
Synthesis, crystal structure, DFT, and photovoltaic studies of BaCeCuS3†
Omair
Shahid
 a,
Sweta
Yadav
a,
Debanjan
Maity
a,
Melepurath
Deepa
a,
Manish K.
Niranjan
b and
Jai
Prakash
*a
a,
Sweta
Yadav
a,
Debanjan
Maity
a,
Melepurath
Deepa
a,
Manish K.
Niranjan
b and
Jai
Prakash
*a
aDepartment of Chemistry, Indian Institute of Technology Hyderabad, Kandi, Sangareddy, Telangana 502284, India. E-mail: jaiprakash@chy.iith.ac.in
bDepartment of Physics, Indian Institute of Technology Hyderabad, Kandi, Sangareddy, Telangana 502284, India
First published on 7th February 2023
Abstract
We report the synthesis of single crystals of the quaternary sulfide, BaCeCuS3, for the first time by heating a polycrystalline sample of BaCeCuS3 with an excess of KCl flux inside a vacuum-sealed fused silica tube. The crystal structure of the compound is determined by a single-crystal X-ray diffraction study which shows that it crystallizes in the primitive orthorhombic crystal system (space group: Pnma) with cell constants of a = 10.6740(13) Å, b = 4.1200(6) Å, and c = 13.409(2) Å. The structure is best described as a pseudo-two-dimensional structure where tetrahedral CuS4 and octahedral CeS6 units serve as the main building blocks. The polyanionic  layers in the structure are separated by the presence of electropositive Ba2+ cations. The optical absorption study on the polycrystalline sample of BaCeCuS3 shows a semiconducting nature with a direct bandgap of 1.8(2) eV consistent with the green color of the sample. The temperature-dependent thermal conductivity (ktot) measurements on the polycrystalline BaCeCuS3 sample reveal an extremely low value of ktot (0.32 W m−1 K−1) at 773 K. DFT calculations were carried out to obtain the electronic structure of the title compound. Our theoretical studies predict a high value of the thermoelectric figure of merit (>1) for BaCeCuS3 by optimization of the charge carriers. To also explore a practical application of BaCeCuS3, a liquid junction solar cell was fabricated: TiO2/CdS/BaCeCuS3/Sn2−/S2−/MWCNTs@Ni, and the cell delivered an efficiency enhanced by ∼45% compared to the one without the sulfide. The performance improvement is affected by the hole conducting properties of BaCeCuS3, which allows facile hole transfer from CdS to the polysulfide, increases the charge separation, and hence increases the efficiency.
layers in the structure are separated by the presence of electropositive Ba2+ cations. The optical absorption study on the polycrystalline sample of BaCeCuS3 shows a semiconducting nature with a direct bandgap of 1.8(2) eV consistent with the green color of the sample. The temperature-dependent thermal conductivity (ktot) measurements on the polycrystalline BaCeCuS3 sample reveal an extremely low value of ktot (0.32 W m−1 K−1) at 773 K. DFT calculations were carried out to obtain the electronic structure of the title compound. Our theoretical studies predict a high value of the thermoelectric figure of merit (>1) for BaCeCuS3 by optimization of the charge carriers. To also explore a practical application of BaCeCuS3, a liquid junction solar cell was fabricated: TiO2/CdS/BaCeCuS3/Sn2−/S2−/MWCNTs@Ni, and the cell delivered an efficiency enhanced by ∼45% compared to the one without the sulfide. The performance improvement is affected by the hole conducting properties of BaCeCuS3, which allows facile hole transfer from CdS to the polysulfide, increases the charge separation, and hence increases the efficiency.
1. Introduction
Quaternary mixed metal chalcogenides of alkali (A)/alkaline-earth (Ak) metals, transition metals (M), and lanthanides (Ln) are well known to show a variety of physical properties, including superconductivity,1 magnetism,2,3 non-linear optics,4 photovoltaics,5 topological quantum science,6 thermoelectric properties,7 and applications like luminescent materials8 and infrared windows.9 Among these quaternary chalcogenides, a family of compounds with a general formula of (A/Ak)MLnQ3 (Q = chalcogens = S, Se, and Te) is recognized for their structural diversity depending on the type of elements present in these structures. These phases are commonly referred to as 1113-phases, and more than 200 structures are reported for this family of compounds.10 These structures are quite flexible in terms of the compositions and oxidation states of metals. Thus, these 1113-phases can accommodate various types of cations. For instance, A or Ak atoms can be substituted by Pb/Th, and Ln atoms can be replaced by transition metals or early actinides (An),11 which prefer octahedral coordination environments. These (A/Ak)MLnQ3 types of structures are mainly categorized into three subtypes based on the formal oxidation state of the constituent elements: Type-I, A+M+M'4+(Q2−)3 [M = Cu+ or Ag+; M' = Zr4+/Hf4+/Np4+],12 Type-II, Ak2+M+Ln3+(Q2−)3,13,14 and Type-III, A2+M+M'3+(Q2−)3 [M' = Sc3+/Y3+].15 The electropositive A+ and Ak2+ cations in these structures mainly serve as electron donors. The main frameworks of these structures are made up of metal-centered tetrahedra and octahedra of chalcogen atoms. The A+ and Ak2+ cations are packed in between the main polyanionic framework of transition metal/lanthanide/actinide atoms and chalcogen atoms.16,17 These quaternary 1113-type chalcogenides crystallize in seven unique structural types: KZrCuS3 (space group: Cmcm), Eu2CuS3 (Pnma), Ba2MnS3 (Pnma), BaCuLaS3 (Pnma), NaCuTiS3 (Pnma), TaCuTiTe3 (P21/m), and BaAgErS3 (C2/m).10In the last twenty years, metal or main-group chalcogenides have been mainly investigated for their potential thermoelectric properties (TE). TE devices are capable of transforming heat energy directly into electricity without any moving parts. Alternatively, these devices can also create temperature gradients by using electricity and thus are used in compact refrigerators for localized cooling. TE materials mainly work on the Seebeck (for electricity generation) and the Peltier effect (for cooling). The efficiency of the thermoelectric material is expressed by the dimensionless figure of merit (zT), which is defined as zT = (S2σ/ktot)T, where the parameters S (Seebeck coefficient), σ (electrical conductivity), and ktot [total thermal conductivity that consists of an electrical contribution (kel) and lattice contribution (klat)] are the functions of the absolute temperature (T). For a thermoelectric material, a high value of zT can be achieved by realizing a high-power factor (S2σ) value and a low thermal conductivity value18 simultaneously at the same temperature. A few examples of the current state-of-the-art TE materials are Bi2Te3,19 doped-AgSbTe2,20 PbTe,21 and SnSe.22
Surprisingly, the TE properties of these 1113 phases were not studied in the past, and these compounds were neglected until recent theoretical studies suggested the possibility of high zT in some of the Cu-containing layered compounds such as TlCuZrSe3, BaCuYTe3, and KZrCuSe3.23 Preliminary studies of the TE properties of polycrystalline BaScCuTe3 are indeed interesting, with low thermal conductivity values.15 The Kanatzidis group recently reported six new compounds of the 1113-family with a general formula of ACuZrQ3 (A = Rb, Cs; Q = S, Se, and Te).12 An ultralow thermal conductivity value of ∼0.35 W m−1 K−1 was observed for a polycrystalline sample of RbCuZrTe3 at 573 K. The ultralow thermal conductivity of this compound was attributed to the rattling of Rb+ cations in the structure at high temperatures.
Apart from the TE properties of quaternary mixed metal chalcogenides, many of these compounds are also explored for possible photovoltaic applications. Examples include Cu(In,Ga)(S,Se)2,24 Cu2ZnSn(S,Se)4,25AkCu2SnS4 (Ak = Ba,Sr), BaCu2SnS4−xSex26etc. These quaternary compounds in thin film form show promising solar power conversion efficiency. Cd-free Cu(In,Ga)(S,Se)2 chalcogenide-based thin film solar cells have been known to be good photovoltaic absorbers having a very high power conversion efficiency (PCE) of 23.35%,27 but due to the toxicity and inadequacy of Cd, In, and Ga, the production of these materials is limited despite them having good PCE. Again, to the best of our knowledge, these 1113-type compounds, which offer a variety of bandgaps ranging from 0.5–2.0 eV, have not yet been subjected to photovoltaic studies.
Motivated by the promising TE and photovoltaic properties of some of the quaternary chalcogenides, we have synthesized single crystals of the quaternary mixed metal sulfide BaCeCuS3 that belongs to the 1113-family to study its structure by single crystal X-ray diffraction methods. The synthesis of the polycrystalline sample of the BaCeCuS3 phase has been reported earlier by Wu et al.28 However, the PXRD pattern is not provided in the publication.28 Thus, it is not known whether the sample was phase pure or whether it contained some secondary phase(s). Only the cell parameters were calculated from the powder X-ray diffraction study of the BaCeCuS3 sample, and its magnetic properties were examined.28 In this article, we also report the synthesis of a phase pure polycrystalline sample of BaCeCuS3 for the first time. This work presents the synthesis, structure, and temperature-dependent thermal conductivity studies of the polycrystalline sample of BaCeCuS3. A detailed DFT study has been carried out to understand the electronic structure, thermoelectric properties, and chemical bonding aspects of the BaCeCuS3 structure. We have also investigated the preliminary photovoltaic (PV) properties of the polycrystalline sample for the first time, which are promising for future investigations of compounds of the 1113-family.
2. Experimental
2.1. Materials used and synthesis of BaCeCuS3
The following starting materials were used for the synthesis of single crystals and a polycrystalline sample of BaCeCuS3: Ba rod (Alfa Aesar, 99%), Ce ingot (Alfa Aesar, 99.8%), Cu shot (Alfa Aesar, 99.9%), S powder (Sigma Aldrich, 99.5%), and KCl powder (SRL, 99.5%). These chemicals were handled inside an Ar-filled glove box. A thin white oxidized layer on the Ba rod was first removed, then small pieces of Ba metal were cut and used in the reactions. Cerium ingots were cut into small pieces for the reactions. KCl powder was dried overnight inside an oven at 373 K before using it in the reactions as a molten flux to grow the single crystals.The heat-treated sealed tube was cracked open under ambient conditions, and an ingot was found as the reaction product. The ingot was then homogenized into a fine powder inside the Ar-filled glove box using an agate mortar and pestle. A circular pellet (diameter = 8 mm) was then made from the powder by applying high pressure (10.3 MP) using a hydraulic press. The pellet was then transferred into a C-coated fused silica tube, evacuated (10−4 Torr), and flame sealed. The sealed tube containing the pellet was reheated inside the muffle furnace with the same heating profile as before. A small part of the pellet was then broken off and homogenized into a fine green-colored powder inside the Ar-filled glove box to collect powder X-ray diffraction data for the phase analysis.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (by weight) of the polycrystalline sample to the flux. The reaction mixture was transferred into a carbon-coated fused silica tube inside an Ar-filled glove box. The tube was then evacuated (10−4 Torr) and flame sealed. The sealed tube was first heated from RT to 1223 K (in 36 h) and was equilibrated at this temperature for 48 h. The reaction mixture was then slowly cooled down to 1023 K at a rate of 4 K h−1. The temperature of the furnace was then further cooled to 673 K at a rate of 5 K h−1 before shutting off the furnace.
:3 (by weight) of the polycrystalline sample to the flux. The reaction mixture was transferred into a carbon-coated fused silica tube inside an Ar-filled glove box. The tube was then evacuated (10−4 Torr) and flame sealed. The sealed tube was first heated from RT to 1223 K (in 36 h) and was equilibrated at this temperature for 48 h. The reaction mixture was then slowly cooled down to 1023 K at a rate of 4 K h−1. The temperature of the furnace was then further cooled to 673 K at a rate of 5 K h−1 before shutting off the furnace.
The morphology of the reaction product was first examined with the help of an optical microscope at RT. A few needle-shaped crystals were manually separated from the white colored KCl flux and placed on a carbon-taped stub. The elemental composition of these needle-shaped crystals was analyzed by energy dispersive X-ray spectroscopic (EDX) studies using an octane elite (EDAX Inc., USA) spectrometer that was attached to a field-emission scanning electron microscope (FE-SEM) (JEOL, Japan; Model: JSM 7800F). The EDX data on the crystals were collected at different spots with an accelerating voltage of 15–20 kV. The average composition of the selected crystals was found to be very close to the loaded composition, i.e., ≈1:1:1:3 with respect to Ba, Ce, Cu, and S, without any noticeable contamination from the KCl flux (Fig. 1a).
 | ||
| Fig. 1 (a) The EDX spectrum collected for a typical crystal of BaCeCuS3. The inset of the figure shows an SEM picture of the BaCeCuS3 crystals. (b) The experimental PXRD pattern of the polycrystalline BaCeCuS3 sample, along with the corresponding simulated pattern obtained from the single crystal X-ray diffraction study. | ||
2.2. Powder X-ray diffraction study (PXRD)
The PXRD data of the finely ground homogeneous powdered sample of BaCeCuS3 was collected at RT using a PAN analytical empyrean diffractometer with a Cu-Kα radiation (λ = 1.5406 Å) source (working voltage and currents were set at 40 kV and 30 mA, respectively). The PXRD data were collected over a 2θ range of 10° to 70° using a θ–2θ geometry for about 30 minutes. The PXRD data were analyzed using Match! version 2 software.302.3. Single crystal X-ray diffraction (SCXRD) study of BaCeCuS3
A Bruker D8 Venture instrument equipped with a Photon III mixed mode detector with graphite monochromatized Mo-Kα radiation (λ = 0.71073 Å) was used for the SCXRD data collection of a BaCeCuS3 crystal at RT. The crystal was coated with a thin layer of N-paratone oil and was then mounted on the tip of an X-ray transparent micro loop that was affixed to a goniometer head. The working voltage and operating current were set to 50 kV and 1.4 mA, respectively, during diffraction data collection for the crystal. The crystal-to-detector distance was held constant at 50 mm, and the exposure time of 3 sec per frame was used for the entire data collection. The intensity data set was collected as a series of 0.5° steps in ω and ϕ-scans. The cell refinements and data reduction were carried out using the APEX3 program.31 The absorption correction for the intensities of the measured reflections was performed using the semiempirical multiscan method of the SADABS program.32The SHELXL-1433 suite of the program was used to solve and refine the crystal structure of BaCeCuS3. The XPREP34 program suggested two space groups based on the extinction conditions: non-centrosymmetric Pna21 (no. 33) and centrosymmetric Pnma (no. 62). The structure was straightforwardly solved and refined in the higher symmetric Pnma space group.
The asymmetric unit of the BaCeCuS3 structure has six independent atomic positions. The full-matrix-least-squares on F2 method33 in the SHELXL35 program was used to refine the crystal structure. The extinction parameter, scale factors, weight corrections, and atomic positions with anisotropic displacement parameters were refined for the structure. The ADDSYM program of PLATON36 was used to validate the symmetry of the structure. Finally, the atomic positions of the structure were standardized using the STRUCTURE TIDY program.37 Further crystallographic and structure refinement details are provided in Tables 1–3, and in the ESI.†
| Chemical formula | BaCeCuS3 |
|---|---|
| a λ = 0.71073 Å, T = 298(2) K. b R(F) = Σ∣∣Fo∣ − ∣Fc∣∣/Σ∣Fo∣ for Fo2 > 2σ(Fo2). c R w(Fo2) = {Σ[w(Fo2 − Fc2)2]/ΣwFo4}1/2. For Fo2 < 0, w = 1/[σ2(Fo2) + (0.0173P)2 + 3.6347P] where P = (Fo2 + 2Fc2)/3. | |
| Space group | D 162h-Pnma |
| a (Å) | 10.6740(13) |
| b (Å) | 4.1200(6) |
| c (Å) | 13.409(2) |
| V (Å3) | 589.69(15) |
| Z | 4 |
| ρ (g cm−3) | 4.924 |
| μ (mm−1) | 18.67 |
| GOF on F2 | 1.068 |
| θ min and θmax | 3.04°–30.72° |
| Completeness to θ = 30.722° | 99.3% |
| R int | 0.033 |
| Largest diff. peak and hole (e Å−3) | 1.58, −1.39 |
| Refinement method | Full-matrix least-squares on F2 |
| R(F)b | 0.021 |
| R w(Fo2)c | 0.049 |
| Atoms | Wyckoff position | Site symmetry | x | y | z | U iso /U eq |
|---|---|---|---|---|---|---|
| a U iso/Ueq is the one-third value of the trace of the orthogonalized Uij tensor. | ||||||
| Ba1 | 4c | .m. | 0.27459(3) | 0.250000 | 0.00562(3) | 0.01562(10) |
| Ce1 | 4c | .m. | 0.01097(3) | 0.250000 | 0.76097(2) | 0.00934(9) |
| Cu1 | 4c | .m. | 0.23815(7) | 0.250000 | 0.28245(6) | 0.01659(16) |
| S1 | 4c | .m. | 0.05409(12) | 0.250000 | 0.37875(10) | 0.0107(2) |
| S2 | 4c | .m. | 0.25670(12) | 0.250000 | 0.68209(10) | 0.0098(2) |
| S3 | 4c | .m. | 0.41059(13) | 0.250000 | 0.39196(10) | 0.0117(2) |
| Atomic pair | Distances (Å) | Atomic pair | Distances (Å) |
|---|---|---|---|
| Ce1–S1 | 2.8698(10) × 2 | Ba1–S2 | 3.1547(12) × 2 |
| Ce1–S2 | 2.8196(13) | Ba1–S3 | 3.2363(11) × 2 |
| 2.8283(13) | |||
| Ce1–S3 | 2.8336(10) × 2 | Ba1⋯Ba1 | 4.1200(6) × 2 |
| Cu1–S1 | 2.3511(15) | Ba1⋯Cu1 | 3.6356(8) × 2 |
| 3.7322(10) | |||
| Cu1–S2 | 2.4614(9) × 2 | Ce1⋯Cu1 | 3.3908(7) × 2 |
| 3.4137(7) × 2 | |||
| Cu1–S3 | 2.3546(15) | Ce1⋯Ce1 | 4.1200(6) × 2 |
| Ba1–S1 | 3.2376(11) × 2 | ||
| 3.3622(14) |
2.4. Solid-state ultraviolet-visible (UV-vis) spectroscopy
The optical bandgap study of the polycrystalline sample of BaCeCuS3 was performed at RT. The polycrystalline sample was mixed with a small amount of dried BaSO4, which acts as a standard reference for the optical absorption study. The reflectance versus wavelength data were collected over the wavelength range of 2400 nm to 400 nm using a Shimadzu UV-3600 instrument. The Kubelka–Munk equation α/S = (1 − R)2/2R (where α, S, and R are the absorption coefficient, scattering coefficient, and reflectance, respectively)38 was used to calculate the bandgap of the sample by converting the reflectance data into absorption data. The value of the direct bandgap of the sample was calculated using a Tauc plot.392.5. Thermal conductivity measurements as a function of temperature
The temperature-dependent thermal diffusivity (D) of the polycrystalline sample of BaCeCuS3 was measured in the 323 K- 773 K range. The finely ground polycrystalline sample of BaCeCuS3 was compacted into a circular pellet (diameter = 8 mm, thickness ∼2 mm) at RT by applying high pressure (10.3 MP) using a hydraulic press. The pellet was then sealed inside a carbon-coated fused silica tube under vacuum (10−4 Torr). To obtain a dense sample for the thermal diffusivity measurements, the circular pellet inside the sealed tube was sintered at 1123 K for 12 h. The sintered pellet was then subjected to the total thermal conductivity (ktot) measurements.The value of ktot (= D × d × Cp) for the sample was calculated by multiplying the D-value with the experimental density (d) and heat capacity (Cp) values.40 The density of the circular pellet was calculated using the Archimedes method and it was found to be 4.63 g cm−3. This density of the circular pellet was compared with the theoretical density and was found to be about 94% of the theoretical density as measured using the Archimedes method.41 The Cp value was determined using the Dulong-Petit law.42 A LINSEIS XFA 500 system that uses a xenon flash lamp to irradiate the sample was used to measure the thermal diffusivity of the circular pellet of the BaCeCuS3 sample. Before placing the pellet into the system, the surface of the pellet was coated with a thin layer of graphite to reduce the heat loss after irradiation. During the measurements, liquid N2 was used to cool the infrared (IR) detector. The Xe flash lamp gradually starts to heat up the pellet from the bottom, and a high-speed IR detector measures the developed temperature over the top surface of the pellet as a function of time. The measurements were performed under vacuum to avoid oxidation of the sample at higher temperatures, and the thermal diffusivity data were obtained at an interval of 50 K. Three thermal diffusivity data sets were collected at each temperature, and an average of the values was taken to calculate the total thermal diffusivity. An experimental error of 10% is associated with the calculated total thermal conductivity values. The microstructure of the pellet was also examined after the thermal diffusivity measurements using a field-emission electron microscope (FE-SEM) (JEOL, Japan; Model: JSM 7800F).
2.6. Fabrication of a photoanode and solar cell using the polycrystalline BaCeCuS3 sample
The following reagents were used to prepare the photoanode and solar cell. A fluorine-doped tin oxide (FTO) coated glass with a sheet resistance of ∼25 Ω cm2 was purchased from Pilkington. The TiO2-P25 powder, fumed silica, and Ni-foam were purchased from Evonik, Cabosil, and Gelon, respectively. Acetyl acetone, Triton X-100, methanol, ethanol, cadmium acetate [Cd(CH3COO)2·2H2O], multi-walled carbon nanotubes (MWCNTs), and sodium sulfide (Na2S) were purchased from Merck. Ultra-pure water with a resistivity of ∼18.2 MΩ cm was obtained through a Millipore Direct-Q3 UV system.A layer of CdS was deposited over the TiO2-coated FTO electrodes by a successive ionic layer adsorption reaction (SILAR) method. In two separate beakers, solutions of 0.1 M cadmium acetate and sodium sulfide in methanol were prepared. At first, the TiO2 electrodes were dipped in a Cd2+ ion-containing beaker and kept there for 2 min. Then the electrodes were allowed to dry at RT. After drying, the electrodes were dipped in a S2− ion-containing beaker for 2 min and then dried at RT. This completes one cycle of SILAR.
Similarly, five more cycles of SILAR were carried out to prepare the CdS/TiO2 electrodes. Then the electrodes were dried at 333 K for 2 days in an oven. The polycrystalline BaCeCuS3 (5 mg) sample was dispersed in 10 mL of toluene. Then 2 drops of the dispersion were drop-cast on the CdS/TiO2 electrode, dried, and the BaCeCuS3/CdS/TiO2 electrode was ready for use.
2.7. Current (I) and voltage (V) measurements of the solar cells
The I–V measurements of the solar cells were recorded on a Newport Oriel 3A solar simulator coupled with a Keithley 2420 source meter. A 450W xenon arc lamp was used as the light source with a light intensity of 100 mW cm−2 and of air mass (AM) 1.5G illumination; the spatial uniformity of the irradiance was determined by calibration with a 2 cm × 2 cm Si reference cell and it was affirmed using a Newport power meter.2.8. Theoretical and computational methodology
The density functional (DFT)43 calculations are performed using the plane-wave expansion formalism as implemented in the VASP package.44 The Kohn-Sham equation is solved by expanding the single-particle wavefunctions based on plane waves using the kinetic energy cutoff of 320 eV (or higher). The electrostatic potentials of the ionic cores are approximated using the projected augmented wave (PAW) pseudopotentials.45 For Ba, Cu, S, and Ce atoms, the (5s2, 5p6, 6s2), (3d10, 4s1), (3s2, 3p4), (5s2, 5p6, 5d1, 6s2) electrons are treated as valence electrons, respectively. In the case of Ce, the 4f electrons are treated as frozen and as the core states. Further, the formal valency of Ce is assumed to be 3. In general, the self-interaction errors in presently available density functionals lead to an incorrect description of f-electrons. However, this shortcoming may be circumvented by treating the 4f electrons as the core electrons. The exchange–correlation (XC) effects are treated using the Perdew-Burke-Ernzerhoff (PBE)46 form of the generalized gradient approximation (GGA) and SCAN (meta-GGA)47 schemes. The integrations in reciprocal space are performed by sampling the Brillouin zone of BaCeCuS3 using (6 × 14 × 5) Monkhorst-Pack k-point meshes, respectively. The atomic Hellmann-Feynman forces are reduced up to ∼0.02 eV Å−1 (or less) in order to obtain optimized crystal unit cells. The total energies converged to 10−6 eV cell−1 to attain the self-consistent field (SCF) in the calculations. The thermoelectric parameters are calculated using the Boltzmann transport theory within the rigid band approach.48 The ESI† file briefly summarizes the methodologies used to compute the optical49 and thermoelectric parameters.3. Results and discussion
3.1. Synthesis and crystal structure
The polycrystalline sample of BaCeCuS3 was synthesized by the stoichiometric reaction of elements at 1273 K using a solid-state sealed tube method. All of the observed reflections in the experimental PXRD pattern of the polycrystalline BaCeCuS3 sample were indexed satisfactorily with the simulated pattern obtained from the single-crystal structure data. Thus, the polycrystalline BaCeCuS3 sample is phase pure (Fig. 1b).The single crystals of BaCeCuS3 were recrystallized by heating the polycrystalline sample in an excess of molten KCl flux. The slow cooling of the reaction mixture from 1223 K to RT resulted in the formation of needle-shaped crystals of the quaternary chalcogenide. The approximate yield of the crystals was about 50%.
The crystal structure of BaCeCuS3 is pseudo-two-dimensional in nature, as evident from Fig. 2a. The crystal structure is made up of six crystallographically independent atoms: Ba1, Ce1, Cu1, S1, S2, and S3, all with the same site symmetry (.m.). Each Ce atom in this structure is bonded to six S atoms having distorted octahedral geometry, whereas the Cu atom is coordinated to four S atoms to form a distorted CuS4 tetrahedron (Fig. 3). The distortion in the geometry of the CuS4 tetrahedra presumably results from steric factors since Cu+ (d10) has a closed cell electronic configuration. Thus, the role of the electronic effect in the distortion of the polyhedra could be ruled out.
 | ||
| Fig. 2 (a) The unit cell structure of BaCeCuS3 and (b) the Ba coordination environment. | ||
 | ||
Fig. 3 The views of (a) the CeS6 octahedral units and a fragment of the (b)  layer along the [010] direction in the BaCeCuS3 structure. layer along the [010] direction in the BaCeCuS3 structure. | ||
The tetrahedron of CuS4 shares its four edges with the four octahedra of CeS6, resulting in the formation of the polyanionic  layers that are extended in the [100] direction. These CuS4 and CeS6 polyhedra are organized in an alternate … tet oct tet oct … fashion along the [010] direction in the
layers that are extended in the [100] direction. These CuS4 and CeS6 polyhedra are organized in an alternate … tet oct tet oct … fashion along the [010] direction in the  layers of the BaCeCuS3 structure, as shown in Fig. 3. The Ba2+ cations act as a space filler and an electron donor in the structure. The S atoms around the Ba2+ in the structure have a mono-capped trigonal prism geometry, as each Ba2+ coordinates with seven S atoms (Fig. 2b).
layers of the BaCeCuS3 structure, as shown in Fig. 3. The Ba2+ cations act as a space filler and an electron donor in the structure. The S atoms around the Ba2+ in the structure have a mono-capped trigonal prism geometry, as each Ba2+ coordinates with seven S atoms (Fig. 2b).
The Ce–S bond distances in BaCeCuS3 (2.8196(13) – 2.8698(10) Å) are comparable to the Ce3+–S distances found in the quaternary sulfide BaCe2MnS5 (2.811(9) – 3.104(1) Å).51 The Cu–S bond distances in BaCeCuS3 (2.3511(15) – 2.4614(9) Å) are also in good agreement with the related quaternary sulfide CsCuCeS3 (2.319(3) – 2.413(2) Å).52
The Ba–S interatomic distances in BaCeCuS3 range from 3.1254(19) Å to 3.612(2) Å. These distances are similar to those found in BaErCuS3 (3.106(2) – 3.407(2) Å) and BaCu3GaS4 (3.2156(19) – 3.2949(16) Å).53
The partition of charges to individual atoms in the BaCeCuS3 structure is straightforward as the structure does not have any homoatomic bonding. The charge-balanced formula of the compound can be written as Ba2+Ce3+Cu+(S2−)3.
3.2. The optical absorption bandgap study of polycrystalline BaCeCuS3
The absorption spectrum of the polycrystalline BaCeCuS3 sample was collected at ambient temperature in the wavelength region starting from 2400 nm to 400 nm. The bandgap of the sample is deduced by extrapolating the linear region of the Tauc plot,39 as shown in Fig. 4. A direct bandgap value of 1.8(2) eV is estimated for the polycrystalline BaCeCuS3 (Fig. 4). Hence, we can conclude that the compound BaCeCuS3 is a narrow bandgap semiconductor. | ||
| Fig. 4 The solid-state direct bandgap Tauc plot for the polycrystalline BaCeCuS3 sample. | ||
3.3. Thermal conductivity study of polycrystalline BaCeCuS3
The total thermal conductivity (ktot) of the sintered cold-pressed polycrystalline sample of BaCeCuS3 was investigated in the temperature range of 323 K to 773 K. The ktot value is the summation of the lattice thermal conductivity (klat) and electrical thermal conductivity (kel) values of a crystalline sample. The BaCeCuS3 sample shows a gradual decrease in the ktot values on raising the temperature, as shown in Fig. 5. The ktot value of the polycrystalline sample decreases from 0.62 W m−1 K−1 (at 323 K) to an extremely low value of 0.32 W m−1 K−1 at 773 K. As the BaCeCuS3 sample has a relatively large bandgap, the contribution of kel to the ktot value is expected to be extremely small. Hence, it is safe to presume that the ktot value for the sample is very close to the klat value. A similar ultralow thermal conductivity value of ∼0.35 W m−1 K−1 at 573 K is reported for the structurally related RbCuZrTe3.12 The rattling of relatively loosely bound Rb+ cations and the distortion of Zr-coordinated octahedra in RbCuZrTe3 are responsible for the ultralow value of the lattice thermal conductivity (klat). On similar lines, it is expected that the rattling of Ba2+ cations in the BaCeCuS3 sample at high temperatures should also lead to low values of klat. However, detailed studies of the thermal parameters of Ba atoms in the BaCeCuS3 sample at high temperatures are required to confirm this conjecture. | ||
| Fig. 5 The temperature-dependent total thermal conductivity (ktot) for the polycrystalline sample of BaCeCuS3. | ||
The extremely small value of 0.32 W m−1 K−1 at 773 K for the BaCeCuS3 sample is promising for thermoelectric applications. However, the relatively larger bandgap of this compound (1.8(2) eV) is not suitable for achieving an excellent thermoelectric figure of merit (zT). A substantial amount of electron/hole doping is required to improve the electrical conductivity of this sample in the future to achieve a reasonable zT value.
The microstructure of the fractured pellet of the BaCeCuS3 sample was examined using FE-SEM to investigate the grain size, density, and packing of the polycrystals in the pellet. The SEM micrographs of the fractured surface of the pellet revealed that the pellet is reasonably dense with no apparent preferential orientation of the microcrystals (Fig. 6).
 | ||
| Fig. 6 The cross-sectional (a–c) FE-SEM images of the BaCeCuS3 pellet used for the thermal conductivity measurements. The surface of the pellet is also visible in (b). | ||
3.4. Solar cell study
The current density (J) versus voltage (V) plot (Fig. 7) of the FTO/TiO2/CdS/Sn2−/S2−/MWCNTs@Ni foam cell under a 1 sun illumination condition showed an open circuit voltage (VOC) = 460 mV, short circuit current density (JSC) = 24.75 mA cm−2, fill factor (FF) = 34.5%, and efficiency (η) = 3.93%. The CdS absorbs blue-green photons, and its bandgap is ∼2.3 eV.54 | ||
| Fig. 7 The J vs. V plot of the FTO/TiO2/CdS/Sn2−/S2−/MWCNTs@Ni, and FTO/TiO2/CdS/BaCeCuS3/Sn2−/S2−/MWCNTs@Ni foam cell. | ||
Under illumination, electronic excitation takes place in CdS. Due to the favorable alignment of the conduction band (CB), the electron moves from the CB of CdS to the CB of TiO2. Then via an external circuit, it reaches the electrocatalytic MWCNTs@Ni foam-based counter electrode (CE), where it reduces the oxidized redox species (Sn2−). The reduced redox species (S2−) reaches the photoanode, captures the holes from the valence band (VB) of CdS, and gets converted to Sn2−. The FTO/TiO2/CdS/BaCeCuS3/Sn2−/S2−/MWCNTs@Ni foam cell showed VOC = 560 mV, JSC = 29.74 mA cm−2, FF = 34.35%, and η = 5.71%, respectively. The VOC, JSC, and η increased by 21.7%, 20.2%, and 45.3%, respectively due to the coating of the polycrystalline BaCeCuS3. This is due to the hole-conducting ability of p-type BaCeCuS3 and its semiconducting nature, which enhances the charge separation and reduces recombination at the photoanode/electrolyte interface.
3.5. Theoretical studies of BaCeCuS3
| a (Å) | b (Å) | c (Å) | V (Å3) | E g (eV) | |||
|---|---|---|---|---|---|---|---|
| BaCeCuS3 | Cal. | GGA | 10.794 | 4.134 | 13.528 | 603.83 | 1.15 |
| SCAN | 10.770 | 4.125 | 13.501 | 603.83 | 1.40 | ||
| Exp. | 10.674 | 4.120 | 13.409 | 589.69 | 1.8(2) |
Next, we analyze the electronic structure of BaCeCuS3. The density of states (DOS) and the band structure plots for BaCeCuS3 are shown in Fig. 8. The valence band maximum (VBM) and the conduction band minimum (CBM) can be seen at 0 and ∼1.2 eV, respectively. The valence band (VB) in the energy window of ∼−5 eV < E < 0 eV, is formed primarily due to the overlap of Cu-3d and S-3p states along with a minor contribution of Ce-5d states. The VB in the −13.5 < E < −11 eV region comprises Ba-5p and S-3s states. The sharp peak in the VB at ∼−17 eV is formed by localized Ce-5p states. The conduction band (CB) in the ∼1.2 eV < E < 5 eV energy region is mainly formed by antibonding Ce-5d and Cu-3d states. The band structure (see Fig. 8b) suggests that the bandgap is direct since both VBM and CBM are located at the Brillouin zone center (Γ-point).
 | ||
Fig. 8 (a) The total and projected density of states (DOS) and the (b) band structure for BaCeCuS3. The dotted line in (a) indicates the valence band maximum. The high symmetry k-points in the Brillouin zone are Γ ≡ (0,0,0),  , ,  , ,  , ,  , ,  , and , and  . . | ||
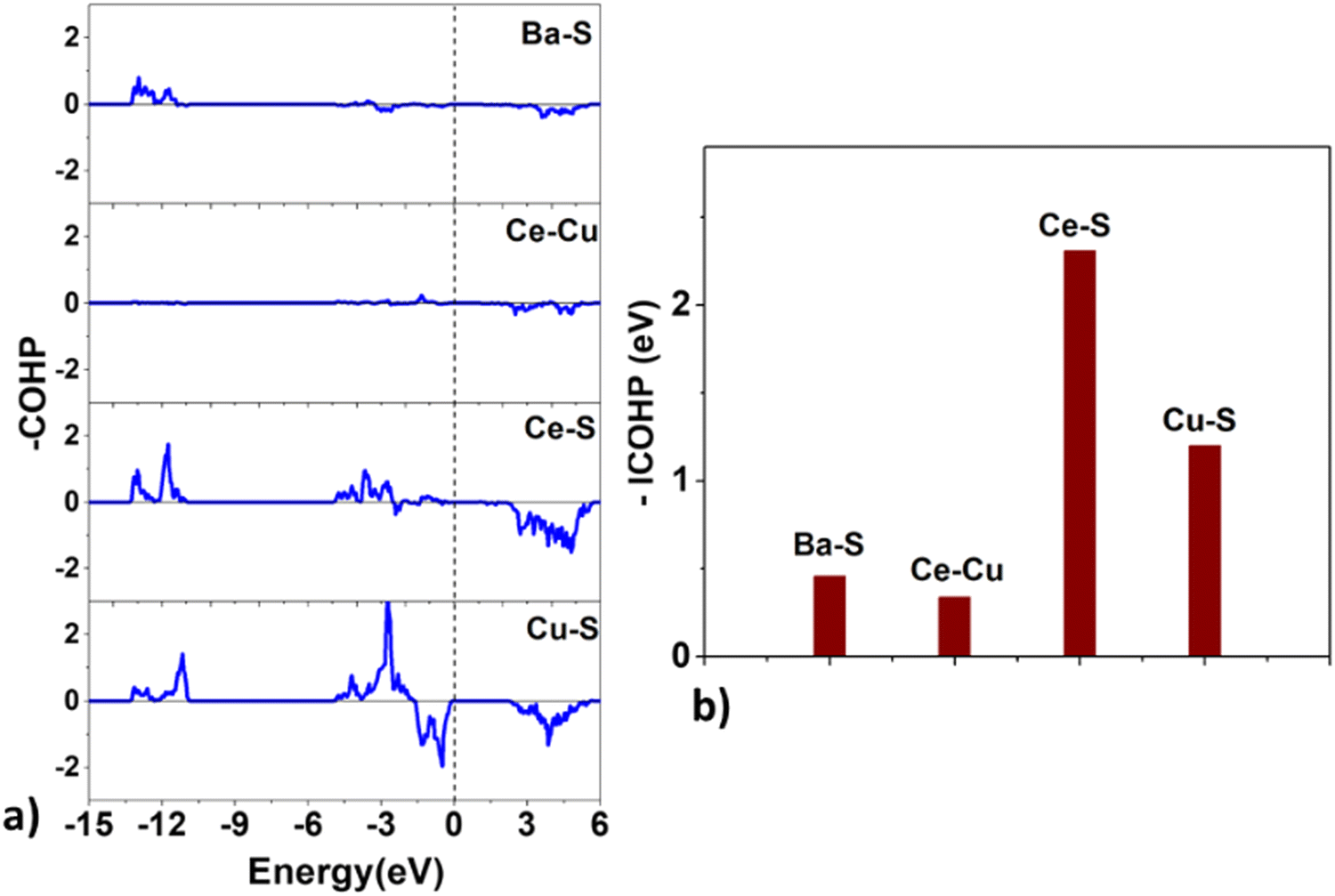 | ||
| Fig. 9 The (a) crystal orbital Hamilton population (COHP) and the (b) integrated COHP for the Ba–S, Ce–Cu, Ce–S, and Cu–S bonds in BaCeCuS3. | ||
Surprisingly, the COHP plot suggests a strong antibonding character at the Fermi level for the Cu–S bonds, and their bond strengths are smaller than those of the Ce–S pair. This deviation could be due to factors that depend on the local chemical environment and/or the overlap of atomic wavefunctions/orbitals. It is well known that the DFT-based description of f-electrons is tricky and challenging due to the presence of self-interaction errors.
Next, we estimate the relative charge transfer between bonding atoms from the computed Bader charges (QB).57 The relative degree of covalent and ionic bonding between atoms as well as their oxidation states may also be estimated from the magnitudes of the ionic Bader charges. In the case of BaCeCuS3, the QB for Ba, Ce, Cu, and S ions are found to be +1.46, +1.83, +0.34, and –1.23, respectively. The QB values indeed show the electron transfer from metals (Ba, Ce, and Cu) to more electronegative S atoms. However, these values are smaller than the formal oxidation states of the ions, i.e., Ba2+Ce3+Cu+(S2−)3 as discussed earlier in Section 3.1. This is suggestive of the presence of some degree of covalent bonding in addition to the predominant ionic bonding in the system.
 | ||
| Fig. 10 The variation of the values of (a) α, (b) (αE), and (c) (αE)2 as a function of energy for BaCeCuS3. The symbols α and E are the absorption and energy, respectively. | ||
The thermoelectric parameters, such as the figure of merit (zT), Seebeck coefficient (S), power factor, etc., are also calculated from the electronic band structure and using the Boltzmann transport theory within the rigid band approach. The relevant equations to calculate these parameters are provided in Section S2 of the ESI† file.
The  where kel and klat are the electronic and lattice (phonon) components of total thermal conductivity, respectively. In our calculation, klat is treated as an external parameter with values taken from the experimental data shown in Fig. 5 in Section 3.3. The thermoelectric parameters are computed as a function of temperature and carrier (hole and electron) concentration. The Seebeck coefficient (S) and zT for p-doped BaCeCuS3 as a function of the carrier (holes) concentration at temperatures 300 K, 600 K, and 800 K are shown in Fig. 11. The magnitude of S is found to vary in the range ∼900 > S > μV K−1 for carrier concentrations ∼1017 < p < 1021 cm−3. As expected, the Seebeck coefficient (S) decreases (increases) with increasing the carrier concentration (temperature). Using a model based on nearly free electrons, a parabolic band, and an energy-independent scattering approximation,58 the S can be shown to depend on temperature (T), carrier concentration (p), and carrier effective mass (m*) as
where kel and klat are the electronic and lattice (phonon) components of total thermal conductivity, respectively. In our calculation, klat is treated as an external parameter with values taken from the experimental data shown in Fig. 5 in Section 3.3. The thermoelectric parameters are computed as a function of temperature and carrier (hole and electron) concentration. The Seebeck coefficient (S) and zT for p-doped BaCeCuS3 as a function of the carrier (holes) concentration at temperatures 300 K, 600 K, and 800 K are shown in Fig. 11. The magnitude of S is found to vary in the range ∼900 > S > μV K−1 for carrier concentrations ∼1017 < p < 1021 cm−3. As expected, the Seebeck coefficient (S) decreases (increases) with increasing the carrier concentration (temperature). Using a model based on nearly free electrons, a parabolic band, and an energy-independent scattering approximation,58 the S can be shown to depend on temperature (T), carrier concentration (p), and carrier effective mass (m*) as  . However, it may be noted that S depends on the complete band structure details and thus may deviate from this expression. Fig. 11a shows such a deviation in S at T = 800 K. zT attains a maximum value for hole concentration of ∼3 × 1020 cm−3 as can be seen in Fig. 11b. Further, zT also increases with temperature as expected. As evident, the computed values of the TE parameters vary significantly with the value of the carrier concentration in the compound. Due to these and other reasons, the theoretical thermoelectric (TE) parameters may not be compared directly with the experimental values. Nevertheless, the ab initio results of the TE parameters may be useful in order to understand the dependence of these parameters on the electronic band structure.
. However, it may be noted that S depends on the complete band structure details and thus may deviate from this expression. Fig. 11a shows such a deviation in S at T = 800 K. zT attains a maximum value for hole concentration of ∼3 × 1020 cm−3 as can be seen in Fig. 11b. Further, zT also increases with temperature as expected. As evident, the computed values of the TE parameters vary significantly with the value of the carrier concentration in the compound. Due to these and other reasons, the theoretical thermoelectric (TE) parameters may not be compared directly with the experimental values. Nevertheless, the ab initio results of the TE parameters may be useful in order to understand the dependence of these parameters on the electronic band structure.
 | ||
| Fig. 11 The variation of the (a) Seebeck coefficient (μV K−1) and the (b) zT value as a function of hole concentration for BaCeCuS3. | ||
4. Conclusions
We have successfully synthesized single crystals and polycrystalline sample of BaCeCuS3 using high-temperature molten flux and solid-state synthesis methods, respectively. The SCXRD study revealed that BaCeCuS3 adopts an orthorhombic Eu2CuS3 structure type (space group Pnma). The main building blocks of the structure are the CeS6 octahedron and CuS4 tetrahedron. The BaCeCuS3 structure is made up of two-dimensional layers that are separated by Ba2+ cations that mainly act as space fillers. The optical absorption study of the polycrystalline sample of BaCeCuS3 establishes its semiconducting behavior with an optical bandgap value of 1.8(2) eV. The total thermal conductivity value (ktot) of the compound is extremely low (0.32 W m−1 K−1) at 773 K. The electronic structure, optical and thermoelectric properties of BaCeCuS3 are explored theoretically using DFT methods. The computed structural parameters of the compound are found to be in good agreement with the experimental data. The nature and relative strength of the chemical bonds are explored by analyzing the computed COHP and Bader atomic charges. The optical and thermoelectric parameters are computed using the electronic band structure. The practical significance of BaCeCuS3 was demonstrated by using it as a hole transport layer in a solar cell (TiO2/CdS/BaCeCuS3/Sn2−/S2−/MWCNTs@Ni), and the cell exhibited a superior efficiency of 5.7%, about 1.5 times greater than that of the cell without this layer. The enhanced performance is attributed to the efficient hole injection from CdS to the polysulfide, via BaCeCuS3, that maximizes the charge separation and increases the efficiency.
layers that are separated by Ba2+ cations that mainly act as space fillers. The optical absorption study of the polycrystalline sample of BaCeCuS3 establishes its semiconducting behavior with an optical bandgap value of 1.8(2) eV. The total thermal conductivity value (ktot) of the compound is extremely low (0.32 W m−1 K−1) at 773 K. The electronic structure, optical and thermoelectric properties of BaCeCuS3 are explored theoretically using DFT methods. The computed structural parameters of the compound are found to be in good agreement with the experimental data. The nature and relative strength of the chemical bonds are explored by analyzing the computed COHP and Bader atomic charges. The optical and thermoelectric parameters are computed using the electronic band structure. The practical significance of BaCeCuS3 was demonstrated by using it as a hole transport layer in a solar cell (TiO2/CdS/BaCeCuS3/Sn2−/S2−/MWCNTs@Ni), and the cell exhibited a superior efficiency of 5.7%, about 1.5 times greater than that of the cell without this layer. The enhanced performance is attributed to the efficient hole injection from CdS to the polysulfide, via BaCeCuS3, that maximizes the charge separation and increases the efficiency.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
JP thanks DST-SERB, the Government of India (GOI), for the financial support under the core research grant (Grant number: CRG/2021/003641) and IIT Hyderabad for the research facilities. OS thanks UGC for the research fellowship. SY thanks the Ministry of Education (MOE), GOI, for the Prime Minister's Research Fellowship (PMRF). The authors also gratefully acknowledge DST-FIST (SR/FST/ETI-421/2016) for the SEM-EDX facility used in this work.References
- C. D. Malliakas, D. Y. Chung, H. Claus and M. G. Kanatzidis, J. Am. Chem. Soc., 2016, 138, 14694–14698 CrossRef CAS PubMed.
- M. Sturza, J. M. Allred, C. D. Malliakas, D. E. Bugaris, F. Han, D. Y. Chung and M. G. Kanatzidis, Chem. Mater., 2015, 27, 3280–3290 CrossRef CAS.
- G. Panigrahi, S. Jana, M. Ishtiyak, S. Narayanswamy, P. P. Bhattacharjee, K. V. Ramanujachary, M. K. Niranjan and J. Prakash, Dalton Trans., 2021, 50, 6688–6701 RSC.
- D. Sahoo and R. Naik, J. Non-Cryst. Solids, 2022, 597, 121934 CrossRef CAS.
- A. Le Donne, V. Trifiletti and S. Binetti, Front. Chem., 2019, 7, 297 CrossRef CAS PubMed.
- H. Zhang, C.-X. Liu, X.-L. Qi, X. Dai, Z. Fang and S.-C. Zhang, Nat. Phys., 2009, 5, 438–442 Search PubMed.
- Y. Shi, A. Assoud, S. Ponou, S. Lidin and H. Kleinke, J. Am. Chem. Soc., 2018, 140, 8578–8585 CrossRef CAS PubMed.
- B. O. Dabbousi, J. Rodriguez-Viejo, F. V. Mikulec, J. R. Heine, H. Mattoussi, R. Ober, K. F. Jensen and M. G. Bawendi, J. Phys. Chem. B, 1997, 101, 9463–9475 CrossRef CAS.
- Y. Zhang, G. Hong, Y. Zhang, G. Chen, F. Li, H. Dai and Q. Wang, ACS Nano, 2012, 6, 3695–3702 CrossRef CAS PubMed.
- L. A. Koscielski and J. A. Ibers, Z. Anorg. Allg. Chem., 2012, 638, 2585–2593 CrossRef CAS.
- J. Prakash, A. Mesbah, J. C. Beard and J. A. Ibers, Z. Anorg. Allg. Chem., 2015, 641, 1253–1257 CrossRef CAS.
- C. C. Laing, B. E. Weiss, K. Pal, M. A. Quintero, H. Xie, X. Zhou, J. Shen, D. Y. Chung, C. Wolverton and M. G. Kanatzidis, Chem. Mater., 2022, 34, 8389–8402 CrossRef CAS.
- N. O. Azarapin, A. S. Aleksandrovsky, V. V. Atuchin, T. A. Gavrilova, A. S. Krylov, M. S. Molokeev, S. Mukherjee, A. S. Oreshonkov and O. V. Andreev, J. Alloys Compd., 2020, 832, 153134 CrossRef CAS.
- K. Eickmeier, R. Poschkamp, R. Dronskowski and S. Steinberg, Eur. J. Inorg. Chem., 2022, e202200360 CAS.
- M. Ishtiyak, S. Jana, R. Karthikeyan, M. Ramesh, B. Tripathy, S. K. Malladi, M. K. Niranjan and J. Prakash, Inorg. Chem. Front., 2021, 8, 4086–4101 RSC.
- Y. Yang and J. A. Ibers, J. Solid State Chem., 1999, 147, 366–371 CrossRef CAS.
- S. Maier, J. Prakash, D. Berthebaud, O. Perez, S. Bobev and F. Gascoin, J. Solid State Chem., 2016, 242, 14–20 CrossRef CAS.
- D. M. Rowe, CRC Handbook of Thermoelectrics, CRC Press, Boca Raton, 1995 Search PubMed.
- T. Witting, T. C. Chasapis, F. Ricci, M. Peters, N. A. Heinz, G. Hautier and G. J. Snyder, Adv. Electron. Mater., 2019, 5, 1800904 CrossRef.
- S. Roychowdhury, T. Ghosh, R. Arora, M. Samanta, L. Xie, N. K. Singh, A. Soni, J. He, U. V. Waghmare and K. Biswas, Science, 2021, 371, 722–727 CrossRef CAS PubMed.
- C.-H. Su, Prog. Cryst. Growth Charact. Mater., 2019, 65, 47–94 CrossRef CAS.
- L.-D. Zhao, C. Chang, G. Tan and M. G. Kanatzidis, Energy Environ. Sci., 2016, 9, 3044–3060 RSC.
- M. F. Mansuetto, P. M. Keane and J. A. Ibers, J. Solid State Chem., 1992, 101, 257–264 CrossRef CAS.
- L. Xu, L.-L. Deng, J. Cao, X. Wang, W.-Y. Chen and Z. Jiang, Nanoscale Res. Lett., 2017, 12, 159 CrossRef PubMed.
- H. Xin, J. K. Katahara, I. L. Braly and H. W. Hillhouse, Adv. Energy Mater., 2014, 4, 1301823 CrossRef.
- D. Shin, T. Zhu, X. Huang, O. Gunawan, V. Blum and D. B. Mitzi, Adv. Mater., 2017, 29, 1606945 CrossRef PubMed.
- M. Nakamura, K. Yamaguchi, Y. Kimoto, Y. Yasaki, T. Kato and H. Sugimoto, IEEE J. Photovolt., 2019, 9, 1863–1867 Search PubMed.
- P. Wu, A. E. Christuk and J. A. Ibers, J. Solid State Chem., 1994, 110, 337–344 CrossRef CAS.
- D. Zhou, J. Dong, Y. Si, F. Zhu and J. Li, Minerals, 2020, 10, 250 CrossRef CAS.
- New Match! version 3.13 (Build 227), https://www.crystalimpact.com/news/20220314b.htm.
- APEX3: Program for Data Collection on Area Detectors, Bruker AXS Inc., Madison, WI, USA, 2016.
- G. M. Sheldrick, A short history of SHELX, Acta Cryst. A, 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct, Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, XPREP Version 2008/2, Bruker AXS Inc., Madison, 2008.
- G. M. Sheldrick, Acta Cryst. A, 2008, 64, 112–122 CrossRef CAS PubMed.
- A. L. Spek, Single-crystal structure validation with the program PLATON, J. Appl. Cryst., 2003, 36, 7–13 CrossRef CAS.
- L. M. Gelato and E. Parthé, J. Appl. Cryst., 1987, 20, 139–143 CrossRef.
- G. Kortüm, Reflectance Spectroscopy, Springer, Berlin, Heidelberg, 1969 Search PubMed.
- A. Z. Johannes, R. K. Pingak and M. Bukit, IOP Conf. Ser. Mater. Sci. Eng., 2020, 823, 012030 CrossRef CAS.
- H. Wang, W. D. Porter, H. Böttner, J. König, L. Chen, S. Bai, T. M. Tritt, A. Mayolet, J. Senawiratne, C. Smith, F. Harris, P. Gilbert, J. Sharp, J. Lo, H. Kleinke and L. Kiss, J. Electron. Mater., 2013, 42, 1073–1084 CrossRef CAS.
- R. Piazza, S. Buzzaccaro, E. Secchi and A. Parola, Soft Matter, 2012, 8, 7112–7115 RSC.
- E. I. Andritsos, E. Zarkadoula, A. E. Phillips, M. T. Dove, C. J. Walker, V. V. Brazhkin and K. Trachenko, J. Phys.: Condens. Matter, 2013, 25, 235401 CrossRef CAS PubMed.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. Sun, A. Ruzsinszky and J. P. Perdew, Phys. Rev. Lett., 2015, 115, 036402 CrossRef PubMed.
- G. K. H. Madsen, J. Carrete and M. J. Verstraete, Comput. Phys. Commun., 2018, 231, 140–145 CrossRef CAS.
- Y. M. Andreev, V. V. Atuchin, G. V. Lanskii, N. V. Pervukhina, V. V. Popov and N. C. Trocenco, Solid State Sci., 2005, 7, 1188–1193 CrossRef CAS.
- P. Lemoine, D. Carré and M. Guittard, Acta Cryst. C, 1986, 42, 390–391 CrossRef.
- M. Wakeshima and Y. Hinatsu, J. Solid State Chem., 2000, 153, 330–335 CrossRef CAS.
- A. C. Sutorik, J. Albritton-Thomas, T. Hogan, C. R. Kannewurf and M. G. Kanatzidis, Chem. Mater., 1996, 8, 751–761 CrossRef CAS.
- Y. Liu, X.-D. Song, R.-C. Zhang, F.-Y. Zhou, J.-W. Zhang, X.-M. Jiang, M. Ji and Y.-L. An, Inorg. Chem., 2019, 58, 15101–15109 CrossRef CAS PubMed.
- H. Chen, W. Fu, H. Yang, P. Sun, Y. Zhang, L. Wang, W. Zhao, X. Zhou, H. Zhao, Q. Jing, X. Qi and Y. Li, Electrochim. Acta, 2010, 56, 919–924 CrossRef CAS.
- M. Probert, Contemp. Phys., 2011, 52, 77 CrossRef.
- V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Phys. Chem. A, 2011, 115, 5461–5466 CrossRef CAS PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- G. J. Snyder and E. S. Toberer, Nat. Mater., 2008, 7, 105–114 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional crystallographic details of the BaCeCuS3 structure, theoretical calculation details, and results. The CIF file of the BaCeCuS3 structure was submitted to the joint CCDC/FIZ Karlsruhe deposition service. CCDC 2125729. For the ESI and crystallographic data in CIF or other electronic formats see DOI: https://doi.org/10.1039/d2nj06301h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |