
Super-resolution imaging of linearized chromatin in tunable nanochannels†
Ji-Hoon
Lee‡
 ab,
Joyce Han-Ching
Chiu‡
ab,
Nicholas J.
Ginga
c,
Tasdiq
Ahmed
ab,
M. D.
Thouless
d,
Yifan
Liu
*e and
Shuichi
Takayama
*ab
ab,
Joyce Han-Ching
Chiu‡
ab,
Nicholas J.
Ginga
c,
Tasdiq
Ahmed
ab,
M. D.
Thouless
d,
Yifan
Liu
*e and
Shuichi
Takayama
*ab
aWallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University School of Medicine, Atlanta, GA 30332, USA. E-mail: takayama@gatech.edu
bThe Parker H. Petit Institute of Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, GA 30332, USA
cDepartment of Mechanical and Aerospace Engineering, University of Alabama in Huntsville, Huntsville, AL 35899, USA
dDepartment of Mechanical Engineering and Department of Materials Science and Engineering, University of Michigan, Ann Arbor, MI 48109, USA
eDepartment of Biochemistry and Molecular Medicine, University of Southern California Keck School of Medicine, Los Angeles, CA 90033, USA. E-mail: yliu3636@usc.edu
First published on 24th May 2023
Abstract
Nanofluidic linearization and optical mapping of naked DNA have been reported in the research literature, and implemented in commercial instruments. However, the resolution with which DNA features can be resolved is still inherently limited by both Brownian motion and diffraction-limited optics. Direct analysis of native chromatin is further hampered by difficulty in electrophoretic manipulation, which is routinely used for DNA analysis. This paper describes the development of a three-layer, tunable, nanochannel system that enables non-electrophoretic linearization and immobilization of native chromatin. Furthermore, through careful selection of self-blinking fluorescent dyes and the design of the nanochannel system, we achieve direct stochastic optical reconstruction microscopy (dSTORM) super-resolution imaging of the linearized chromatin. As an initial demonstration, rDNA chromatin extracted from Tetrahymena is analyzed by multi-color imaging of total DNA, newly synthesized DNA, and newly synthesized histone H3. Our analysis reveals a relatively even distribution of newly synthesized H3 across two halves of the rDNA chromatin with palindromic symmetry, supporting dispersive nucleosome segregation. As a proof-of-concept study, our work achieves super-resolution imaging of native chromatin fibers linearized and immobilized in tunable nanochannels. It opens up a new avenue for collecting long-range and high-resolution epigenetic information as well as genetic information.
New conceptsWe report on the use of nano-thin h-PDMS films to fabricate normally-closed, tunable nanochannels for multi-color super-resolution imaging of native chromatin fiber via direct stochastic optical reconstruction microcopy (dSTORM). Currently, most nanofluidic-based systems are developed for DNA linearization. Chromatin linearization is intrinsically more challenging, due to complex interactions among its protein and DNA components. Chromatin fibers are also more difficult to load into nanochannels by electrophoresis, due to much reduced net charges compared to naked DNA molecules. The key achievement of this work that the collapsed nanochannel system not only efficiently linearizes, but can also immobilize native chromatin fibers and sufficiently eliminate Brownian motion to enable sub-diffraction optical resolution. Immobilization is critical as dSTORM requires iterative imaging of the same chromatin fiber multiple times with different dye molecules activated stochastically between images. We successfully demonstrate long-range epigenetic information analysis at the individual chromatin fiber level, providing evidence of dispersive histone inheritance from mother to daughter chromatin during DNA replication. In short, our novel approach based on tunable nanochannels provides an efficient means of studying of epigenetic mechanisms at single chromatin level in its native environment. |
Introduction
In the eukaryotic nucleus, DNA is hierarchically organized into chromatin. The basic structural unit of chromatin is the nucleosome, in which 147 bp of DNA is wrapped around core histones.1 Myriad enzymatic modifications of these histones serve as important epigenetic mechanisms for regulating genetic activity.2 Significant progress over the past 20 years in mapping epigenetic marker distribution has been achieved with techniques such as chromatin immunoprecipitation (ChIP), where antibodies targeting histones or modifying enzymes are used to isolate short (∼500 bp) chromatin fragments, thus identifying marker presence.3 Emerging from these analyses is a paradigm: specific epigenetic marks are often strategically arrayed at key genomic landmarks, including transcriptional enhancers and promoters.4 However, this paradigm is derived from the synthesis of information at the ensemble level. As critical long-range information concerning the connectivity between neighboring epigenetic marks is not preserved, distribution patterns at the single molecule level remain elusive.Native chromatin fibers can be linearized and imaged, providing an alternative for mapping their associated epigenetic marks at the single-molecule level and over the long range.5,6 A conventional approach for biopolymer linearization is molecular combing. DNA molecules can be uniformly stretched by meniscus retraction, which is routinely performed by commercially available DNA combing systems. It has been adapted for stretching chromatin fibers, though with less consistency.3,4 The minimum length of chromatin needed for linearization by meniscus retraction, as determined by the evaporation rate, is tens of kilobase pairs (kbp), severely limiting the sample range.7 Though high-throughput in theory, analysis of DNA/chromatin combing data is slow and labor-intensive in practice. Other strategies to linearize DNA and chromatin include end-tethering to beads, and stretching with optical or magnetic tweezers. These have been used to investigate the mechanical properties and structures of DNA and chromatin.8–10 However, these methods are also low-throughput in nature, and the set-up necessary to implement these techniques poses limitations on imaging.
Alternatively, DNA and chromatin fibers can be linearized by using nanochannels.7,11–15 Through self-avoidance interactions, polymers will stretch out when confined in space below their persistence lengths.16,17 The nanoconfinement technique has long been employed in DNA mapping,18–21 but has yet to be widely tested for chromatin stretching.5,22 The reduced net charge on the chromatin, where the negatively-charged DNA is neutralized by the positively-charged histones, precludes electrophoretic loading of the nanochannel array with chromatin fibers, for which less efficient hydraulic loading is often used. Chromatin fibers are also stickier than naked DNA posing a challenge for linearization by nanoconfinement. State-of-the-art nanochannel arrays, such as those commercially available from Bionano Genomics inc., offer high-throughput and reproducible optical mapping of megabase pair (Mbp) lengths of DNA.21–33 However, epigenetic marks on adjacent nucleosomes can only be resolved by sub-diffraction super-resolution imaging (e.g., the expected 20–30 nm lateral resolution of direct stochastic optical reconstruction microscopy: dSTORM). Moreover, such confinement methods present challenges for coordinate-stochastic super-resolution microscopy, as Brownian motion of biopolymers within the nanochannels interferes with the calculation of molecular positions.6
We have previously demonstrated a high degree of control over nanochannel localization and cross-sectional size through fracture-based fabrication of high-density nanochannel arrays.34,35 Recently, we improved on the previous work,13 by developing tunable, normally-closed nanochannels that are fabricated in thin films of hard polydimethylsiloxane (h-PDMS) with nanoscale thickness. Previous demonstrations of h-PDMS nanochannels were restricted to h-PDMS films with a micron-scale thickness owing to limitations in the fabrication process. Our h-PDMS with nanoscale thickness permits tuning of the nanochannel physical dimensions (i.e., width and height) such that native chromatin fibers can now be successfully linearized and immobilized, providing opportunities for epigenomic analysis with super-resolution microscopy (Fig. 1). However, before these h-PDMS nanochannels could be used with super-resolution microscopy, the inherent oxygen permeability of PDMS material had to be considered, because oxidation of fluorescent dyes quenches triplet states and increases the risk of photobleaching,36,37 thereby limiting dSTORM effectiveness while using h-PDMS/PDMS devices. This was overcome by first screening a panel of self-blinking dyes to obtain optimal combinations compatible with our h-PDMS nanochannel device. This allowed multi-color super-resolution imaging of linearized Tetrahymena rDNA mini-chromosomes. Based on the Fourier ring correlation (FRC) method,38 the achievable lateral resolution was computed to be ∼20 nm, which is in line with the expected dSTORM resolution. The super-resolution images revealed distributions of newly-synthesized histones and both newly-synthesized and total DNA along the ∼20 kbp chromatin fiber stretched to its contour length. The results shed light on the mode of histone transmission during replication. The following sections of this paper provide details for this approach, which achieves high-throughput linearization and super-resolution imaging of native chromatin fibers with h-PDMS tunable nanochannels. Our proof-of-concept study provides a novel alternative for collecting long-range epigenetic information at the single-molecule level.
 | ||
| Fig. 1 Graphical illustration of the workflow of using tunable nanochannels for super-resolution imaging of linearized native chromatin. (A) The use of soft lithography methods to fabricate h-PDMS-based tunable nanochannels. (B) Using an automated stretcher system to effectively control widths of nanochannels. (C) Linearization and immobilization of chromatin fibers in tunable nanochannels. (D) dSTORM imaging of linearized chromatin fibers in closed nanochannels. | ||
Results and discussion
Overview
Fig. 1 shows the overall workflow of using h-PDMS nanochannels for linearization and super-resolution imaging of native chromatin fibers. First, using an automated stretcher system that we recently developed,39 the h-PDMS device was stretched uniaxially to initiate cracks for suitable nanochannels. Subsequently, the automated stretcher allowed these crack-based nanochannels to be opened to appropriate widths. Then, fluorescently-labelled chromatin fibers extracted from Tetrahymena were loaded into the microfluidic inlet channels feeding into the open nanochannels. The chromatin fibers entered the open nanochannels through capillary flow. Next, the applied strain from the automated stretcher was slowly removed to progressively narrow the nanochannels, until they were fully closed. Note that it has been previously shown that step-wise closing (with a manual stretching device) of the nanochannels yields greater linearization.13 Our automated stretcher system39 has the ability to take advantage of this phenomenon with much finer control over relevant parameters, including unloading rate, compared to manual stretcher systems previously used. In the small conduits formed when the channel diameters fall below the persistence length of DNA/chromatin, nanoconfinement restricts the conformational degrees of freedom and favors the stretched conformation. Chromatin fibers were, therefore, linearized to near their full contour lengths in the nanochannels and eventually immobilized. This approach with the tunable nanochannel device allowed super-resolution imaging by dSTORM.Fabrication and optimization of h-PDMS nanochannels
Fig. S1 in the ESI† illustrates the fabrication of the tunable h-PDMS crack-based nanochannels. Briefly, h-PDMS was diluted to a specific concentration determined by the desired nanoscale thickness, and then spin-coated onto a master mold. The master mold was composed of an SU-8 photoresist pattern on a silicon wafer containing micron-scale inlet and outlet channels that are unconnected until later in the fabrication process. After coating the master mold with h-PDMS, regular PDMS was cast on top of it, allowed to cure, and then peeled off from the master mold (Fig. 2A and B). Subsequently, the h-PDMS/PDMS complex was uniaxially stretched to an appropriate amount of strain to initiate the desired density of nanocracks in the h-PDMS (Fig. 2C), which then connected the inlet and outlet microchannels. Lastly, a thin PDMS membrane was plasma-bonded on top of the h-PDMS side of the pre-cracked device to create a layer that covers the nanocracks in the surface to form enclosed nanochannels (Fig. 2D). Determining optimal conditions for the nanochannel fabrication required iterations of design modifications (Fig. S2, ESI†) and a series of characterizations (Fig. S3–S5, ESI†). The notch patterns were implemented to provide improved control over the spatial arrangement of nanocracks. These notches shield intrinsic flaws in the material from the applied stress field, leaving only the flaws at their tips to be active and available to initiate a crack.35,40,41 The number of active flaws increases with strain, until there is a one-to-one match with the patterns. Empirical testing of different designs (Fig. S2A–C, ESI†) resulted in a final configuration containing notches on only one side, with a 25 μm pitch (Fig. S2D, ESI†). The cracks were initiated on the side with the notch patterns and then propagated to connect the inlet microchannel to the outlet microchannel and create a series of regularly spaced parallel nanochannels.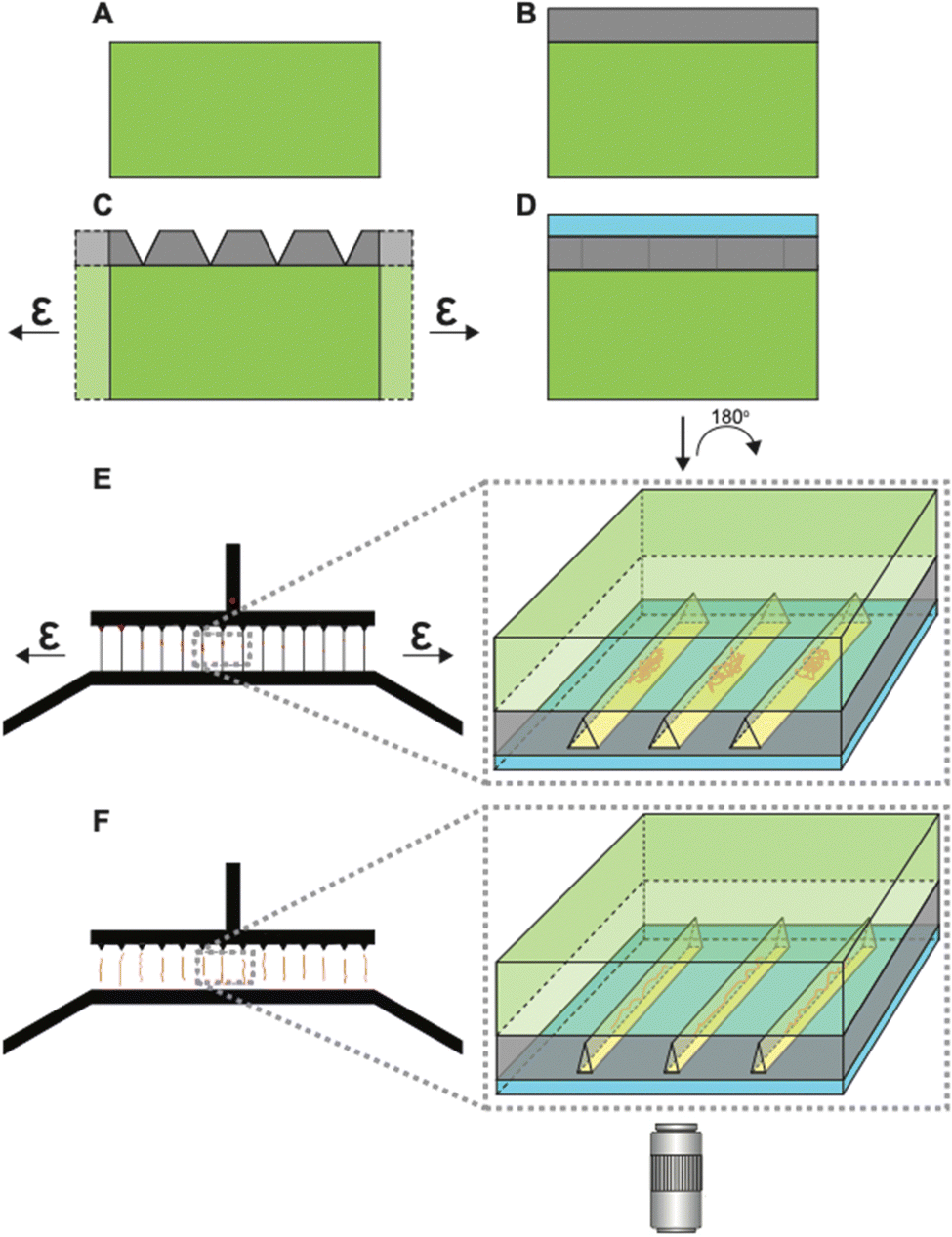 | ||
| Fig. 2 Tunable nanochannel schematics (not drawn to scale). (A) PDMS substrate. (B) Thin h-PDMS film layer on the PDMS substrate. (C) Application of strain generates cracks in the h-PDMS layer. (D) Sealing of the h-PDMS crack layer with a thin PDMS membrane creates enclosed nanochannels from the surface cracks. This complete device is flipped 180° for super-resolution microscopy. (E) Top-down view of microscale inlet and outlet channels connected by crack-based nanochannels. Applying strain opens the nanochannels, which allows coiled chromatin fibers to flow into open nanochannels. Inset shows the frontal section of the device showing open channels with coiled chromatin fibers. (F) Removing the strain causes closing of the channels, thereby linearizing trapped chromatin fibers to contour lengths. Inset shows a snapshot of closing channels with chromatin fibers being linearized. Linearized chromatin fibers are then imaged using dSTORM. | ||
The coating of h-PDMS on the SU-8 mold was found to be affected by the type of SU-8 used. Controlling the thickness of the h-PDMS film situated between the SU-8 patterns of the inlet and outlet microchannels was crucial for determining the nanocrack cross-section best suited for chromatin fiber uptake and linearization. When SU-8-2002 was used to fabricate the 2 μm-thick master mold patterns, the h-PDMS film did not have a uniform thickness, with the film being thickest near the inlet and outlet SU-8 patterns and becoming thinner at increasing distances from those patterns. The resulting h-PDMS film was thinnest at the halfway point between the inlet and outlet microchannels, displaying a concave surface profile (Fig. S3A and B, ESI†). Conversely, for the h-PDMS film coated on 20 μm-thick features fabricated with SU-8-2025, the opposite behavior was observed, with a convex surface profile (Fig. S3C and D, ESI†). Differing chemical formulations of various SU-8, leading to a difference in surface hydrophobicity, may provide explanations for such behaviors (Table S1, ESI†). In turn, these different thickness profiles affected the crack morphology and the dynamic behavior of crack opening and closing (Fig. S4, ESI†). When the SU-8-to-h-PDMS thickness ratio was low (i.e., concave h-PDMS), the cracks initiated from the inlet side at notches where the h-PDMS was thickest. Additionally, the cracks were wider near the ends and narrower in the mid-sections when opened (Fig. S4A, ESI†). The opposite was observed with convex h-PDMS films (Fig. S4B, ESI†), where the crack was the widest in the thicker mid-section of the cracks when opened. This was consistent with the conventional understanding of the mechanics of cracked layers.42 We postulate that the concave h-PDMS layer-generating nanocracks with wider openings (Fig. S3E and F, ESI†)-was more favorable for sample loading, and so the final systems were designed accordingly.
To further characterize the nanochannel cross-sections, the thickness of the h-PDMS and the crack depths were measured using atomic force microscopy (AFM) and a stage-compatible manual stretcher (Fig. S5A, ESI†). These measurements showed that there was a proportional relationship between the concentration of precursor h-PDMS and the thickness of the resulting film (Fig. S5B and Table S2, ESI†). This relationship was useful in designing nanochannels for linearization of DNA/chromatin. In addition to measuring the film thickness of the h-PDMS, it was also necessary to measure the depth of the nanocracks, since the cracks could potentially propagate into the bulk of the PDMS substrate (Fig. S5C and Table S3, ESI†). The data on crack depth as a function of applied strain also confirmed the normally-closed nature and tunability of the nanochannel width, ranging from fully closed to fully opened. While the crack depths were slightly deeper than the h-PDMS film thickness, the cracks generally stayed within the thin film (Table S2, ESI†). This observation is consistent with the previous analysis on the mechanical property difference between h-PDMS and bulk PDMS, such that the crack depth does not go into the bulk substrate significantly.43 From these AFM measurements of the crack depth (Fig. S5C and Table S2, ESI†), an h-PDMS concentration (%w/w) of ∼1.5%, corresponding to 300 nm-thick h-PDMS film, was found to be suitable for efficient chromatin linearization (Fig. S5D, ESI†), taking into account both the size of chromatin in its native coiled form and the penetration depth for dSTORM Z-resolution.
Next, the widths of the nanochannels were then controlled precisely by applying desired levels of strain with our automated stretcher system.39 Moreover, the complete nanochannel system was optimized for easy loading of chromatin fibers by capillary and hydrodynamic flow, instead of an electric field. Having a series of parallel and uniform nanochannels increased the probability of capturing chromatin fibers, and increased the potential throughput of the system. Low chromatin concentration ensured that no more than one chromatin fiber was trapped in each channel. Owing to the unique tunable feature of our stretcher-assisted system, closing the nanochannels allowed us to effectively confine, stretch, and immobilize the chromatin.
Since these crack-based nanochannels are fully closed when no strain is applied, they are classified as “normally-closed”. This contrasts with “normally-open” channels that are fully open when no strain is applied, and then close when a strain is applied and maintained. Previously demonstrated “normally-open” devices,34,44 including the CLiC scope45 require equipment to maintain the closed nature of the channel and linearization during the imaging process. An advantage of “normally-closed” nanochannels used here, is that they allow chromatin/DNA to be linearized using the stretching device, but then, once the strain from the device is removed, the chromatin/DNA remains immobilized in the closed channel, solving the thermal fluctuation issues.6 The h-PDMS/PDMS device can then be removed from the stretcher and mounted for super-resolution imaging. This eliminates concerns or restrictions of fitting a stretching device on the super-resolution microscope stage, and broadens potential applications.
Optimal dSTORM dye combination for h-PDMS system
The oxygen-permeable nature of PDMS, while advantageous for microfluidic cell cultures,36,46,47 is undesirable for imaging acquisition by dSTORM, which requires oxygen scavengers to remove oxygen in solution to prevent photobleaching and triplet state quenching.48 Moreover, multi-color imaging in dSTORM is often challenging and requires careful consideration and optimization of dyes.49,50 Therefore, matching an appropriate fluorophore and buffer system is crucial in PDMS-based devices. By trapping the fluorophores in the fluid pockets of oxidized PDMS (see ESI† for detailed information), we were able to recreate similar environment experienced by immobilized chromatin in closed nanochannels for dSTORM dye optimization. Briefly, widely used fluorescent dyes, Alexa Fluor 647 (AF 647) and CF 568, showed minimal dSTORM localizations in conventional dSTORM blinking buffers (Fig. 3B and C). When oxygen scavenging solution was supplemented to the blinking buffers, AF 647 and CF 568 showed immense improvement compared to blinking buffers alone (Fig. 3D and E), but with rapid photobleaching, the dyes were still impractical for proper multi-color imaging (Table S4, ESI†). Instead, self-blinking dyes SaraFluor 650B (SF650B, HMSiR), SaraFluor 488B (SF488B, HEtetTFER), FLIP565 and CAGE635 (data not shown) showed promising results in the oxygen-rich and low-buffer-volume environment of the PDMS nanochannels (Fig. 3F–H). Note that four of the dyes (AF 647, CF 568, HEtetTFER, CAGE 635) were tested in their unconjugated forms, while the two remaining dyes (HMSiR and FLIP 565) were pre-conjugated with secondary antibodies. SaraFluor, FLIP, and CAGE are rhodamine-based fluorophores. SaraFluor uses intramolecular spirocyclization to convert between triplet and ground states spontaneously,51,52 whereas FLIP requires UV activation,53 and the CAGE dye is released by photoactivation.54 The chemistry of these dyes allows them to avoid UV damage and remain shielded within the harsh environments of imaging buffers needed for coordinate-stochastic super-resolution imaging,55 and the dyes have been shown to be suitable for the nanochannel PDMS system.56 With appropriate combinations of the self-blinking dyes and target molecules (Table S4, ESI†), we were able to perform multi-color super-resolution imaging of linearized chromatin fibers. | ||
| Fig. 3 Dye and buffer selection for dSTORM imaging of chromatin fibers in PDMS nanochannels. Different combinations of fluorescent dyes and imaging buffers have been tested to optimize the labelling system. (A) Optical reconstruction by dSTORM relies on stochastic, continuous, and reversible activation of fluorophores, hence the need for dye blinking. (B) AF 647 in blinking buffer. (C) CF 568 in blinking buffer. (D) AF 647 in OxEA solution. (E) CF 568 in OxEA solution. (F) HMSiR-goat anti-mouse IgG in water. (G) FLIP 565-goat anti-rabbit IgG in water. (H) HEtetTFER in water. Regular fluorescent dyes (AF 647 and CF 568) showed poor localizations in either the blinking buffer or OxEA solution in the nanochannel PDMS device. Self-blinking dyes showed better results, and therefore were chosen for our labeling system. | ||
Linearization and immobilization of Tetrahymena chromatin fibers in h-PDMS nanochannels
Most prior studies have used reconstituted chromatin to demonstrate chromatin linearization capability,11,14 while a few have tested chromatin purified from HeLa cells, with minimal biological implications.12,13,15 Here, we chose to measure the Tetrahymena rDNA mini-chromosome as a standard reference (Fig. 4 and Fig. S6, ESI†). It is uniform in size (a palindromic dimer of 21 kb) (Fig. S7, ESI†), with consistent and well-characterized chromatin organization, and it is abundantly present in, and can be readily purified from, Tetrahymena macronuclei.57,58 Its small full contour length, estimated to be ∼3 μm, makes it very difficult to stretch by combing, but the full contour length is readily achievable by nanoconfinement.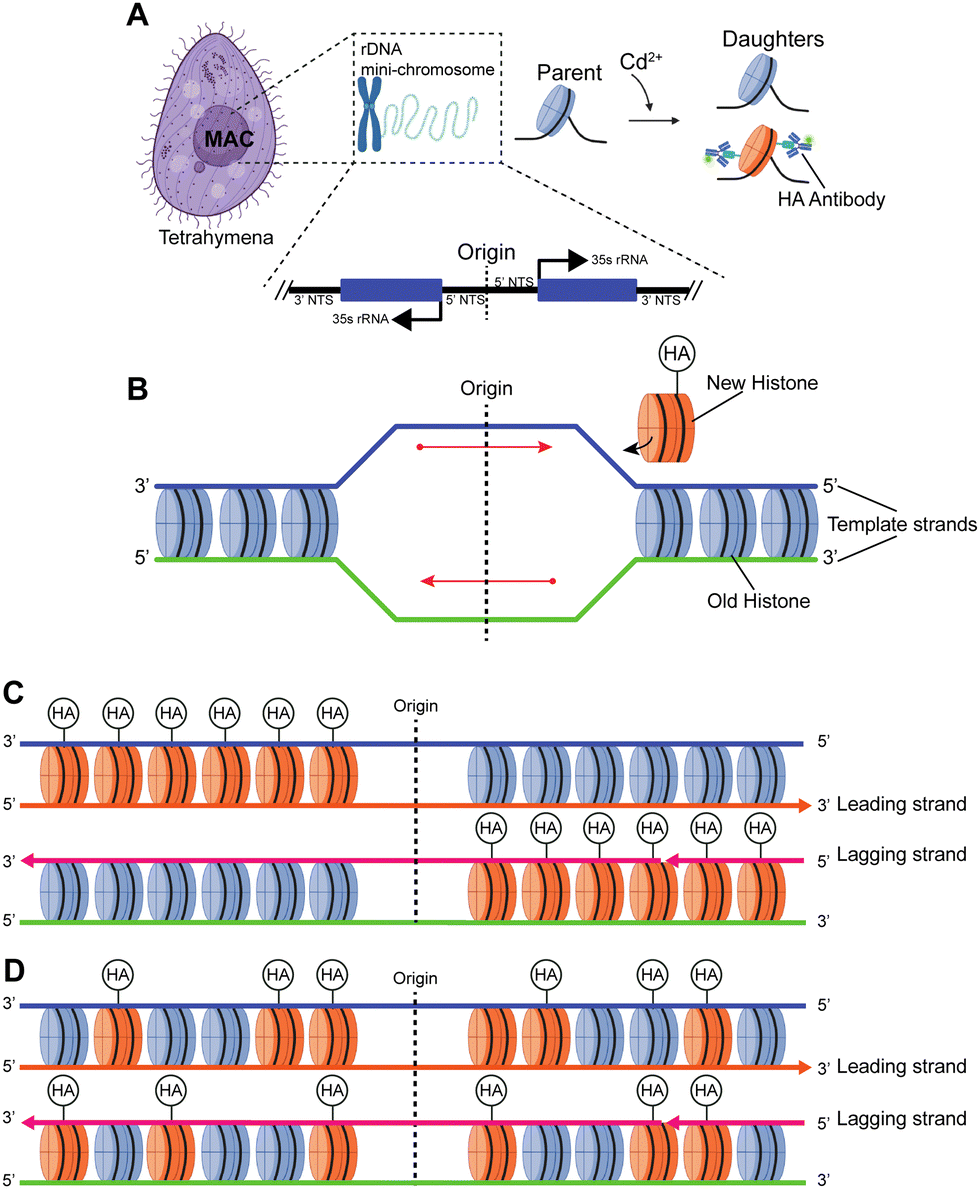 | ||
| Fig. 4 Studying histone transmission in the Tetrahymena model system. (A) The macronucleus (MAC) of Tetrahymena contains the rDNA mini-chromosome, which is a 21 kb palindromic dimer with centrally localized replication origins. Genetic engineering of Tetrahymena model allows Cd2+-inducible expression of hemagglutinin (HA)-tagged canonical H3. Labeling HA-H3 with fluorescent antibody allows efficient localization of newly synthesized histones. (B) DNA replication is initiated from the centrally localized replication origin, during which newly synthesized histones are added to two new daughter DNA molecules (detectable by BrdU incorporation). There are two alternative modes for histone transmission during replication: conservative (C) and dispersive transmission (D). (C) In the conservative transmission of histones, distributions of old and new histones on leading and lagging strands of DNA replication are biased, leading to asymmetrically distributed HA-H3 on the two halves of the palindromic rDNA chromosome. (D) In the dispersive transmission of histones, distributions of old and new histones on leading and lagging strands of DNA replication are NOT biased, leading to symmetrically distributed HA-H3 on the two halves of the palindromic rDNA chromosome. | ||
Fig. 5A demonstrates how trapped chromatin in a narrow fixed-dimension conduit can still wiggle around. Significant movements were observed within just 10 seconds, and as dSTORM requires thousands of image acquisitions over a few minutes, such mobility prevents localizing fluorophores for image reconstruction (Fig. 5B). The situation only worsens in multi-color imaging, as different color channels are imaged sequentially and must be perfectly aligned. In our h-PDMS-based nanochannels, the closing of the nanochannels eliminated the movement of trapped chromatin fibers, solving these critical problems (Fig. 5C).
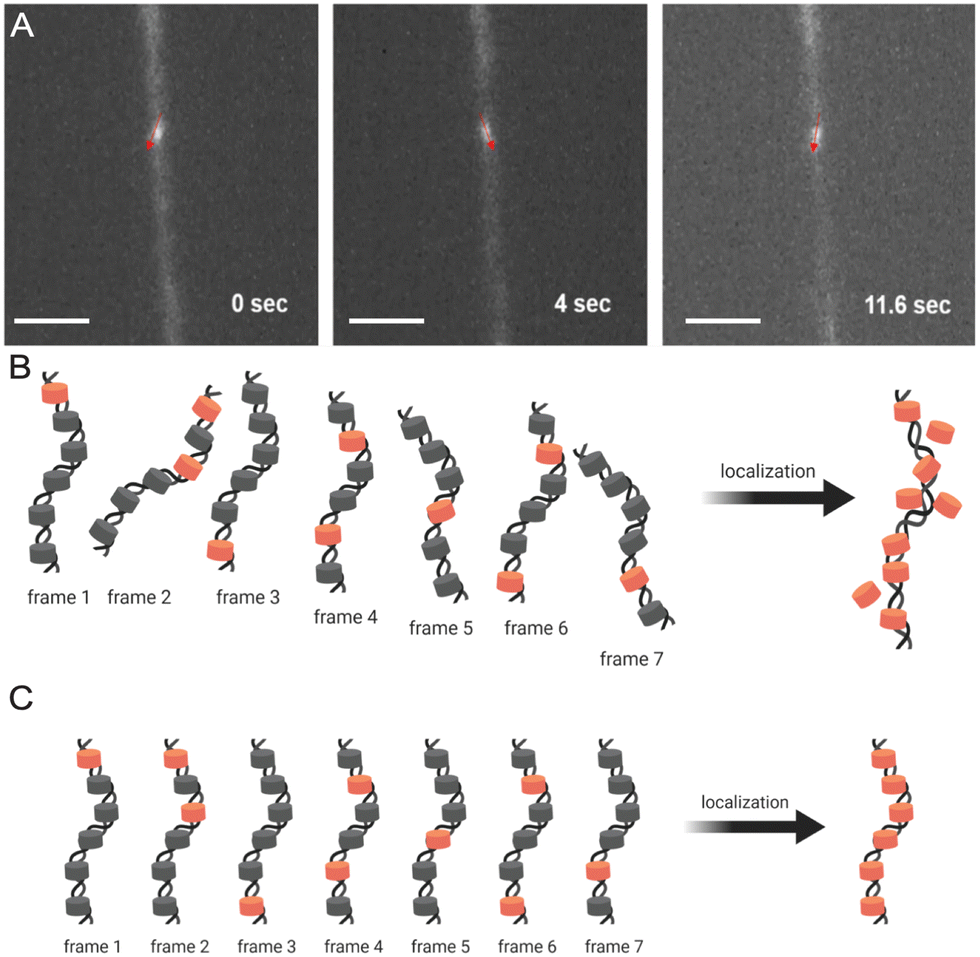 | ||
| Fig. 5 Super-resolution image reconstruction from dSTORM requires full immobilization of chromatin fiber in the closed nanochannels. (A) A chromatin fiber in an incompletely closed nanochannel can still wiggle around due to Brownian motion. (B) Accurate localization of fluorophores in reconstructed images is not possible due to the wiggling movements of the chromatin. (C) Complete immobilization of a chromatin fiber allows accurate spatial localization of fluorophores. Scale bar in (A), 200 μm. | ||
The results demonstrated that the tunable nanochannels were capable of immobilization of biopolymers, allowing confinement and reconstruction of fluorophores signals. Fig. 6A and B illustrate the predicted images for the two models of histone segregation. BrdU is a thymidine analog that can be quickly incorporated into newly synthesized DNA when introduced into culture media.59,60 Anti-HA and anti-BrdU antibodies labeled the newly synthesized histone and newly synthesized DNA, respectively (Fig. 6C and D). Further analyses on the distributions (Fig. 6G) of the markers were performed to collect the long-range epigenetic information on individual chromatin fibers.
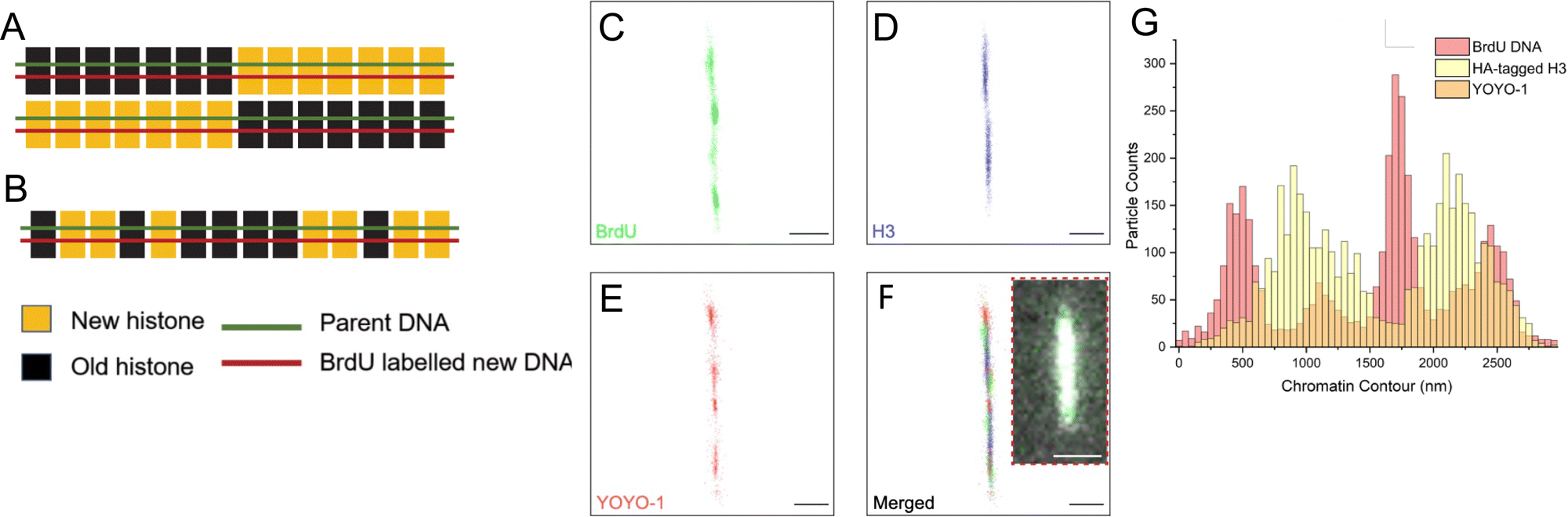 | ||
| Fig. 6 Super-resolution imaging results favor dispersive histone transmission. (A) With the conservative model, H3 histones should be localized only at one-half of the linearized rDNA chromatin fiber. (B) With the dispersive model, H3 histones should be randomly localized across the chromatin fiber. With the nanochannel system and appropriate dye selection, dSTORM successfully resolved BrdU (C), H3-HA (D) and YOYO-1 (E), on a linearized rDNA chromatin fiber. (F) Merged super-resolution image of the labeled chromatin fiber, with the inset showing the same fiber in wide-field microscopy. (G) Distribution of fluorescent labels along the linearized chromatin fiber in (F) showed that distribution of newly synthesized H3 (HA-H3) across both halves were not significantly different from random distribution, supporting dispersive transmission. Scale bars in (C)–(F), 500 nm. Inset scale bar in (F), 30 μm. | ||
A histogram reporting extensions of all 39 trapped chromatin fibers is shown in Fig. 7. They are divided into three classes: (I) 17 were extended to less than 50% of the contour length, indicating that the chromatin remained in the random coil state, possibly due to entanglement (Fig. S8A–C, ESI†), (II) 19 were extended to between 50 and 150%, representing the expected chromatin state with efficient extension and limited dechromatinization (Fig. 6F and Fig. S9, S10, ESI†), and (III) 3 were extended well beyond 150%, probably in a state of substantial dechromatinization (Fig. S8D–F, ESI†). The wide dispersion of the rDNA chromatin lengths reflects the intrinsic difficulty in consistent chromatin stretching, as the applied force needs to fall within a narrow range that is strong enough to overcome interactions between nucleosomes to extend the chromatin, but weak enough to preserve protein–DNA interactions within the nucleosome to prevent dechromatinization and over-stretching. We focused our subsequent analysis on class II, representing the well-stretched rDNA chromatin fibers.
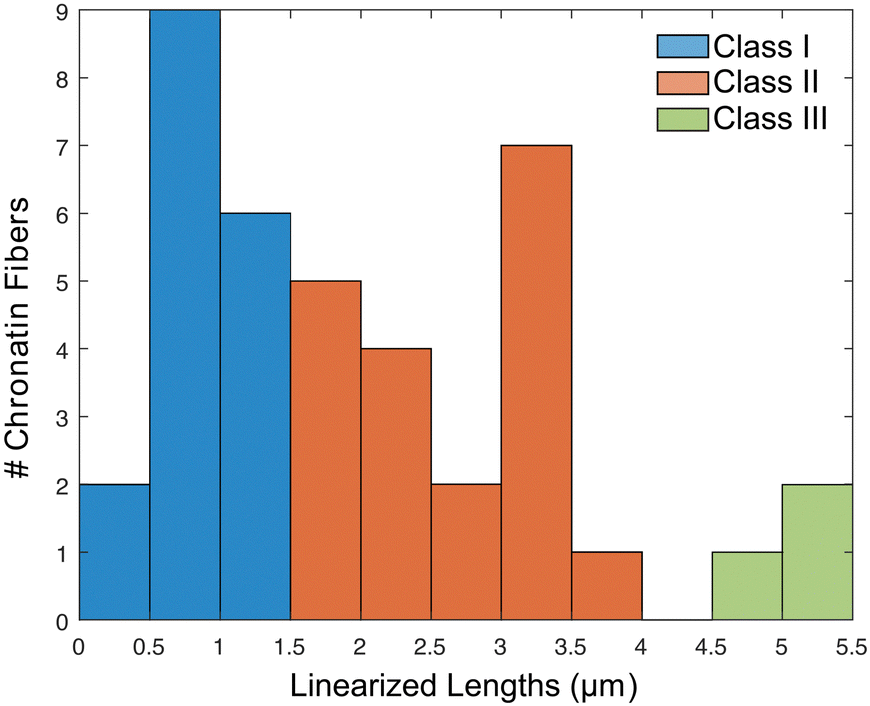 | ||
| Fig. 7 Histogram of observed linearized chromatin fibers along the nanochannels. Fibers falling between [1.5, 4.5] m were considered a single chromatin fiber properly linearized to near its full contour lengths. | ||
Chromatin super-resolution microscopy
The resolution of conventional fluorescence microscopy (>200 nm) is not sufficient for resolving the nucleosome distribution of the fully extended 11 nm chromatin fiber. Recent progress in various technologies for super-resolution microscopy allows us to gain increasingly detailed information about chromatin, both in vivo and in vitro. In contrast to the standard fixed-dimension nanochannels, chromatin linearization in the tunable nanochannels proved to be more compatible with super-resolution microscopy. Significant improvement in resolution can be seen in Fig. 6F, which shows the same chromatin fiber imaged by dSTORM and diffraction-limited widefield. The image resolution resulting from our experiments is approximately 20 nm in the XY-plane, as reported previously,61 and further confirmed with FRC analysis on the two-color images.62,63Collecting long-range epigenetic information on individual chromatin fibers
There is a long-standing controversy concerning conservative vs. dispersive segregation of nucleosomes:64 the former argues that old histones reassemble preferentially on one arm of DNA replication forks with new histones on the other (Fig. 4C), while the latter argues for random reassembly (Fig. 4D). To address this question, we developed procedures to examine the segregation of canonical histone H3 in Tetrahymena rDNA chromatin. Expression of hemagglutinin (HA)-tagged canonical H3 was put under the control of the MTT3 promoter, and was readily induced by Cd2+ in a dosage-dependent manner (Fig. 4 and Fig. S6, ESI†). As the pool for non-nucleosomal H3 in Tetrahymena is very small and actively turned over, we could quickly introduce tagged H3 into chromatin and effectively differentiate the new tagged H3 from the old untagged H3. Cd2+ dosage was optimized to ensure sufficient HA-tagged H3 expression without cell death from cadmium toxicity. The Cd2+ treatment was limited to 1 hour, which is substantially shorter than a full cell cycle, thereby limiting HA-tagged H3 to only newly synthesized DNA (Fig. 6A and B). Successful rDNA extraction was verified by electrophoresis (Fig. S7, ESI†). The two replication origins are very close to the palindromic center of the rDNA, and one is randomly chosen to fire in each cell cycle.65,66 Therefore, the direction of replication would be fixed for the rDNA, as the replication bubble expands from the center to the terminus. Essentially, one half of rDNA is replicated by leading strand synthesis, while the other is by lagging strand. There will be an even distribution of old H3 on both halves in the case of dispersive segregation, while there will be a strong bias in the case of conservative segregation.With multi-color super-resolution imaging by dSTORM, we were able to investigate histone inheritance during replication. The chromatin fibers classified in class II in Fig. 7, representing well-stretched rDNA mini-chromosomes, were used for categorizing conservative or dispersive distribution. The distribution of HA-H3 signals along the left and the right half of the linearized chromatin fibers were tested using the two-sample Kolmogorov–Smirnov statistical test. Failure to reject the null hypothesis allows us to conclude that the two distributions were similar, thus categorizing the fiber as dispersive. A total of 13 out of 19 linearized chromatin fibers were classified as potentially dispersive, supporting the dispersive transmission model (Fig. 6F and Fig. S9, ESI†). Table S5 (ESI†) summarizes the statistical analyses of the representative chromatin fibers. Notably, there were a few fibers that were categorized as conservative, as shown in Fig. S10 (ESI†). In short, our method proved to be an effective method of collecting long-range epigenetic information on individual chromatin fibers. The results suggested that the dispersive histone segregation theory is indeed favored over the conservative counterpart, as shown previously.4,67–69
Conclusions
We have used tunable nanochannels to achieve efficient loading, linearization, immobilization, and super-resolution imaging of chromatin fibers. In the open state, tunable nanochannels facilitate the hydraulic loading of chromatin fibers, which resist electrophoretic loading due to their nominal net charges. Gradual closing of tunable nanochannels improves chromatin linearization, as complex interactions among protein and DNA components of the chromatin are given time to play out.13 In the normally closed state, tunable nanochannels near completely immobilize chromatin fibers, allowing super-resolution imaging for which stability over time is essential. Closed h-PDMS nanochannels are in an oxygen-rich environment with limited access to imaging buffers, incompatible with the conventional dye blinking mechanism. We have screened and identified self-blinking dyes, enabling multi-color dSTORM imaging of linearized and immobilized chromatin fibers. It is worth noting that super-resolution imaging entails long exposure time and a small field-of-view, and therefore has an inherent conflict with high throughput. In the future, we may further exploit tunable nanochannels by combining a fast scan by conventional microscopy to search for areas of interest with open nanochannels and then a zoom-in by super-resolution microscopy to reveal their details with closed nanochannels.Our newly developed method opens the door to collecting long-range genetic and epigenetic information contained in native chromatin fibers. This kind of connectivity information is generally lost in chemical methods based on short-read sequencing.70,71 As a proof-of-concept, we have examined histone transmission in the Tetrahymena rDNA mini-chromosome. By assessing the distribution of newly synthesized histone H3, we provide evidence supporting the dispersive model, rather than the conservative model. Our method can be readily extended to study interactions between multiple epigenetic marks and chromatin-templated events over a wide range of multi-kbs to hundreds of kbs.
Experimental
h-PDMS fabrication
Expanding from our previously published micro-channel h-PDMS system,35 we further diluted h-PDMS to downsize dimensions to sub-micron-scale. h-PDMS was prepared with 1.7 g of (7.0–8.0% vinylmethylsiloxane)-dimethylsiloxane copolymer, trimethylsiloxyl terminated (VDT-731; Gelest Inc.), 9 μL of platinum-divinyltetramethyldisiloxane complex (SIP6831.2; Gelest Inc.) as a catalyst, and 12 μL of 2,4,6,8-tetramethyl-tetravinylcyclotetrasiloxane as moderator (396281; Sigma-Aldrich), followed by two-step dilutions with n-hexane (H303-1; Fisher) to between 1.4% to 1.8%. Slow-speed spin coating of the diluted solution was used to achieve a thickness of 200 nm. Bulk PDMS at a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio was cast on top of the h-PDMS thin film to create the two-layer system. The h-PDMS cracks were fabricated using the same strategy of stretcher-induced cracking followed by plasma bonding to a thin PDMS membrane (∼40 μm) under air plasma exposure at 100 W for 30 seconds (Fig. S1, ESI†). The detailed protocol is described in the ESI.†
:1 ratio was cast on top of the h-PDMS thin film to create the two-layer system. The h-PDMS cracks were fabricated using the same strategy of stretcher-induced cracking followed by plasma bonding to a thin PDMS membrane (∼40 μm) under air plasma exposure at 100 W for 30 seconds (Fig. S1, ESI†). The detailed protocol is described in the ESI.†
Super resolution dyes and imaging buffers
dSTORM imaging buffer was prepared by mixing 20 μL of 1 M cysteamine (30070; Sigma), 10 μL of 2-mercaptoethanol (63689; Sigma), 20 μL of 50× Gloxy mixture and 950 μL of 50 mM Tris-HCl pH 8.0, 10 mM NaCl, 10% (w/v) glucose. The Gloxy mixture is comprised of 8440 AU glucose oxidase (G2133; Sigma) and 70200 AU catalase (C40; Sigma) in 1 mL of 50 mM Tris pH 8.0 and 10 mM NaCl buffer.72 The OxEA solution was prepared using a protocol modified from Nahidiazar et al.49: 50 mM cysteamine, 3% (v/v) OxyFlour™ (Oxyrase Inc.), 20% (v/v) of sodium DL-lactate solution (L1375; Sigma) in PBS, pH 8.2.
Popular super-resolution dyes Alexa 647 (ThermoFisher) and CF 568 (Biotium) were chosen as model super-resolution fluorophores. SaraFluor 650B (HMSiR)-goat anti-mouse IgG (A202-01; Goryo Chemical), SaraFluor 488B (HEtetTFER)-NHS (A218-01; Goryo Chemical), FLIP 565-goat anti-rabbitIgG (FL565-1002; Abberior) and CAGE 635-NHS (CA635; Abberior) were chosen as self-blinking dyes. AF 647 and CF 567 were imaged in either imaging buffer or OxEA solution, while the self-blinking dyes were imaged in distilled water (15230, Gibco).
Tetrahymena strain and culture condition
A Tetrahymena strain, MTT3 HHT1 CHA-CHX, was generated with Cd2+-inducible HA-tagged H3 expression. The Tetrahymena strain was cultured overnight to its mid-logarithmic phase (∼1 × 105 cells per mL) in SPP media. The SPP media was prepared by mixing 10 g protease peptone (211684; VWR), 2 g dextrose (G7528; Sigma), 1 g yeast extract (BP1422; Fisher), 0.03 g ethylenediaminetetraacetic acid ferric sodium salt (E6760; Sigma) in 1 L of distilled water.57,73Chromatin extraction
The Tetrahymena strain was incubated with 10 μM thymidine-analogue BrdU for 1 hour to label newly synthesized DNA. CdCl2 (final concentration of 1 μg mL−1) was added to the Tetrahymena culture for 1 hour to induce HA-H3 expression. The nuclear isolation procedure was adapted from the octanol-skin method. In brief, Tetrahymena cells were spun down and resuspended with 200 mL of medium A, which constituted of 0.1 M sucrose (84097; Sigma), 5 mM magnesium chloride (M1028; Sigma), 4% gum arabic (G9752; Sigma), 10 mM Tris pH 7.5, and 10 mM butyric acid (B103500; Sigma) in distilled water (Gibco, 15230), pH-adjusted to 6.75. The cells were sheared mechanically in a blender along with 1.25 mL 1-ocatanol (8209310100; Sigma), 2 mL of 100 mM phenylmethanesulfonyl fluoride (P7626; Sigma) protease inhibitor, and 2 mL of 0.5 M EDTA (BMA51201; Fisher) for minimizing DNA degradation by DNases. The nuclei were collected at 2000 × g centrifugation for 10 min and resuspended in a wash buffer composed of 50 mM Tris pH 7.5 and 2 mM MgCl2. This was followed by further centrifugation at 3000 × g for 8 min to again collect the nuclei for resuspension in nucleoli extraction buffer (10 mM Tris pH 7.5, 10% glycerol, 0.05 M NaCl, 0.5 mM MgCl2, 1 mM 2-mercaptoethanol, 1% Nonidet P-40 and 2 mM PMSF). Nuclear membranes were then removed via mechanical shearing with a Dounce homogenizer with a tight-fitting pestle in the presence of nucleoli extraction buffer. The nucleoli (in the supernatant) were separated from the bulkier nuclei (in the pellet) by spinning at 500 × g for 10 min. Nucleoli were subsequently treated with RNase A for 1 hour at 37 °C to remove RNA components.Chromatin labeling
Prior to fluorescent labeling, the chromatin extract was treated with ammonium sulfate to solubilize non-histone proteins before undergoing buffer exchange facilitated with a centrifugal filter unit to remove undesirable constituents in the nucleoli extraction buffer. The purified extract was then incubated for 1 hour with anti-BrdU (B8434; Sigma) and anti-HA (3724S; Cell Signaling) primary antibodies targeting the newly synthesized DNA strands and the HA-tagged histone H3, respectively. Excess antibodies were removed using the aforementioned centrifugal filter units. Secondary antibodies labeled with SaraFluor 650B and FLIP 565 were used to detect the anti-BrdU and anti-HA antibodies, respectively. The immunolabeled extracts were again filtered and washed to remove excess secondary antibodies before final incubation with YOYO-1. A final filter and wash were necessary to remove excess antibodies and dye molecules. The labeled chromatin was stored at −20 °C.Chromatin stretching and dye loading
Chromatin fibers were diluted at least 100-fold from purified stock with 1% Triton X-100 (Sigma-Aldrich, T8787) in solution to assist loading. 5 μL of chromatin solution was deposited into the top inlet port, and to control the hydraulic pressure difference, 3 μL of distilled water was added into the inlet port of the bottom microchannel. The h-PDMS device was mounted onto an automated stretcher,39 and stretched to ∼50 to 70% strain until the crack channels were visible under brightfield microscopy. The crack channels were then closed, which allowed wetting of the crack channels. Subsequent stretching to induce a channel opening state caused chromatin fibers to enter the crack channels. All stretcher motor actions were performed at a fixed 1 mm s−1 actuator speed in this study. Once the strain was released from the stretcher, the nanochannels collapsed, linearizing and immobilizing the trapped chromatin fibers. The device was then ready for super-resolution imaging.The dye-only solutions (for dye optimization; see ESI†) were similarly loaded into the ox-PDMS nanochannels. The trapping of dye solutions often occurred when nanochannels collapsed too fast, or due to incomplete cracks (those failing to reach the bottom microchannel during the strain-induced cracking process). Both the oxidized device containing fluorophore bubbles and the h-PDMS device with the trapped chromatin fibers were imaged by dSTORM.
dSTORM imaging of h-PDMS device
dSTORM imaging was performed using a Bruker Vutara 352 with a 63× NA 1.2 Plan-Apochromat water immersion objective. Before each experiment, the instrument was calibrated using 100 nm-diameter TetraSpeck microspheres in order to produce point spread functions the native software deemed to be optimal. The h-PDMS device was placed on top of a no. 0 glass coverslip to support the PDMS slab device and to keep the device free from bending by gravity, as any strain on the PDMS may induce channel opening, disrupting dSTORM acquisition. All images were collected and analyzed using Vutara SRX software. Denoising was applied on the images, with the value set to 0.2.Surface and crack profile measurement
The relationship between the thickness of the spin coat-deposited h-PDMS thin film and the dilution factor was measured with atomic force microscopy (Veeco AFM) and contact profilometry (Dektak 150 Profilometer). 20 μL droplets of standard PDMS are carefully deposited on top of the h-PDMS-coated mold, partially covering the SU-8 features. The system is cured at 120 °C for 10 minutes after which the PDMS is peeled off, removing both h-PDMS and PDMS simultaneously while leaving the silicon substrate bare. In doing so, the h-PDMS layer becomes accessible for analysis, particularly the section between SU-8 features. This area of the layer is also where nanocracks are located, and thus crack depths could also be measured with AFM. Pre-cracked h-PDMS/PDMS slabs were mounted onto a manual stretcher and stretched with approximately 32%, 45% and 59% strain for AFM measurements.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to acknowledge the core facilities at the Parker H. Petit Institute for Bioengineering and Bioscience at the Georgia Institute of Technology for the use of their shared equipment, services, and expertise. We thank the NIH (R01 GM123517) for funding. This work was also performed in part at the Georgia Tech Institute for Electronics and Nanotechnology, a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (Grant ECCS-2025462).Notes and references
- T. Jenuwein and C. D. Allis, Science, 2001, 293, 1074–1080 CrossRef CAS PubMed.
- B. D. Strahl and C. D. Allis, Nature, 2000, 403, 41–45 CrossRef CAS PubMed.
- M. Wooten, Z. Nizami, X. Yang, J. Snedeker, R. Ranjan, J. M. Kim, E. Urban, V. Tran, J. Buss, J. Gall, J. Xiao and X. Chen, SSRN Electron. J. DOI:10.2139/SSRN.3155700.
- J. Xie, M. Wooten, V. Tran and X. Chen, Trends Cell Biol., 2017, 27, 527–540 CrossRef CAS PubMed.
- C. Heck, Y. Michaeli, I. Bald and Y. Ebenstein, Curr. Opin. Biotechnol, 2019, 55, 151–158 CrossRef CAS PubMed.
- J. Jeffet, S. Margalit, Y. Michaeli and Y. Ebenstein, Essays Biochem., 2021, 65, 51–66 CrossRef CAS PubMed.
- A. Cerf, H. C. Tian and H. G. Craighead, ACS Nano, 2012, 6, 7928–7934 CrossRef CAS PubMed.
- M. L. Bennink, S. H. Leuba, G. H. Leno, J. Zlatanova, B. G. De Grooth and J. Greve, Nat. Struct. Biol., 2001, 8, 606–610 CrossRef CAS PubMed.
- Y. Cui and C. Bustamante, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 127–132 CrossRef CAS PubMed.
- A. Kaczmarczyk, T. B. Brouwer, C. Pham, N. H. Dekker and J. van Noort, Methods Mol. Biol., 2018, 1814, 297–323 CrossRef CAS PubMed.
- D. E. Streng, S. F. Lim, J. Pan, A. Karpusenka and R. Riehn, Lab Chip, 2009, 9, 2772–2774 RSC.
- B. R. Cipriany, R. Zhao, P. J. Murphy, S. L. Levy, C. P. Tan, H. G. Craighead and P. D. Soloway, Anal. Chem., 2010, 82, 2480–2487 CrossRef CAS PubMed.
- T. Matsuoka, B. C. Kim, J. Huang, N. J. Douville, M. D. Thouless and S. Takayama, Nano Lett., 2012, 12, 6480–6484 CrossRef CAS PubMed.
- S. F. Lim, A. Karpusenko, A. L. Blumers, D. E. Streng and R. Riehn, Biomicrofluidics, 2013, 7, 064105 CrossRef PubMed.
- J. W. Yeh and K. Szeto, Biomicrofluidics, 2017, 11, 044108 CrossRef PubMed.
- A. B. Bhandari, J. G. Reifenberger, H. M. Chuang, H. Cao and K. D. Dorfman, J. Chem. Phys., 2018, 149, 104901 CrossRef PubMed.
- J. O. Tegenfeldt, C. Prinz, H. Cao, S. Chou, W. W. Reisner, R. Riehn, Y. M. Wang, E. C. Cox, J. C. Sturm, P. Silberzan and R. H. Austin, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10979–10983 CrossRef CAS PubMed.
- S. K. Das, M. D. Austin, M. C. Akana, P. Deshpande, H. Cao and M. Xiao, Nucleic Acids Res., 2010, 38, e177 CrossRef PubMed.
- H. Cao, J. O. Tegenfeldt, R. H. Austin and S. Y. Chou, Appl. Phys. Lett., 2002, 81, 3058–3060 CrossRef CAS.
- W. Reisner, N. B. Larsen, A. Silahtaroglu, A. Kristensen, N. Tommerup, J. O. Tegenfeldt and H. Flyvbjerg, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13294–13299 CrossRef CAS PubMed.
- R. Riehn, M. Lu, Y.-M. Wang, S. F. Lim, E. C. Cox and R. H. Austin, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10012–10016 CrossRef CAS PubMed.
- W. Reisner, J. N. Pedersen and R. H. Austin, Rep. Prog. Phys., 2012, 75, 106601 CrossRef PubMed.
- S. Bocklandt, A. Hastie and H. Cao, Adv. Exp. Med. Biol., 2019, 1129, 97–118 CrossRef CAS PubMed.
- H. Cao, A. R. Hastie, D. Cao, E. T. Lam, Y. Sun, H. Huang, X. Liu, L. Lin, W. Andrews, S. Chan, S. Huang, X. Tong, M. Requa, T. Anantharaman, A. Krogh, H. Yang, H. Cao and X. Xu, GigaScience, 2014, 3, 2047-217X-3-34 CrossRef PubMed.
- E. T. Lam, A. Hastie, C. Lin, D. Ehrlich, S. K. Das, M. D. Austin, P. Deshpande, H. Cao, N. Nagarajan, M. Xiao and P.-Y. Kwok, Nat. Biotechnol., 2012, 30, 771–776 CrossRef CAS PubMed.
- N. Douville, D. Huh and S. Takayama, Anal. Bioanal. Chem., 2008, 391, 2395–2409 CrossRef CAS PubMed.
- W. Reisner, K. J. Morton, R. Riehn, Y. M. Wang, Z. Yu, M. Rosen, J. C. Sturm, S. Y. Chou, E. Frey and R. H. Austin, Phys. Rev. Lett., 2005, 94, 196101 CrossRef PubMed.
- K. Jo, D. M. Dhingra, T. Odijk, J. J. de Pablo, M. D. Graham, R. Runnheim, D. Forrest and D. C. Schwartz, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2673–2678 CrossRef CAS PubMed.
- L. Dai, J. van der Maarel and P. S. Doyle, Macromolecules, 2014, 47, 2445–2450 CrossRef CAS.
- R. Chantiwas, S. Park, S. A. Soper, B. C. Kim, S. Takayama, V. Sunkara, H. Hwang and Y.-K. Cho, Chem. Soc. Rev., 2011, 40, 3677–3702 RSC.
- S. Howorka and Z. Siwy, ACS Nano, 2016, 10, 9768–9771 CrossRef CAS PubMed.
- M. Alizadehheidari, E. Werner, C. Noble, M. Reiter-Schad, L. K. Nyberg, J. Fritzsche, B. Mehlig, J. O. Tegenfeldt, T. Ambjörnsson, F. Persson and F. Westerlund, Macromolecules, 2015, 48, 871–878 CrossRef CAS.
- H. Sharim, A. Grunwald, T. Gabrieli, Y. Michaeli, S. Margalit, D. Torchinsky, R. Arielly, G. Nifker, M. Juhasz, F. Gularek, M. Almalvez, B. Dufault, S. S. Chandra, A. Liu, S. Bhattacharya, Y.-W. Chen, E. Vilain, K. R. Wagner, J. Pevsner, J. Reifenberger, E. T. Lam, A. R. Hastie, H. Cao, H. Barseghyan, E. Weinhold and Y. Ebenstein, Genome Res., 2019, 29, 646–656 CrossRef CAS PubMed.
- D. Huh, K. L. Mills, X. Zhu, M. A. Burns, M. D. Thouless and S. Takayama, Nat. Mater., 2007, 6, 424–428 CrossRef CAS PubMed.
- B. C. Kim, C. Moraes, J. Huang, T. Matsuoka, M. D. Thouless and S. Takayama, Small, 2014, 10, 4020–4029 CrossRef CAS PubMed.
- H. Shiku, T. Saito, C. C. Wu, T. Yasukawa, M. Yokoo, H. Abe, T. Matsue and H. Yamada, Chem Lett., 2006, 35, 234–235 CrossRef CAS.
- A. Renn, J. Seelig and V. Sandoghdar, Mol. Phys., 2006, 104, 409–414 CrossRef CAS.
- N. Banterle, K. H. Bui, E. A. Lemke and M. Beck, J. Struct. Biol., 2013, 183, 363–367 CrossRef CAS PubMed.
- N. J. Ginga, J. H.-C. Chiu, J.-H. Lee, M. D. Thouless and S. Takayama, Microfluid. Nanofluid., 2022, 26, 27 CrossRef CAS.
- B. C. Kim, T. Matsuoka, C. Moraes, J. Huang, M. D. Thouless and S. Takayama, Sci. Rep., 2013, 3, 3027 CrossRef PubMed.
- J. Huang, B. C. Kim, S. Takayama and M. D. Thouless, J. Mater. Sci., 2014, 49, 255–268 CrossRef CAS PubMed.
- M. S. Hu, M. D. Thouless and A. G. Evans, Acta Metall., 1988, 36, 1301–1307 CrossRef CAS.
- M. D. Thouless, Z. Li, N. J. Douville and S. Takayama, J. Mech. Phys. Solids, 2011, 59, 1927–1937 CrossRef CAS PubMed.
- M. Yu, Y. Hou, R. Song, X. Xu and S. Yao, Small, 2018, 14, 1800229 CrossRef PubMed.
- D. J. Berard, M. Shayegan, F. Michaud, G. Henkin, S. Scott and S. Leslie, Appl. Phys. Lett., 2016, 109, 033702 CrossRef.
- E. Leclerc, Y. Sakai and T. Fujii, Biomed. Microdev., 2003, 5, 109–114 CrossRef CAS.
- Z. Ye, W. Yang, Y. Zheng, S. Wang, X. Zhang, H. Yu, S. Li, C. Luo, X. Peng and Y. Xiao, Angew. Chem., Int. Ed., 2022, 61, e202211540 CAS.
- S. Van De Linde, S. Aufmkolk, C. Franke, T. Holm, T. Klein, A. Löschberger, S. Proppert, S. Wolter and M. Sauer, Chem. Bio, 2013, 20, 8–18 CrossRef CAS PubMed.
- L. Nahidiazar, A. V. Agronskaia, J. Broertjes, B. D. Van Broek and K. Jalink, PLoS One, 2016, 11, e0158884 CrossRef PubMed.
- Y. Zheng, Z. Ye, X. Zhang and Y. Xiao, J. Am. Chem. Soc., 2023, 145, 5125–5133 CrossRef CAS PubMed.
- S. N. Uno, M. Kamiya, T. Yoshihara, K. Sugawara, K. Okabe, M. C. Tarhan, H. Fujita, T. Funatsu, Y. Okada, S. Tobita and Y. Urano, Nat. Chem., 2014, 6, 681–689 CrossRef CAS PubMed.
- S. N. Uno, M. Kamiya, A. Morozumi and Y. Urano, Chem. Commun., 2017, 54, 102–105 RSC.
- B. Roubinet, M. Bischoff, S. Nizamov, S. Yan, C. Geisler, S. Stoldt, G. Y. Mitronova, V. N. Belov, M. L. Bossi and S. W. Hell, J. Org. Chem., 2018, 83, 6466–6476 CrossRef CAS PubMed.
- J. Fölling, V. Belov, R. Kunetsky, R. Medda, A. Schönle, A. Egner, C. Eggeling, M. Bossi and S. W. Hell, Angew. Chem., Int. Ed., 2007, 46, 6266–6270 CrossRef PubMed.
- K. Grußmayer, T. Lukes, T. Lasser and A. Radenovic, ACS Nano, 2020, 14, 9156–9165 CrossRef PubMed.
- C. E. Boott, R. F. Laine, P. Mahou, J. R. Finnegan, E. M. Leitao, S. E. D. Webb, C. F. Kaminski and I. Manners, Chem. – Eur. J., 2015, 21, 18539–18542 CrossRef CAS PubMed.
- M. A. Gorovsky, J. Protozoology, 1973, 20, 19–25 CrossRef CAS PubMed.
- J. Engberg, G. Christiansen and V. Leick, Biochem. Biophys. Res. Commun., 1974, 59, 1356–1365 CrossRef CAS PubMed.
- J. Loidl and H. Scherthan, J. Cell Sci., 2004, 117, 5791–5801 CrossRef CAS PubMed.
- C. Yu, H. Gan, A. Serra-Cardona, L. Zhang, S. Gan, S. Sharma, E. Johansson, A. Chabes, R. M. Xu and Z. Zhang, Science, 2018, 361, 1386–1389 CrossRef CAS PubMed.
- P. M. Tiwari, D. Vanover, K. E. Lindsay, S. S. Bawage, J. L. Kirschman, S. Bhosle, A. W. Lifland, C. Zurla and P. J. Santangelo, Nat. Commun., 2018, 9, 3999 CrossRef PubMed.
- R. P. J. Nieuwenhuizen, K. A. Lidke, M. Bates, D. L. Puig, D. Grünwald, S. Stallinga and B. Rieger, Nat. Methods, 2013, 10, 557–562 CrossRef CAS PubMed.
- X. Zhang, P.-Y. Lin, K. Liakath-Ali and T. C. Südhof, Nat. Commun., 2022, 13, 2297 CrossRef CAS PubMed.
- A. T. Annunziato, Biochim. Biophys. Acta Gene Regul. Mech., 2012, 1819, 196–210 CrossRef CAS PubMed.
- D. M. MacAlpine, Z. Zhang and G. M. Kapler, Mol. Cell. Biol., 1997, 17, 4517–4525 CrossRef CAS PubMed.
- S. Gao, J. Xiong, C. Zhang, B. R. Berquist, R. Yang, M. Zhao, A. J. Molascon, S. Y. Kwiatkowski, D. Yuan, Z. Qin, J. Wen, G. M. Kapler, P. C. Andrews, W. Miao and Y. Liu, Genes Dev., 2013, 27, 1662–1679 CrossRef CAS PubMed.
- V. Jackson and R. Chalkley, Biochemistry, 1985, 24, 6930–6938 CrossRef CAS PubMed.
- G. Russev and R. Hancock, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 3143–3147 CrossRef CAS PubMed.
- V. Jackson and R. Chalkley, Cell, 1981, 23, 121–134 CrossRef CAS PubMed.
- T. Matsuoka, B. Choul Kim, C. Moraes, M. Han and S. Takayama, Biomicrofluidics, 2013, 7, 041301 CrossRef PubMed.
- N. Gilat, D. Fridman, H. Sharim, S. Margalit, N. R. Gassman, Y. Michaeli and Y. Ebenstein, Biophys. Rep., 2021, 1, 100017 CAS.
- E. Alonas, A. W. Lifland, M. Gudheti, D. Vanover, J. Jung, C. Zurla, J. Kirschman, V. F. Fiore, A. Douglas, T. H. Barker, H. Yi, E. R. Wright, J. E. Crowe and P. J. Santangelo, ACS Nano, 2014, 8, 302–315 CrossRef CAS PubMed.
- M. T. Sweet and C. D. Allis, Cold Spring Harb. Protoc., 2006 DOI:10.1101/pdb.prot4502.
Footnotes |
| † Electronic supplementary information (ESI) available: ox-PDMS discussion, detailed h-PDMS methods, Tables S1–S5, and Fig. S1–S10. See DOI: https://doi.org/10.1039/d3nh00096f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |