DOI:
10.1039/D3NA00466J
(Paper)
Nanoscale Adv., 2023,
5, 4735-4751
Synergistic redox enhancement: silver phosphate augmentation for optimizing magnesium copper phosphate in efficient energy storage devices and oxygen evolution reaction†
Received
28th June 2023
, Accepted 4th August 2023
First published on 7th August 2023
Abstract
The implementation of battery-like electrode materials with complicated hollow structures, large surface areas, and excellent redox properties is an attractive strategy to improve the performance of hybrid supercapacitors. The efficiency of a supercapattery is determined by its energy density, rate capabilities, and electrode reliability. In this study, a magnesium copper phosphate nanocomposite (MgCuPO4) was synthesized using a hydrothermal technique, and silver phosphate (Ag3PO4) was decorated on its surface using a sonochemical technique. Morphological analyses demonstrated that Ag3PO4 was closely bound to the surface of amorphous MgCuPO4. The MgCuPO4 nanocomposite electrode showed a 1138 C g−1 capacity at 2 A g−1 with considerably improved capacity retention of 59% at 3.2 A g−1. The increased capacity retention was due to the fast movement of electrons and the presence of an excess of active sites for the diffusion of ions from the porous Ag3PO4 surface. The MgCuPO4–Ag3PO4//AC supercapattery showed 49.4 W h kg−1 energy density at 550 W kg−1 power density and outstanding capacity retention (92% after 5000 cycles). The experimental findings for the oxygen evolution reaction reveal that the initial increase in potential required for MgCuPO4–Ag3PO4 is 142 mV, indicating a clear Tafel slope of 49 mV dec−1.
1. Introduction
Nowadays, the most commonly used energy resources are fossil fuels, which lead to the emission of hazardous gases into the environment. Moreover, the increasing demand for energy is resulting in the quick depletion of these energy resources. All of these circumstances require renewable energy resources to fulfill energy requirements. However, due to the intermittent behavior of wind and solar energy resources, the use of energy storage devices (ESDs) has become more prominent. An ESD balances the demands and supply of electricity. Two similar kinds of electrode are used in electric double-layer capacitors (EDLCs) that deliver quick bursts of energy, long cycle life, and exceptional power density. However, they have low energy density, limiting their ability to meet increasing technological needs.1,2 Lithium-ion batteries have high energy but have a disadvantage in delivering quick bursts of energy, which significantly lowers their life span compared to an EDLC. Furthermore, if overloaded or overheated, an LIB can explode.3,4 There have been numerous efforts to broaden the EDLC working potential and increase its energy while maintaining its power density. A new asymmetric device known as a supercapattery has recently received a lot of attention because it combines capacitive-graded materials (electrostatic storage) and other battery-graded materials (faradaic storage). RuO2, a well-known outstanding pseudocapacitor electrode, demonstrated an exceptional capacitance of 1340 F g−1. But its high expense and ecotoxicity prevent it from being used commercially. Other oxide-based metals, such as NiO and Co3O4, show promise, but their electrical resistance is low. As a result, it is critical to identify materials for a supercapattery that are inexpensive but have similar effectiveness to current materials.5,6 The catalysts used in the oxygen electrode for the oxygen evolution reaction (OER) have garnered significant attention due to their crucial role in these systems. There is an urgent need to develop highly efficient catalysts, free of noble metals, that exhibit superior catalytic activity for the OER. This need arises from the desire to replace expensive and scarce RuO2 and IrO2 catalysts.7
Scientists are presently focusing on tertiary phosphides as encouraging electrodes in supercapacitors. For example, Lei et al.8 used a simple hydrothermal process to make a ZnNiCoP–NF nanostructure, which showed 1111 C g−1 capacity at 10 A g−1. They demonstrated that the fabricated electrochemical design is ideal for energy-storage uses. Yang et al.9 produced Ni–Co–Mo ternary phosphide formed on carbon cloth fibers, which had a specific capacitance of 433 F g−1 at 1 A g−1 current density.
Silver is undoubtedly a viable substitute for carbon-based materials. Moon et al.10 developed a translucent and flexible supercapacitor with silver nanowires coated with gold. Despite their widespread use in bendable and wearable electrical devices, silver nanowire networks have high contact resistance among the nanowires.11–13 Silver nanoparticles with graphene, metal oxides, carbon nanotubes, and conducting polymers were tried earlier. A matrix of Ag with carbon in a composite form shows no pseudocapacitance. However, due to the increased electrical activity, its EDLC performance improves. The Ag/PANI nanocomposite synthesized by Patil et al.14 and Tang et al.15 showed that the presence of Ag in a PANI heterostructure makes the movement of ions faster. The Ag coating on PANI produces high specific energy due to quicker electron transport and electrolyte access via a porous network.16 Copper phosphate (Cu2PO4) is thought to be a perfect alternative to conventional metallic phosphates since Cu has significantly greater conductance. Cu2PO4 exhibits further intriguing dielectric and thermal characteristics. The efficacy of Cu2PO4 in supercapacitors has received little attention. Its electrochemical performance was improved by incorporating metal ions, which increases the conductivity. Thus, additional attempts to enhance the electrical conductivity of such electrode materials are required. The addition of another metal ion with similar properties can significantly improve the electrical characteristics. Thus, the movement of ions between the two metal phosphates becomes easier and the interfacial impedance of the composite substance can be reduced. Additionally, a large useful surface area is accessible for a redox reaction, increasing the capacity and power density of the supercapacitor.17
Herein, precipitation followed by calcination was used to produce silver phosphate nanoparticles (Ag3PO4 NPs) on amorphous MgCuPO4. Because of its high electrical conductivity, Ag was selected to improve the electrical conductivity of amorphous MgCuPO4. To prevent the recombination of particles, an amorphous MgCuPO4 and silver complex (Ag(NH3)2+) mixture was made using a sonochemical technique. MgCuPO4–Ag3PO4 composites were developed to raise the energy density, reduce resistance, and expand the supercapattery cycle life. The presence of low-Eg metal ions in the amorphous structure of MgCuPO4 is still a novel way to increase the supercapacitive performance. Furthermore, the application of the oxygen evolution reaction (OER) was also examined in the context of MgCuPO4–Ag3PO4 composites. The impact of different MgCuPO4 to Ag3PO4 weight ratios on the overall storage capability was investigated. A comprehensive charge transfer procedure between MgCuPO4 and solid Ag3PO4 was observed.
2. Experimental work
2.1. Materials
Carbon black, magnesium nitrate hexahydrate (Mg(NO3)2·6H2O), hydrochloric acid (HCl), copper(II) nitrate tri-hydrate (Cu(NO3)2·3H2O), NMP, and silver nitrate (AgNO3) were received from Friendemann Schmidt. Tri-sodium phosphate (Na3PO4) and potassium hydroxide pellets (KOH) were bought from Duksan Pure Chemicals. All of the materials used were of analytical quality and were used without purification. Deionized water (DIW) was used throughout the experiments.
2.2. Synthesis of MgCuPO4–Ag3PO4
A hydrothermal and sonochemical technique was used to synthesize MgCuPO4–Ag3PO4 composites with various weight ratios of MgCuPO4 and Ag3PO4. The as-prepared MgCuPO4 (set A) was made using the methods described in the literature.18 First, 0.8 M magnesium nitrate hexahydrate (Mg(NO3)2·6H2O) solution was mixed with a 0.8 M solution of copper(II) nitrate tri-hydrate (Cu(NO3)2·3H2O). The mixture was stirred continuously for 25 min and transferred into an autoclave and heated at 140 °C. The centrifuge machine (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm Model RST-10M) was used to eliminate impurities from the solution. After drying at 40 °C the prepared MgCuPO4 was collected. The addition of NH3 into a 0.5 M solution of AgNO3 reduces Ag+. A horn sonicator was used for sonication until a diaminesilver(I) complex was produced. The complex solution of diaminesilver(I) was then combined with 0.02 g of MgCuPO4 solution and continuously stirred (3–4 h). The mixture was rinsed many times with DIW before drying for 4 h at 40 °C. Finally, the desiccated materials were calcined for 3 h at 300 °C and labeled S1. Two more MgCuPO4–Ag3PO4 nanocomposites were made for optimization reasons by altering the wt% ratio of as-prepared MgCuPO4 to 0.03 and 0.05 g and they were designated S2 and S3, respectively. Fig. 1 shows the synthetic process for MgCuPO4–Ag3PO4 nanocomposites. Table 1 shows different wt% ratios in three different composites of MgCuPO4–Ag3PO4 (S1, S2, and S3).
000 rpm Model RST-10M) was used to eliminate impurities from the solution. After drying at 40 °C the prepared MgCuPO4 was collected. The addition of NH3 into a 0.5 M solution of AgNO3 reduces Ag+. A horn sonicator was used for sonication until a diaminesilver(I) complex was produced. The complex solution of diaminesilver(I) was then combined with 0.02 g of MgCuPO4 solution and continuously stirred (3–4 h). The mixture was rinsed many times with DIW before drying for 4 h at 40 °C. Finally, the desiccated materials were calcined for 3 h at 300 °C and labeled S1. Two more MgCuPO4–Ag3PO4 nanocomposites were made for optimization reasons by altering the wt% ratio of as-prepared MgCuPO4 to 0.03 and 0.05 g and they were designated S2 and S3, respectively. Fig. 1 shows the synthetic process for MgCuPO4–Ag3PO4 nanocomposites. Table 1 shows different wt% ratios in three different composites of MgCuPO4–Ag3PO4 (S1, S2, and S3).
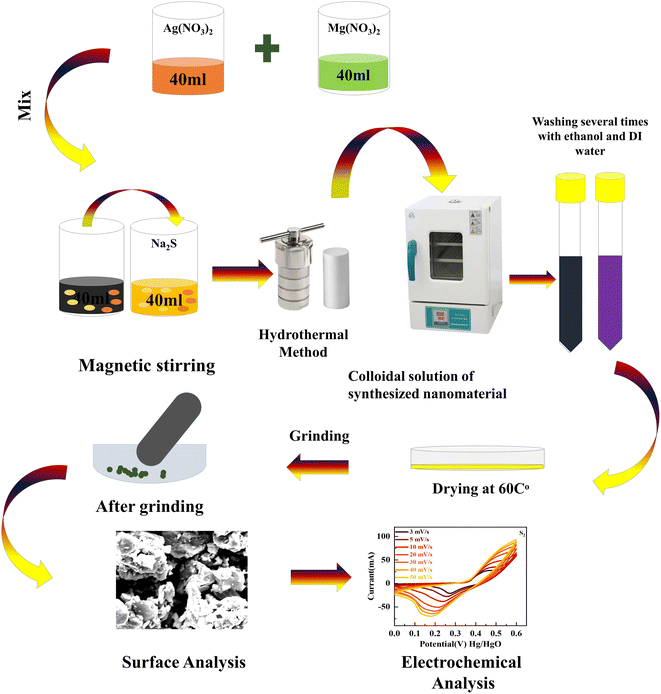 |
| | Fig. 1 Systematic illustration of the hydrothermal technique to synthesize MgCuPO4–Ag3PO4 nanocomposites. | |
Table 1 Different weight ratios of Ag to MgCuPO4 for the synthesis of MgCuPO4–Ag3PO4 nanocomposites
| Sample |
MgCuPO4 mass |
Ag mass |
Weight ratio (%) |
| S1 |
20 mg |
0.4 mg |
0.083:1 |
| S2 |
30 mg |
0.4 mg |
0.145:1 |
| S3 |
50 mg |
0.4 mg |
0.232:1 |
2.3. Characterization
2.3.1. Structural and surface analysis.
The crystalline phases of the materials (MgCuPO4, S1, S2, and S3) were analyzed using XRD (D5000, Siemens). Kα radiation with 1.54 Å wavelength and 0.02 s−1 scan rate was used to determine the crystallinity of S1, S2, and S3. FTIR (Thermo Scientific Nicolet IS10 Smart ITR) was used to investigate the purity of the samples (S1, S2, and S3) which were scanned at 1 cm−1 resolution. X-ray photoelectron spectroscopy (XPS) measurements were taken at binding energies from 0 to 1000 eV, with 1 eV resolution. Single XPS spectra were performed at a resolution of 0.1 eV. Origin Pro 8.1 was used to fit the spectra with numerous Gaussian curves. Scanning electron microscopy (SEM) was applied to analyze the morphological characteristics of the MgCuPO4–Ag3PO4 nanocomposites. Jenway's 6800 design was used to determine the optical absorbance characteristics over a spectral range of 200 to 800 nm.
2.3.2. Electrochemical studies.
The active materials (S1, S2, and S3) were coated onto nickel foam (NF) and employed as a working electrode in an electrochemical research setup. In a three-cell setup, the counter and reference electrodes were platinum wire and Hg/HgO, respectively. Throughout the electrochemical studies in a typical cell, a 1 M potassium hydroxide (KOH) solution was kept constant. The slurry for the working electrode was prepared by blending 75% active material (MgCuPO4–Ag3PO4) and 15% carbon black, which were bonded together with the help of PVDF binder (10%). This slurry was mixed continuously for 5–6 h to produce a homogeneous suspension. This suspension was then easily applied to NF. Before starting the experiment, the NF was thoroughly cleaned with HCl, ethanol, acetone, and DIW. The NMP slurry was coated on a 1 × 1 cm2 area of NF. The total weight of the active material was roughly 5.5 mg. Furthermore, in the supercapattery application, activated carbon (AC) and the best composite were used as the positive and negative electrodes. The electrochemical properties of all of the materials were studied using a workstation CS300 potentiostat. A multi-channel Autolab, PGSTAT30 potentiostat was used for the Mott–Schottky (MS) measurements.
3. Results and discussion
3.1. Characterization of materials
3.1.2. Structural analysis of MgCuPO4–AgPO4.
The crystallinities of MgCuPO4, S1, S2, and S3 were studied by XRD and the outputs are depicted in Fig. 2. First, the XRD of MgCuPO4 was studied, as indicated by Fig. 2(a). There were three different XRD patterns represented by three different colors assigned to Mg3(PO4)2, Cu3(PO4)2, and MgCuPO4. The diffraction spikes at 2θ = 12°, 22°, 23.2°, 26°, 28.32°, 31.3°, 36.5°, 38°, 48.23°, 52.8°, and 57.1° belong to the (110), (101), (−111), (210), (021), (121), (−221), (031), (330), (−331), and (150) planes, which match JCPDS 33-0876.19 The XRD peaks for Cu3(PO4)2 showed diffraction spikes at 2θ = 15.32°, 21.2°, 25°, 31.33°, 35°, 37°, 40°, 44.3°, and 56.5° belonging to (001), (110), (−101), (001), (121), (210), (200), (013), and (−221), which matches JCPDS 01-080-0991.20 The XRD pattern for MgCuPO4 indicated almost similar diffraction patterns to Mg3(PO4)2 and Cu3(PO4)2. The width of these spikes was evidence of the amorphous nature of MgCuPO4. The incorporation of Ag3PO4 is indicated in Fig. 2(b). Three different XRD patterns for S1, S2, and S3 are represented in Fig. 2(b). There were multiple reflections associated with S1, S2, and S3. The XRD pattern shows the crystalline nature of Ag3PO4. The strong peaks for Ag3PO4 at 2θ = 29.1°, 33.3°, 36.6°, 47.5°, 52.1°, 55.0°, and 57.5° correspond to the (200), (210), (211), (310), (222), (320), and (321) planes. This XRD pattern correlated with JCPDS 06-0505, which shows the formation of Ag3PO4.21 As the amount of MgCuPO4 decreased, the intensity of XRD spikes for Ag3PO4 increased. The lack of a positional shift in the Ag3PO4 peaks indicates that Ag3PO4 was not incorporated into the MgCuPO4 lattice, but rather developed on the MgCuPO4 surface.22Table 2 shows the lattice parameters of the samples.
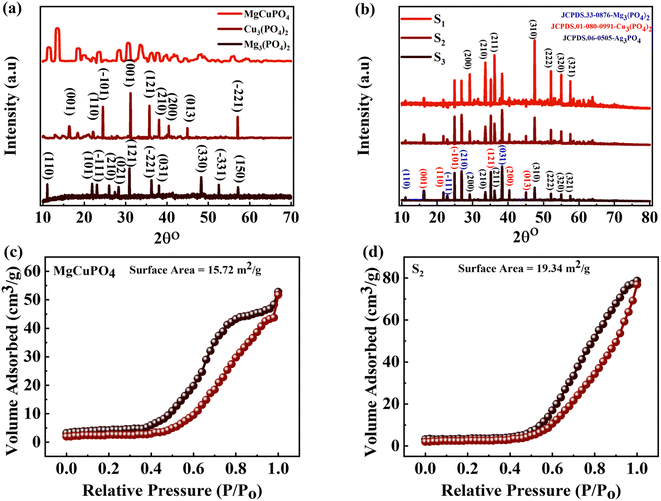 |
| | Fig. 2 (a) The XRD pattern for Mg3(PO4), Cu3(PO4), and MgCuPO4. (b) The XRD pattern for MgCuPO4–Ag3PO4 (S1, S2, S3) nanocomposites. (c and d) BET measurement for MgCuPO4 and the S2 nanocomposite. | |
Table 2 Structural parameters for the analysis of MgCuPO4 and MgCuPO4–Ag3PO4 nanocomposites
| Sample |
Lattice parameters |
Volume (nm)3 |
Particle size (nm) |
Strain (εr) (10−3) |
Density (g cm−3) |
Dislocation density (1015 m−2) |
|
α
|
β
|
Γ
|
| Standard |
7.599 |
8.235 |
5.076 |
|
|
|
|
|
| MgCuPO4 |
7.597 |
8.237 |
5.075 |
0.38 |
40–50 |
0.65 |
5.4087 |
0.55 |
| S1 |
7.600 |
8.240 |
5.075 |
0.41 |
51–56 |
0.71 |
5.4093 |
0.58 |
| S2 |
7.613 |
8.243 |
5.074 |
0.43 |
53–58 |
0.69 |
5.4098 |
0.63 |
| S3 |
7.615 |
8.247 |
5.074 |
0.44 |
52–59 |
0.70 |
5.4100 |
0.71 |
The specific surface areas (SSAs) of MgCuPO4 and the MgCuPO4–Ag3PO4 (S2) nanocomposite were calculated using the Brunauer–Emmett–Teller (BET) technique, because S2 demonstrated the best performance compared to S1 and S3, as described in the following part. The N2 adsorption–desorption isotherms of MgCuPO4 and S2 are shown in Fig. 2(c) and (d). The BET graphs for MgCuPO4 and S2 exhibit an IV-type hysteresis loop, showing their porous structure. The porous behavior of MgCuPO4 and S2 resulted in decreases in electrolyte ion diffusion time. The SSA of MgCuPO4 and S2 were 15.72 and 19.34 m2 g−1, respectively. The total pore volumes for MgCuPO4 and S2 estimated through BET calculations were 0.04928 and 0.052596 cm3 g−1, respectively. The smaller SSA and pore volume of MgCuPO4 compared to S2 were due to the absence of Ag3PO4 in the MgCuPO4–Ag3PO4 nanocomposite. Furthermore, this may be due to the distribution of Ag3PO4 NPs into MgCuPO4.23
Fig. 3(a) and (b) show the FTIR spectra of amorphous MgCuPO4, S1, S2, and S3. The band for the O–P–O bond (v4 PO43−) for the triply degenerate asymmetric mode occurred at 563 cm−1. While the band at 1036 cm−1 belongs to the P–O bond (v3 PO43−) stretching mode. The above-mentioned bands are evidence of the presence of phosphate (PO43−). The bands moved to a lower wavenumber as the amount of Ag3PO4 increased in the MgCuPO4–Ag3PO4 nanocomposites.24 The band appearing at 1645 cm−1 belongs to v1 (A1) H2O and those at 3200–3800 cm−1 belong to v2 (A1) H2O. After the incorporation of Ag3PO4 (S1, S2, and S3), the band intensities rose considerably, showing that the percentages of phosphorous and water on the MgCuPO4–Ag3PO4 surface increased.25 The adsorbed water resulted in increased storage performance due to an enhanced inter-particle path.26,27
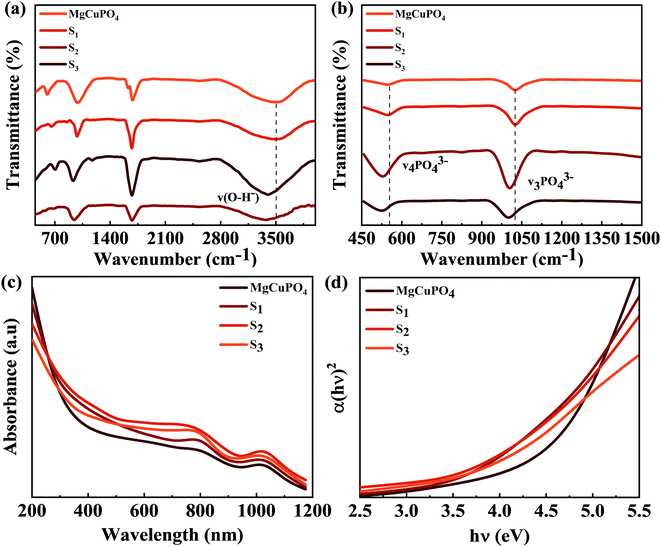 |
| | Fig. 3 (a) FTIR spectra for MgCuPO4, S1, S2, and S3 at 500–4000 cm−1. (b) FTIR spectra for MgCuPO4, S1, S2, and S3 at 450–1500 cm−1. (c) UV-vis spectra for MgCuPO4, S1, S2, and S3. (d) The band gap determination for MgCuPO4, S1, S2, and S3. | |
3.1.3. Optical properties of MgCuPO4–AgPO4.
UV-vis absorption spectroscopy was applied to check the impact of Ag3PO4 on the optical performance of MgCuPO4. The spectra are shown in Fig. 3(c). In a deformed octahedral coordinated environment, MgCuPO4 has an absorption edge at 808 nm and a second absorption edge at 1000 nm, which are due to Cu+ and Mg+, respectively.28,29 Because of the decreasing fractions of MgCuPO4 found in MgCuPO4–Ag3PO4 nanocomposites, the absorptivity of MgCuPO4 progressively declines from S1 to S3. Furthermore, as the proportions of MgCuPO4 decreased, the absorption edge red shifted towards the visible region. The existence of more Ag3PO4 NPs than MgCuPO4, which absorbs in the visible region, causes the peaks to redshift. This redshift may also be due to narrowing of the band gap, as evidenced by the altered Kubelka–Munk function of light energy (αhν)2versus photon energy (hν), as shown in Fig. 3(d). The Tauc equation was used to determine the band gap (Eg) values of the samples.30,31
The absorption coefficient is denoted by α and the proportionality constant by K. K was determined from K = (2.303 × 103)(A)/L, where absorbance is represented by A and optical path by L (1 cm). The Eg value was estimated by extrapolating the straight line of the image shown in Fig. 3(d). Eg values for S1, S2, and S3 and amorphous MgCuPO4 were 3.9, 3.6, 3.5, and 4.3 eV, respectively. The electrical insulating properties of amorphous MgCuPO4 were reflected in its high Eg value. The gradual drop in Eg in MgCuPO4–AgPO4 nanocomposites was due to the combined impact between the two elements. The overlapping of energy levels causes a reduction in the band gap according to energy band theory. This indicates that Ag3PO4 were well developed on the MgCuPO4 surface, exhibiting the combined electronic characteristics.32,33
3.1.4. XPS spectrum for MgCuPO4–AgPO4.
Because of the dispersion of Ag3PO4 NPs on the MgCuPO4 surface, XPS was used to analyze the surface composition of MgCuPO4 and MgCuPO4–AgPO4 nanocomposites. The XPS spectrum of S2 showed the presence of several distinct peaks due to Mg, P, C 1s, Ag, O 1s, and Cu, as indicated in Fig. 4(a). Fig. 4(b) depicts the deconvoluted core level spectrum of Mg 2p, which includes two bands at 49 and 51 eV binding energy corresponding to Mg2+ and Mg0, respectively.34 The XPS spike for Cu 2p3/2 located at 932.5 eV correlates to Cu2+ possibly reacting with a phosphate ion. The peak for Cu 2p1/2 appeared at 952.5 eV, as indicated in Fig. 4(c).35 The XPS spectrum for P 2p is represented in Fig. 4(d). The presence of P 2p1/2 at 135 eV and P 2p3/2 at 133 eV confirm the presence of phosphorous. The deconvoluted XPS spectrum of P 2p showed that the first peak at 133 eV accounts for 75–80% of the P 2p signal. This can be ascribed to the oxidation of the MgCuPO4 surface metal phosphate molecule in PO43−.36 The second spike at 135 eV showed the presence of 10–20% residual metaphosphate.37 S2 demonstrated the presence of symmetrical spin–orbit components. The XPS spectra for Ag showed two distinct peaks which were separated by 6 eV. The Ag 3d5/2 peak appeared at 368 eV and Ag 3d3/2 peak appeared at 374 eV, which corresponds to Ag+, as indicated in Fig. 4(e).38
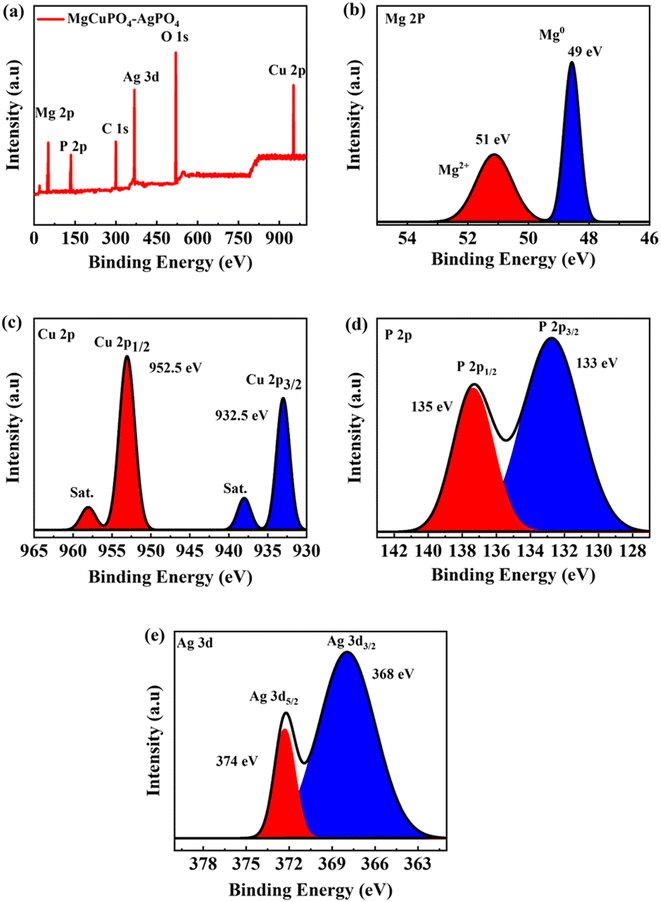 |
| | Fig. 4 (a) XPS survey spectrum for S2. (b) XPS spectrum for Mg. (c) XPS spectrum for Cu 2p. (d) XPS spectrum for P 2p. (e) XPS spectrum for Ag 3d. | |
3.1.5. Morphological study of MgCuPO4–AgPO4.
SEM analysis was used to examine the shape of MgCuPO4 and its three composites with Ag3PO4 (S1, S2, and S3). Fig. 5(a)–(d) depict an uneven form of amorphous MgCuPO4 and its composites with Ag3PO4 with 40–50 nm particle size. Fig. 5 shows that as the amorphous MgCuPO4 content of the nanocomposite decreased, more Ag3PO4 NPs were found on the surface of the amorphous MgCuPO4. However, for S1 and S3, the recombination of ions could restrict the diffusion of ions. The recombination of ions could be attributed to an overabundance of Ag3PO4, which restricts the movement of MgCuPO4 ions.39,40
 |
| | Fig. 5 (a–d) SEM images for S1, S2, S3, and S4 composites. | |
3.2. Electrochemical results
The synthesized samples (MgCuPO4, S1, S2, and S3) were then electrochemically tested using CV measurements, which were determined in the potential window (PW) of 0–0.6 V at a scan rate of 3–50 mV s−1, as depicted in Fig. 6. For all samples, faradaic behavior was observed from the presence of redox peaks. This was because of the faster movement of ions within the materials, causing a redox reaction at the surface of the electrodes.41,42 S2 had the greatest redox current strength at 3 mV s−1 scan rate compared to the other MgCuPO4–AgPO4 nanocomposites. This shows that the addition of Ag3PO4 increased the redox current due to the synergetic impact of MgCuPO4 and Ag3PO4. But further addition of Ag3PO4 influenced the redox potentials, as can be seen from the CV plot of S3 (Fig. 6(d)). The finding suggested that electrolyte ions engage extensively with the numerous electro-active sites of the materials when the amount of Ag3PO4 increases. With higher loadings of Ag3PO4 in MgCuPO4, the electrical transmission route and electrolyte ion diffusion rate of the nanocomposites differ. This indicated that the number of phosphate groups in the MgCuPO4–Ag3PO4 composite, as well as metal ion concentrations, affect electron and ion motion.43 Furthermore, the variations in anodic and cathodic potentials can be linked to the presence of the redox process.44 The CV curve of MgCuPO4 changes its shape at 100 mV s−1 (Fig. 6(a)) because the electrolyte ions did not reach the MgCuPO4 inner sites.45 S2 had a greater CV area (and thus a greater specific capacity) than MgCuPO4 at a low scan rate. S2 also maintained its form at higher scan rates due to its stable behavior.46,47Fig. 6(a)–(d) illustrate the CV curves for S2 and the other samples (MgCuPO4, S1, and S3) that were gradually enhanced with rising scan rates (3–50 mV s−1). Fig. S1† shows the CV curves for MgCuPO4, S1, and S3 at an operating potential of 0–0.8 V.
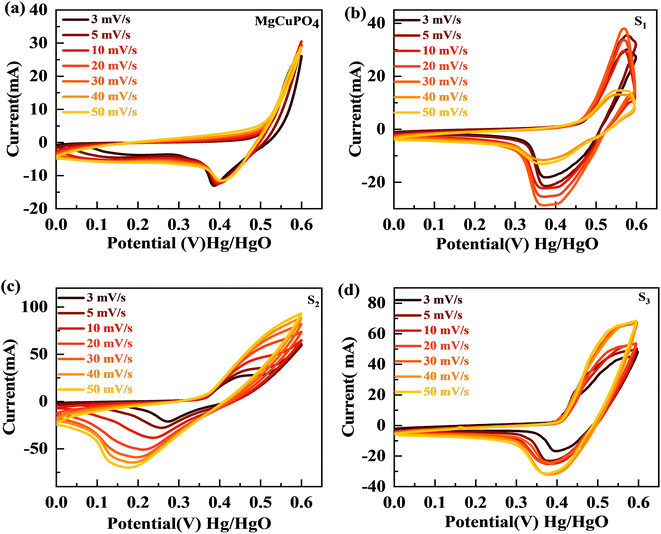 |
| | Fig. 6 (a–d) The CV curves for MgCuPO4, S1, S2, and S3, respectively. | |
For battery-type materials, the most suitable word to determine the storage capability is the specific capacity (C g−1). However, some researchers in the literature also use capacitance (F g−1) which is not correct. Table 3 shows the values of specific capacity at all scan rates. Eqn (8) can be used to determine the specific capacity.48
| | 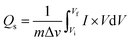 | (8) |
In
eqn (8),
Qs indicates the specific capacity,
m is the active mass, Δ
v represents the change in scan rate; the current is denoted by
I, and the operating potential by
V. The specific capacities calculated for MgCuPO
4, S
1, S
2, and S
3 at various scans are represented in
Fig. 7(a). The specific capacities for MgCuPO
4–Ag
3PO
4 nanocomposites were higher than that for MgCuPO
4. The specific capacity for S
2 at 3 mV s
−1 was 839 C g
−1, which was higher than that for MgCuPO
4 (335 C g
−1), S
1 (442 C g
−1), or S
3 (671 C g
−1). The specific capacities for all samples (MgCuPO
4, S
1, S
2, and S
3) at 3 mV s
−1 are represented in
Fig. 7(b). When an amount of Ag
3PO
4 is added into the heterostructure of MgCuPO
4, the specific capacity increases due to the combined impact of MgCuPO
4–Ag
3PO
4, with a large number of accessible pores and faster movement of ions. A further increase in the amount of Ag
3PO
4 resulted in a decrease in its capacity. An excess amount of Ag
3PO
4 also results in blockage of a number of pores. Therefore, the specific capacity for S
3 decreases.
Table 3 Specific capacity for MgCuPO4, S1, S2, and S3 through CV and GCD
| Scan rate (mV s−1) |
Specific capacity (C g−1) |
| MgCuPO4 |
S1 |
S2 |
S3 |
| CV |
GCD |
CV |
GCD |
CV |
GCD |
CV |
GCD |
| 3 |
336.16 |
317 |
442 |
330 |
839 |
1138 |
671 |
512 |
| 5 |
264.60 |
269 |
379.71 |
269 |
682.04 |
933 |
583.42 |
478 |
| 10 |
200.12 |
245 |
305.08 |
251 |
571.73 |
891 |
447.96 |
447 |
| 20 |
129.15 |
215 |
212.86 |
236 |
333.16 |
832 |
257.97 |
426 |
| 30 |
71.05 |
192 |
122.49 |
225 |
194.76 |
745 |
184.12 |
398 |
| 40 |
40.49 |
183 |
83.07 |
210 |
126.813 |
682 |
119.18 |
355 |
| 50 |
20.88285 |
169 |
49.40 |
192 |
86.44 |
663 |
70.54 |
313 |
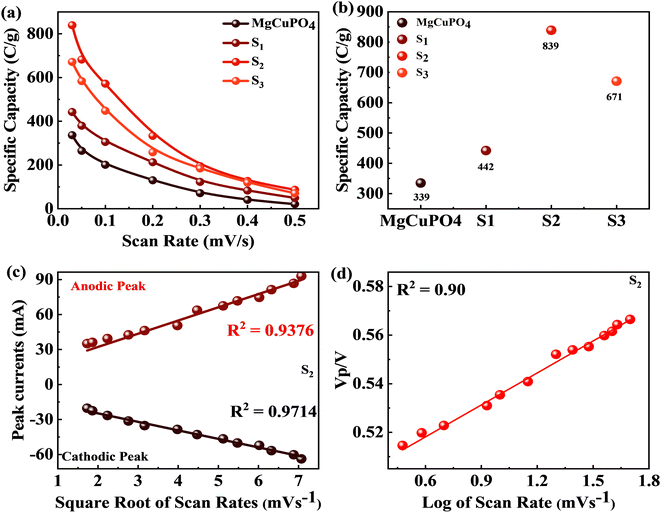 |
| | Fig. 7 (a) Specific capacity for MgCuPO4, S1, S2, and S3 at various scan rates. (b) Specific capacity for MgCuPO4, S1, S2, and S3 at 3 mV s−1 scan rate. (c) Anodic and cathodic peaks calculated for S2. (d) Peak voltage plotted against log of scan rate for S2. | |
The positive and negative current spikes measured through CV readings of S2 gradually increased with the square root of the scan rate. The value of the Ipa/Ipc fraction was almost 1. This indicated that the reversible faradaic processes were responsible for charge storage, as shown in Fig. 7(c).49 The linear relation between anodic and cathodic currents was evidence of the battery nature of the MgCuPO4–Ag3PO4 (S2) nanocomposite. Fig. 7(d) indicates a straight-line relationship between voltage and log of scan rate, which proves the diffusion-controlled process for S2. The chemical equations below can be used to describe redox reactions.50
| | 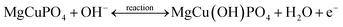 | (9) |
| |  | (10) |
The discharge profiles of MgCuPO4 and MgCuPO4–Ag3PO4 nanocomposites obtained using the GCD method are shown in Fig. 8(a)–(d). The shape of the GCD curves was non-linear for MgCuPO4, S1, S2, and S3 because of deep ion interaction, showing that charge storage contribution comes mainly from the redox reactions. At a current density of 2 A g−1, S2 had the greatest discharge time compared to the other MgCuPO4–AgPO4 nanocomposite electrodes. At 3.2 A g−1 current, the GCD trajectory of MgCuPO4 was also relatively shorter than that of S2. This showed that the performance of MgCuPO4 decreases with current density more frequently than the other MgCuPO4–Ag3PO4 nanocomposites, as illustrated by Fig. 8(a)–(d). The GCD comparison at 2.0 A g−1 for MgCuPO4, S1, S2, and S3 is represented in Fig. 9(a). As can be seen from Fig. 9(a), S2 had the largest plateau region compared to the other samples. This may be due to the larger surface area exposed to electrolytic ions, faster transportation of ions, and higher conductivity of S2. The specific capacity, Qs, of the electrodes was determined using eqn (11) from the galvanostatic discharge data.
| |  | (11) |
in
eqn (11),
Qs represents the specific capacity,
I is the current, Δ
t indicates the discharge time, and
m is the active mass.
Fig. 9(b) depicts a plot of specific capacity for MgCuPO
4, S
1, S
2, and S
3 against current density derived from the GCD curves. At all present current densities, the specific capacity for MgCuPO
4 had reduced from 316 (2 A g
−1) to 169 C g
−1 (3.2 A g
−1), with 53% capacity retention. S
1 had a capacity retention of 58% with a capacity of 330 C g
−1 at 2 A g
−1 and 192 C g
−1 at 3.2 A g
−1. Again S
2 outperforms the other composite materials, showing 1138 C g
−1 capacity at 2 A g
−1. S
2 can retain 663.8 C g
−1 capacity at 3.2 C g
−1 showing 59% capacity retention. Furthermore, an increase in the wt% ratio of Ag
3PO
4 decreases the overall performance. The specific capacities decrease for S
3 to 512 C g
−1 at 2 A g
−1 and 313.6 C g
−1 at 3.2 A g
−1, demonstrating 61% capacity retention.
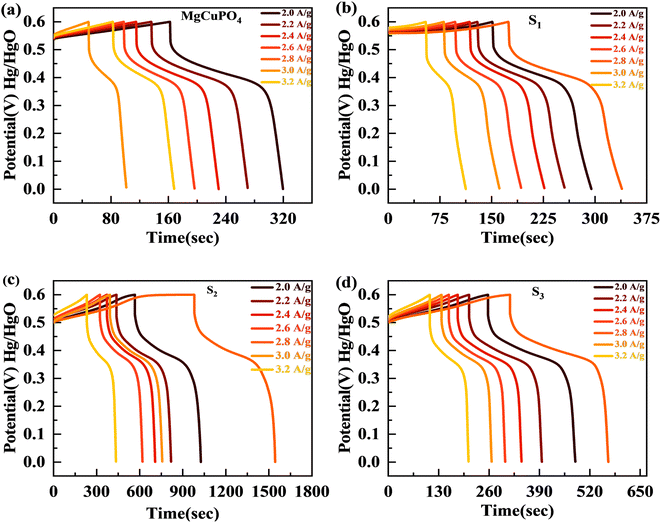 |
| | Fig. 8 (a–d) The GCD plots for MgCuPO4, S1, S2, and S3 at different currents. | |
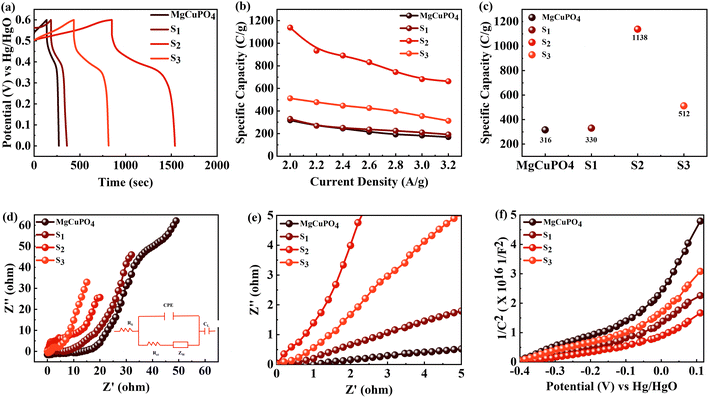 |
| | Fig. 9 (a) GCD comparison for MgCuPO4, S1, S2, and S3 at 2.0 A g−1 current density. (b) Specific capacity computed for MgCuPO4, S1, S2, and S3 at varied current densities. (c) Specific capacity computed for MgCuPO4, S1, S2, and S3 at 2 A g−1 current density. (d) EIS graph for MgCuPO4, S1, S2, and S3. (e) High-magnification EIS images for MgCuPO4, S1, S2, and S3. (f) MS plot for MgCuPO4, S1, S2, and S3. | |
This increase in rate capacity was due to the presence of Ag3PO4 NPs, which decreased the internal impedance of MgCuPO4.51 Among all the MgCuPO4–Ag3PO4 (S1, S2, and S3) nanocomposites studied, S2 had the highest capacity. The increased electrochemical performance for S2 was due to two conflicting effects: raising the quantity of Ag3PO4 increases the conductivity of MgCuPO4–Ag3PO4 nanocomposites. However, too many Ag3PO4 nanoparticles degrade the performance of the S3 electrode due to agglomerations produced, which reduces the contact area between the electrode and electrolytic solution. Another cause was the lack of amorphous MgCuPO4 in S3, as amorphous MgCuPO4 was responsible for increased active sites for redox reactions.52 Therefore, owing to the structural stability of crystalline Ag3PO4 NPs, the rate capacity of S2 remains greater than that of MgCuPO4. Fig. 9(c) indicates the comparative calculated specific capacities for MgCuPO4, S1, S2, and S3 at 2.0 A g−1.
The electrochemical impedance spectra (EIS) of the electrode materials were measured to determine charge and ion transport. The Nyquist graphs of amorphous MgCuPO4, S1, S2, and S3 are shown in Fig. 9(d). Because of the roughness of the electrode surface, uneven electric field, and varying electrochemical activity, the form of the EIS plots departs from the semicircular shape.53 The corresponding series resistance was calculated using the contact of the EIS graphs with the real part of the impedance spectrum. At high frequencies, the semicircle width is attributed to charge transfer impedance (Rct).54 In Fig. 9(d) (inset), the circuit diagram for the fitted Nyquist plot is also represented. The semicircle can be seen from the ZCPE and Rct parallel connection in the circuit diagram. According to the EIS diagram, the Rs values for S1, S2, S3, and MgCuPO4 were 0.72, 0.65, 0.81, and 1.05 U, respectively, as shown in Fig. 9(d). The lower Rs values of all MgCuPO4–AgPO4 nanocomposites compared to amorphous MgCuPO4 nanocomposites suggest higher conductance. Moreover, the semicircle diameter of MgCuPO4 was considerably bigger than that of the MgCuPO4–AgPO4 nanocomposites in the Nyquist plots, which results in greater charge transfer resistance. In all the MgCuPO4–Ag3PO4 nanocomposites studied, S2 had the shortest diameter, indicating the lowest internal resistance. Furthermore, the straight line for S2 was the sharpest, indicating the lowest ion diffusion resistance. The high-resolution EIS spectra for MgCuPO4, S1, S2, and S3 are represented in Fig. 9(e). A potential-dependent capacity test was used to further explore the effect of Ag3PO4 modification on the electrical characteristics of MgCuPO4. MS plots were made from the capacitances obtained from the imaginary portion of the impedance. Fig. 9(f) depicts the MS plots for MgCuPO4, S1, S2, and S3. The dominant charge carriers in all MgCuPO4, S1, S2, and S3 materials were electrons, as indicated by the positive slope of the straight lines (n-type semiconductors). The slope obtained from the straight line plotted between 1/C2 and V can be used to evaluate charge carrier density among electrode materials. The charge carrier has a negative relationship with the MS inclination.55 The S1, S2, and S3 slopes were lower than those of MgCuPO4, implying that the MgCuPO4–Ag3PO4 composites had a greater charge carrier density. The S2 curve had the lowest slope, demonstrating its increased electron density. These findings suggest that the incorporation of Ag3PO4 into MgCuPO4 results in a higher density of charge carriers.56
The electron transmission rate of crystalline Ag3PO4 NPs differs from that of diffuse MgCuPO4. The number of pores provided by MgCuPO4 was used to store the electrolyte ions that were able to penetrate the inner surface of MgCuPO4. Most of the amorphous MgCuPO4 surfaces were engaged in the faradaic reactions. Additionally, the electrolyte could not reach internal surfaces at a higher scan rate/current density, thus producing a low specific capacity.57 In contrast, the electrolytic ions interact with the topmost surface layer of Ag3PO4 NPs as well as the inner surface of MgCuPO4. Thus, Ag3PO4 adds useful surface area to unutilized amorphous MgCuPO4 surfaces and maintains a large number of oxidation/reduction reactions at higher currents.58 Despite the fact that amorphous MgCuPO4 offers a porous structure that helps to store more electrolytic ions, it still had low conductance.59 In this study, the high electrical conductivity of Ag3PO4 NPs fixed on the weakly conductive amorphous MgCuPO4 accelerates the electron transfer mechanism and lowers the impedance of the MgCuPO4–Ag3PO4 composites. Furthermore, the Ag3PO4 nanoparticles reduce the distance between electron transport routes and the collector, resulting in increased electrochemical performance.60
3.3. Supercapattery
The broad PW results in increased energy density for SCs. The supercapattery (S2//AC) was designed with S2 and activated carbon (AC). AC due to its permeable and porous structure allows more electrolyte ions to be stored.61 The individual CV graphs were originally measured in a three-cell setup for S2 and AC to measure the overall voltage of the supercapattery (Fig. 10(a)). The working PW of AC was 0 to −1 V and that of S2 was 0–0.6 V. Thus, the S2//AC supercapattery was stable over 1.6 V. No redox peak appeared at 0–0.5 V, which showed that the dominant storage mechanism was provided by the capacitive nature of AC. At higher potential, >0.6 V, redox peaks emerged, indicating the faradaic reaction of S2. Fig. 10(b) shows that the shape of the CV graphs for the S2//AC supercapattery was not altered at a higher scan (100 mV s−1), which proves the high stability of the S2//AC device.
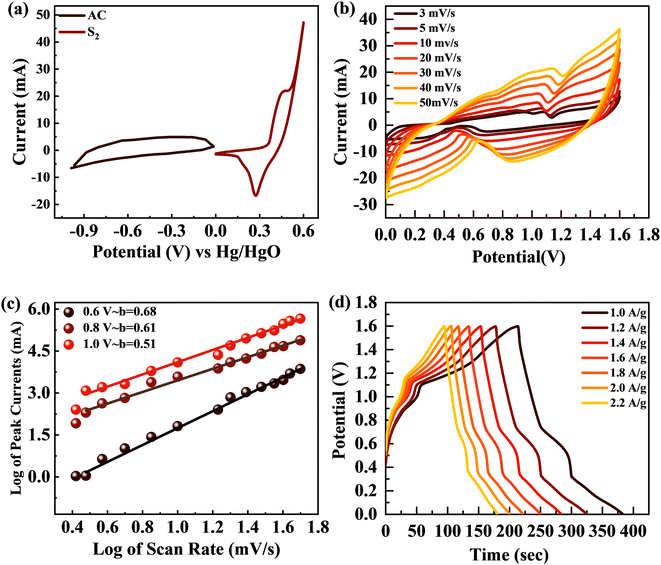 |
| | Fig. 10 (a) CV curves for AC and S2 at 3 mV s−1. (b) CV curves for the S2//AC supercapattery at 3–50 mV s−1. (c) b-fitting computed for the S2//AC device. (d) GCD curves for the S2//AC supercapattery at 1–2.2 A g−1 current density. | |
The power law, which connects current density I with scan rate (v), was used for theoretical study of the electrochemical behavior of the asymmetric device. The charge storage process in a supercapattery is investigated using b-fitting. The b-value was calculated using the slope drawn between the log of scan rate and the log of peak current.
The adjustable factors are represented by k and b, as stated in the equations, while v is the scan rate and I is the current. The exponent b is critical in deciding the charge accumulation in a supercapattery. For a b-value of 0–0.5, the dominant charge storage process is diffusion regulated. For a b-value of 0.8–1.0, the dominant charge storage process is adsorption/desorption. For a b-value of 0.5–0.8, the storage process is due to both faradaic and adsorption/desorption processes.62 The b-fitting for this supercapattery (S2//AC) was between 0.5 and 0.8, as shown in Fig. 10(c). This proves the formation of a supercapattery.
The GCD measurements for the S2//AC supercapattery were performed at various densities (1.0 to 2.2 A g−1), as depicted in Fig. 10(d). The GCD curves of the S2//AC device were nearly symmetrical at all currents, which demonstrates its higher stability.63 Furthermore, the GCD contours were non-linear due to the faradaic response of S2. The specific capacity for the S2//AC supercapattery was also calculated from eqn (8) and (11). At a current intensity of 1.0 A g−1, the specific capacity of S2//AC was 193 C g−1, as indicated in Fig. 11(a). The S2//AC device can retain 60% of its initial capacity at 2.2 A g−1 current. While the capacity through a CV for the S2//AC supercapattery was 183 C g−1 (at 3 mV s−1), as indicated in Fig. 11(b).
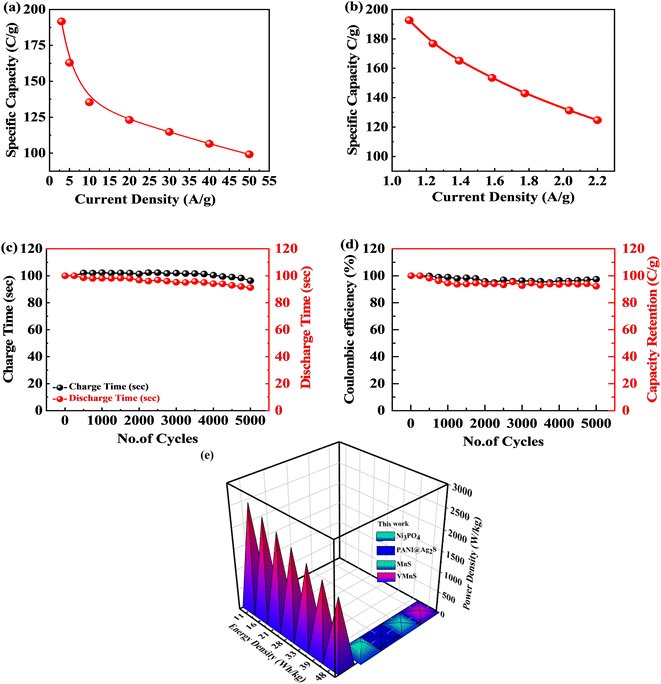 |
| | Fig. 11 (a and b) Specific capacity for the S2//AC supercapattery computed through CV and GCD. (c and d) Charge/discharge and capacity retention for the S2//AC supercapattery after 5000 cycles. (e) Energy and power density for the S2//AC supercapattery. | |
Fig. 11(c) and (d) represent the stability test for the S2//AC supercapattery device against 5000 cycles. The capacity was increased marginally to 110% at the start, owing to the wettability of the electrode surface and slow activation of the electrode.64 After 5000 cycles, the capacity declined slowly to 92%, showing that the device was stable. The coulombic efficiency after 5000 cycles was retained at 98%. The decay in capacity was due to structural degradation of the electrode materials. The nanocomposite agglomeration as well as separation of the electrode material from the electrolyte are also responsible for the decrease in capacity.65,66
Fig. 11(e) depicts the Ragone plot of the estimated energy density (E) and power density (P) of the S2//AC supercapattery using the following formulae.
| | 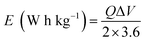 | (14) |
| | 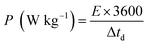 | (15) |
where
Q denotes specific capacity,
V denotes the PW, and
td denotes discharge duration. S
2//AC provided a high energy density as well as an outstanding power density (49.4 W h kg
−1 at 550 W kg
−1 and 19.5 W h kg
−1 at 6382 W kg
−1). These results outperformed previous studies, as indicated in
Table 4.
Table 4 Energy vs. power density for the MgCuPO4–Ag3PO4//AC device and comparison with the literature
| Previous study |
Energy density |
Power density |
| Ni–Co–S70 |
42 |
750 |
| Ni–V–S2 (ref. 71) |
12 |
900 |
| MnS72 |
10.55 |
294.35 |
| Sr-based materials73 |
21.8 |
224 |
| Co/WS74 |
42.2 |
1047 |
| 2D VS2/P75 |
28.22 |
596 |
| MnO2@CoS nanosheets76 |
34 |
597.24 |
|
This work
|
49.4
|
550
|
4. Oxygen evolution reaction (OER)
Linear sweep voltammetry (LSV) was employed to evaluate the electrocatalytic performance of MgCuPO4, S1, S2, S3, and S4 composites, as well as the bare stainless steel (SS) substrate. To conduct the investigation, 1 M KOH electrolyte was utilized. The primary objective was to assess their ability to generate a current density of 10 mA cm−2 with minimal overpotential, aiming to match the widely employed anode materials RuO2 and IrO2 in various industrial applications. The LSV data revealed distinctive behavior among the tested materials.67
The MgCuPO4 composite exhibited an onset overpotential of 340 mV, whereas the S1 and S3 composites displayed starting overpotentials of 263 and 221 mV, respectively. In contrast, the S2 composite showcased an even lower overpotential of 142 mV, further underscoring its electrocatalytic capabilities. Conversely, the bare SS substrate necessitated a higher overpotential of 388 mV to achieve a current density of 10 mA cm−2 (Fig. 12(a)).
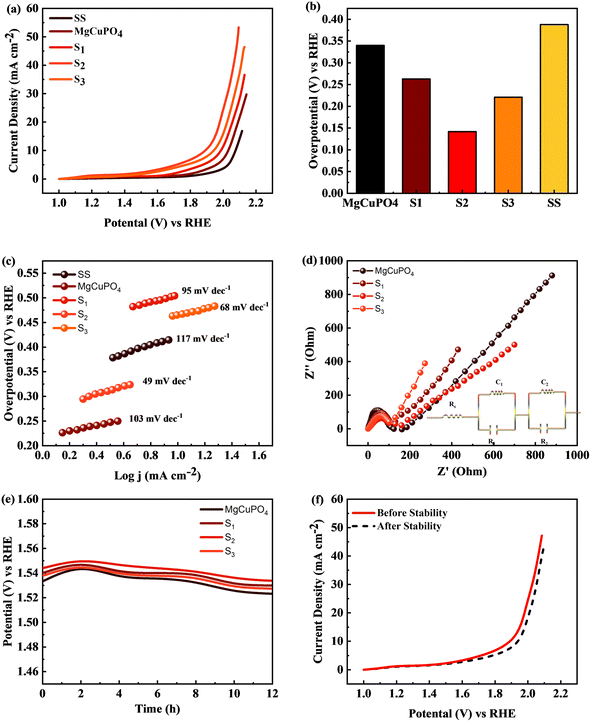 |
| | Fig. 12 (a) Linear sweep voltammograms (LSV) of MgCuPO4, S1, S2, and S3 in comparison to SS. (b) Overpotential needed to achieve 10 mA cm−2 current for all composites. (c) Tafel plots for SS, MgCuPO4, S1, S2, and S3. (d) EIS measurements for MgCuPO4, S1, S2, and S3 composites. (e) Chronopotentiometric study for MgCuPO4, S1, S2, and S3. (f) Electrocatalytic analysis of S2 composite after OER stability. | |
The bare SS substrate was included in the assessment as a reference point to evaluate the effectiveness of the composites. The higher overpotential required by the bare SS substrate suggests low electrocatalytic activity and inadequate efficiency in the oxygen evolution reaction (OER). Conversely, the significantly lower overpotentials observed for the MgCuPO4, S1, S2, and S3 composites indicate superior performance and potential for diverse applications, particularly in renewable energy conversion and storage devices.
Moreover, Fig. 12(b) illustrates comparable overpotentials at j = 10 mA cm−2, a typical measure of OER activity. A lower overpotential implies significantly better performance in the OER.68,69 These findings highlight the exceptional electrocatalytic capabilities of the MgCuPO4, S1, S2 and S3 composites compared to the bare SS substrate. Their reduced onset overpotentials and improved catalyst utilization suggest the potential to enhance energy storage technologies and enable sustainable energy conversion processes. The effectiveness of the electrochemical processes can be improved by these materials (MgCuPO4, S1, S2, and S3), taking us one step forward to a more environmentally friendly and sustainable energy density.
Tafel plots were used to analyze the reaction kinetics of the MgCuPO4, S1, S2, and S3 materials, as shown in Fig. 12(c). The Tafel slopes reveal information about the reaction response of the materials.
| | | η = a + blni | (17) |
The MgCuPO4 composite exhibits a Tafel slope of 103 mV dec−1; the S1 composite shows a Tafel slope of 95 mV dec−1; S2 shows a Tafel slope of 49 mV dec−1 (see Table 5); and S3 shows a Tafel slope of 68 mV dec−1. The reduced Tafel slope of S2 (117 mV dec−1) in comparison to the other composites and bare SS points to quicker reaction rates. As a result, the oxygen evolution process (OER) can occur at greater speeds in the S2 composite due to its more effective catalytic activity.
Table 5 Comparison of Tafel slope and overpotential of this work with previous studies
| Literature |
Tafel slope (mV dec−1) |
Overpotential (mV) |
Ref. |
| Cobalt sulfide thin films |
57 |
300 |
77
|
| Ni12P5 nanocapsule |
49.8 |
152 |
7
|
| NixPy |
72.2 |
157 |
78
|
| C@Ni–P |
64 |
153 |
79
|
|
MgCuPO
4
–Ag
3
PO
4
(S
2
)
|
49
|
142
|
This work
|
The charge transport process inside the MgCuPO4, S1, S2, and S3 materials, along with the bare SS substrate, was examined using electrochemical impedance spectra (EIS), in conjunction with Tafel studies. Significant features were discovered from the Nyquist plots, as illustrated in Fig. 12(d). Surprisingly, the semicircle diameter of the S2 composite was less than those of the MgCuPO4, S1, S2, and S3 composites. This suggests that the S2 composite is more conductible. As shown in the insets of Fig. 12(d), analogous circuits were used to analyze the EIS data. These circuits included elements that represented the impedance of the solution (Rs), the inherent resistance (R1) of the catalyst, and the impedance of the electrocatalytic activity (R2). Notably, the electrocatalytic resistance of the S2 composite was determined to be 659 Ω cm2, significantly lower than the resistances of the MgCuPO4 (732 Ω cm2), S1 composite (788 Ω cm2) or the S3 composite (914 Ω cm2). Table S1† presents the impedance of MgCuPO4, S1, S2, and S3 in the oxygen evolution reaction.
To evaluate the long-term electrochemical stability of the MgCuPO4, S1, S2, and S3 composites, a chronopotentiometric mode was employed. Fig. 12(e) demonstrates a minor potential drop over time, attributed to an improvement in catalytic efficiency. Continuous gas liberation was observed during the stability investigation, indicating the ongoing oxygen evolution process. Fig. 12(f) presents the LSV graphs of S2.
The results of the study indicate that the MgCuPO4, S1, S2, and S3 composites exhibit favorable reaction kinetics and long-term electrochemical stability specifically for the oxygen evolution reaction (OER). The lower Tafel slope observed for the S2 composite suggests faster reaction rates, while the slight reduction in potential during the stability test signifies improved catalytic efficiency. These characteristics are crucial for ensuring the sustained performance and durability of electrochemical devices, underscoring the significance of these composites in applications related to energy storage and conversion.
5. Conclusions
The sonochemical and hydrothermal technique followed by calcination treatment effectively synthesizes MgCuPO4 and MgCuPO4–Ag3PO4 nanocomposites with various MgCuPO4–Ag3PO4 content ratios. XRD phase structure analysis verified that the crystalline structure of Ag3PO4 NPs increases with decreasing amorphous MgCuPO4 content. The FTIR spectra revealed that phosphate and hydroxyl groups were chemisorbed on the MgCuPO4–Ag3PO4 surface. The UV-vis spectroscopic analysis revealed that MgCuPO4–Ag3PO4 composites were more conductive than MgCuPO4. The SEM analysis showed that Ag3PO4 nanoparticles were firmly fixed on the amorphous surface of MgCuPO4. The MgCuPO4–Ag3PO4 nanocomposite-based electrode demonstrated increased rate capability from 53% (MgCuPO4) to 59% at 3.2 A g−1. The rapid electron movement and the large number of active sites given by MgCuPO4 and Ag3PO4 composites enhanced the rate capacity. Because amorphous MgCuPO4 has a low electrical conductivity, modifying amorphous MgCuPO4 with crystalline Ag3PO4 NPs offers a potential method to increase conductivity and rate capability. The constructed S2//AC supercapattery had an energy density of 49.4 W h kg−1 at 550 W kg−1 power density. The S2//AC supercapattery showed 92% capacity retention after 5000 cycles. Investigation of the OER application demonstrated that S2 exhibited the lowest Tafel slope, measuring 49 mV dec−1. The results obtained from this study suggest that MgCuPO4–Ag3PO4 composites hold great promise as potential candidates for future energy storage devices and further investigation into the oxygen evolution reaction (OER).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to thank Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2023R9), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia, for supporting this project.
References
- M. F. El-Kady, M. Ihns, M. Li, J. Y. Hwang, M. F. Mousavi, L. Chaney, A. T. Lech and R. B. Kaner, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4233–4238 CrossRef CAS PubMed.
- R. Kötz and M. Carlen, Electrochim. Acta, 2000, 45, 2483–2498 CrossRef.
- F. Larsson and B.-E. Mellander, J. Electrochem. Soc., 2014, 161, A1611 CrossRef CAS.
- F. S. Omar, A. Numan, S. Bashir, N. Duraisamy, R. Vikneswaran, Y.-L. Loo, K. Ramesh and S. Ramesh, Electrochim. Acta, 2018, 273, 216–228 CrossRef CAS.
- G. Z. Chen, Int. Mater. Rev., 2017, 62, 173–202 CrossRef CAS.
- A. Numan, N. Duraisamy, F. S. Omar, D. Gopi, K. Ramesh and S. Ramesh, Prog. Nat. Sci.: Mater. Int., 2017, 27, 416–423 CrossRef CAS.
- H. Wan, L. Li, Y. Chen, J. Gong, M. Duan, C. Liu, J. Zhang and H. Wang, Electrochim. Acta, 2017, 229, 380–386 CrossRef CAS.
- X. Lei, S. Ge, Y. Tan, Z. Wang, J. Li, X. Li, G. Hu, X. Zhu, M. Huang and Y. Zhu, ACS Appl. Mater. Interfaces, 2020, 12, 9158–9168 CrossRef CAS PubMed.
- Y. Yang, Y. Zhou, Z. Hu, W. Wang, X. Zhang, L. Qiang and Q. Wang, J. Alloys Compd., 2019, 772, 683–692 CrossRef CAS.
- H. Moon, H. Lee, J. Kwon, Y. D. Suh, D. K. Kim, I. Ha, J. Yeo, S. Hong and S. H. Ko, Sci. Rep., 2017, 7, 41981 CrossRef CAS PubMed.
- L. Tang, F. Duan and M. Chen, RSC Adv, 2016, 1244, 65012–65019 RSC.
- Y. Shao, H. Wang, Q. Zhang and Y. Li, J. Mater. Chem. C, 2013, 1, 1245–1251 RSC.
- D. S. Patil, S. A. Pawar, J. H. Kim, P. S. Patil and J. C. Shin, Electrochim. Acta, 2016, 213, 680–690 CrossRef CAS.
- D. S. Patil, J. Shaikh, S. Pawar, R. Devan, Y. Ma, A. Moholkar, J. Kim, R. Kalubarme, C. Park and P. Patil, Phys. Chem. Chem. Phys., 2012, 14, 11886–11895 RSC.
- L. Tang, F. Duan and M. Chen, J. Mater. Sci.: Mater. Electron., 2017, 28, 7769–7777 CrossRef CAS.
- B. Pandit, V. Devika and B. R. Sankapal, J. Alloys Compd., 2017, 726, 1295–1303 CrossRef CAS.
- K. M. Lee, C. W. Lai, K. S. Ngai and J. C. Juan, Water Res., 2016, 88, 428–448 CrossRef CAS PubMed.
- H. Hassan, M. W. Iqbal, S. Gouadria, A. M. Afzal and H. Hegazy, J. Mater. Sci.: Mater. Electron., 2023, 34, 439 CrossRef CAS.
- R. Mahajan, R. Prakash, S. Kumar, V. Kumar, R. Choudhary and D. Phase, Optik, 2021, 225, 165717 CrossRef CAS.
- W. Zhao, G. Zhong, M. J. McDonald, Z. Gong, R. Liu, J. Bai, C. Yang, S. Li, W. Zhao and H. Wang, Nano energy, 2016, 27, 420–429 CrossRef CAS.
- J.-K. Liu, C.-X. Luo, J.-D. Wang, X.-H. Yang and X.-H. Zhong, CrystEngComm, 2012, 14, 8714–8721 RSC.
- R. J. V. Michael, B. Sambandam, T. Muthukumar, M. J. Umapathy and P. T. Manoharan, Phys. Chem. Chem. Phys., 2014, 16, 8541–8555 RSC.
- Y. Lv, H. Wang, X. Xu, J. Shi, W. Liu and X. Wang, Chem. Eng. J., 2017, 326, 401–410 CrossRef CAS.
- P. Benedek, N. Wenzler, M. Yarema and V. C. Wood, RSC Adv., 2017, 7, 17763–17767 RSC.
- Y. Wang, K. Wang and X. Wang, J. Colloid Interface Sci., 2016, 466, 178–185 CrossRef CAS PubMed.
- E. Uchaker, Y. Zheng, S. Li, S. Candelaria, S. Hu and G. Cao, J. Mater. Chem. A, 2014, 2, 18208–18214 RSC.
- J. Zheng and C. Huang, J. New Mater. Electrochem. Syst., 2002, 5, 41–46 CAS.
- V. Stefov, V. Koleva, M. Najdoski, A. Cahil and Z. Abdija, Maced. J. Chem. Chem. Eng., 2020, 39, 239–249 CrossRef CAS.
- Z. Hua, B. Li, L. Li, X. Yin, K. Chen and W. Wang, J. Phys. Chem. C, 2017, 121, 60–69 CrossRef CAS.
- S. Saha, M. Jana, P. Khanra, P. Samanta, H. Koo, N. C. Murmu and T. Kuila, ACS Appl. Mater. Interfaces, 2015, 7, 14211–14222 CrossRef CAS PubMed.
- P. Reunchan, A. Boonchun and N. Umezawa, Phys. Chem. Chem. Phys., 2016, 18, 23407–23411 RSC.
- S. Saha, M. Jana, P. Khanra, P. Samanta, H. Koo, N. C. Murmu and T. Kuila, RSC Adv., 2016, 6, 1380–1387 RSC.
- I. W. Hwang, C. Soci, D. Moses, Z. Zhu, D. Waller, R. Gaudiana, C. J. Brabec and A. J. Heeger, Adv. Mater., 2007, 19, 2307–2312 CrossRef CAS.
- H.-H. Huang, W.-C. Shih and C.-H. Lai, Appl. Phys. Lett., 2010, 96, 193505 CrossRef.
- P. Mondal, A. Sinha, N. Salam, A. S. Roy, N. R. Jana and S. Islam, RSC Adv., 2013, 3, 5615–5623 RSC.
- B. Wang, J. Liu, Q. Sun, R. Li, T.-K. Sham and X. Sun, Nanotechnology, 2014, 25, 504007 CrossRef PubMed.
- G. Yun, Q. Guan and W. Li, RSC Adv., 2017, 7, 8677–8687 RSC.
- V. A. Kumar, T. Uchida, T. Mizuki, Y. Nakajima, Y. Katsube, T. Hanajiri and T. Maekawa, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2016, 7, 015002 Search PubMed.
- C. A. Amarnath, N. Venkatesan, M. Doble and S. N. Sawant, J. Mater. Chem. B, 2014, 2, 5012–5019 RSC.
- S. Nagamuthu, S. Vijayakumar and K.-S. Ryu, J. Phys. Chem. C, 2016, 120, 18963–18970 CrossRef CAS.
- N. Duraisamy, A. Numan, S. O. Fatin, K. Ramesh and S. Ramesh, J. Colloid Interface Sci., 2016, 471, 136–144 CrossRef CAS PubMed.
- P. Jaraba, L. Agüı, P. Yanez-Sedeno and J. Pingarron, Electrochim. Acta, 1998, 43, 3555–3565 CrossRef CAS.
- C. Chen, X. Li, F. Deng and J. Li, RSC Adv., 2016, 6, 79894–79899 RSC.
- J. Yan, T. Wei, Z. Fan, W. Qian, M. Zhang, X. Shen and F. Wei, J. Power Sources, 2010, 195, 3041–3045 CrossRef CAS.
- Z. Wu, Y. Lv, Y. Xia, P. A. Webley and D. Zhao, J. Am. Chem. Soc., 2012, 134, 2236–2245 CrossRef CAS PubMed.
- H. U. Shah, F. Wang, A. M. Toufiq, A. M. Khattak, A. Iqbal, Z. A. Ghazi, S. Ali, X. Li and Z. Wang, Int. J. Electrochem. Sci., 2016, 11, 8155–8162 CrossRef CAS.
- A. Numan, N. Duraisamy, F. S. Omar, Y. Mahipal, K. Ramesh and S. Ramesh, RSC Adv., 2016, 6, 34894–34902 RSC.
- H. u. Hassan, M. W. Iqbal, A. M. Afzal, T. Abbas, A. Zaka, A. Yasmeen, N. A. Noor, S. Aftab and H. Ullah, Int. J. Energy Res., 2022, 1–13 Search PubMed.
- N. Neghmouche and T. Lanez, Int. Lett. Chem., Phys. Astron., 2013, 4, 37–45 Search PubMed.
- H. Pang, Y.-Z. Zhang, Z. Run, W.-Y. Lai and W. Huang, Nano Energy, 2015, 17, 339–347 CrossRef CAS.
- R.-Z. Li, R. Peng, K. D. Kihm, S. Bai, D. Bridges, U. Tumuluri, Z. Wu, T. Zhang, G. Compagnini and Z. Feng, Energy Environ. Sci., 2016, 9, 1458–1467 RSC.
- L. Ma, X. Shen, Z. Ji, G. Zhu and H. Zhou, Chem. Eng. J., 2014, 252, 95–103 CrossRef CAS.
- B. Wang, X. He, H. Li, Q. Liu, J. Wang, L. Yu, H. Yan, Z. Li and P. Wang, J. Mater. Chem. A, 2014, 2, 12968–12973 RSC.
- N. Nadiah, F. S. Omar, A. Numan, Y. Mahipal, S. Ramesh and K. Ramesh, Int. J. Hydrogen Energy, 2017, 42, 30683–30690 CrossRef CAS.
- Z. H. Bakr, Q. Wali, J. Ismail, N. K. Elumalai, A. Uddin and R. Jose, Electrochim. Acta, 2018, 263, 524–532 CrossRef CAS.
- R. Imani, M. Pazoki, A. Tiwari, G. Boschloo, A. P. Turner, V. Kralj-Iglič and A. Iglič, Nanoscale, 2015, 7, 10438–10448 RSC.
- Z. Gao, X. Wang, J. Chang, D. Wu, L. Wang, X. Liu, F. Xu, Y. Guo and K. Jiang, RSC Adv., 2015, 5, 48665–48674 RSC.
- S. Vadivel, D. Maruthamani, M. Kumaravel, B. Saravanakumar, B. Paul, S. S. Dhar, K. Saravanakumar and V. Muthuraj, J. Taibah Univ. Sci., 2017, 11, 661–666 CrossRef.
- S. Xu, X. Li, Z. Yang, T. Wang, W. Jiang, C. Yang, S. Wang, N. Hu, H. Wei and Y. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 27868–27876 CrossRef CAS PubMed.
- D. S. Patil, S. A. Pawar, R. S. Devan, M. G. Gang, Y.-R. Ma, J. H. Kim and P. S. Patil, Electrochim. Acta, 2013, 105, 569–577 CrossRef CAS.
-
M. S. Halper and J. C. Ellenbogen, Supercapacitors: A Brief Overview, The MITRE Corporation, McLean, Virginia, USA, 2006, p. 34 Search PubMed.
- M. Z. Iqbal, M. M. Faisal, S. R. Ali, S. Farid and A. M. Afzal, Electrochim. Acta, 2020, 346, 136039 CrossRef CAS.
- Y. Ma, Y. Jia, L. Wang, M. Yang, Y. Bi and Y. Qi, J. Mater. Chem. A, 2016, 4, 10414–10418 RSC.
- G. K. Veerasubramani, K. Krishnamoorthy and S. J. Kim, J. Power Sources, 2016, 306, 378–386 CrossRef CAS.
- Y. He, W. Chen, X. Li, Z. Zhang, J. Fu, C. Zhao and E. Xie, ACS Nano, 2013, 7, 174–182 CrossRef CAS PubMed.
- R. Kumar, A. Agrawal, R. K. Nagarale and A. Sharma, J. Phys. Chem. C, 2016, 120, 3107–3116 CrossRef CAS.
- C. Wang, A. Schechter and L. Feng, Nano Research Energy, 2023, 2, e9120056 CrossRef.
- J. Xu, X. Zhong, X. Wu, Y. Wang and S. Feng, J. Energy Chem., 2022, 71, 129–140 CrossRef CAS.
- K. Ham, J. Lee, K. Lee and J. Lee, J. Energy Chem., 2022, 71, 580–587 CrossRef CAS.
- J. Cao, Y. Hu, Y. Zhu, H. Cao, M. Fan, C. Huang, K. Shu, M. He and H. C. Chen, Chem. Eng. J., 2021, 405, 126928 CrossRef CAS.
- M. Z. Iqbal, M. M. Faisal, S. R. Ali and M. Alzaid, Solid State Ionics, 2020, 354, 115411 CrossRef CAS.
- S. Kumar, S. Riyajuddin, M. Afshan, S. K. T. Aziz, T. Maruyama and K. Ghosh, J. Phys. Chem. Lett., 2021, 12, 6574–6581 CrossRef CAS PubMed.
- M. Z. Iqbal, A. Khan, A. Numan, S. S. Haider and J. Iqbal, Ultrason. Sonochem., 2019, 59, 104736 CrossRef CAS PubMed.
- Y. Wang and N. Herron, J. Phys. Chem., 1987, 91, 5005–5008 CrossRef CAS.
- A. Sharma, S. Kapse, A. Verma, S. Bisoyi, G. K. Pradhan, R. Thapa and C. S. Rout, ACS Appl. Energy Mater., 2022, 5, 10315–10327 CrossRef CAS.
- Q. Hu, M. Tang, M. He, N. Jiang, C. Xu, D. Lin and Q. Zheng, J. Power Sources, 2020, 446, 227335 CrossRef CAS.
- S. B. Kale, A. C. Lokhande, R. B. Pujari and C. D. Lokhande, J. Colloid Interface Sci., 2018, 532, 491–499 CrossRef CAS PubMed.
- J. Li, X. Zhou, Z. Xia, W. Gao, Y. Ma and Y. Qu, ACS Appl. Mater. Interfaces, 2016, 8, 10826–10834 CrossRef CAS PubMed.
- Z. Yu, L. Tetard, L. Zhai and J. Thomas, Energy Environ. Sci., 2015, 8, 702–730 RSC.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Nora Hamad
Al-Shaalan
*a,
Nora Hamad
Al-Shaalan























