
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtein engineering of multi-enzyme virus-like particle nanoreactors for enhanced chiral alcohol synthesis†
Taotao
Feng‡
a,
Jiaxu
Liu‡
a,
Xiaoyan
Zhang‡
a,
Daidi
Fan
 b and
Yunpeng
Bai
*a
b and
Yunpeng
Bai
*a
aState Key Laboratory of Bioreactor Engineering, Shanghai Collaborative Innovation Center for Biomanufacturing, East China University of Science and Technology, Shanghai 200237, China. E-mail: ybai@ecust.edu.cn
bShaanxi R&D Centre of Biomaterials and Fermentation Engineering, School of Chemical Engineering, Northwest University, Xi'an, Shaanxi 710069, China
First published on 18th October 2023
Abstract
In the past decade, virus-like particles (VLPs) that can encapsulate single or multiple enzymes have been studied extensively as typical nanoreactors for biocatalysis in vitro, yet their catalytic efficiencies are usually inadequate for real applications. These biocatalytic nanoreactors should be engineered like their free-enzyme counterparts to improve their catalytic performance for potential applications. Herein we engineer biocatalytic VLPs for the enhanced synthesis of chiral alcohols. Different methods including directed evolution were applied to the entire bacteriophage P22 VLPs (except the coat protein), which encapsulated a carbonyl reductase from Scheffersomyces stipitis (SsCR) and a glucose dehydrogenase from Bacillus megaterium (BmGDH) in their capsids. The best variant, namely M5, showed an enhanced turnover frequency (TOF, min−1) up to 15-fold toward the majority of tested aromatic prochiral ketones, and gave up to 99% enantiomeric excess in the synthesis of chiral alcohol pharmaceutical intermediates. A comparison with the mutations of the free-enzyme counterparts showed that the same amino acid mutations led to different changes in the catalytic efficiencies of free and confined enzymes. Finally, the engineered M5 nanoreactor showed improved efficiency in the scale-up synthesis of chiral alcohols. The conversions of three substrates catalyzed by M5 were all higher than those catalyzed by the wild-type nanoreactor, demonstrating that enzyme-encapsulating VLPs can evolve to enhance their catalytic performance for potential applications.
Introduction
Compartmentalization is the basis of cells and has evolved to control specific biological activities in isolated and confined cellular environments. Recent studies have shown that many nanostructures can promote enzyme catalysis in cells, e.g., nanoscale dual-enzyme proximity,1 nanocluster of enzyme molecules,2 and nanoporous gel states.3 Nanoscale macromolecular complexes are formed to ensure the rapid transfer of metabolic intermediates between the active sites of multiple enzymes. Ordered nanostructures, therefore, provide the structural basis for efficient catalysis in vivo. In addition to nano-compartments surrounded by lipid membranes, compartments assembled from purely proteinaceous materials have attracted much attention because of their genetic programmability, structural diversity, and functional controllability. There is a large diversity of protein-based compartments with a variety of biological functions.4–6 Protein compartments that incorporate catalytic species in their interiors have evolved as nanoreactors. These separate them from the rest of the cell, facilitate some specific biochemical activities and play a key role in cellular metabolism.7–9Inspired by nature, many artificial protein cages have been constructed in vitro to localize and control specific chemical reaction processes (Scheme 1).10–12 These proteinaceous compartments can mimic the catalytic environments in cells, and thereby significantly improve the efficiencies of enzyme-catalyzed reactions by enhancing the mass transfer inside the protein cages.13 For example, virus-like nanoparticles (VLPs) derived from the bacteriophage P22 have emerged as a mature and robust platform for nanoreactor design, self-assembly, and cargo packaging.14–16 By genetically programming the scaffold protein (SP), fused enzymes, and coat protein (CP), self-assembled P22 VLPs can be easily controlled to act as nanoreactors with different catalytic activities. Various single and multiple enzymes have been encapsulated in P22 VLPs for single and cascade enzymatic reactions.17–19 VLP-based biocatalysis can be used in biomanufacturing and health-care applications, e.g., in chiral compound synthesis, hydrogen production,8 and cell metabolism.9
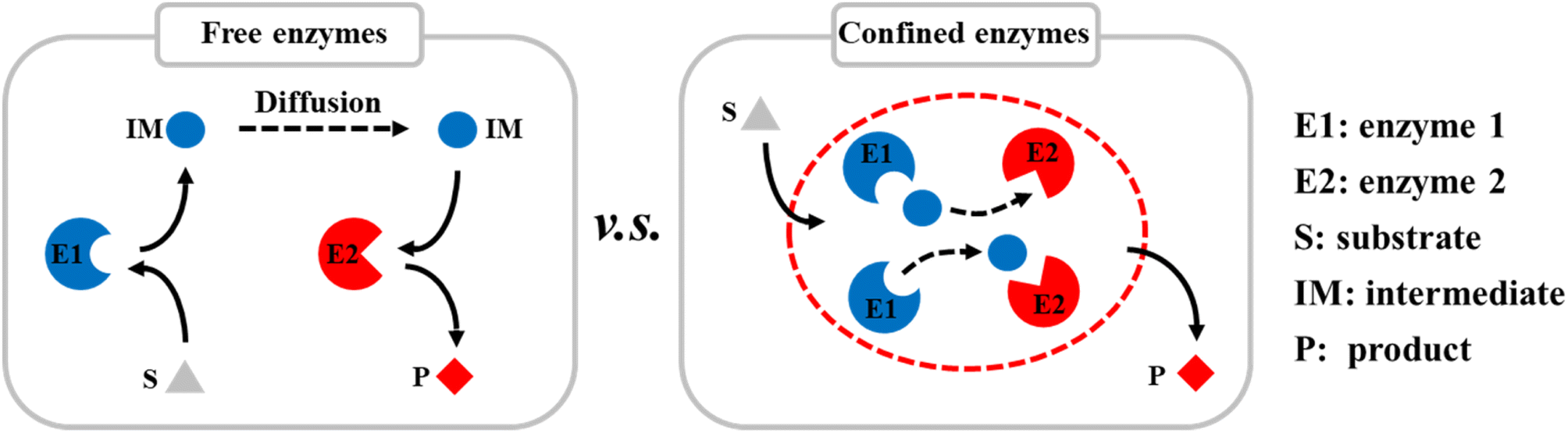 | ||
| Scheme 1 Diagram of encapsulation of multiple enzymes in protein cages in vitro to improve the catalytic efficiencies of reactions by enhancing mass transfer. | ||
Previously, we successfully encapsulated SsCR (Protein Data Bank, PDB: 5GMO) and BmGDH (PDB: 1GCO) in single P22 VLPs to construct a self-sufficient system for chiral alcohol synthesis with enhanced cofactor recycling efficiency (Scheme 2a).20 We found that the catalytic activity of BmGDH was lower than that of SsCR; therefore BmGDH became the rate-limiting enzyme in the dual-enzyme catalytic system and limited the total catalytic efficiency of the nanoreactor toward ketone substrates. However, to the best of our knowledge, it has not yet been determined whether the whole P22 biocatalytic nanoreactor can be engineered to give higher activity, similar to the protein engineering of the free-enzyme counterparts.21–25 Laboratory protein engineering is usually performed to enhance the catalytic performance of enzymes, in which free enzyme variants dissolved in a buffer solution are screened in microplate wells; this takes no account of the effects of the physical environment on protein engineering. Thus, the evolved enzyme shows a higher activity in a free environment instead of a confined environment. It has been reported that physical effects such as the confinement effect and crowded cellular environment may cause conspicuous changes in the structures of enzymes and influence their catalysis,13,26–30 and we guess this may also influence enzyme engineering in vitro. Therefore, to obtain VLPs with higher catalytic performances, a more effective method is needed to engineer enzymes in confined nanoreactors.
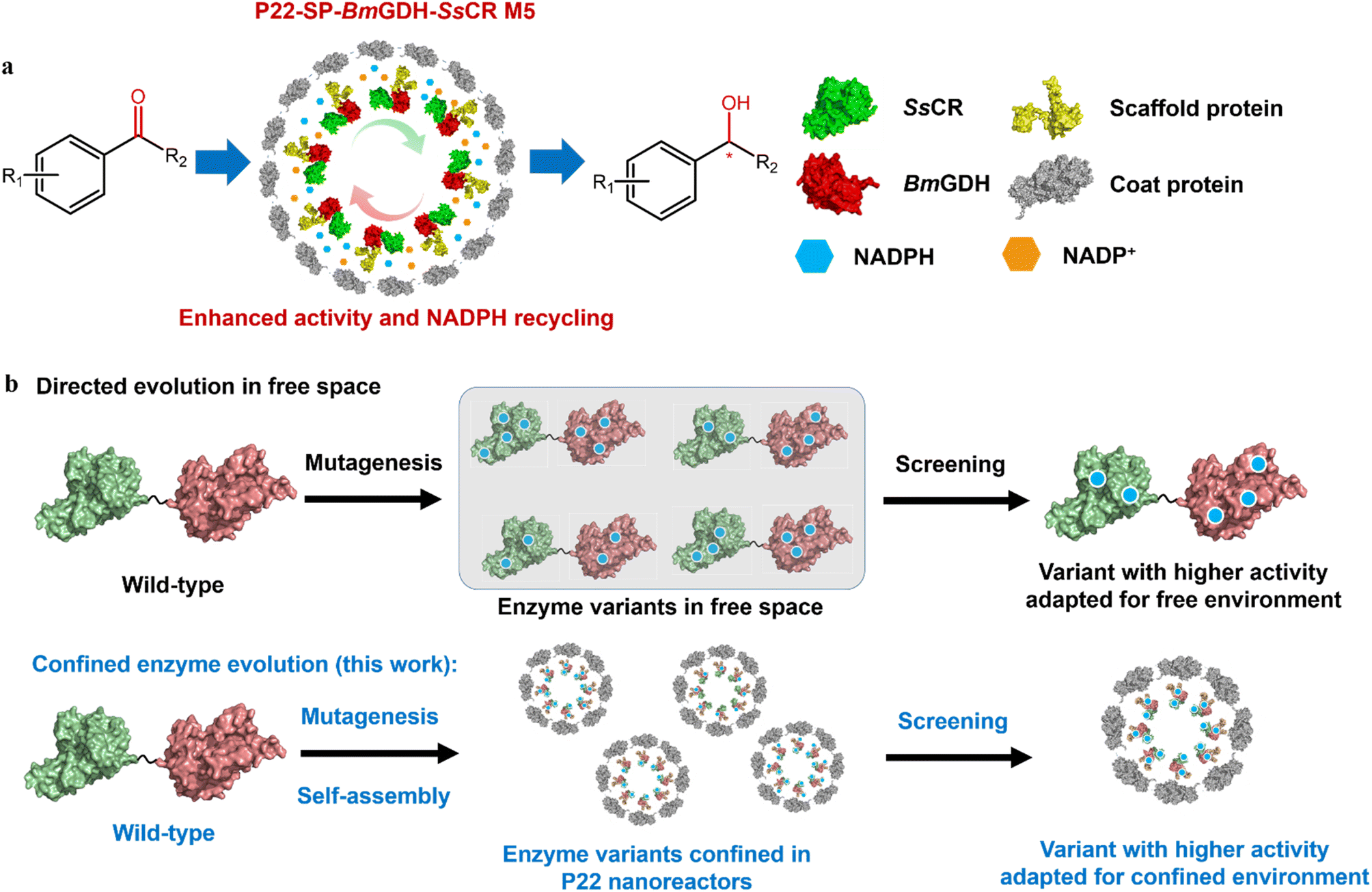 | ||
| Scheme 2 Diagram of spatially confined enzyme engineering. (a) Asymmetric reduction of aromatic prochiral ketones to the corresponding chiral alcohols by engineered P22-SP-BmGDH-SsCR M5. (b) Comparison of protein engineering in free and confined-space. | ||
To this end, we developed a strategy that involves enzyme engineering of all the enzyme components encapsulated in a P22 nanoreactor, except the CP, to obtain P22-SP-BmGDH-SsCR M5, an engineered biocatalyst with an improved catalytic activity toward ketones (Scheme 2b). This method used a combination of mutagenesis methods and self-assembly of protein variants to produce libraries of P22-based mutants. Subsequent screening with a model substrate, namely 2,2′,4′-trichloroacetophenone (1a) for ketone reduction activity and glucose for glucose dehydrogenation activity, yielded the best-adapted variant for confined environments after iterative mutation (Scheme 2b). In addition, we introduced the same mutations obtained from P22-SP-BmGDH-SsCR variants into the free SP-BmGDH-SsCR enzyme and free single enzymes, respectively, and compared the changes in their catalytic performances to investigate the effect of confinement on protein engineering.
Results and discussion
Construction of P22-SP-BmGDH-SsCR nanoreactors
First, a single plasmid (pRSFDuet-1) containing a CP gene, a fusion protein gene (SP-BmGDH-SsCR), and an independent BmGDH gene was employed in E. coli BL21 (DE3) for protein expression (Fig. 1a). The protein components were expressed together with the induction of isopropyl-β-D-thiogalactopyranoside (IPTG) and self-assembled into P22-SP-BmGDH-SsCR nanoreactors (Fig. 1b). The nanoreactor showed a Vmax of 5.0 μmol min−1 mg−1 with substrate 1a, but severe substrate inhibition occurred at higher substrate concentrations (KI = 0.1 mM) (Fig. S1a†). For glucose, the Vmax is 0.29 μmol min−1 mg−1 without substrate inhibition (Fig. S1b†). A previous report suggested that the order of protein expression is crucial for nanoreactor assembly, and enzyme maturation before encapsulation is required for retaining activity.14 Accordingly, we prepared nanoreactors by first expressing the fusion protein and the independent BmGDH from pRSFDuet-1, and then inducing the CP expression from pBAD-hisB with L-arabinose (Fig. 1c and d). The Vmax of this nanoreactor increased to 6.7 μmol min−1 mg−1 for 1a and 0.68 μmol min−1 mg−1 for glucose (Fig. S1c and d†), and it was selected as M0 for further engineering to improve its catalytic efficiency. However, the substrate inhibition for 1a was not eliminated (KI = 0.1 mM). In addition to nanoreactors, free enzymes, such as the fused SP-BmGDH-SsCR (Fig. S2†), SsCR (Fig. S3†), and BmGDH (Fig. S4†), were also prepared and purified for the subsequent studies of enzyme engineering in different environments. | ||
| Fig. 1 Expression and characterization of the wild-type P22-SP-BmGDH-SsCR nanoreactors. (a) Single plasmid (pRSFDuet-1) containing the genes of the fusion protein, GDH, and CP was employed in E. coli BL21 (DE3), and protein components were expressed together after the induction of IPTG. (b) TEM of nanoreactors assembled from proteins expressed from a single plasmid. (c) Two plasmids (pRSFDuet-1 and pBAD-hisB) separately containing the genes of protein cargo (the fusion protein and GDH) and CP was employed. The expression of CP was inducted by L-arabinose after the expression of protein cargo induced by IPTG. (d) TEM of nanoreactors assembled from proteins expressed from double plasmids. | ||
Protein engineering of P22-SP-BmGDH-SsCR nanoreactors
The turnover frequency (TOF, 3.8 × 102 min−1) of M0 (the wild-type P22-SP-BmGDH-SsCR) was determined with 1 mM 1a, 10 mM glucose, and 1 mM NADP+ at 30 °C (Table 1, entry 1). The first libraries containing 6000 nanoreactor variants were constructed by introducing mutations into both the fusion protein gene (SP-BmGDH-SsCR) and the independent BmGDH gene through error-prone polymerase chain reaction (ep-PCR). First, we quickly scanned the reduction and dehydrogenate activities of the mutated nanoreactor library toward 1a and glucose (Fig. S5†) in 96 microplates and obtained 24 better variants which showed higher activity. Subsequently, to further confirm their activity, we expressed and purified these 24 variants, and then measured their reduction and dehydrogenate activities. After a comprehensive evaluation of these two activities of the 24 variants (Fig. S6†), we selected 8 variants from them for sequencing and identified the mutation V356A, which was located in the BmGDH part of the nanoreactor P22-SP-BmGDH-SsCR and this variant was named M1 (V11A in BmGDH and V356A in SP-BmGDH-SsCR). This mutation increased the catalytic efficiency (kcat/Km) of SsCR and BmGDH by 1.9- and 1.7-fold, respectively (Table S1†). The introduction of V356A increased the TOF of M1 toward 1a to 6.3 × 102 min−1 (Table 1, entry 2), but substrate inhibition was not alleviated. To reduce substrate inhibition and improve the nanoreactor catalytic performance at higher substrate concentrations, we introduced the mutation L211H (L211H in SsCR and L211H of SP-BmGDH-SsCR) into SsCR to obtain M2.31 The kinetic analysis of M2 showed that substrate inhibition (1a) was removed (Fig. S7†), with an increased TOF of 2.3 × 103 min−1 (Table 1, entry 3), which was 6.2 times higher than that of M0.| Nanoreactor | Mutation | Conv.b [%] | TOFc [min−1] | Fold |
|---|---|---|---|---|
| a Reaction conditions: 1 mM 1a, 10 mM glucose, 1 mM NADP+, 0.1 mg P22-SP-BmGDH-SsCR, 10 mL PBS (100 mM, pH 6.0), and 20% DMSO at 30 °C. b Conversion of 1a was determined with chiral GC by measuring the peak signal of 2a at 1 h. c TOF (mol substrate converted per mol P22-SP-BmGDH-SsCR per min) was calculated according to the substrate conversion at 1 h. | ||||
| M0 | — | 0.60 | 3.8 × 102 | 1.0 |
| M1 | V356A | 1.0 | 6.3 × 102 | 1.6 |
| M2 | V356A/L211H | 3.9 | 2.3 × 103 | 6.2 |
| M3 | V356A/L211H/S743G | 5.6 | 3.4 × 103 | 8.9 |
| M4 | V356A/L211H/S743G/S445H | 8.8 | 5.3 × 103 | 14 |
| M5 | V356A/L211H/S743G/S445H/V193T | 9.5 | 5.8 × 103 | 15 |
To further enhance the TOF with 1a, M2 was engineered with a second round of random mutagenesis to obtain M3. The newly introduced mutation S129G (S129G in SP and S743G SP-BmGDH-SsCR) was located in the SP part of the fusion protein, which is far from SsCR in the gene sequence. It is interesting that the introduction of this mutation increased the catalytic efficiency of SsCR and increased the TOF to 3.4 × 103 min−1 (Table 1, entry 4). We speculated that this phenomenon may occur because the two sequentially distant domains are spatially close to each other after protein folding and encapsulation.32,33 More importantly, recent research results have shown that SP can have a significant impact on enzyme activity, which can be alleviated through encapsulation and the interaction between SP and the interior surface of VLPs.34,35 Our result supports these findings because the mutation of SP can influence the activity of encapsulated enzymes. Notably, the mutation in SP did not change the number of encapsulated enzymes by comparing the SDS-PAGE of M0 (ref. 20) and M5 (Fig. 4a), indicating that the enhanced activity of SsCR is more likely attributed to structural changes caused by SP mutation.
Alanine scanning was performed on M3 for the amino acids around the BmGDH substrate pocket and C-terminal amino acids,36 which are considered to be critical for BmGDH activity. The scanning results indicate that S100 is a potential site; saturation mutagenesis was performed and alanine was finally replaced with histidine to generate M4 (S100H in BmGDH and S445H in SP-BmGDH-SsCR). The TOF of M4 was 5.3 × 103 min−1 (Table 1, entry 5), which is 13.9-fold higher than that of M0. Similarly, saturation mutagenesis libraries (NNK degenerate codon) of residues located around the SsCR substrate binding pocket were constructed and screened, which contributed to the discovery of V193T (V193T in SsCR and V193T in SP-BmGDH-SsCR). The Km of SsCR was significantly decreased by the introduction of this mutation, and this increased the catalytic efficiency of SsCR (Table S3 and Fig. S7†). The TOF of M5, i.e., 5.8 × 103 min−1 (Table 1, entry 6), is 15.0-fold higher than that of M0, and the product (R)-2-chloro-1-(2,4-dichlorophenyl) ethanol (2a) was obtained with 99% ee, indicating that the stereoselectivity was not affected by engineering.
Investigation of the confinement effect on enzyme engineering in VLPs
First, the effects of mutations on the catalytic efficiencies of enzymes in confined nanoreactors and free enzymes were compared. To this end, we constructed the free fusion enzyme SP-BmGDH-SsCR (Fig. S2†), free single enzyme SsCR (Fig. S3†) and BmGDH (Fig. S4†), and introduced the same mutations as those in M5 into them to obtain the corresponding variants. These mutations were classified into two groups. V356A and S445H occurred only at BmGDH in M5; therefore they were introduced into the BmGDH part of the fusion enzyme SP-BmGDH-SsCR (Fig. 2a, left panel; Table S4†) and the free BmGDH (Table S5†). Similarly, L211H and V193T were only introduced into the SsCR part of SP-BmGDH-SsCR (Fig. 2a, right panel; Tables S4†) and the free SsCR (Table S6†). For each group, the catalytic efficiencies (kcat/Km) of the variants were determined and compared with those of their corresponding wild types (baseline) at each mutation. We found that the introduction of the same mutations affected the enzymes in free forms and confined nanoreactors differently. The effects were also different for BmGDH and SsCR. For example, when V193T occurred at SsCR, the catalytic efficiencies of the enzymes (the nanoreactor, fused enzyme, and free SsCR) all increased, although their fold changes were different (Fig. 2a, right panel). Likewise, L211H caused similar decreases in the catalytic efficiencies of the three enzymes. In contrast, when the mutations took place on BmGDH, the changes in the catalytic efficiency showed clear differences. For example, V356A increased the catalytic efficiency of BmGDH in the nanoreactor 1.7-fold (Tables S3†), whereas the catalytic efficiency was almost unchanged when V356A was introduced into free SP-BmGDH-SsCR (Tables S4†) or free BmGDH (Tables S5†). S445H increased the catalytic efficiencies of the nanoreactor (Tables S3†) and free BmGDH (Tables S5†) but did not affect that of the fused enzyme (Table S4†).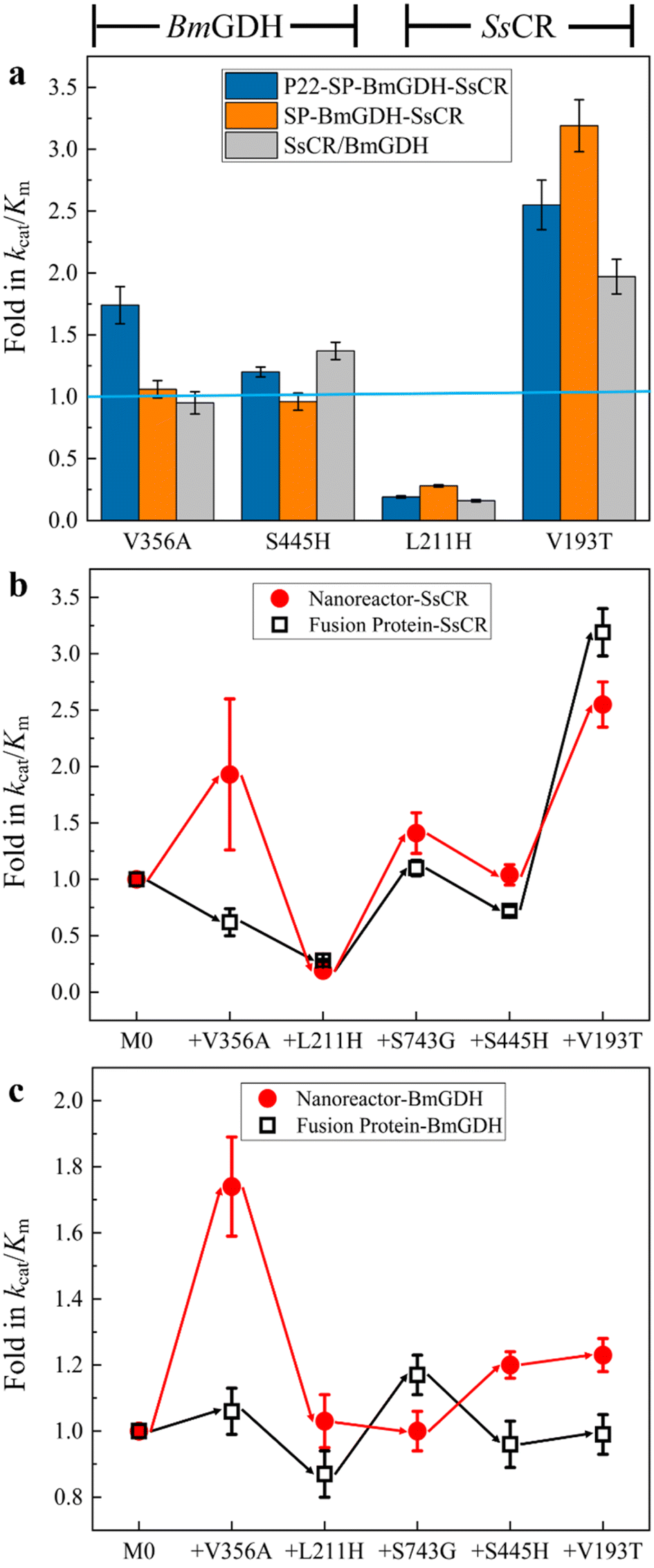 | ||
| Fig. 2 Confinement effect on the engineering of enzymes in free and confined environments. (a) Comparison of the effects of mutations on the catalytic efficiencies of BmGDH and SsCR in nanoreactors, free fusion proteins, and free single enzymes. Comparison of the influence of mutations on nanoreactors and fusion proteins for ketone reduction (b) and glucose dehydrogenation (c). | ||
We then investigated the effects of confinement on the engineering process of the dual-enzyme system in the confined nanoreactor and in the fused enzyme. Because SsCR and BmGDH formed a dual-enzyme system, each mutation obtained in the nanoreactor variants was introduced into the fused enzyme (SP-BmGDH-SsCR) in the same order, which ensured that they shared the same iterative engineering process. Because the addition of one mutation caused changes in kcat/Km simultaneously for ketone reduction (Fig. 2b) and glucose oxidation (Fig. 2c), both catalytic efficiencies of the new variants were determined and compared with that of the parent generation to clarify the effect of confinement on enzymes (Tables S3 and S4, Fig. S7 and S8†). As shown in Fig. 2b, the iterative addition of the same mutations shaped two different processes for the dual-enzyme systems. In particular, V356A increased the reductive activity of the nanoreactor but decreased that of the free fusion enzyme. However, the rest of the mutations generated similar trends in catalytic efficiencies. Furthermore, the differences between the glucose dehydrogenation activities of the dual enzymes in confined and free environments were more obvious (Fig. 2c). After each mutation, the change direction (red and black arrows) and the fold in the catalytic efficiency were generally different. It is reasonable to attribute these differences to the different physical environments (confined vs. free) in which the dual enzymes reside. It should be noted that the confinement effect had a greater influence on BmGDH. BmGDH is located at the middle position in the fusion protein composed of SP, BmGDH and SsCR, which means that the SP and SsCR on both sides will affect its structure, although the truncated SP is still predominantly disordered and flexible. Moreover, the crowded space caused by the high packing density (29 BmGDH and 176 SsCR) inside the P22 particles significantly reduces the flexibility of BmGDH.20 Of course, high packing density and fusion expression can also affect SsCR, which is restricted to a relatively smaller sub-shell of VLP. However, SsCR is a single-subunit enzyme and located at one end of the fusion protein, enabling it to have a relatively higher flexibility. In addition, considering that BmGDH requires the assembly of four identical subunits to form a complete catalytic structure in order to perform its catalytic function,37 its complete spatial structure is much larger than that of SsCR, which makes it more sensitive to changes in spatial hindrance.
The TOFs of the wild type (V0) and variants of the free fusion protein SP-BmGDH-SsCR (V1–V5) were determined with 1 mM 1a, 10 mM glucose, and 1 mM NADPH at 30 °C (Table S2†), and compared with those of the nanoreactors under the same conditions (Fig. 3a). As mutations accumulated, the TOF of the nanoreactor toward 1a gradually increased to a value 15.0-fold higher than that of M0; however, the TOF of V5 was only 1.8-fold higher than that of V0. Then, the time-course reduction reactions of 1a by nanoreactors and fusion proteins were performed to compare their real catalytic performances (Fig. 3b). The results show that the reaction rate for M5 was significantly higher than that for M0. M5 achieved 100% conversion of 1 mM 1a in 8 h, whereas M0 converted only approximately 20% of 1a at the same time, and the final conversion was only 50% after 24 h (Fig. 3b). When the cofactor NADPH concentration was decreased to 10 μM, the catalytic performance of M5 was still significantly better than that of M0 (Fig. S9a†). This indicates that the engineered nanoreactor can still catalyze the reaction more efficiently at low cofactor concentrations. In contrast, when the fusion enzymes were used for the reaction with 1a, there was a much smaller difference between the results with the wild-type V0 and mutant V5 (Fig. 3c). When the substrate concentration was increased to 10 mM, the catalytic rates for V0 and V5 differed (Fig. S9b†). This was the result of substrate inhibition of V0, which limited its catalytic rate at relatively high substrate concentrations. To sum up, the conversion results correlate well with the TOF changes in the two systems. This indicates that protein engineering in spatially confined VLPs not only reshaped the nanoreactor (Fig. 2) but also generated enzyme variants that were more powerful than those generated by free-enzyme engineering, although the same protein engineering strategies (the same mutations and same mutation sequences) were applied.
 | ||
| Fig. 3 Substrate conversion by variants of nanoreactors and free fusion enzymes. (a) TOFs of P22-SP-BmGDH-SsCR and SP-BmGDH-SsCR. (b) Time-course conversion of 1 mM 1a by P22-SP-BmGDH-SsCR with 1 mM NADPH. (c) Time-course conversion of 1 mM 1a by SP-BmGDH-SsCR with 1 mM NADPH. | ||
Characterization of the nanoreactors P22-SP-BmGDH-SsCR M0 and M5
Nanoreactors M0 and M5 were characterized to determine whether the protein engineering affected protein assembly and the sizes of nanoreactors. The analysis of purified P22-SP-BmGDH-SsCR M5 by SDS-PAGE showed bands corresponding to SP-BmGDH-SsCR (∼84.5 kDa), CP (∼46.7 kDa), and the independent BmGDH subunit (∼28.1 kDa), with no evidence of single-enzyme degradation products or additional bands (Fig. 4a). Transmission electron microscopy showed that M5 was monodisperse, like M0 (Fig. 4b). The diameters of M0 and M5 measured by TEM were both ∼68.0 ± 3.0 nm. The particle sizes of M5 and M0 were also determined by multi-angle laser light scattering; the diameters of M5 and M0 both showed a normal distribution, and the average diameters of M0 and M5 were approximately 68.1 ± 1.0 nm and 69.7 ± 1.0 nm, respectively (Fig. 4c). These results correlated well and indicated that these several mutations did not affect the size of the nanoreactors after protein engineering.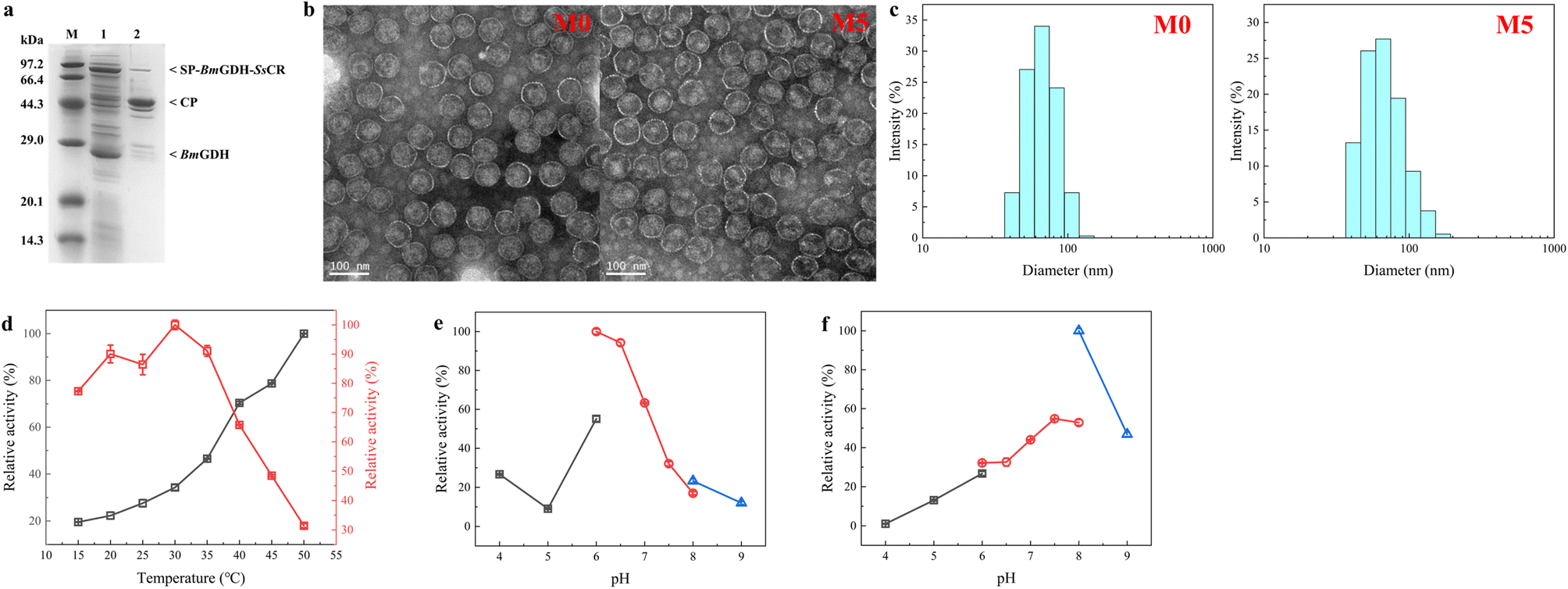 | ||
| Fig. 4 Characterization of P22-SP-BmGDH-SsCR M0 and M5. (a) SDS-PAGE analysis of P22-SP-BmGDH-SsCR M5. Lanes: M, protein marker; 1, supernatant obtained from cell lysis; 2, purified enzymes. (b) Transmission electron microscopy image of P22-SP-BmGDH-SsCR particles M0 and M5 (∼68 ± 3 nm) and scale bar 100 nm. (c) Size-exclusion chromatography coupled with multi-angle laser light scattering for the analysis of P22-SP-BmGDH-SsCR. (d) Determination of optimum reaction temperatures for SsCR (red) and BmGDH (black) encapsulated in M5. (e) Determination of optimum reaction pH for SsCR encapsulated in M5. (f) Determination of optimum reaction pH for BmGDH encapsulated in M5. Buffers used in (e) and (f): black, sodium citrate buffer; red, sodium phosphate buffer; blue, Tris–HCl buffer. | ||
The optimum temperatures and pH values for the catalytic reaction with SsCR and BmGDH encapsulated in M5 and M0 were determined (Fig. 4d and S10†). The optimum temperatures and pH values for the two enzymes did not change after engineering. For M5 and M0, SsCR gave the highest reaction activity at 30 °C. When the temperature exceeded 35 °C, the enzymatic activity decreased rapidly. For BmGDH, the catalytic activity of BmGDH increased in the temperature range 15–50 °C. We therefore chose 30 °C as the reaction temperature for subsequent experiments.
For both M5 and M0, SsCR gave the highest activity in PBS buffer at pH 6.0 (Fig. 4e and S10b†), and BmGDH gave the highest activity in Tris–HCl buffer at pH 8.0 (Fig. 4f and S10c†). However, in Tris–HCl buffer at pH 8.0, the activity of SsCR was less than 20% of its maximum, and therefore PBS at pH 6.0 was chosen as the buffer system for subsequent experiments.
Asymmetric reductions of substrates 1a–1j were performed to further evaluate the substrate scope of M5; the TOFs and product enantiomer excesses (ee values) were used as benchmarks of activity and stereoselectivity (Fig. 5). For the 10 tested substrates, M5 showed enhanced activities toward seven substrates, namely 1a, 1d, 1e–1h, and 1j, the same activity toward 1c, and slightly decreased activities toward two substrates, namely 1b and 1i, compared with those achieved with M0. Clearly, protein engineering generated mutations that were beneficial for most substrates as well as the model substrate 1a. For substrates 1a, 1e, and 1f, the TOFs increased 15.0-, 11.7-, and 4.8-fold, respectively. In addition, M5 showed weaker substrate inhibition, mainly because of the L211H mutation. The stereoselectivities of M5 toward the five substrates 1a, 1c, 1d, 1f, and 1h were unchanged. However, considering that the screening method aims to pick up variants with higher activities, it is not unusual to obtain hits with reduced stereoselectivities (e.g.1b, 1e, 1g, 1i and 1j). The clarification of the underlying mechanism is still a challenge because of the lack of effective tools for observing the structures of enzymes confined in P22 nanoparticles.
 | ||
| Fig. 5 Substrate scopes of M0 and M5. Assays were performed with P22-SP-BmGDH-SsCR (0.1 mg), 1 mM substrate, PBS buffer (100 mM, 10 mL), and 20 vol% DMSO at pH 6.0 and 30 °C. Substrate conversions were determined at 60 min and TOFs (mol substrate converted per mol P22-SP-BmGDH-SsCR per min) were then calculated. Chiral gel chromatography was used to determine ee values (Table S7†). | ||
Scaled-up synthesis of chiral alcohols
Optically pure aromatic chiral alcohols are essential chiral scaffolds for the production of pharmaceuticals. For example, (R)-2-chloro-1-(2,4-dichlorophenyl)ethanol (2a) is used in the synthesis of many antifungal agents such as miconazole, econazole, and sertaconazole,38,39 and (R)-2-chloro-1-(2,4-difluorophenyl)ethanol (2h) is used in the synthesis of several compounds that prevent thrombosis. The corresponding prochiral haloketones were therefore selected as model substrates for testing the potential of M5.As shown in the conversion profiles, the catalytic performances of M5 were better than those of M0 toward all substrates. The conversion of 1a reached over 95% within 6 h when M5 was used as the catalyst. In the presence of M0, a lower conversion (21.2%) was achieved within 6 h, and the conversion reached only 81.8% after 24 h (Fig. 6a). In the reduction of 10 mM 2-chloro-1-(2,4-difluorophenyl)ethanone (1h), M5 achieved >99.0% conversion within 6 h, whereas M0 afforded only 37.6% conversion within 6 h and 89.5% after 24 h (Fig. 6b). In the reduction of 2′-chloroacetophenone (1g) with M5, 50.2% conversion was achieved in 24 h. In contrast, 11.8% conversion was obtained after 24 h with M0 as the catalyst (Fig. 6c).
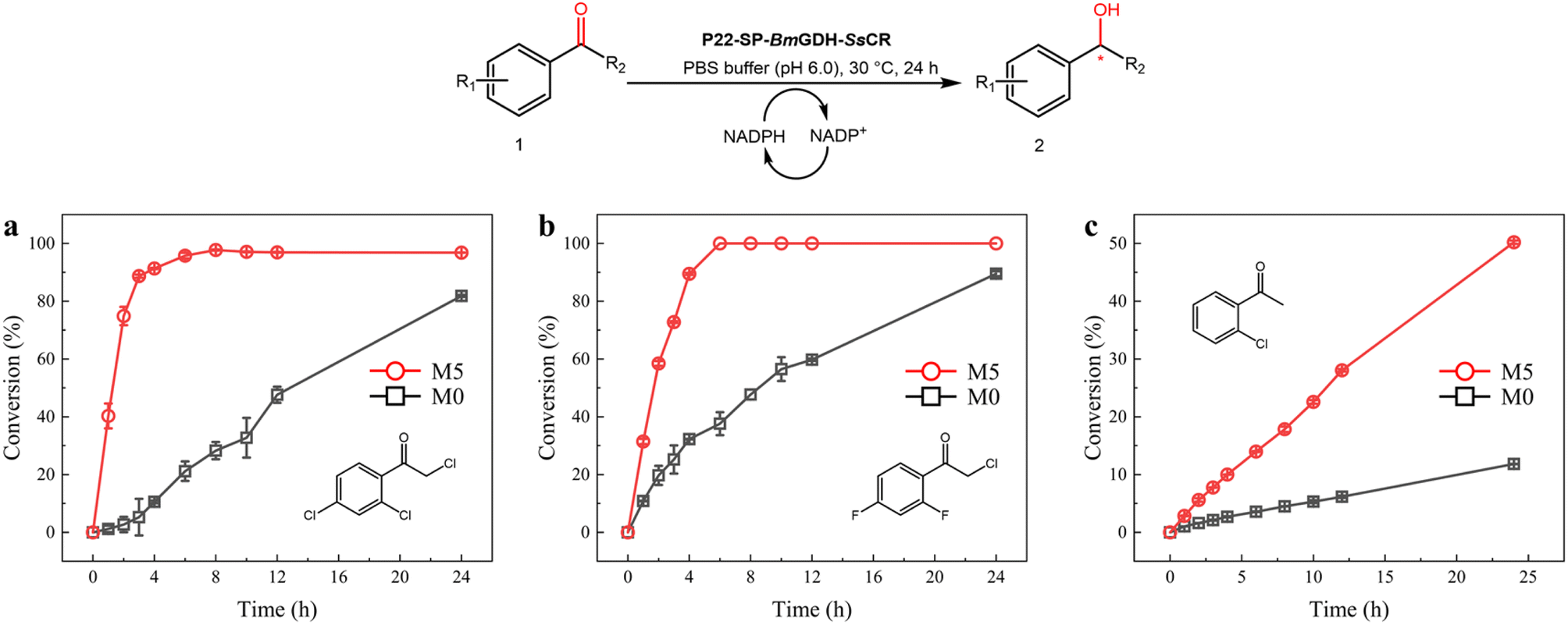 | ||
| Fig. 6 Scaled-up synthesis of chiral alcohols with P22-SP-BmGDH-SsCR M0 (black square) and M5 (red circle). Reactions were performed with 10 mM 1a (a), 1f (b), and 1h (c), and 1 mg of purified M0 and M5, in a sodium phosphate buffer (100 mM, pH 6.0). | ||
Conclusions
In this work, the confined interior spaces of P22 VLPs were used to assemble a carbonyl reductase–glucose dehydrogenation dual-enzyme system. The entire nanoreactor was then subjected to protein engineering. The TOFs achieved with an engineered nanoreactor, namely P22-SP-BmGDH-SsCR M5, in catalyzing a series of aromatic prochiral ketones were significantly better than those obtained with the wild-type nanoreactor. By comparing the catalytic efficiencies of enzymes evolved in confined P22 nanoreactors with those of enzymes evolved in free environments, we showed that the confinement effect influenced the catalytic performance of the evolved enzymes. This led to a 15.0-fold TOF enhancement in the conversion of the model substrate 1a. However, although the same mutations were applied, only a 1.8-fold enhancement was observed for the free dual-enzyme system. In conclusion, we reported an effective method that various enzyme engineering tools including directed evolution can be used to generate more powerful biocatalysts with fine-tuned nanostructures. This work shows that enzymes behave differently in different contexts and provides insights into the roles that physical effects play in protein engineering.Experimental
Materials
Escherichia coli BL21 strains harboring SsCR, BmGDH, SP, and CP genes were constructed previously and stored in our laboratory. DNA primers were purchased from Generay Biotech Co., Ltd (Shanghai, China). All chemicals were purchased from TCI (Japan), and Aladdin and Shaoyuan (Shanghai, China), and used without further treatment unless otherwise indicated. NADPH and NADP+ were purchased from Bontac Bioengineering (Shenzhen, China). Luria–Bertani (LB) medium was used for the culture of E. coli cells. Gas chromatographic analysis was performed with a Shimadzu GC-2010 Pro system with a CP-Chirasil-Dex CB column (25 m × 0.25 mm × 0.39 mm, Varian) and a flame ionization detector.Random mutagenesis and site-saturation mutagenesis
A recombination plasmid containing the P22-SP-BmGDH-SsCR M0 gene was used as the template for random mutagenesis. MnCl2 (0.075 mM for SsCRM0-BmGDHM0-SP or 0.2 mM for BmGDHM0) was used to obtain the desired mutagenesis rate (approximately one or two amino acid substitutions). The amplified PCR products were extracted, digested with Nde I and Xho I (for SsCRM0-BmGDHM0-SP) or Sal I and Not I (for BmGDHM0), ligated with pRSFDuet-1, and then transformed into chemically competent E. coli BL21 (DE3) cells. The recombination plasmid containing the P22-SP-BmGDH-SsCR M3 gene was amplified by PCR with NNK codon degeneracy. The resulting PCR products were digested with Dpn I (20 U) at 37 °C for 3 h, transformed into chemically competent E. coli BL21 (DE3) cells, and plated on LB agar plates containing 50 μg mL−1 kanamycin and 100 μg mL−1 ampicillin. The colonies were picked with sterile toothpicks to inoculate LB medium (300 mL) containing 50 μg mL−1 kanamycin in 96-well plates. The cultures were grown overnight at 37 °C prior to inoculating LB medium (600 mL) into new 96-well plates. The plates were incubated at 37 °C for 3 h. Protein expression was induced by addition of IPTG (0.1 mM) and L-arabinose (0.02 mg/100 mL) at 16 °C and incubation was continued for another 24 h. The cells were lysed by adding a buffer (400 mL) containing lysozyme (0.75 mg mL−1) and DNase I (0.01 mg mL−1) at 37 °C for 2 h. The plates were centrifuged at 3420×g for 20 min at 4 °C. A sample (100 μL) from each well was transferred to a microtiter plate, and then the reaction was initiated by adding a mixture of 100 mM phosphate buffer (pH 6.0, 100 μL), 0.2 mM NADPH, and substrate 1a (4 mM), or a mixture of 100 mM phosphate buffer (pH 6.0, 150 mL), 1 mM NADP+, and 10 mM glucose. The activity of the mutant was determined by recording the change in the NADPH/NADP+ absorbance at 340 nm for 10 min at 30 °C by using a microplate spectrophotometer (BioTek, USA). Mutants with higher activities were chosen for rescreening in a 96-deep-well plate, and the top hits were grown on a 100 mL scale. The best mutants were selected for sequencing and purification for further characterization.Expression and purification of P22-SP-BmGDH-SsCR
E. coli strains containing both plasmids were grown at 37 °C in LB medium with 50 μg mL−1 kanamycin and 100 μg mL−1 ampicillin. The expressions of SP-BmGDH-SsCR and independent BmGDH were induced by addition of IPTG to a final concentration of 40 μM at OD600 = 0.6. Cultures were grown for 16 h at 16 °C after addition of IPTG, and expression of the pBAD-HisB vector containing the CP gene was then induced with l-arabinose (0.02 g/100 mL). Growth was continued for 8 h at 16 °C. Cells were harvested by centrifugation at 7000×g for 10 min (Eppendorf centrifuge 5430R). Cell pellets were suspended in 100 mM phosphate buffer (pH 6.0), and the cell slurry was sonicated on ice. Bacterial cell debris was removed via centrifugation at 8000×g for 60 min. The supernatant was concentrated by ultrafiltration. The concentrate was loaded on a 35% sucrose cushion and centrifuged at 181![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×g for 90 min with an ultracentrifuge (50.2Ti ultracentrifuge rotor). The resulting virus pellet was suspended in 100 mM phosphate buffer (pH 6.0). The protein was further purified on an S-500 70 HR Sephacryl size-exclusion column (GE Healthcare Life Sciences), and its concentration was quantified by using a NanoDrop 2000c instrument (Thermo Scientific).
000×g for 90 min with an ultracentrifuge (50.2Ti ultracentrifuge rotor). The resulting virus pellet was suspended in 100 mM phosphate buffer (pH 6.0). The protein was further purified on an S-500 70 HR Sephacryl size-exclusion column (GE Healthcare Life Sciences), and its concentration was quantified by using a NanoDrop 2000c instrument (Thermo Scientific).
Expression and purification of SP-BmGDH-SsCR, SsCR, and BmGDH
All variants were grown at 37 °C in LB medium with 50 μg mL−1 kanamycin. Protein expression was induced by the addition of IPTG to a final concentration of 40 μM at OD600 = 0.6. Cells were harvested by centrifugation at 7000×g for 10 min (Eppendorf centrifuge 5430R). Cell pellets were suspended in 100 mM phosphate buffer (pH 6.0) and the cell slurry was sonicated on ice. Bacterial cell debris was removed via centrifugation at 8000×g for 60 min. The supernatant was applied to a Ni-NTA affinity chromatography column that had been equilibrated with a binding buffer (20 mM imidazole, 0.5 M NaCl, and 20 mM Tris–HCl buffer, pH 7.4). The proteins were eluted and collected in different tubes, according to an increasing imidazole gradient from 10 to 500 mM. Based on SDS-PAGE results, the purified enzymes were collected for ultrafiltration and their concentrations were quantified by using a NanoDrop 2000c instrument (Thermo Scientific). They were stored at −80 °C after liquid-nitrogen flash freezing.Enzyme assay and kinetic analysis
The activity was assayed at 30 °C by monitoring the changes in the NADPH absorbance at 340 nm with a UV spectrophotometer (Shimadzu UV-1900). The standard assay mixture (1 mL) consisted of sodium phosphate buffer (100 mM, pH 7.0, 970 μL), the substrate (10 μL), NADPH/NADP+ (10 μL), and the pure enzyme (10 μL) at an appropriate concentration. One unit of enzyme activity (U) is defined as the amount of enzyme that can catalyze the oxidation of 1 μmol of NADPH or reduction of 1 μmol of NADP+ per minute under the above conditions. The substrates were dissolved to different final concentrations in DMSO, and the specific activities were determined by using a UV spectrophotometer. The data were processed using Origin 9.0 software, based on the Michaelis–Menten equation.Stereoconfiguration assay of products
The stereoconfigurations of the various products were determined by using a reaction solution (1 mL), which was produced by mixing 10 mM substrate, 20 mM glucose, 0.1 mM NADP+, and an appropriate amount of P22-SP-BmGDH-SsCR at 30 °C under stirring at 1000 rpm. After 24 h, the reaction was terminated by adding 1.0 M sulfuric acid solution (pH 2.0). The reaction mixture was extracted with ethyl acetate. The enantioselectivity was determined by chiral gas chromatography analysis.Transmission electron microscopy
A sample (10 μL) containing P22-enzyme particles (0.1 mg mL−1 protein) was applied to a copper grid with a carbon and Formvar coating, incubated (1 min), and dried at room temperature. After negative staining, images were obtained with a JEM-1400 transmission electron microscope at an accelerating voltage of 200 kV. The electron microscopy images were processed using ImageJ software (NIH).Author contribution
Y. P. B. conceived and supervised the project. T. T. F. and J. X. L. designed and conducted experiments with the help of Y. P. B. and X. Y. Z. T. T. F. wrote the manuscript and all authors edited the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially sponsored by the National Key Research and Development Program of China (2021YFC2102804) and the National Natural Science Foundation of China (No. 22078096).References
- H. J. Cheng, L. Zhang, J. He, W. J. Guo, Z. Y. Zhou, X. J. Zhang, S. M. Nie and H. Wei, Anal. Chem., 2016, 88, 5489–5497 CrossRef CAS PubMed.
- S. G. An, R. Kumar, E. D. Sheets and S. J. Benkovic, Science, 2008, 320, 103–106 CrossRef CAS PubMed.
- O. Medalia, I. Weber, A. S. Frangakis, D. Nicastro, G. Gerisch and W. Baumeister, Science, 2002, 298, 1209–1213 CrossRef CAS PubMed.
- W. Bonacci, P. K. Teng, B. Afonso, H. Niederholtmeyer, P. Grob, P. A. Silver and D. F. Savage, Proc. Natl. Acad. Sci. U.S.A., 2012, 109, 478–483 CrossRef CAS PubMed.
- A. Chessher, R. Breitling and E. Takano, ACS Biomater. Sci. Eng., 2015, 1, 345–351 CrossRef CAS PubMed.
- Y. Diekmann and J. B. Pereira-Leal, J. Biol. Chem., 2013, 449, 319–331 CAS.
- C. K. Weiss and K. Landfester, Catalysts, 2013, 3, 401–417 CrossRef CAS.
- C. M. Agapakis, P. M. Boyle and P. A. Silver, Nat. Chem. Biol., 2012, 8, 527–535 CrossRef CAS PubMed.
- A. H. Chen and P. A. Silver, Trends Cell Biol., 2012, 22, 662–670 CrossRef CAS PubMed.
- C. Lindbladh, M. Rault, C. Hagglund, W. C. Small, K. Mosbach, L. Bulow, C. Evans and P. A. Srere, Biochemistry, 1994, 33, 11692–11698 CrossRef CAS PubMed.
- J. L. Lin, L. Palomec and I. Wheeldon, ACS Catal., 2014, 4, 505–511 CrossRef CAS.
- S. Schoffelen and J. C. M. van Hest, Soft Matter, 2012, 8, 1736–1746 RSC.
- A. Kuchler, M. Yoshimoto, S. Luginbuhl, F. Mavelli and P. Walde, Nat. Nanotechnol., 2016, 11, 409–420 CrossRef CAS.
- P. C. Jordan, D. P. Patterson, K. N. Saboda, E. J. Edwards, H. M. Miettinen, G. Basu, M. C. Thielges and T. Douglas, Nat. Chem., 2016, 8, 179–185 CrossRef CAS PubMed.
- E. Edwards, R. Roychoudhury, B. Schwarz, P. Jordan, J. Lisher, M. Uchida and T. Douglas, J. Mater. Chem. B, 2016, 4, 5375–5384 RSC.
- Y. Wang, M. Uchida, H. K. Waghwani and T. Douglas, ACS Synth. Biol., 2020, 9, 3298–3310 CrossRef CAS PubMed.
- D. P. Patterson, B. Schwarz, R. S. Waters, T. Gedeon and T. Douglas, ACS Chem. Biol., 2014, 9, 359–365 CrossRef CAS PubMed.
- W. M. Aumiller, M. Uchida and T. Douglas, Chem. Soc. Rev., 2018, 47, 3433–3469 RSC.
- D. P. Patterson, P. E. Prevelige and T. Douglas, ACS Nano, 2012, 6, 5000–5009 CrossRef CAS PubMed.
- Y. Q. Zhang, T. T. Feng, Y. F. Cao, X. Y. Zhang, T. Wang, M. R. H. Nina, L. C. Wang, H. L. Yu, J. H. Xu, J. Ge and Y. P. Bai, ACS Catal., 2021, 11, 10487–10493 CrossRef CAS.
- Z. X. Xu, W. D. Liu, C. C. Chen, Q. Li, J. W. Huang, T. P. Ko, G. Z. Liu, W. T. Liu, W. Peng, Y. S. Cheng, Y. Chen, J. Jin, H. Z. Li, Y. Y. Zheng and R. T. Guo, ACS Catal., 2016, 6, 7657–7663 CrossRef CAS.
- X. M. Gong, Z. Qin, F. L. Li, B. B. Zeng, G. W. Zheng and J. H. Xu, ACS Catal., 2019, 9, 147–153 CrossRef CAS.
- S. Duewel, L. Schmermund, T. Faber, K. Harms, V. Srinivasan, E. Meggers and S. Hoebenreich, ACS Catal., 2020, 10, 1272–1277 CrossRef CAS.
- B. M. Su, Z. H. Shao, A. P. Li, M. Naeem, J. Lin, L. D. Ye and H. W. Yu, ACS Catal., 2020, 10, 864–876 CrossRef CAS.
- M. Wehrmann, E. M. Elsayed, S. Kobbing, L. Bendz, A. Lepak, J. Schwabe, N. Wierckx, G. Bange and J. Klebensberger, ACS Catal., 2020, 10, 7836–7842 CrossRef CAS.
- G. H. Wei, W. H. Xi, R. Nussinov and B. Y. Ma, Chem. Rev., 2016, 116, 6516–6551 CrossRef CAS PubMed.
- R. J. Ellis, Trends Biochem. Sci., 2001, 26, 597–604 CrossRef CAS PubMed.
- T. E. Sintra, S. P. M. Ventura and J. A. P. Coutinho, J. Mol. Catal. B: Enzym., 2014, 107, 140–151 CrossRef CAS.
- I. M. Kuznetsova, K. K. Turoverov and V. N. Uversky, Int. J. Mol. Sci., 2014, 15, 23090–23140 CrossRef PubMed.
- A. P. Minton, Curr. Opin. Struct. Biol., 2000, 10, 34–39 CrossRef CAS PubMed.
- Y.-P. Shang, Q. Chen, A.-T. Li, S. Quan, J.-H. Xu and H.-L. Yu, J. Biotechnol., 2020, 308, 141–147 CrossRef CAS PubMed.
- K. A. Dill and J. L. MacCallum, Science, 2012, 338, 1042–1046 CrossRef CAS PubMed.
- D. S. Marks, L. J. Colwell, R. Sheridan, T. A. Hopf, A. Pagnani, R. Zecchina and C. Sander, PLoS One, 2011, 6, 12 CrossRef PubMed.
- L. Esquirol, D. McNeale, T. Douglas, C. E. Vickers and F. Sainsbury, ACS Synth. Biol., 2022, 11, 2709–2718 CrossRef CAS PubMed.
- D. McNeale, L. Esquirol, S. Okada, S. Strampel, N. Dashti, B. Rehm, T. Douglas, C. E. Vickers and F. Sainsbury, ACS Appl. Mater. Interfaces, 2023, 15, 17705–17715 CrossRef CAS PubMed.
- T. Nishioka, Y. Yasutake, Y. Nishiya and T. Tamura, FEBS J., 2012, 279, 3264–3275 CrossRef CAS PubMed.
- K. Yamamoto, G. Kurisu, M. Kusunoki, S. Tabata, I. Urabe and S. Osaki, J. Biochem., 2001, 129, 303–312 CrossRef CAS PubMed.
- F. Giraud, C. Loge, F. Pagniez, D. Crepin, P. Le Pape and M. Le Borgne, Bioorg. Med. Chem. Lett., 2008, 18, 1820–1824 CrossRef CAS PubMed.
- J. Mangas-Sanchez, E. Busto, V. Gotor-Fernandez, F. Malpartida and V. Gotor, J. Org. Chem., 2011, 76, 2115–2122 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Protein expression and purification of nanoreactors, free fusion enzymes and free single enzymes, protein gel electrophoresis, conversion of 1a by nanoreactors and fusion proteins, transmission electron microscopy, enzyme assay, kinetic analysis of enzyme variants, and gas chromatography spectra of products. See DOI: https://doi.org/10.1039/d3na00515a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |