
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRobust 2 nm-sized gold nanoclusters on Co3O4 for CO oxidation†
Quanquan
Shi
*ae,
Zhiwen
Li
bd,
Changhai
Cao
c,
Gao
Li
 *bd and
Sami
Barkaoui
*b
*bd and
Sami
Barkaoui
*b
aCollege of Science, Inner Mongolia Agricultural University, Hohhot 010018, China. E-mail: qqshi@imau.edu.cn
bState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: gaoli@dicp.ac.cn; samibarkaoui501@gmail.com
cKey Laboratory of Biofuels and Biochemical Engineering, SINOPEC Dalian Research Institute of Petroleum and Petrochemicals Co., Ltd, Dalian 116045, China
dUniversity of Chinese Academy of Sciences, Beijing 100049, China
eInner Mongolia Key Laboratory of Soil Quality and Nutrient Resource & Key Laboratory of Agricultural Ecological Securi-ty and Green Development at Universities of Inner Mongolia Autonomous, Hohhot 010018, China
First published on 5th September 2023
Abstract
In this study, gold nanoparticles were dispersed on Co3O4 nanoplates, forming a specific Au–Co3O4 interface. Upon calcination at 300 °C in air, aberration-corrected STEM images evidenced that the gold nanoclusters (NCs) on Co3O4{111} were maintained at ca. 2.2 nm, which is similar to the size of the parent Au colloidal particles, demonstrating the stronger metal-support interaction (SMSI) on Co3O4{111}. Au/Co3O4{111} showed good catalytic activity (a full CO conversion achieved at 80 °C) and durability (over 10 hours) in CO oxidation, which was mainly due to the promotion by the surface oxygen vacancies and intrinsic defects of Co3O4{111} for activating O2 and by Au0, Auδ+, and Au+ species on the surface of gold NCs for CO activation, as evidenced by Raman and Fourier-transform infrared (FT-IR) spectroscopy analysis. Au/Co3O4 catalyzed CO oxidation obeyed the Langmuir–Hinshelwood mechanism at low temperatures.
Introduction
Carbon monoxide (CO), emitted from mobile and stationary combustion sources, is one of the major air pollutants, and its presence even in traceable amounts may cause serious environmental and health problems. Therefore, the elimination of CO, via oxidation by air, acts as one of the important technologies for air cleanup. Gold nanoparticles (NPs) at the size range of 2–5 nm, supported on reducible oxides, have been demonstrated to be extremely active for CO oxidation even at room temperature.1,2 However, the chemical nature of gold particle sizes and the identification of active sites is still being debated. There are several parameters, including the chemical state of Au, the size of the Au-NPs, and the gold–support interface, that can intensely affect the activity of CO oxidation on gold catalysts.3–7 To date, the interfacial perimeter of the Au NPs and oxide supports is popularly proposed as the active site for CO oxidation, where the reaction occurs via a redox mechanism. For example, on Au/TiO2 (ref. 4 and 5) and Au/Fe2O3,7 CO is proposed to be chemically adsorbed on gold NPs and the O2 gas is activated on the oxygen vacancy at the interface. Therefore, identification of the atomic structure of the active gold-oxide is of critical importance for the design of highly active gold catalysts. The geometrical structure of the gold-supported interface at the atomic scale is less studied.Reducible oxides, such as Fe2O3, TiO2, CeO2, and Co3O4, often exhibit exceptionally high activity for CO oxidation, when they are used to support gold nanoparticles.8–14 This is primarily because these oxides could create oxygen vacancies on their surfaces close to the Au particles, forming strong metal-support interactions (SMSI). In this context, Au/Co3O4 catalyst has attracted particular attention because of their exceptionally high activities for low-temperature CO oxidation.15 The key function of cobalt oxide is to disperse and stabilize gold NPs through its surface oxygen vacancies that strongly depend on the size and shape of cobalt oxide crystallites. The dispersion of gold NPs on high surface area support is expected to promote catalytic activity and stability, as it not only increases the gold loading but also improves its dispersion, which would be beneficial to avoid the sintering of Au NPs.16,17
Herein, we prepared a nanoplate-shaped Co3O4 oxide with the preferential exposure of Co3O4{111} as the support to disperse gold nanoclusters (NCs) with a uniform size of ∼2 nm. The atomic arrangements of Au{111} were obtained by aberration-corrected STEM technology, which formed a strong metal-support interaction. The resulting Au/Co3O4 catalysts, exhibiting surface oxygen vacancies and intrinsic defects of Co3O4{111} and the Au0, Auδ+, and Au+ species on the surface of gold NCs, showed significant activities in the CO oxidation at low reaction temperatures. The catalyzed CO oxidation over Au/Co3O4 obeyed the Langmuir–Hinshelwood mechanism at low temperatures.
Experimental
Synthesis of Co3O4 oxides
Co3O4 was synthesized following typical procedures reported in the literature.18,19 CoCl2·6H2O was dissolved in deionized water, and triethylamine was added. After 10 min stirring, the solution was transferred and reacted at 180 °C for 20 h in an autoclave. The precipitates were filtered and washed with deionized water, followed by calcination at 350 °C for 3 h, which was then used to support the gold particles.Preparation of Au/Co3O4 catalysts
Au/Co3O4 catalysts were prepared by an immobilization method of Co3O4 oxides with Au:PVP (polyvinyl alcohol) colloids of ∼2 nm, which were prepared by a typical procedure.7 Typically, Co3O4 oxides were added into the aqueous solution containing the fresh Au:PVP colloids, which were stirred at room temperature for 3 h. The solid was collected by filtration and washed with distilled water. The solid sample was dried at 80 °C and calcined at 300 °C for 2 h in air to remove all the PVP ligands. Furthermore, the analysis of inductively coupled plasma mass spectroscopy (ICP-MS) showed that the actual loading of Au was 0.65 wt% in the prepared Au/Co3O4.Characterization
X-ray powder diffraction patterns were recorded on a Rigaku D/MAX-2500PC with Cu Kα radiation (λ 1.5418 Å) at 40 kV and 200 mA. Transmission electron microscopy (TEM) images were recorded on a Hitachi 7700 microscope operated at 120 kV. Aberration-corrected scanning transmission electron microscopy (AC-STEM) images were obtained on a JEM-ARM200F at 200 kV. The specimen was prepared by ultrasonically dispersing the sample powder into ethanol, and drops of the suspension were deposited on a carbon-coated copper grid and dried at room temperature. Raman spectra were collected using a Renishaw inVia Raman microscope with a laser wavelength of 785 nm and a laser power of 3 mW and were taken under the same conditions after 60 seconds of exposure. TPO analysis was performed with an AutoChem II 2920 instrument (Micromeritics) and analyzed with a thermal conductor detector (TCD). Before analysis, 50 mg of the samples were pre-treated at 200 °C for 1 h in He gas to clean the surface of the samples. After cooling to room temperature, 5% of O2/He (30 mL min−1) was used as a feed gas to perform TPO from RT to 600 °C. The infrared Fourier transform spectra were recorded using a Vertex 70v spectrometer (Bruker). Spectra were recorded at 4 cm−1 spectral resolution and 60 kHz scanning velocity. Au/Co3O4 samples were pretreated at 300 °C in an Ar atmosphere for one hour. After the system was cooled to room temperature, the flow was switched to a feed gas of 1% CO/Ar, and the infrared spectra were in situ recorded. Visible Raman spectra were collected on a Jobin-Yvon U1000 scanning double monochromator with a 532 nm single-frequency laser.Catalytic test
CO oxidation was performed in a flow reactor. Preliminary reactions were carried out with ∼0.200 g Au/Co3O4 or pristine Co3O4 catalysts were taken in the form of 40–60 mesh particles. Before the reactions, the samples were activated at 300 °C for 1 h in the presence of 20% O2/N2 gas. When the reactor was cooled to room temperature, a feed gas (1% CO/20% O2/N2) was passed over the catalysts at a flow rate of 30 mL min−1. The amounts of CO, CO2, and O2 in the inlet and outlet streams were analyzed using an online gas chromatograph. The CO conversion (XCO, %) was calculated on the basis of CO consumption and CO2 formation by eqn (1):| XCO = (C0 − C1)/C0 × 100% | (1) |
Results and discussion
Characterization of Au/Co3O4
The XRD pattern of the as-prepared Au/Co3O4 catalyst is shown in Fig. 1. The diffraction peaks of Co3O4 were observed at 19.0°, 31.3°, 36.8°, 44.8°, 55.7°, 59.4°, and 65.2°, which can be assigned to the (111), (220), (311), (222), (400), (422), (511), and (440) planes of the Co3O4 phase (JPDS No. 00-042-1467), respectively, with a space group of Fd3m.20 The high peak intensities confirmed the high crystallization of the Co3O4 support. Another XRD peak at ∼38.5° corresponds to the Au(111) phase, clearly suggesting the existence of gold NCs in the Au/Co3O4 sample.21 | ||
| Fig. 1 Powder XRD of the Au/Co3O4 sample. | ||
Furthermore, the morphology of gold NCs and Co3O4 oxides and the size of Au particles were characterized by TEM methods. TEM images showed that the Co3O4 oxide had a hexagonal shape, with an average edge length of ∼160 nm and thickness of ∼25 nm. As shown in the AC-STEM image, Au NCs of the Au/Co3O4 showed a particle size of 2.2 ± 0.6 nm (Fig. 2a). Au particles were uniformly loaded onto the surface of the Co3O4{111} plane, based on the lattice fringes with an inter-planar spacing of 0.46 nm, which can be attributed to the Co3O4{111} of Co3O4 (Fig. 2b).18,22
 | ||
| Fig. 2 (a) AC-STEM images of Au/Co3O4 sample. (b) The Au particle on Co3O4{111}. Statistics in (a) were performed by counting 202 particles. | ||
To study the chemical state of the surface gold atoms on the gold nanoparticles, Fourier-transform infrared (FT-IR) spectroscopy for the CO adsorption on the Au/Co3O4 catalyst was employed at room temperature. As shown in Fig. 3a, the CO adsorption on Au/Co3O4 results in the appearance of three bands in the carbonyl stretching region, with their maxima located at 2106 cm−1, 2120 cm−1, and 2142 cm−1. The adsorption band at 2106 cm−1 is assigned to CO absorbed linearly on metallic gold particles (Au0–CO).23,24 It can be predicted that this band characterizes weakly adsorbed species because it easily disappeared even after a short time of 2 min. The band observed at 2120 cm−1 can be attributed to Auδ+–CO (0 < δ < 1). The 2142 cm−1 band frequency region can be assigned to CO adsorbed on gold exposed at the surface of the particles where gold sites are to some extent, made electropositive by the adsorbed oxygen (Au+–CO).23 Thus, Auδ+ and Au+ species coexist on the surface of the gold particles, which facilitates the nanogold-catalyzed CO oxidation for the adsorption and activation of CO molecules.
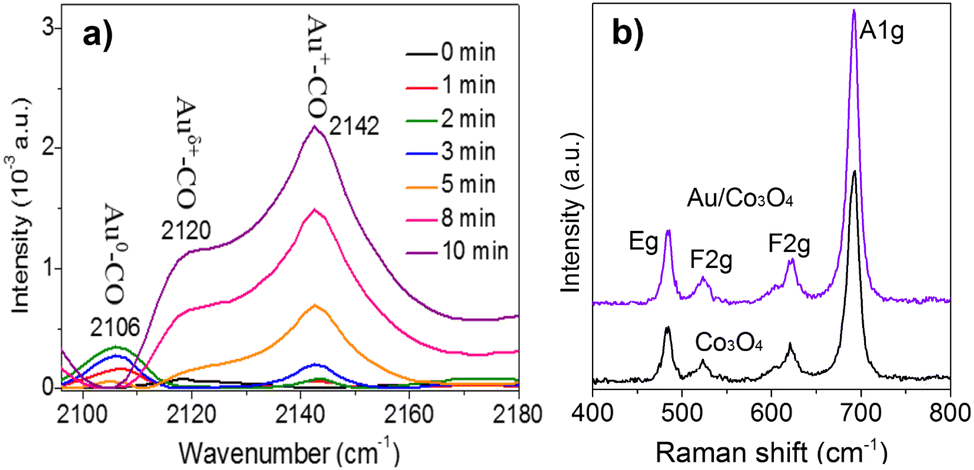 | ||
| Fig. 3 (a) CO-FTIR of Au/Co3O4 catalysts. The adsorption bands at 2105 cm−1, 2120 cm−1, and 2142 cm−1 are assigned to the Au0–CO, Auδ+–CO (0 < δ < 1), and Au+-CO species, respectively. (b) Raman spectra of the bare Co3O4 oxides and the as-prepared Au/Co3O4 catalysts. | ||
Furthermore, Raman analysis was carried out to clearly identify the surface oxygen vacancies of the bare Co3O4 oxides and Au/Co3O4 catalyst. As presented in Fig. 3b, the bare Co3O4 and Au/Co3O4 displayed similar Raman features: four vibration peaks (i.e., A1g + Eg + 2 F2g) in the range of 400–800 cm−1. The Raman peaks at 484 cm−1, 523 cm−1, and 622 cm−1 are assigned to the Eg, F2g(2), and F(1)2g symmetry, respectively. Another peak at 691 cm−1 with A1g symmetry is attributed to the characteristics of the octahedral CoO6 sites, corresponding to the unique characteristics of the spinel-type cubic Co3O4 phase, in good agreement with the reported literature25,26 Furthermore, the ratio of the surface oxygen vacancies to surface lattice oxygen was calculated to be 0.46 on the Co3O4 support and 0.52 ion Au/Co3O4, based on the full width at half-maximum of A1g peaks.25 Thus, Au/Co3O4 exhibits obviously high defective structures, induced by the anchored Au nanoclusters, which can promote the oxidation, as the defective structures can largely promote the adsorption and activation of oxygen species during the catalytic CO oxidation.27
Catalytic performance
Furthermore, Au/Co3O4 catalysts were evaluated in the CO oxidation reactions. As shown in Fig. 4a, the bare Co3O4 oxide was inactive at low reaction temperatures of 20–60 °C and showed a 31% CO conversion at 80 °C and a full conversion at 120 °C. While, the Au/Co3O4 catalyst gave a promising CO conversion (35%) at a low temperature of 20 °C, and the full CO conversion was achieved at 80 °C, Fig. 4a, green line. Therefore, the introduction of Au NCs largely promoted the catalytic activity in CO oxidation. The activation energy for CO oxidation was calculated to be 15.49 kJ mol−1 for Au/Co3O4, in good agreement with that reported in the early literature.28 Therefore, we deduce that the Au/Co3O4-catalyzed CO oxidation should obey the Langmuir–Hinshelwood mechanism at low temperatures (20–60 °C), as the oxygen molecules and CO should be adsorbed and activated over the surface oxygen vacancies and gold particles, respectively, rather than on the surface lattice oxygen sites.29,30 | ||
| Fig. 4 (a) Catalytic activity of Co3O4 oxides and Au/Co3O4 for CO oxidation as the function of temperature. Reaction conditions: the gas flow was composed of 1% CO/20% O2/N2 (vol.) with a flow rate of 30 mL min−1, and 50 mg catalysts were used. (b) Arrhenius plots of the rate vs.(1/T) for CO oxidation reaction over Au/Co3O4 catalysts. (c) Durability tests of Au/Co3O4 catalysts in the CO oxidation at 25 °C (using 50 mg of catalyst) and 60 °C (40 mg of catalyst). Tests conditions: 1% CO/20% O2/N2 with a flow rate of 30 mL min−1. The CO conversion were measured in the range of 25% to 45%. | ||
Furthermore, the durability of the Au/Co3O4 catalysts was investigated at 25 and 60 °C. As depicted in Fig. 4c, the activity of the Au/Co3O4 catalysts was dropped from 2.92 to 1.87 molreacted CO gAu−1 s−1 during the 12 h reaction in the CO oxidation (red line). While, Au/Co3O4 gave a stable activity performance of 5.26–5.39 molreacted CO gAu−1 s−1 during the test (9 h, black line), which is higher than the reported value over the oxide-supported gold NCs catalysts, as shown in Table S1.†8–12 Overall, the results indicated that Au/Co3O4 catalysts exhibited excellent durability for CO oxidation. No apparent sintering of Au NCs was observed in TEM analysis of the spent Au/Co3O4 catalysts (Fig. 5a), demonstrating that the slight activity loss was not caused by the aggregation of gold particles. The CO2 species were clearly observed in the TPO tests of the spent Au/Co3O4 catalysts, compared with the pristine one, as shown in Fig. 5b. Hence, the slight deactivation at low reaction temperature (e.g., 25 °C) should be associated with the formation and accumulation of carbonate-like and H2O/OH− species at the support interface, resulting in the blockage of active sites during the CO oxidation reaction.31–34
 | ||
| Fig. 5 (a) TEM image and (b) TPO analysis of the spent Au/Co3O4 after CO oxidation. The statistical analysis was performed by counting over 100 gold particles. | ||
Conclusions
In summary, gold nanoclusters of ca. 2.2 nm immobilized on the nanoplate-shaped Co3O4 oxides were synthesized by a simple impregnation of gold colloids and oxides. STEM analysis identified that the Au particles of Au{111} and Au{100} exposures interacted with Co3O4{111}. Au/Co3O4{111} exhibited good catalytic activity and durability in the CO oxidation, which may be due to the strong metal-support interaction. FT-IR and Raman spectroscopy analyses confirmed that the surface oxygen vacancies and intrinsic defects on Co3O4{111} and Auδ+ and Au+ species on the surface of gold NCs are responsible for the high catalytic behavior in the CO oxidation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support from the fund of National Natural Science Foundation of China (22065029 and 22172167), the Natural Science Foundation of Inner Mongolia Autonomous Region (2022MS02002), and central guidance for Local Scientific and Technological Development funds (2022ZY0081).Notes and references
- G. C Bond and D. T. Thompson, Catal. Rev.: Sci. Eng., 1999, 41, 319–388 CrossRef.
- Z. Li, X. Zhang, Q. Shi, X. Gong, H. Xu and G. Li, Nanoscale Adv., 2021, 3, 7002–7006 RSC.
- Z. Jin, Y. Y. Song, X. P. Fu, Q. S. Song and Ch.-J. Jia, Chin. J. Chem., 2018, 36, 639–643 CrossRef CAS.
- X. Wei, B. Shao, Y. Zhou, Y. Li, Ch. Jin, J. Liu and W. Shen, Angew. Chem., 2018, 57, 11289–11293 CrossRef CAS PubMed.
- X. Du, Y. Huang, X. Pan, B. Han, Y. Su, Q. Jiang, M. Li, H. Tang, G. Li and B. Qiao, Nat. Commun., 2020, 11, 5811 CrossRef CAS PubMed.
- Y. Wang, D. Widmann, F. Lehnert, D. Gu, F. Schuth and R. Jurgen Behm, Angew. Chem., 2017, 56, 1–7 CrossRef CAS.
- B. Shao, J. Zhang, J. Huang, B. Qiao, Y. Su, Sh. Miao, Y. Zhou, D. Li, W. Huang and W. Shen, Small Methods, 2018, 2, 1800273 CrossRef.
- Y. Zhang, Z. Li, J. Zhang, L. Xu, Z. Han, A. Baiker and G. Li, Nano Res., 2023, 16, 8919–8928 CrossRef CAS.
- M. Chen and D. W. Goodman, Acc. Chem. Res., 2006, 39, 739–746 CrossRef CAS PubMed.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331–1335 CrossRef CAS PubMed.
- W. Li, C. Liu, H. Abroshan, Q. Ge, X. Yang, H. Xu and G. Li, J. Phys. Chem. C, 2016, 120, 10261–10267 CrossRef CAS.
- Y. Denkwitz, Z. Zhao, U. Hormann, U. Kaiser, V. Plzak and R. J. Behm, J. Catal., 2007, 251, 437–442 CrossRef.
- Z. Li, W. Li, H. Abroshan, Q. Ge, Y. Zhou, C. Zhang, G. Li and R. Jin, Nanoscale, 2018, 10, 6558–6565 RSC.
- H. Atout, A. Bouguettouch, D. Chebli, J. Crespo, J. Dupin, J. M. López-de-Luzuriaga, H. Martínez, M. Monge, M. Elena Olmos and M. Rodríguez-Castillo, New J. Chem., 2021, 45, 11727–11736 RSC.
- Z. Zhou, S. Kooi, M. Flytzani-Stephanopoulos and H. Saltsburg, Adv. Funct. Mater., 2008, 18, 2801–2807 CrossRef CAS.
- Y. Liu, H. Dai, J. Deng, S. Xie, H. Yang, W. Tan, Y. Jiang and G. Guo, J. Catal., 2014, 309, 408–418 CrossRef CAS.
- Q. Shi, X. Zhang, Z. Li, A. Raza and G. Li, ACS Appl. Mater. Interfaces, 2023, 15, 30161–30169 CrossRef CAS PubMed.
- T. Barakat, J. C. Rooke, E. Genty, R. Cousin, S. Siffert and B. I. Su, Energy Environ. Sci., 2013, 6, 371–391 RSC.
- X. Wei, S. Barkaoui, J. Chen, Q. Fang, G. Cao, Z. Wu, F. Wang and G. Li, Nanoscale Adv., 2021, 3, 1741–1746 RSC.
- U. Sengupta, A. Mukherjee, M. P. S. Mukhopadhyay, K. J. Jenkinson, A. E. H. Wheatley and A. Kar, New J. Chem., 2023, 47, 11425–11443 RSC.
- Y. Guo, Q. Zhou, X. Chen, Y. Fu, S. Lan, M. Zhu and Y. Du, J. Mater. Sci. Technol., 2022, 119, 53–60 CrossRef CAS.
- H. Chen, M. Yang, S. Tao and G. Chen, Appl. Catal., B, 2017, 209, 648–656 CrossRef CAS.
- F. Boccuzzi, A. Chiorino and M. Manzoli, Surf. Sci., 2000, 454–456, 942–946 CrossRef CAS.
- X. Zhang, Z. Li, W. Pei, G. Li, W. Liu, P. Du, Z. Wang, Z. Qin, H. Qi, X. Liu, S. Zhou, J. Zhao, B. Yang and W. Shen, ACS Catal., 2022, 12, 3634–3643 CrossRef CAS.
- G. Zhai, J. Wang, Z. Chen, W. An and Y. Men, Chem. Eng. J., 2018, 337, 488–498 CrossRef CAS.
- S. Barkaoui, M. Haddaoui, H. Dhaouadi, N. Raouafi and F. Touati, J. Solid State Chem., 2015, 228, 226–231 CrossRef CAS.
- Y. Cao, L. Zhao, T. Gutmann, Y. Xu, L. Dong, G. Buntkowsky and F. Gao, J. Phys. Chem. C, 2018, 122, 20402–20409 CrossRef CAS.
- M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M. J. Genet and B. Delmon, J. Catal., 1993, 144, 175–192 CrossRef CAS.
- Y. Chen, Y. Li, W. Chen, W. Xu, Z. Han, A. Waheed, Z. Ye, G. Li and A. Baiker, Nano Res., 2022, 15, 1366–1374 CrossRef CAS.
- Y. Wang, Q. Jiang, L. Xu, Z. Han, S. Guo, G. Li and A. Baiker, ACS Appl. Mater. Interfaces, 2021, 13, 61078–61087 CrossRef CAS PubMed.
- H. Wang, H. Zhu, Z. Qin, F. Liang, G. Wang and J. Wang, J. Catal., 2009, 26, 154–162 CrossRef.
- C. H. Kim and L. T. Thompson, J. Catal., 2005, 230, 66–74 CrossRef CAS.
- Q. Guo, S. Chen, Y. Liu and Y. Wang, Chem. Eng. J., 2010, 165, 846–850 CrossRef CAS.
- Q. Shi, Y. Zhang, Z. Li, Z. Han, L. Xu, A. Baiker and G. Li, Nano Res., 2023, 16, 6951–6959 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00561e |
| This journal is © The Royal Society of Chemistry 2023 |