
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanocomposites of Cu2O with plasmonic metals (Au, Ag): design, synthesis, and photocatalytic applications
Enrico Daniel R.
Legaspi
ab and
Michelle D.
Regulacio
 *a
*a
aInstitute of Chemistry, University of the Philippines Diliman, Quezon City, 1101, Philippines. E-mail: mdregulacio@up.edu.ph
bMaterials Science and Engineering Program, University of the Philippines Diliman, Quezon City, 1101, Philippines
First published on 25th September 2023
Abstract
Metal–semiconductor nanocomposites have been utilized in a multitude of applications in a wide array of fields, prompting substantial interest from different scientific sectors. Of particular interest are semiconductors paired with plasmonic metals due to the unique optical properties that arise from the individual interactions of these materials with light and the intercomponent movement of charge carriers in their heterostructure. This review focuses on the pairing of Cu2O semiconductor with strongly plasmonic metals, particularly Au and Ag. The design and synthesis of Au–Cu2O and Ag–Cu2O nanostructures, along with ternary nanostructures composed of the three components, are described, with in-depth discussion on the synthesis techniques and tunable parameters. The effects of compositing on the optical and electronic properties of the nanocomposites in the context of photocatalysis are discussed as well. Concluding remarks and potential areas for exploration are presented in the last section.
 Enrico Daniel R. Legaspi | Enrico Daniel R. Legaspi received his MS in Materials Science and Engineering from the University of the Philippines Diliman, where he is presently pursuing his PhD under the mentorship of Dr Michelle D. Regulacio. His research revolves around the design and synthesis of metal–semiconductor nanocomposites with properties that are useful in catalytic and sensing applications. |
 Michelle D. Regulacio | Michelle D. Regulacio earned her PhD in Chemistry from Georgetown University and trained as a postdoctoral researcher at Princeton University. She was a research scientist at A*STAR Singapore before joining the University of the Philippines Diliman as an associate professor. Her research focuses on the design and synthesis of inorganic nanomaterials (metals, semiconductors, and their hybrids), with applications in the fields of catalysis, sensing, biomedicine, textile functionalization, and energy conversion and storage. |
1. Introduction
Nanomaterials have been the subject of intensive study in recent decades due to the structural and size effects that bestow them with unique properties that are not seen in bulk materials. Of notable interest are their optical properties that arise from their interactions with electromagnetic radiation or light. Given that nanomaterials possess particle dimensions in the nanometer range, they are smaller than the wavelengths of light they interact with, resulting in optical properties that are different from those of bulk materials. Two prime classes of materials that display interesting optical behavior at the nanoscale are metal oxide semiconductors and plasmonic metals. Nanomaterials of metal oxide semiconductors (e.g. TiO2, ZnO, and Cu2O) possess tunable band structures that generate valuable electron–hole pairs when illuminated with light.1,2 Meanwhile, the interaction of plasmonic metal (e.g. Au, Ag, and Cu) nanomaterials with light gives rise to localized surface plasmon resonance (LSPR), which dictates the optical activity of these materials.3,4 With their remarkable optical properties, both classes of nanomaterials can be used in a plethora of applications that cut across a wide range of scientific disciplines. However, maximizing the use of metal oxide semiconductors and plasmonic metals is challenging due to the drawbacks associated with each material. To attenuate these drawbacks, these materials can be composited with each other to create metal–semiconductor nanocomposites.5–11 The coupling takes advantage of the individual properties of each component and even possibly elicits synergistic effects while mitigating the negative aspects.Numerous review articles have been published on metal–semiconductor nanocomposites, but many of these existing reviews are more general in terms of the materials that are coupled together to form the composite.5–8,12–14 This review gives particular attention to the pairing of semiconducting Cu2O with the plasmonic metals (PM) Au and Ag. The growing body of literature on this specific pairing over the last decade is a clear indication that research on this topic is rapidly progressing. A review article dedicated to this hybrid combination is therefore timely and relevant. In this review, the design and synthesis of PM–Cu2O nanocomposites are thoroughly described to provide a detailed overview of how these hybrids are formed. Their use in photocatalytic applications are also examined, especially their effectiveness in the photocatalytic degradation of organic dyes.
1.1 Copper(I) oxide (Cu2O) nanomaterials
Cuprous oxide (Cu2O) is a semiconductor that exhibits tunable optical properties in its nanostructured form. It is highly attractive for practical applications because it is made up of elements that are inexpensive, earth-abundant, and nontoxic. Synthesis of their nanostructures is relatively simple. They are usually prepared through facile chemical reduction procedures at mild temperatures and basic pH conditions. Numerous Cu2O nanostructures, such as nanocubes, nanooctahedra, nanospheres, nanowires, nanotubes, and nanoplates, have been synthesized, with nanocubes being the most common morphology due to the cubic crystal structure of Cu2O.15–19 The type of exposed crystal facets in Cu2O nanostructures has been shown to affect their photocatalytic behavior.20,21 Thus, modification of their photocatalytic properties can be readily achieved through careful morphological control.Cu2O is a p-type semiconductor with a theoretical band gap of 2.2 eV. Similar to other leading metal oxide semiconductors like TiO2 and ZnO, Cu2O generates electron–hole pairs when irradiated with photons. Electrons are excited from the valence band to the conduction band when the energy of incident photons exceeds the band gap energy. The wavelengths of light that correspond to this energy for TiO2 and ZnO are in the ultraviolet (UV) spectral range. For Cu2O, its band gap energy lies in the more desirable visible region. This enables Cu2O to take advantage of solar radiation, which is rich in visible photons. The excitation of electrons by light leaves behind positively charged holes in the valence band. These photogenerated electron–hole pairs and the subsequent associated decay processes grant Cu2O its photocatalytic properties. Unfortunately, the electron–hole pairs generated have a high recombination rate in pure Cu2O particles, which limits its potential applications.19
1.2 Au and Ag nanomaterials
Au and Ag are regarded as plasmonic metals due to their unique interactions with electromagnetic radiation, specifically radiation with wavelengths that correspond to their surface plasmon resonance.3,4 When incident light reaches the surface of the plasmonic metal nanoparticle, coupling occurs between the electrons in the metal and the oscillating light wave, resulting in the coherent oscillation of electrons. The incoming electric field causes excitation in the nanoparticle, while the out-of-equilibrium surface charges produce a restoring electric field. When the external field oscillates at the frequency of the particles' resonant mode, the incoming and restoring electric fields sync and cause an increase in the amplitude of the electric field surrounding the plasmonic metal particle. This phenomenon is termed localized surface plasmon resonance and is known to induce near-field enhancements in highly localized regions just a few nanometers around the particle.The LSPR frequency is highly dependent upon several factors, such as the size and morphology of the nanomaterial, the identity or composition of the metal, and the index of refraction of the surrounding medium.22–25 Changing any of these variables through various synthesis procedures allows for the tuning of the position of the LSPR peak, which consequently dictates the material's potential applications. The extinction spectra of spherical Au nanoparticles typically show an LSPR band peaking at around 520 nm.24 This peak was found to redshift with increasing particle size and with aggregation, as well as in high-refractive index environments. For anisotropic Au nanostructures (e.g. nanorods and nanostars), multiple LSPR bands that extend to longer wavelengths of light are observed as a consequence of multiple resonance modes.26,27 In the case of Ag, the LSPR peak for simple spherical nanoparticles is typically found at shorter wavelengths of approximately 400 nm.28,29 Similar to Au, this LSPR peak shifts to longer wavelengths when the particle size is increased, when aggregated, or when surrounded with a high-refractive index material. Additionally, shape anisotropy gives rise to several Ag LSPR bands that cover a wider spectral range.
Although Au and Ag exhibit similar optical effects, there are a number of considerations to be made when deciding whether Au or Ag will be used for particular applications. Au nanoparticles are stable and relatively easy to synthesize in diverse shapes and sizes.24,26 Au nanoparticles are also biocompatible and can directly conjugate and interact with various biomolecules (e.g. proteins, antibodies, and nucleic acids), which make them ideal for use in biomedical applications.30,31 However, a significant drawback that limits the widespread application of Au nanoparticles is the high cost of Au. Ag nanoparticles have diverse uses for a number of reasons. Ag has the highest electrical and thermal conductivity among all metals and exhibits the strongest plasmon response, even greater than Au.25,32 In terms of cost, Ag is substantially cheaper than Au. However, unlike Au, Ag cannot be haphazardly used in biomedical applications due to its cytotoxicity.33 Additionally, Ag reacts with sulfur in the air and forms a silver sulfide film on its surface.34
1.3 PM–Cu2O nanocomposites
Individually, Cu2O, Au, and Ag exhibit exceptional properties, but they are not without issues, as noted in the preceding subsections. Constructing nanocomposites of Cu2O with Au or Ag or both plasmonic metals can help address the downsides of the individual components. For instance, the high recombination rate of photogenerated electron–hole pairs in Cu2O can be mitigated by compositing it with a metal, which creates a Schottky junction at the metal–semiconductor interface.35 This heterojunction is a potential energy barrier that hinders the recombination of charge carriers, effectively promoting electron–hole separation that can then lead to an enhancement in photocatalytic activity. Aside from the Schottky junction effect, there are other photocatalytic enhancement mechanisms that come into play when the metal used is strongly plasmonic. These plasmon-mediated mechanisms are described in detail in Section 3.In terms of cost, pairing precious Au with inexpensive Cu2O produces a more economical material. In the case of Ag, compositing with Cu2O can result in a more durable material. Moreover, coupling these plasmonic metals with Cu2O, which has a higher refractive index than water, presents a critical variable that can be controlled to tune their LSPR frequency. For example, when Au nanoparticles are encased in a Cu2O shell, the Au LSPR band redshifts and broadens with increasing thickness of the Cu2O shell, as shown in Fig. 1.36 Because the LSPR frequency can also be modulated by controlling the size and morphology of the plasmonic metal component, the creation of rationally designed PM–Cu2O nanocomposites can allow for absorption of light over a wider range of wavelengths. This is especially beneficial for solar-driven applications, as being restricted to a narrow spectral range is detrimental to the practicality of using these materials.
 | ||
| Fig. 1 (a) TEM image of Au–Cu2O nanocomposites with a core–shell structure. (b) UV-vis spectra and photograph (inset) of colloidal dispersions of Au–Cu2O core–shell nanocomposites with increasing Cu2O shell thickness (from left to right), clearly showing the redshifting of the LSPR band. Reproduced with permission from ref. 36. Copyright 2012 American Chemical Society. | ||
2. Design and synthesis of PM–Cu2O nanocomposites
Nanocomposites of Cu2O with PMs come in a variety of hybrid configurations, which depend on the synthesis technique, the relative amount of each component, and the reaction conditions. The structures of these materials have a significant effect on their photocatalytic properties, which emphasizes the necessity of understanding how these structures are formed. In line with this, this section details the design and synthesis of various Au–Cu2O and Ag–Cu2O nanocomposites, including ternary structures that contain all three materials. A summary of the various PM–Cu2O nanocomposites that have been reported in the literature is presented in Table 1.| Hybrid configuration | Morphology, size | Modified feature/s | Synthesis method | Ref |
|---|---|---|---|---|
| (A) Core–satellite | ||||
| Au-decorated Cu2O | Cu2O: porous sphere, 150–200 nm | Au size | Two-pot | 37 |
| Au satellite size: tuned, 5–25 nm | ||||
| Au-decorated Cu2O | Cu2O: cube, 200 nm; octahedron, 500 nm; rhombic dodecahedron, 370 nm | Cu2O morphology | Two-pot | 40 |
| Au satellite size: ≤10 nm | ||||
| Au-decorated Cu2O | Cu2O: octahedron, 136 nm (edge length) | Au position | Two-pot | 44 |
| Au satellite size: 6.8 nm | Hybrid configuration | |||
| Au-decorated Cu2O | Cu2O: sphere, 800 nm; cube, 700 nm (edge length); 3D flower with elongated petals | Cu2O morphology | Two-pot | 45 |
| Au satellite size: ∼10 nm | ||||
| Au-decorated Cu2O | Cu2O: different polyhedral shapes, 100–150 nm | Cu2O morphology | Two-pot | 42 |
| Au satellite size: 20 nm | Hybrid configuration | |||
| Metal component (Au, Pd) | ||||
| Au-decorated Cu2O | Cu2O: octahedron, 300–500 nm (edge length) | Metal component (Au, Ag) | Two-pot, with light irradiation | 39 |
| Au satellite size: 20–50 nm | ||||
| Ag-decorated Cu2O | Cu2O: same as above | |||
| Ag satellite size: 10–30 nm | ||||
| Ag-decorated Cu2O | Cu2O: truncated cube, 700 nm (edge length) | Ag amount/density | One-pot | 38 |
| Ag satellite size: 25–30 nm | ||||
| Ag-decorated Cu2O | Cu2O: cube, 1.2 μm (edge length) | Ag amount/density | One-pot | 43 |
| Ag satellite size: 10 nm | ||||
| Ag-decorated Cu2O | Cu2O: different polyhedral shapes, 1.6–2.5 μm (diagonal size) | Cu2O morphology | One-pot | 41 |
| Ag satellite size: tuned, 10–20 nm | Ag size, amount/density | |||
| Cu2O-decorated Au | Au: hexoctahedron, 130 nm | Hybrid configuration | Two-pot | 46 |
| Cu2O satellite size: 90 nm | ||||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| (B) Core–shell | ||||
| Au@Cu2O | Au and Cu2O: different anisotropic and faceted shapes (e.g. cuboctahedron, truncated icosahedron, rod, plate) | Au morphology | Two-pot | 50 |
| Cu2O morphology | ||||
| Au@Cu2O | Au and Cu2O: different shapes (e.g. octahedron, cube, rod), tunable size | Au size, morphology | Two-pot | 57 |
| Cu2O size, morphology | ||||
| Au@Cu2O | Au: rhombic dodecahedron, truncated octahedron, icosahedron, trisoctahedron | Au morphology | Two-pot | 60 |
| Cu2O: face-raised cube, cuboctahedron, octahedron, truncated icosahedron | Cu2O morphology | |||
| Au@Cu2O | Au: octahedron, tunable size (50–70 nm) | Au size | Two-pot | 56 |
| Cu2O: cube, octahedron, cuboctahedron, tunable size (90–220 nm) | Cu2O size, thickness, morphology | |||
| Au@Cu2O | Au: octahedron, 90 nm | Cu2O morphology | Two-pot | 54 |
| Cu2O: cube and octahedron, 350–420 nm | ||||
| Au@Cu2O | Au: sphere, 60 nm | Cu2O shell thickness | Two-pot | 36 |
| Cu2O: sphere to ellipsoid, tunable thickness (2–21 nm) | ||||
| Au@Cu2O | Au: rod, 78 × 26 nm | Au position | Two-pot | 44 |
| Cu2O: octahedron, 136 nm (edge length) | Hybrid configuration | |||
| Au@Cu2O | Au: rod | Cu2O shell thickness | Two-pot | 53 |
| Cu2O: octahedron, tunable edge length (96–250 nm) | ||||
| Au@Cu2O | Au: rod, 40–60 nm (length), 10–15 nm (width) | Au morphology | Two-pot | 52 |
| Cu2O: brick, 40 nm (thickness) | ||||
| Au@Cu2O | Au: rod, 112 × 37 nm (aspect ratio = 3) | Cu2O shell thickness | Two-pot | 61 |
| Cu2O: tunable thickness (15–30 nm) | ||||
| Au@Cu2O | Overall morphology: flower with Au core and Cu2O petals | Cu2O shell thickness | Two-pot | 58 |
| Au: sphere, 16.3 nm | ||||
| Cu2O: tunable thickness (2–40 nm) | ||||
| Au@Cu2O | Overall morphology: flower with Au cluster core and cube-like Cu2O petals | Cu2O shell coverage | One-pot | 59 |
| Overall size: 54 nm | ||||
| Au@Cu2O | Au: sphere, 9 nm | Metal component (Au, Ag, Pd) | Two-pot, microwave-assisted | 51 |
| Cu2O: cube, 30–50 nm (edge length) | ||||
| Ag@Cu2O | Ag: sphere, 13 nm | |||
| Cu2O: cube, 60–80 nm (edge length) | ||||
| Ag@Cu2O | Ag: sphere, tunable (20–100 nm) cube, tunable edge length (40–150 nm) | Ag size, morphology | Two-pot | 49 |
| Cu2O: tunable thickness | Cu2O shell thickness | |||
| Ag@Cu2O | Overall morphology: flower with Au spherical core and Cu2O petals | Cu2O shell uniformity | Two-pot | 64 |
| Overall size: 120 nm | ||||
| Ag@Cu2O | Ag: wire | Cu2O shell thickness | Two-pot, with pulsed laser irradiation | 65 |
| Cu2O: aggregated particles, tunable thickness (20–140 nm) | ||||
| Ag@Cu2O | Ag: wire, 10 μm × 100 nm sphere, 100–200 nm | Ag morphology | Two-pot | 66 |
| Cu2O: aggregated particles, 30 nm thick | ||||
| Ag@Cu2O | Ag: sphere, 30 nm | Cu2O shell thickness | Two-pot | 67 |
| Cu2O: tunable thickness (11.4–40 nm) | ||||
| Ag@Cu2O | Ag: sphere, 11 nm | Cu2O shell thickness | One-pot | 63 |
| Cu2O: tunable thickness (5.8–11 nm) | ||||
| Cu2O@Au | Cu2O: octahedron, 1.4 μm | Hybrid configuration | Two-pot, GRR | 62 |
| Au: aggregated particles, 10–50 nm | ||||
| Cu2O@Ag | Cu2O: octahedron, 1 μm (edge length) | Hybrid configuration | Two-pot, GRR | 69 |
| Ag: aggregated particles, 80–100 nm | ||||
| Cu2O@Ag | Cu2O: sphere, 400–500 nm | — | Two-pot, thermal decomposition | 71 |
| Ag: aggregated particles | ||||
| Cu2O@Ag | Cu2O: hollow sphere, 300 nm | Ag size | One-pot | 70 |
| Ag: aggregated particles, 20–50 nm | ||||
| Cu2O@Ag | Overall morphology: cube, submicron | — | Hydrothermal | 72 |
| Cu2O@Ag | Cu2O: aggregated particles, 150–170 nm | Ag size | Thermal oxidation, magnetron sputtering | 68 |
| Ag: flowers, tunable size | ||||
|
||||
| (C) Yolk–shell | ||||
| Au@Cu2O | Au: sphere, 22 nm | Void size | Two-pot | 78 |
| Cu2O: porous hollow sphere, 140 nm | ||||
| Au@Cu2O | Au: sphere, 15 and 63 nm | Hybrid configuration | Two-pot; void formed via Ostwald ripening | 80 |
| Cu2O: porous hollow sphere | Au size | |||
| Overall size: tuned, 98–183 nm | Overall size | |||
| Au@Cu2O | Au: bipyramid, rod (70 × 18 nm) | Au morphology | Two-pot; void formed via Ostwald ripening | 81 |
| Cu2O: porous hollow ellipsoid, 30 nm (shell thickness) | ||||
| Overall size: 260 nm | ||||
| Ag@Cu2O | Ag: cube, 70 nm | Void size | Two-pot; void formed via Ostwald ripening | 82 |
| Cu2O: porous hollow cube | ||||
| Overall size: tuned, 146–313 nm | Overall size | |||
| Ag@Cu2O | Overall size: ∼100 nm | — | Thermal treatment of Cu@Ag; void formed via Kirkendall effect | 83 |
| Cu2O@Au | Cu2O: etched sphere, submicron | Hybrid configuration | Two-pot; void formed via etching | 76 |
| Au: aggregated particles, 5 nm | ||||
| Cu2O@Au | Cu2O: etched octahedron | Hybrid configuration | Two-pot; void formed via etching | 84 |
| Au: aggregated particles, 5–10 nm | ||||
|
||||
| (D) Janus | ||||
| Au–Cu2O | Au: sphere, 60 nm | Au embedment depth | Two-pot | 87 |
| Cu2O: cube | Hybrid configuration | |||
| Ag–Cu2O | Ag: cube, 60–70 nm (edge length) | Ag embedment depth | Film-mediated embedment | 90 |
| Cu2O: pyramidal, rectangular | ||||
|
||||
| (E) Dumbbell | ||||
| Cu2O–Au–Cu2O | Au: rod (center), 80 × 16 nm | Hybrid configuration | Two-pot | 92 |
| Cu2O: cuboid (ends), 48 × 28 nm | Au aspect ratio | |||
| Au embedment depth: 14 nm | Cu2O size | |||
|
||||
| (F) Ternary | ||||
| Au@Ag@Cu2O core–shell–shell | Au: rod, tuned aspect ratio (1–5) | Au aspect ratio | Three-pot | 93 |
| Ag thickness: fixed at 3 nm | ||||
| Cu2O thickness: tuned, 5–30 nm | Cu2O shell thickness | |||
| Au@Ag@Cu2O core–shell–shell | Au: octahedron, 35 nm | Ag size | Three-pot | 94 |
| Ag: cube, tunable edge length (38–50 nm) | ||||
| Cu2O: different polyhedral shapes, tunable size (121–257 nm) | Cu2O size, morphology | |||
| Ag@Cu2O–Au core–shell–satellite | Ag: sphere, 35 nm | Cu2O shell thickness | Three-pot | 95 |
| Cu2O: porous sphere | ||||
| Au satellite size: 5 nm | Au amount/density | |||
| Au@Cu2O–Ag core–shell–satellite | Au: sphere | Ag amount/density | Three-pot | 96 |
| Cu2O: porous sphere | ||||
| Overall hybrid size: 80–100 nm | ||||
| Au@Cu2O–Ag core–shell–satellite | Au: sphere, 13 nm | Cu2O shell thickness | Three-pot | 97 |
| Cu2O: porous sphere, tunable thickness (32–80 nm) | ||||
2.1 Core–satellite structure
The core–satellite structure (also termed decorated structure or planet–satellite structure) is a nanocomposite configuration that consists of a singular core or “planet” of one component with smaller structures of the other component scattered along its surface like “satellites.” A key feature of core–satellite structures is that both components have exposed surfaces, which allow for direct interaction with possible substrates or targets. The properties of these materials are dependent not only on the overall structure itself but also on the individual structure of each component. For core–satellite structures of PM–Cu2O, the satellite component is usually the plasmonic metal, of which small nanoparticles decorate the surface of the larger Cu2O core.37–45 Adorning the Cu2O core with PM satellites instead of encapsulating it in a full PM shell minimizes the consumption of the costly noble metal.Zhao et al. fabricated Au-decorated Cu2O nanostructures using a two-pot synthesis procedure that involves simple reduction at room temperature.37 Porous Cu2O nanospheres were first synthesized by preparing an ammoniacal Cu2+ solution and subsequently reducing it to Cu2O using ascorbic acid. The product was isolated through centrifugation and dried to be used as the starting material for the composite. To obtain the core–satellite structure, varying concentrations of the Au precursor (0.5–5 wt% HAuCl4) were added to the Cu2O nanospheres dispersed in water. NaBH4 was then added to reduce HAuCl4 to metallic Au. The SEM images of the product confirmed the formation of a core–satellite configuration, where the Cu2O nanospheres (diameter = 150–200 nm) are decorated with smaller Au nanoparticles (diameter = 5–25 nm). The study highlighted the importance of the loading amount of HAuCl4, which was found to directly influence the size of Au satellites in the final structure. Higher concentrations of HAuCl4 resulted in larger Au satellites. Meanwhile, Qin et al. utilized a facile one-pot synthesis procedure to synthesize Ag-decorated Cu2O nanocubes at 55 °C.38 The Cu2O nanocubes were synthesized by first preparing Cu(OH)2 and then reducing it to Cu2O through the addition of ascorbic acid. To form Ag satellites on the surface of the nanocubes, AgNO3 was subsequently added, and the mixture was left to react for 30 min. The SEM images in Fig. 2 show the successful formation of tiny Ag nanoparticles on the surface of the Cu2O nanocubes. Increasing the Ag precursor concentration increased the number of Ag satellites; however, the average Ag particle size did not change significantly, which indicates that precursor concentration is not fully predictive of satellite size. Luo et al. also prepared Ag-decorated Cu2O nanocomposites via a one-pot synthesis protocol.41 Six types of polyhedral shapes were synthesized for the Cu2O core by using different amounts of the PVP surfactant. The Ag coverage was also varied by changing the loading amount of AgNO3. Increasing the loading amount resulted in more and larger satellites.
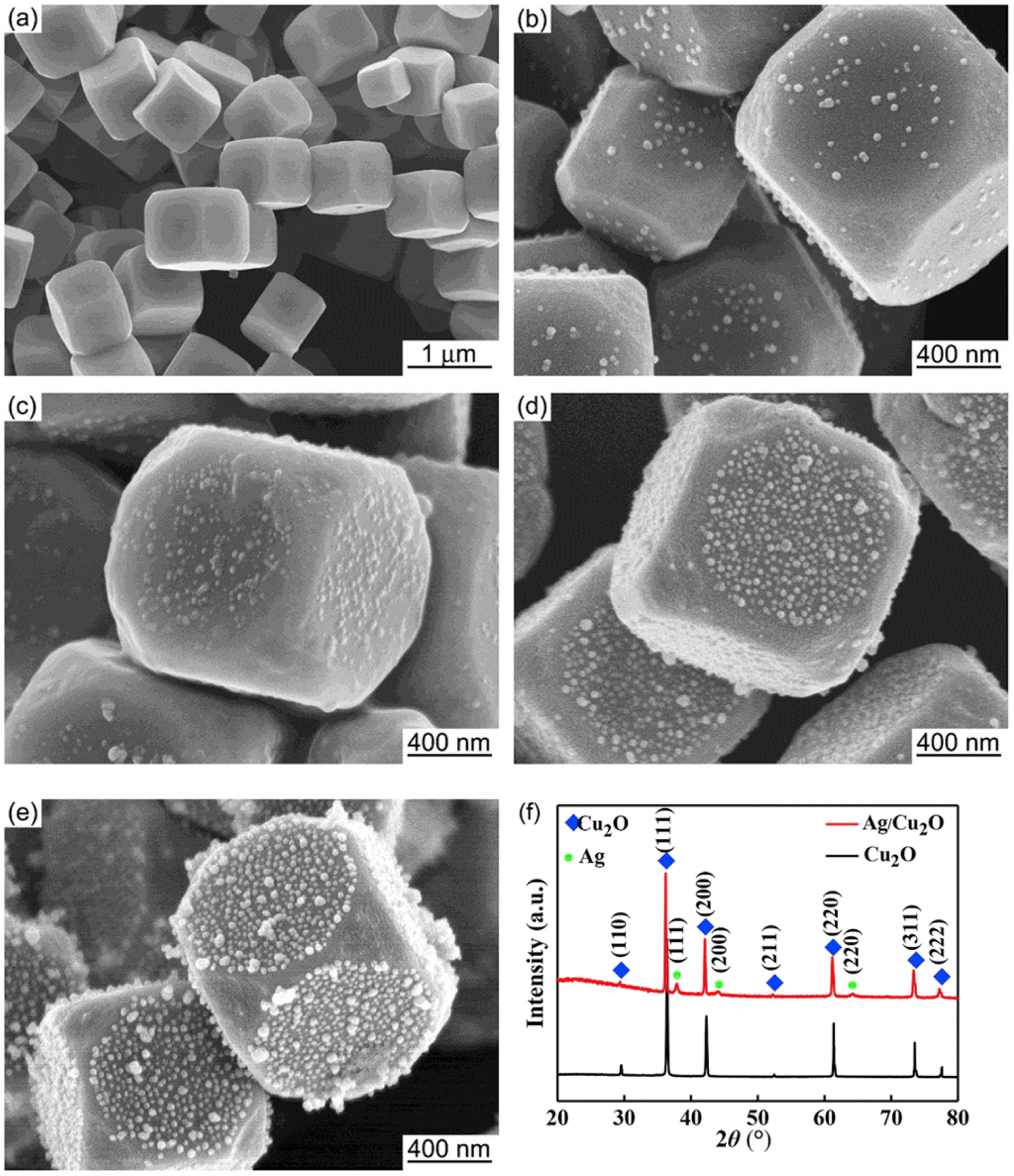 | ||
| Fig. 2 SEM images of (a) pristine Cu2O and (b–e) Ag-decorated Cu2O nanostructures, representing a core–satellite structure. The number of Ag satellites increased with increasing concentration of AgNO3 (from (b) to (e)). The XRD patterns for (a) and (e) are shown in (f). Reproduced with permission from ref. 38. Copyright 2019 Elsevier. | ||
Wang et al. prepared PM-decorated structures of Cu2O through a light-mediated deposition method.39 In brief, octahedral Cu2O nanocrystals were first obtained by adding N2H4·H2O (reductant) to a basic CuCl2 solution. The isolated nanocrystals were then dispersed in water, sonicated, and irradiated with visible light using a 500 W column-like iodine tungsten lamp from a distance of 20 cm. Depending on the desired hybrid material, either AgNO3 or KAuCl4 was added, and the resulting mixture was subjected to irradiation. Visible-light irradiation of semiconducting Cu2O produces electrons and holes, and the photogenerated electrons were believed to cause the reduction of the metal precursors to their corresponding PM nanoparticles. A temperature of 30 °C was maintained throughout the procedure. It was noted that significant etching of Cu2O occurs when the temperature is lowered to 20 °C.
Hong et al. were able to create a unique core–satellite architecture where hexoctahedral-shaped Au nanocrystals are decorated with Cu2O.46 In this structure, the Cu2O satellites are specifically situated at the Au hexoctahedron's eight sharp vertices pointed toward the <111> direction. The high anisotropy of the hexoctahedron morphology, which is enclosed by high-indexed facets and rich in high-curvature sites, and the use of a suitable stabilizing agent were viewed as key factors in the site-selective nucleation and growth of Cu2O. PVP was found to be an appropriate stabilizer for the creation of this distinct architecture because it preferentially adsorbs onto low-curvature sites, leaving the sharp vertices exposed for the deposition of Cu2O. When the synthesis was performed using SDS instead of PVP, isotropic growth of Cu2O on the entire surface of Au occurred, resulting in a core–shell structure.
2.2 Core–shell structure
Core–shell structures have one component at the center (the core) that is entirely encapsulated in the other component (the shell). For PM–Cu2O nanocomposites, two possible core–shell configurations exist: (i) PM@Cu2O where the core is made up of the plasmonic metal and the shell comprises Cu2O and (ii) Cu2O@PM where Cu2O is the core and the metal serves as the shell. Core–shell structures offer some advantages over core–satellite structures. One specific advantage for Ag@Cu2O structures is that they address the relatively low stability of Ag by limiting its interactions with reactants and the surrounding medium. In general, the encapsulation of plasmonic metals prevents their corrosion and dissolution.47 The core–shell architecture also allows for maximum metal–support interaction due to the three-dimensional contact between the metal and the semiconductor. The type of core–shell configuration (i.e., whether it is PM@Cu2O or Cu2O@PM) is crucial for certain applications. For instance, the Cu2O@PM configuration is preferred for SERS-based detection to maximize the contact of the plasmonic metal with the analyte, as the enhancement of Raman signals is largely influenced by the LSPR property of the PM component. In the case of photocatalysis, the PM@Cu2O configuration with an optimal Cu2O shell thickness is desirable to maximize the photocatalytic enhancement effects.The formation of a core–shell configuration is strongly favored when the materials that are combined have similar crystal symmetry and lattice constants.48 Cu2O, Au, and Ag are structurally compatible as they all crystallize in the face-centered cubic crystal system. The small lattice mismatch (f) between Cu2O and Au (f = 4.5%)36 and between Cu2O and Ag (f = 4.2%)49 allows for the creation of their core–shell hybrids. Core–shell structures of Au@Cu2O,36,44,50–61 Cu2O@Au,42,62 Ag@Cu2O,49,51,63–67 and Cu2O@Ag68–72 have been reported in the literature. They are most commonly prepared using two-pot colloidal synthesis procedures, where the core component is synthesized and isolated first before being fully encapsulated in the shell component, usually through simple reduction in solution. An advantage of a two-pot synthesis is that there is full control over the shape and size of the core. Other synthesis methods, such as one-pot co-reduction,63 microwave-assisted,51 pulsed laser irradiation,65 and magnetron sputtering techniques,68 have been employed as well.
Huang's group has published numerous studies on Au@Cu2O core–shell nanostructures.50,54–57 Precise control of morphology was attained by using Au nanoparticles of different shapes (e.g. plates, rods, and octahedra) as the structure-directing core for the overgrowth of the Cu2O shell.50 A two-pot synthesis method was employed, where the Au cores were synthesized first through reduction of HAuCl4 at elevated temperatures in the presence of reducing (sodium citrate, NaBH4) and capping (CTAB, CTAC) agents. The isolated Au cores were redispersed in water, and the dispersion was added to a solution containing CuCl2 and SDS. The Cu2O shell was grown with the subsequent addition of NaOH and hydroxylamine (NH2OH), followed by 2 h of aging. Fig. 3a–c show representative TEM images of the synthesized Au@Cu2O core–shell nanostructures with different morphologies.50 In their ensuing studies, Huang et al. noted that the LSPR absorption properties of Au@Cu2O core–shell nanostructures are largely facet-dependent, allowing for the tuning of the plasmonic band through precise morphological control.55,56 Jing et al. also used the two-pot synthesis method to fabricate Ag@Cu2O core–shell nanostructures with different morphologies (Fig. 3d–f).49 Starting with pre-synthesized Ag nanospheres and nanocubes as core materials, Cu2O shells were grown onto the surfaces of the Ag cores through controlled reduction of Cu(NO3)2 by N2H4 under basic conditions. The LSPR frequency of the Ag core was tuned across a wide spectral range by changing the size and morphology of the Ag core and by modulating the thickness of the Cu2O shell. Different shell thicknesses were obtained by varying the molar ratio of Cu(NO3)2 to the Ag core.
 | ||
| Fig. 3 TEM images of (a–c) Au@Cu2O and (d–f) Ag@Cu2O core–shell nanocomposites with different morphologies. Reproduced with permission from (a–c) ref. 50, copyright 2009 American Chemical Society, and (d–f) ref. 49, copyright 2014 American Chemical Society. | ||
One-pot procedures provide a facile, scalable, and economical synthesis method for core–shell structures. These typically involve the co-reduction of precursors to form the hybrid, where the species with a more positive reduction potential is reduced first and forms the core, followed by the reduction of the other species on the core surface to form the shell. Lee et al. obtained Ag@Cu2O core–shell nanostructures through this approach.63 In a three-neck flask, Cu(acac)2 and AgNO3 were mixed in oleylamine, and the flask was then heated to 230 °C for 3 h under inert atmosphere to produce the hybrid. According to their previous work,73 in oleylamine solutions, AgNO3 is reduced to Ag at 80 °C, while Cu(acac)2 is reduced to Cu2O at 180 °C, which means that Ag will form first when the reaction mixture containing both precursors is heated up. As expected, increasing the amount of Cu(acac)2 resulted in increased shell thickness and overall particle size. The Ag core diameter was similar regardless of the amount of Cu(acac)2 added, which indicates that all of the AgNO3 was reduced to Ag before the formation of Cu2O. Legaspi et al. produced Au@Cu2O core–shell nanostructures with a unique nanoflower hybrid design using a one-pot synthesis procedure at ambient conditions.59 The core consists of aggregated small Au nanoparticles, whereas the shell is composed of larger Cu2O nanoparticles that surround the core in a petal-like arrangement. It was noted that the formation of a hybrid structure is more favored if the precursors for both the Cu2O and Au are already present in the reaction mixture prior to the addition of the reducing agent.
Microwave synthesis is advantageous for nanoparticle synthesis as it allows for the heating of precursor materials in a quick and homogeneous manner. This consequently leads to uniform nucleation, rapid crystal growth and thus, narrow size distribution.74 Torras and Roig presented a novel microwave-assisted synthesis procedure for both Ag@Cu2O and Au@Cu2O nanocomposites with spherical cores and cubic shells.51 The spherical metal cores were first made through a polyol synthesis performed in a microwave synthesizer at 120 °C. To form the Cu2O coating, a benzyl alcohol dispersion of the metal cores was mixed with Cu(acac)2 and PVP and was then subjected to microwave heating at 200 °C. This microwave-assisted technique was also successful in producing Pd@Cu2O nanocomposites.
In creating the reverse core–shell configuration (i.e., Cu2O@PM), the galvanic replacement reaction (GRR) strategy has proven to be useful. GRR-mediated synthesis is driven by the difference in the reduction potential of the redox couples involved.75 In the case of Cu2O@PM, the precursors of the plasmonic noble metal are reduced by pre-synthesized Cu2O particles. This was demonstrated by Bakthavatsalam and Kundu in their synthesis of octahedral Cu2O@Ag core–shell structures.69Fig. 4a displays a schematic illustration of the formation process and the SEM images of the obtained structures. First, the Cu2O surface was etched with an acid to release Cu+ ions, which can spontaneously react with the Ag+ ions in solution via GRR. The Cu+ ions get oxidized to Cu2+ ions as they reduce Ag+ ions to Ag, which are deposited as nanoparticles onto the Cu2O surface. Chen et al. fabricated octahedral Cu2O@Au core–shell structures following a similar process (Fig. 4b).62 It must be stressed that to achieve the Cu2O@PM core–shell configuration, the loading amount of the metal precursor should be carefully controlled to ensure the formation of a continuous metal shell layer. An insufficient loading amount can result in a core–satellite structure where the Cu2O core is only partially decorated with metal nanoparticles. On the other hand, an excessively high loading amount can lead to extensive GRR that dissolves the Cu2O core. Partial dissolution of the core produces a yolk–shell structure (see Section 2.3), whereas complete dissolution yields a hollow metal structure as the final product.76
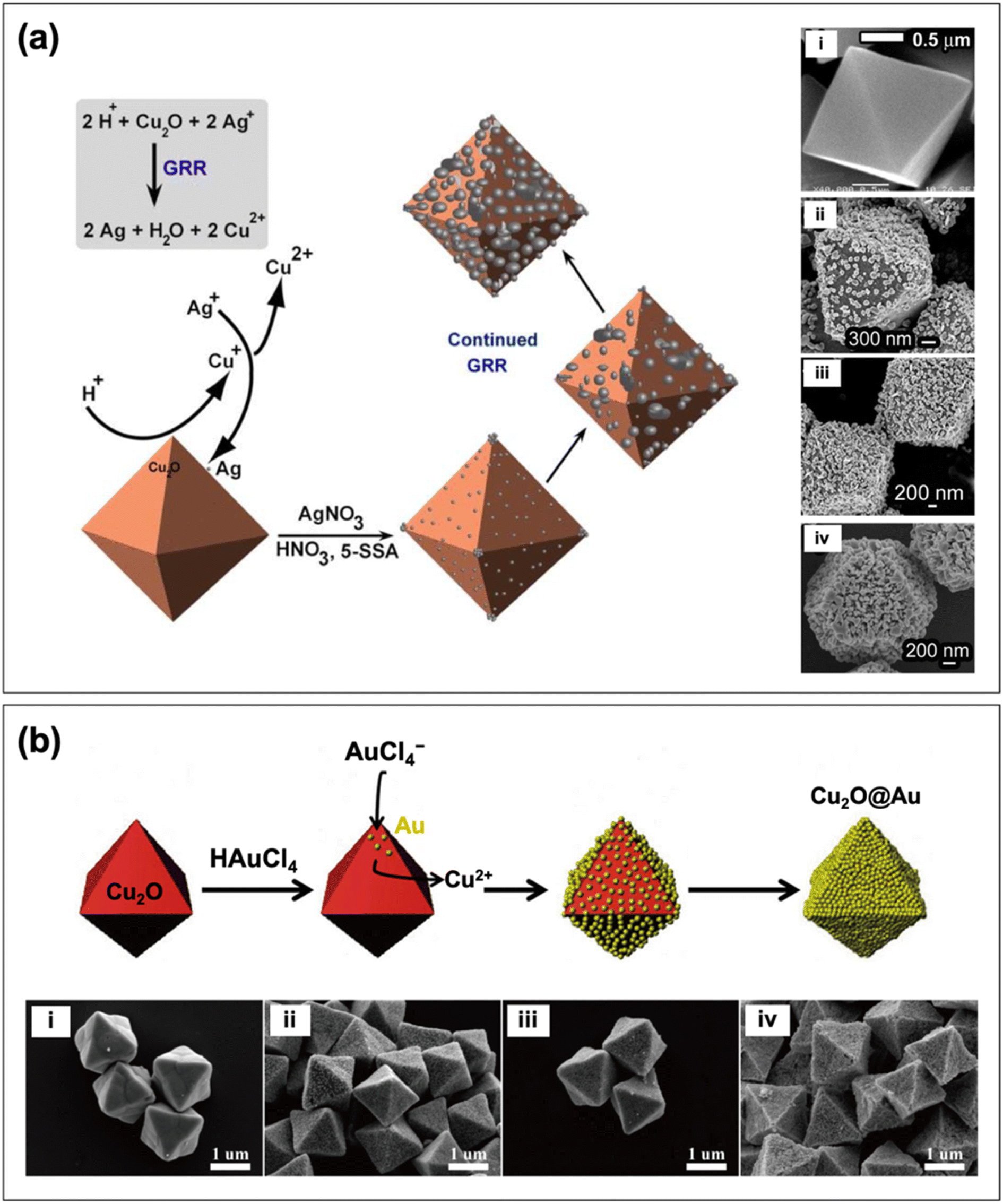 | ||
| Fig. 4 Schematic depiction of the formation of (a) Cu2O@Ag and (b) Cu2O@Au core–shell nanocomposites through GRR. Also shown are the SEM images of the structures produced with (i) no metal and ((ii) to (iv)) increasing loading amount of metal. Reproduced with permission from (a) ref. 69, copyright 2017 the Royal Society of Chemistry, and (b) ref. 62, copyright 2016 Elsevier. | ||
2.3 Yolk–shell structure
Yolk–shell (or rattle-like) structures are similar to core–shell structures in that a shell component surrounds a core component. The key difference is the presence of a void in between the core and the shell in the yolk–shell structure. Yolk–shell structures have a number of properties that are advantageous for photocatalysis. The void contributes another variable that can be used to tune the catalytic efficiency, stability, and recyclability of the hybrid material.77,78 The presence of a void also allows for the scattering of incident light after it penetrates the shell, which contributes to a greater light-harvesting property of the hybrid.79Yolk–shell structures can be fabricated through a variety of means, such as the Kirkendall effect, Ostwald ripening, chemical etching, and thermal treatment.76,77,80–84 In the same study as their Au@Cu2O core–shell synthesis, Zhang et al. synthesized Au@Cu2O yolk–shell structures by simply extending the reaction time from 2 min to 60 min.80 With the prolonged reaction time, the polycrystalline Cu2O shell of the original core–shell structure underwent a symmetric hollowing process, which formed the characteristic void. Fig. 5a shows a schematic illustration of this hollowing process along with TEM images of the structures. The core–shell structure is maintained at a reaction time of 5 min. At 30 min, slight hollowing occurs, and voids begin to form around the Au cores. At 60 min, significantly larger voids are present, and the desired rattle-like formation was obtained. Extending the reaction time further to 90 min resulted in the collapse of the shell structure, which appears as fragmented crystals in the image. Two key observations here are the inside-out nature of the hollowing process and the maintained outer radius of the Cu2O shell. A similar hollowing process was described by the authors in their previous work, and they attributed the symmetric hollowing to Ostwald ripening.85 This approach is differentiated from other yolk–shell synthesis methods because the core structure is maintained as the void is formed. Xiong et al. obtained Ag@Cu2O yolk–shell structures also through an Ostwald ripening process.82 Core–shell structures were first synthesized by coating Ag nanocubes with a polycrystalline Cu2O shell. The solution was aged for 13 min to allow for the hollowing of the shell. The inner region of the Cu2O shell has a higher surface energy compared to its outer surface, which makes it more susceptible to dissolution. As a result, Cu2O dissolves from the inside and recrystallizes on the surface as Ostwald ripening progresses.
 | ||
| Fig. 5 Schematic representation of the formation of (a) Au@Cu2O and (b) Cu2O@Au yolk–shell nanocomposites. The TEM images of the structures produced are also shown. Adapted with permission from (a) ref. 80, copyright 2011 American Chemical Society, and (b) ref. 84, copyright 2017 Springer Nature. | ||
Chen et al. detailed a self-generated acid etching method for the formation of Cu2O@Au yolk–shell structures. Octahedral Cu2O cores were first formed through the reduction of CuCl2 in a basic solution using N2H4·H2O.84 The octahedral cores were then dispersed in ethanol, and PVP was added. Varying volumes of HAuCl4 were introduced followed by NaBH4. Depending on the amount of HAuCl4 added, core–satellite, core–shell, or yolk–shell structures were formed (in order of increasing HAuCl4). Fig. 5b presents the progression of the synthesis reaction as more HAuCl4 is added. In contrast to the hollowing presented by Zhang et al. (Fig. 5a),80 the void in the Cu2O@Au yolk–shell structures stemmed from the etching of the Cu2O core rather than the shell. Still, Cu2O was the component that was removed from the structure to form the void. In the self-generated acid etching synthesis, the Cu2O core is etched by HCl and H3BO3, which are generated during the reduction of HAuCl4 to Au by NaBH4.76 This method provides a means of producing Cu2O@Au yolk–shell structures with tunable void and core sizes through control of the amount of HAuCl4.
2.4 Janus and dumbbell structures
Janus structures (or heterodimers) possess two faces of differing components that each have a specific shape and size.86 In Janus-type nanocomposites, the constituent materials are joined at a heterojunction with the rest of their structures being exposed to the environment and open to interactions. Unlike the previously discussed hybrid configurations, there are no core or shell/satellite components in Janus structures. They have been shown to be better than core–shell structures in terms of promoting charge separation and preventing charge accumulation.87 Additionally, due to their asymmetric structure, Janus-type systems have been shown to function as photocatalytic nanomotors that can modulate mass transfer behavior for highly efficient photocatalysis.88Janus structures can be created through lattice mismatching, which takes advantage of the difference in the lattice constants of the two components.89 However, due to the relatively small lattice mismatch (f < 5%) between Cu2O and the two plasmonic metals (Au and Ag), lattice mismatching cannot be utilized to construct their Janus structures, given that core–shell structures are usually formed. This has led scientists to devise different strategies for their fabrication. For example, Gale-Mouldey et al. developed a partial embedment technique to synthesize Janus-type Ag–Cu2O nanocomposites, where Ag is only partially coated with Cu2O.90 Their approach involves the following steps: (i) depositing pre-synthesized Ag nanocubes on a polystyrene (PS) film substrate, (ii) embedding the Ag nanocubes into the PS film via heating, (iii) growing Cu2O on the exposed area of the Ag nanocubes, and (iv) dissolving the PS film to release the Ag–Cu2O Janus hybrid. The degree of exposure of the Ag domains in the Janus structure can be tailored by adjusting the embedment depth through controlled heating of the substrate.
Xu et al. were able to produce Au–Cu2O Janus structures using 5-amino-2-mercaptobenzimidazole (AMBI) as a modifying ligand.87 Pre-synthesized Au nanospheres were incubated with an ethanolic solution of AMBI at 60 °C for 2 h. This incubation step was performed to coat the surface of the Au nanospheres with AMBI ligands. Cu2O was then allowed to grow onto the Au nanospheres to form the heterodimers. The effect of AMBI concentration on the synthesized structures was observed through SEM (Fig. 6). In the absence of AMBI, cubic Cu2O fully coated the Au cores, forming a core–shell structure (Fig. 6a). When AMBI was present, the Au nanospheres became only partially coated with Cu2O cubes (Fig. 6b–d). The height of the exposed Au domain (i.e., the degree of exposure) was found to increase with AMBI concentration. AMBI is a strong thiol ligand that serves to control the interfacial energy of Au seeds to allow for the formation of a Janus structure rather than a core–shell one. Basically, the presence of AMBI on the Au surface weakens the Au–Cu2O interaction and disrupts lattice matching, causing interfacial strain that deters the complete encapsulation of the Au nanospheres.
 | ||
| Fig. 6 Schematic illustration of the tunable synthesis of Janus-type Au–Cu2O nanocomposites through control of interfacial energy using AMBI as the modifying ligand. Also shown are the SEM images of the structures obtained (a) without AMBI and (b–d) with increasing AMBI concentration. Reproduced with permission from ref. 87. Copyright 2020 Wiley-VCH. | ||
Another type of heterostructure design is the dumbbell structure, aptly named from its resemblance to a dumbbell. In a dumbbell configuration, one component is a nanorod, while the other component caps both ends of the nanorod.91 It is crucial that the second component selectively nucleates and grows on the tips of the nanorod, leaving the rest of the structure exposed to the environment. With their successful synthesis of Au–Cu2O Janus nanostructures, Xu et al. also employed their AMBI-mediated approach to prepare Au–Cu2O nanocomposites with a dumbbell morphology (Fig. 7a).92 In this case, they used Au nanorods instead of Au nanospheres as their starting material. When no AMBI was present, Cu2O was able to completely encapsulate the Au nanorods, producing a core–shell configuration (Fig. 7b). In the presence of AMBI, Cu2O coated only the ends of the nanorods, leading to a well-defined dumbbell structure (Fig. 7c).
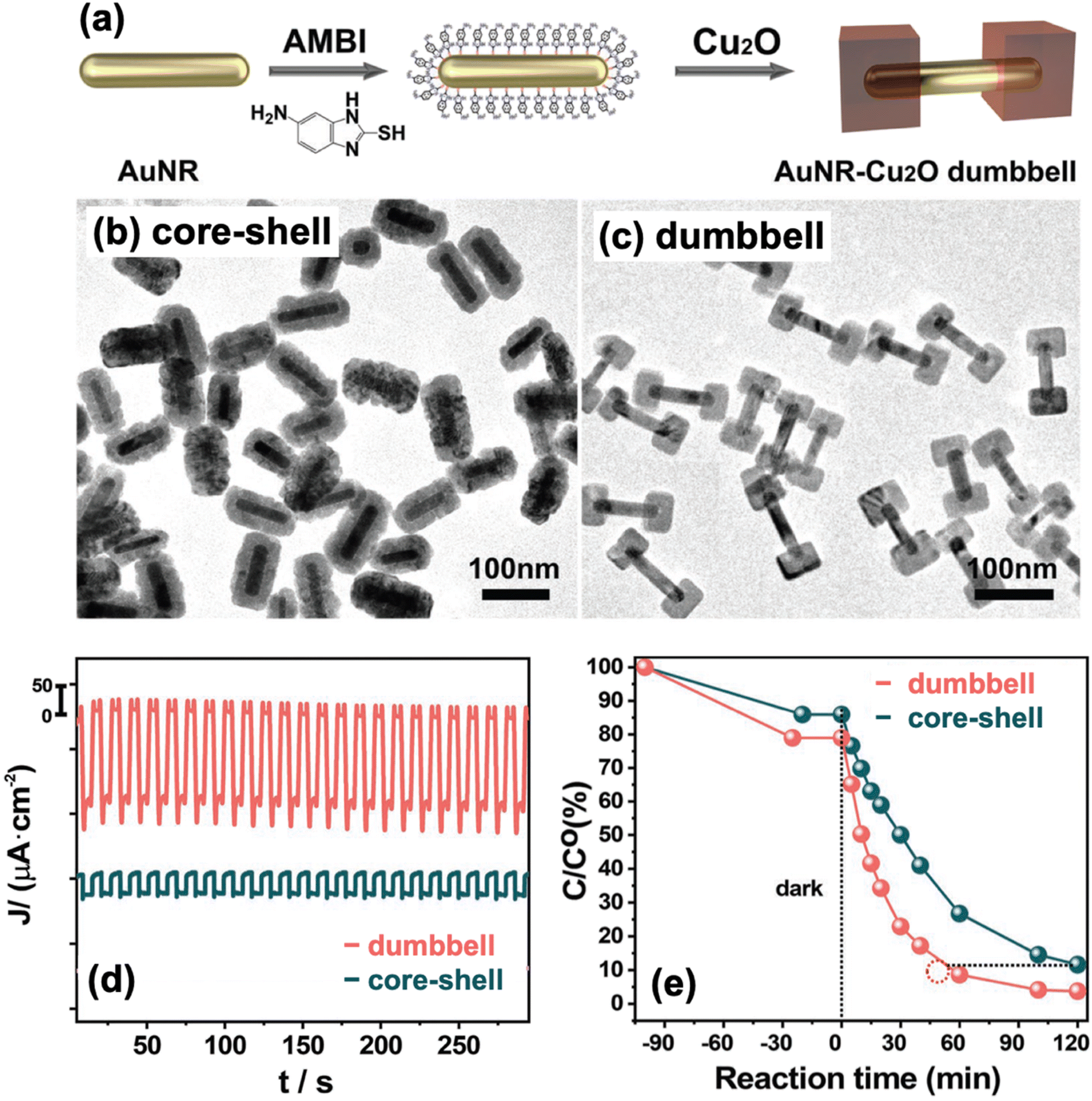 | ||
| Fig. 7 (a) Schematic diagram of the AMBI-mediated synthesis of dumbbell-type Au–Cu2O nanocomposites. (b and c) TEM images of the structures obtained (b) without AMBI and (c) in the presence of AMBI. (d and e) Comparison of the (d) photocurrent response and (e) photocatalytic activity for MO degradation of dumbbell and core–shell structures. Reproduced with permission from ref. 92. Copyright 2023 Wiley-VCH. | ||
2.5 Ternary structure
Nanocomposites are not limited to the combination of only two components. It is also possible to create multinary structures such as ternary structures consisting of Cu2O, Au, and Ag. The synthesis of these structures are often amalgamations of previously discussed techniques. Compared to binary structures, nanostructures consisting of three components take more resources—not just reagents but also time and effort—to synthesize. Thus, the benefit of compositing three different materials must be weighed against the extra resources expended.In their preparation of ternary structures, Hu et al. used a three-pot procedure where Au nanorods were synthesized in the first pot and then coated with Ag in the second pot. In the third pot, the Au@Ag core–shell nanorods were coated with Cu2O, yielding Au@Ag@Cu2O core–shell–shell nanorods.93 In forming the Cu2O shell, the Au@Ag nanorods were mixed with SDS, ascorbic acid, and CuCl2 as the precursor to Cu2O. The mixture was placed in an ice bath, and NaOH was added to initiate the shell formation. The low temperature was crucial as it prevented the self-nucleation of Cu2O. The LSPR features of the ternary structure were tuned by modulating the Au nanorod aspect ratio and the Cu2O shell thickness. Yang et al. also prepared Au@Ag@Cu2O core–shell–shell nanostructures but with different faceted morphologies (i.e., rhombic dodecahedron, truncated octahedron, and cuboctahedron).94 They started with the hydrothermal synthesis of octahedral Au nanocrystals, which they subsequently enclosed within Ag nanocubes. These Au@Ag nanocubes were then encased in a polyhedral Cu2O shell. By modulating the amount of the metal cores, they were able to control the overall size and morphology of the encapsulating Cu2O shell, which resulted in tunable optical properties.
Another synthesis pathway for ternary structures is to synthesize a PM@Cu2O core–shell structure and then decorate it with a different plasmonic metal to form a core–shell–satellite architecture, where Cu2O is sandwiched between the two PMs. Both Ag and Au have been used as cores and satellites for this type of structure.95–97 To create Au-decorated Ag@Cu2O, Chen et al. first prepared the Ag core through simple reduction of AgNO3 with sodium citrate under reflux.95 This was then added to a solution containing PVP and Cu(NO3)2 under constant stirring. N2H4·H2O was added to induce the formation of the Cu2O shell on the Ag surface, yielding a Ag@Cu2O core–shell structure. A ternary structure was synthesized by simply adding HAuCl4 to an aqueous dispersion of the core–shell structure. A schematic depiction of the synthesis and the TEM images of the products are shown in Fig. 8. The synthesized ternary structure consists of a uniform spherical Ag core that is fully coated with an Au-decorated Cu2O shell. As the added HAuCl4 concentration increased, so did the density of Au decoration on the surface of the Ag@Cu2O structures. Following a similar protocol, Wu et al. produced the reverse configuration, where Au@Cu2O is decorated with Ag satellites.96
 | ||
| Fig. 8 Schematic depiction of the synthesis of Au-decorated Ag@Cu2O nanocomposites, an example of a ternary structure. Also shown are the TEM images of the structures obtained (a) without Au and (b–g) with increasing loading amount of Au. Reproduced with permission from ref. 95. Copyright 2018 Elsevier. | ||
3. Photocatalytic applications
Literature on the photocatalytic applications of PM–Cu2O nanocomposites has mostly focused on the degradation of organic dyes, which can be conveniently monitored using UV-vis spectroscopy. When irradiated with light, the PM–Cu2O photocatalysts are able to accelerate the degradation of organic dyes into environmentally benign compounds, presenting a viable solution for the elimination of harmful pollutant dyes in wastewater. For example, Zhao et al. conducted photocatalytic degradation studies using Au-decorated Cu2O nanocomposites as photocatalysts for the following organic dyes under visible light: methylene blue (MB), methyl orange (MO), rhodamine B (RhB), and congo red (CR).37 For all four dyes, it was found that the hybrid showed superior photocatalytic activity relative to pristine Cu2O. The authors stated two reasons for the improved photocatalytic properties: (i) the Schottky junction effect enhances the electron–hole separation rate and (ii) the plasmonic effect of Au boosts the absorption of visible light. Meanwhile, Yan et al. tested their synthesized Ag@Cu2O core–shell nanowires for the photocatalytic degradation of RhB under visible light irradiation.65 They observed a dramatically enhanced photocatalytic behavior for the Ag@Cu2O nanowires relative to uncoated Ag nanowires and pristine Cu2O nanoparticles (Fig. 9a). This underscores the immense value that can be derived from compositing the two materials. Similar to the previous example, the enhancement was credited to both the Schottky junction effect and the plasmonic effect. The mechanisms behind these photocatalytic enhancement effects are discussed below.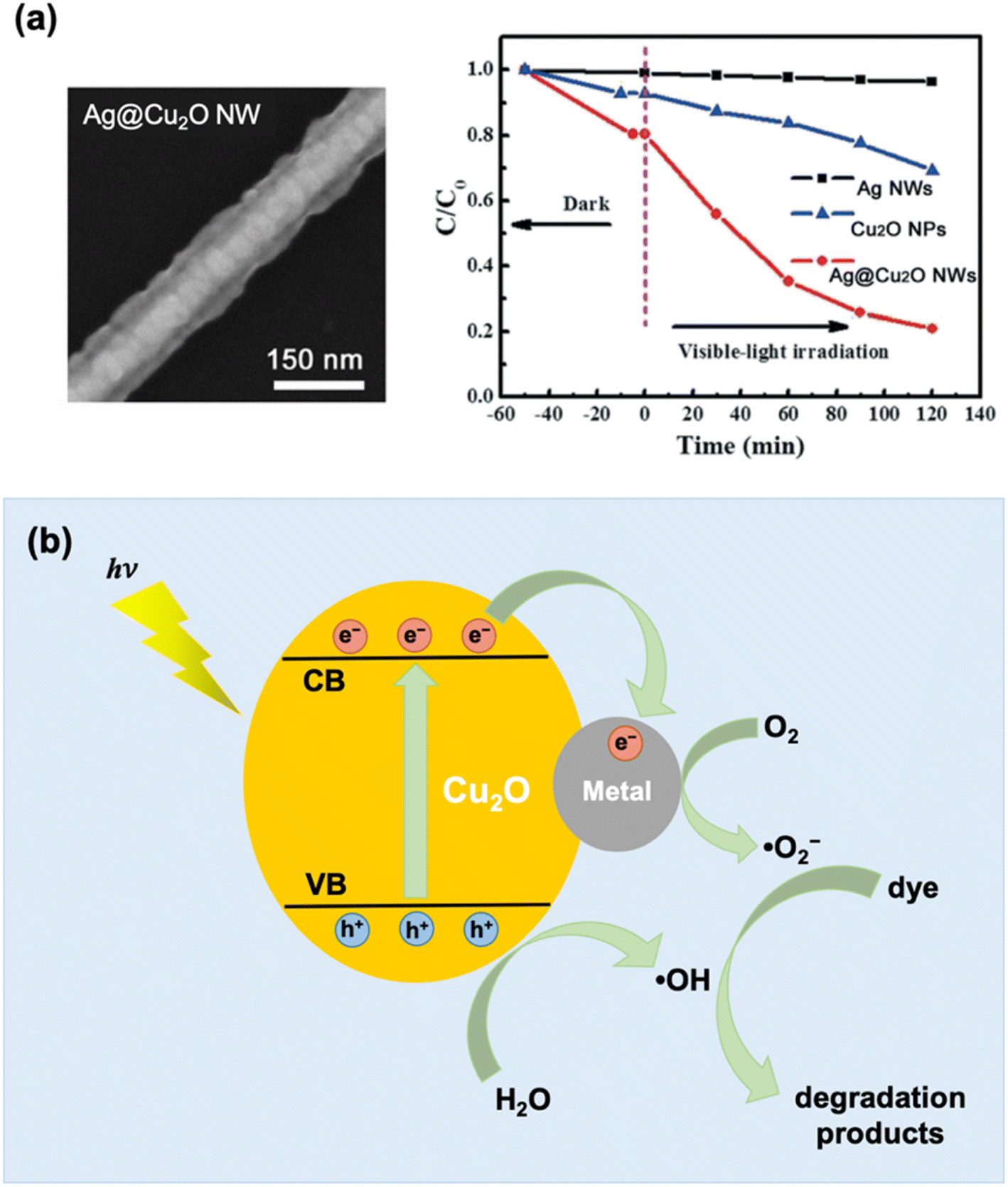 | ||
| Fig. 9 (a) SEM image and photocatalytic degradation activity of Ag@Cu2O core–shell nanowires (NWs) under visible light using RhB as target dye. The photocatalytic activity of uncoated Ag NWs and pristine Cu2O nanoparticles are also shown in the plot for comparison. Reproduced with permission from ref. 65. Copyright 2021 the Royal Society of Chemistry. (b) Schematic illustration of photocatalytic dye degradation mechanism based on the Schottky junction effect, which results in enhanced charge separation at the metal–Cu2O interface. | ||
3.1 Mechanisms of photocatalytic enhancement
 | ||
| Fig. 10 Schematic depiction of the three primary mechanisms for plasmon-enhanced photocatalysis by plasmonic metal–semiconductor nanocomposites: (a) hot electron injection (HEI), (b) plasmon-induced near-field interaction, and (c) plasmonic light scattering. | ||
The plasmon-induced near-field interaction mechanism (Fig. 10b) involves the generation of strong electromagnetic fields that are localized at the metal nanoparticle surface (referred to as near-fields).105 Under light irradiation, the LSPR induced on a plasmonic metal nanoparticle gives rise to near-fields that are spatially non-homogeneous, being more intense at the nanoparticle surface and decreasing exponentially with distance. A semiconductor that is positioned close to the photoexcited plasmonic metal will experience these fields, promoting interband transitions that create electron–hole pairs in the semiconductor. It has been determined that the rate of electron–hole pair generation is directly proportional to the square of the local intensity of the electric field.106 Thus, the strong plasmon-induced near-fields can induce the formation of a large number of electron–hole pairs in semiconductor regions close to the metal. These fields can also mediate in a non-radiative energy transfer process between the plasmonic metal (donor) and the semiconductor (acceptor) in the form of plasmon-induced resonance energy transfer (PIRET).107 PIRET leads to direct excitation of the electron–hole pairs in the semiconductor via the relaxation of the LSPR dipole. A spectral overlap between the LSPR absorption of the plasmonic metal and the band gap absorption of the semiconductor is necessary for the near-field effects to be observable. Unlike the HEI mechanism, intimate contact between the metal and the semiconductor is not a requirement. The near-field enhancement effects can be realized even when the plasmonic metal and the semiconductor are separated by an insulating layer. For example, Wu's group observed enhanced photocatalytic efficiency for Au@SiO2@Cu2O nanocomposites, even when Au and Cu2O are separated by an insulating SiO2 layer.107 However, the SiO2 layer should not be too thick as the enhancement was found to decrease with increasing metal–semiconductor separation distance.108
The third plasmon-mediated mechanism is based on the light scattering effect of plasmonic metal nanoparticles. When irradiated with light, plasmonic metal nanoparticles are able to spread out the incident resonant photons, resulting in a longer average photon path for the metal–semiconductor hybrid compared to the pristine semiconductor (Fig. 10c).101,109 Resonant photons that are not initially absorbed by the semiconductor can be scattered by the metal domains, allowing these photons to pass several times through the semiconductor, thereby increasing their chance for absorption.102 This leads to a higher rate of electron–hole pair generation in the semiconductor. The size of the metal nanoparticles strongly influence their light scattering ability.110,111 As the scattering-to-absorption ratio of metal nanoparticles increases with increasing particle size, the plasmonic light scattering mechanism becomes significant only when the size of the metal domains is sufficiently large (>50 nm).
3.2 Tunable photocatalytic properties
There are different ways by which the photocatalytic properties of PM–Cu2O nanocomposites can be optimized. For PM@Cu2O core–shell structures, one parameter that can be readily adjusted is the thickness of the Cu2O shell. Li et al. synthesized Ag@Cu2O core–shell nanostructures with different shell thicknesses and studied their photocatalytic activity toward MO degradation under visible light illumination (Fig. 11a).67 The core–shell hybrids exhibited better photocatalytic performance than pristine Cu2O, and the authors attributed this to plasmon-mediated effects. The enhancement was maximized by tuning the LSPR absorption through modulation of the Cu2O shell thickness. Moreover, the highest photocatalytic efficiency was achieved when the wavelength of incident light corresponds to the LSPR wavelength (Fig. 11b), confirming that the photocatalytic enhancement is plasmon-driven. The size of the Ag core for all samples was kept identical at 30 nm, which is too small for the light scattering mechanism to be significant. Transient absorption spectroscopy data revealed that both HEI (or DET) and PIRET mechanisms contribute to the enhancement of photocatalytic activity (Fig. 11c).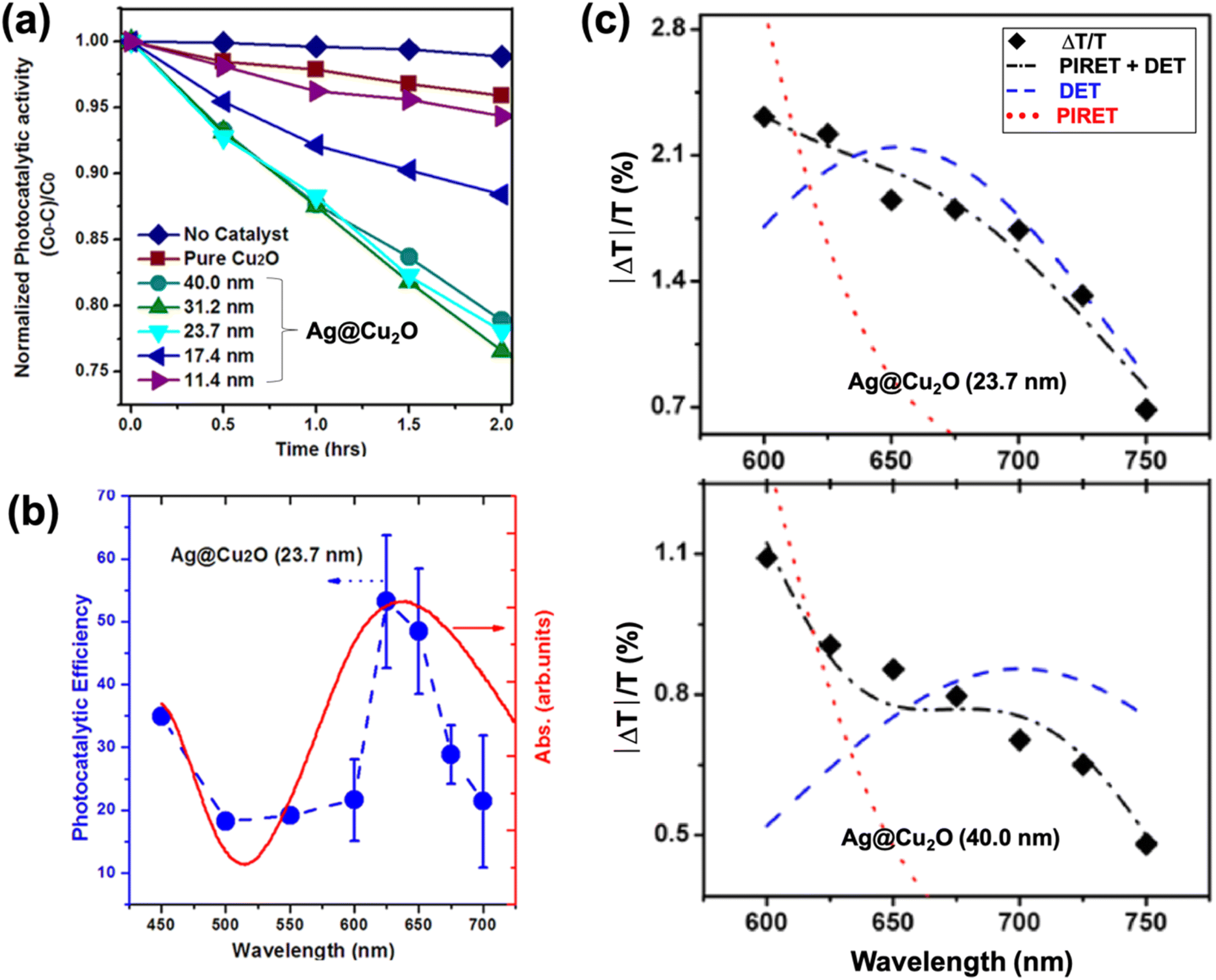 | ||
| Fig. 11 (a) Photocatalytic MO degradation rate for Ag@Cu2O core–shell nanostructures with different shell thicknesses. (b) Absorption spectrum (red) and apparent photocatalytic efficiency as a function of the wavelength of monochromatic light (blue) for Ag@Cu2O hybrid with shell thickness of 23.7 nm. (c) Transient absorption spectra (♦) and mechanism determination for Ag@Cu2O hybrids with shell thickness of 23.7 nm (top) and 40.0 nm (bottom). Each shell thickness is fit by a model for PIRET, DET, and PIRET + DET to show that both mechanisms are responsible for generation of charge carriers in Cu2O. Reproduced with permission from ref. 67. Copyright 2013 American Chemical Society. | ||
The effect of the Cu2O shell morphology on the photocatalytic activity of Au@Cu2O core–shell hybrids was examined by Wang et al.60 Two types of faceted morphology were investigated: cubic and octahedral. The researchers synthesized various samples, including pristine Cu2O cubes and octahedra, Au@Cu2O core–shell cubes and octahedra, and Au@Cu2O core–shell face-raised cubes and octahedra, and evaluated their activity toward the photocatalytic degradation of MO dye under irradiation from a Xe lamp. The results showed that only the octahedral structures effectively catalyzed the degradation reaction, with the Au@Cu2O face-raised octahedral structure displaying the highest photocatalytic efficiency. This was attributed to the presence of more {111} facets in the face-raised octahedral geometry. Strong photocatalytic interactions occur with {111} facets (i.e., the exposed faces of octahedra) because Cu2O crystals bound by these facets possess Cu atoms with dangling bonds, which increase surface energy and endow the surface with a more positive charge.112 This promotes interaction with negatively charged molecules, such as MO, the anionic dye used in the experiment. All the cubic structures were found to be photocatalytically inactive as Cu2O cubes are bound by low-energy {100} faces. The authors also stated that the cubic Cu2O structures that showed photocatalytic activity in a previous study were actually truncated cubes that contain {110} edges and {111} corners.54 This highlights the significance of controlling the surface facets of PM–Cu2O hybrids and provides some considerations for the optimization of their photocatalytic properties.
For PM-decorated Cu2O hybrid designs (i.e. core–satellite structures), an optimal loading amount of PM is crucial to maximize the photocatalytic efficiency. Qin et al. used their Ag-decorated Cu2O hybrid material as a catalyst in a photodegradation experiment with MB as the target dye.38 They found that there is a loading amount “sweet spot,” in which a moderate amount of Ag satellites results in the highest photocatalytic activity. The results of this study comport with those of Li et al., who reported that an intermediate loading amount of Ag is more beneficial to the photodegradation of MO compared to low or high loading amounts.43 Low amounts of Ag decoration do not allow the plasmonic effects to be maximized. Meanwhile, excessive amounts can result in Ag blocking the photon absorption of Cu2O and covering the Cu2O active sites. Wang et al. compared the photocatalytic properties of Cu2O–Ag and Cu2O–Au core–satellite structures, which they prepared using the same synthetic protocol.39 The photocatalytic properties of the two hybrid materials were tested through the photodegradation of pyronine B. The Cu2O–Au hybrid was found to degrade pyronine B at a higher rate than the Cu2O–Ag hybrid, albeit the difference is very minimal with both facilitating approximately 75% degradation over the course of 150 min compared to 36% using Cu2O alone. The authors attributed this slight difference to the relative oxidative stability of Au compared to Ag.
The type of hybrid configuration can also influence the photocatalytic activity of the composite material. Janus and dumbbell structures are better than core–shell structures in avoiding charge buildup and in facilitating charge separation.87,92 A major downside of a core–shell structure is that the encapsulating shell causes charge carriers to accumulate in the core component, and this negatively impacts the photocatalytic efficiency as only the charge carriers on the surface can partake in photocatalytic reactions. A full shell also limits the core in absorbing incoming photons. In Janus and dumbbell structures, large sections of both components are exposed to the environment and are therefore accessible to interactions. Moreover, the distal separation of the metal and semiconductor domains in such structures allows for a more efficient charge separation. Xu et al. compared the photocurrent generation capability of Au–Cu2O Janus and Au@Cu2O core–shell structures and found that the Janus-type hybrid performed better, demonstrating its superior ability to promote charge separation.87 In a separate study, they compared Au–Cu2O dumbbell and Au@Cu2O core–shell structures and observed a greater photocurrent generation for the dumbbell structure (Fig. 7d).92 Consequently, the dumbbell structure exhibited better photocatalytic performance than the core–shell structure when tested for MO degradation (Fig. 7e).
In a recent work by Kovács et al., the position of Au was found to dictate the photocatalytic properties of Au–Cu2O nanocomposites.44 They compared two types of configuration: core–satellite where Au nanoparticles decorate the surface of octahedron-shaped Cu2O and core–shell where an Au nanorod is embedded within an octahedral Cu2O shell. The overall size, shape (octahedral), and composition (Cu:Au ratio) of the hybrids were kept identical. Their study focused on the charge separation efficiency from the semiconductor standpoint. To exclude the contribution from plasmonic effects, they performed the photodegradation experiments using a 396 nm UV LED as the light source. This excitation wavelength is off-resonant with the LSPR of Au; hence, the electron–hole pairs were generated solely in Cu2O. The performance of the Au-decorated Cu2O system was superior to that of the Au@Cu2O core–shell structure, implying that a more improved charge separation can be achieved when Au is exposed rather than buried. Moreover, the core–satellite structure generates more easily accessible charge carriers at the particle surface where photocatalytic reactions take place.
The benefits of a yolk–shell structure as a photocatalyst were described in a paper by Chen and co-workers. Using MO as the test dye, they observed an improved photocatalytic degradation rate for their Cu2O@Au yolk–shell hybrid compared to pristine Cu2O and Au-decorated Cu2O.84 The authors described their yolk–shell structure as a cavity micro-reactor in which pollutant degradation can take place. The porous Au shell allows light to reach the Cu2O component and also enables the dye molecules to enter the cavity and react with the charge carriers on the Cu2O surface. The cavity in a yolk–shell structure also enables strong scattering of light, which improves the light-harvesting capability of the material.79
The morphology of the plasmonic metal component also exerts a substantial influence on the photocatalytic capacity of the hybrid. Ma et al. prepared Au@Cu2O yolk–shell nanostructures with different Au core morphologies, such as sphere, rod, and bipyramid.81 Due to shape anisotropy, the Au nanorods and nanobipyramids exhibited strong longitudinal LSPR absorption at the near-infrared (NIR) region. As a consequence, the yolk–shell hybrids that contain these Au cores displayed a broader absorption range, which is desirable for maximum utilization of solar light. The photocatalytic behaviors of the different samples, including pure hollow Cu2O (i.e., no Au core), were examined for visible-light-driven MO degradation and for NIR-light-activated hydrogen production. As expected, the yolk–shell hybrids with anisotropic Au cores performed better than both the hollow Cu2O and the yolk–shell hybrid with spherical Au cores. Hu et al. fabricated nanorods of ternary Au@Ag@Cu2O and used them for the visible-light-mediated photodegradation of MO.93 These ternary nanorods were found to be photocatalytically superior to the corresponding ternary nanospheres due to the multiple plasmon resonances arising from the anisotropy of the Au@Ag nanorod interior coupled with the dielectric property of the Cu2O shell. The multiplasmon modes expanded the spectral overlap between the LSPR absorption band of the PM core and the absorption band edge of Cu2O, which is necessary for PIRET-based plasmonic enhancement effect. Further optimization of the optical properties was done through tuning of the Au aspect ratio and the Cu2O shell thickness.
The advantages of creating a ternary nanocomposite were seen in a recent study by Wu et al.96 The Ag-decorated Au@Cu2O hybrid outperformed the binary Au@Cu2O system (i.e., without Ag decorations) in degrading malachite green (MG) under visible light illumination. As Cu2O is sandwiched between the two plasmonic metals in the ternary system, a Schottky barrier is formed at both the Au–Cu2O and Ag–Cu2O interfaces, prolonging the lifetime of the photogenerated charge carriers. The near-field effects are also heightened in this type of configuration, generating more electron–hole pairs, which leads to further enhancement in photocatalytic efficiency.
4. Summary and outlook
This review has gone over the key points related to nanocomposites of Cu2O with the plasmonic metals Au and Ag. These materials have been used in conjunction with each other to attenuate the drawbacks of the individual components and to take advantage of the positive effects that arise from compositing. The benefits include the suppression of electron–hole recombination through the formation of a Schottky barrier at the PM–Cu2O interface, the expanded light absorbance range, and the increased electron–hole pair generation rate resulting from plasmonic effects, which can be manipulated through precise control of size, morphology, and shell thickness. Various configurations have been discussed along with their potential impact on the photocatalytic properties of the resulting materials. Included here are core–satellite, core–shell, yolk–shell, Janus, dumbbell, and ternary structures. These structures have been obtained using a plethora of synthesis techniques, and the tuning points of these techniques have been detailed as well.The potential use of PM–Cu2O nanocomposites in photocatalysis was explored in depth in this review, citing numerous studies that have been published over the last decade. Several mechanisms have been proposed to explain the observed enhancement in photocatalytic behavior, but further research is needed to build on the current knowledge base. A deeper understanding of the various possible contributing mechanisms and the interplay between them can aid us better in designing photocatalyst systems that can fully maximize the enhancement effects. It must also be noted that much of the current research on the photocatalytic applications of PM–Cu2O nanocomposites has delved mainly into the degradation of organic dyes. There are other important photocatalytic reactions, such as hydrogen production from water and reduction of CO2, but literature reports on these are scarce for photocatalytic PM–Cu2O systems. Moreover, recent reports have shown that aside from photocatalysis, PM–Cu2O nanocomposites are similarly promising in electrocatalysis,45,82 sonocatalysis,113 organic transformation catalysis,114 and borohydride-mediated catalysis,59,61 but the mechanisms behind their favorable performance in these applications have yet to be thoroughly examined. In addition, there have been a couple of studies showing that these composite materials exhibit the photothermal effect,57,94 but their potential in photothermal catalysis is a topic that is overlooked and needs to be taken into consideration when studying their photo-activated catalytic activity. All these present a possible road forward with regard to research on PM–Cu2O nanocomposites. It would be interesting to know how the different PM–Cu2O hybrid designs discussed in Section 2 can be utilized for these different types of catalytic applications. Emerging hybrid designs, such as the inter-embedded heterostructure configuration,115 should be explored as well.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the University of the Philippines Enhanced Creative Work and Research grant (ECWRG-2020-2-15R) for funding support.References
- I. Ali, M. Suhail, Z. A. Alothman and A. Alwarthan, RSC Adv., 2018, 8, 30125–30147 RSC.
- M. S. S. Danish, L. L. Estrella, I. M. A. Alemaida, A. Lisin, N. Moiseev, M. Ahmadi, M. Nazari, M. Wali, H. Zaheb and T. Senjyu, Metals, 2021, 11, 80 CrossRef CAS.
- X. Lu, M. Rycenga, S. E. Skrabalak, B. Wiley and Y. Xia, Annu. Rev. Phys. Chem., 2009, 60, 167–192 CrossRef CAS PubMed.
- H. Kang, J. T. Buchman, R. S. Rodriguez, H. L. Ring, J. He, K. C. Bantz and C. L. Haynes, Chem. Rev., 2019, 119, 664–699 CrossRef CAS PubMed.
- S. K. Dutta, S. K. Mehetor and N. Pradhan, J. Phys. Chem. Lett., 2015, 6, 936–944 CrossRef CAS PubMed.
- Y. Fu, J. Li and J. Li, Nanomaterials, 2019, 9, 359 CrossRef CAS PubMed.
- S. Liu, M. D. Regulacio, S. Y. Tee, Y. W. Khin, C. P. Teng, L. D. Koh, G. Guan and M.-Y. Han, Chem. Rec., 2016, 16, 1965–1990 CrossRef CAS PubMed.
- N. Wu, Nanoscale, 2018, 10, 2679–2696 RSC.
- A. B. Djurišić, Y. H. Leung and A. M. Ching Ng, Mater. Horiz., 2014, 1, 400 RSC.
- K. Y. Tang, J. X. Chen, E. D. R. Legaspi, C. Owh, M. Lin, I. S. Y. Tee, D. Kai, X. J. Loh, Z. Li, M. D. Regulacio and E. Ye, Chemosphere, 2021, 265, 129114 CrossRef CAS PubMed.
- J. Z. X. Heng, K. Y. Tang, M. D. Regulacio, M. Lin, X. J. Loh, Z. Li and E. Ye, Nanomaterials, 2021, 11, 856 CrossRef CAS PubMed.
- M. M. Abouelela, G. Kawamura and A. Matsuda, J. Cleaner Prod., 2021, 294, 126200 CrossRef CAS.
- Y. Ben-Shahar, D. Stone and U. Banin, Chem. Rev., 2023, 123, 3790–3851 CrossRef CAS PubMed.
- Y. Lv, S. Duan and R. Wang, Prog. Nat. Sci.: Mater. Int., 2020, 30, 1–12 CrossRef CAS.
- S. Sun, X. Zhang, Q. Yang, S. Liang, X. Zhang and Z. Yang, Prog. Mater. Sci., 2018, 96, 111–173 CrossRef CAS.
- F. Mohammadparast, S. B. Ramakrishnan, N. Khatri, R. T. A. Tirumala, S. Tan, A. K. Kalkan and M. Andiappan, ACS Appl. Nano Mater., 2020, 3, 6806–6815 CrossRef CAS.
- W.-H. Ke, C.-F. Hsia, Y.-J. Chen and M. H. Huang, Small, 2016, 12, 3530–3534 CrossRef CAS PubMed.
- S. Thoka, A.-T. Lee and M. H. Huang, ACS Sustainable Chem. Eng., 2019, 7, 10467–10476 CrossRef CAS.
- B. A. Koiki and O. A. Arotiba, RSC Adv., 2020, 10, 36514–36525 RSC.
- Q. Su, C. Zuo, M. Liu and X. Tai, Molecules, 2023, 28, 5576 CrossRef CAS PubMed.
- A. Kumar Sahu, M. Pokhriyal, S. Upadhyayula and X. S. Zhao, J. Phys. Chem. C, 2022, 126, 13094–13104 CrossRef CAS.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- Z. Starowicz, R. Wojnarowska-Nowak, P. Ozga and E. M. Sheregii, Colloid Polym. Sci., 2018, 296, 1029–1037 CrossRef CAS PubMed.
- M. Hu, J. Chen, Z.-Y. Li, L. Au, G. V. Hartland, X. Li, M. Marquez and Y. Xia, Chem. Soc. Rev., 2006, 35, 1084 RSC.
- M. Rycenga, C. M. Cobley, J. Zeng, W. Li, C. H. Moran, Q. Zhang, D. Qin and Y. Xia, Chem. Rev., 2011, 111, 3669–3712 CrossRef CAS PubMed.
- C. L. Nehl and J. H. Hafner, J. Mater. Chem., 2008, 18, 2415 RSC.
- F. Hao, C. L. Nehl, J. H. Hafner and P. Nordlander, Nano Lett., 2007, 7, 729–732 CrossRef CAS PubMed.
- N. G. Bastús, J. Piella and V. Puntes, Langmuir, 2016, 32, 290–300 CrossRef PubMed.
- M. A. Mahmoud and M. A. El-Sayed, J. Phys. Chem. Lett., 2013, 4, 1541–1545 CrossRef CAS PubMed.
- L. C. Kennedy, L. R. Bickford, N. A. Lewinski, A. J. Coughlin, Y. Hu, E. S. Day, J. L. West and R. A. Drezek, Small, 2011, 7, 169–183 CrossRef CAS PubMed.
- S. J. Amina and B. Guo, Int. J. Nanomed., 2020, 15, 9823–9857 CrossRef CAS PubMed.
- K. E. Fong and L.-Y. L. Yung, Nanoscale, 2013, 5, 12043 RSC.
- P. V. AshaRani, G. Low Kah Mun, M. P. Hande and S. Valiyaveettil, ACS Nano, 2009, 3, 279–290 CrossRef CAS PubMed.
- J. L. Elechiguerra, L. Larios-Lopez, C. Liu, D. Garcia-Gutierrez, A. Camacho-Bragado and M. J. Yacaman, Chem. Mater., 2005, 17, 6042–6052 CrossRef CAS.
- L. V. A. Sayson and M. D. Regulacio, ChemNanoMat, 2022, 8, e202200052 CrossRef CAS.
- D.-Y. Liu, S.-Y. Ding, H.-X. Lin, B.-J. Liu, Z.-Z. Ye, F.-R. Fan, B. Ren and Z.-Q. Tian, J. Phys. Chem. C, 2012, 116, 4477–4483 CrossRef CAS.
- C. Zhao, H. Fu, X. Yang, S. Xiong, D. Han and X. An, Appl. Surf. Sci., 2021, 545, 149014 CrossRef CAS.
- H. Qin, Q. Wei, J. Wu, F. Yang, B. Zhou, Y. Wang and S. Tian, Mater. Chem. Phys., 2019, 232, 240–245 CrossRef CAS.
- Z. Wang, S. Zhao, S. Zhu, Y. Sun and M. Fang, CrystEngComm, 2011, 13, 2262 RSC.
- G.-Z. Yuan, C.-F. Hsia, Z.-W. Lin, C. Chiang, Y.-W. Chiang and M. H. Huang, Chem.–Eur. J., 2016, 22, 12548–12556 CrossRef CAS PubMed.
- H. Luo, J. Zhou, H. Zhong, L. Zhou, Z. Jia and X. Tan, RSC Adv., 2016, 6, 99105–99113 RSC.
- M. D. Susman, R. Popovitz-Biro, A. Vaskevich and I. Rubinstein, Small, 2015, 11, 3942–3953 CrossRef CAS PubMed.
- J. Li, L. Sun, Y. Yan and Z. Zhu, Micro Nano Lett., 2016, 11, 363–365 CrossRef CAS.
- D. Kovács, A. Deák, G. Z. Radnóczi, Z. E. Horváth, A. Sulyok, R. Schiller, O. Czömpöly and D. Zámbó, J. Mater. Chem. C, 2023, 11, 8796–8807 RSC.
- A. Zhang, J. Wu, L. Xue, C. Li, S. Zeng, D. Caracciolo, S. Wang and C.-J. Zhong, ACS Appl. Mater. Interfaces, 2021, 13, 46577–46587 CrossRef CAS PubMed.
- J. W. Hong, D. H. Wi, S.-U. Lee and S. W. Han, J. Am. Chem. Soc., 2016, 138, 15766–15773 CrossRef CAS PubMed.
- T. Hirakawa and P. V. Kamat, J. Am. Chem. Soc., 2005, 127, 3928–3934 CrossRef CAS PubMed.
- J. Liu and J. Zhang, Chem. Rev., 2020, 120, 2123–2170 CrossRef CAS PubMed.
- H. Jing, N. Large, Q. Zhang and H. Wang, J. Phys. Chem. C, 2014, 118, 19948–19963 CrossRef CAS.
- C.-H. Kuo, T.-E. Hua and M. H. Huang, J. Am. Chem. Soc., 2009, 131, 17871–17878 CrossRef CAS PubMed.
- M. Torras and A. Roig, Cryst. Growth Des., 2021, 21, 5027–5035 CrossRef CAS.
- S. M. Majhi, P. Rai, S. Raj, B.-S. Chon, K.-K. Park and Y.-T. Yu, ACS Appl. Mater. Interfaces, 2014, 6, 7491–7497 CrossRef CAS PubMed.
- L. Kong, W. Chen, D. Ma, Y. Yang, S. Liu and S. Huang, J. Mater. Chem., 2012, 22, 719–724 RSC.
- C.-H. Kuo, Y.-C. Yang, S. Gwo and M. H. Huang, J. Am. Chem. Soc., 2011, 133, 1052–1057 CrossRef CAS PubMed.
- M. H. Huang, S. Rej and C.-Y. Chiu, Small, 2015, 11, 2716–2726 CrossRef CAS PubMed.
- Y.-C. Yang, H.-J. Wang, J. Whang, J.-S. Huang, L.-M. Lyu, P.-H. Lin, S. Gwo and M. H. Huang, Nanoscale, 2014, 6, 4316 RSC.
- H.-J. Wang, K.-H. Yang, S.-C. Hsu and M. H. Huang, Nanoscale, 2016, 8, 965–972 RSC.
- S. Zhu, D. Deng, M. T. Nguyen, Y. R. Chau, C.-Y. Wen and T. Yonezawa, Langmuir, 2020, 36, 3386–3392 CrossRef CAS PubMed.
- E. D. R. Legaspi, M. D. Regulacio, L. A. E. Pineda, L. V. A. Sayson, W. Jiang, J. Z. X. Heng, W. Wu and E. Ye, ChemistrySelect, 2023, 8, e202300904 CrossRef CAS.
- W.-C. Wang, L.-M. Lyu and M. H. Huang, Chem. Mater., 2011, 23, 2677–2684 CrossRef CAS.
- Y.-Q. Dou, T.-S. Deng, Q. Zhang, X. Zhao, J. Liu and Z. Cheng, J. Mater. Sci., 2023, 58, 7583–7593 CrossRef CAS.
- L. Chen, Y. Zhao, Y. Zhang, M. Liu, Y. Wang, X. Qu, Y. Liu, J. Li, X. Liu and J. Yang, Colloids Surf., A, 2016, 507, 96–102 CrossRef CAS.
- C. Lee, K. Shin, Y. J. Lee, C. Jung and H. M. Lee, Catal. Today, 2018, 303, 313–319 CrossRef CAS.
- H. Feng, W. Wang, W. Wang, M. Zhang, C. Wang, C. Ma, W. Li and S. Chen, J. Colloid Interface Sci., 2021, 601, 531–543 CrossRef CAS PubMed.
- S. Yan, Q. Yue and J. Ma, CrystEngComm, 2021, 23, 24–29 RSC.
- J. Xiong, Z. Li, J. Chen, S. Zhang, L. Wang and S. Dou, ACS Appl. Mater. Interfaces, 2014, 6, 15716–15725 CrossRef CAS PubMed.
- J. Li, S. K. Cushing, J. Bright, F. Meng, T. R. Senty, P. Zheng, A. D. Bristow and N. Wu, ACS Catal., 2013, 3, 47–51 CrossRef CAS.
- J. Zou, W. Song, W. Xie, B. Huang, H. Yang and Z. Luo, Nanotechnology, 2018, 29, 115703 CrossRef PubMed.
- R. Bakthavatsalam and J. Kundu, CrystEngComm, 2017, 19, 1669–1679 RSC.
- W. Zhang, X. Yang, Q. Zhu, K. Wang, J. Lu, M. Chen and Z. Yang, Ind. Eng. Chem. Res., 2014, 53, 16316–16323 CrossRef CAS.
- Z. Yang, C. Ma, W. Wang, M. Zhang, X. Hao and S. Chen, J. Colloid Interface Sci., 2019, 557, 156–167 CrossRef CAS PubMed.
- R. Ji, W. Sun and Y. Chu, RSC Adv., 2014, 4, 6055 RSC.
- C. Lee, N. R. Kim, J. Koo, Y. J. Lee and H. M. Lee, Nanotechnology, 2015, 26, 455601 CrossRef PubMed.
- Y.-J. Zhu and F. Chen, Chem. Rev., 2014, 114, 6462–6555 CrossRef CAS PubMed.
- H. Cheng, C. Wang, D. Qin and Y. Xia, Acc. Chem. Res., 2023, 56, 900–909 CrossRef CAS PubMed.
- M. Pang, Q. Wang and H. C. Zeng, Chem.–Eur. J., 2012, 18, 14605–14609 CrossRef CAS PubMed.
- X. Sun, J. Han and R. Guo, Front. Chem., 2020, 8, 606044 CrossRef CAS PubMed.
- B.-B. Zhang, Y.-H. Wang, S.-M. Xu, K. Chen, Y.-G. Yang and Q.-H. Kong, RSC Adv., 2020, 10, 19192–19198 RSC.
- M. Xiao, Z. Wang, M. Lyu, B. Luo, S. Wang, G. Liu, H. Cheng and L. Wang, Adv. Mater., 2019, 31, 1801369 CrossRef PubMed.
- L. Zhang, D. A. Blom and H. Wang, Chem. Mater., 2011, 23, 4587–4598 CrossRef CAS.
- Y. Ma, X. Liu, X. Wei, J. Le, Y. Fu, Q. Han, H. Ji, Z. Yang and H. Wu, Langmuir, 2021, 37, 4578–4586 CrossRef CAS PubMed.
- L. Xiong, X. Zhang, L. Chen, Z. Deng, S. Han, Y. Chen, J. Zhong, H. Sun, Y. Lian, B. Yang, X. Yuan, H. Yu, Y. Liu, X. Yang, J. Guo, M. H. Rümmeli, Y. Jiao and Y. Peng, Adv. Mater., 2021, 33, 2101741 CrossRef CAS PubMed.
- Y. Lu, H. S. Kang, Y. S. Lim, B. C. Lee, S.-H. Kim and L. Piao, Bull. Korean Chem. Soc., 2015, 36, 2150–2153 CrossRef CAS.
- R. Chen, J. Lu, S. Liu, M. Zheng and Z. Wang, J. Mater. Sci., 2018, 53, 1781–1790 CrossRef CAS.
- L. Zhang and H. Wang, ACS Nano, 2011, 5, 3257–3267 CrossRef CAS PubMed.
- N. Safaie and R. C. Ferrier, J. Appl. Phys., 2020, 127, 170902 CrossRef.
- W. Xu, J. Jia, T. Wang, C. Li, B. He, J. Zong, Y. Wang, H. J. Fan, H. Xu, Y. Feng and H. Chen, Angew. Chem., Int. Ed., 2020, 59, 22246–22251 CrossRef CAS PubMed.
- B. Dai, Y. Zhou, X. Xiao, Y. Chen, J. Guo, C. Gao, Y. Xie and J. Chen, Advanced Science, 2022, 9, 2203057 CrossRef CAS PubMed.
- J. Qiu, Q. N. Nguyen, Z. Lyu, Q. Wang and Y. Xia, Adv. Mater., 2022, 34, 2102591 CrossRef CAS PubMed.
- A. Gale-Mouldey, E. Jorgenson, J. P. Coyle, D. Prezgot and A. Ianoul, J. Mater. Chem. C, 2020, 8, 1852–1863 RSC.
- M. Wang, A. Hoff, J. E. Doebler, S. R. Emory and Y. Bao, Langmuir, 2019, 35, 16886–16892 CrossRef CAS PubMed.
- W. Xu, R. Xiao, S. An, C. Li, J. Ding, H. Chen, H. B. Yang and Y. Feng, Small, 2023, 19, 2300587 CrossRef CAS PubMed.
- Z. Hu, Y. Mi, Y. Ji, R. Wang, W. Zhou, X. Qiu, X. Liu, Z. Fang and X. Wu, Nanoscale, 2019, 11, 16445–16454 RSC.
- K.-H. Yang, S.-C. Hsu and M. H. Huang, Chem. Mater., 2016, 28, 5140–5146 CrossRef CAS.
- L. Chen, M. Liu, Y. Zhao, Q. Kou, Y. Wang, Y. Liu, Y. Zhang, J. Yang and Y. M. Jung, Appl. Surf. Sci., 2018, 435, 72–78 CrossRef CAS.
- T. Wu, H. Zheng, Y. Kou, X. Su, N. R. Kadasala, M. Gao, L. Chen, D. Han, Y. Liu and J. Yang, Microsyst. Nanoeng., 2021, 7, 23 CrossRef CAS PubMed.
- T. Wu, Y. Kou, H. Zheng, J. Lu, N. R. Kadasala, S. Yang, C. Guo, Y. Liu and M. Gao, Nanomaterials, 2019, 10, 48 CrossRef PubMed.
- B. Dai, J. Guo, C. Gao, H. Yin, Y. Xie and Z. Lin, Adv. Mater., 2023, 35, 2210914 CrossRef CAS PubMed.
- B. Dai, J. Fang, Y. Yu, M. Sun, H. Huang, C. Lu, J. Kou, Y. Zhao and Z. Xu, Adv. Mater., 2020, 32, 1906361 CrossRef CAS PubMed.
- A. Ayati, A. Ahmadpour, F. F. Bamoharram, B. Tanhaei, M. Mänttäri and M. Sillanpää, Chemosphere, 2014, 107, 163–174 CrossRef CAS PubMed.
- M. R. Khan, T. W. Chuan, A. Yousuf, M. N. K. Chowdhury and C. K. Cheng, Catal. Sci. Technol., 2015, 5, 2522–2531 RSC.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- Y. Wang, J. Zhang, W. Liang, H. Yang, T. Guan, B. Zhao, Y. Sun, L. Chi and L. Jiang, CCS Chem., 2022, 4, 1153–1168 CrossRef CAS.
- C. Clavero, Nat. Photonics, 2014, 8, 95–103 CrossRef CAS.
- M. Thangamuthu, T. V. Raziman, O. J. F. Martin and J. Tang, J. Electrochem. Soc., 2022, 169, 036512 CrossRef CAS.
- P. Anger, P. Bharadwaj and L. Novotny, Phys. Rev. Lett., 2006, 96, 113002 CrossRef PubMed.
- S. K. Cushing, J. Li, F. Meng, T. R. Senty, S. Suri, M. Zhi, M. Li, A. D. Bristow and N. Wu, J. Am. Chem. Soc., 2012, 134, 15033–15041 CrossRef CAS PubMed.
- J. Li, S. K. Cushing, F. Meng, T. R. Senty, A. D. Bristow and N. Wu, Nat. Photonics, 2015, 9, 601–607 CrossRef CAS.
- A. Amirjani, N. B. Amlashi and Z. S. Ahmadiani, ACS Appl. Nano Mater., 2023, 6, 9085–9123 CrossRef CAS.
- P. K. Jain, K. S. Lee, I. H. El-Sayed and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 7238–7248 CrossRef CAS PubMed.
- A. Tcherniak, J. W. Ha, S. Dominguez-Medina, L. S. Slaughter and S. Link, Nano Lett., 2010, 10, 1398–1404 CrossRef CAS PubMed.
- J.-Y. Ho and M. H. Huang, J. Phys. Chem. C, 2009, 113, 14159–14164 CrossRef CAS.
- S. Yadav, M. Chauhan, M. Jacob and P. Malhotra, New J. Chem., 2022, 46, 9685–9694 RSC.
- G. Singh, M. Kumar and V. Bhalla, ACS Sustainable Chem. Eng., 2018, 6, 11466–11472 CrossRef CAS.
- B. Ma, J. Bi, J. Lv, C. Kong, P. Yan, X. Zhao, X. Zhang, T. Yang and Z. Yang, Chem. Eng. J., 2021, 405, 126709 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |