
Green electro-organic synthesis of a novel catechol derivative based on o-benzoquinone nucleophilic addition†
Mohamed
Abd-Elsabour
a,
Hytham F.
Assaf
a,
Ahmed M.
Abo-Bakr
a,
Abdulrahman G.
Alhamzani
 *b,
Mortaga M.
Abou-Krisha
ab,
Aamal A.
Al-Mutairi
b and
Hesham M.
Alsoghier
*a
*b,
Mortaga M.
Abou-Krisha
ab,
Aamal A.
Al-Mutairi
b and
Hesham M.
Alsoghier
*a
aChemistry Department, Faculty of Science, South Valley University, 83523 Qena, Egypt
bChemistry Department, College of Science, Imam Mohammad Ibn Saud Islamic University (IMSIU), Riyadh 11623, Saudi Arabia. E-mail: agalhamzani@imamu.edu.sa
First published on 30th November 2022
Abstract
In this work, a green-electrochemical synthesis was applied to catechol oxidation (1) to o-benzoquinone (2) using cyclic voltammetry and potential controlled coulometry. Encouraging results were obtained in the presence of semicarbazide (3) as a nucleophile in an ethanol/phosphate buffer mixture (pH 7.0). The pH, number of cycles, and scan rate were studied. The results indicate that the generated o-benzoquinone (2) participates in a Michael addition reaction with nucleophilic hydrazide via the ECEC mechanism to form a new triazinone adduct. The obtained results showed that a novel heterocyclic triazinone (9) product functionalized with hydroxyl and amide groups had been synthesized with a 68% yield. After purification, the product has been characterized by IR, UV-visible, GC/MS, 1H, and 13C NMR. The green-electrochemical synthesis approach produced novel organics with excellent purities and yields while being environmentally friendly and free of hazardous reagents.
1. Introduction
Green-electrochemical synthesis is a recent direction for many chemists in recent years for the construction of novel heterocyclic molecules.1–13 The synthesis of these organic compounds using a traditional method has several drawbacks that requires toxic reagents and expensive catalysts and is time-consuming.2,4,7 Thus, there is an urgent need to focus on green electrochemical chemistry for organic synthesis to avoid these obstacles.2,4,14,15 The great advantage of this clean method is producing organic compounds without using toxic and hazardous reagents, a small number of steps to reach the target molecule, and efficiency and economy with high-quality products.2,4,12 Catechol is considered one of the essential aromatic compounds that widely exist in nature.7,16–21 It is used as a raw material in many industries such as photography, dye, cosmetics, chemical, and pharmaceutical industries.7,8,10,13,17,19,22,23 In addition, catechol has biological importance and acts as an antioxidant and antiradical24,25 due to its high electron transfer. Many drugs used in cancer therapy, such as adriamycin, carbazilquinone, doxorubicin, daunorubicin, and mitomycin C contain catechol,7,18,20,26,27 while various other quinones have different applications in the industry.7,28,29Furthermore, many quinones are involved in enzyme inhibition and DNA cross-linking.11,30,31 Among these, catechol and hydroquinone can be electrochemically oxidized to ortho and para-benzoquinones by reversible two-electron and two-proton processes.8,32,33 The formed benzoquinones are quite reactive intermediates and are quickly attacked by a variety of nucleophiles through various reactions such as Michael's addition.1,8,10,13,14,32,34,35 Many studies were interested in nucleophilic Michael's addition to catechols based on electrochemical oxidation in aqueous solutions. In the case of thiosemicarbazide as a nucleophile, the yield of the new cyclized product of catechol was 72%,8 but with different thiol nucleophiles, the yields of catechol products were 60–82%.13 Moreover, Michael's addition of 2-acetylcyclopentanone as a nucleophile to different catechol derivatives produced new catechol products with a high economy, without toxic reagents and solvents, good yields (60–80%) and high purity via the ECEC mechanism pathway.8,32,36,37
Hydrazides constitute an important class of organic compounds due to their diverse biological properties.38 They exhibit good antitumor and anti-diabetic activities.8,10,23,39,40 In addition, some hydrazide derivatives were found to possess anti-inflammatory and analgesic activities.41,42 To date, different procedures have been investigated for the synthesis of carbohydrazide. Hydrazinolysis of esters in alcoholic solutions is the basic method for preparing carboxylic acid hydrazides.43 However, it is rare for esters to produce significant amounts of diacylhydrazine.44 Therefore, it is necessary to develop rapid and green methods to synthesize new carbohydrazides. In this study, we benefited from the easy oxidation of catechol to o-quinone (stable quinoid form) in the presence of a hydrazide derivative as a nucleophile to form a new triazinone as a promising heterocyclic compound, which may have biological activity with anticancer and antiulcer properties and anti-inflammatory effects.
Herein, we employed a straightforward and uncomplicated evolution of the green electrochemical method to synthesize a new heterocyclic compound functionalized with a phenolic group. To understand the mechanism of catechol's electrochemical oxidation in the presence of a semicarbazide, some parameters such as cycling numbers, nucleophile concentrations, and scan rates will be scanned. These voltammetry parameters revealed that the electro-spawned o-benzoquinone and semicarbazide–diazenecarboxamide equilibrium are quite reactive intermediates that facilitate rapid Michael-addition cyclization in an aquatic medium to produce the novel triazinone adduct. The FT-IR, UV-Visible, GC/MS, 1H, and 13C NMR spectra were monitored to characterize the novel heterocyclic triazinone cyclo-adduct.
2. Materials and methods
2.1. Chemicals and reagents
A stock solution of 5 mM catechol (1) (Aldrich Chemical Co. Ltd, 99.8% purity) and 5 mM semicarbazide (3) (Sigma-Aldrich ≥99% purity) was prepared daily by dissolving the appropriate weight of reagents with deionized water/ethanol (50/50) as a base. A 0.10 M phosphate buffer solution (Na2HPO4, El Nasr Pharm. Chem. Co (ADWIC), 98% purity) was used as a supporting electrolyte. Furthermore, 0.10 M NaOH (El Nasr Pharm. Chem. Co (ADWIC), 96% purity) was used to adjust the pH to 7.0 before the electrolysis experiment. The other reagents and solvents in our experiments were of analytical grade and without any further purification. All the electrochemical measurements were carried out at room temperature (25 ± 1 °C).2.2. Electrochemical equipment
Voltammetric experiments were performed using an EG&G Princeton applied research potentiostat/galvanostat model 263 in a three-electrode electrochemical cell. A saturated Ag/AgCl, a high purity Pt wire (0.6 mm thick), and a glassy carbon electrode (GCE Cat. No. 002012, OD: 6 mm ID: 3 mm diameter, ALS Co., Ltd) served as the reference, counter, and working electrodes, respectively. A pH meter (CyberScan 500, Eutech-India) was used to measure the pH value. Before the experiment, the GCE was hand polished successfully with a 3 μm alumina slurry to a mirror finish and rinsed with deionized water. Later on, it was sonicated in a mixed solution of nitric acid and acetone (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for 5 minutes. A clean and active GCE surface can be checked by cycling the electrode potential between −1.0 and 1.0 V in a 1.0 M H2SO4 solution until a stable voltammogram is obtained. Finally, the GCE surface was washed with deionized water and dried in the air.
:1) for 5 minutes. A clean and active GCE surface can be checked by cycling the electrode potential between −1.0 and 1.0 V in a 1.0 M H2SO4 solution until a stable voltammogram is obtained. Finally, the GCE surface was washed with deionized water and dried in the air.
2.3. Procedures for the electro-organic synthesis of 6,7-dihydroxy-1,4-dihydrobenzo[e][1,2,4]triazin-3(2H)-one (9)
In a typical procedure, 100 mL of a water/ethanol mixture (50/50 v/v) containing a phosphate buffer (0.10 M, pH 7.0) was pre-electrolyzed in an undivided (≈25 cm3) glass cell at 0.50 V vs. Ag/AgCl. Then, 5 mM of catechol (1) and 5 mM of semicarbazide hydrochloride (3) were added to the electrochemical cell (Scheme 2). The reaction mixture was electrolyzed at a constant positive potential of 0.50 V vs. Ag/AgCl for some hours (≈12 h) in the air at room temperature. The electrosynthesis was terminated when the anodic current decreased by more than 95% of the initial current value. Due to the deposition and adsorption of reactants and product on the surface of the working electrode, the process was interrupted several times during the electrolysis, and the GCE was washed in acetone repeatedly for reactivation. At the end of electrolysis, a few drops of acetic acid were added to the solution, and the cell was poured into a Petri dish and left at room temperature for a few days until completely dry. The solid residue that was formed was heated in 10 mL of absolute ethanol and then filtered immediately. After cooling, the formed solid powder was filtered and washed thoroughly with petroleum ether 80–100 and crystallized from ethanol to form the novel triazinone 9 brown powder with a 68% yield. (Scheme 3).2.4. FT-IR and UV-visible spectroscopy measurements
The JASCO (FT/IR-4100) FOURIER TRANSFORM INFRARED SPECTROMETER was employed to measure the reactants' infrared spectra and the products (1, 3, and 9). Electronic UV-Visible absorption spectra were monitored on a Shimadzu 2401PC spectrophotometer (equipped with a Julabo F30 ultra thermostat with an accuracy of ± 0.5 °C) within the wavelength range of 200–800 nm using a thermostat (T = 25.0 ± 0.2 °C) 1 cm matched quartz cells. The reactants and products (1, 3, and 9) were prepared using a double-distilled aqueous phosphate buffer solution (0.10 M Na2HPO4). The concentrations of UV-Visible measured aqueous solutions of reactants and products (1, 3, and 9) were 3.63 × 10−4 M, 2.15 × 10−3 M, and 8.58 × 10−4 M, respectively.2.5. NMR spectroscopy measurements
The 1H and 13C NMR spectra were recorded on a Bruker AvanceII + 400 MHz, 9.4 T. Ultrashielded 1H frequency: 400 MHz spectrometers operating at 400 MHz for 1H and 101 MHz for 13C. Product sample 9 was dissolved in DMSO. The 1H and 13C chemical shifts (δ) are expressed in ppm and referenced to the central peak of the solvent DMSO (δ = 2.50 (1H) and 39.5 (13C)). All 1H and 13C experiments were performed using the manufacturer's software (TOPSPIN 2.1). All the measurements were achieved at room temperature (≈25 °C) in the NMR Unit, Faculty of Pharmacy, Mansoura University.2.6. Gas chromatography/mass spectrometry (GC/MS) measurements
The GC/MS were recorded on a PerkinElmer® Clarus® 600 GC/MS (USA) with a programmable split/splitless injector. Analyses were performed with helium as a carrier gas at a constant pressure mode (65.2 kPa). The separation was carried out in a gradient temperature program.3. Results and discussion
Catechol (1) to o-benzoquinone (2) electro oxidation–reduction equilibrium and semicarbazide·HCl (3) oxidation are well-recognized electrochemical reactions but are not still clear yet.7,8 A further electrochemical study for these reactions is performed at different pH values to clarify and confirm the suggested interaction mechanism between catechol and hydrazides.8–10,13,15,17,19,233.1. Effect of pH on the transformation of catechol (1) to o-benzoquinone (2)
To attain an accurate pH value, preliminary electrochemical measurements were carried out on a GCE using different pH values. Fig. 1A shows the cyclic voltammograms of 5 mM catechol (1) (in the absence of 3) in a 0.10 M phosphate buffer solution (pH range of 3.0–11.0) at a scan rate of 100 mV s−1. It is clear that as the pH of the solution increases, the peak potentials shift negatively. Moreover, at higher pH values, the peak current ratio (IClp/IAlp) for catechol (1) rapidly decreases because of unwanted side reactions such as dimerization or hydroxylation.34,39 It must be noted that the minor potential difference between anodic and cathodic peaks (ΔEp = EpA − EpC) occurs at pH 3.0. However, the anion of hydrazide nucleophiles (3 and 4) (Scheme 1) can be protonated and inactivated toward the Michael addition reaction with o-benzoquinone (2) under acidic conditions.41 Additionally, in primary conditions, the peak current ratio (IClp/IAlp) is less than unity and decreases with an increasing pH (Fig. 1B) because of the dimerization reaction.10,45 Therefore, a phosphate buffer solution (0.10 M, pH 7.0) was subsequently chosen as an optimal pH value to obtain a high yield of purity products.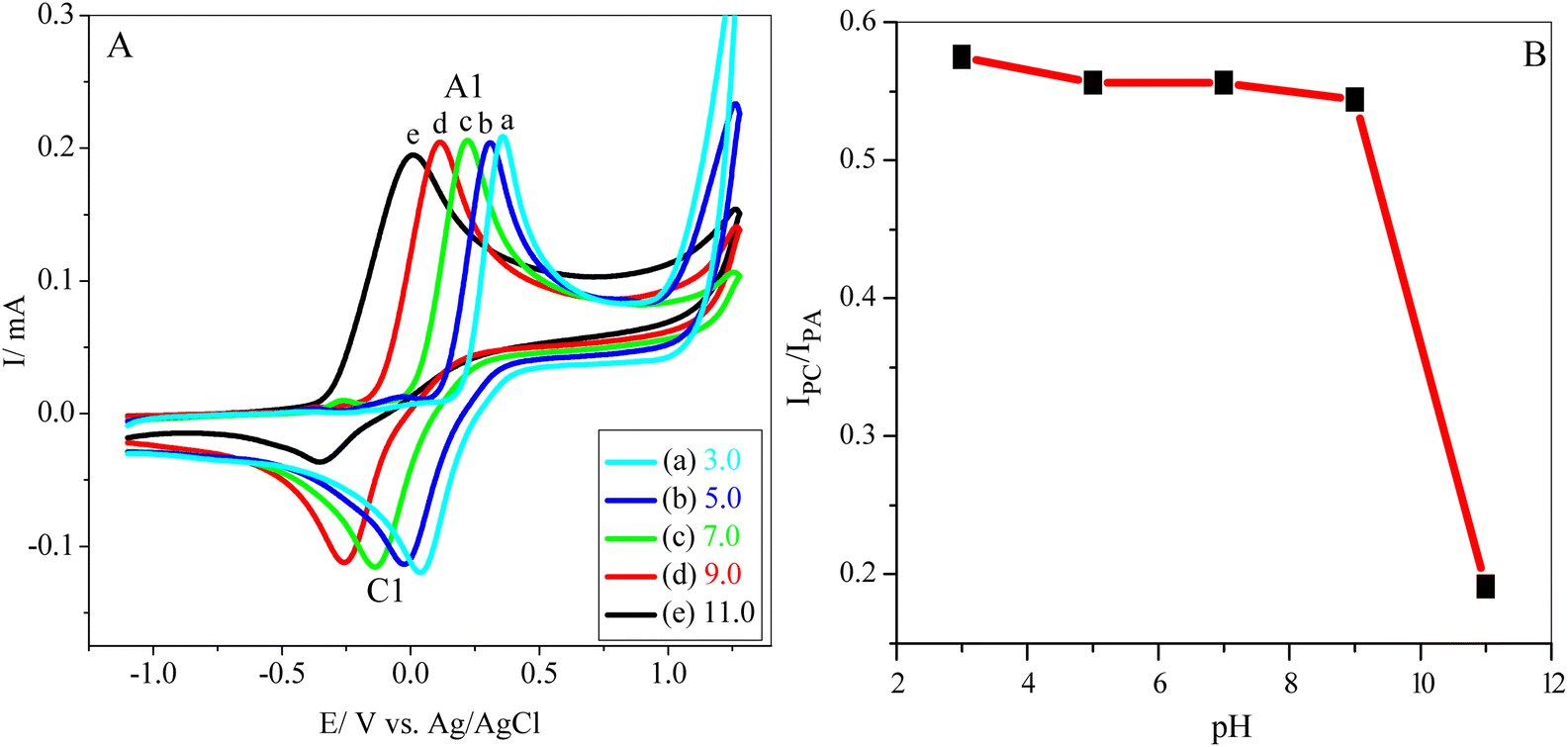 | ||
| Fig. 1 (A) CVs of 5 mM catechol (1) in a 0.10 M phosphate buffer solution with various pHs (a to e are 3.0, 5.0, 7.0, 9.0, and 11.0). (B) Variation of peak current ratio (IClp/IAlp) vs. pH at a scan rate 100 mV s−1, T = 25 ± 1 °C. | ||
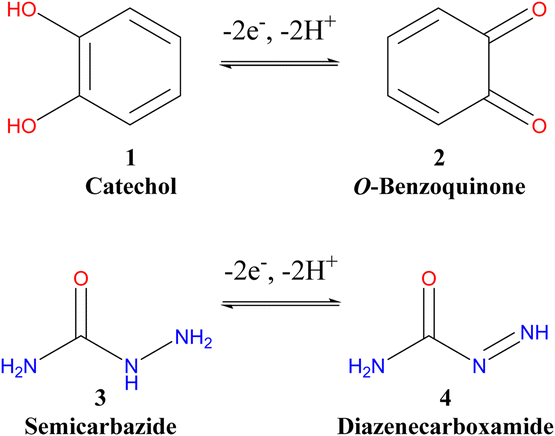 | ||
| Scheme 1 The suggested oxidation–reduction equilibriums of catechol (1) and semicarbazide (3) in the absence of each other.8–11,13,15 | ||
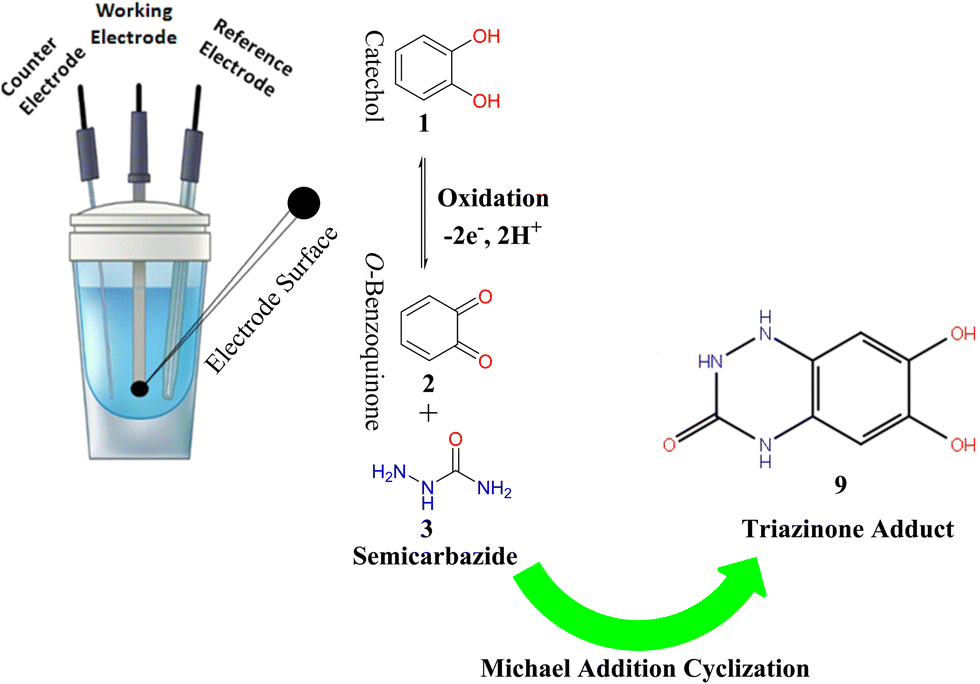 | ||
| Scheme 2 The graphical summary of the electrochemical synthesis of the triazinone (9). | ||
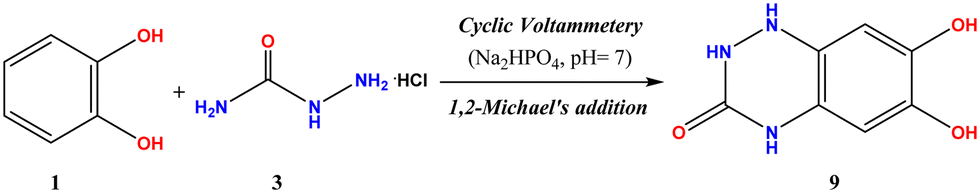 | ||
| Scheme 3 Cyclic voltammetry of catechol (1) and semicarbazide. HCl to form the triazinone (9). | ||
3.2. Electro-oxidation of catechol (1) in the presence of semicarbazide (3)
Our cyclic voltammetry (CV) was employed to investigate the electrochemical behavior of catechol (1) in the absence and presence of semicarbazide (3) as a nucleophile. Fig. 2 shows the typical CVs of a 5 mM solution of catechol (1) in water/ethanol (50/50) containing phosphate buffer (0.10 M, pH 7.0) in the presence and absence of 5 mM of semicarbazide (3). As can be seen in Fig. 2, the CV of 1 in the absence of 3 (curve a) shows one anodic peak (A1) at 0.22 V (vs. Ag/AgCl), and a corresponding cathodic peak (C1) at −0.14 V. These well-defined peaks belong to the electrochemical oxidation of catechol (1) to o-benzoquinone (2) and vice versa (Scheme 1) within a quasi reversible two-electron process.44 Furthermore, the peak current ratio (IClp/IAlp) is nearly equal to unity, thus confirming the relative stability of o-benzoquinone produced on the GCE surface under optimum conditions.7,15,46,47 The electro-oxidation of a 5 mM solution of 1 and 5 mM of 3 as the nucleophile was studied in detail.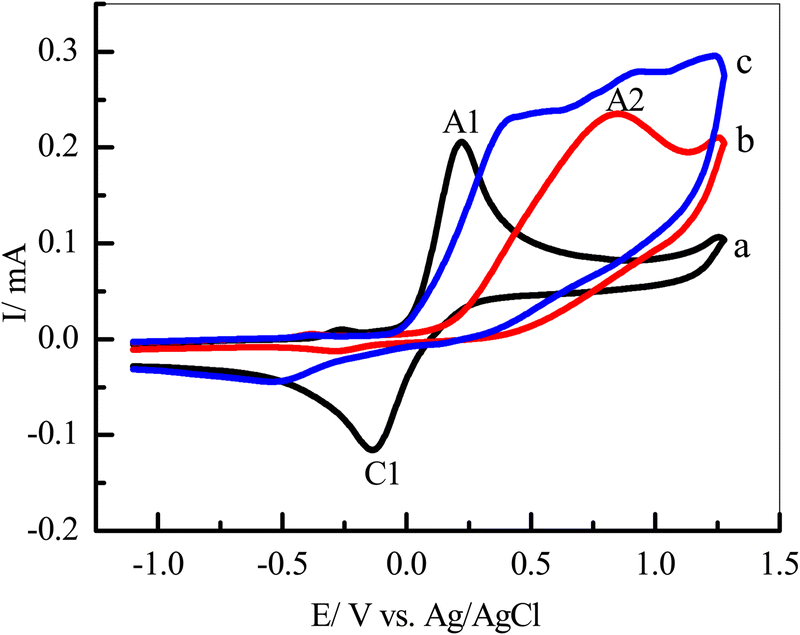 | ||
| Fig. 2 CVs of (a) 5 mM catechol (1) in the absence of semicarbazide (3), (b) 5 mM of 3 in the absence of 1 and (c) 5 mM of 1 with 5 mM of 3 in 0.10 M of phosphate buffer/ethanol (50/50, v/v), pH 7.0 on the GCE at a scan rate 100 mV s−1, T = 25 ± 1 °C. | ||
In Fig. 2, curve b is the CV of 3 in the absence of 1 with the same range of potential, respectively; only one anodic peak, A2, which appeared at 0.85 V (Fig. 2, curve b), was related to the oxidation of 3 to 4 (Scheme 1). Comparing between anodic peaks (A1, 0.22 V and A2 0.85 V) potentials, we can observe that oxidation of catechol 1 is more favourable than oxidation of semicarbazide 3.
In the end, the CVs were obtained for a 5 mM solution of 1 in the presence of 5 mM of 3 (Fig. 2, curve c). We have observed that the current of the anodic peak (A1) considerably increased while the cathodic counterpart of the anodic peak, relating to the reduction of o-quinone, rapidly decreased. This indicates a chemical reaction between the electrochemically generated oxidized o-benzoquinone (2) at A1 and nucleophile 3. The potential of peak A1 shifted toward a more positive value, and peak C1 shifted toward a more negative value (Fig. 2, curves c) in the presence of 3. These changes are probably due to a thin film of product formed at the surface of the GC electrode, inhibiting to some extent the performance of the electrode process.7,15,46,47
3.3. Effect of the number of cycles
The multi-CVs of a 5 mM solution of 1 in water/ethanol (50/50) containing phosphate buffer (0.10 M, pH 7.0) in the presence of 5 mM of 3 were also recorded (Fig. 3). It is observed that no new anodic peak related to the oxidation of the catechol intermediates 6 and 7 appeared in the consecutive cycles (Scheme 4). This is a result of the oxidation potentials 6 and 7 being very close to the oxidation potential of the starting catechol (1).1,11,45,48,49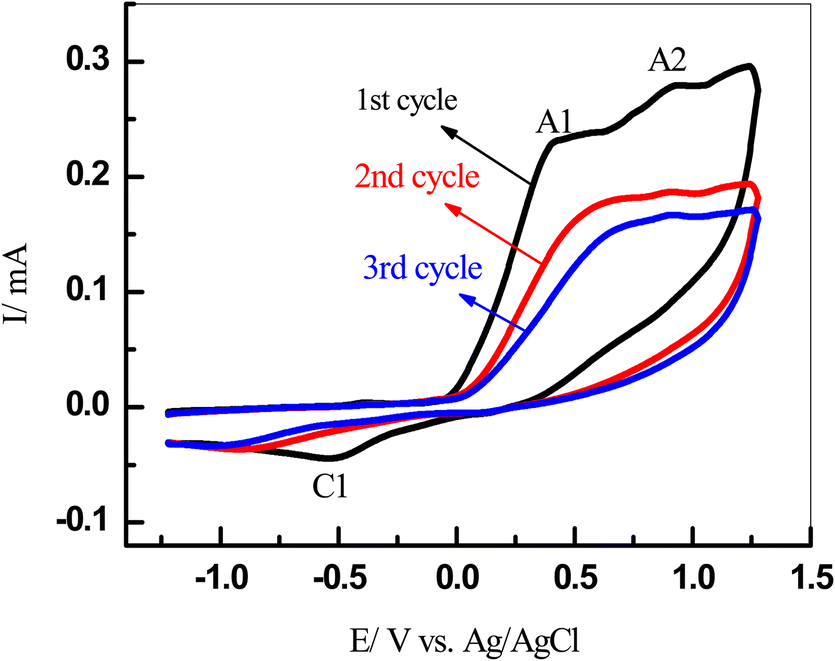 | ||
| Fig. 3 Multi-CVs of a 5 mM solution of 1 in water/ethanol (50/50) containing a phosphate buffer (0.10 M, pH 7.0) in the presence of 5 mM of 3 at the GCE at a scan rate of 100 mV s−1, T = 25 ± 1 °C. | ||
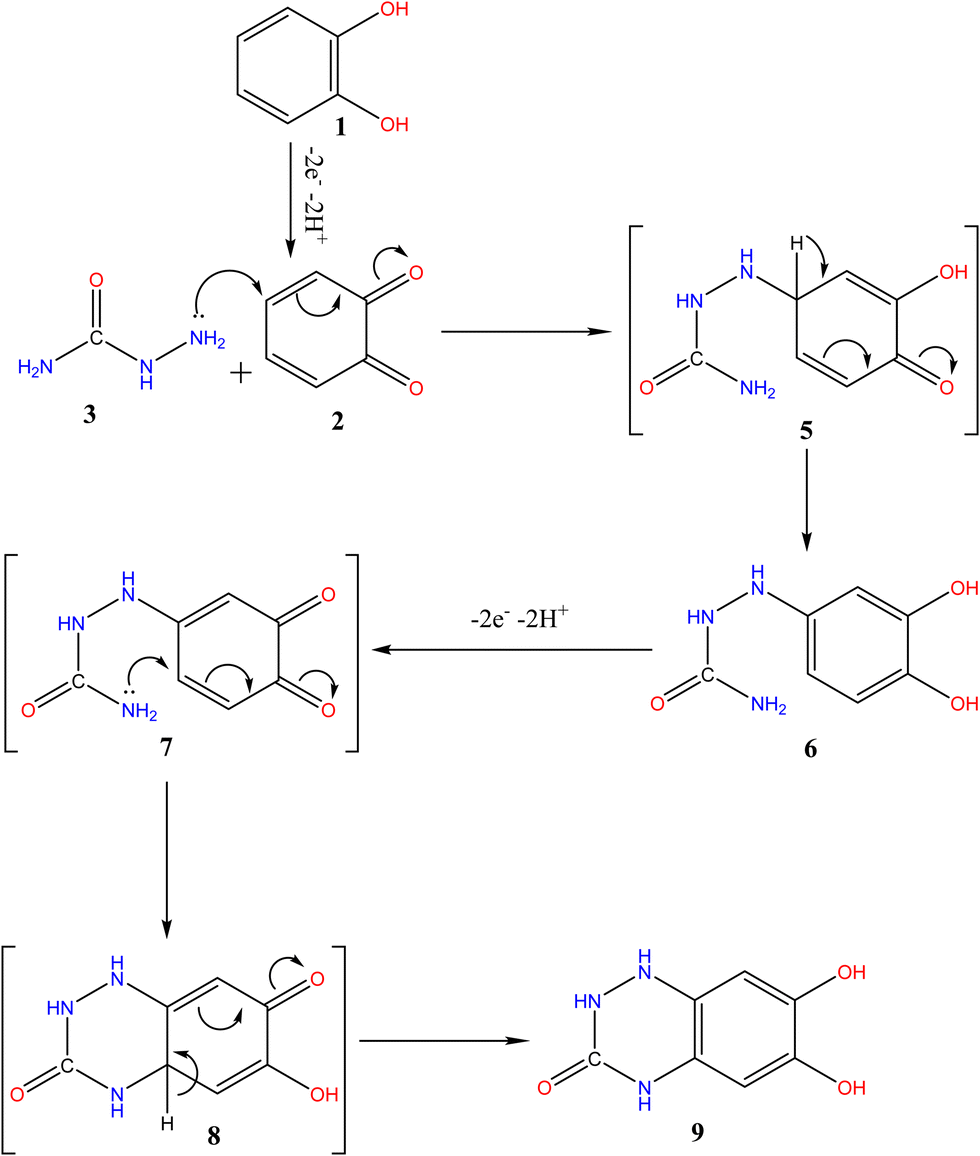 | ||
| Scheme 4 The proposed ECEC mechanism of 1,2-Michael's addition reaction between o-benzoquinone (2) and semicarbazide (3) to form the triazinone 9.1,11,45,48,49 | ||
Thus, the intermediates 6 and 7 could be oxidized with an oxidation peak close to that of catechol. On the other hand, repetitive cycling leads to a decrease in the current peak. Moreover, their oxidation potential shifted to more positive values and vice versa for the reduction peak. This is probably because of the formation of a thin film of product at the electrode surface, inhibiting to a certain extent the performance of the electrode process8,10,18,19,45
3.4. Effect of scan rate
The electrochemical behaviour of 5 mM of 1 in water/ethanol (50/50) containing phosphate buffer (0.10 M, pH 7.0) in the presence of 5 mM of 3 at various scan rates was also studied (Fig. 4). It is apparent that the anodic peak (A1) current increases proportionally with the increase in the scan rate while in the presence of nucleophile 3, the cathodic peak (C1) of catechol was not observed even at higher scan rates (Fig. 4). These behaviours can be attributed to 3's high reactivity in terms of 1,4-Michael addition reaction. To prove this mechanism, a plot of the peak current function for the anodic peak A1 (Ilp/v1/2) versus the scan rate was shown in Fig. 5. It is clear that the peak current function was decreased at a lower scan rate. Such behaviour is adopted as indicative of an electron transfer followed by a chemical reaction (EC) mechanism.8,19,45,50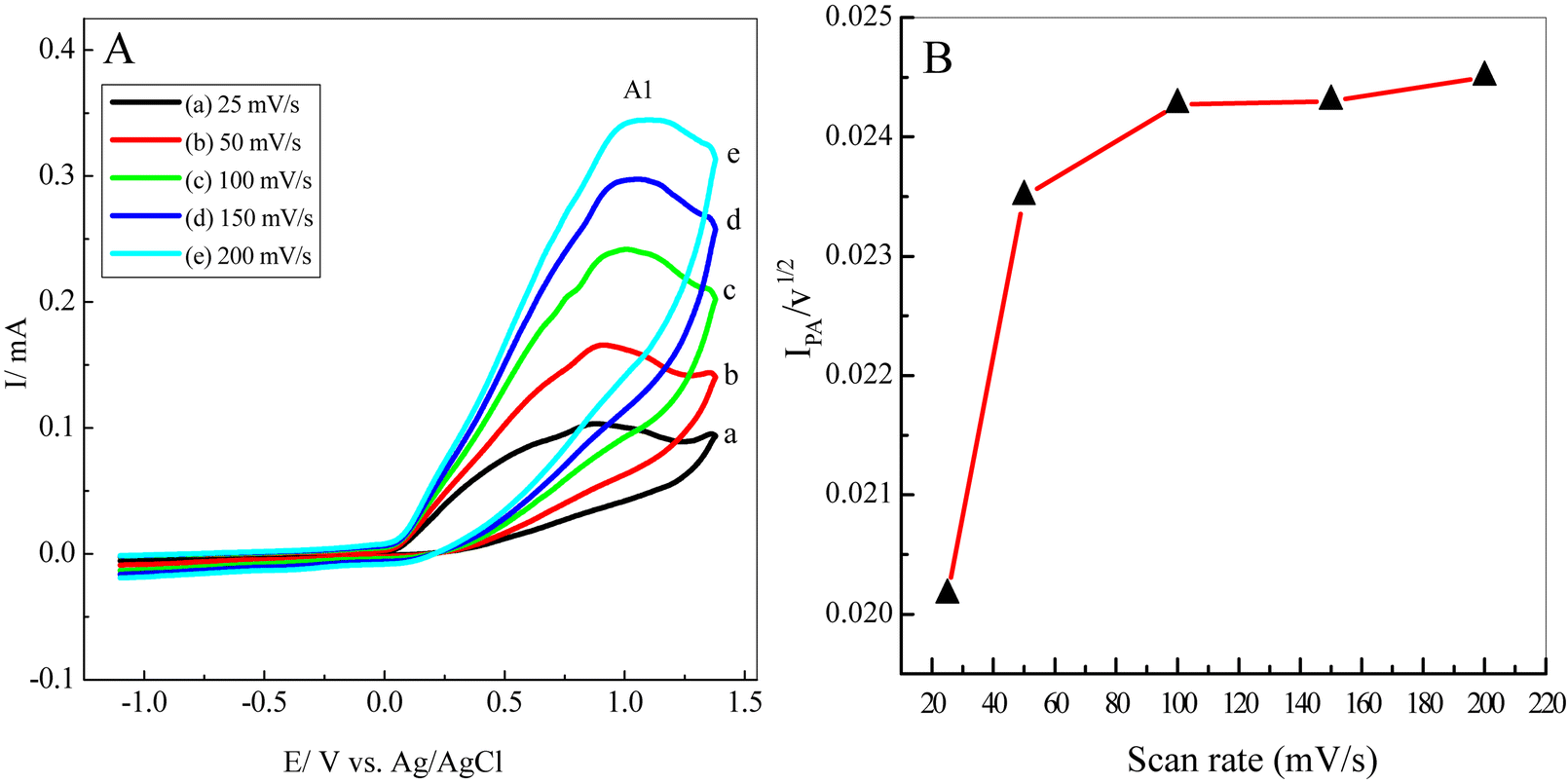 | ||
| Fig. 4 (A) Typical CVs of a 5 mM solution of catechol (1) in water/ethanol (50/50) containing phosphate buffer (0.10 M, pH 7.0) in the presence of 5 mM of semicarbazide. HCl (3) at different scan rates (25, 50, 100, 150, and 200 mV s−1) at the GCE, T = 25 ± 1 °C. (B) Variation of (Ilp/v1/2) vs. scan rate. | ||
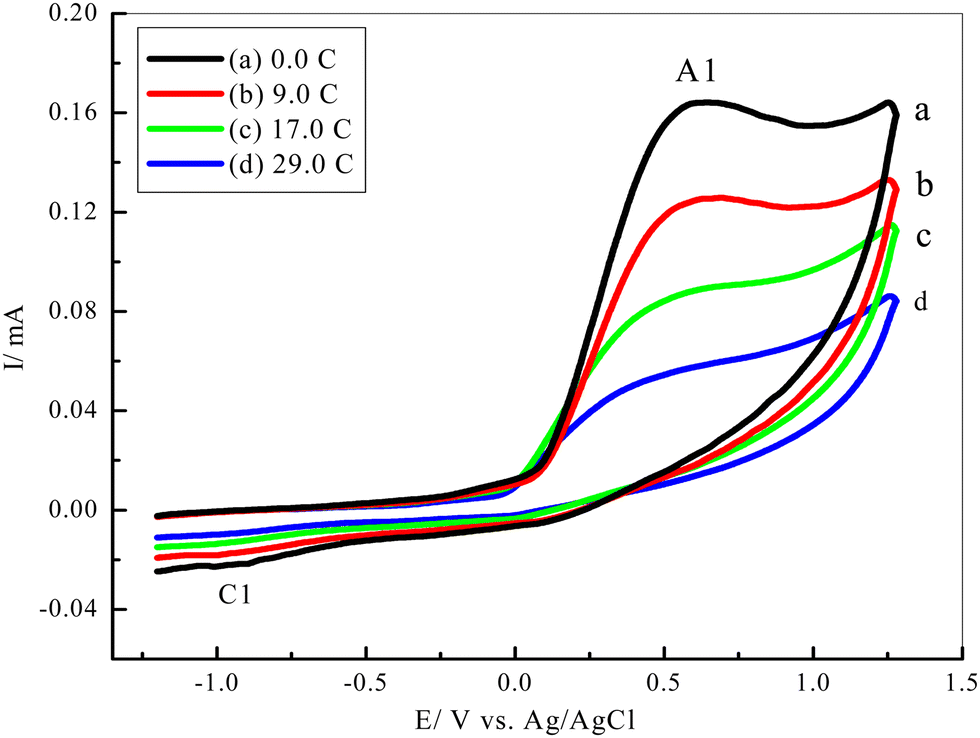 | ||
| Fig. 5 CVs of a 5 mM solution of 1 in water/ethanol (50/50) containing phosphate buffer (0.10 M, pH 7.0) in the presence of 5 mM of 3 at the GCE during controlled-potential coulometry at 0.35 V vs. Ag/AgCl, after the consumption of coulomb charge (a to d are 0.0, 9.0, 17.0 and 29.0 C) at a scan rate 100 mV s−1, T = 25 ± 1 °C. | ||
3.5. Controlled-potential electrolysis of catechol (1) in the presence of semicarbazide (3)
For more mechanistic studies, controlled potential coulometry was performed in a solution of ethanol-phosphate buffer (0.10 M, pH 7.0) containing 5 mM of catechol (1) and 5 mM of semicarbazide. HCl (3). The progress of coulometry was monitored using the cyclic voltammetry method at a scan rate of 100 mV s−1, as seen in (Fig. 5). The anodic (A1) and cathodic (C1) current peaks decreased and finally almost disappeared as the coulometric process proceeded.3.6. Reaction mechanism
Based on the above results, We noted that the first EC mechanism8,36 of the 1,2-Michael addition reaction of 3 and o-benzoquinone (2) (Scheme 4) is much faster than other side reactions, leading presumably to intermediate 6.1,11,45,48,49 Additionally, compound 6 oxidation is easier than the oxidation of the parent starting molecule 1 because of the existence of an electron-donating group,45,49,51 with the formation of an intermediate 7. Following this, the nucleophilic NH2 quickly attacks via the second EC mechanism,8,36,37 in a second 1,2-Michael addition, through the formation of intermediate 8, which is reduced to form product 9. The overall proposed mechanism in (Scheme 4) can be routed via the ECEC mechanism pathway.8,32,36,373.7. Spectroscopic characterizations of electro-synthesized 6,7-dihydroxy-1,4-dihydrobenzo[e][1,2,4]triazin-3(2H)-one (9)
The chemical structure of the electro-synthesized product 9 was characterized by FT-IR, UV-Visible, 1H NMR, 13C-NMR, and GC/MS spectra. After crystallization from ethanol, compound 9 was obtained as a brown powder, with m.p. 185–186 °C and 68% yield.The FT-IR spectra of reactants (catechol and semicarbazide) and the product are represented in Fig. 6. The FT-IR spectrum of semicarbazide. HCl showed (NH2 and NH) stretching vibration bands at 3433, 3252, and 3315 cm−1.
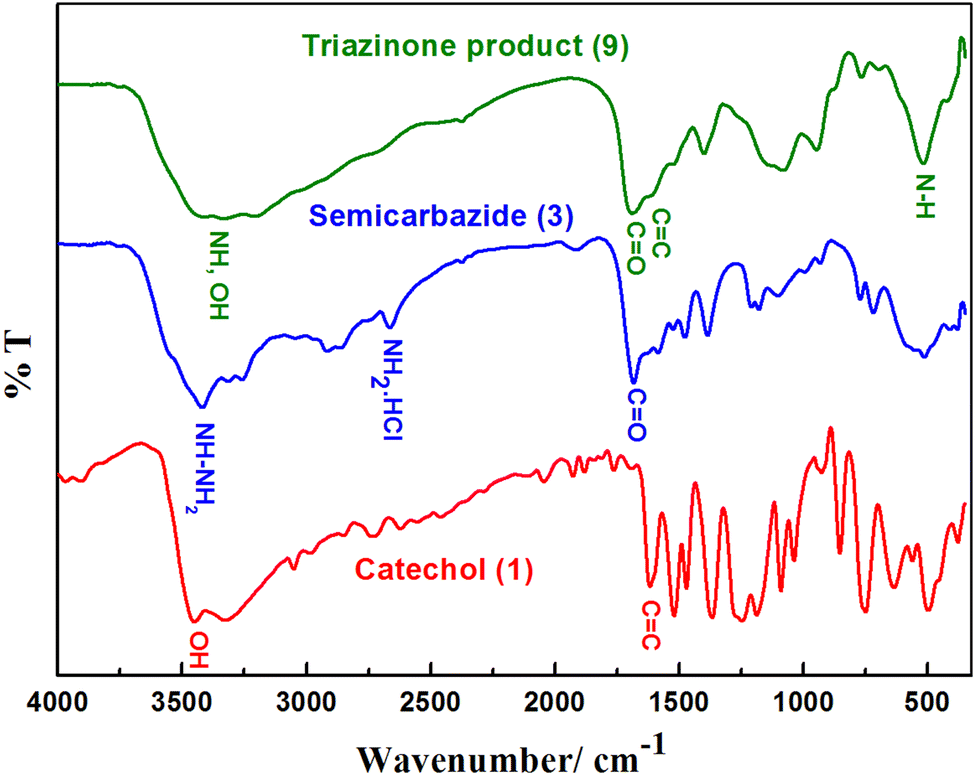 | ||
| Fig. 6 FT-IR spectra of catechol (1), semicarbazide (3), and triazinone (9). | ||
However, bands at 2920 cm−1, 2868 cm−1, and 2670 cm−1 are assigned to N–H of (NH2·HCl) amine hydrochloride 3. Furthermore, a specific peak at 1682 cm−1 originates from C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of the primary amide overlapping with N–H bending and bands at 1209 and 1181 cm−1 for N–C–N Sym, 770 and 512 cm−1 for NH2 deformation, and 716 cm−1 for NH deformation.
O stretching vibrations of the primary amide overlapping with N–H bending and bands at 1209 and 1181 cm−1 for N–C–N Sym, 770 and 512 cm−1 for NH2 deformation, and 716 cm−1 for NH deformation.
The FT-IR of catechol (Fig. 6) showed stretching vibrations of the OH groups at 3455 cm−1 and 3322 cm−1, as well as C–H stretching oscillation of the aromatic ring of catechol at 3050 cm−1. Moreover, bands between 1622 cm−1 and 1472 cm−1 are attributed to the aromatic ring of CC vibration. Absorption bands at 1259 cm−1 and 1182 cm−1 are assigned to the C–O vibration of catechol. The other absorption bands at 852 cm−1 and 748 cm−1 are ascribed to the out-of-plane bending of the –C–H bonds of the ortho-substituted aromatic ring of catechol.
Finally, the FT-IR of the triazinone product 9 (Fig. 6) showed broad bands of both OH and NH groups at 3422 cm−1 and 3197 cm−1. The shift to a short wavenumber (red-shift) and the broadness of these bands confirmed the formation of a cyclized product between catechol and semicarbazide.7,8,10,13,17,19,22,23 Furthermore, the disappearance of semicarbazide and catechol bands at 2920 cm−1, 2868 cm−1, and 2670 cm−1 in the spectrum of product 9 is referred to as the cyclization between catechol and semicarbazide to form the new product.8–10,13,15,17,19,23
Moreover, the broadness of the bands at 1685 cm−1 is attributed to the CO stretching vibration of semicarbazide's secondary amide overlapping with the aromatic ring of the CC vibration bands of catechol. This confirms the formation of a novel cyclized product. Additionally, the bands at 1521 cm−1 and 1400 cm−1 are shifted to short wavenumbers compared with the 1585 cm−1 N–H bending of the secondary amide of semicarbazide, and the 1470 cm−1 C–N and CC stretching band of semicarbazide and catechol confirm, without doubt, the formation of a cyclized product between catechol and semicarbazide.
The broadness and the red-shifts of the peaks at 1137 cm−1 and 1087 cm−1 are assigned to the cyclized novel product's C–O vibration bands compared with the catechol C–O vibration bands at 1259 cm−1 and 1182 cm−1. The disappearance of specific ortho out-of-plane –C–H strong bending bands of catechol at 852 cm−1, 748 cm−1 and the presence of new, weak broad bending bands at 872 cm−1 and 765 cm−1 are ascribed to the out-of-plane bending of the –C–H bonds of the tetrasubstituted aromatic ring of the cyclized product. The shift to long wavenumber and sharpness of the band at 519 cm−1 could be assigned to the N–H deformation of the cyclized ring formed in the product (9).8–10,13,15,17,19,23
Furthermore, the UV-Visible study of the reactants (catechol (1) and semicarbazide (3)) and triazinone 9 were investigated in a phosphate buffer solution (Fig. 7). The appearance of a wide broad peak at 387 nm accompanied by a broad peak at 275 nm confirms the formation of the novel cyclized product (9), despite the existence of a sharp peak at 275 nm in catechol (1) and the disappearance of a characteristic peak in the range 250–600 nm of semicarbazide (3).
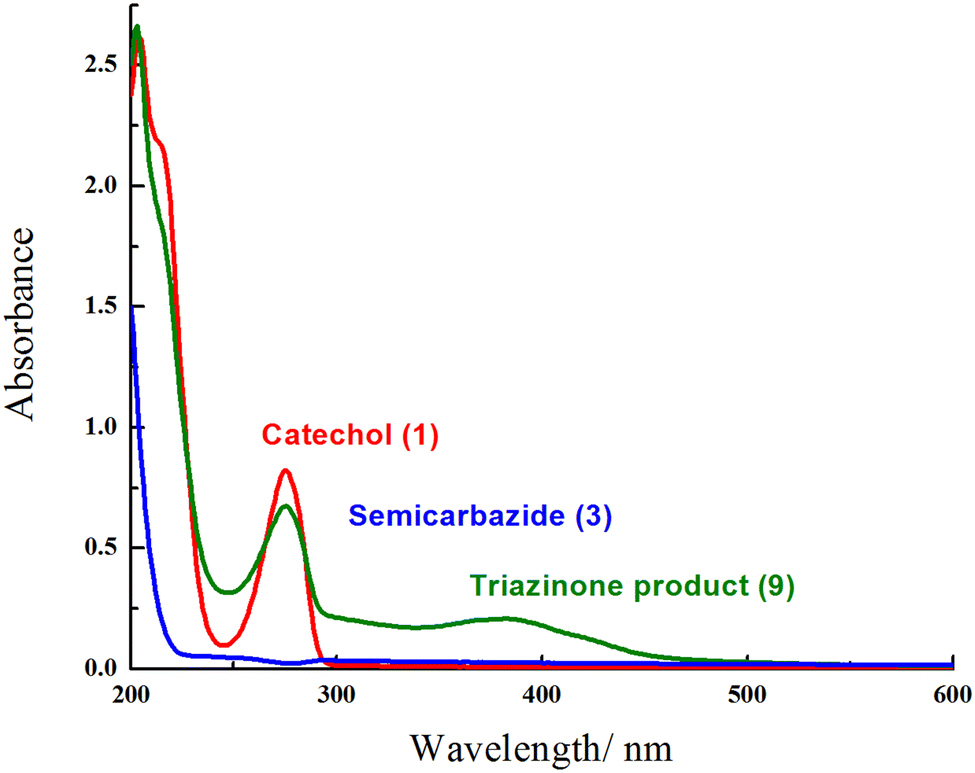 | ||
| Fig. 7 UV-Visible spectra of catechol (1), semicarbazide (3), and the triazinone product (9) in a phosphate buffer solution. | ||
1H and 13C NMR are potent tools for characterizing our electro-synthesized product. The 1H and 13C NMR spectra of product 9 in DMSO-d6 are represented in Fig. 8 and 9. The proton NMR spectrum of adduct 9 (Fig. 8) showed accompanying broad bands at chemical shift (δ 5.93–6.03 ppm) could be assigned to two NH protons23,52–56 of the novel triazinone ring. Similarly, 2 accompanied OH protons exist as a broad peak at chemical shift (δ 7.50 ppm) formed merged with a third amidic NH proton at δ 7.80 ppm.
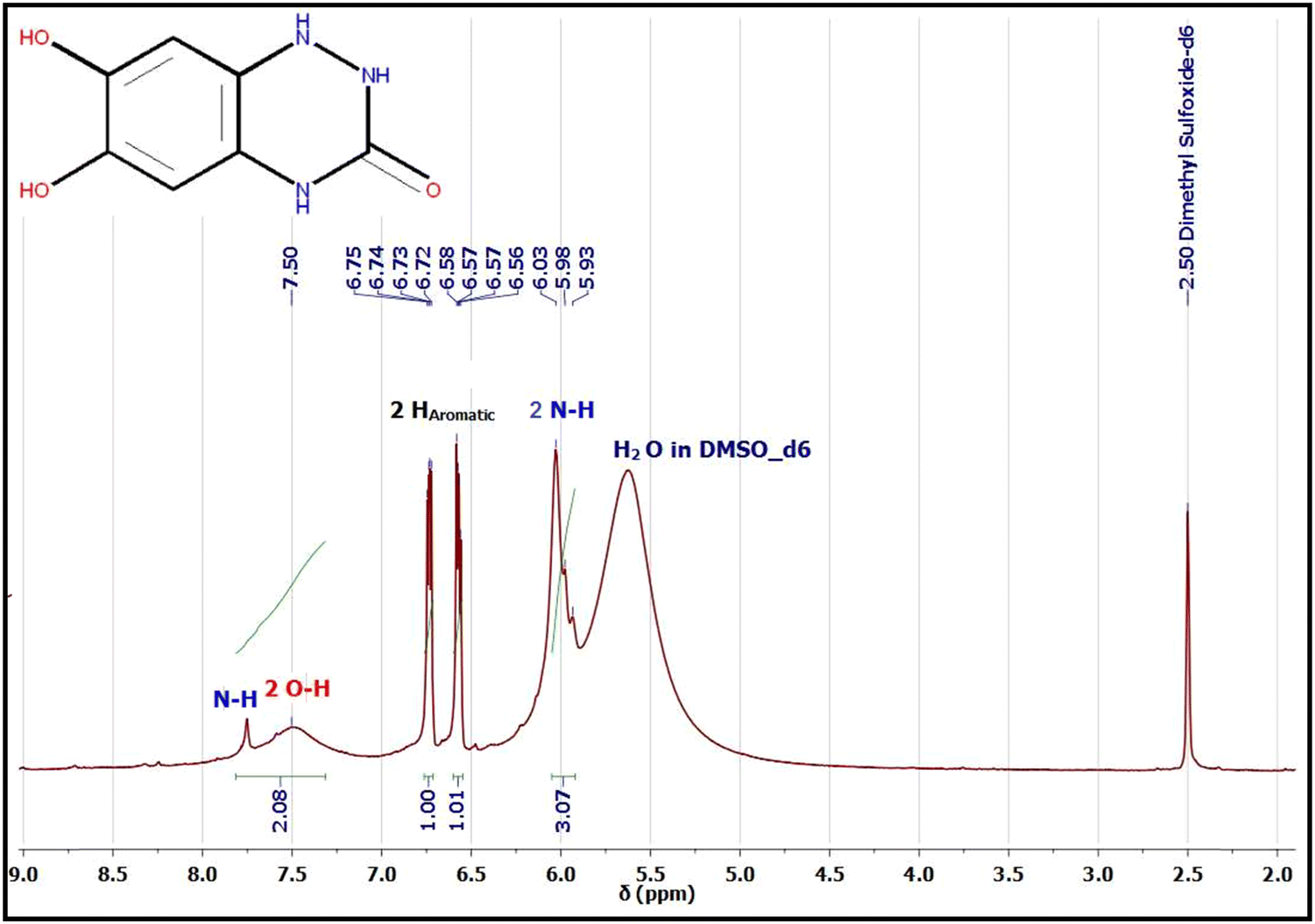 | ||
| Fig. 8 1H NMR spectrum of electro-synthesized product 9 in DMSO-d6. | ||
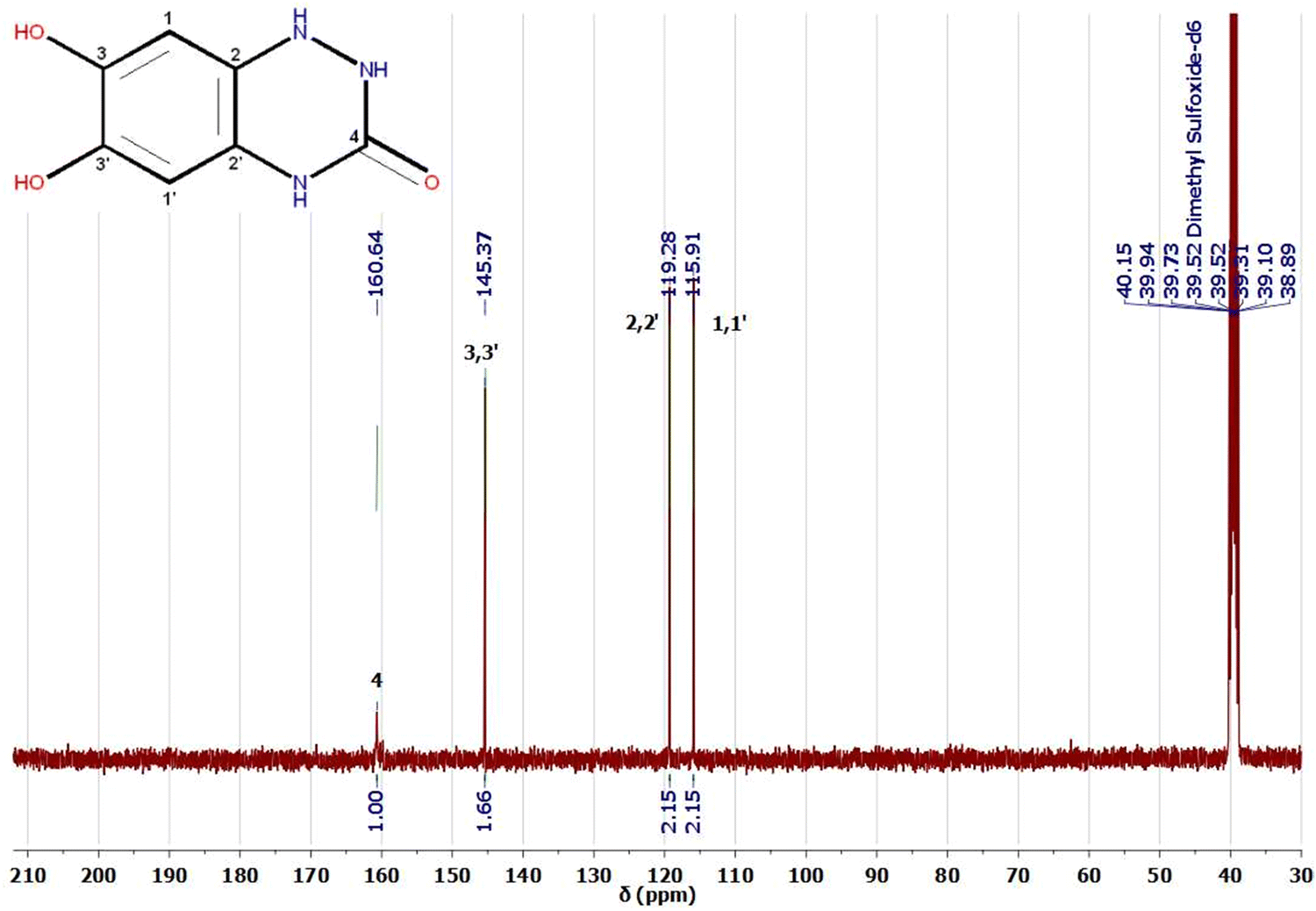 | ||
| Fig. 9 13C NMR spectrum of the electro-synthesized product 9 in DMSO-d6. | ||
On the other hand, the appearance of only two sharp bands of the aromatic protons at chemical shifts δ 6.73 ppm (dd, J = 5.7, 3.6 Hz, 1H) and δ 6.57 ppm (dd, J = 5.7, 3.6 Hz, 1H) confirmed without any doubt the formation of the novel triazinone ring in conjugation with the catechol ring.11,53 These two aromatic protons appear as double doublets due to the long-range coupling between OH and NH groups. However, 1H NMR in D2O, the exchangeable protons (OH and NH) disappear,23,52–55 and the aromatic protons of triazinone product 9 show two singlet peaks at chemical shifts δ 7.00 ppm and δ 7.07 ppm as shown in the ESI† (Fig. S1). Moreover, 13C NMR data in DMSO-d6 (Fig. 9) support the results of 1H NMR, IR, and UV, where the existence of sharp peaks at δ 160.64 ppm (for CO novel triazinone ring),11,53–56δ 145.37 ppm (for overlapped57,58 two aromatic carbon atoms connected to OH groups), δ 119.28 ppm (for two overlapped57–59 aromatic carbon atoms connected to N–H groups), and at δ 115.91 ppm (for two superimposed57–59 aromatic carbon atoms). Also, the 13C NMR of triazinone product 9 in D2O in the ESI† (Fig. S2) is inconsistent with our 13C NMR data in DMSO-d6. Furthermore, product sample 9 shows a sharp GC peak (100% intensity) at a retention time of 34.69 minutes, confirming the high purity of this product and the m/z [M + H+] = 182.0, m/z [M] = 181, m/z [M − H+] = 180.0. (More details about GC/MS fragmentation of the product 9 are achieved in ESI† (Fig. S5 and S6.)
Finally, our IR, UV-Visible, NMR, and GC/MS spectra established without doubt the formation of the novel cyclized triazinone ring connected to the catechol ring (product 9).
4. Conclusions
In this work, the electro-organic synthesis of catechol (1) followed by a 1,2-Michael addition reaction with semicarbazide (3) via an ECEC mechanism was carried out. The electron transfer rate was faster in a phosphate buffer solution with a pH value of 7.0. The results showed that o-benzoquinone (2) could be used as a mediator in the synthesis of many organic compounds using the green and facile electrochemical method at room temperature. The study of the novel product, using FT-IR, UV-Visible, GC/MS, and NMR spectroscopic techniques, revealed the formation of a novel triazinone heterocyclic ring (9). Under eco-friendly conditions, this simple way was found to be particularly effective in producing a new catechol derivative, 9, with a 68% yield.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors extend their appreciation to the Deputyship for Research and Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project Number IFG-IMAMU-0701. Moreover, The authors extend their grateful to Chemistry Department, Faculty of Science, South Valley University, Egypt.References
- M. Ameri, A. Asghari, A. Amoozadeh, H. Daneshinejad and D. Nematollahi, Chin. Chem. Lett., 2014, 25, 797–801 CrossRef CAS.
- M. A. Chamjangali, H. Daneshinejad, M. Bakherad and M. Ameri, Cheminf., 2016, 47, 5562 Search PubMed.
- M. Tranchant, A. Serrà, C. Gunderson, E. Bertero, J. García-Amorós, E. Gómez, J. Michler and L. Philippe, Appl. Catal., A, 2020, 602, 117698 CrossRef CAS.
- Y. Yuan and A. Lei, Nat. Commun., 2020, 11, 802 CrossRef CAS PubMed.
- S. Bornemann and S. T. Handy, Molecules, 2011, 16, 5963–5974 CrossRef CAS PubMed.
- S. S. H. Davarani, N. S. Fumani, H. Arvin-Nezhad and F. Moradi, Tetrahedron Lett., 2008, 49, 710–714 CrossRef CAS.
- H. Filik and A. A. Avan, Arabian J. Chem., 2020, 13, 6092–6105 CrossRef CAS.
- H. Ghadimi, A. S. Mohamed Ali, N. Mohamed and S. Ab Ghani, J. Electrochem. Soc., 2012, 159, E127–E131 CrossRef CAS.
- D. Nematollahi, A. Amani and E. Tammari, J. Org. Chem., 2007, 72, 3646–3651 CrossRef CAS.
- D. Nematollahi, A. Afkhami, E. Tammari, T. Shariatmanesh, M. Hesari and M. Shojaeifard, Chem. Commun., 2007, 162–164 RSC.
- D. Nematollahi and H. Shayani-jam, J. Org. Chem., 2008, 73, 3428–3434 CrossRef CAS PubMed.
- H. J. Schäfer, C. R. Chim., 2011, 14, 745–765 CrossRef.
- A. Toghan, A. M. Abo-Bakr, H. M. Rageh and M. Abd-Elsabour, RSC Adv., 2019, 9, 13145–13152 RSC.
- R. Rekik, M. Hamza, M. Jaziri and R. Abdelhedi, Arabian J. Chem., 2020, 13, 357–365 CrossRef CAS.
- S. M. Sayyah and R. E. Azooz, Arabian J. Chem., 2016, 9, S576–S586 CrossRef CAS.
- F. Melak, M. Redi, M. Tessema and E. Alemayehu, Nat. Sci., 2013, 05, 888–894 Search PubMed.
- Y. H. Huang, J. H. Chen, L. J. Ling, Z. B. Su, X. Sun, S. R. Hu, W. Weng, Y. Huang, W. B. Wu and Y. S. He, Analyst, 2015, 140, 7939–7947 RSC.
- R. Abdel-Hamid, M. K. Rabia and E. F. Newair, Arabian J. Chem., 2016, 9, 350–356 CrossRef CAS.
- A. Karthika, V. Ramasamy Raja, P. Karuppasamy, A. Suganthi and M. Rajarajan, Arabian J. Chem., 2020, 13, 4065–4081 CrossRef CAS.
- Z. You, Y. Fu, A. Xiao, L. Liu and S. Huang, Arabian J. Chem., 2021, 14, 102990 CrossRef CAS.
- M. Palaniandavar and R. Mayilmurugan, C. R. Chim., 2007, 10, 366–379 CrossRef CAS.
- N. Al-Qasmi, M. Tahir Soomro, I. M. I. Ismail, E. Y. Danish and A. A. Al-Ghamdi, Arabian J. Chem., 2019, 12, 946–956 CrossRef CAS.
- F. Hasanpour, M. Nekoeinia, A. Semnani and R. Shirazinia, Chem. Pap., 2019, 73, 2081–2089 CrossRef CAS.
- I. V. Smolyaninov, G. K. Fukin, N. T. Berberova and A. I. Poddel’sky, Molecules, 2021, 26(8), 2171 CrossRef CAS.
- İ. Polatoğlu and D. Karataş, J. Mol. Struct., 2020, 1202, 127192 CrossRef.
- H. D. Beall and S. I. Winski, Front. Biosci., 2000, 5, 639–648 Search PubMed.
- A. J. Shuhendler, R. Y. Cheung, J. Manias, A. Connor, A. M. Rauth and X. Y. Wu, Breast Cancer Res. Treat., 2009, 119, 255 CrossRef PubMed.
- K. Kaleem, F. Chertok and S. Erhan, Prog. Org. Coat., 1987, 15, 63–71 CrossRef.
- M. N. Akhtar, M. Shahid, M. S. Ahmad, W. Zierkiewicz, M. Michalczyk, M. B. Taj, M. Khalid and M. A. Hanif, J. Mol. Struct., 2022, 1252, 131685 CrossRef CAS.
- C. J. Cramer, Essentials of Computational Chemistry, Theories and Models, Wiley, Chichester, England, 2002, p. 562 Search PubMed.
- B. Prabhu, A. Sivakumar, D. Balakrishnan and S. Sundaresan, J. Exp. Ther. Oncol., 2015, 11, 139–416 CAS.
- D. Nematollahi, M. Alimoradi and S. W. Husain, Electrochim. Acta, 2006, 51, 2620–2624 CrossRef CAS.
- C. Belle, K. Selmeczi, S. Torelli and J.-L. Pierre, C. R. Chim., 2007, 10, 271–283 CrossRef CAS.
- S. Amidi, F. Kobarfard, A. B. Moghaddam, K. Tabib and Z. Soleymani, Iran. J. Pharm. Res., 2013, 12, 89–101 Search PubMed.
- S. S. H. Davarani, N. S. Fumani, H. Arvin-Nezhad and F. Moradi, Tetrahedron Lett., 2008, 49, 710–714 CrossRef CAS.
- D. Nematollahi and Z. Forooghi, Electroanalysis, 2003, 15, 1639–1644 CrossRef CAS.
- A. S. Kumar, R. Shanmugam, N. Vishnu, K. C. Pillai and S. Kamaraj, J. Electroanal. Chem., 2016, 782, 215–224 CrossRef CAS.
- K. V. N. Esguerra and J.-P. Lumb, Chem. – Eur. J., 2017, 23, 8596–8600 CrossRef CAS.
- T. L. Smalley, A. J. Peat, J. A. Boucheron, S. Dickerson, D. Garrido, F. Preugschat, S. L. Schweiker, S. A. Thomson and T. Y. Wang, Bioorg. Med. Chem. Lett., 2006, 16, 2091–2094 CrossRef CAS PubMed.
- M. Pitucha, Z. Karczmarzyk, M. Swatko-Ossor, M. Drozd, W. Wysocki, G. Ginalska, Z. Urbanczyk-Lipkowska, D. Kowalczuk and M. Morawiak, J. Mol. Struct., 2020, 1219, 128552 CrossRef CAS.
- S. M. Sondhi, N. Singh, M. Johar and A. Kumar, Bioorg. Med. Chem., 2005, 13, 6158–6166 CrossRef CAS.
- M. İhsan Han, U. İnce, M. G. Gündüz, G. P. Coşkun, K. Birgül, Ş. D. Doğan and Ş. G. Küçükgüzel, J. Mol. Struct., 2022, 1261, 132907 CrossRef.
- X. Zhang, M. Breslav, J. Grimm, K. Guan, A. Huang, F. Liu, C. A. Maryanoff, D. Palmer, M. Patel, Y. Qian, C. Shaw, K. Sorgi, S. Stefanick and D. Xu, J. Org. Chem., 2002, 67, 9471–9474 CrossRef CAS PubMed.
- R. F. M. Elshaarawy and C. Janiak, Z. Naturforsch., B: J. Chem. Sci., 2011, 66, 1202–1208 CrossRef CAS.
- D. Nematollahi, M. Rafiee and L. Fotouhi, J. Iran. Chem. Soc., 2009, 6, 448–476 CrossRef CAS.
- A. Abdel, M. S. El and A. O. Abdelhamid, Eur. J. Chem., 2012, 3, 455–460 CrossRef.
- M. M. Abdou, Arabian J. Chem., 2018, 11, 1061–1071 CrossRef CAS.
- D. Nematollahi, H. Hesari, H. Salehzadeh, M. Hesari and S. Momeni, C. R. Chim., 2016, 19, 357–362 CrossRef.
- D. Nematollahi, H. Shayani-Jam, M. Alimoradi and S. Niroomand, Electrochim. Acta, 2009, 54, 7407–7415 CrossRef CAS.
- D. Nematollahi and V. Hedayatfar, J. Chem. Sci., 2011, 123, 709–717 CrossRef CAS.
- D. Nematollahi, A. Afkhami, F. Mosaed and M. Rafiee, Res. Chem. Intermed., 2004, 30, 299–309 CrossRef CAS.
- H. Zhang, J. Liu and Q. Xu, Front. Chem. China, 2007, 2, 164–168 CrossRef.
- D. Cai, T. Li, Q. Xie, X. Yu, W. Xu, Y. Chen, Z. Jin and C. Hu, Molecules, 2020, 25(18), 4279 CrossRef.
- S. M. Gomha and H. M. E. Hassaneen, Molecules, 2011, 16, 6549–6560 CrossRef CAS PubMed.
- H. A. Saad, M. M. Youssef and M. A. Mosselhi, Molecules, 2011, 16, 4937–4957 CrossRef CAS PubMed.
- H. A. Saad and A. H. Moustafa, Molecules, 2011, 16, 5682–5700 CrossRef CAS PubMed.
- R. Slegeris, B. A. Ondrusek and H. Chung, Polym. Chem., 2017, 8, 4707–4715 RSC.
- A. Alshargabi, G.-Y. Yeap, W. A. K. Mahmood and R. Samikannu, J. Mol. Struct., 2013, 1039, 189–196 CrossRef CAS.
- J. S. González-González, R. Jiménez-López, D. Ortegón-Reyna, G. Gonzalez-Carrillo and F. J. Martínez-Martínez, Appl. Sci., 2021, 11(9), 3810 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nj04530c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |