halogenated nitroarenes, which demonstrates three advantages. First, the Pt@KIT-6 nanocomposite has a stable mesoporous nanostructure, which effectively enhances the active site and hydrogen adsorption capacity. The uniformly distributed pore structure and large specific surface area were confirmed by electron microscopy and N2 physisorption, respectively. In addition, the aggregation of the loaded metal was avoided, which facilitated the maintenance of high activity and selectivity. The conversion and selectivity reached 99% within 5.0 minutes at room temperature (20 °C). Furthermore, the continuous-flow microreactor allows precise control and timely transfer of the reaction system, reducing the impact of haloid acids. The activity and selectivity of the Pt@KIT-6 nanocomposite showed virtually no degradation after 24 hours of continuous operation of the entire continuous-flow system. Overall, the Pt@KIT-6 nanocomposite showed good catalysis for the hydrogenation of halogenated nitroarenes in the continuous-flow microreactor. This work provides insights into the rational design of a highly active and selective catalyst for selective hydrogenation systems.
Introduction
Halogenated arylamines are an important class of chemical intermediates used in the synthesis of pesticides, veterinary drugs, and dyes.1–5 The preparation of halogenated arylamines by hydrogenation with halogenated nitroarenes is the most popular method today because of its low cost and environmental friendliness.6–8 However, during the hydrogenation of the halogenated nitroarenes, there is competition between the carbon–halogen bond and the nitro group, which may lead to poor selectivity of the hydrogenation.9,10 Wang et al. prepared a Ni–CeO2/SiO2 heterojunction catalyst with a “raisin-bun” structure.10 Improved activity and selectivity of hydrogenating halogenated nitroarenes were achieved by the cooperation of the built-in electric field and oxygen vacancies. Yue et al. performed highly selective hydrogenation of halogenated nitroarenes on Ru/CN nanocomposites by in situ pyrolysis.11 Although these new catalysts showed good selectivity, many of them showed decreases in catalytic activity and selectivity during long-term operation.12 In addition, the cumbersome preparation processes and harsh application conditions of some catalysts were not conducive to the efficient synthesis of halogenated arylamines.12 On the other hand, mesoporous nanocomposites are an ideal class of catalysts or catalyst carriers with great potential for application in selective hydrogenation reactions due to their abundant active sites, good gas adsorption properties, and stable structures.13–17Since the invention of mesoporous materials, various mesoporous silica materials such as MCM-41, SBA-15, and KIT-6 have been used as catalyst carriers for hydrogenation reactions.18–20 Compared with one-dimensionally arranged MCM-41 and two-dimensionally arranged SBA-15, KIT-6 has ordered 3D mesoporous channels and thus is superior to catalysts with low-order channels in terms of mass transfer characteristics.21 Based on this advantage, the KIT-6-based catalysts outperform the MCM-41-based and SBA-15-based catalysts in terms of active phase dispersion, reactant and product transport, and catalytic activity.22,23 Yu et al. prepared KIT-6 mesoporous silica-supported copper by the ammonia evaporation method and applied it to the catalytic hydrogenation of dimethyl oxalate to ethylene glycol.24 The high specific surface area and interconnected mesoporous channels of KIT-6 facilitated the dispersion of copper. Neeli et al. synthesized a Rh/ED-KIT-6 catalyst consisting of Rh nanoparticles embedded in KIT-6, which was functionalized by N1-[3-(trimethoxysilyl)propyl]ethane-1,2-diamine via Rh3+ adsorption and reduction in the liquid phase.25 Under mild reaction conditions, Rh/ED-KIT-6 nanocatalysts exhibited a high turnover frequency in the transfer hydrogenation of furfural using formic acid. However, less attention has been paid to the application of KIT-6 in the selective hydrogenation of halogenated nitroarenes.
For the preparation of halogenated arylamines by hydrogenation of halogenated nitroarenes, the risk factor of conventional batch reactors is high due to hydrogen leakage, high reaction temperatures, and high pressures.26 In addition, trace amounts of haloid acids are inevitably produced during the hydrogenation process.9 These acids are highly acidic, and the intense hydrogenation conditions cause them to continuously corrode conventional batch reactors, further increasing the risk to the hydrogenation system.10 In addition, the concentration of acids gradually increases as the reaction proceeds, leading to catalyst deactivation in the conventional batch reactor.27,28 On the other hand, continuous-flow microreactors have been increasingly used for hydrogenation due to their intrinsic safety.29–33 In addition, continuous-flow reactors enable the timely transfer of the reaction solution, inhibit dehalogenation, and greatly reduce the impact of haloid acids on the reactor and catalyst, making them ideal for preparing halogenated arylamines.34–36 Duan et al. developed an H-flow system with a micro-packed bed reactor for the selective hydrogenation of halogenated nitroarenes.37 Compared with batch reactors, the dehalogenation effects of this H-flow system could be negligible. However, highly active and selective catalysts are still required to optimize the hydrogenation reactions.38–40
In this work, we designed a micro fixed bed reactor (MFBR) for the continuous-flow selective hydrogenation of halogenated nitroarenes (Fig. 1). The efficient selective hydrogenation catalyst filled in the MFBR was a Pt@KIT-6 nanocomposite synthesized by impregnation with the mesoporous material KIT-6 as a carrier. The mesoporous nanocomposite catalyst was systematically characterized in terms of both microstructure and elemental composition. After optimization of the reaction condition parameters, the hydrogenation system was evaluated for different substitution sites and different substituted halogens and non-halogenated nitroaromatics to verify the generality of the hydrogenation system. In addition, the stability of the Pt@KIT-6 nanocomposite was tested in long-term experiments.
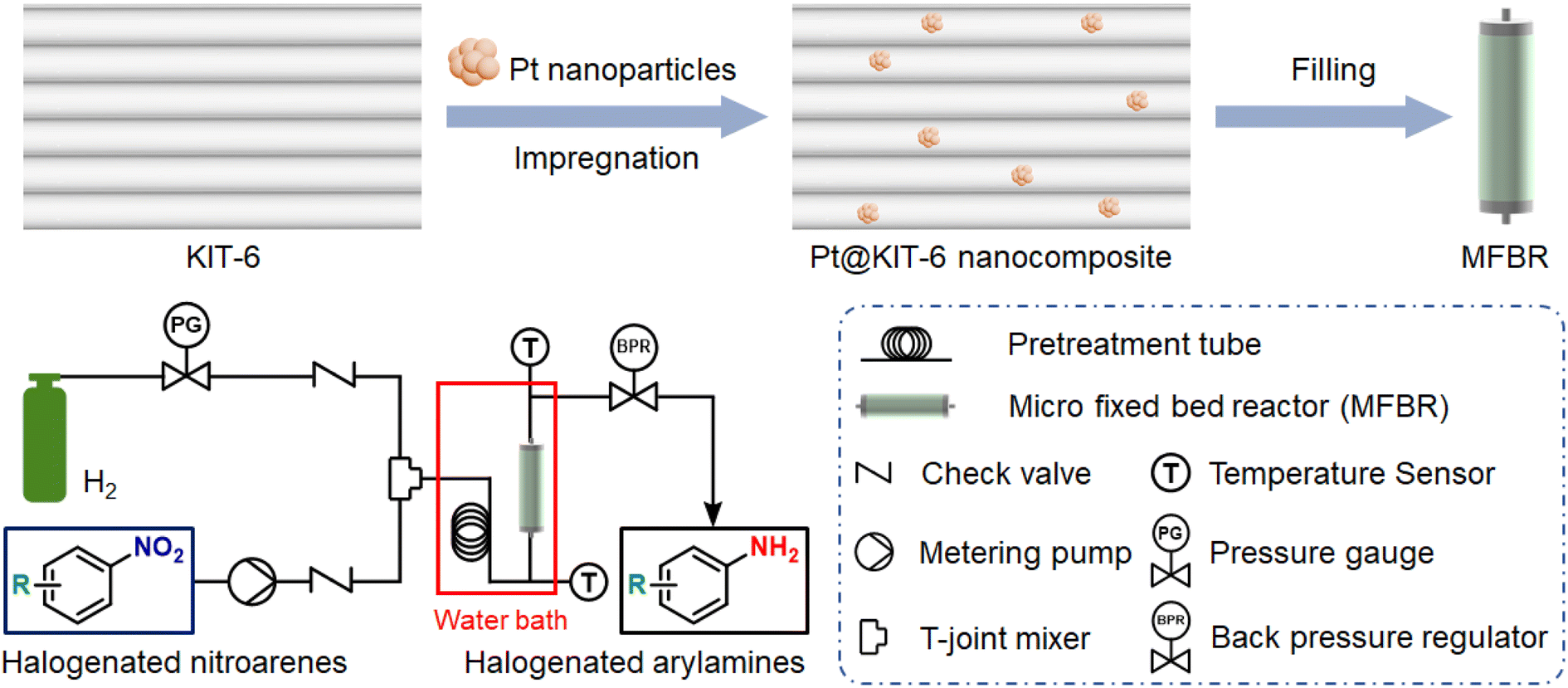 | ||
| Fig. 1 The MFBR of continuous-flow selective hydrogenation with Pt@KIT-6 nanocomposite. | ||
Materials and methods
Materials
All chemical reagents were of analytical grade and purchased from Sinopharm Chemical Reagent Co., Ltd. (SCRC) without further purification. They are poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (average Mn ∼ 5800) (Pluronic®P-123), tetraethyl orthosilicate (TEOS) (28.4% SiO2), hydrochloric acid (HCl) (12 M), n-butanol (AR), platinum tetrachloride (98%, Pt ≥57%), platinum/carbon (Pt/C, 5% (w/w) on carbon), ethanol (EtOH, AR), methanol (MeOH, AR), dimethylformamide (DMF, AR), ethyl acetate (EA, AR), acetonitrile (MeCN, AR), tetrahydrofuran (THF, AR), silica (100–200 mesh), 1-fluoro-2-nitrobenzene (99%), 1-fluoro-3-nitrobenzene (98%), 4-fluoronitrobenzene (98%), 2-chloronitrobenzene (99%), 1-chloro-3-nitrobenzene (98%), 4-chloronitrobenzene (99%), 1-bromo-2-nitrobenzene (99%), 3-bromonitrobenzene (98%), 4-nitrobromobenzene (99%), 1-iodo-2-nitrobenzene (98%), 1-iodo-3-nitrobenzene (98%), 4-iodonitrobenzene (99%), nitrobenzene (99%), 4-nitroanisole (98%), 4-nitrobenzoic acid (99%), 2-nitrobenzonitrile (98%), 4-nitrobenzamide (98%), 2-chloro-5-nitropyridine (98%), 2-methoxy-5-nitropyridine (98%), and 5-nitropyridine-2-carboxylic acid (97%). H2 (99.999%) was purchased from Hangzhou Jingong Special Gas Co., Ltd.Synthesis of the Pt@KIT-6 nanocomposite
Mesoporous material KIT-6 was synthesized following the conventional method reported in the literature (see “Synthesis of KIT-6” in the ESI†).41 0.1 g of PtCl4 was added to 12 g of HCl (12 M) to dissolve completely. 1 g of KIT-6 was then added to the above solution and stirred continuously at 80 °C until the liquid was completely evaporated and the solid was obtained. The solid was calcined in a preheated tube furnace at 400 °C for 5 h under a hydrogen atmosphere (flow rate = 100 mL min−1). After cooling, the mesoporous catalyst Pt@KIT-6 nanocomposite was obtained. The obtained Pt@KIT-6 was immediately stored in a vacuum drying oven to protect it from the effects of water and oxygen in the environment.Hydrogenation in a micro fixed bed reactor (MFBR)
An HPLC column (5 mm in diameter and 50 mm long) was pre-loaded with the synthesized mesoporous catalyst Pt@KIT-6 nanocomposite and filled with silica in its remaining space. The ends of the column were then fitted with sieve plates as the MFBR. Halogenated nitroarenes were dissolved in solvent and pumped into the microreactor through a metering pump. The pressure of the hydrogen involved in the reaction was controlled by adjusting the pressure gauge. The gas and liquid flow tubes were each fitted with a check valve to avoid backflow. The hydrogen and the reaction solution dissolved in halogenated nitroarenes were mixed in the T-joint mixer and then flowed into a 1.0 m pretreatment tube for reaching the reaction temperature. Next, the gas–liquid mixture undergoes hydrogenation in the MFBR. The MFBR was placed vertically in a water bath, which facilitated full contact of reactants with catalyst and achieved precise control of the reaction temperature. All tubes were 1/16′′ outside diameter stainless steel tubes. The entire system controlled the pressure by adjusting the manual back pressure regulator. The check valve and back pressure regulator ensure that the continuous-flow system is isolated from air. The entire hydrogenation was influenced by factors such as reaction temperature, flow rate, catalyst and solvent.Purification and yield determination
The halogenated nitroarenes conversion and the halogenated arylamines selectivity were determined by high-performance liquid chromatography (HPLC) using an external standard method. HPLC detection conditions: HPLC column (XB-C18, dim 4.6 × 250 mm, 10 μm), mobile phase (isocratic elution, H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH = 40:60), flow rate (1.00 mL min−1), chromatographic column temperature (30 °C), ultraviolet wavelength (254 nm). The halogenated nitroarenes conversion and the halogenated arylamines selectivity were calculated using eqn (1) and (2), respectively:
:MeOH = 40:60), flow rate (1.00 mL min−1), chromatographic column temperature (30 °C), ultraviolet wavelength (254 nm). The halogenated nitroarenes conversion and the halogenated arylamines selectivity were calculated using eqn (1) and (2), respectively: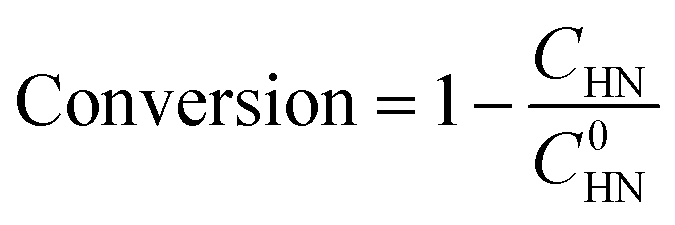 | (1) |
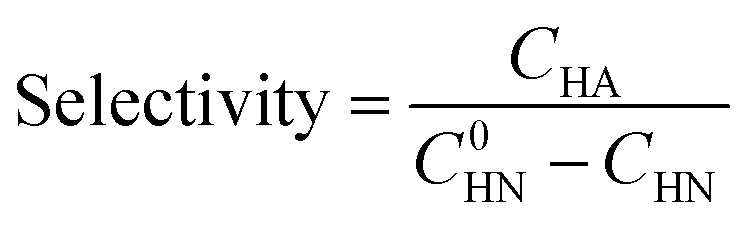 | (2) |
After purification by silica gel column chromatography, the isolated halogenated arylamines were obtained. Then, the isolated yield and the production rate of the halogenated arylamines were calculated using eqn (3) and (4), respectively:
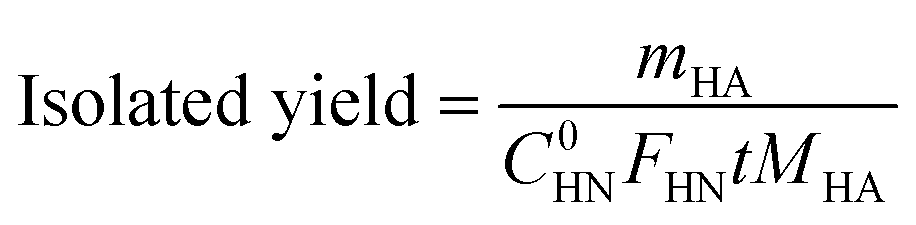 | (3) |
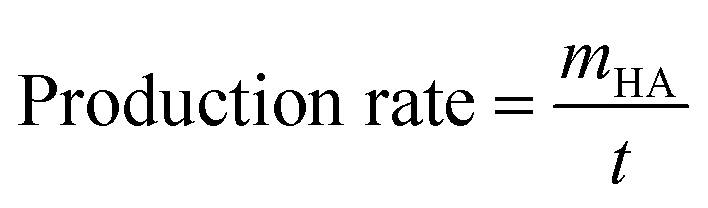 | (4) |
Characterization
X-ray diffraction (XRD) characterization was recorded with a PANalytical Empyrean powder diffractometer (PANalytical) using Cu Kα radiation (λ = 0.1541 nm). The working voltage was 40 kV and the working current was 40 mA. Field emission scanning electron microscopy (FE-SEM) and energy-dispersive X-ray spectroscopy (EDS) measurements were performed using a HITACHI Regulus 8100 instrument (Hitachi) and an Oxford Ultim Max 65 instrument (Oxford), respectively. Transmission electron microscopy (TEM) characterization was performed using an HT-7700 apparatus (Hitachi). The high-resolution TEM (HRTEM) images with the selected area electron diffraction (SAED) patterns were obtained using an FEI Talos F200X instrument (FEI). N2 physisorption isotherms were recorded using a Micromeritics 3Flex instrument (Micromeritics). X-ray photoelectron spectroscopy (XPS) spectra were obtained using a Kratos AXIS Ultra DLD apparatus (Kratos) with a monochromatic Al Kα X-ray source. Inductively coupled plasma optical emission spectrometer (ICP-OES) analysis and inductively coupled plasma mass spectrometry (ICP-MS) analysis were performed using an Agilent 5110 instrument (Agilent) and a PerkinElmer NexION 1000 instrument (PerkinElmer), respectively. High-performance liquid chromatography (HPLC) characterization was recorded on an Agilent 1100 Series instrument (Agilent). Nuclear magnetic resonance (NMR) characterization was performed using a Bruker Avance III HD 600 MHz instrument (Bruker).Results and discussion
Characterization of the Pt@KIT-6 nanocomposite
After the preparation of mesoporous material KIT-6, the hydrogenation catalyst Pt@KIT-6 nanocomposite was synthesized by impregnating metallic platinum (Pt) on KIT-6. Fig. 2 shows the XRD patterns of the prepared mesoporous KIT-6 and Pt@KIT-6 nanocomposite. The small-angle XRD patterns (Fig. 2a) of KIT-6 and Pt@KIT-6 nanocomposite show four clear diffraction peaks at 1.18°, 1.32°, 1.97°, and 2.05° in the region of 2θ = 0.5–5.0°, which can be assigned to the (211), (220), (420), and (332) planes of the mesoporous material with highly ordered 2D P6mm hexagonal symmetry.42,43 The similar small-angle XRD patterns of KIT-6 and Pt@KIT-6 nanocomposite indicate that the porous structure of the mesoporous material KIT-6 is preserved after strong acid immersion and high-temperature calcination. The wide-angle XRD patterns (Fig. 2b) of KIT-6 and Pt@KIT-6 nanocomposite show a weak and broad diffraction peak at a 2θ value of about 23°, indicating that the prepared silica is in an amorphous state.44,45 On the other hand, the wide-angle XRD pattern (Fig. 2b) of the Pt@KIT-6 nanocomposite shows four clear diffraction peaks at 40.1°, 46.6°, 68.0°, and 81.8°, which can be assigned to the (111), (200), (220), and (311) planes of Pt0 (JCPDS card 00-087-0640).46 This indicates that Pt4+ has been reduced to Pt0 with good crystallinity.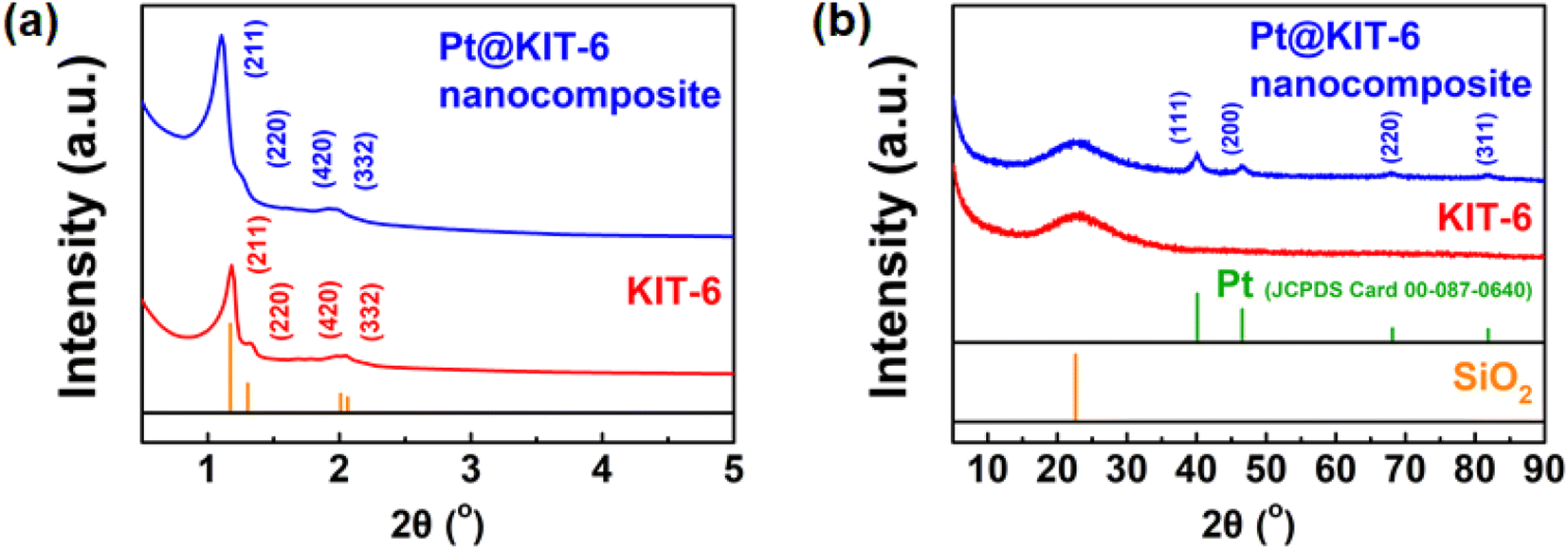 | ||
| Fig. 2 (a) Small-angle XRD pattern of Pt@KIT-6 nanocomposite and KIT-6; (b) wide-angle XRD pattern of Pt@KIT-6 nanocomposite and KIT-6. | ||
To probe the microstructure of the Pt@KIT-6 nanocomposite, electron microscopy images and N2 physisorption isotherms were collected. The SEM image (Fig. 3a) shows that the external appearance of the synthesized Pt@KIT-6 nanocomposite is a bulk structure with folds. In addition, there are no obvious solid particles on the surface of the Pt@KIT-6 nanocomposite. Meanwhile, the TEM image (Fig. 3b) shows that the cross-section of the Pt@KIT-6 nanocomposite still maintains a highly regular honeycomb structure without obvious Pt aggregation, which is beneficial to improve the catalytic activity of Pt nanoparticles.47 The lattice of the loaded Pt can be clearly observed by further HRTEM characterization. The HRTEM image (Fig. 3c) shows an interlayered spacing of 0.2258 nm, which corresponds to the d spacing of the (111) crystal planes of Pt, indicating that the loaded Pt is well crystallized in the form of nanoparticles.48 Simultaneously, the HRTEM image (Fig. S1a†) shows a uniform distribution of Pt nanoparticles. The particle size distribution of Pt nanoparticles is shown in Fig. S1b,† with an average particle size of 2.51 ± 0.3 nm. This further indicates that there is no obvious aggregation of Pt nanoparticles, which may be due to the confinement effect of the ordered mesoporous structure.49,50 The diffraction rings of the Pt@KIT-6 nanocomposite in the SAED pattern (Fig. 3d) indicate that it is polycrystalline, which facilitates the catalytic activity of Pt.51,52 In addition, diffraction rings on the (111), (200), (220) and (311) crystal planes of Pt0 can be seen, which is consistent with the XRD analysis and further confirms that Pt nanoparticles are loaded on the mesoporous material KIT-6.53 The Pt loading content on KIT-6 detected by ICP-OES (Table S1†) was around 5% (w/w).
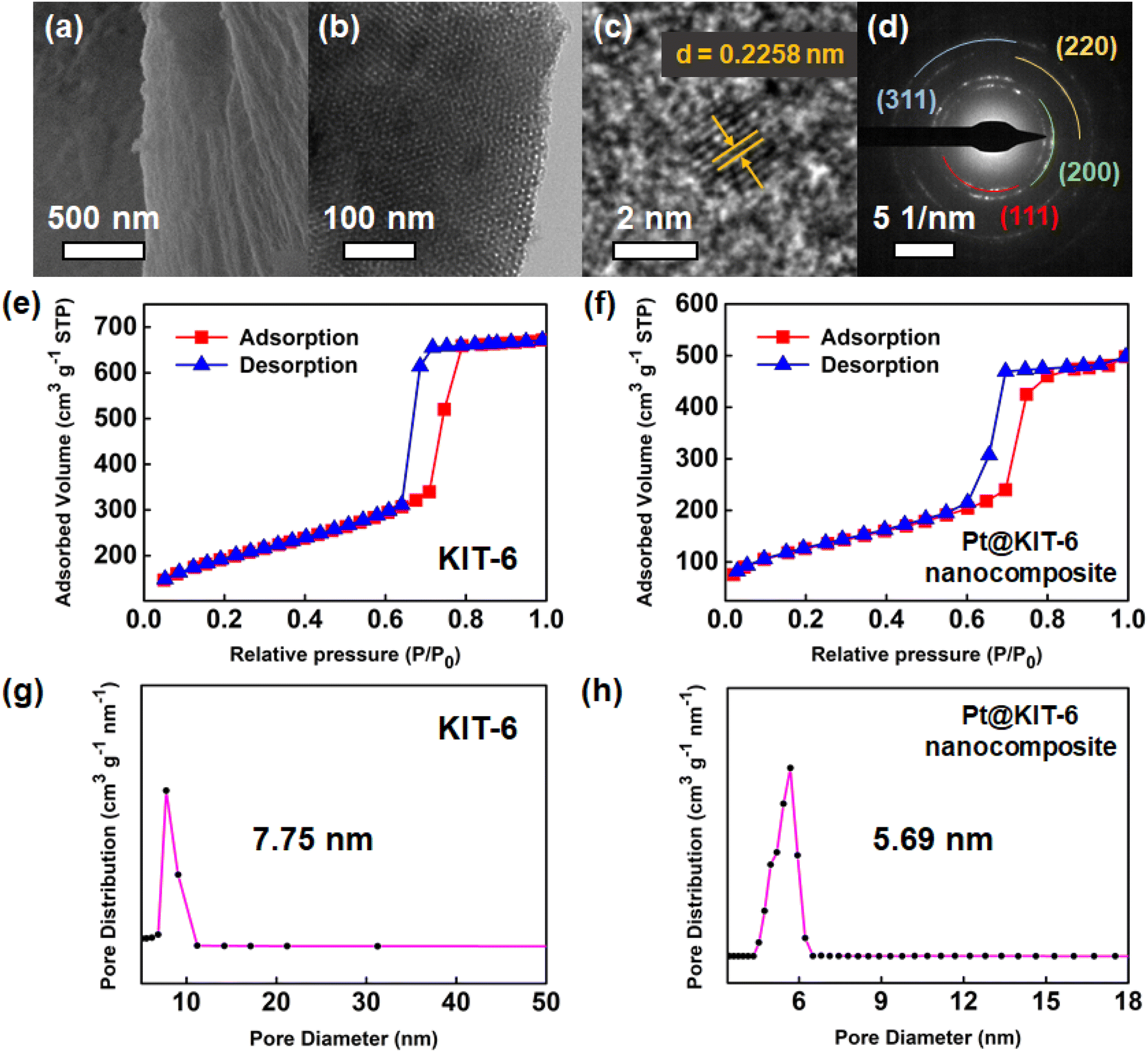 | ||
| Fig. 3 Pt@KIT-6 nanocomposite: (a) SEM image, (b) TEM image, (c) HRTEM image, (d) SAED pattern; N2 adsorption–desorption isotherms for (e) KIT-6 and (f) Pt@KIT-6 nanocomposite; pore size distributions of (g) KIT-6 and (h) Pt@KIT-6 nanocomposite. | ||
Conventional commercial Pt/C simply loads Pt particles onto the surface of bulk carbon (Fig. S1c†), which often leads to Pt aggregation and may affect the activity and selectivity of the Pt catalyst.54,55 In contrast, the porous structure of the Pt@KIT-6 nanocomposite facilitates increased adsorption of hydrogen and its stable loading structure is beneficial to further maintain the activity and selectivity of the catalyst. The N2 adsorption–desorption isotherms of KIT-6 and Pt@KIT-6 nanocomposite (Fig. 3e–h) show typical type IV isotherms, where the H1 hysteresis loop is associated with the capillary condensation step, further indicating the presence of a homogeneous mesostructure in addition to the results of electron microscopic characterization.56
The specific surface area, mean pore size, and total pore volume of KIT-6 are 665 m2 g−1, 7.75 nm, and 1.033 cm3 g−1, respectively, while these parameters for the Pt@KIT-6 nanocomposite are 451 m2 g−1, 5.69 nm, and 0.770 cm3 g−1, respectively. To further investigate the reasons for the decrease in these parameters, KIT-6 was subjected to same loading treatment as the Pt@KIT-6 nanocomposite without the addition of Pt. The three parameters were 623 m2 g−1, 7.69 nm, and 0.960 cm3 g−1 (Fig. S2†), indicating that the significant decreases in surface area, mean pore size, and total pore volume are mainly caused by the loading of Pt nanoparticles on the pores.
The composition and elemental distribution were investigated by EDS analysis. The EDS elemental mappings (Fig. 4a) show that the Pt@KIT-6 nanocomposite consists of only three elements, namely Pt, Si, and O, with no other elements, indicating a good purity of the catalyst. In addition, Pt is uniformly distributed in the catalyst system. Then, the chemical state of the Pt@KIT-6 nanocomposite was characterized by XPS. The spectra were analyzed using XPSPEAK (Version 4.1) and the background response of the peaks was eliminated using the Shirley algorithm. The binding energies of XPS spectra were calibrated according to the C 1s (C–C bonds of adventitious carbon) at 284.8 eV. The Pt 4f spectrum (Fig. 4b) shows two dominant peaks at 74.35 eV and 71.00 eV with a spacing of 3.35 eV, which is consistent with the Pt 4f5/2 and Pt 4f7/2 of Pt0, demonstrating the presence of Pt0 in the mesoporous catalyst.57 In addition, the Pt 4f spectrum shows two weak Pt2+ peaks (Pt0/Pt2+ = 3.4), which may be caused by the surface oxidation of Pt.58 The spectrum of Si 2p (Fig. 4c) shows a peak at a binding energy of 103.03 eV, which corresponds to the Si4+ state.59 Furthermore, a strong peak at 532.38 eV can be observed in the O 1s spectrum (Fig. 4d), which can be described as the O2− state.59
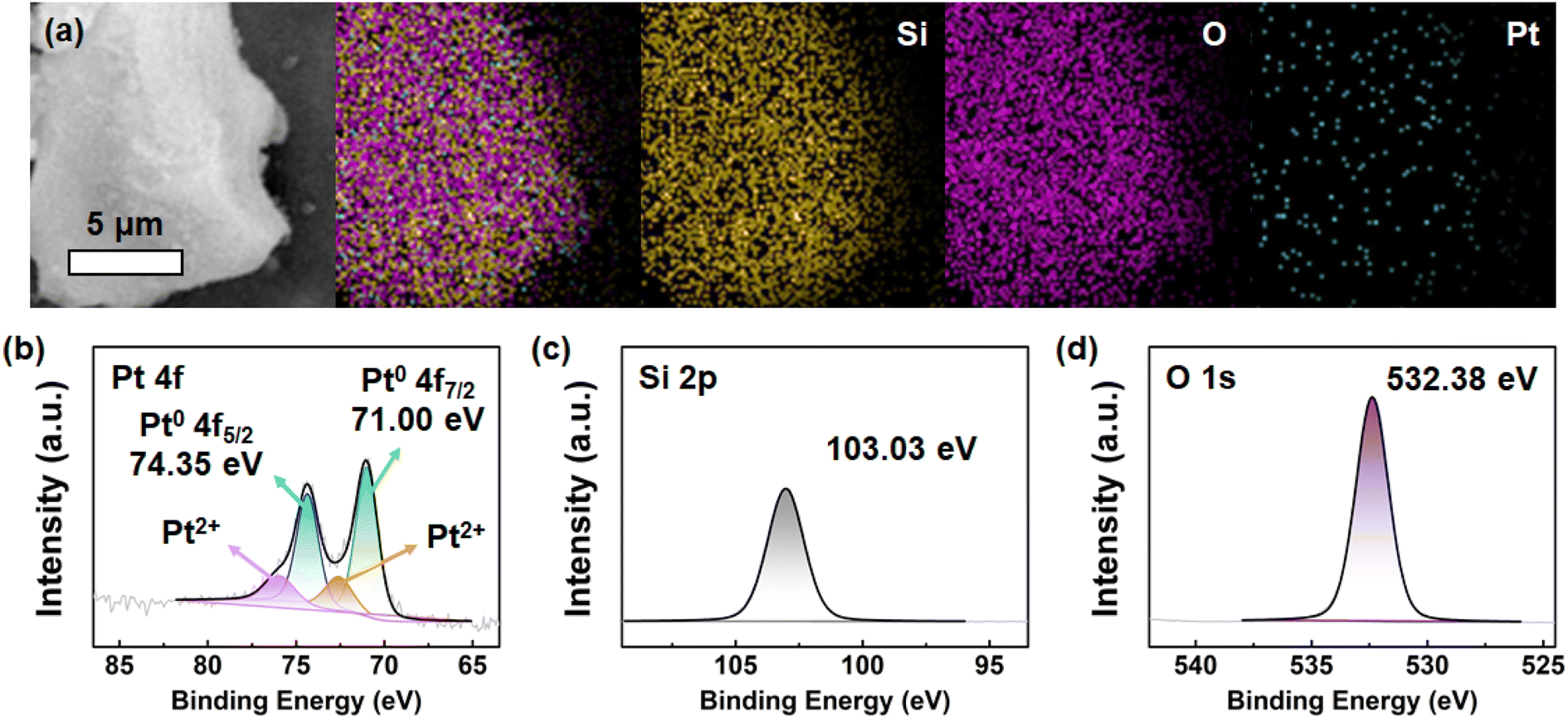 | ||
| Fig. 4 (a) EDS elemental mappings of Pt@KIT-6 nanocomposite; XPS spectra of Pt@KIT-6 nanocomposite: (b) Pt 4f, (c) Si 2p, (d) O 1s. | ||
Continuous-flow hydrogenation
The effects of flow rate, reaction temperature, hydrogen pressure, and solvent during the continuous hydrogenation of halogenated nitroarenes catalyzed by the Pt@KIT-6 nanocomposite were investigated (Table 1). When the flow rate of 4-chloronitrobenzene solution was 0.02, 0.05, 0.10, and 0.15 mL min−1 at 20 °C, the conversion of 4-chloronitrobenzene reached 99%, 99%, 99%, and 97%, and the selectivity of 4-chloroaniline was 97%, 98%, 99%, and 99% (Table 1, entries 1–4). This indicated that longer residence time enhanced dehalogenation, but shorter residence time led to inadequate hydrogenation. In addition, increasing the reaction temperature from 10 °C to 30 °C resulted in higher conversion but lower selectivity, and further increasing the temperature to 40 °C resulted in a further decrease in selectivity (Table 1, entries 5–7). Furthermore, increasing the H2 pressure from 1.0 to 3.0 MPa had no significant effect on the conversion or selectivity (Table 1, entries 3, 8, and 9). Regarding the effect of solvent, the conversion of 4-chloronitrobenzene was 68%, 40%, 19%, and 11% when the solvent was dimethylformamide, ethanol, acetonitrile, and ethyl acetate, respectively (Table 1, entries 10–13), while the hydrogenation reaction barely took place in tetrahydrofuran (Table 1, entry 14). As reported in the literature, hydrogen is more soluble in methanol, which leads to a higher activity factor for hydrogenation.60,61 In summary, the optimized flow rate, residence time, reaction temperature, H2 pressure, and solvent were 0.10 mL min−1, 5.0 min, 20 °C, 1.0 MPa, and methanol.|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Flow rate (mL min−1) | H2 pres. (MPa) | Temp. (°C) | Solvent | Conv. (%) | Selec. (%) |
| a Reaction conditions: substrate (0.1 mol L−1), catalyst (Pt@KIT-6 nanocomposite, 50 mg), back-pressure (0.9 MPa). b The catalyst was commercial bulk Pt/C (50 mg). | ||||||
| 1 | 0.02 | 1.0 | 20 | MeOH | 99 | 97 |
| 2 | 0.05 | 1.0 | 20 | MeOH | 99 | 98 |
| 3 | 0.10 | 1.0 | 20 | MeOH | 99 | 99 |
| 4 | 0.15 | 1.0 | 20 | MeOH | 97 | 99 |
| 5 | 0.10 | 1.0 | 10 | MeOH | 95 | 99 |
| 6 | 0.10 | 1.0 | 30 | MeOH | 99 | 97 |
| 7 | 0.10 | 1.0 | 40 | MeOH | 99 | 93 |
| 8 | 0.10 | 2.0 | 20 | MeOH | 99 | 99 |
| 9 | 0.10 | 3.0 | 20 | MeOH | 99 | 99 |
| 10 | 0.10 | 1.0 | 20 | DMF | 68 | 99 |
| 11 | 0.10 | 1.0 | 20 | EtOH | 40 | 99 |
| 12 | 0.10 | 1.0 | 20 | MeCN | 19 | 99 |
| 13 | 0.10 | 1.0 | 20 | EA | 11 | 99 |
| 14 | 0.10 | 1.0 | 20 | THF | Trace | — |
| 15b | 0.10 | 1.0 | 20 | MeOH | 99 | 91 |
The commercial bulk 5% (w/w) Pt/C, purchased from Sinopharm Chemical Reagent Co. without further purification, was used as a control group. Under the optimal reaction conditions of Pt@KIT-6 (Table 1, entry 3), the selectivity of 4-chloroaniline was only 91% when the catalyst was substituted with Pt/C (Table 1, entry 15). As is shown in Table S1,† the Pt loading of our Pt@KIT-6 nanocomposite was also around 5% (w/w) by ICP-OES. Therefore, the difference in activity and selectivity between Pt@KIT-6 and Pt/C was caused by the difference in carrier structure or the loading form of Pt.62
To investigate the compatibility of this continuous selective hydrogenation system with different substrates, nitroarenes with different functional groups were used as reactants, especially halogenated nitroarenes (Table 2). When common halogen groups, such as –F, –Cl, –Br, and –I, were present in the ortho, meta, or para-positions of nitroarenes, the hydrogenation system showed higher than 98% activity and selectivity (Table 2, entries 1–12). The study by Zhang et al. using the Pt single-atom catalysts for the selective hydrogenation of halogenated nitroarenes showed similar results.63 A high conversion could also be achieved for nitroarenes without any other functional group or with conventional electron-withdrawing and electron-donating groups (Table 2, entries 13–17). Meanwhile, unsaturated groups other than nitro in these substrates were retained, further demonstrating the high selectivity of the catalytic system. Furthermore, when the substrate was a nitroheterocyclic compound, the hydrogenation system still remained high conversion and selectivity, regardless of whether the substituent on the heterocycle was a halogen, electron-withdrawing, or electron-donating group (Table 2, entries 18–20). This indicated that the synthesized Pt@KIT-6 nanocomposite was suitable for the selective hydrogenation of both the phenyl ring and heterocyclic substrates. Notably, the synthesis of 4-chloroaniline at high concentration showed a conversion of 99% and selectivity of 98%, demonstrating the practical value of this continuous-flow hydrogenation system (Table 2, entry 21). The isolated yields and the production rates were then calculated and listed in Scheme S1.† The molecular structures of all arylamines were further analyzed by NMR, and are listed in the ESI.†
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Substrate | Product | No. | Conv. (%) | Selec. (%) |
| a Reaction conditions: substrate (0.1 mol L−1, methanol as solvent), catalyst (Pt@KIT-6 nanocomposite, 50 mg), flow rate (0.10 mL min−1), H2 (1.0 MPa), back-pressure (0.9 MPa), 20 °C, 5.0 min. b The reaction temperature was 50 °C. c The highly concentrated reaction conditions: substrate (2.0 mol L−1, methanol as solvent), catalyst (Pt@KIT-6 nanocomposite, 1.0 g), flow rate (0.10 mL min−1), H2 (1.0 MPa), back-pressure (0.9 MPa), 20 °C, 5.0 min. | |||||
| 1 |
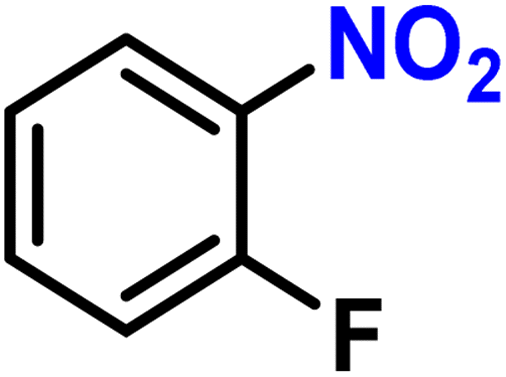
|
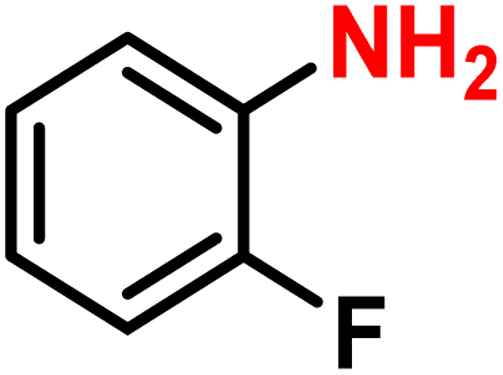
|
2a | 99 | 99 |
| 2 |
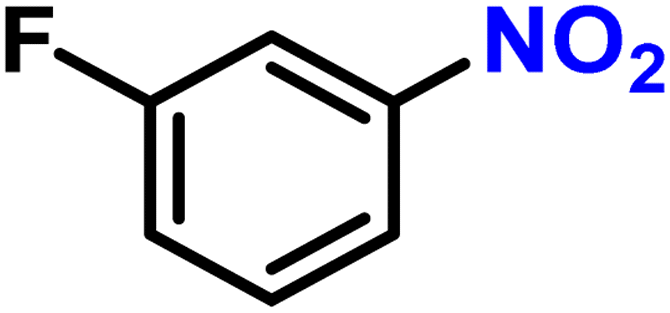
|
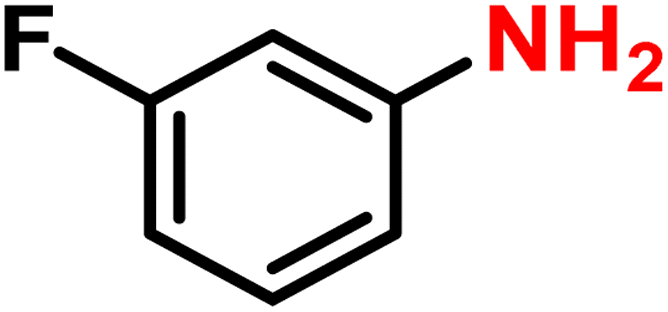
|
2b | 99 | 99 |
| 3 |
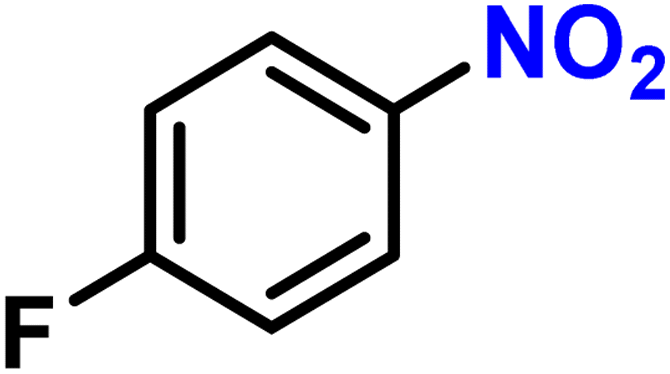
|
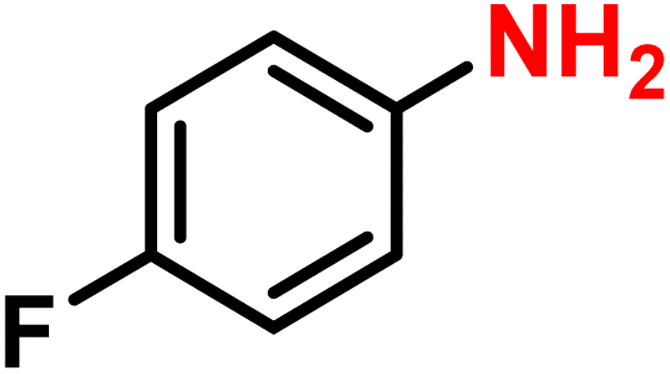
|
2c | 99 | 99 |
| 4b |
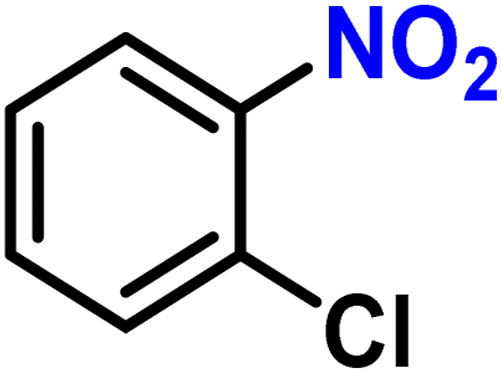
|
|
2d | 98 | 99 |
| 5 |
|
|
2e | 99 | 99 |
| 6 |
|
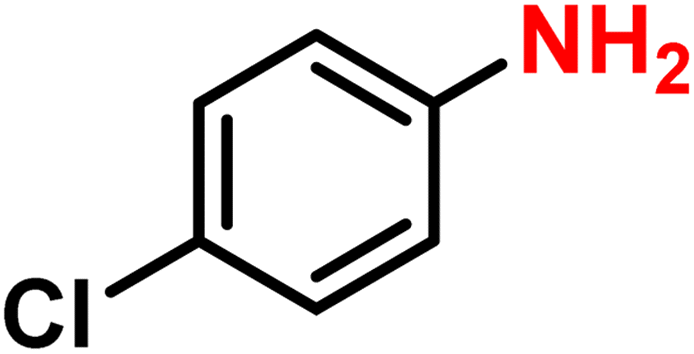
|
2f | 99 | 99 |
| 7b |
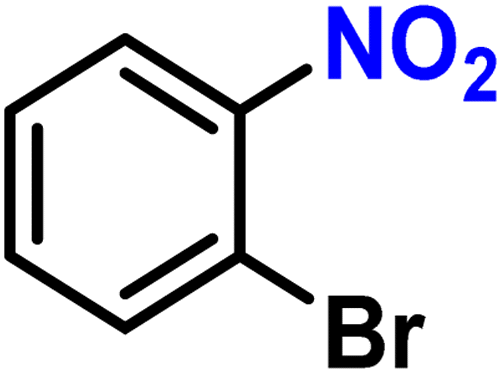
|
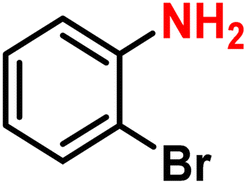
|
2g | 98 | 99 |
| 8 |
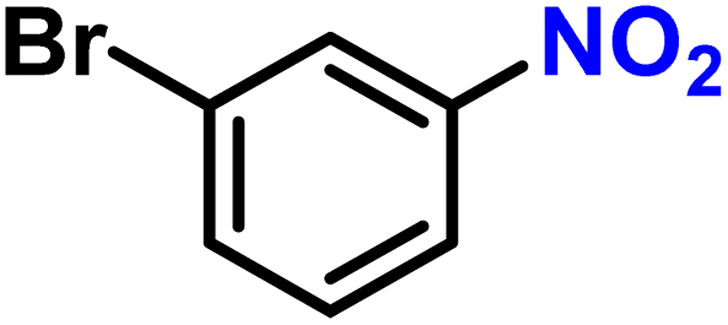
|
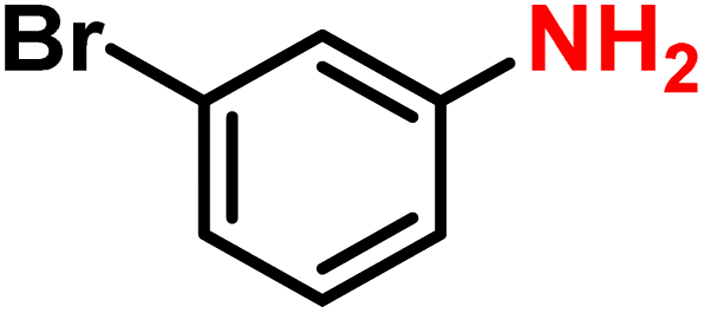
|
2h | 99 | 99 |
| 9 |
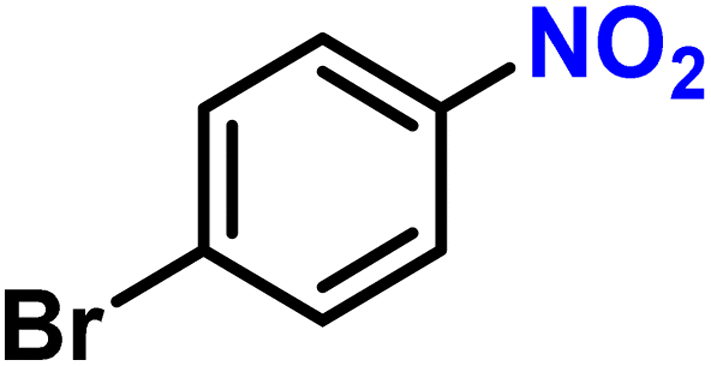
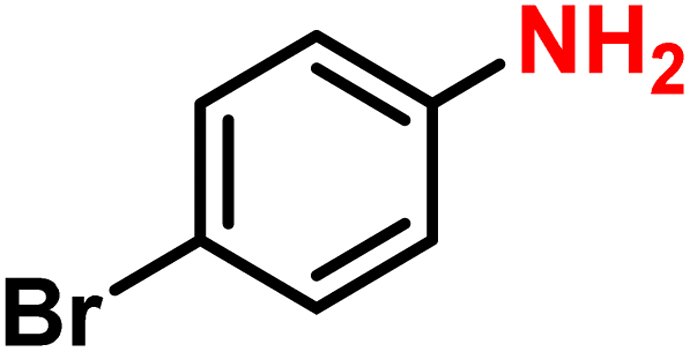
|
|
2i | 99 | 99 |
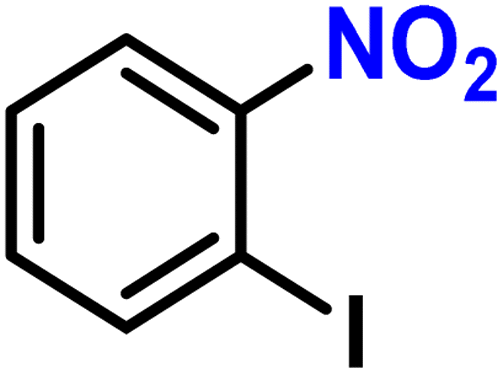
| 10b |
|
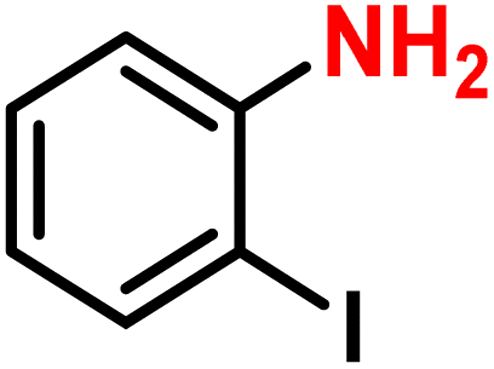
|
2j | 98 | 98 |
| 11b |
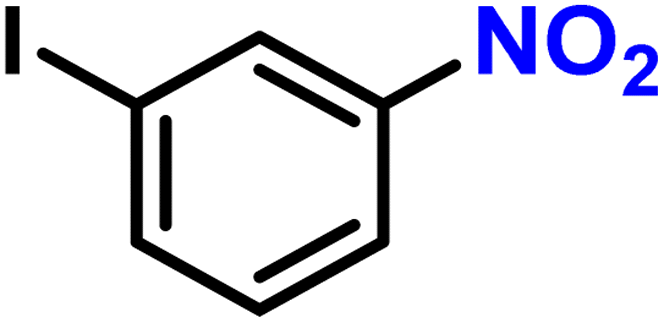
|
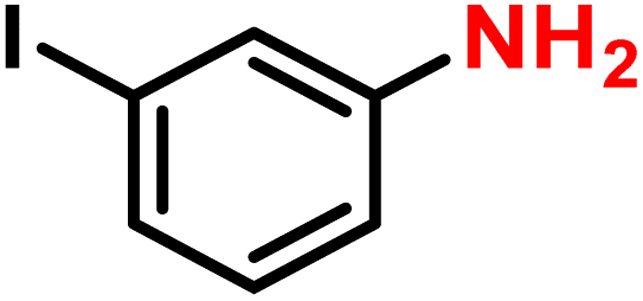
|
2k | 98 | 99 |
| 12b |
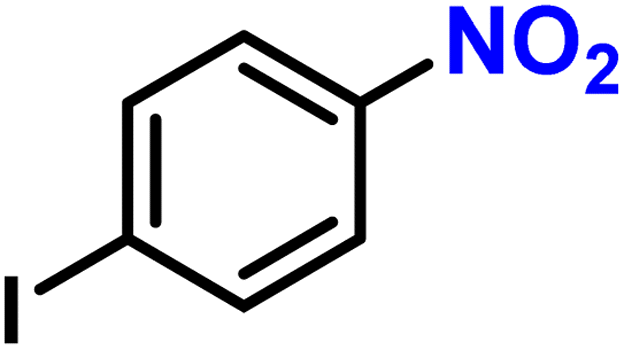
|
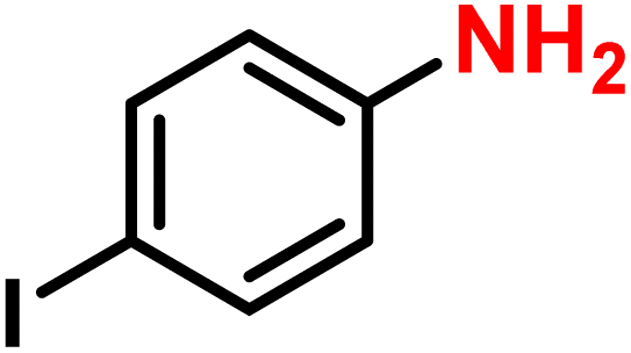
|
2l | 99 | 98 |
| 13 |
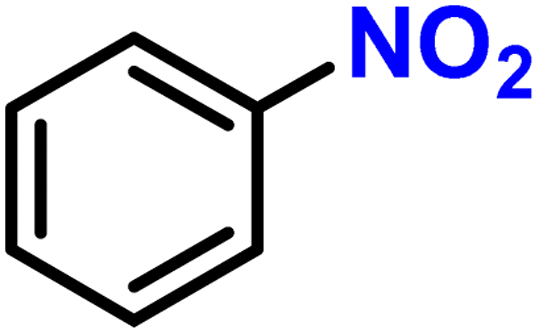
|
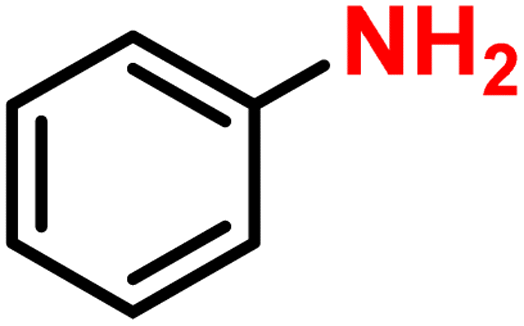
|
2m | 99 | 99 |
| 14 |
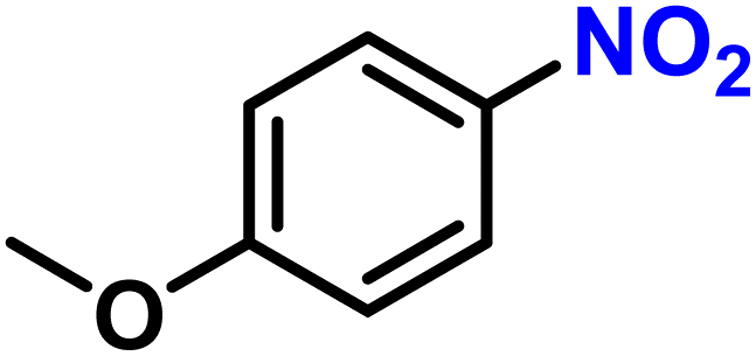
|
|
2n | 99 | 99 |
| 15 |
|
|
2o | 99 | 99 |
| 16 |
|
|
2p | 98 | 99 |
| 17 |
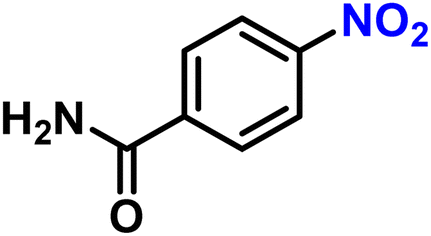
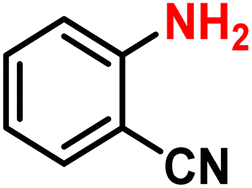
|
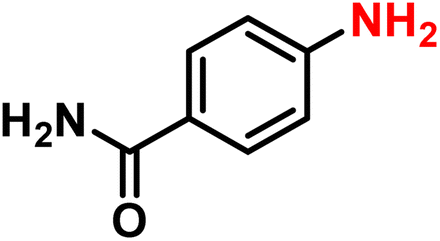
|
2q | 98 | 99 |
| 18 |
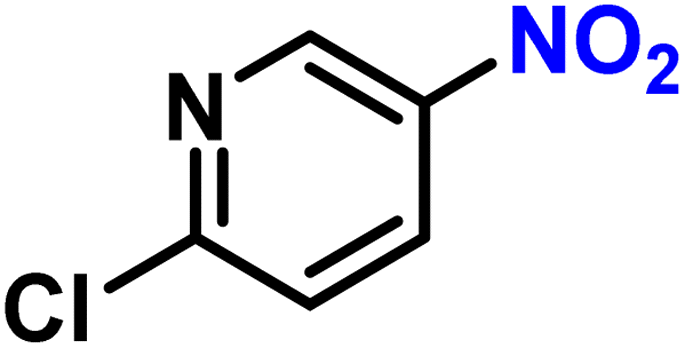
|
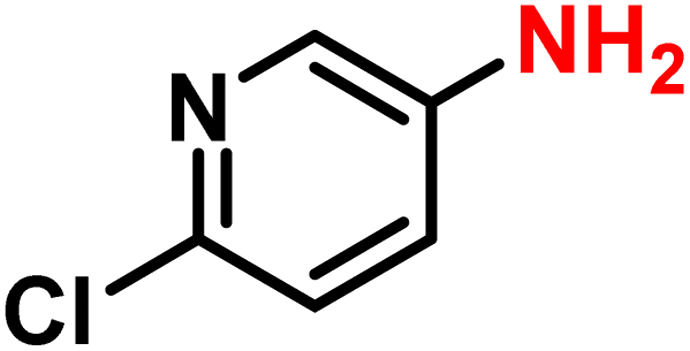
|
2r | 99 | 99 |
| 19 |
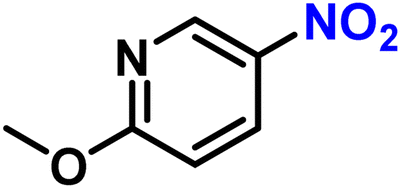
|
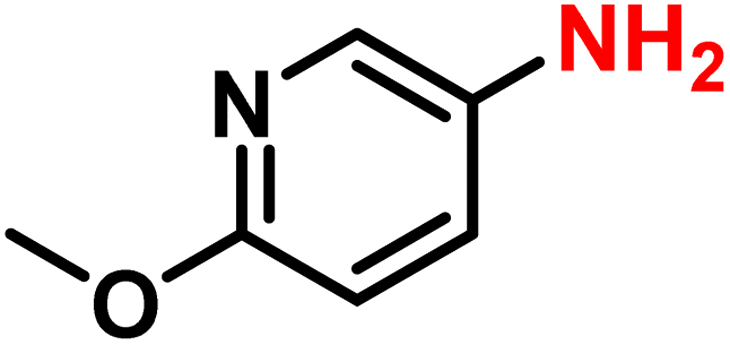
|
2s | 99 | 99 |
| 20 |
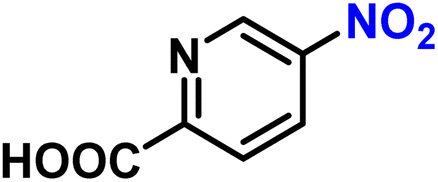
|
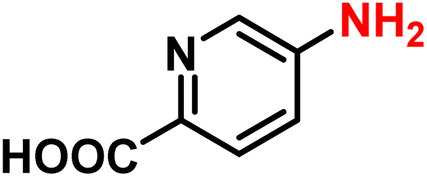
|
2t | 99 | 99 |
| 21c |
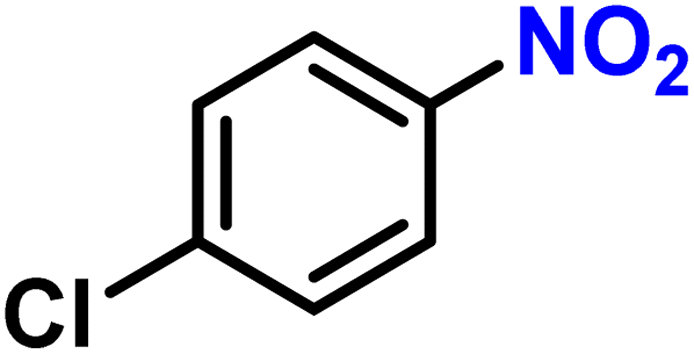
|
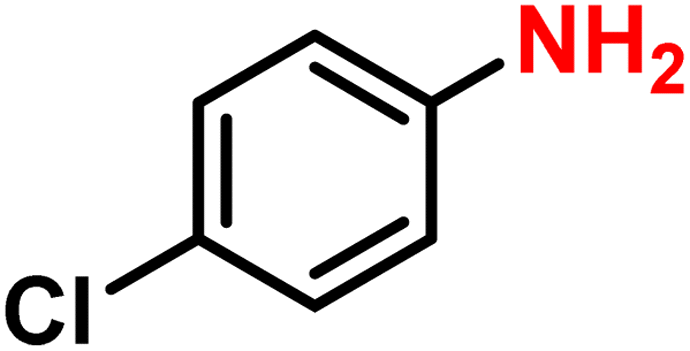
|
2f′ | 99 | 98 |

Stability of the catalyst
This continuous-flow hydrogenation system shows slightly reduced conversion and selectivity after 24 hours of continuous operation, demonstrating the excellent and stable catalytic performance of the Pt@KIT-6 nanocomposite (Fig. 5).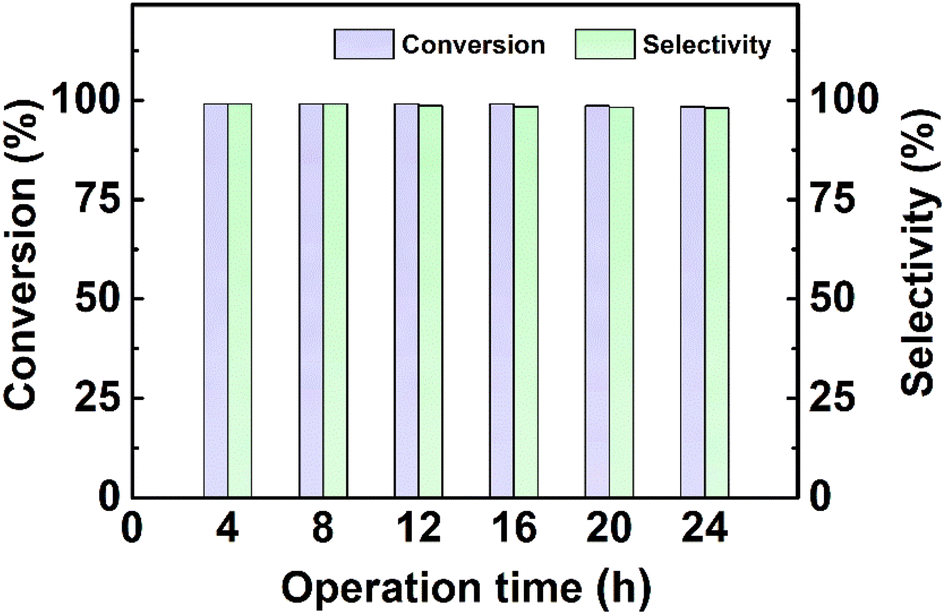 | ||
| Fig. 5 Operation time of continuous-flow hydrogenation in a microreactor. Reaction conditions: 4-chloronitrobenzene (0.1 mol L−1, methanol as solvent), catalyst (Pt@KIT-6 nanocomposite, 50 mg), flow rate (0.10 mL min−1), H2 (1.0 MPa), back-pressure (0.9 MPa), 20 °C, 5.0 min. | ||
In addition, the ICP-MS analysis shows that Pt metal is almost absent from the reaction solution after 24 h, indicating that barely any leaching of Pt from the catalyst occurs (Table S1†). In addition, the Pt@KIT-6 nanocomposite is characterized again after 24 h of reaction. The small-angle XRD pattern (Fig. S3†) of the Pt@KIT-6 nanocomposite shows four clear diffraction peaks at 0.96°, 1.11°, 1.78°, and 1.86°, which are indexed to the (211), (220), (420), and (332) planes of the mesoporous material with highly ordered 2D P6mm hexagonal symmetry.42,43 Meanwhile, the TEM image (Fig. S4†) of the Pt@KIT-6 nanocomposite demonstrates the regular honeycomb pore structure. Both small-angle XRD and TEM characterization indicate that the Pt@KIT-6 nanocomposite retains a mesoporous structure after 24 h of reaction. Moreover, the TEM image shows almost no Pt metal aggregation. The EDS elemental mappings (Fig. S5†) demonstrate that the distribution of Pt metal remains uniform in the Pt@KIT-6 nanocomposite and the ICP-OES analysis (Table S1†) further demonstrates that the content of Pt loading on KIT-6 is still around 5% (w/w) after 24 h of reaction. The EDS and ICP-OES analysis indicate that the loading form of Pt metal is stable. The wide-angle XRD pattern (Fig. S6†) shows diffraction peaks at a 2θ value of approximately 23°, which are indexed to amorphous state SiO2, and 2θ = 40.0°, 46.8°, 67.8°, and 81.7°, which are indexed to the (111), (200), (220), and (311) planes of Pt0 (JCPDS card 00-087-0640).44–46 The obtained XPS spectra of Pt 4f, Si 2p, and O 1s (Fig. S7†) are almost identical to the ones obtained before the reaction.57–59 The wide-angle XRD and XPS characterizations further demonstrate the chemical state stability of the Pt@KIT-6 nanocomposite.
To further test the stability and catalytic performance of the catalyst, the Pt@KIT-6 nanocomposite was continuously operated in the continuous-flow hydrogenation system for 72 hours under the conditions optimized above. The test results showed that the conversion of halogenated nitroarenes and the selectivity of halogenated arylamines could still reach 98% and 97%, respectively, which indicated that the Pt@KIT-6 nanocomposite had good catalytic stability (Fig. S8†). After the selective hydrogenation test, the Pt@KIT-6 nanocomposite was characterized by XRD and TEM to further verify their stability. The small-angle XRD and the wide-angle XRD patterns (Fig. S9a and b†) demonstrate the diffraction peaks assigned to the crystal planes of KIT-6 and Pt0, respectively, which are almost the same as those detected before the reaction. The TEM image (Fig. S9c†) still shows little to no Pt metal aggregation. The above results further illustrate the excellent stability of the Pt@KIT-6 nanocomposite.
In order to further analyze the catalytic performance of the Pt@KIT-6 nanocomposite, the experimental results were compared with the data reported in the literature.39,40,49,50,64,65 The conversion of 4-nitrobromobenzene and the selectivity of 4-bromoaniline catalyzed by different Pt-based catalysts under optimized conditions are listed in Table S2.† It is clear that the Pt@KIT-6 nanocomposite developed in this work had excellent conversion (>99%) and selectivity (>99%) under mild conditions. Notably, the catalytic hydrogenation time of the Pt@KIT-6 nanocomposite was only 5.0 minutes and the catalytic stability could be maintained for over 72 hours, which was considerably better than that for most of the previously reported Pt-based catalysts. Based on the above comparative analysis, the Pt@KIT-6 nanocomposite has excellent catalytic activity and stability, and is promising for the selective hydrogenation of halogenated nitroarenes.
Reaction mechanism
There is general agreement in the literature that two routes exist for the hydrogenation mechanism of nitroarenes involving Pt metal-based catalysts, namely the direct hydrogenation route and the indirect hydrogenation route (Fig. 6).66–68 The direct route is a reduction of nitroarene (I) to a nitroso (II) intermediate followed by a further rapid reduction to a hydroxylamine (III) intermediate and finally a direct reduction to arylamine (VII). The indirect route is the condensation of nitroso and hydroxylamine intermediates, formed in the direct route, to the azoxy (IV) intermediate, and then multi-step hydrogenation to obtain arylamine undergoing azo (V) and hydrazo (VI) intermediates. Based on the results of catalyst characterization and continuous-flow synthesis, the high activity of the Pt@KIT-6 nanocomposite is mainly attributed to the porous structure that facilitates hydrogen adsorption, which has been verified by N2 physisorption and electron microscopy. Meanwhile, the high selectivity is mainly due to the stable nanocomposite structure, which avoids aggregation of the loaded metals, and the continuous-flow system, which allows the timely transfer of the product to avoid dehalogenation. Furthermore, adsorption tests of 4-bromoaniline in methanol over the Pt@KIT-6 nanocomposite and commercial bulk Pt/C were performed (Fig. S10†).69 As shown in Fig. S10a,† commercial bulk Pt/C shows a faster adsorption rate and higher saturated adsorption capacity compared to the Pt@KIT-6 nanocomposite. Moreover, the adsorption reaction is a first-order reaction as indicated by plotting the fitting lines of −ln(1 − X) vs. t (Fig. S10b†), where X and t refer to the conversion of 4-bromoaniline and the adsorption time.69 The adsorption rate constants k for the Pt@KIT-6 nanocomposite and the commercial bulk Pt/C are 0.00187 min−1 and 0.0385 min−1, respectively, which is a difference of more than 20-fold. This suggests that 4-bromoaniline is difficult to adsorb on the Pt@KIT-6 nanocomposite, which greatly inhibits the dehalogenation. The above results further demonstrate that the Pt@KIT-6 nanocomposite has better selectivity.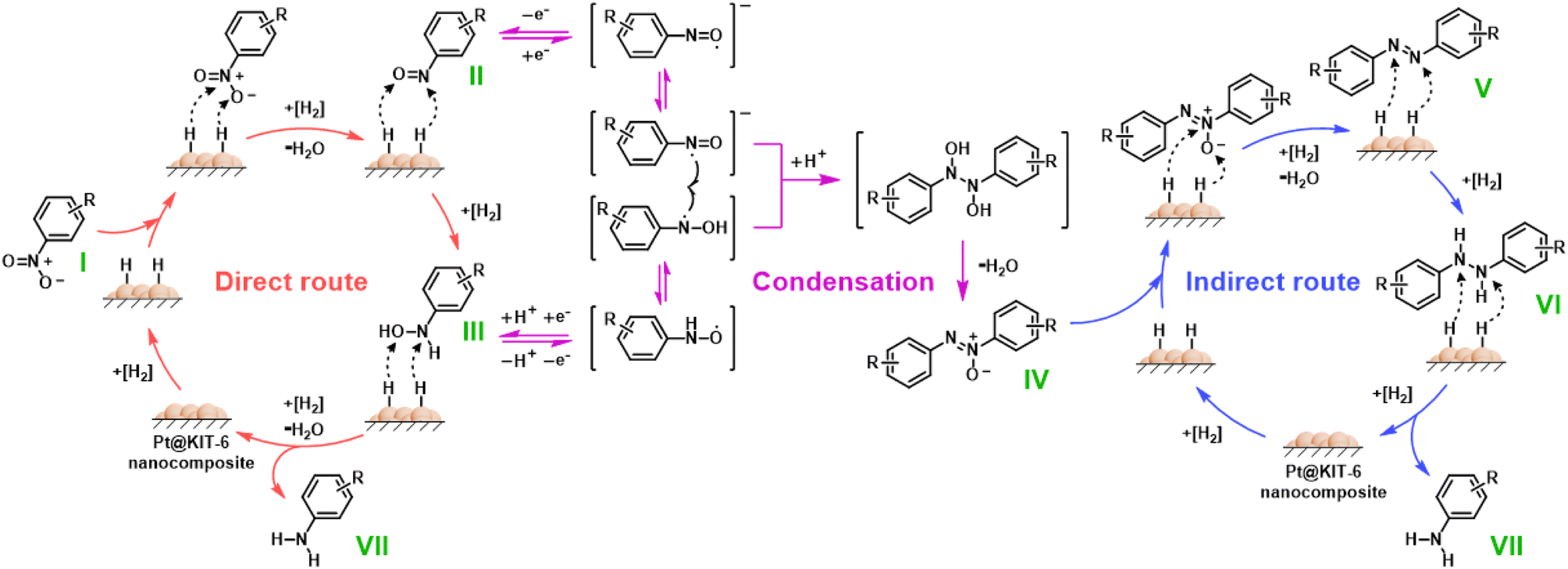 | ||
| Fig. 6 General hydrogenation mechanism of nitroarenes. | ||
Conclusions
A mesoporous Pt@KIT-6 nanocomposite, created by loading Pt metal nanoparticles onto the mesoporous material KIT-6, was applied to the continuous-flow selective hydrogenation of halogenated nitroarenes. The characterization of the Pt@KIT-6 nanocomposite demonstrates the stable structure and large specific surface area, which facilitate hydrogen adsorption, avoid aggregation of metal nanoparticles, and ensure stable high activity and selectivity over time. The Pt@KIT-6 nanocomposite shows excellent conversion and selectivity beyond that of commercial bulk Pt/C in the preparation of halogenated arylamines in a continuous-flow hydrogenation system, both reaching 99%. More importantly, the conversion and selectivity of the Pt@KIT-6 nanocomposite barely decrease after 24 hours of continuous-flow microreactor operation, effectively avoiding the cumbersome recovery of the catalyst and the effect of trace haloid acids on the reaction system. The high hydrogenation activity of the Pt@KIT-6 nanocomposite and the rapid transfer of the reaction solution by the continuous-flow system function simultaneously to inhibit dehalogenation effectively and improve conversion and selectivity. This work has important implications for the creation of novel catalysts for application in continuous-flow selective hydrogenation.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research was supported by the Joint Funds of the Zhejiang Provincial Natural Science Foundation of China under Grant No. LHDMZ23B060001 and Zhejiang Province Science and Technology Plan Project under Grant No. 2022C01179.References
- I. Sorribes, L. C. Liu and A. Corma, ACS Catal., 2017, 7, 2698–2708 CrossRef CAS.
- Y. Markushyna, C. Teutloff, B. Kurpil, D. Cruz, I. Lauermann, Y. B. Zhao, M. Antonietti and A. Savateev, Appl. Catal., B, 2019, 248, 211–217 CrossRef CAS.
- S. Zhang, C. R. Chang, Z. Q. Huang, J. Li, Z. M. Wu, Y. Y. Ma, Z. Y. Zhang, Y. Wang and Y. Q. Qu, J. Am. Chem. Soc., 2016, 138, 2629–2637 CrossRef CAS PubMed.
- W. Zhang, W. Wu, F. Guo and X. W. Dong, J. Colloid Interface Sci., 2023, 640, 1068–1079 CrossRef CAS PubMed.
- Y. J. Song, H. Wang, Z. T. Wang, B. B. Guo, K. G. Jing, Y. J. Li and L. Wu, ACS Catal., 2018, 8, 9656–9664 CrossRef CAS.
- W. Zhang, W. Wu, Y. Long, F. S. Wang and J. T. Ma, J. Colloid Interface Sci., 2018, 522, 217–227 CrossRef CAS PubMed.
- T. Song, P. Ren, Y. A. Duan, Z. Z. Wang, X. F. Chen and Y. Yang, Green Chem., 2018, 20, 4629–4637 RSC.
- Y. N. Duan, T. Song, X. S. Dong and Y. Yang, Green Chem., 2018, 20, 2821–2828 RSC.
- L. Huang, Y. Lv, S. Liu, H. S. Cui, Z. Y. Zhao, H. Zhao, P. L. Liu, W. Xiong, F. Hao and H. A. Luo, Ind. Eng. Chem. Res., 2020, 59, 1422–1435 CrossRef CAS.
- J. S. Wang, Y. Zhang, X. N. Xu and M. Bao, ACS Appl. Mater. Interfaces, 2023, 15, 8149–8156 CrossRef CAS PubMed.
- S. N. Yue, X. G. Wang, S. T. Li, Y. Sheng, X. J. Zou, X. G. Lu and C. L. Zhang, New J. Chem., 2020, 44, 11861–11869 RSC.
- C. S. Couto, L. M. Madeira, C. P. Nunes and P. Araujo, Appl. Catal., A, 2016, 522, 152–164 CrossRef CAS.
- G. Consolati, R. Ferragut, A. Galarneau, F. Renzo and F. Quasso, Chem. Soc. Rev., 2013, 42, 3821–3832 RSC.
- J. Wei, Z. K. Sun, W. Luo, Y. H. Li, A. A. Elzatahry, A. M. Al-Enizi, Y. H. Deng and D. Y. Zhao, J. Am. Chem. Soc., 2017, 139, 1706–1713 CrossRef CAS PubMed.
- A. Bhat, A. J. Hill, G. B. Fisher and J. W. Schwank, Appl. Catal., B, 2021, 297, 120476 CrossRef CAS.
- S. S. Park and C. S. Ha, Adv. Funct. Mater., 2018, 28, 1703814 CrossRef.
- B. Szczesniak, J. Choma and M. Jaroniec, Microporous Mesoporous Mater., 2018, 261, 105–110 CrossRef CAS.
- B. Chen, B. W. Hui, Y. T. Dong, Q. Sheng, X. Li, Q. L. Hao and C. J. Liu, Fuel, 2022, 324, 124405 CrossRef CAS.
- X. S. Cheng, D. X. Wang, J. C. Liu, X. Kang, H. J. Yan, A. P. Wu, Y. Gu, C. G. Tian and H. G. Fu, Nanoscale, 2018, 10, 22348–22356 RSC.
- T. Chen, Z. P. Shi, G. H. Zhang, H. C. Chan, Y. J. Shu, Q. S. Gao and Y. Tang, ACS Appl. Mater. Interfaces, 2018, 10, 42475–42483 CrossRef CAS PubMed.
- W. J. Son, J. S. Choi and W. S. Ahn, Microporous Mesoporous Mater., 2008, 113, 31–40 CrossRef CAS.
- W. F. Peng, S. W. Liu, X. G. Li, G. Feng, J. H. Xia and Z. H. Lu, Chin. Chem. Lett., 2022, 33, 1403–1406 CrossRef CAS.
- A. D. Delgado, L. Alvarez-Contreras, K. A. Beltran, P. S. Cardenas, C. Leyva-Porras and A. Aguilar-Elguezabal, Microporous Mesoporous Mater., 2020, 306, 110436 CrossRef CAS.
- X. B. Yu, M. Burkholder, S. G. Karakalos, G. L. Tate, J. R. Monnier, B. F. Gupton and C. T. Williams, Catal. Sci. Technol., 2021, 11, 2403–2413 RSC.
- C. K. P. Neeli, Y. M. Chung and W. S. Ahn, ChemCatChem, 2017, 9, 4570–4579 CrossRef CAS.
- D. J. Sui, F. Mao, H. P. Fan, Z. L. Qi and J. Huang, Chin. J. Chem., 2017, 35, 1371–1377 CrossRef CAS.
- K. Lee, M. Y. Kim and M. Choi, ACS Sustainable Chem. Eng., 2018, 6, 13035–13044 CrossRef CAS.
- L. G. Aguiar and A. F. Siqueira, Ind. Eng. Chem. Res., 2022, 61, 8708–8713 CrossRef CAS.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed.
- H. Kreissl, J. Jin, S. H. Lin, D. Schuette, S. Stortte, N. Levin, B. Chaudret, A. J. Vorholt, A. Bordet and W. Leitner, Angew. Chem., Int. Ed., 2021, 60, 26639–26646 CrossRef CAS PubMed.
- C. M. Cova, A. Zuliani, R. Manno, V. Sebastian and R. Luque, Green Chem., 2020, 22, 1414–1423 RSC.
- L. A. S. Cavaca, J. A. S. Coelho, S. D. Lucas, R. M. S. Loureiro, R. F. A. Gomes and C. A. M. Afonso, React. Chem. Eng., 2023, 8, 482–489 RSC.
- F. Brandi, M. Baumel, V. Molinari, I. Shekova, I. Lauermann, T. Heil, M. Antonietti and M. Al-Naji, Green Chem., 2020, 22, 2755–2766 RSC.
- M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC.
- N. C. Neyt and D. L. Riley, React. Chem. Eng., 2021, 6, 1295–1326 RSC.
- A. Tanimu, S. Jaenicke and K. Alhooshani, Chem. Eng. J., 2017, 327, 792–821 CrossRef CAS.
- X. N. Duan, J. B. Yin, A. X. Feng, M. M. Huang, W. S. Fu, W. F. Xu, Z. F. Huang and J. S. Zhang, J. Flow Chem., 2022, 12, 121–129 CrossRef CAS.
- Y. X. Geng, C. Chen, Z. M. Gao, X. J. Feng, W. Liu, Y. H. Li, T. N. Jin, Y. T. Shi, W. Zhang and M. Bao, ACS Appl. Mater. Interfaces, 2021, 13, 23655–23661 CrossRef CAS PubMed.
- K. J. Li, R. X. Qin, K. L. Liu, W. T. Zhou, N. Liu, Y. Z. Zhang, S. J. Liu, J. Chen, G. Fu and N. F. Zheng, ACS Appl. Mater. Interfaces, 2021, 13, 52193–52201 CrossRef CAS PubMed.
- S. Iihama, S. Furukawa and T. Komatsu, ACS Catal., 2016, 6, 742–746 CrossRef CAS.
- W. M. Xu, K. J. Chai, Y. W. Jiang, J. B. Mao, J. Wang, P. F. Zhang and Y. F. Shi, ACS Appl. Mater. Interfaces, 2019, 11, 17670–17677 CrossRef CAS PubMed.
- Y. F. Shi, Y. Wan, R. L. Liu, B. Tu and D. Y. Zhao, J. Am. Chem. Soc., 2007, 129, 9522–9531 CrossRef CAS PubMed.
- Y. F. Shi, Y. Meng, D. H. Chen, S. J. Cheng, P. Chen, T. F. Yang, Y. Wan and D. Y. Zhao, Adv. Funct. Mater., 2006, 16, 561–567 CrossRef CAS.
- D. Y. Zhao, Q. S. Huo, J. L. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- K. J. Chai, Y. F. Shi, Y. Q. Wang, P. J. Zou, Q. R. Yuan, W. M. Xu and P. F. Zhang, Nanoscale, 2020, 12, 21869–21878 RSC.
- C. L. Qin, A. X. Fan, X. Zhang, X. P. Dai, H. Sun, D. H. Ren, Z. Dong, Y. Wang, C. L. Luan, J. Y. Ye and S. G. Sun, Nanoscale, 2019, 11, 9061–9075 RSC.
- F. F. Cai, J. J. Ibrahim, Y. Fu, W. B. Kong, J. Zhang and Y. H. Sun, Appl. Catal., B, 2020, 264, 118500 CrossRef.
- A. Casu, M. Dalmases, M. X. Lin, Y. Wang, N. Homs, P. R. de la Piscina, J. Llorca, A. Figuerola and A. Falqui, Nanoscale, 2020, 12, 16627–16638 RSC.
- J. F. Liang, X. M. Zhang, L. Y. Jing and H. Q. Yang, Chin. J. Catal., 2017, 38, 1252–1260 CrossRef CAS.
- W. J. Yu, L. L. Lou, S. S. Li, T. Y. Ma, L. Z. Ouyang, L. Feng and S. X. Liu, RSC Adv., 2017, 7, 751–757 RSC.
- A. Alinezhad, T. M. Benedetti, L. Gloag, S. Cheong, J. Watt, H. S. Chen, J. J. Gooding and R. D. Tilley, ACS Appl. Nano Mater., 2020, 3, 5995–6000 CrossRef CAS.
- S. Watzele, J. Fichtner, B. Garlyyev, J. N. Schwammlein and A. S. Bandarenka, ACS Catal., 2018, 8, 9456–9462 CrossRef CAS.
- F. Xie, M. Y. Gan and L. Ma, Nanoscale, 2021, 13, 18226–18236 RSC.
- S. B. Tian, B. X. Wang, W. B. Gong, Z. Z. He, Q. Xu, W. X. Chen, Q. H. Zhang, Y. Q. Zhu, J. R. Yang, Q. Fu, C. Chen, Y. X. Bu, L. Gu, X. M. Sun, H. J. Zhao, D. S. Wang and Y. D. Li, Nat. Commun., 2021, 12, 3181 CrossRef CAS PubMed.
- Y. Xu, M. Y. Chu, F. F. Liu, X. C. Wang, Y. Liu, M. H. Cao, J. Gong, J. Luo, H. P. Lin, Y. Y. Li and Q. Zhang, Nano Lett., 2020, 20, 6865–6872 CrossRef CAS PubMed.
- J. Xu, Y. Hong, M. J. Cheng, B. Xue and Y. X. Li, Microporous Mesoporous Mater., 2019, 285, 223–230 CrossRef CAS.
- A. M. Motin, T. Haunold, A. V. Bukhtiyarov, A. Bera, C. Rameshan and G. Rupprechter, Appl. Surf. Sci., 2018, 440, 680–687 CrossRef CAS.
- R. Vakili, E. K. Gibson, S. Chansai, S. J. Xu, N. Al-Janabi, P. P. Wells, C. Hardacre, A. Walton and X. L. Fan, ChemCatChem, 2018, 10, 4238–4242 CrossRef CAS PubMed.
- Y. D. Ma, S. S. Li, T. H. Zhang, Y. Y. Zhang, X. Y. Wang, Y. H. Xiao, Y. Y. Zhan and L. L. Jiang, Nanoscale, 2021, 13, 5026–5032 RSC.
- R. A. Rajadhyaksha and S. L. Karwa, Chem. Eng. Sci., 1986, 41, 1765–1770 CrossRef CAS.
- J. Wang, C. Q. Lv, J. H. Liu, R. R. Ren and G. C. Wang, Int. J. Hydrogen Energy, 2021, 46, 1592–1604 CrossRef CAS.
- J. B. Wu, L. K. Xiong, B. T. Zhao, M. L. Liu and L. Huang, Small Methods, 2020, 4, 1900540 CrossRef CAS.
- H. Zhang, X. C. Zhang, Q. D. Sun, Q. He, H. B. Ji and X. H. He, Chem. Eng. J., 2023, 455, 140808 CrossRef CAS.
- W. Shi, B. S. Zhang, Y. M. Lin, Q. Wang, Q. Zhang and D. S. Su, ACS Catal., 2016, 6, 7844–7854 CrossRef CAS.
- W. C. Du, S. X. Xia, R. F. Nie and Z. Y. Hou, Ind. Eng. Chem. Res., 2014, 53, 4589–4594 CrossRef CAS.
- P. M. Uberman, C. S. Garcia, J. R. Rodriguez and S. E. Martin, Green Chem., 2017, 19, 739–748 RSC.
- X. N. Duan, X. P. Wang, X. K. Chen and J. S. Zhang, Org. Process Res. Dev., 2021, 25, 2100–2109 CrossRef CAS.
- Y. Y. Guo, Z. H. Li, T. Y. Xia, Y. L. Du, X. M. Mao and Y. Q. Li, Nat. Commun., 2019, 10, 4420 CrossRef PubMed.
- F. Zhang, C. Liang, M. Z. Chen, H. B. Guo, H. Y. Jiang and H. X. Li, Green Chem., 2013, 15, 2865–2871 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00437f |
| This journal is © The Royal Society of Chemistry 2023 |