
Current status of secondary metabolite pathways linked to their related biosynthetic gene clusters in Aspergillus section Nigri
Xinhui
Wang
 ,
Scott A.
Jarmusch
,
Jens C.
Frisvad
and
Thomas O.
Larsen
*
,
Scott A.
Jarmusch
,
Jens C.
Frisvad
and
Thomas O.
Larsen
*
DTU Bioengineering, Technical University of Denmark, DK-2800, Kgs. Lyngby, Denmark. E-mail: tol@bio.dtu.dk
First published on 19th May 2022
Abstract
Covering: up to the end of 2021
Aspergilli are biosynthetically ‘talented’ micro-organisms and therefore the natural products community has continually been interested in the wealth of biosynthetic gene clusters (BGCs) encoding numerous secondary metabolites related to these fungi. With the rapid increase in sequenced fungal genomes combined with the continuous development of bioinformatics tools such as antiSMASH, linking new structures to unknown BGCs has become much easier when taking retro-biosynthetic considerations into account. On the other hand, in most cases it is not as straightforward to prove proposed biosynthetic pathways due to the lack of implemented genetic tools in a given fungal species. As a result, very few secondary metabolite biosynthetic pathways have been characterized even amongst some of the most well studied Aspergillus spp., section Nigri (black aspergilli). This review will cover all known biosynthetic compound families and their structural diversity known from black aspergilli. We have logically divided this into sub-sections describing major biosynthetic classes (polyketides, non-ribosomal peptides, terpenoids, meroterpenoids and hybrid biosynthesis). Importantly, we will focus the review on metabolites which have been firmly linked to their corresponding BGCs.
 Xinhui Wang | Xinhui Wang received her Master's degree in Pharmacognosy from Tianjin University, supervised by Prof. Robert P. Borris in 2018, and obtained her PhD in fungal natural product discovery from Technical University of Denmark (DTU) in 2022. She is currently a Postdoc researcher at the DTU with Prof. Thomas O. Larsen. Her research focuses on mass spectrometry analysis and bioguided discovery of fungal natural products and fish killing toxins, including characterization of complex structures of ladder-frame polyethers by advanced NMR techniques. |
 Scott A. Jarmusch | Scott A. Jarmusch received a B.S. in Biochemistry from the University of North Carolina at Greensboro, a M.A. Ancient History from University of Liverpool and a PhD in Chemistry from the University of Aberdeen under the supervision of Prof Marcel Jaspars and Dr Rainer Ebel. After a one year postdoc at Uppsala University, he is currently a postdoc in the Center for Microbial Secondary metabolites (CeMiSt) at the Technical University of Denmark. His research interests include RiPP ecology, microbial chemical ecology and MS-based metabolomics applied to micro-organisms. |
 Jens C. Frisvad | Jens C. Frisvad was born in Kgs. Lyngby, Denmark in 1952. He studied chemical engineering at DTU with focus on organic chemistry and mycology. He received his PhD in 1982 and defended his Dr techn. thesis on “Secondary metabolites and species concepts in Penicillium and Aspergillus” in 1998. He became full professor in Industrial Mycology at DTU in 2001. In 1994, he stayed for six months with late Professor Martha Christensen, Dept. of Botany, University of Wyoming and has been collaborating extensively for many years with Dr Robert A. Samson at the Westerdijk Fungal Biodiversity Institute, Utrecht, the Netherlands. |
 Thomas O. Larsen | Thomas O. Larsen was born in Slagelse, Denmark in 1963. He studied Chemical Engineering at DTU with focus on Organic Chemistry and Microbiology. After a job at Danisco A/S working with aroma synthesis he returned to DTU to study fungal volatile production and received his PhD in 1994. He then joined the Marine Chemistry Group at the University of Copenhagen, headed by Reader Carsten Christophersen where he was trained as a classical natural product chemist, before returning to DTU to take up a permanent position. Currently he holds a position as full Professor in Microbial Natural Product Chemistry at Department of Biotechnology and Biomedicine at DTU. |
1 Introduction
1.1 Chemodiversity within genus Aspergillus
Fungal species within genus Aspergillus are able to produce a large number of secondary metabolites including polyketides, non-ribosomal peptides, terpenoids and numerous hybrids of these aforementioned classes. It has been well documented that the combined profile of fungal secondary metabolites in a given species is very specific and can therefore be used in chemotaxonomy.1 In line with this there is a high degree of metabolite consistency from isolate-to-isolate within a species even though certain metabolite biosynthetic gene clusters (BGCs) might be silenced in some isolates.2 On the other hand individual secondary metabolites may occur in other species, even those phylogenetically and ecologically distantly related to Aspergillus. Consequently, correct identification of species in genus Aspergillus, based partly on chemotaxonomy, has notoriously been challenging, since closely related species often share many phenotypic features including metabolite production. This has led to the majority of the series in Aspergillus being invalidly described or named as clades, where a new valid series classification was introduced by Houbraken et al.3 By using a phylogenetic approach, including data on secondary metabolite production, Aspergillus was subdivided in six subgenera, 27 sections and 75 series, altogether including a list of 446 Aspergillus species.3To further investigate both inter- and intraspecies genomic variation as well as the chemical potential of species in genus Aspergillus, a large sequencing project including one representative strain of >350 known Aspergillus spp. has been initiated using the Joint Genome Institute (JGI) fungal genome pipeline.4,5 One of the first major outcomes from this initiative was a comprehensive bioinformatic analysis of all detected BGCs from species in section Nigri.6 In line with the above mentioned studies, a major conclusion from this work was that secondary metabolism is unique to individual species. The authors reported a total of 2717 encoding secondary metabolite BGCs from a total of 37 genomes and found that on average, each species contains 8.75 unique BGCs.6 Similar to previous studies,7,8 the authors also identified and linked groups of BGCs, that are functionally closely related and encode the production of the same or very similar secondary metabolites, into 455 biosynthetic gene cluster families.6 This large number is in sharp contrast to the relatively few secondary metabolites that have so far been linked to their corresponding BGCs, indicating the overall potential for discovery in Nigri.
1.2 Biosystematics and genomes of Aspergillus subgenus Circumdati section Nigri
Species in Aspergillus section Nigri are among the most important filamentous fungi used in biotechnology and are also common in many natural habitats. Most species in section Nigri are asexual, producing a large number of asexual (mitotic) spores (conidia), however, a few species have been shown to have a sexual state.7–11 Interestingly, the two mating types needed for sexual reproduction have been found in different Aspergillus section Nigri species.11,12 A large number of species can produce sclerotia (large, firm, frequently rounded masses of hyphae serving as propagules of new growth or as long-lasting resting structures) under certain conditions,13–19 and sclerotium production is important as certain indoloterpenes are only produced in the sclerotia, and indoloterpenes may go unnoticed if sclerotia are not formed.8 Section Nigri is most closely related to sections Flavi, Candidi, and Circumdati in subgenus Circumdati of Aspergillus according to phylogeny,19 morphology and secondary metabolite production.20,21 Notably, section Nigri seems to be an outlier as full genome sequencing indicates that section Nigri is phylogenetically more closely related to subgenus Nidulantes than to subgenus Circumdati.6,22 Furthermore members of section Nigri differ from these groups of Aspergillus subgenera by having the ubiquinone Q-9 rather than the Q-10 (and/or Q-10 (H2)), as do species in sections Flavi, Candidi, Circumdati and subgenus Nidulantes. Visagie et al. placed biseriate section Nigri species as related to the subgenus Circumdati sections Terrei and Flavipedes, and uniseriate section Nigri species close to subgenus Circumdati section Petersoniorum, leaving section Nigri as a polyphyletic taxon, based on ITS and calmodulin sequences.23 Here we follow the phylogeny and taxonomy suggested by Houbraken et al.,3 except that we provisionally accept the species A. fijiensis, A. violaceofuscus and A. lacticoffeatus.The species in the A. niger group (now called Aspergillus subgenus Circumdati section Nigri) have been treated by Mosseray (1934), and Raper and Fennell (1965).24,25 Similarly, very detailed morphological and physiological taxonomic studies were done by Murakami et al.26–36 with an emphasis on industrial black Kuro koji strains. Later new taxonomies and phylogenies were suggested, based on morphology, physiology, household gene sequences and secondary metabolite profiles.3,6,13,15–17,20,22,37–72
Aspergillus subgenus Circumdati section Nigri presently consists of 5 formal series: ser. Nigri, ser. Carbonarii, ser. Heteromorphi (biseriate species, i.e. species with both metulae and phialides on the globose conidial Aspergillus head (vesicle)), in addition to ser. Japonici (uniseriate species, i.e. species with phialides only on the globose conidial Aspergillus head (vesicle)) and ser. Homomorphi (biseriate).3,46 Ser. Nigri includes 14 species; Ser. Carbonarii includes 4 species; series Heteromorphi includes 2 species; series Japonici includes 15 species, and finally ser. Homomorphi only has one member (Table 1). Many other names have been published and frequently used, especially A. acidus, A. aureus var. pallidus, A. awamori, A. coreanus, A. inuii, A. kawachii and A. nakazawai (all synonyms of A. luchuensis) and A. pulverulentus, A. saitoi and A. saitoi var. kagoshimaensis (synonyms of A. tubingensis), and A. aureus, A. batatae, A. miyakoensis, A. usamii and A. usamii var. shirousamii (all synonyms of A. niger).61,62Aspergillus awamori is a name often used, and was originally a name for a domesticated form of A. niger, later it was regarded as cryptic phylogenetic species in A. niger based on a non-domesticated strain from the plant Welwitschia mirabilis.54 This interpretation of A. awamori was abandoned and the name A. welwitschiae given to this taxon, while the domesticated forms were named A. luchuensis, which is the most commonly used Kuro koji fungus.61,62 The names A. phoenicis and A. ficuum have been rejected in order to conserve the well-known name A. niger.73 The number of accepted species in Aspergillus section Nigri is therefore currently 36. Additionally, many of the species of Aspergillus section Nigri have been genome sequenced (Table 1). Thus there are good opportunities for mining these genomes for BGCs encoding for secondary metabolites.
| Series | Species |
|---|---|
| Nigri | A. brasiliensis, A. chiangmaiensis, A. costaricensis, A. eucalypticola, A. lacticoffeatus, A. luchuensis, A. neoniger, A. niger, A. piperis, A. pseudopiperis, A. pseudotubingensis, A. tubingensis, A. vinaceus, and A. welwitchiae |
| Carbonarii | A. carbonarius (=A. fonsecaeus), A. ibericus, A. sclerotiicarbonarius, and A. sclerotioniger |
| Heteromorphi | A. ellipticus and A. heteromorphus |
| Japonici | A. aculeatinus, A. aculeatus, A. brunneoviolaceus, A. fijiensis, A. floridensis, A. hydei, A. japonicus, A. labruscus, A. oxumae, A. saccharolyticus, A. serratalhadensis, A. siamensis, A. trinidadensis, A. uvarum and A. violaceofuscus |
| Homomorphi | A. homomorphus 3,18 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|
| The species of Aspergillus section Nigri that have been genome sequenced | |
| A. niger,6,11,74–80A. luchuensis,75,81–85A. tubingensis,6,75,86,87A. aculeatus,6,88A. aculeatinus in addition to A. brasiliensis, A. brunneoviolaceus, A. carbonarius, A. costaricensis, A. ellipticus, A. eucalypticola, A. fijiensis, A. heteromorphus, A. homomorphus, A. ibericus, A. indologenus, A. japonicus, A. lacticoffeatus, A. neoniger, A. piperis, A. saccharolyticus, A. sclerotiicarbonarius, A. sclerotioniger, A. uvarum, A. vadensis, A. violaceofuscus, and A. welwitschiae6,89 | |
Some species in Aspergillus section Nigri can produce mycotoxins, notably ochratoxin A (A. carbonarius, A. lacticoffeatus, A. niger, A. sclerotioniger, A. welwitschiae),40,47,90–96 and fumonisins B2, B4 and B6 (A. lacticoffeatus, A. niger and A. welwitschiae).46,95–100 Secalonic acid, only found in the uniseriate Aspergillus section Nigri species, has also been mentioned as a mycotoxin.101 Oxalic acid is a widespread weak mycotoxin of Aspergillus niger and related species in series Nigri and Carbonarii. It has been linked to kidney failure after growth and in human lungs and transport of calcium oxalate, via the blood to the kidneys.102 Oxalic acid, citric acid and gluconic acid are produced by many species in section Nigri. These natural products should be regarded as secondary metabolites when they are accumulated, secreted and regulated by the general regulator LaeA, in addition to being taxonomically restricted,103–105 but as primary metabolites when they are in flux in the TCA cycle (and therefore not secreted and accumulated and furthermore present in all fungi and many other organisms). Altogether, a large number of secondary metabolites have been reported from Aspergillus niger and similar species,102,106–109 and preliminary genome scale models for Aspergillus niger metabolic networks have been published.110,111
2 Secondary metabolites and their biosynthetic pathway from Aspergillus section Nigri
As of June 2021, a total of 176 compounds have been isolated and characterized from the species of Aspergillus section Nigri. Table 2 highlights some metabolites discovered from 2009 to 2021 not included in our previous review paper,109 and Table 3 highlights the secondary metabolites for which their biosynthetic pathway have been characterized.| Metabolites | Producing organism | References |
|---|---|---|
| Fumonisin Py | A. welwitschiae | 112 |
| Fumonisin La | A. welwitschiae | 112 |
| Ergosterimide B | A. tubingensis YP-2 | 113 |
| Demethylincisterol A5 | A. tubingensis YP-2 | 113 |
| Sclerolizine | A. sclerotiicarbonarius | 114 |
| Emindole SC | A. sclerotiicarbonarius | 114 |
| Carbonarins I | A. sclerotiicarbonarius | 114 |
| Carbonarins J | A. sclerotiicarbonarius | 114 |
| Aculenes A–C | A. aculeatus | 115 |
| Okaramine S | A. aculeatus | 115 |
| Epi-10,23-dihydro-24,25-dehydroaflavinine | A. aculeatus | 115 |
| Acu-dioxomorpholine | A. aculeatus | 115 |
| Acucalbistrin A | A. aculeatus | 115 |
| Acucalbistrin B | A. aculeatus | 115 |
| Dihydrocarolic acid | A. niger | 116 |
| Penitricin D | A. niger | 116 |
| New diketopiperazine dimer | A. niger | 117 |
| Sclerotionigrin A | A. sclerotioniger | 118 |
| Sclerotionigrin B | A. sclerotioniger | 118 |
| Asperpyrone D | A. tubingensis | 119 |
| Methyl protodioscin | A. niger | 120 |
| Aurasperone H | A. niger | 121 |
| Secalonic acid F and D | A. aculeatus | 122 |
| Gibberellic acid | A. niger | 123 |
| Aspiperidine oxide | A. indologenus | 124 |
| Pre-aurantiamin | A. brunneoviolaceus, A. japonicus and A. aculeatus | 65 |
| Homopyrone A | A. homomorphus | 125 |
| Homopyrone B | A. homomorphus | 125 |
| Name of metabolites | BGC names or key genes | Producer organism | Linkage between metabolite and BGC | Key reference(s) |
|---|---|---|---|---|
| Naphtho-γ-pyrones | albA (fwnA) | A. niger | The function of albA was confirmed by gene deletion in auxotrophic A. niger strain ATCC 11414 (KB1001) | 126 and 127 |
| In another study, the loci involved in spore pigmentation of A. niger were identified. Metabolic profiling of the fawn mutant revealed the production of naphtho-γ-pyrones depends on fwnA (albA) | ||||
| aun and bfo (tailoring genes) | A. niger | The tailoring enzymes included in a gene cluster that is separate from PKS core gene albA were identified by gene deletions in A. niger FGSC A1180 (aun) | 128 | |
| A. aculeatus | Heterologous expression of aunB and bfoB in A. aculeatus to explore the substrate specificity of the dimerization | |||
| Kotanin | ktnS | A. niger | Bioinformatics analysis was used for searching NR-PKS containing BGCs and gene deletion experiments were performed for functional analysis in the NHEJ-deficient A. niger strain FGSC A1180 | 129 |
| TAN-1612 | adaA | A. niger | Transcriptional factor was replaced by promoter and the gene cluster was identified using a targeted genome mining approach A. niger ATCC 1015. The function of bifunctional MβL-TE was identified by heterologous pathway reconstitution in Saccharomyces cerevisiae | 130 |
| Azanigerones | azaA and azaB | A. niger | The silent gene was activated by overexpression of transcriptional factor (azaR). Two convergent PKSs in A. niger ATCC 1015 were identified by transcriptional analysis and deletion of a key PKS gene (azaB) | 131 |
| Nigerpyrones | epaA | A. niger | The epigenetic regulator gcnE was deleted in a highly mutated industrial strain, A. niger FGSC A1279. The function of key gene epaA was confirmed by gene deletion by homologous recombiation and complementary experiments | 132 |
| 2,4-Dihydroxy-3,5,6-trimethylbenzaldehyde | dtbA | A. niger | The function of dtbA was demonstrated by heterologous expression in A. nidulans. R domain was replaced by a TE release domain in the DtbA to expand the structural diversity | 133 |
| Funalenone | phnA | Penicillium herquei | A. niger is a funalenone producer with several homologous genes to those found in P. herquei. The phnA was deleted using homologous recombination and hygromycin as selection marker. The function of phnA was further confirmed by heterologous expression in S. cerevisiae. Point mutation of PhnA was used to investigate the roles of PT and TE/CLC of PhnA | 134 |
| Aculinic acid and aculin A | acuA | A. aculeatus | The function of core gene was verified by integration into the IS1 locus in A. nidulans (heterologous expression) and AMA1-based episomal expression and 13C labeling experiments in A. aceleatus | 115 and 135 |
| Fumonisins | fum5 | A. niger and A. welwitschiae | The fumonisin biosynthesis have been extensively studied in the different species in Fusarium. The fumonisin production and biosynthesis in black aspergilli were explored mainly with homologous genes analysis | 136–138 |
| Homopyrone A and B | AhpA | A. homomorphus | Retro-biosynthesis, genome mining, and gene deletions were used to successfully link to the biosynthetic gene cluster | 125 |
| Aculeacin A | EcdA | A. aculeatus | Aculeacin biosynthesis is very closely related to echinocandin B biosynthesis. The gene cluster of aculeacin A was identified by investigating the evolution of chemical diversity in echinocandin lipopeptide metabolites. The proline hydroxylase, Aa-HtyE, was characterized through heterologous expression in E. coli | 139 and 140 |
| Malformins | MlfA | A. brasiliensis | The function of core gene mlfA was confirmed by targeted gene deletion in A. brasiliensis and genetic complementation experiments | 107 |
| Atrofuranic acid | — | A. niger | The compound was discovered when atromentin synthetases were heterologously expressed in A. niger in order to explore the reaction mechanism of the thioesterase domain | 141 |
| Ergosterimide | — | A. niger | This compound was proposed to be a Diels–Alder adduct of ergosteroid and maleimide | 142 |
| Taxol | — | A. aculeatinus | The biosynthesis pathway of taxol formed from endophytic fungi was not clear | 143 |
| Pyranonigrins E–J | pynA | A. niger | The pyn gene cluster was activated by expressing the transcriptional regulator pynR under control of the arginase (aga) promotor. The biosynthetic pathway was identified by targeted gene deletion in A. niger, heterologous in vivo bioconversion in S. cerevisiae SCKW12 and in vitro assays | 144 and 145 |
| Pyranonigrins A, S | pyrA | P. thymicola | The metabolites were first reported from A. niger. The biosynthetic pathway was elucidated by genome mining and heterologous expression in A. nidulans | 146 |
| Pyranoviolin A | pyvA | A. violaceofuscus | The BGC was identified via bioinformatics analysis and comparison with the homologous genes to those of pyranonigrins. The PKS–NRPS was confirmed through targeted gene deletion | 147 |
| Ochratoxin A | otaA and otaB | A. carbonarius | The NRPS was verified by targeted gene disruption | 148 and 149 |
| Acurin A | acrA and acrB | A. aculeatus | The functions of genes were identified through gene deletion experiments | 150 |
| Yanuthones | yanA and yanG | A. niger | The PKS was confirmed by heterologous expression in A. nidulans. The biosynthetic pathway was investigated by individually deleting all genes | 151 and 152 |
| Aculene A–C | aneB and aneC | A. aculeatus | The biosynthetic pathway was investigated through gene inactivation, feeding experiments and heterologous reconstitution approaches | 153 |
| Alkylcitric acids | akcA, akcB and akcD | A. niger | The gene cluster was revealed by genomic data analysis, overexpression of transcriptional factor and targeted gene deletion in A. niger | 154 |
2.1 Non-reduced polyketides
Fungal polyketides are synthesized by multidomain polyketide synthases (PKSs), which can be classified as nonreducing (NR), partially reducing (PR), and highly reducing (HR) PKSs. NR-PKSs synthesize a diverse array of polycyclic compounds that contain several important domains at minimum, including: ACP transacylase (SAT), β-ketoacyl synthase (KS), acyl transferase (AT), product template (PT), and acyl carrier protein (ACP). Additional domains that may be present include: C-methyltransferase (C-MeT), thioesterase (TE), Claisen cyclase (CLC) or reductase (R) domains. This section summarizes the secondary metabolites of NR-PKS origin in Aspergillus section Nigri, including azanigerone A (1), kotanin (2), naphtho-γ-pyrones, TAN-1612 (3), 2,4-dihydroxy-3,5,6-trimethylbenzaldehyde (4), nigerpyrone (5) and funalenone (6) (Table 4 and Fig. 1). The PT domains of NR-PKSs can be categorized phylogenetically into eight major groups (group I–VIII), each of which corresponds to a unique polyketide length or cyclization regioselectivity. The NR-PKS derived secondary metabolites in black aspergilli described here belong to group III, group V and group VII. Group III catalyze a single C2–C7 cyclization and multicyclization and product release is TE/CLC mediated. Group VII also catalyze C2–C7 monocyclization and most contain a C-MeT domain. Furthermore, all NR-PKSs in this group release the polyketide product via a reductive release mechanism (terminal R domain). Group V catalyze C6–C11 cyclization most frequently and C4–C13 positions for the secondary cyclization. Group V NR-PKSs are unique due to the fact they all lack the C-terminal release domain. Alternatively, they use a stand-alone metallo-β-lactamase-type TE (MβL-TE).155| Final product | Gene name | NR-PKS domain organization | |
|---|---|---|---|
| a The biosynthetic pathway of phenalenone was identified in Penicillium herquei, which can provide the comparative analysis for the biosynthesis of funalenone. b Putatively assigned to a group based on their domain organization and closest characterized homologue. | |||
| NR-PKS | Azanigerone A (1) | azaA (group VII) |

|
| Kotanin (2) | ktnS (group V) |

|
|
| Naphtho-γ-pyrones | AlbA (fwnA) (group III) |

|
|
| TAN-1612 (3) | adaA (group V) |

|
|
| 2,4-Dihydroxy-3,5,6-trimethylbenzaldehyde (4), 6-ethyl-2,4-dihydroxy-3,5-dimethylbenzaldehyde (39) | dtbA (group VII) |

|
|
| Nigerpyrone (5) | epaA (group VII) |

|
|
| Phenalenone (42)a | phnA (group III)b |

|
|
| PR-PKS | Aculinic acid (48) | acuA |

|
| Ochratoxin A (50) | otaA |

|
|
| Yanuthones | yanA |

|
|
| HR-PKS | Fumonisins | fum 1 |

|
| Azanigerone A (1) | azaB |

|
|
| Homopyrone A and B (70, 71) | ahpA |
|
|
| Pyranonigrins E–J (79–84) | pynA (PKS–NRPS) |

|
|
| Pyranonigrin A (85) | pyrA (PKS–NRPS) |

|
|
| Pyranoviolin A (87) | pyvA (PKS–NRPS) |

|
|
 | ||
| Fig. 1 Examples of NR-PKS products from Aspergillus section Nigri. | ||
 | ||
| Fig. 2 Proposed biosynthetic pathway of DHN-melanin and naphthopyrone dimers in black aspergilli.128 | ||
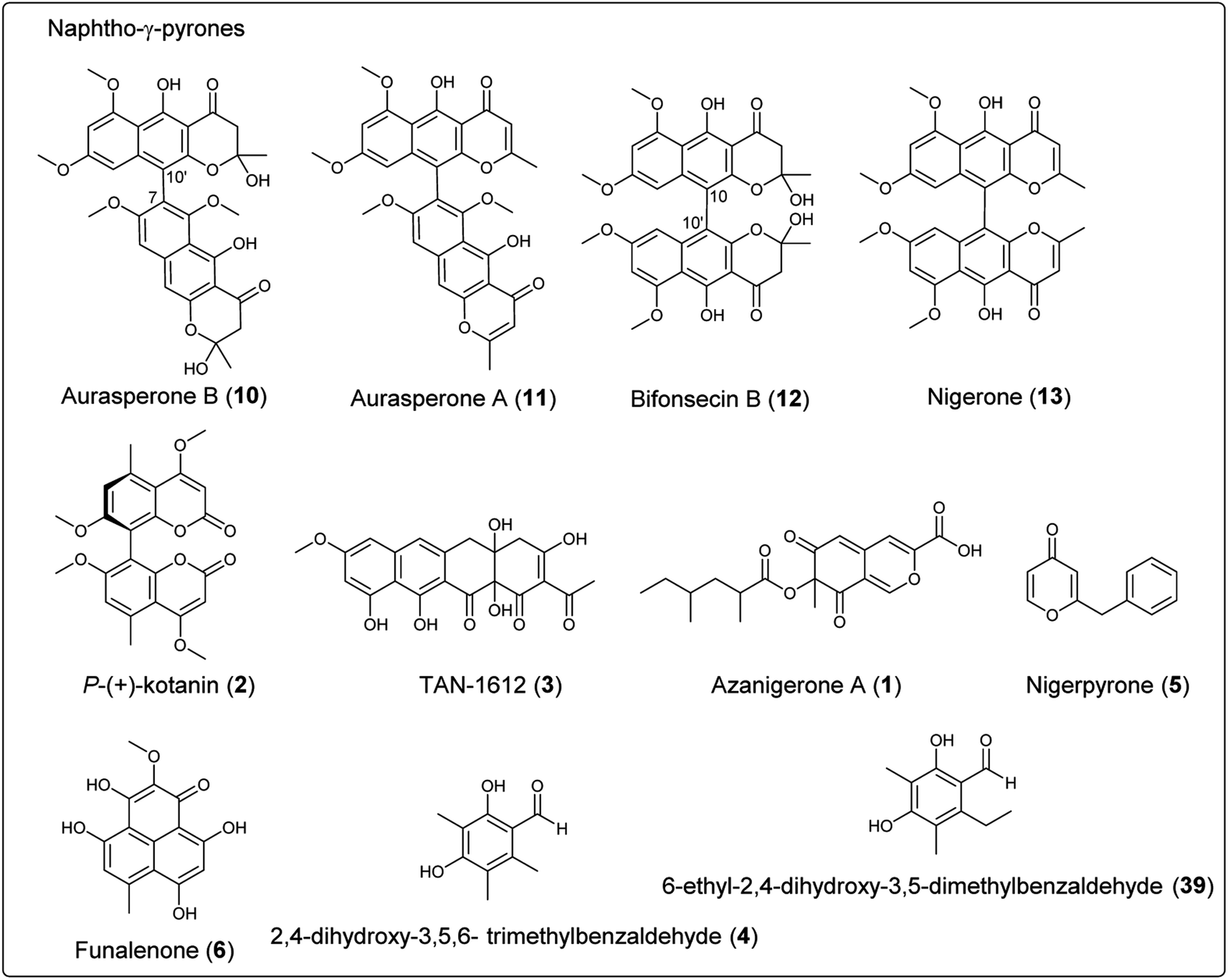
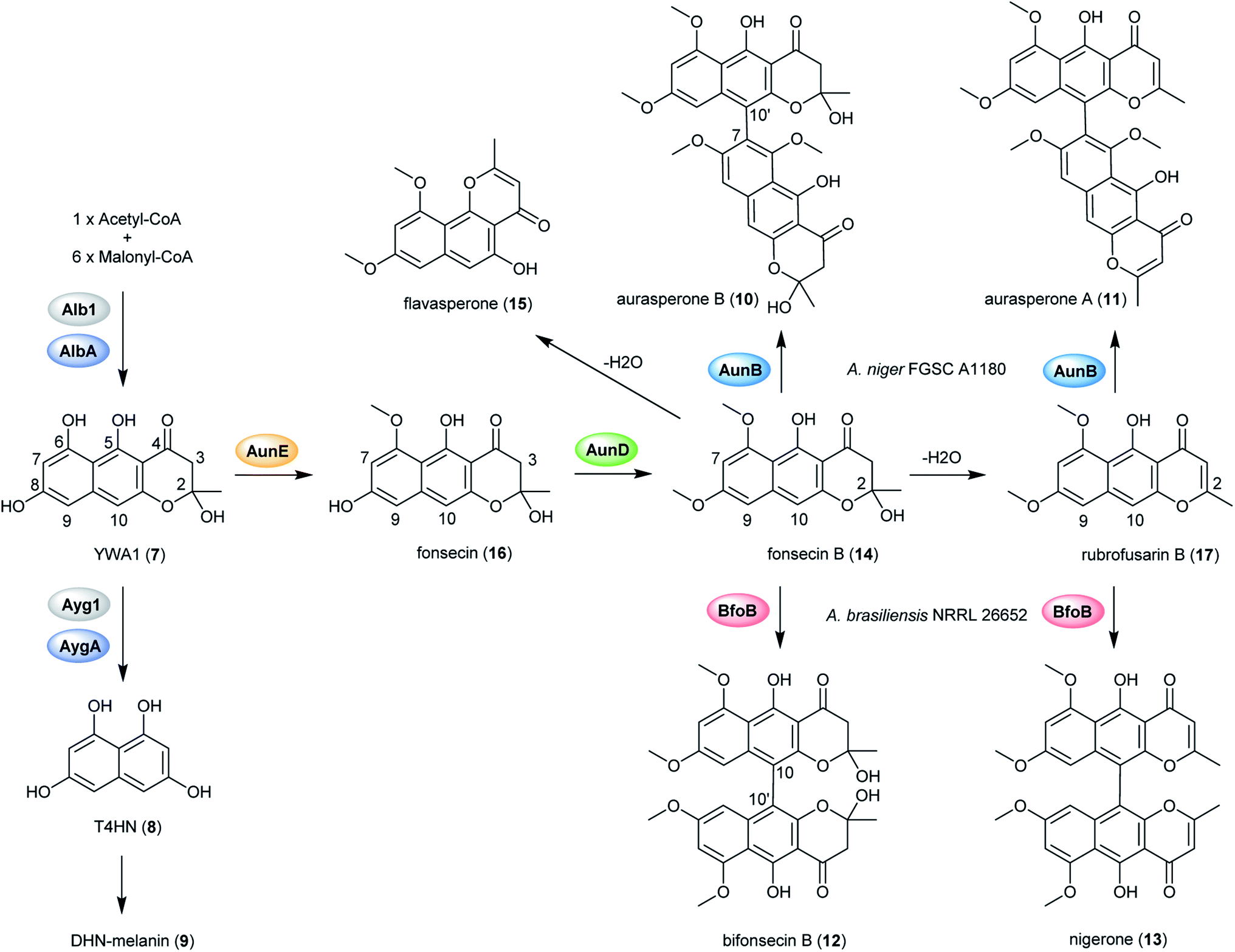
In order to explore the naphtho-γ-pyrones biosynthesis in A. niger, albA, a homolog of PKS alb1 originating in A. fumigatus was deleted and led to the abolishment of production of all naphtho-γ-pyrones.126 A contemporary study showed that the naphtho-γ-pyrones polyketides were specifically dependent on fwnA (albA) using UV mutagenesis.127 In addition, both studies showed that albA in A. niger is responsible for the biosynthesis of melanin, which is consistent with the prediction that A. niger DHN-melanin (9) production is similar to the pathway elucidated for A. fumigatus.126,127,158,159 Moreover, a putative ortholog of A. fumigatus ayg1, aygA, was identified in A. niger. When aygA was deleted, melanin production was blocked but naphtho-γ-pyrones were still observed, indicating aygA is not involved in the naphtho-γ-pyrone biosynthetic pathway but only in the biosynthesis of 9 (Fig. 2).126 However, since the downstream biosynthetic genes responsible for naphtho-γ-pyrones are not clustered with the PKS gene albA, researchers were not able to identify the enzymes responsible for methylation and dimerization. It has recently been discovered that dimerization of naphtho-γ-pyrones monomers is catalyzed by cytochrome P450 enzymes in black aspergilli and they are not in the same gene cluster as the polyketide synthase gene.128 To identify the genes involved in naphthopyrone biosynthesis, the dimeric derivatives in five lineages of black aspergilli were re-evaluated by Obermaier and Müller.128 They identified aurasperone B (10) and aurasperone A (11) in A. niger (FGSC A1180), A. tubingensis and A. carbonarius. Bifonsecin B (12) and nigerone (13) were found in A. niger (ATCC 36626) and one strain of A. brasiliensis. The genes encoding tailoring enzymes form a gene cluster that is separate from the polyketide synthase encoding gene. It was therefore proposed that the transcription of these genes is co-regulated with the expression of albA (PKS gene involved in the biosynthesis of YWA1).128 Because YWA1 (7) is also the precursor of the spore pigment melanin, the upregulation most likely coincide with sporulation.158
In order to explore the oxidative enzyme for dimerization from a sporulating culture, publicly available transcriptomes of A. carbonarius obtained from both static and shaken liquid cultures were screened. This identified a CYP enzyme (aunB) clustered with two O-MT genes (aunD, aunE). Other producers of dimeric naphtho-γ-pyrones, such as A. brasiliensis (bfo), A. niger (aun), and A. tubingensis (aun), also had homologous clusters.128 To investigate the function of these enzymes in the biosynthesis of naphthopyrone dimers, an aunB deletion strain was generated in A. niger FGSC A1180 and led to the accumulation of the monomeric compounds fonsecin B (14) and flavasperone (15). This indicated the aunB gene to be responsible for dimerization of the monomeric precursor.128 Additionally, deletion of the O-MT gene aunD resulted in an accumulation of fonsecin (16), which identified that aunD methylates fonsecin. Another methyltransferase gene, aunE, was proposed to be involved in methylation of the hydroxyl group at position 6. To explore the substrate specificity of the CYP enzymes for catalyzing monomeric naphthopyrones into dimers, aunB from A. niger FGSC A1180 and bfoB from A. brasiliensis NRRL 26652 were heterologously expressed in A. aculeatus, which has not been reported to produce dimeric naphthopyrones. AunB was shown to catalyze the regioselective dimerization of fonsecin B (14) and rubrofusarin B (17) to aurasperone B (10) and A (11), respectively, while bfoB catalyze dimerization of bifonsecin B (12) and nigerone (13) (Fig. 2).128 Additional monomeric metabolites, fonsecin (16) and flavasperone (15), were not converted into dimers. Further exploration showed the substrate dependency of the atroposelectivity for the CYP enzymes. (M)-Nigerone was produced by a BfoB catalyzed dimerization of the monomers fonsecin B (14) (enantiomeric excess >95%). However, feeding with rubrofusarin B (17) resulted in an excess of (P)-nigerone. The exact mechanisms leading to atropselectivity is complex and future research will need to explain how the enzymes interact with the various monomeric substrates.128
The BGC responsible for biosynthesis of kotanin was identified using a combination of bioinformatics and gene deletion experiments (Fig. 3);129 searching only for BGCs containing NR-PKS encoding genes as no reduction steps were predicted to be involved in formation of the core dihydroxy coumarin structure from a linear pentaketide. Candidate clusters were further restricted to include only those containing cytochrome P450s, monooxygenases and methyltransferases, all of which would be required for kotanin (2) biosynthesis. KtnS was identified as the NR-PKS responsible for kotanin (2) biosynthesis and further confirmed by targeted gene deletion in the NHEJ-deficient A. niger strain FGSC A1180, resulting in complete loss of 4,7-dihydroxy-5-methylcoumarin (19) production. A functional analysis of the genes involved in the same cluster was also performed by targeted gene deletions, which confirmed the involvement of the O-methyltransferase KtnB in 7-demethylsiderin (20) production and the cytochrome P450 monooxygenase KtnC in kotanin (2) biosynthesis.
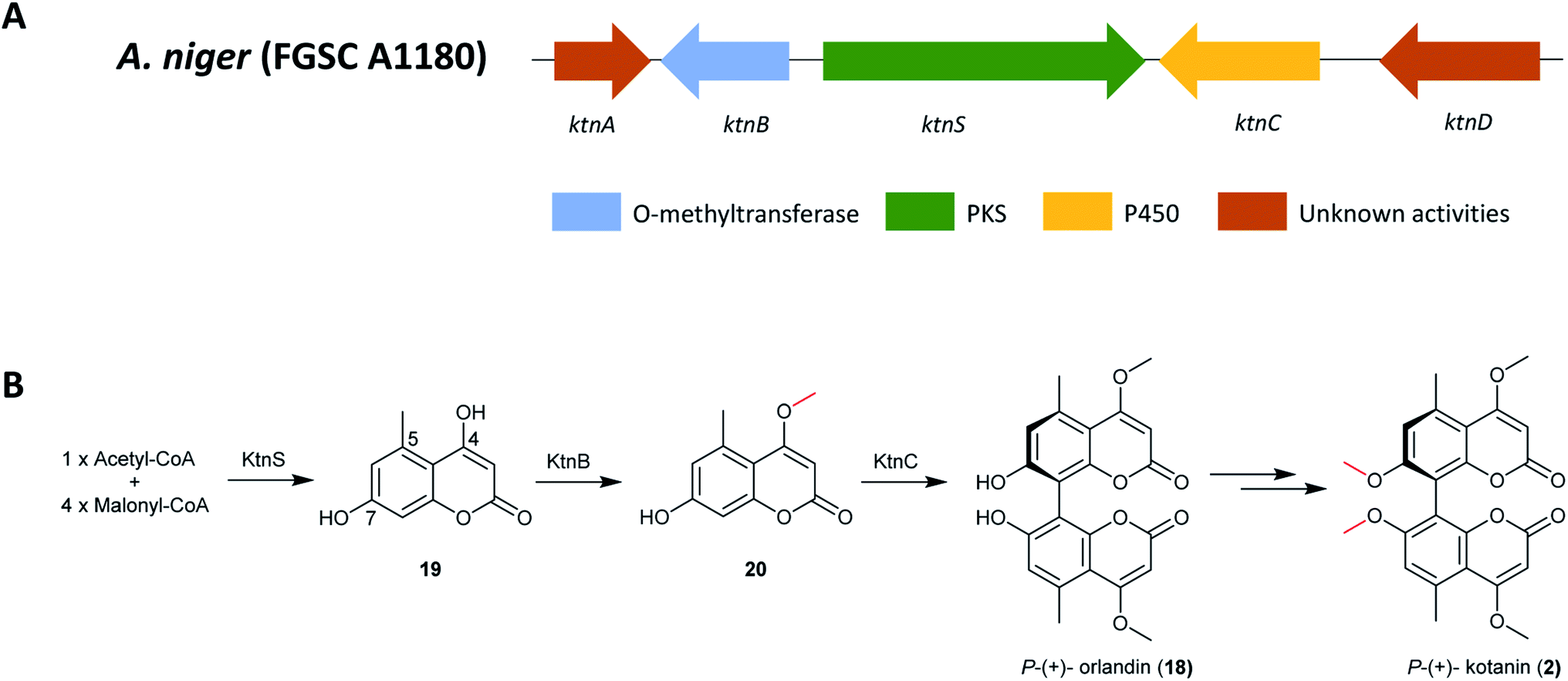 | ||
| Fig. 3 (A) The gene cluster ktn in A. niger and (B) the proposed biosynthetic pathway of P-(+)-kotanin (2).129,163 | ||
Furthermore, an oxidative phenol coupling catalyzed by the cytochrome P450 monooxygenase KtnC leads to formation of P-(+)-orlandin (18). Docking experiments were performed to gain insights into the mechanism of regio- and stereoselectivity, which revealed that it is likely dependent on substrate orientation in the active site of cytochrome P450 monooxygenase KtnC. Subsequent research also showed that KtnC selectivity catalyzes the intermolecular coupling of 7-demethylsiderin (20), regioselectively to the 8,8′-dimer and stereoselectively to the P-atropisomer.163 Finally, P-(+)-kotanin (2) is then generated following O-methylation of P-(+)-orlandin (Fig. 3).
The PT domain of this compound belongs to group V NR-PKSs, which utilize a trans-acting metallo-β-lactamase-type TE (MβL-TE) instead of a C-terminal release domain; this MβL-TE PT domain would be responsible for catalyzing the C6–C11 regioselective cyclization. However, no MβL-TE or similar enzymes for product release was found in ktn, suggesting a spontaneous (or other) release mechanism of the final polyketide. In addition, the cyclization pattern of the core polyketide structure by ktnS is C2–C7 and not the typical C6–C11 cyclization pattern seen for the group V NR-PKSs. This might be because of the short pentaketide chain or this branch represents a separate group of cyclization encoding PT domains.
Because of the similarities between the ada gene cluster for naphthacenedione 3 and the known apt cluster linked to the biosynthesis of anthraquinone asperthecin, the biosynthesis of these chemically different metabolites were done in parallel via heterologous pathway reconstitution in Saccharomyces cerevisiae.130 This revealed that a novel α-hydroxylation-dependent Claisen cyclization cascade, which involves a flavin-dependent monooxygenase that hydroxylates the α-carbon of an acyl carrier protein-bound polyketide and a bifunctional metallo-β-lactamase-type thioesterase (MβL-TE). The bifunctional MβL-TE catalyzes the cyclization of the fourth ring to afford the naphthacenedione scaffold upon α-hydroxylation, whereas it performs hydrolytic release of an anthracenone product (21) in the absence of α-hydroxylation. The MβL-TE is the first example of a thioesterase in polyketide biosynthesis that catalyzes a Claisen-like condensation without an α/β hydrolase fold. These mechanistic features should be general to the biosynthesis of tetracyclic naphthacenedione compounds in fungi.130
The pathway begins with the production of decaketide (C20) backbone 22 synthesized and cyclized by AdaA, followed by the hydroxylation of the C-2 position by the monoxygenase Ada C to produce 23. Next, the final cyclization in 24 production is mediated by the MβL-TE, AdaB. Finally, AdaD then performs the C-9 O-methylation to complete the biosynthesis of 3 (Fig. 4).
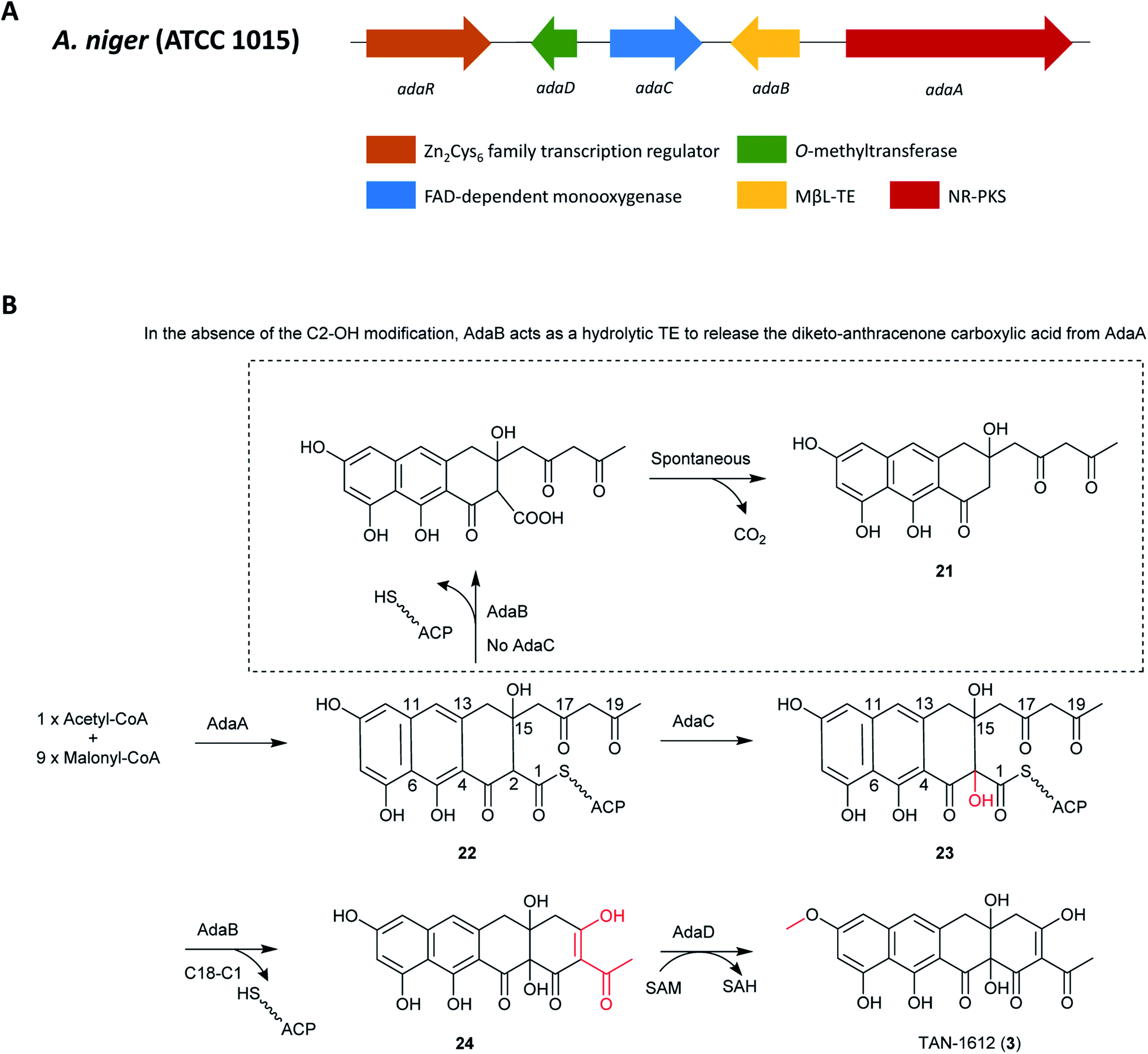 | ||
| Fig. 4 (A) Organization of the ada gene cluster. (B) Biosynthesis of TAN-1612 (3) in A. niger ATCC 1015.130 | ||
Azaphilones represent a class of metabolites that consist of a highly-oxygenated bicyclic core and a chiral quaternary center. This family is structurally diverse and includes numerous bioactive compounds.170 Studies have scanned the sequenced genome of A. niger ATCC 1015 for dual PKS gene clusters and located a single gene cluster containing a HR-PKS azaA and a NR-PKS azaB. These appeared to be homologs of afoE and afoG in the A. nidulans afo cluster.131,168,171 Analysis of the nearby genes in the dual PKS cluster revealed the presence of a Zn(II)2Cys6 zinc finger transcription factor azaR. Further investigation revealed that several tailoring enzymes were likely involved in modification of a final polyketide product. Interestingly, most of these genes have a corresponding homolog in the asperfuranone gene cluster.168 The extensive overlap between the aza and afo BGCs clearly indicated that the aza cluster encoded for the production of an asperfuranone-like compound (Fig. 5).131
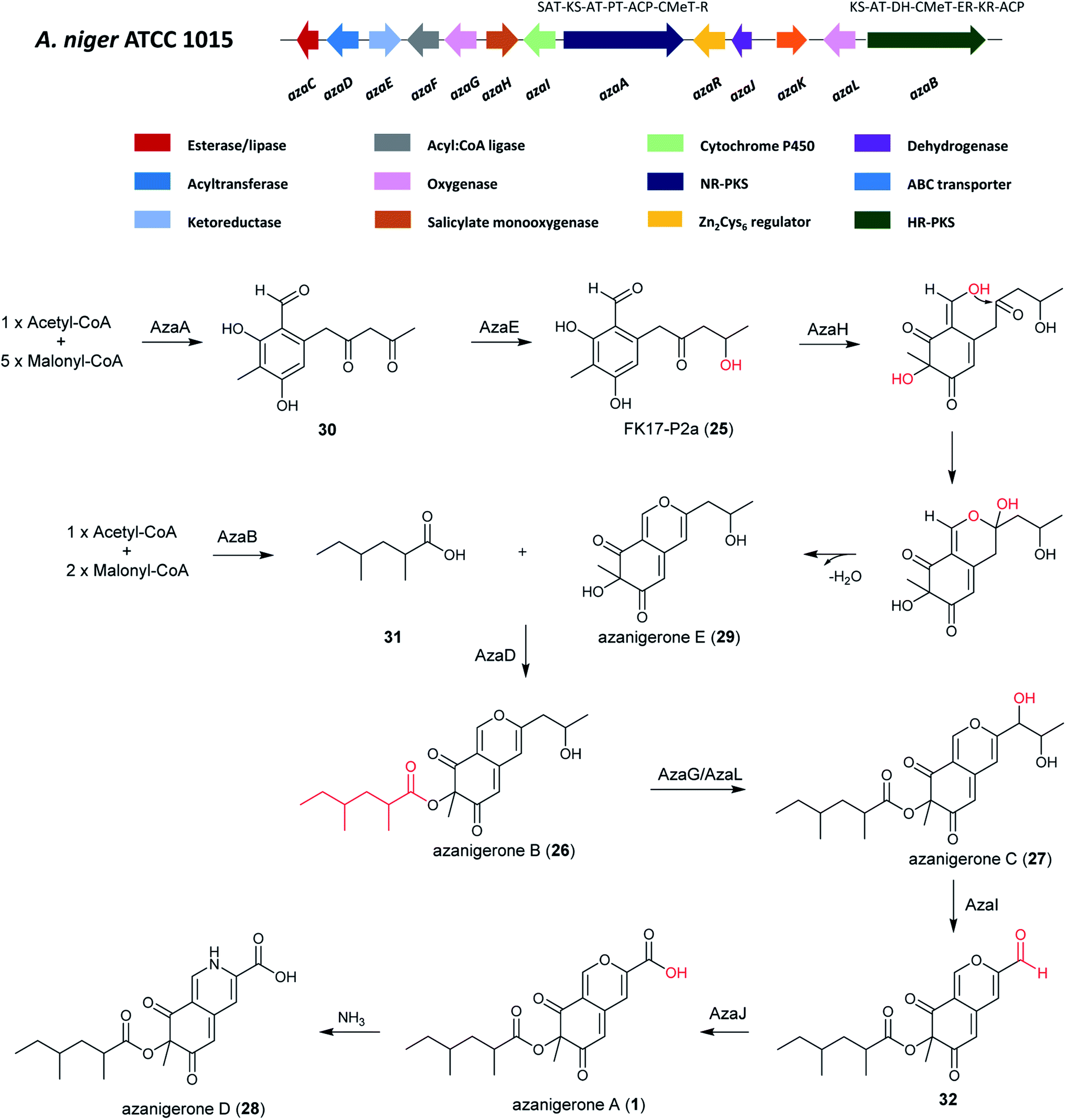 | ||
| Fig. 5 The A. niger aza cluster and the proposed biosynthetic pathway of azanigerones from the aza cluster.131 | ||
RT-PCR transcriptional analysis indicated there were 12 genes in the aza cluster were under control of the pathway-specific regulator azaR.131 After the overexpression of azaR, azanigerone A (1) was obtained as the most abundant product. Several other related compounds were observed at earlier time points, including FK17-P2a (25), azanigerones B (26) and C (27). In addition, azanigerone D (28) was observed to replace azanigerone A as the dominant product at later time points.131
In order to investigate the collaborative mechanism of NR-PKS AzaA and HR-PKS AzaB in azanigerone biosynthesis, an azaB-disruption strain was generated, which upon culturing led to the accumulation of two new compounds, identified as azanigerones E (29) and F (the O4-acetylated version of 29). This suggests a convergence synthesis model, in which AzaA and AzaB biosynthesize two discrete polyketide products that are combined at a later step in the pathway. Importantly, this is the first report of convergent collaboration between a NR-PKS and a HR-PKS in secondary metabolite biosynthesis.131 Furthermore, through enzyme expression and a series of in vitro assays, the critical role of AzaH was confirmed in morphing the benzaldehyde intermediate (FP17-2Pa) into the bicyclic, pyran-containing azaphilone core structure. Altogether, these findings enabled a pathway for biosynthesis of the azanigerones in A. niger to be proposed.131
The biosynthesis of azanigerone A (1) begins with the polyketide assembly by AzaA, forming the hexaketide precursor (31) from successive condensations of five malonyl-CoA units. Then the reduction of the terminal ketone is catalyzed by the ketoreductase AzaE resulting in the formation of 25. Subsequently, the monooxygenase AzaH was demonstrated to be the only enzyme to hydroxylate 25, which facilitates formation of the pyran-ring and generation of 29 after dehydration. In parallel, the 2,4-dimethylhexanoyl chain (31) is synthesized by the HR-PKS AzaB and is combined with 29 to generate 26 in a reaction facilitated by the acyltransferase AzaD. Next, 26 is hydroxylated by the FAD-dependent monooxygenase AzaG or AzaL to yield 27, followed by C–C oxidative cleavage by cytochrome P450 AzaI to obtain 32, and oxidation of the aldehyde to a carboxylic acid by AzaJ, yielding 1. 1 can then undergo addition of NH3 to generate 28, which was observed to replace 1 production at later time points (Fig. 5).
Since the structures of 5, 33 and 34 all contain one phenylacetate and two acetate units, this suggests that they may originate from a polyketide biosynthetic pathway. The PhlB gene (phenylacetyl-CoA ligase) of Penicillium chrysogenum is responsible for the incorporation of phenylacetate,173 a homologous BLAST search was thus performed resulting in the identification of ORF An09g01820 (epaB) in A. niger. Analysis of nearby genes in the epa cluster revealed the presence of a core hybrid NR-PKS enzyme EpaA, an acyl-CoA transferase EpaC, and three genes orf 1–3 encoding an oxidoreductase, a 3-hydroxybenzoate 4-hydro-xylase, and a salicylate hydroxylase, respectively.172
The NR-PKS responsible for the production of 5, 33 and 34 was confirmed to be epaA through gene deletion by homologous recombination, which led to the abolishment of the production of all three metabolites. The complementary strain (epaA-com) was able to produce them all, which further confirmed that the identified BGC is responsible for the synthesis of 5, 33 and 34. In addition, feeding purified 5 and 33 into cell-free extracts (including all biosynthetic enzymes and cofactors for the biosynthesis of 34) of the ΔgcnE mutant did not produce 34, suggesting that 5 and 33 were by-products in the biosynthesis of 34.172
Based on these results, the biosynthetic pathway of 5, 33 and 34 was proposed (Fig. 6).172 The pathway starts with the biosynthesis of phenylacetyl triketide (35) by the NR-PKS EpaA. This prompted the researchers to hypothesize the presence of a phenylacetyl-CoA initiator unit, followed by releasing of a polyketide chain as an aldehyde via the R-domain. Next, the cyclization and dehydration by nonenzymatic rearrangement likely generates 5.172 For the biosynthesis of 33 and 34, an additional methyl group is added through the C-methyltransferase domain before the final chain release, yielding 36. Subsequent, reduction to generate an aldehyde product, oxidation and transamination to produce an intermediate amide (37), followed by enolization, cyclization and water loss to generate 33.172 The researchers proposed that EpaC (An09g01800), an acyl-CoA transferase can additionally catalyze the transfer of 2-methylsuccinyl-CoA, a common intermediate in the ethylmalonyl-CoA pathway, to generate an alternative product 38 and then form 34 by cyclization and water loss.172
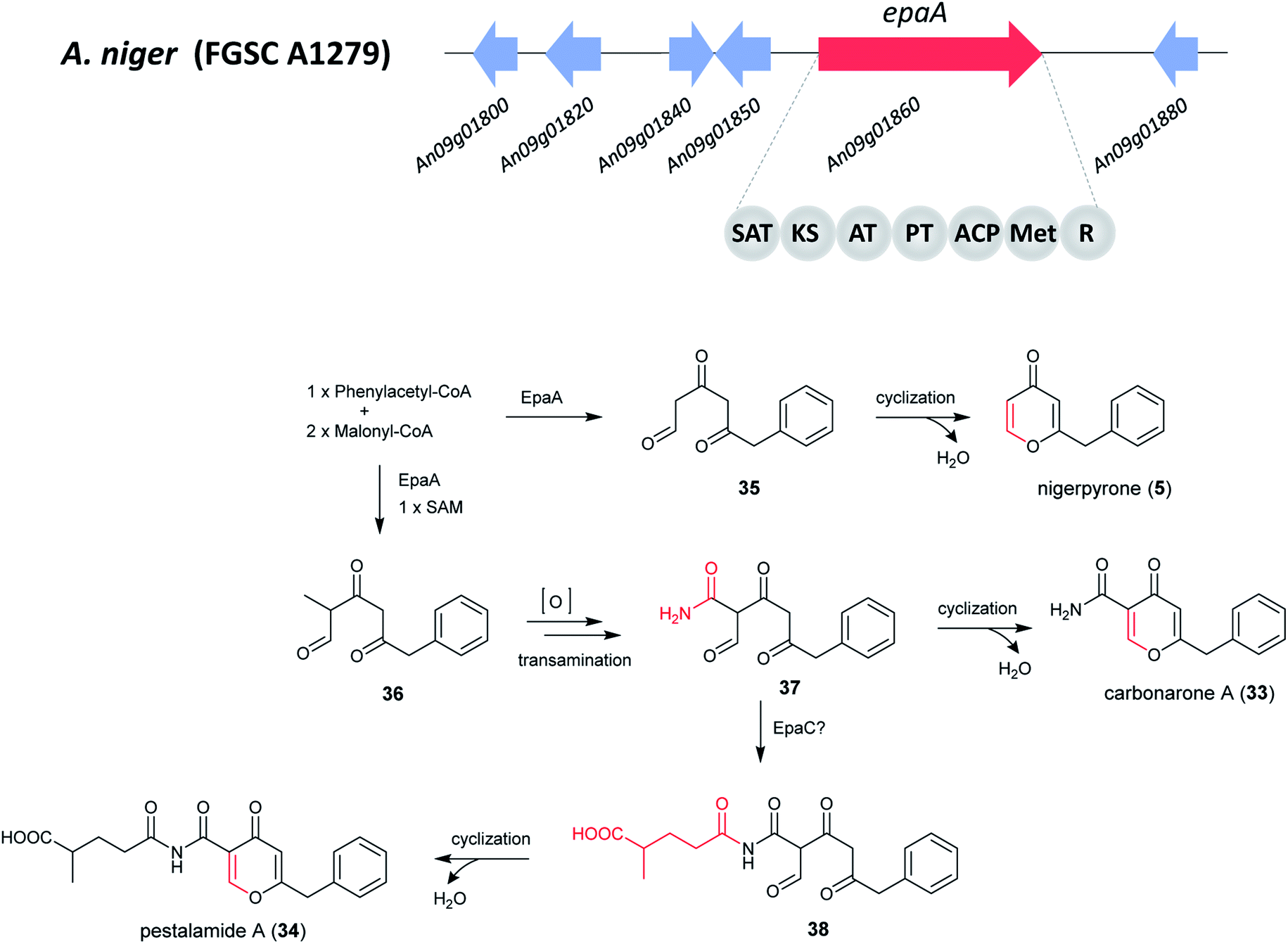 | ||
| Fig. 6 Putative gene cluster and the proposed biosynthetic pathway of nigerpyrone, carbonarone and pestalamide in A. niger.172 Adapted from Wang et al. | ||
Heterologous expression of dtbA in A. nidulans demonstrated that DtbA catalyzes production of two polyketides, 2,4-dihydroxy-3,5,6- trimethylbenzaldehyde (4) and 6-ethyl-2,4-dihydroxy-3,5-dimethylbenzaldehyde (39) (Fig. 7). In order to further expand the structural diversity of products produced by fungal NR-PKSs, this BGC was engineered with a TE release domain instead of the R-domain. By replacing the DtbA R domain with the AusA (austinol biosynthesis) or ANID_06448 TE domain, DtbAΔR+TE chimeric PKSs were formed allowing the development of two metabolites with carboxylic acids replacing the corresponding aldehydes (40, 41). The TE domains were selected based on phylogenetic relatedness to DtbA, as well as NR-PKS with a similar CMeT domain location. This indicates that the release domains are selective catalysts that co-evolved with their cognate CMeT domains, rather than being simple non-selective and slow hydrolases.133
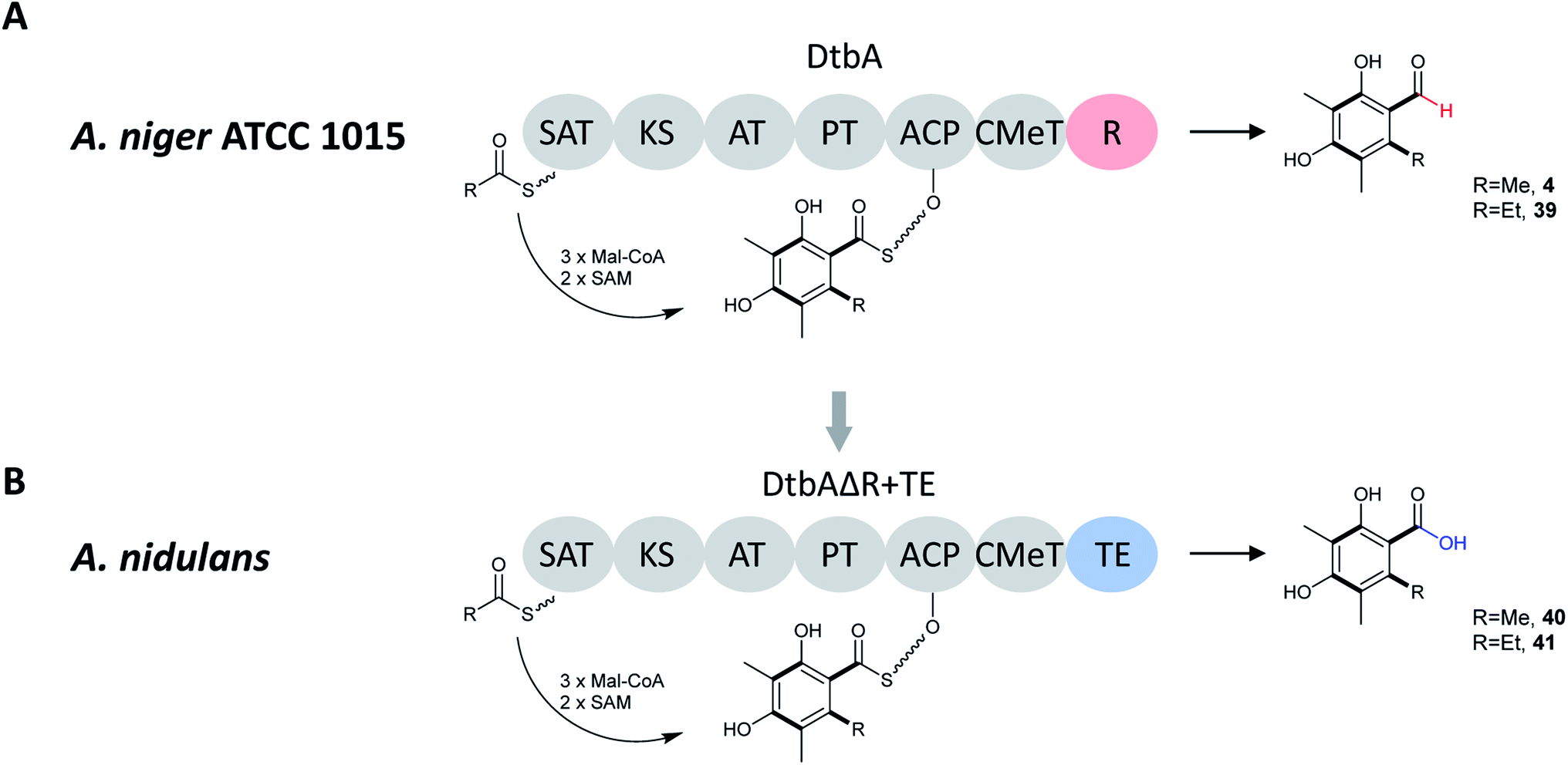 | ||
| Fig. 7 (A) Domain architecture, putative starter units, unreleased intermediates, and released polyketides for DtbA in A. niger ATCC 1015. (B) Domain architecture, putative starter units, unreleased intermediates, and released polyketides for hybrid DtbAΔR+TE(AusA or ANID_06448) in A. nidulans.133 | ||
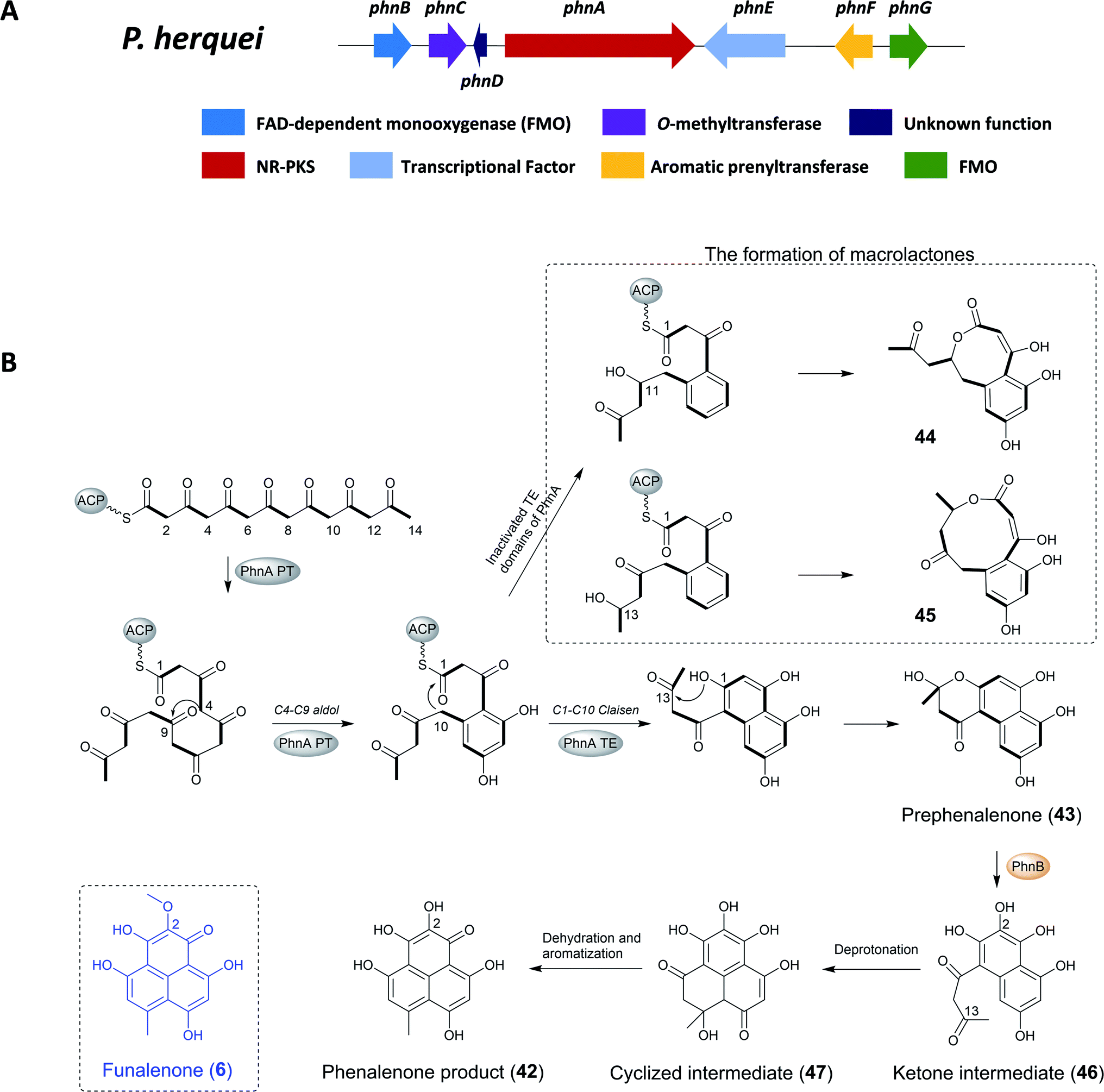 | ||
| Fig. 8 (A) The gene cluster phn and (B) the proposed biosynthetic pathway of phenalenone in P. herquei.134 | ||
The previous study found that the core of phenalenone polyketides is synthesized by the combined functions of a NR-PKS (PhnA) and a flavin-dependent monooxygenase (FMO) (PhnB) that catalyze an unusual aromatic hydroxylation of the NR-PKS product prephenalenone (43). PhnA synthesizes a novel angular naphtha-γ-pyrone product that is oxidatively morphed into the phenalenone structure by a FMO PhnB. Importantly, the combined activities of PhnA and PhnB represent a new strategy of polyketide cyclization in nature. In addition, the neighboring genes encode FMO (PhnB and PhnG), O-MT (PhnC), and prenyltransferase (PrT, PhnF) that are consistent with the structural features of herqueinone.134
To confirm the involvement of phnA in the biosynthesis of herqueinone, the KS region of phnA was deleted using homologous recombination and hygromycin as selection marker, leading to the complete loss of the production of herqueinone. To identify the polyketide product of PhnA and the functions of its cyclization domains, phnA was heterologous expressed in Saccharomyces cerevisiae strain BJ5464-NpgA, yielding the naphtha-γ-pyrone 43. Feeding experiments with purified 43 to the ΔphnA-blocked mutant restored the production of herqueinone, which further confirmed 43 as a key intermediate in the biosynthesis of herqueinone.134
To investigate the role of the PT domain, as well as the TE/CLC domain, the researchers constructed an inactivated TE domain of PhnA either by point mutation or domain deletion (PhnA S2009A and ΔTE), which led to the production of two macrolactones (44, 45) instead of 43. The formation of 44 and 45 in this process requires ketone reduction of the C11 and C13 carbonyl groups, respectively, and subsequent attack by the hydroxyl groups on the C1 thioester. It should be noted that the ketone reduction steps are catalyzed by endogenous yeast ketoreductase and not the domains inside PhnA. Therefore, although the TE/CLC mutation abolished the second aromatic ring derived from the C1–C10 Claisen condensation in 43, whereas first aromatic ring fixed from the C4–C9 aldol condensation remained intact. This confirmed that the PT domain in PhnA is only responsible for directing C4–C9 cyclization while the TE/CLC domain catalyzes C1–C10 cyclization.134
To identify the enzymes responsible for the pyrone opening and C8–C13 cyclization for the formation of 42, targeted gene deletion of selected phn genes was carried out. The inactivation of phnB led to the abolishment of production of herqueinone and the accumulation of 43, which indicated its important role in transforming 43 into a phenalenone structure. Further investigation showed that PhnB was responsible for converting the γ-pyrone 43 into the tricyclic phenalenone as well as C2 hydroxylation.
Thus, following release of the polyketide product from PhnA and rapid formation of the hemiketal 43, the active site of PhnB catalyzed the C2-hydroxylation through deprotonation of the hemiketal C13-OH to yield a ketone intermediate (46). Subsequently, tautomerization of the C3 enol group leads to rapid cyclization to yield the cyclized intermediate, which then aromatizes through dehydration and keto–enol tautomerization to yield the phenalenone product. The remaining enzymes encoded in the gene clusters, including PhnC (O-MT), PhnF (PrT) and PhnG (FMO), have been confirmed to transform prephenalenone to herqueinone.134 Funalenone is the C2-methoxy derivative of phenalenone, and the structural features are reflected in the A. niger gene cluster, which only contains homologues to PhnA, PhnB, PhnC (O-MT), and PhnD (Table 5).
| Proposed function of encoded enzyme | A. niger | |
|---|---|---|
| a In the clusters listed here, each contains a PhnB homolog with >60% sequence identity. b CAK38307 contains two conserved domains, corresponded to PhnB and PhnD, respectively. | ||
| PhnA | Nonreducing polyketide synthase (SAT-KS-AT-PT-ACP-ACP-TE/CLC) | CAK38306 |
| PhnB | FAD-dependent hydroxylase | CAK38307 |
| PhnC | O-Methyltransferase | CAK38305 |
| PhnD | Unknown function (Dabb superfamily protein) | CAK38307b |
| PhnE | Transcriptional factor | No homolog |
| PhnF | Aromatic prenyltransferase | No homolog |
| PhnG | FAD dependent pyridine nucleotide-disulfide oxidoreductase | No homolog |
2.2 Partially reduced polyketides
In section Nigri, the secondary metabolites of PR-PKSs origin that have so far been linked to their related gene cluster include aculinic acid (48), yanuthone D (49) and ochratoxin A (50) (Table 4). The biosynthetic pathway of 48 further downstream products are described in this section, whereas the biosynthetic pathways of 49 and 50 are shown in the section describing hybrid compounds. PR-PKSs typically possess a ketoreductase (KR) domain, introducing hydroxyl groups at specific sites of the growing polyketide chain thereby setting the structural basis for subsequent cyclization reactions, including lactone formation of otherwise aromatic compounds. In line with this they usually lack dehydratase (DH) or enoyl reductase (ER) domains for further reductive processing as found in the HR-PKSs. These enzymes catalyze the production of small molecules, such as 6-methylsalicylic acid (51, 6-MSA) and mellein.176Next, the acu gene cluster, including the acuA (51 synthase encoding gene) and eight surrounding genes acuB–I, was defined (Fig. 9), leading to the proposed biosynthetic pathway for the formation of 48 and 52 from 51 (Fig. 10). The pathway initiates the biosynthesis of 51 by the PKS AcuA, followed by decarboxylation by AcuB to produce 57, similar to what is seen in 49 biosynthesis, followed by oxidation catalyzed by AcuC to produce 58. Next, AcuD and AcuE catalyzes two oxidations of the aromatic ring in the gentisyl alcohol (59) and 60. Inspired by how 51 is used as the starting point in biosynthesis of patulin, the researchers proposed that the following aromatic ring opening is catalyzed through a Baeyer–Villiger type of oxidation, catalyzed by AcuF, with the carboxylic acid at C1 subsequently being reduced to an aldehyde by AcuG, yielding 61. Next, as also proposed for the patulin biosynthetic pathway, a hemiacetal is formed,178 followed by the reduction of the double bond in the side chain by a putative dehydrogenase AcuH. Finally, by non-enzymatic hemiacetal formation, keto–enol tautomerism results in the production of 48, which exists as two diastereomers (both R/S configurations at C1).
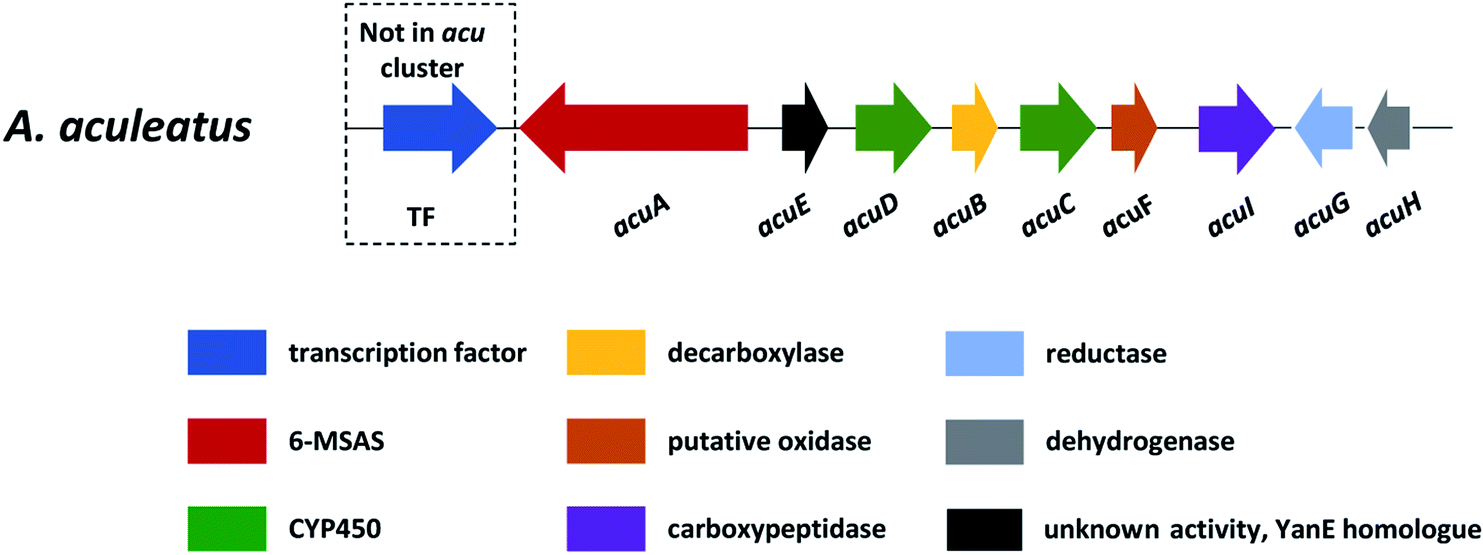 | ||
| Fig. 9 The acu cluster in A. aculeatus. | ||
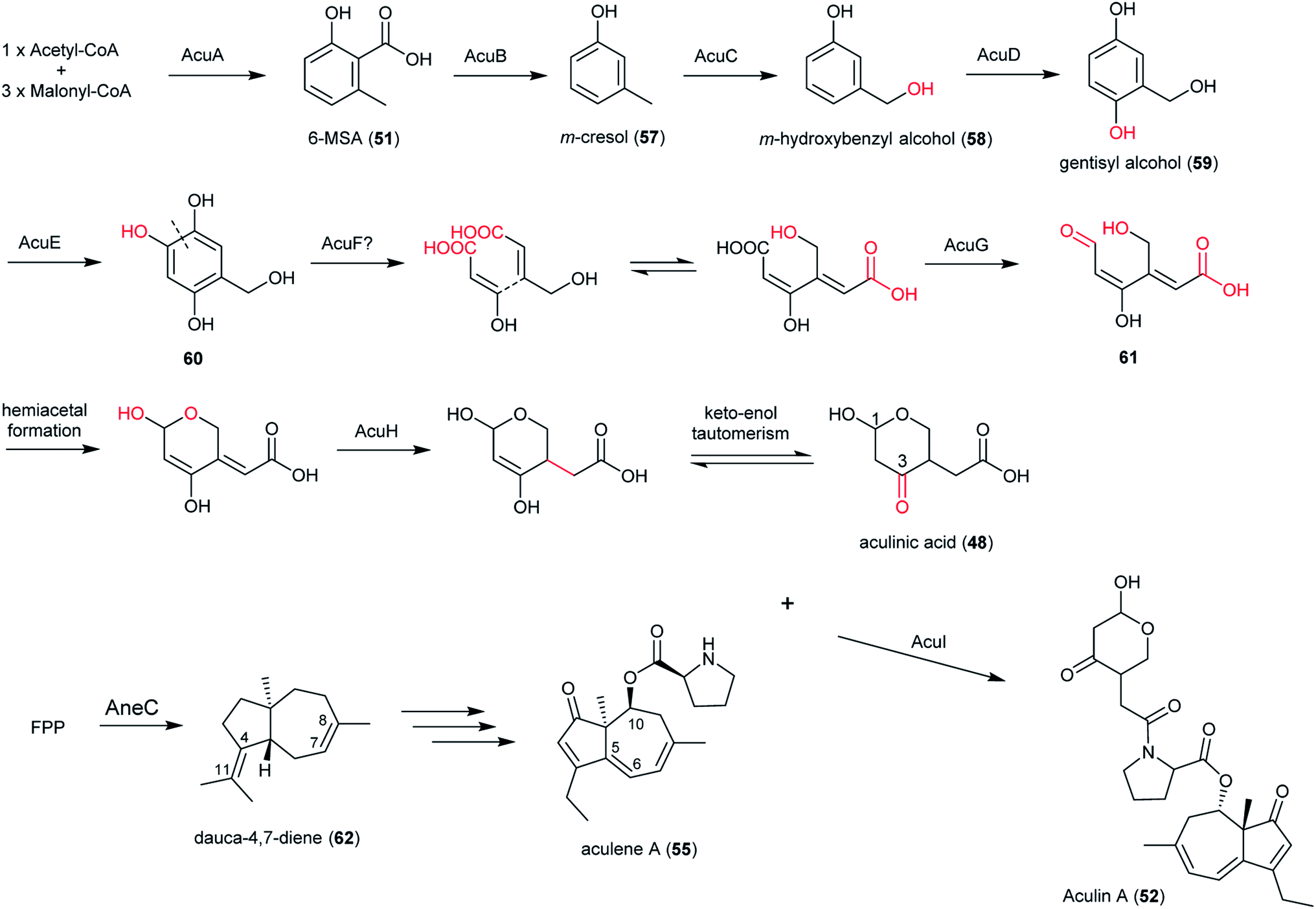 | ||
| Fig. 10 Proposed biosynthetic pathway of aculinic acid and aculin A in A. aculeatus.52 | ||
In addition, the 55 moiety was proposed from the terpene pathway and recently confirmed it is derived from a 62 precursor through demethylation and will be further described in this review in Section 6.2.153 However, the linking of 48 and 55 is still unclear but it was hypothesized that AcuI is involved in the attachment of proline to 48 to form 52-54. Therefore, 6-MSA is incorporated into 48, which then incorporates into 52–54. Ultimately, further exploration is needed to elucidate the intermediate biosynthetic pathway steps and confirm the function of enzymes involved in acu gene cluster.
2.3 Highly reduced polyketides
Highly-reducing PKSs (HR-PKSs) possess the complete or almost complete ensemble of β-keto processing domains (Table 4). Fumonisins (fum1) and azanigerone A (1) (azaB) are secondary metabolites produced from section Nigri derived from HR-PKSs. In addition to the minimal PKS domains for all fungal iterative PKSs (ACP, MAT, KS), HR-PKSs also have a ketoreductase (KR) domain that reduces the β-ketone to the corresponding β-alcohol and a dehydratase (DH) domain that subsequently removes the β-alcohol to obtain an enoyl thioester. An enoyl reductase (ER) domain is also found in some HR-PKSs, which further reduces the enoyl thioester to a saturated acyl thioester. Many HR-PKSs also have a C-methyltransferase (CMeT) domain that may methylate the α position of the growing polyketide chain before the β-ketone is reduced.179Fumonisins, along with host-specific toxins (AAL), are known as sphinganine-analog mycotoxins (SAMT) due to their structural similarity to the early precursors of sphingolipid pathways. Fumonisins consist of a linear polyketide-derived backbone with two methyl, one amine, one to four hydroxyls, and two tricarboxylic ester groups located at different positions along the backbone (Fig. 11). Because this cluster was first discovered and identified in Fusarium species, we first describe the FUM cluster in Fusarium and then the homologous gene cluster fum in Aspergillus species.
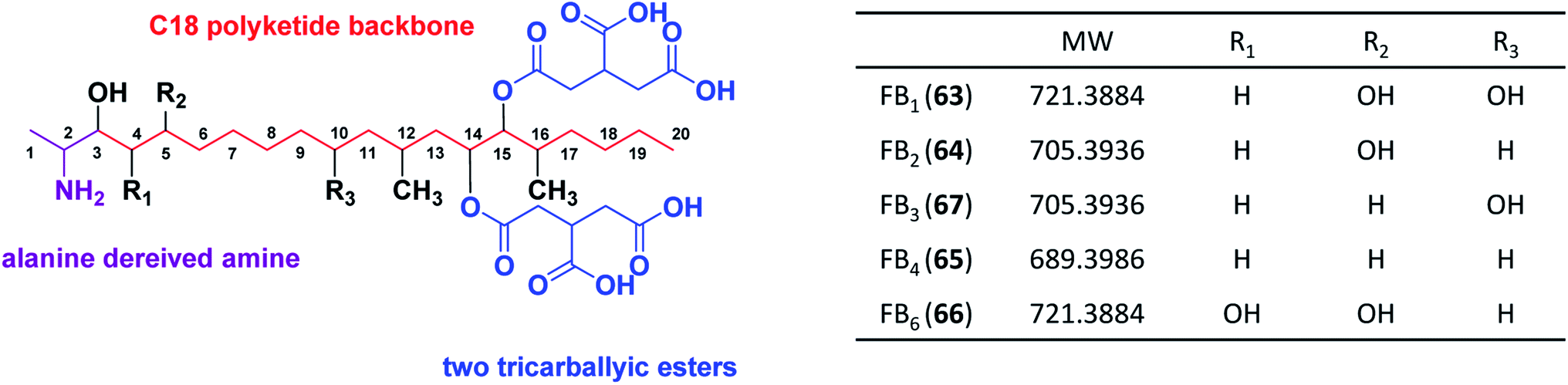 | ||
| Fig. 11 Structure of FB1, B2, B3, B4 and B6. | ||
A cluster of 17 genes required for fumonisin biosynthesis and regulation was identified in Fusarium using heterologous expression which include: FUM1, FUM6–8, FUM3 (FUM9), FUM10–11, FUM2 (FUM12), FUM13–19, FUM 20–21.138 These genes can be categorized into four different groups: (a) backbone gene encoding the dimethylated polyketide that undergoes condensation with alanine (PKS FUM1 and aminotransferase FUM8), (b) decorating post-PKS steps of biosynthesis (FUM3 (FUM9), FUM2 (FUM12), FUM6, FUM13, FUM7, FUM10, FUM14), (c) regulatory genes or transcription factors (FUM21) and (d) unassigned or non-essential genes (FUM11, FUM19, FUM15-18 and FUM20). In the B series fumonisins that consist of a linear 20-carbon backbone, the biosynthetic pathway starts with linear dimethylated polyketide formation (C3–C20), followed by the polyketide condensation with alanine (C1–C2), and finally by carbonyl reduction, oxygenation and esterification with two propane-1,2,3-tricarboxylic acids.185
The fum cluster in A. niger includes 11 homologue genes to Fusarium spp.: the polyketide synthase (fum1), three hydroxylases (fum3, fum6, fum15), a dehydrogenase (fum7), an aminotransferase (fum8), an acyl-CoA synthase (fum10), a carbonyl reductase (fum13), a condensation-domain protein (fum14), an ABC transporter (fum19), and transcription factor (fum21) (Fig. 12). However, the Aspergillus cluster does not contain FUM11, 2, 16, 17, and 18. The absence of FUM2, which catalyzes C-10 hydroxylation, is consistent with the fact that A. niger produces 64, 65 and 66, but not 63 and FB3 (67) (Fig. 13).137 In addition, the absence of FUM11, 16, 17, and 18 in A. niger also supports the hypothesis that these genes are not essential for fumonisin production.138 In contrast, the BGC in A. niger includes a short-chain of dehydrogenase gene (designated here as sdr1) that is not present in the Fusarium cluster for which the function has yet to be characterized.
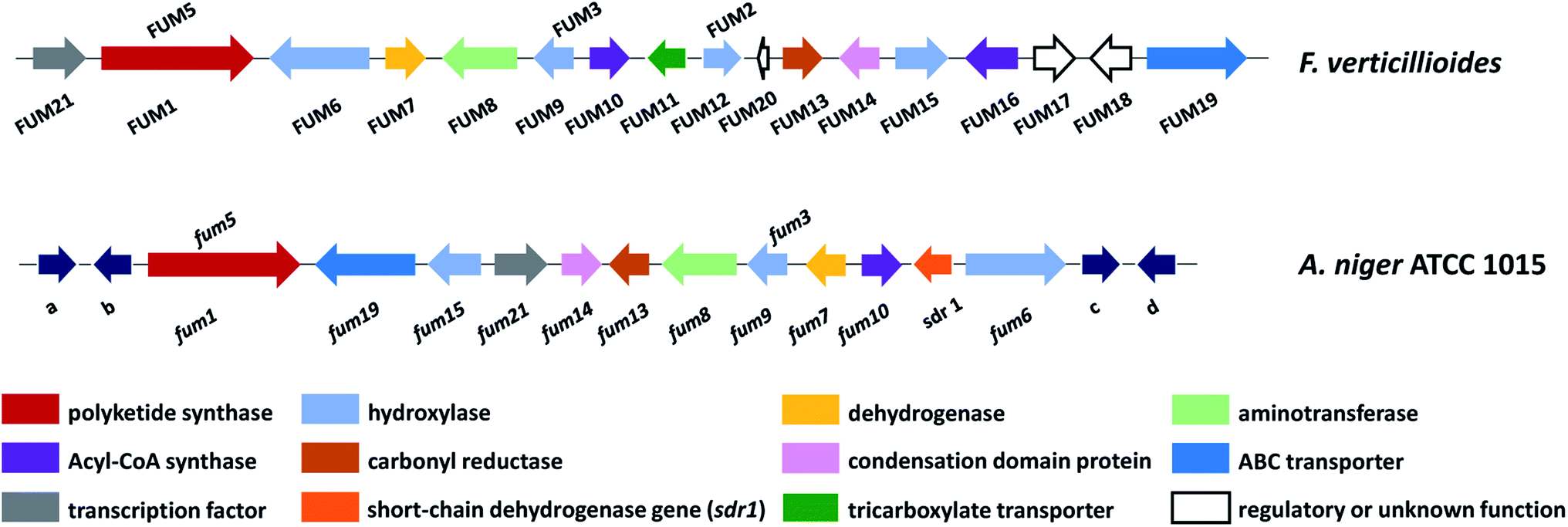 | ||
| Fig. 12 Fumonisin gene cluster in F. verticillioides and A. niger, abbreviated as FUM in Fusarium and fum in Aspergillus. Genes a–d (dark blue arrows) are predicted to encode: a – polyketide synthase; b – major facilitator superfamily transport protein; c – 5-methyltetrahydropteroyltriglutamate-homocysteine methyltransferase/methionine synthase; d – alkaline phytoceramidase.136 | ||
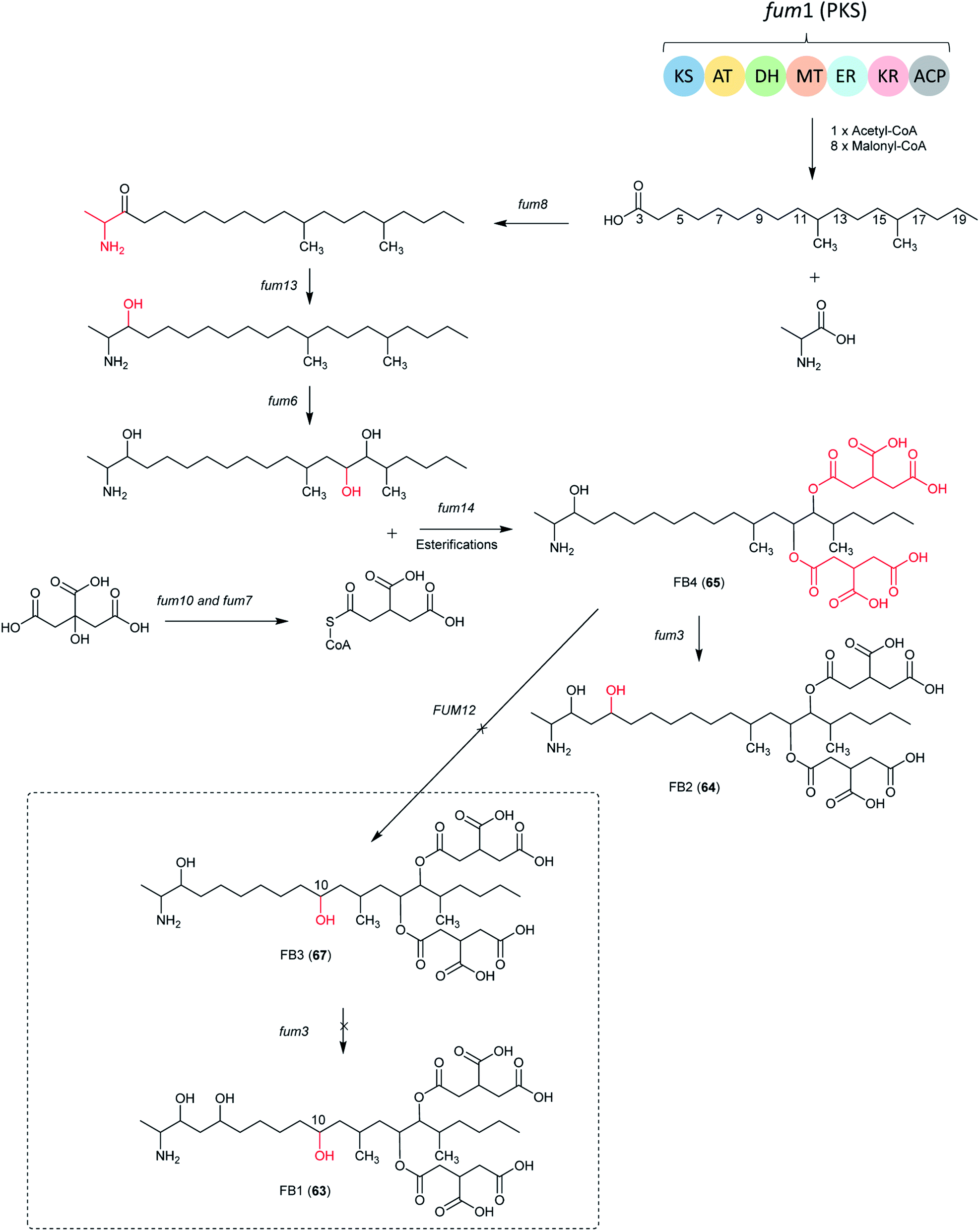 | ||
| Fig. 13 The biosynthetic pathway of FB1, B2, B3 and B4. The structures in dotted frame cannot be produced by Aspergillus species because of the absence of FUM12.138 | ||
In Aspergillus spp., the B series fumonisin biosynthetic pathway starts with formation of a linear dimethylated polyketide by fum1 and polyketide condensation with alanine by fum8, followed by carbonyl reduction by fum13. Two hydroxyls are introduced at C-14 and C-15 by fum 6, followed by esterification with two propane-1,2,3-tricarboxylic acids by fum 7, 10 and 14 to form FB4. Next, the C-5 hydroxylation is catalyzed by fum3 to produce 64.
Additionally, one study investigated the involvement of the fum8 in 64 (biosynthesis in A. niger). The disruption of fum8, coding for an α-oxoamine synthase, resulted in the loss of 64 biosynthesis in A. niger indicating that this gene is involved in 64 biosynthesis in A. niger. The study also showed that 64 is not necessary for vegetative growth and UV tolerance in this fungus.186 Moreover, a study of the black aspergilli from California grapes showed that fumonisin-nonproduction was associated with the absence of six fum genes in A. welwitschiae but not in A. niger.100,136
Recently, two classes of non-aminated fumonisins named FPy and Fla (68 and 69), were isolated from A. welwitschiae, making them the first reported naturally occurring fumonisins lacking an amine.112 These non-aminated metabolites showed significantly less toxicity in the Lemna minor (duckweed) bioassay than the fumonisin B type, providing new insights into the toxicity mechanism of fumonisins. Further exploration with 13C and 15N stable isotope incorporation suggested a post-biosynthetic oxidative deamination of 65. This work shows that certain Aspergillus species can produce an enzyme that naturally detoxifies fumonisins. These enzymes may have many practical applications in food production systems.112
The proposed biosynthetic pathway of 70 and 71 start with the generation of cinnamic acid (Fig. 14), which was catalyzed from phenylalanine by the phenylalanine ammonia-lyase, AhpB. The putative ligase, AhpE, might ligate the cinnamic acid to produce cinnamoyl-CoA, which is the activated starter unit in the biosynthesis. Next, AhpA catalyzed the biosynthesis of the backbone of the homopyrones by the addition of three malonyl-CoA extender units to offer a 15-carbon chain.125 Additionally, the KR and DH in PKS AhpA were predicted to catalyze the reduction at C-9 and the subsequent water loss, respectively. A freestanding thioesterase (TE) named AhpC was proposed to be responsible for the hydrolytic release and the following spontaneous lactonization. After the pyrone formation, the products are proposed to be methylated on C-13 by the O-methyltransferase ahpD.125
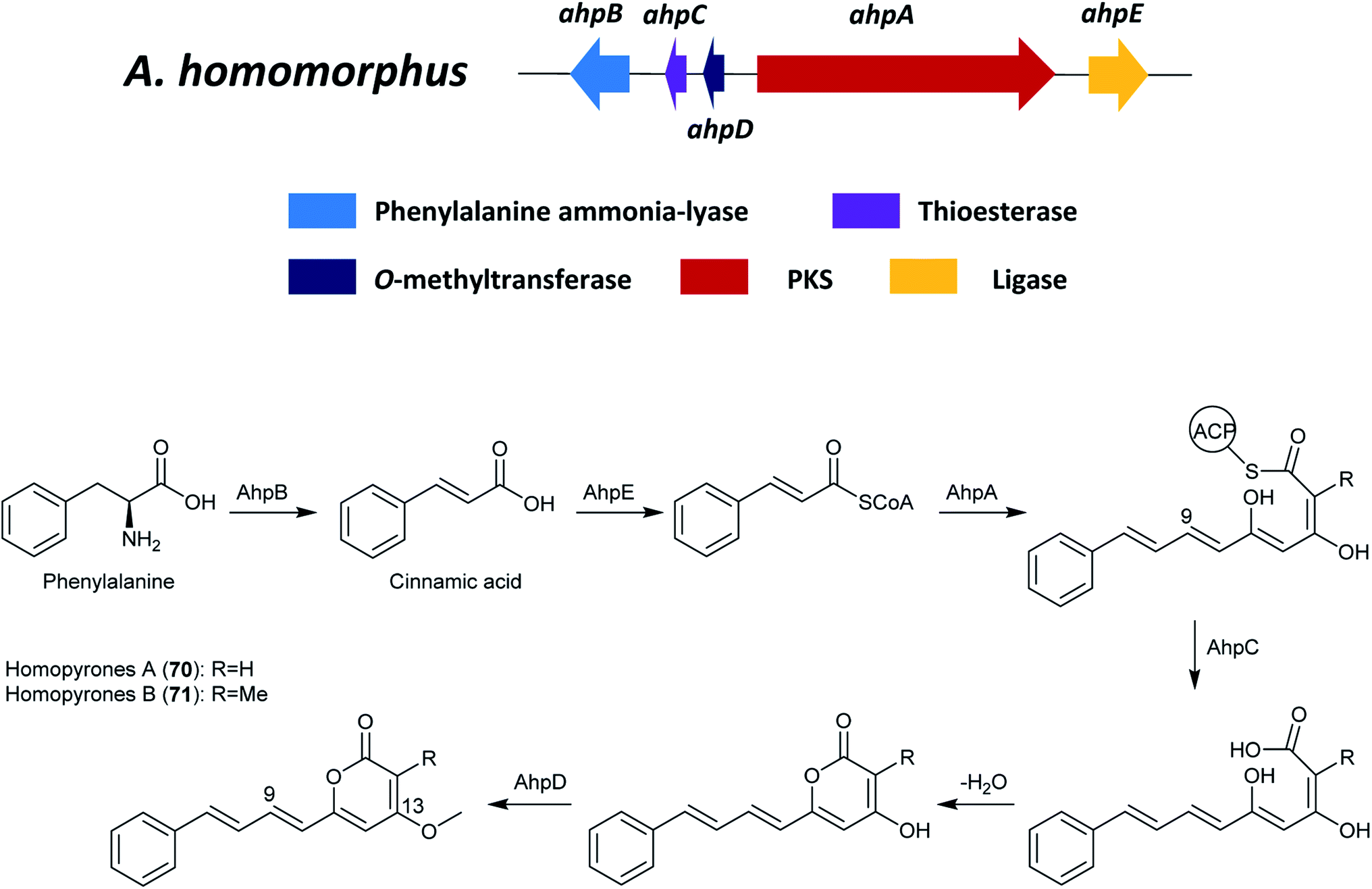 | ||
| Fig. 14 The gene cluster ahp and the proposed biosynthetic pathway of homopyrone A and B in A. homomorphus.125 | ||
3 Linear and cyclic non-ribosomal peptides
Nonribosomal peptides (NRPs) are produced by dedicated nonribosomal peptide synthetases (NRPSs) that are mainly found in bacteria and fungi.188 NRPSs are large modular multi-functional enzymes that contain at least three domains: an adenylation (A) domain for selection, activation, and loading of the amino acid, a thiolation (T) domain, or peptidyl carrier protein (PCP) domain, for binding the activated substrate to a 4′-phosphopantetheine cofactor via a thioester bond and a condensation (C) domain for the peptide formation between two amino acids from the adjacent T domains.188,189Although the canonical function of C domains is to catalyze the formation of a peptide bond between the growing peptide, the CT domain (a subset of C domains) can be recruited by fungal NRPS to perform the equivalent reaction of a thioesterase domain. To date, all fungal NRPSs that assemble macrocyclic peptides terminate with a condensation-like CT domain that catalyzes the cyclization reaction, and thus, is a near universal macro-cyclization strategy used by fungal NRPSs.188,190 Optional domains include epimerization (E), formylation (F), methylation (M), heterocyclization (Cy), reduction (R), and oxidation (Ox) domains for additional modifications. In this section, aculeacin A (72)/echinocandin B (73) (ecd and hty) and malformins (mlfA) are secondary metabolites produced from section Nigri derived from NRPSs. Additionally, a putative BGC for asperazine biosynthesis was proposed in the A. tubingensis G131 and this cluster shows homology with the fumitremorgin biosynthesis cluster identified in A. fumigatus Af293 and the ditryptophenaline production in A. flavus.191,192 However, genetic experiments should be performed in the future to confirm the involvement of this gene cluster in biosynthesis of asperazine.
3.1 Aculeacin A
Aculeacin A (72) is an antifungal lipopeptide isolated from A. aculeatus that contains palmitic acid as the lipophilic side chain.193 It relates to the echinocandin class, which consists of cyclic non-ribosomal hexapeptides with a lipophilic side chain that exhibits high antifungal bioactivity. Furthermore, 72 includes the same six amino acids as echinocandin B (73), while aculeacin biosynthesis is very homologous to 73 biosynthesis.194,195 This gene cluster responsible for the synthesis of 73 (ecd and hty) in A. pachycristatus NRRL 11440 (previously named as Emericella rugulosa) was characterized via genome sequencing and bioinformatics analysis and was confirmed by functional knock-out experiments and heterologous expression of selected pathway genes.140 This gene cluster contains genes encoding for a six-module nonribosomal peptide synthetase EcdA, an acyl-AMP ligase EcdI, and oxygenases EcdG, EcdH, and EcdK. The hty subcluster containing htyA, htyB, htyC, and htyD genes has been shown to be responsible for L-homotyrosine (L-homoTyr) biosynthesis (Fig. 15).140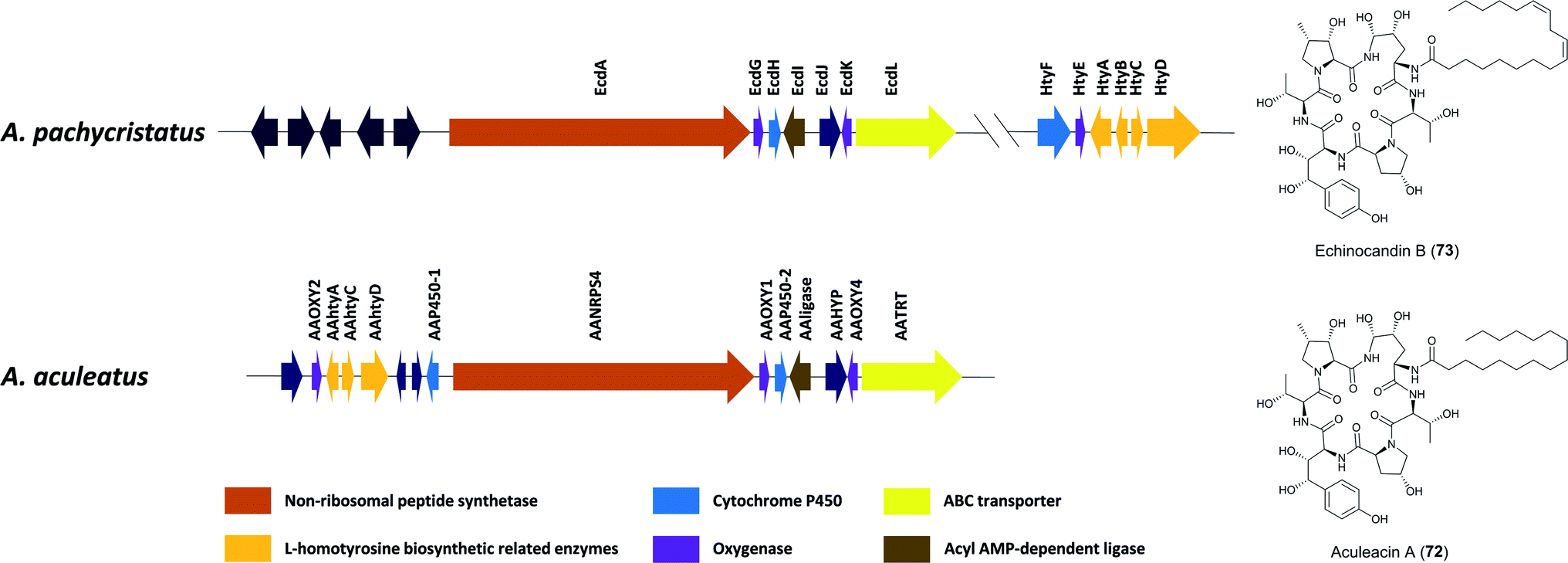 | ||
| Fig. 15 Gene clusters of echinocandin B (73) in A. pachycristatus and aculeacin A (72) in A. aculeatus.139 Genes in dark blue are of unknown function or believed not to participate in the biosynthesis. | ||
The researchers proposed one possible pathway for the biosynthesis of 73 (Fig. 16): linoleoyl-AMP, produced by EcdI from linoleate, is transferred to the initiation T0 (first thiolation domain) of EcdA. The linoleoyl-S-phosphopantetheinyl-T0 is sequentially extended with L-ornithine, L-threonine, L-proline, L-homotyrosine, L-threonine, and 4R-methyl-L-proline to form the linear hexapeptide. Thereafter, the terminal condensation (CT) performs macrocyclization of the NRPS product and the cyclic scaffold is released from EcdA. In addition, an intriguing structural feature of echinocandins is the incorporation of hydroxylated amino acids. Later research identified five oxygenation events by these three iron dependent enzymes, EcdG, EcdH and EcdK.195 EcdH, a predicted P450 type hemeprotein monoxygenase, has been shown to iteratively hydroxylate C5 and C4 of L-ornithine after the formation of the cyclic peptide, while the ecdG gene encodes α-ketoglutarate (α-KG)-dependent mononuclear, nonheme iron oxygenase that has been shown to hydroxylate free L-homoTyr to generate 3-hydroxyl-L-homoTyr. Furthermore, another α-KG-dependent nonheme iron oxygenase EcdK is involved in hydroxylating C5 of methyl-proline, to produce 4R-methyl-proline (4-Me-Pro).195 However, the last echinocandin oxygenation event is still to be characterized.
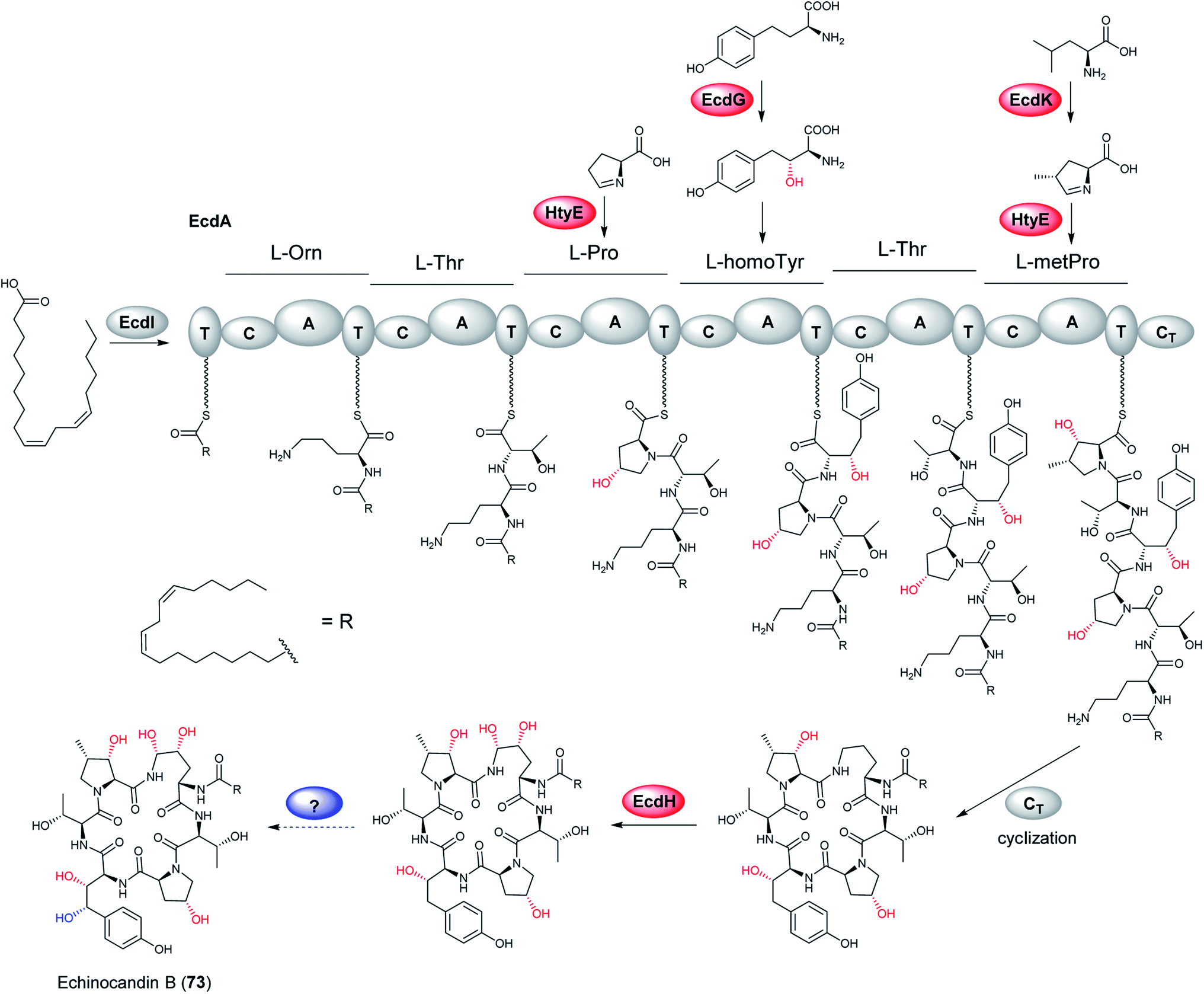 | ||
| Fig. 16 Proposed biosynthetic pathway of echinocandin B (73). Confirmed hydroxylation steps are highlighted in red, while proposed one is in blue.195 | ||
Moreover, to investigate the evolution of chemical diversity in echinocandin lipopeptide metabolites, the gene cluster responsible for 72 synthesis was characterized via DNA sequences for additional echinocandin-type pathway gene clusters.139 In the gene cluster of 72, the hty genes are located immediately upstream of and are separated by, two unknown genes and a cytochrome P450 from the core NRPS gene. Recently, Zhang et al. characterized the α-ketoglutarate/Fe2+-dependent proline hydroxylase (HtyE) from A. pachycristatus and A. aculeatus in the respective 73 and 72 BGCs through heterologous expression in Escherichia coli.196,197 This study showed that both Ap- and Aa-HtyE converted L-proline to trans-4- and trans-3-hydroxyproline but at different ratios.
3.2 Malformins
Malformins are a group of cyclic pentapeptide compounds produced by several species in section Nigri,198 which exhibit antibacterial activity and anti-cancer properties in mouse and human carcinoma cells.199,200 Recently, researchers have conducted a workflow of genome analysis to predict the gene cluster responsible for biosynthesis of malformin A2 (74) and C (75) (Fig. 17).107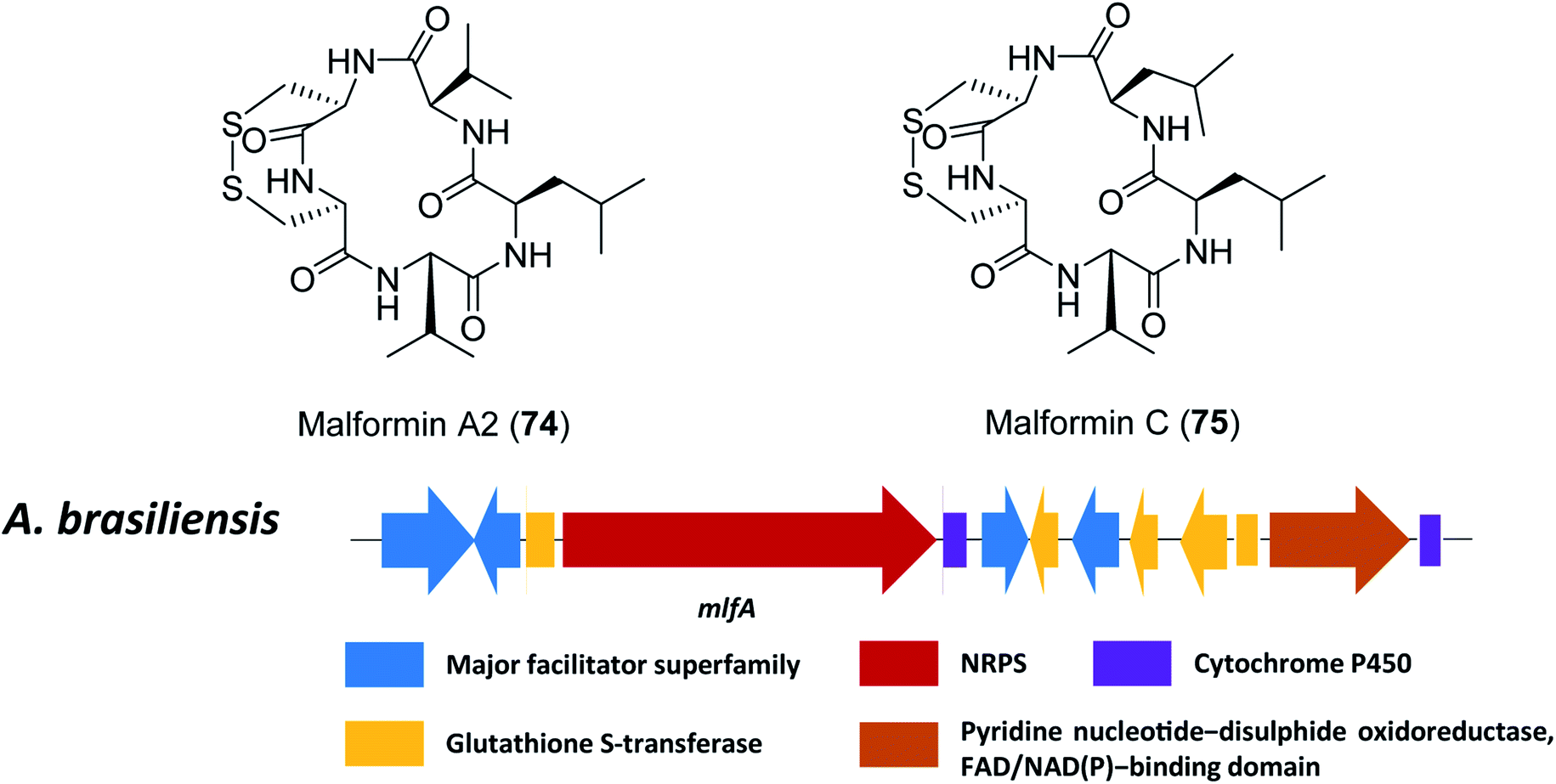 | ||
| Fig. 17 The structures of malformin A2 (74) and malformin C (75), and predicted gene cluster for malformin production in A. brasiliensis.107 | ||
Since malformins are pentapeptides that contain a disulphide bond, only one gene cluster (mlf) containing NRPS genes and disulphide bond-associated genes were included as search criteria. This search led to the identification of the likely related gene cluster responsible for biosynthesis of malformin in 18 fungal strains, importantly, including previous reported malformin producing species.107,109 The NRPS responsible for malformin biosynthesis was confirmed to be mlfA through targeted gene deletion in A. brasiliensis, which led to a total abolishment of 74 and 75 production. Furthermore, a genetic complementation by a constitutively expressed mlfA in the mlfAΔ strain restored 74 and 75 production, to further confirm the function of mlfA. Based on these results, it was hypothesized that some of the individual NRPS adenylation domains are less selective since multiple malformins disappear after deletion of the NRPS.
3.3 Atrofuranic acid (NRPS-like)
Single modular NRPS-like enzymes can be considered as a special group of NRPS, which contain a thioesterase (TE) or reductase (R) domain instead of the peptide-forming C domain. NRPS-like enzymes with A–T–TE architectures recognize and activate α-keto acids by their A domains. The activated substrates are tethered to the 4′-phosphopantetheine arm of the T domain. After the transfer to the TE domain, a second α-keto acid molecule is activated by the A domain and bound to the T domain. The two building blocks are then linked by the TE domain and released as condensation products with different ring systems.201 The condensation of aromatic α-keto acids produces a variety of interconnecting core ring structures, which are proposed to be performed by non-reducing NRPS-like enzymes.When the researchers heterologously expressed atromentin synthetases in A. niger in order to explore the reaction mechanism of the thioesterase domain, they discovered the metabolite atrofuranic acid (76), as well as metabolite X (77), gyroporin (78) and p-hydroxybenzoic acid (decomposed product of atrofuranic acid). However, atromentin synthetases produce atromentin in A. oryzae, which implied that the physiology of A. niger initiates a cross-specificity and cross-chemistry event on the NRPS-like enzyme. When feeding with 2-13C-labeled L-tyrosine, which was incorporated into 76 as a precursor, the biosynthesis pathway for metabolites produced in A. niger could be proposed (Fig. 18). Biosynthesis of 78 was suggested to be proceed via an uncharacterized metabolite, 77, proposed to be derived by a Piancatelli-type rearrangement from 76,202,203 which subsequently by decarboxylation and oxidation leads to formation of 78.
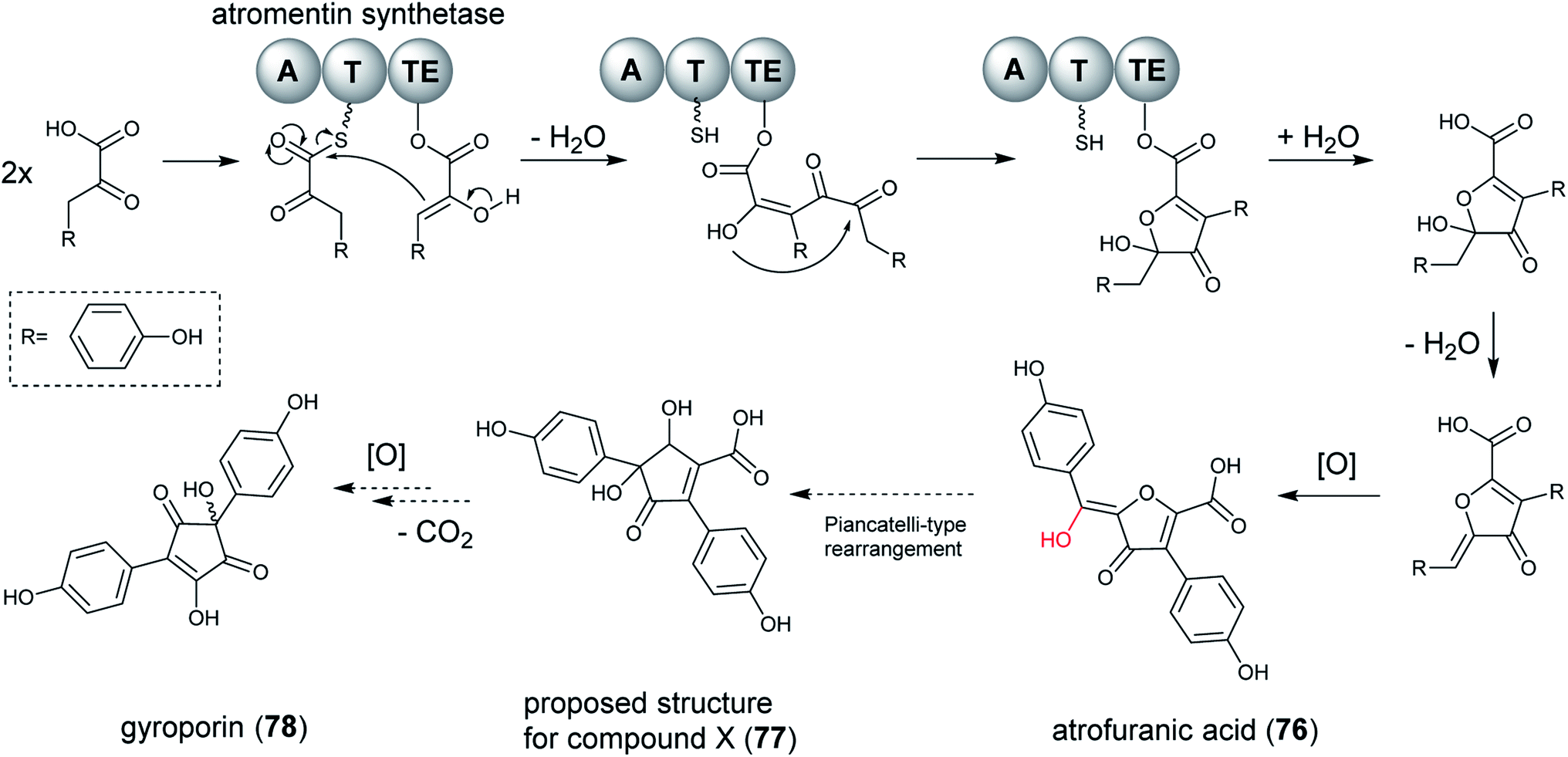 | ||
| Fig. 18 Proposed biosynthetic pathway of atrofuranic acid (76) in Aspergillus species from section Nigri. A subsequent Piancatelli-type rearrangement and decarboxylation results in the formation of gyroporin (78).141 | ||
Further exploration showed that this cross-chemistry was not limited to A. niger but also occurred in A. brasiliensis, which contains an intrinsic atromentin synthetase, indicating that modifications of benzoquinone core structures are common to Aspergillus species of the section Nigri. Therefore, characterization of products from NRPS-like enzymes may be hampered by cross-chemistry occurring on metabolite core structures during heterologous expression.141 It appears that using different heterologous expression platforms in parallel to elucidate the true nature of products generated by uncharacterized NRPS-like enzymes is critical.
4 Terpenoids
Despite being an incredibly structurally diverse class of natural products in fungi, all terpenoids are derived from the common five carbon precursor molecules dimethylallyl diphosphate (DMAPP) and isopentenyl diphosphate (IPP), which are produced from acetyl-CoA via the mevalonate pathway. Successive head-to-tail condensation of one to three IPP extender units to DMAPP results in monoterpenes (C10), sesquiterpenes (C15) and diterpenes (C20). Longer chains (C30 and C40) are usually formed by a head-to-head condensation of two sesquiterpenes (C15) or diterpenes (C20) molecules.204 Then these linear hydrocarbons undergo a dephosphorylation and cyclization cascade to produce terpenes. This highly complex reaction is catalyzed by enzymes known as terpene synthases. Two distinct classes of terpene synthases exist, defined according to their substrate activation mechanism. Class I terpene synthases catalyze an ionization-dependent cyclization of substrate, while class II terpene synthases catalyze a protonation-dependent cascade.205 Further biosynthetic pathway enzymes such as cytochrome P450 monooxygenases, oxidoreductases, and different groups of transferases modify this initial terpene scaffold, producing the final terpenoid.2054.1 Ergosterimide
Ergosterimide was characterized from the culture extract of Aspergillus niger EN-13, an endophytic fungus isolated from the marine brown alga Colpomenia sinuosa.142 Its synthetic steroidal derivative, 3β-acetoxy-7α,15-ethanocholest-8(14)-ene-1′,2′- dicarboxylic acid anhydride, was formed in the Diels–Alder addition of maleic anhydride to 3β-acetoxy-ethanocholest-7,14-diene under mild condition.206 Based on this information, the researchers proposed that ergosterimide is a natural Diels–Alder adduct of ergosteroid and maleimide, which is probably enzymatically catalyzed. Additionally, this is the first reported cycloadduct of a steroid and maleimide so far.142 However, more genetic experiments are needed in the future.4.2 Taxol
Taxol is an extremely effective but rare antitumor agent extracted from Taxus spp. barks. Many taxol-producing endophytic fungi have been reported,207 including A. aculeatinus Tax-6 that was isolated from Taxus chinensis var. mairei.208 In addition, a mutant strain, A. aculeatinus BT-2 was obtained from the original strain, A. aculeatinus Tax-6, using fungicidin as the mutagenic agent, which produces higher quantities of taxol than the A. aculeatinus BT-2.143 The biosynthetic pathway of taxol in Taxus spp. has been revealed,209 yet the biosynthesis pathway of taxol formed from endophytic fungi is not clear. One study explored the genes involved in taxol synthesis in A. aculeatinus Tax-6 to understand the molecular mechanisms of producing fungal taxol. The results showed that genes involved in the mevalonate (MVA) pathway and non-mevalonate (MEP) pathway were expressed, including isopentenyl pyrophosphate transferase, geranyl pyrophosphate transferase, and geranylgeranyl pyrophosphate synthetase. In addition, to identify the mechanism of the difference in taxol production, the transcriptomes of the two taxol-producing fungi were compared and the changes in the gene expression between them were explored. Most genes related to the MVA pathway in the mutant strain BT-2 showed upregulation. However, the key downstream genes related to taxol biosynthesis, i.e., taxadiene synthetase (TS) were not found.1435 Hybrid PKS–NRPS
5.1 Pyranonigrins E–J and pyranonigrins A, S
Pyranonigrins are a family of antioxidative compounds isolated from A. niger, which all feature a unique pyrano[2,3-c]pyrrole bicyclic skeleton.145 The biosynthetic pathway of pyranonigrins E–J (79–84) was elucidated in A. niger, while the pyranonigrins A (85) and S (86) were in Penicillium thymicola (Fig. 19).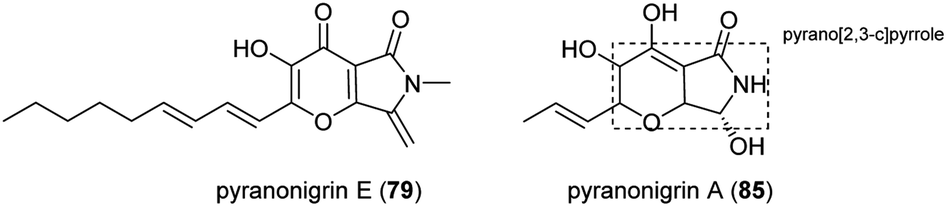 | ||
| Fig. 19 Structures of pyranonigrins E (79) and pyranonigrins A (85). | ||
The BGC which is related to production of 79 (not to be confused with pyranonigrin E reported in another study210) was induced and the metabolites was isolated in A. niger ATCC 1015 as a new PKS–NRPS hybrid secondary metabolite after the pyn gene cluster was activated by the expression of the pathway-specific Zn2Cys6 transcriptional regulator pynR under control of the arginase (aga) promoter.144 Furthermore, cluster activation resulted in induced transcriptions of the pyranonigrin encoding genes pynA, pynB, pynC, pynD, and pynE. PynA encodes a PKS–NRPS hybrid enzyme, PynB encodes a FAD/FMN-containing dehydrogenase, PynC encodes a methyltransferase, PynD encodes a cytochrome P450 oxidase, and PynE encodes an NAD-binding protein.144
In subsequent research, Yamamoto et al. found more pyranonigrins through activating the pyn cluster in high yields by replacing of the pynR promoter with the robust glaA promoter. Subsequently, the complete pathway of pyranonigrins was identified by targeted gene deletion, heterologous in vivo bioconversion and in vitro assays. Furthermore, this study identified three extra genes, including the protease-like protein PynH that performs an exo-methylene formation, the stand-alone thioesterase PynI that is capable of concomitant tetramic acid formation and chain release from the PKS–NRPS hybrid enzyme PynA, and FAD-containing monooxygenase (FMO) PynG that catalyzes an epoxidation-mediated cyclization to form the dihydro-γ-pyrone moiety.145
Altogether, the proposed biosynthetic pathway initiates with biosynthesis of the PKS–NRPS product by PynA, followed by release of the intermediate from PynA by PynI, generating 84. Next, PynC catalyzes N-methylation of 84, resulting in 83. Then PynG catalyzes an epoxidation-mediated cyclization to form the dihydro-γ-pyrone moiety, which is followed by PynD-catalyzed alcohol oxidation and enolization to form 82. PynH then catalyzes the formation of 79, which is dimerized to form 80. Alternatively, PynE can catalyze the conversion of 79 to 81, which can be reversed by PynB.144,171
A very notable discovery in this pathway was PynH, a predicted protease that performs dehydration of a serine side chain to form an exo-methylene in pyranonigrin E. It is highly unusual to find an exo-methylene-forming enzyme that is involved in the modification of a fungal polyketide-nonribosomal peptide hybrid compound. Another important finding is that the stand-alone thioesterase PynI is capable of tetramic acid formation and chain release from the PKS–NRPS hybrid enzyme PynA (Fig. 20).
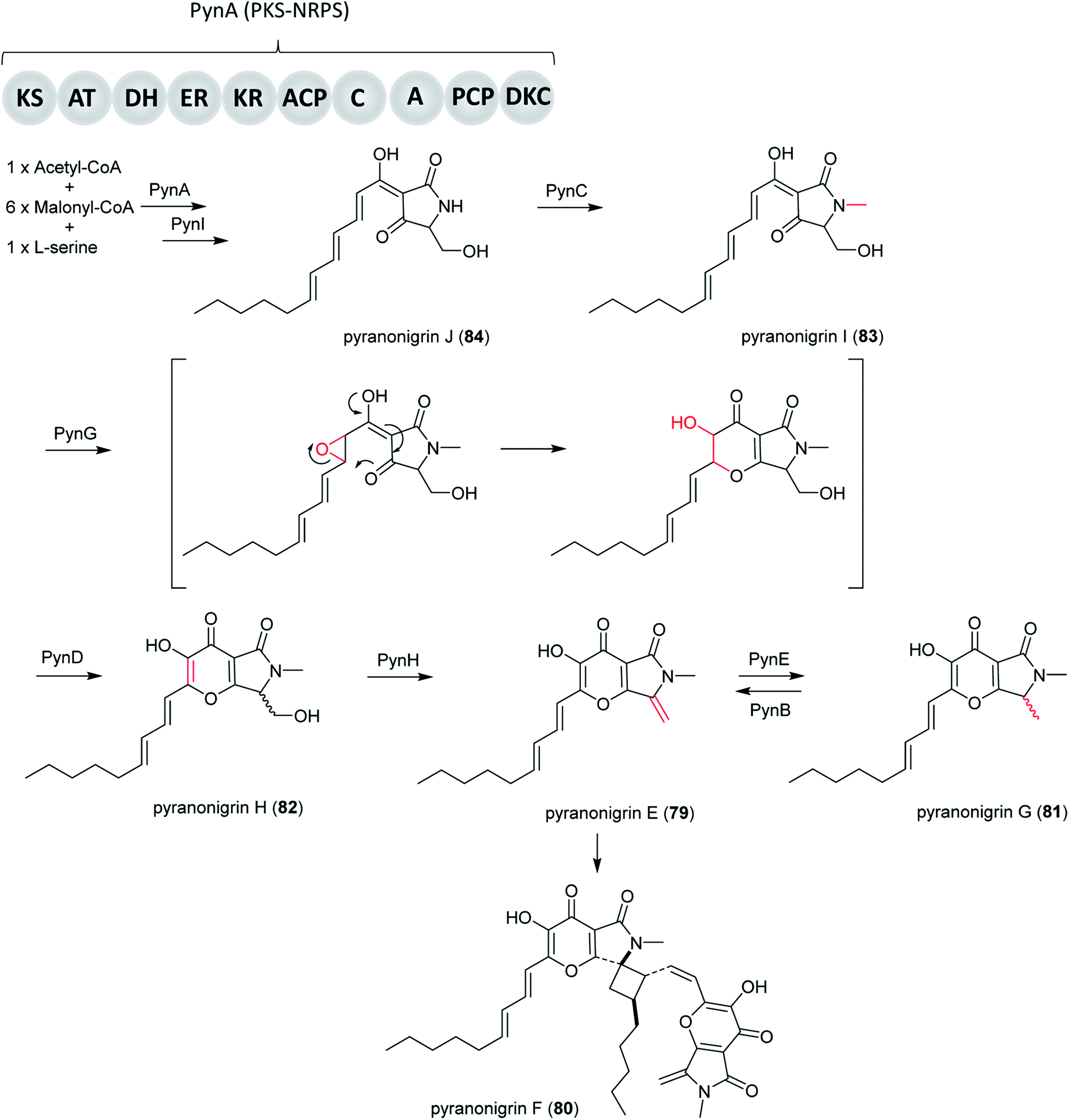 | ||
| Fig. 20 Biosynthesis pathway of pyranonigrins E–J (79–84) in A. niger.145 | ||
In addition, the potent antioxidant pyranonigrin A (85), which was first reported in A. niger,211,212 was found to be biosynthesized by a PKS–NRPS gene cluster pyr in P. thymicola, which is different from the pyn cluster for 79. Its biosynthetic pathway is elucidated in P. thymicola by genome mining and heterologous expression.146
Basic Local Alignment Search Tool (BLAST) search results revealed a PKS–NRPS (PyrA) from P. thymicola IBT 5891 that exhibits moderate sequence similarity to PynA (50% identity/67% similarity) and An18g00520 (61% identity/75% similarity), both of which are from A. niger A1179.213 In addition, An18g00520 was shown to be responsible for the production of tetramic acid type compound, proposed to be the precursor to 86.214 Based on these information, the heterologous expression of pyrA in A. nidulans was carried out, resulting in the production of the product of An18g00520. This indicated the product encoded by pyr cluster shares the same biosynthetic precursor with 86.146 Subsequent heterologous expression of all the genes involved in the entire pyr gene cluster in A. nidulans led to the production of 85. Based on the findings from both in vivo and in vitro studies, only four genes, pyrA–D, are required for the biosynthesis of 85.146
Therefore, the biosynthetic pathway of 85 in P. thymicola initiates the biosynthesis of the PKS–NRPS product by the PyrA, followed by product release catalyzed by either a Dieckmann cyclase (DKC) and/or the hydrolase PyrD. Then, FAD-binding monooxygenase PyrC may catalyze epoxidation followed by subsequent ring closure to form the pyrano[2,3-c]pyrrole core, followed by conversion to 86 and finally the α-carbon of the glycine part of 85 is oxidized by cytochrome P450 PyrB (Fig. 21).
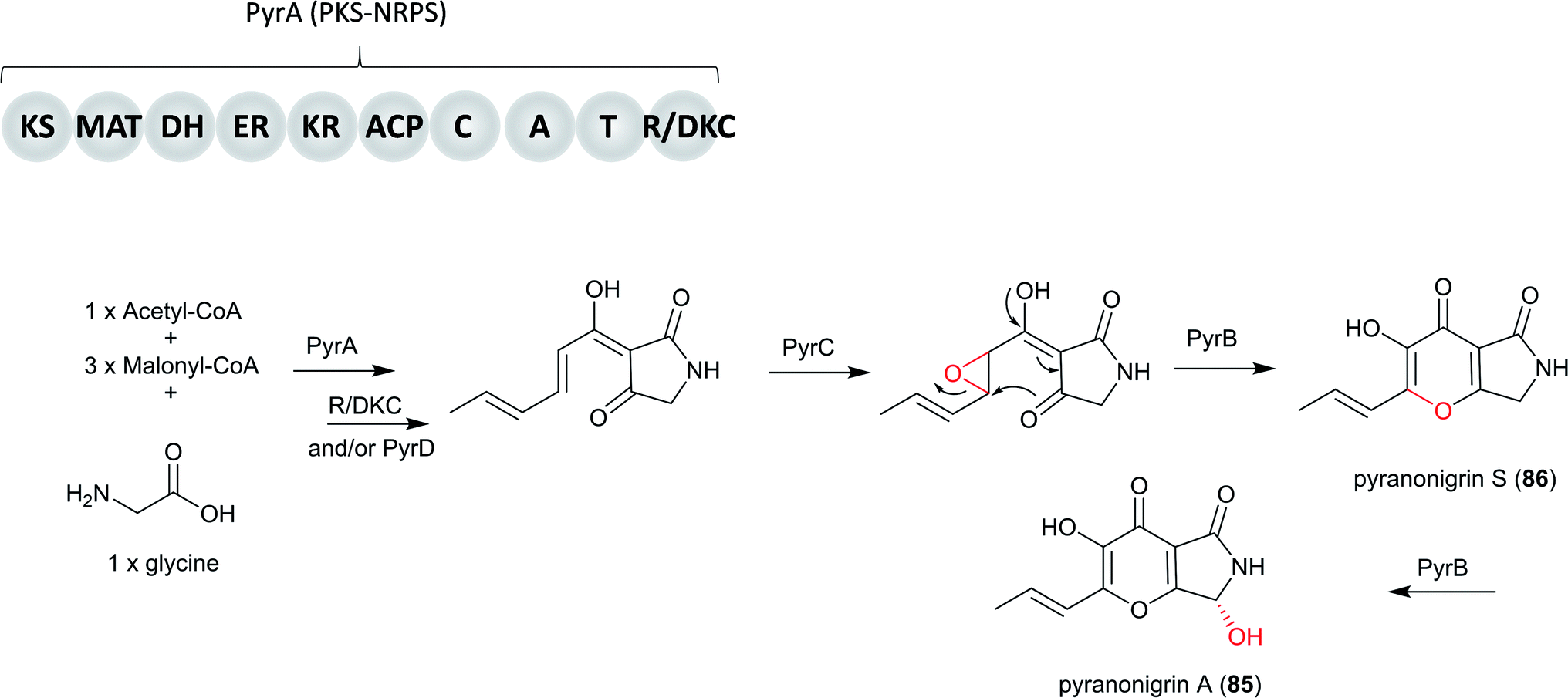 | ||
| Fig. 21 The proposed biosynthesis of pyranonigrin A (85) in P. thymicola IBT 5891.146 | ||
5.2 Pyranoviolin A
Pyranoviolin A (87) was discovered from the genome-sequenced fungus A. violaceofuscus CBS 115571 and it is the first pyranonigrin analogue containing the C-3 methoxy group.147The BGC pyv of 87 was identified via bioinformatics analysis and comparison to the homologous genes of pyranonigrins (Fig. 22).144,146,214 This revealed the pyv cluster encodes a PKS–NRPS hybrid PyvA, a cytochrome P450 monooxygenase PyvB, two flavin-dependent monooxygenases (FMOs) PyvC and PyvE, an α/β hydrolase PyvD, a glucose-methanol-choline (GMC) family of oxidoreductase PyvF, and O-methyltransferase PyvH. The PKS–NRPS responsible for 87 was confirmed to be PyvA through targeted gene deletion, which led to complete abolishment of 87 production. Based on the bioinformatic analysis and previous studies on pyranonigrins, the biosynthetic pathway leading to 87 was proposed (Fig. 23).
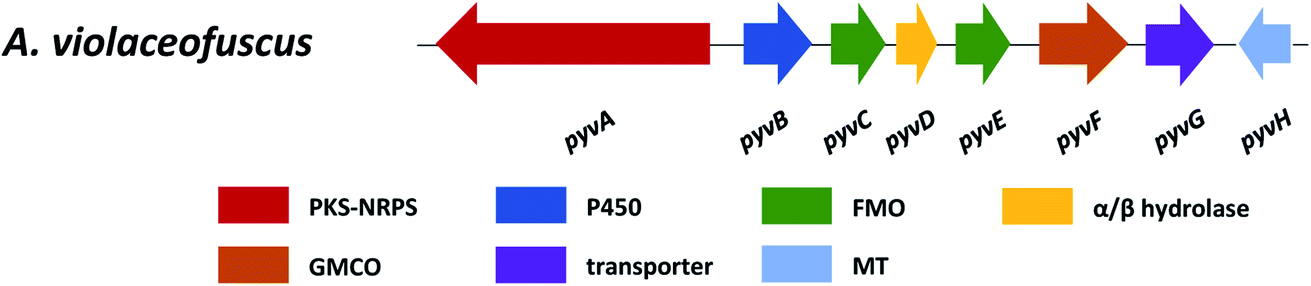 | ||
| Fig. 22 The pyv gene cluster in A. violaceofuscus. PKS–NRPS, polyketide synthase-non-ribosomal peptide synthetase; P450, cytochrome P450 monooxygenase; FMO, flavin-dependent monooxygenase; GMCO, glucose–methanol–choline family of oxidoreductase; MT, methyltransferase. | ||
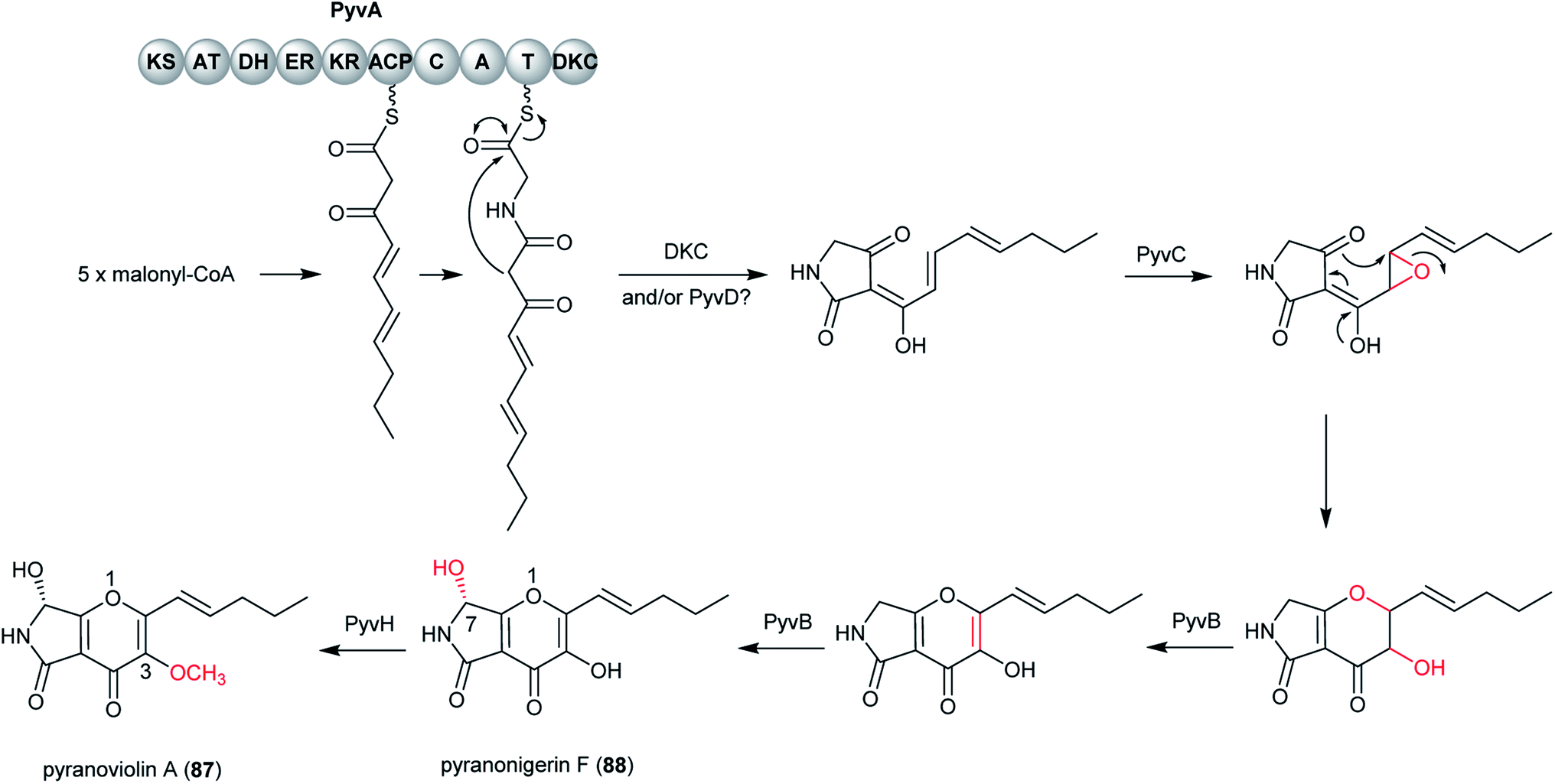 | ||
| Fig. 23 Proposed biosynthetic pathway of pyranoviolin A (87) in A. violaceofuscus.147 | ||
The pathway initiates by the PKS PyvA to synthesize C10 backbone from one acetyl-Co unit and four malonyl-CoA units, followed by the condensation with the thiolation (T) domain-bound glycine activated by the adenylation (A) domain. Then the product release is catalyzed by Dieckmann condensation (DKC) and/or the α/β hydrolase PyvD, yielding the tetramic acid moiety. Next, the FMO PyvC may catalyze the epoxidation of one of the olefins on the polyketide chain, followed by the epoxide ring-opening and the dihydro-γ-pyrone ring formation. Next, the P450 PyvB maybe catalyze two consecutive reactions, in which the dihydro-γ-pyrone is oxidized to γ-pyrone and C-7 is hydroxylated to yield pyranonigrin F (88) [note that two distinct natural products are individually named “pyranonigrin F”].145,215 Finally, the O-methyltransferase PyvH, which is specifically found in the 87 pathway, catalyze the methylation for the C-3 hydroxy group to produce 87.
5.3 Ochratoxin A
Ochratoxin A (50) is a potent mycotoxin which can be found in a wide variety of food products, produced by among others the species of A. carbonarius and A. niger in section Nigri. Structurally, 50 comprises a dihydrocoumarin moiety linked via an amide bond to a molecule of L-phenylalanine, derived from the shikimic acid pathway. In ochratoxigenic Penicillium species, the majority of the molecular characteristics of 50 biosynthesis have been identified, which includes genes encoding a PKS (otapksPN), an NRPS (otanpsPN) and other tailoring genes.216–218 The 50 PKS was recently identified in Penicillium verrucosum, and its involvement in 50 biosynthesis was verified by targeted gene disruption.219In Aspergillus species, a PKS required for 50 biosynthesis was identified in A. ochraceus and partially characterized in Aspergillus westerdijkiae.220,221 In addition, in A. carbonarius, the AcOTAnrps gene encoding the NRPS responsible for the ligation of the phenylalanine to the polyketide group, was identified by gene disruption.148 In the same study, the order of reactions in the biosynthetic pathway of 50 in A. carbonarius was clarified demonstrating that the amide bond between the phenylalanine and the polyketide dihydroisocoumarin is catalyzed by the synthetase and that it precedes the chlorination step.148 Furthermore, a flavin-halogenase in the biosynthesis of 50 in A. carbonarius was identified by gene deletion and confirmed that the chlorination step which converts ochratoxin B (OTB, 89) to 50 represents the final stage of the biosynthetic pathway.
Recently, in order to study the regulatory mechanism of 50 biosynthetic pathway, Wang et al. sequenced and assembled the complete genome of A. ochraceus fc-1 and used that for comparative genomic analyses with five other sequenced ochratoxin A-producing fungi (A. carbonarius, A. niger, A. steynii, A. westerdijkiae, and P. nordicum).149 By comparing the genomic analysis of these six 50 producers, a conserved ochratoxin A (50) BGC was identified, including a PKS (otaA), an NRPS (otaB), a cytochrome P450 monooxygenase (otaC), a halogenase (otaD), and a basic leucine zipper (bZIP) transcription factor (otaR1), which are all required for 50 biosynthesis. A GAL4-like Zn2Cys6 binuclear cluster DNA-binding protein (otaR2) and a FAD-dependent oxidoreductase (otaE) were also identified but they are not essential to the 50 production (Fig. 24). Ochratoxin A (50) biosynthesis begins with the formation of 7-methylmellein catalyzed by OtaA utilizing acetyl-CoA and four malonyl-CoA units, followed by the oxidation to OTβ by OtaC and OtaE. Next, OTβ and L-phenylalanine are combined by the NRPS OtaB to form an amide bond to synthesize 89. Finally, 89 is chlorinated by a halogenase OtaD to give 50 (Fig. 25).
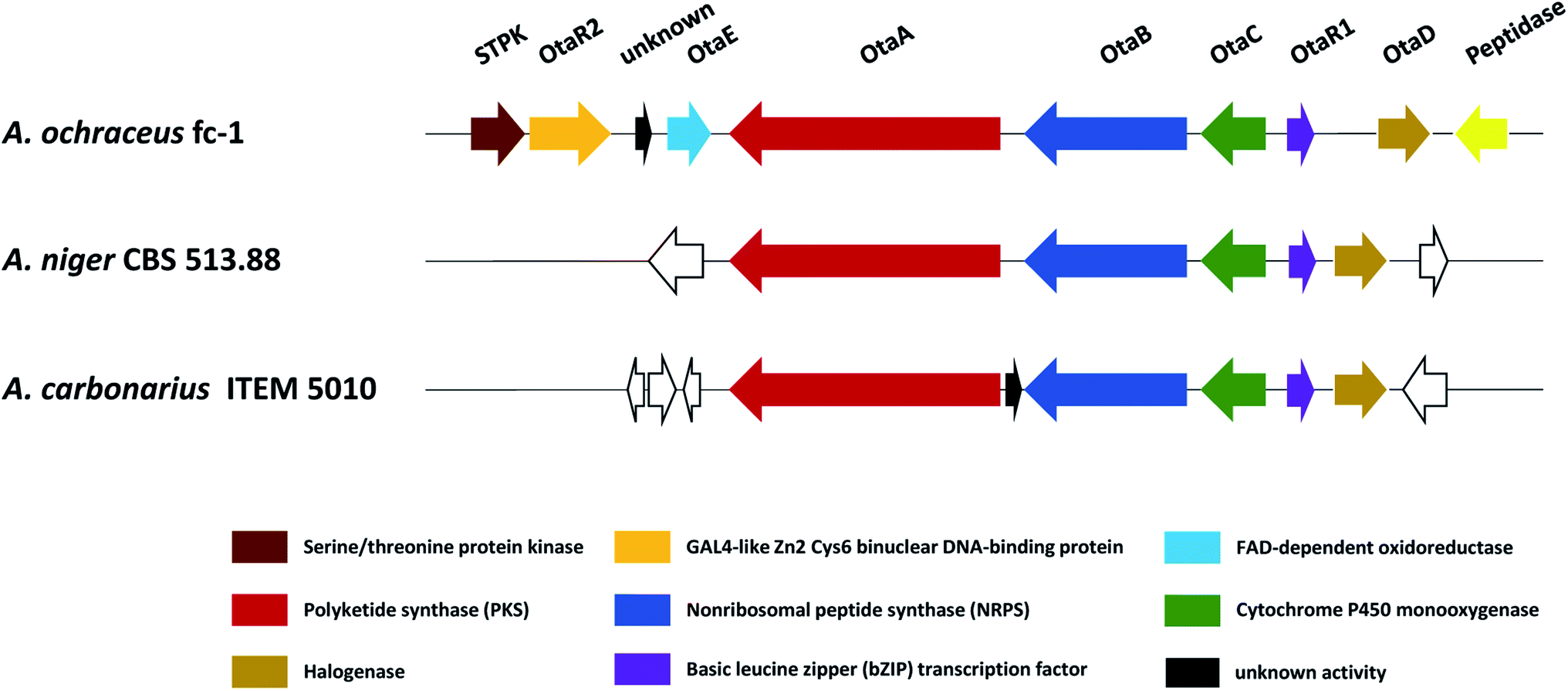 | ||
| Fig. 24 Proposed ochratoxin A (50) BGC family in A. ochraceus, A. niger and A. carbonarius. Genes shown in the same color displayed high similarity with each other. | ||
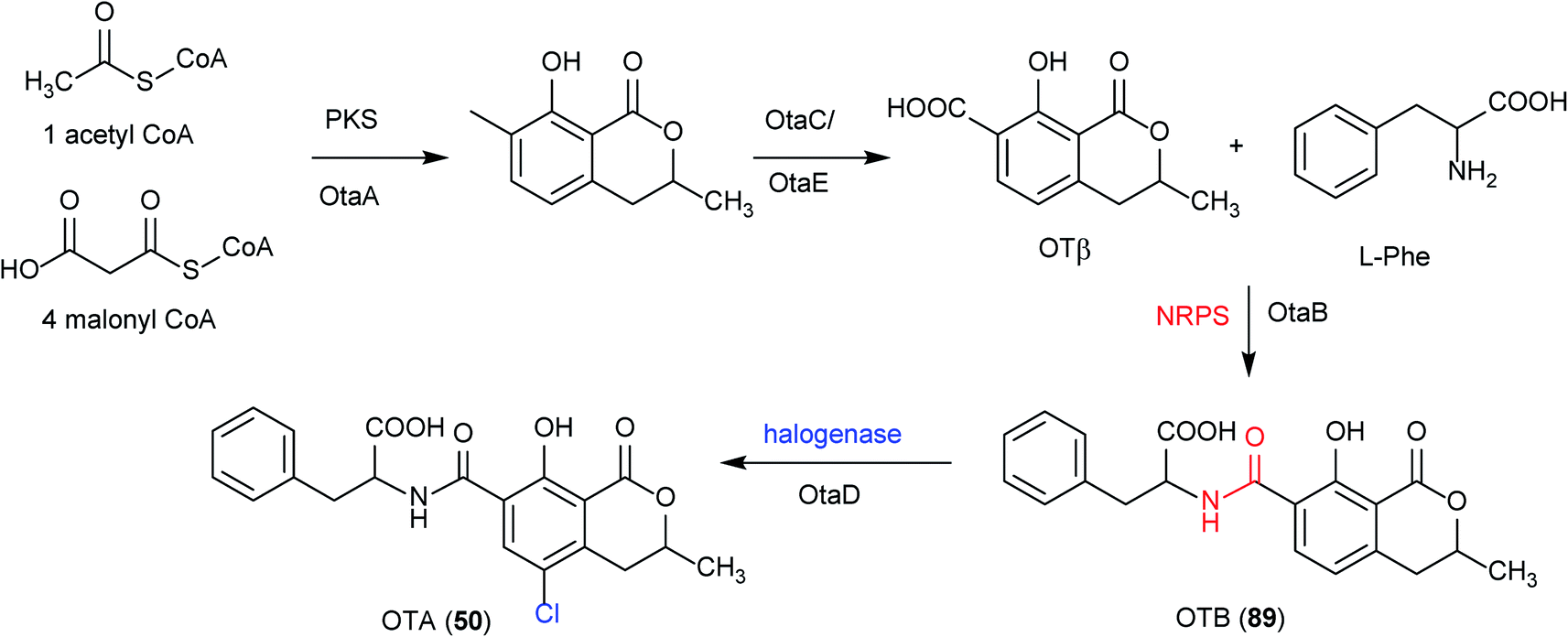 | ||
| Fig. 25 Proposed ochratoxin A (50) biosynthetic pathway, including intermediate metabolites and catalytic enzymes.148 | ||
5.4 Acurin A
Acurin A (90) was isolated from an extract of A. aculeatus. The BGC was identified based on bioinformatics, RT-qPCR experiments and gene-deletion analysis and revealed that at least eight genes (acrA-G and acrR) are vital for the biosynthesis of 90.150Since the structure of 90 is very similar to that of fusarin C, for which the biosynthesis is partly characterized, it was hypothesized that 90 originated from a polyketide and an amino acid fusion. The candidate clusters were further narrowed down to identify the putative PKS and NRPS genes for the production of 90. Intriguingly, bioinformatics analysis indicated that the scaffold for 90 formation potentially could be synthesized by unlinked PKS and NRPS enzymes. In addition, the researchers also found that uncoupled PKS and NRPS encoding genes are conserved in a fungal clade in section Nigri. Moreover, the PKS didn't appear to have a release domain but instead the predicted NRPS contained a reductase domain for the potential release of 90 backbone. Examination of genes encoding the PKS and NRPS responsible for 90 biosynthesis were identified to be acrA and acrB through gene deletion experiment in A. aculeatus, which led to the abolishment of 90 production. Further bioinformatics analyses showed a lack of an epimerase domain is present in the NRPS indicating L-serine is incorporated into 90. In addition to the PKS and NRPS encoding genes of 90, there are six other surrounding genes (acrC–G) including a regulatory transcription factor (acrR). Next, a functional analysis of the genes involved in the formation of 90 was also performed by targeted gene deletions. This confirmed the involvement of the transcription factor acrR and O-methyltransferase (acrG). However, no accumulation of potential intermediates were found in acrC (hydrolase), acrD (oxidoreductase), acrE (CYP450) and acrF (CYP450) deletion strains, which might be because of the unstable intermediate products or a scenario where more than two enzymes interact with each other during the reactions. Altogether, these results enabled Wolff et al. to propose a pathway for biosynthesis of 90.150
The pathway starts with the polyketide assembly by the PKS AcrA forming a heptaketide backbone from successive condensations of six malonyl-CoA units (Fig. 26). Then the fusion of an amino-acid moiety is catalyzed by the NRPS AcrB. The functions of AcrC–F are still unclear, however the researchers hypothesized that the PK-NRP hybrid product release from the enzyme by reductive release to set up the formation of the lactam ring by Aldol condensation, followed by a water loss to generate a double bond followed by reduction. Finally, an O-methyltransferase AcrG is needed for the final step in the biosynthesis of 90.
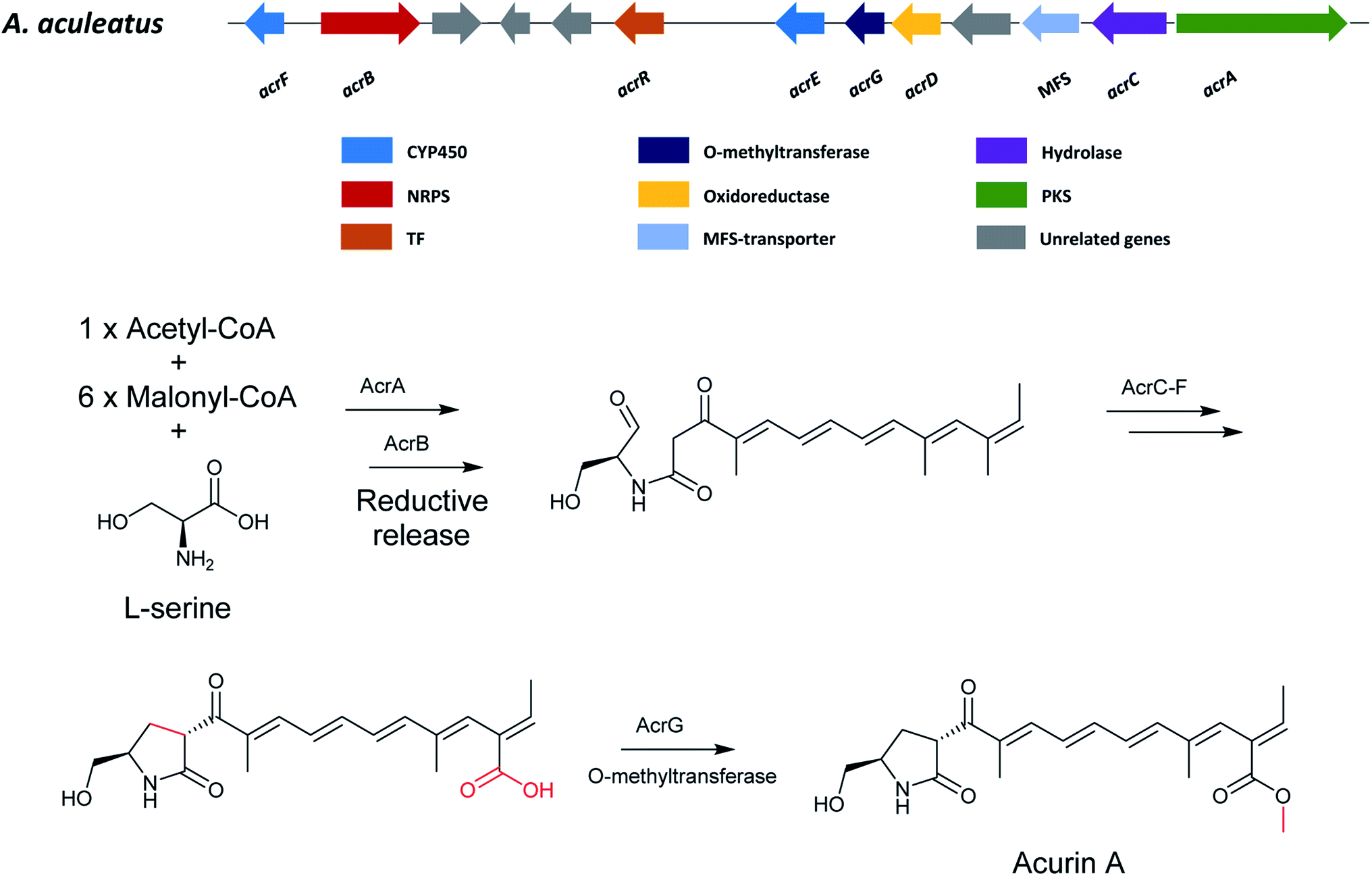 | ||
| Fig. 26 The gene cluster in A. aculeatus and the proposed biosynthetic pathway of acurin A (90).150 | ||
6 Meroterpenoids
6.1 Yanuthones
The yanuthones comprise a group of compounds containing a core structure constituted of an epoxylated six-membered ring with a sesquiterpene chain at C-13 and varied side chains at C-15 and C-16.171,222 Many yanuthones have shown antifungal bioactivity, and among them yanuthone D (49) displayed the most potent antifungal activity against Candida albicans.152The yan BGC for 49 contains ten genes in the genome of A. niger. The PKS-encoding YanA was heterologously expressed in A. nidulans to confirm its involvement in the biosynthesis of the initial polyketide product 6-MSA (51). The biosynthetic pathway of 49 was further investigated by individually deleting all genes within the yan cluster (Fig. 27) which resulted in the accumulation of numerous yanuthone intermediates and enabled a biosynthetic pathway to be proposed (Fig. 28).
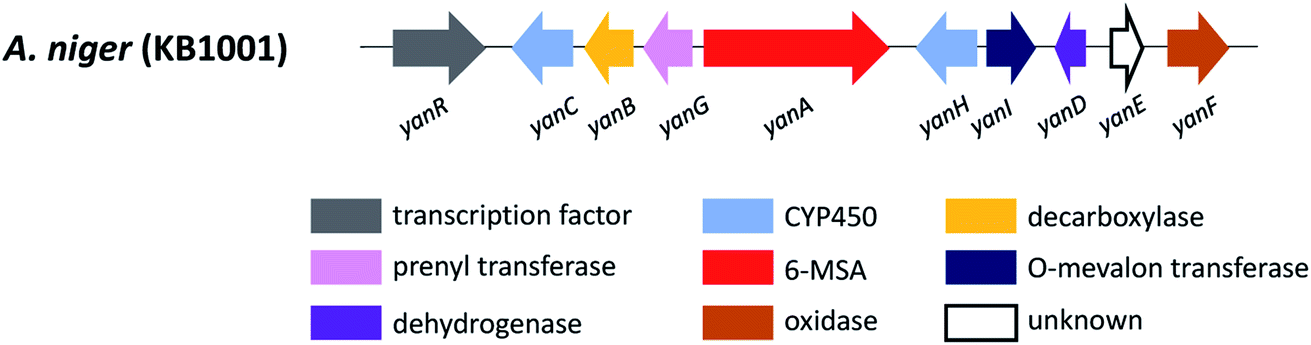 | ||
| Fig. 27 The yan cluster. The yanA 6-MSA synthase-encoding gene is flanked by nine cluster genes (yanB, yanC yanD, yanE, yanF, yanG, yanH, yanI, and yanR) whose products contain all necessary activities for conversion of 6-MSA into yanuthone D (49).151 | ||
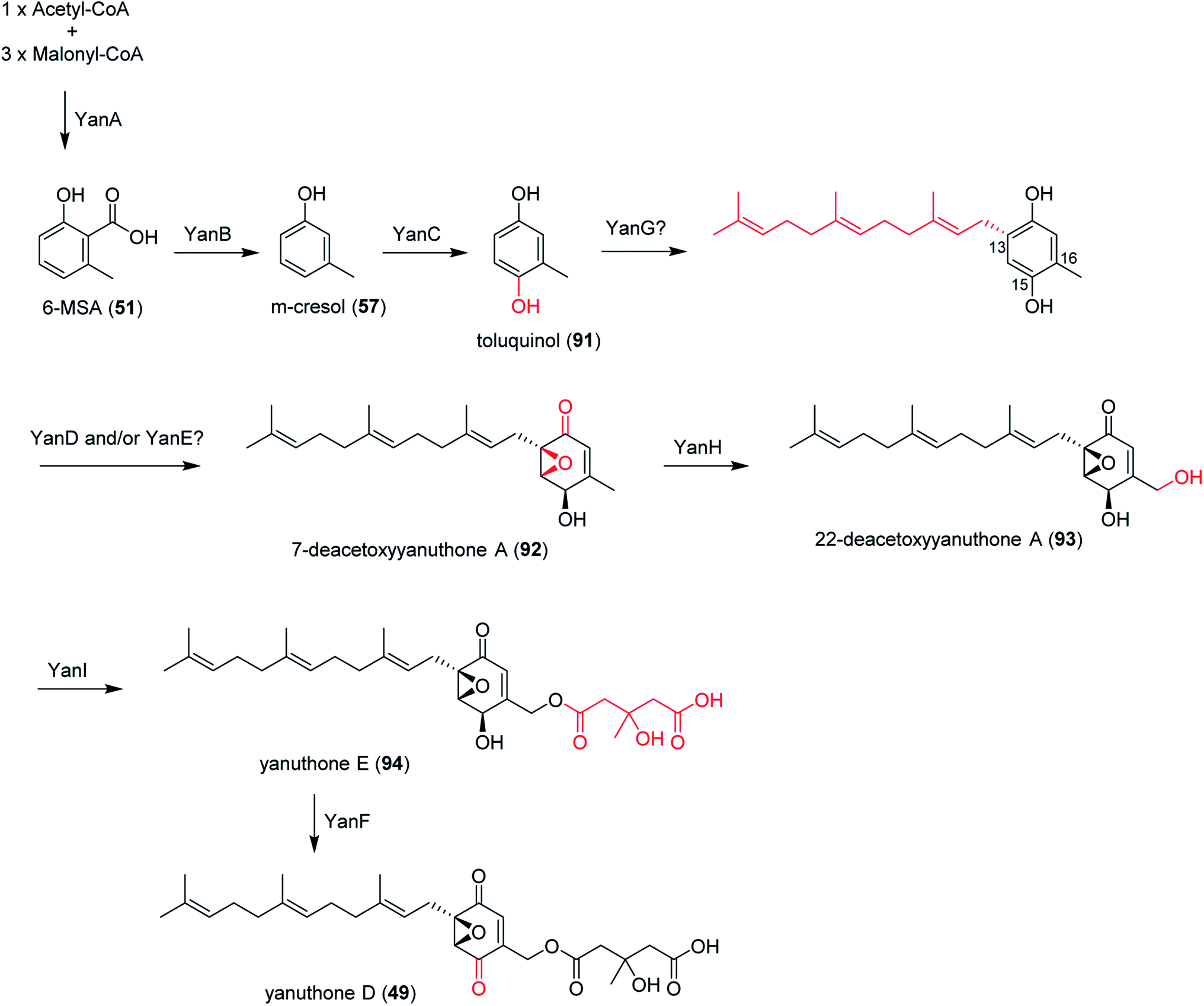 | ||
| Fig. 28 Biosynthesis of yanuthone D (49) in A. niger.151,223 | ||
The pathway starts with the biosynthesis of 6-MSA (51) catalyzed by the PKS YanA, followed by decarboxylation by YanB producing m-cresol (57), and hydroxylation by the cytochrome P450 YanC, generating toluquinol (91). The latter two steps for epoxidation and prenylation are still not very clear since no biosynthetic intermediate has been obtained from the targeted gene deletions. It has been proposed that the farnesylation likely precede epoxidation since the C-prenylation reaction generally requires the π-electron of a prenyl acceptor to attack the allylic cation generated on the prenyl donor. Therefore, 91 may also be accepted by YanG, followed by epoxidation by YanD/YanE generating 7-deacetoxyyanuthone (92), as epoxidized 91 loses a π-electron at the prenylation site due to the epoxide formation.151,223 Next, cytochrome P450 YanH catalyzes the conversion to 22-deacetylyanuthone A (93), which is then converted to yanuthone E (94) by the O-mevalon transferase YanI. Importantly, this is the first time that O-mevalon transferase activity has been molecularly characterized. Finally, the oxidase YanF catalyzes the formation of 49 from 94.171 Deletion of the individual genes in the BGC also led to formation of not only hydroxylated metabolites but also a glycosylated yanuthone.50
In addition to 49 and its analogues with a C7 scaffold defined as class I yanuthones, yanuthone X1 and X2 were also discovered, which instead contain a C6-core scaffold oxygenated at C-16 defined as class II yanuthones and derived from an unknown precursor.108,222
6.2 Aculene A–C
A. aculeatus is an industrially important black filamentous fungus. After the prioritization of the growing conditions and the dereplication by UV- and HRMS-data to identify known compounds, a unique type of norsesquiterpenes (C14), named aculenes A and B (55, 56), were isolated and structurally characterized from A. aculeatus IBT 21030.11555 and 56 are of mixed biosynthetic origin containing the amino acid L-proline moiety but also a nordaucane skeleton that the researchers proposed to be derived from an ent-daucane precursor through demethylation (loss of a methyl group on the C5n terpene scaffold) (Fig. 31). Lee et al. used gene inactivation, feeding experiments, and heterologous reconstitution approaches in order to understand the demethylation (C-12) process that converts the ent-daucane-containing asperaculane into the nordaucane aculene scaffold.153Because the L-proline moiety in 55 was likely introduced by a nonribosomal peptide synthase (NRPS), the researchers accessed the genome sequence from JGI of A. aculeatus ATCC16872 (=BCRC32190) and scanned for all adenylation domain encoding genes and their neighboring genes.224 A candidate BGC, ane, was found containing one single-module NRPS (AneB) with adenylation and thiolation domains as well as a Class I terpene cyclase (AneC). Further investigation revealed several genes nearby aneB and aneC with putative activities consistent with the structure of 55, including a phytanoyl-CoA dioxygenase (aneA), three cytochrome P450 monooxygenases (aneD, aneF and aneG) that could perform oxidative modifications on the terpene scaffold, a α,β-hydrolase (aneE) that may transfer or release L-proline from AneB to the sesquiterpene moiety, and a proline iminopeptidase (aneH) (Fig. 29).
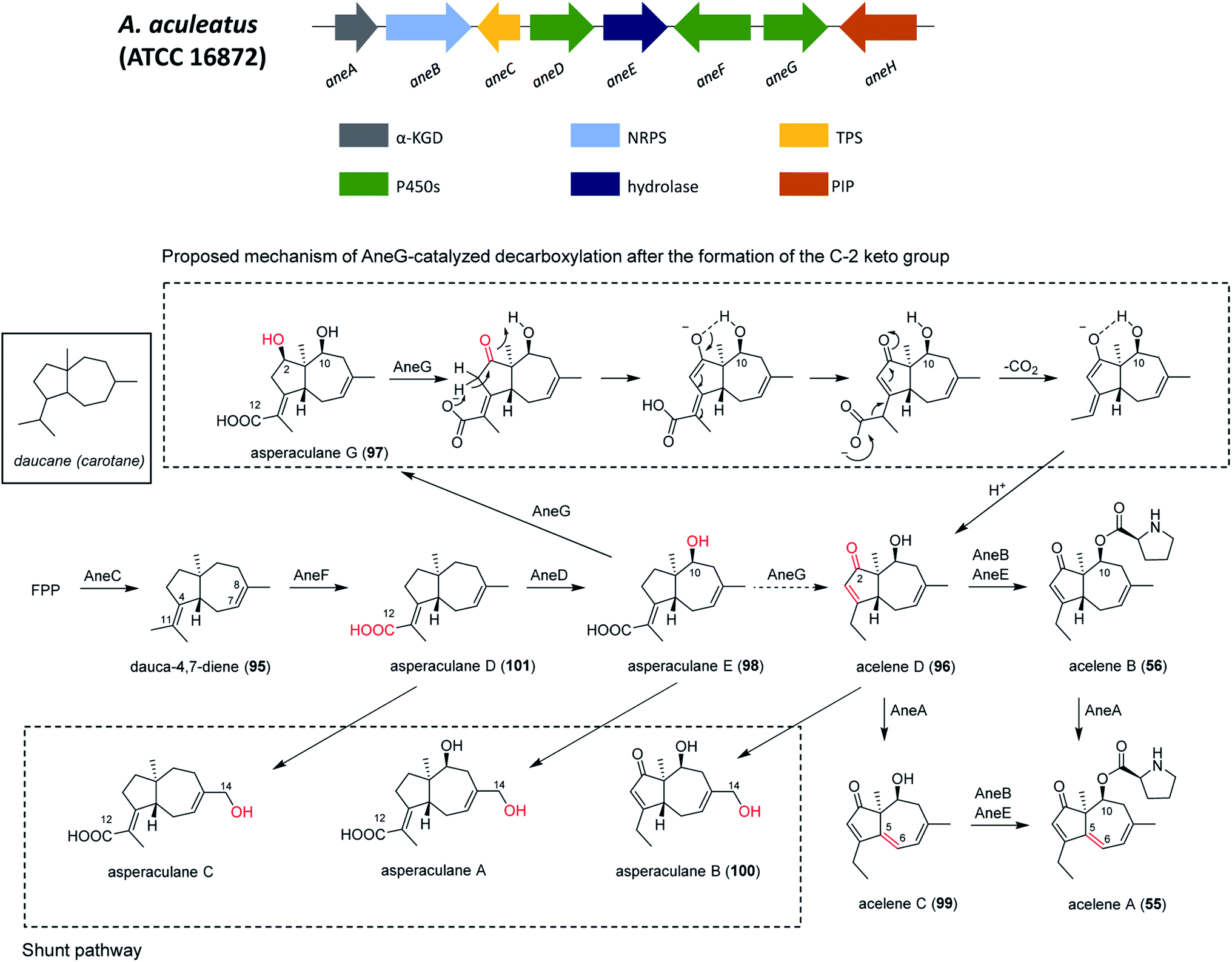 | ||
| Fig. 29 The gene cluster ane in A. aculeatus and the biosynthetic pathway of aculene A.153 | ||
To further explore the functions of those enzymes, genetic inactivation on aneC was performed which produced no aculene products and showed no accumulation of any intermediates. Next, intron-free aneC from A. aculeatus was cloned and transformed into Saccharomyces cerevisiae strain BJ5464-NpgA, which showed the production of dauca-4, 7-diene (95) as a sole product. These results confirmed that AneC is a terpene cyclase that produce 95 with an ent-daucane skeleton for the biosynthesis of 55.
Each of the three P450 monooxygenase genes (aneG, aneF and aneD) were then inactivated. The results indicated the involvement of oxidation by AneG, AneD and AneF at C-2, C-10 and C12 respectively. In addition, the researchers hypothesized that the P450s could also play a role in the demethylation process. To further verify the function of the three P450 monooxygenases (ane G, D, F), heterologous co-expression of these enzymes with AneC using a S. cerevisiae strain harboring a single copy of the cytochrome P450 reductase gene from A. terreus (AtCPR) were performed first.224 They confirmed the role of aneF, aneD, aneG when they coexpressed aneF with aneC, aneC/F/D and aneC/G/F. However, the enzyme involved in the second oxidation at C-2 to generate a ketone functionality from a C-2 hydroxy group and decarboxylation at C-12 was still unknown because of the decreasing amounts of downstream metabolites when coexpressing aneC/F/D/G. This might be because of degradation by endogenous enzymes in S. cerevisiae of the aculenes homologues. Therefore, the coexpression of aneC/F/D/G in A. oryzae was performed, which led to the production of aculene D (96) as a major product together with asperaculane G (97). Thus, AneG performs C-H activation at the C-2 position of asperaculane E (98) and catalyzes a double oxidation to install a C-2 ketone group. The involvement of 97 as pathway intermediate was validated, as the feeding of 97 to ΔaneC restored the production of 55. Overall, the researchers proposed that all the three P450s are necessary for this stepwise demethylation process. AneF oxidizes C-12 to a carboxylate, AneG installs a necessary electro-withdrawing carbonyl group on C-2, while AneD introduces a10-hydroxy group to assist the tautomerization.
Next, the modification enzymes involved in the gene cluster was investigated. Deletion of AneA, which encodes an α-ketoglutarate-dependent dioxygenase, led to the abolishment of the production of 55 and aculene C (99), and accumulation of 56 and 96, and a minor component asperaculane B (100) (shunt product). This suggest that the unsaturation is catalyzed by AneA.
Deletion of aneB and aneE both led to the absence of 55 and 56 and the accumulation of 96 and 100. This result suggests that AneB adenylates L-proline and AneE transfers this activated L-proline derivative to 96 to form 56.
Compared to the downstream pathway products, there are several upstream intermediates containing a 14-hydroxy group. To confirm that the 14-hydroxylated compounds are shunt products, the researchers supplied 96 and 100 to aneC-deleted strain. The former restores the production of aculene but the latter does not, which suggest that the metabolites with 14-hydroxy group are shunt products in the biosynthesis of 55.
Overall, the biosynthetic pathway of 55 initiates with the biosynthesis of 95 by AneC, followed by C-12 carboxylation by AneF to produce asperaculane D (101), and C-10 hydroxylation by AneD, yielding asperaculane E (98). Then AneG installs an electron-withdrawing carbonyl group at the C-2 position, which triggers C-12 decarboxylation to yield the nordaucane skeleton in 96. Next, the unsaturation between C5 and C6 are catalyzed by AneA, yielding 55 and 99. Finally, the adenylation is catalyzed by AneB, followed by transferring the activated L-proline derivatives to 96, yielding 56.
7 Fatty acid derived secondary metabolites
7.1 Alkylcitric acids
Alkylcitric acids comprise a saturated alkyl “tail” and a citrate-derived “head”. Many of these compounds have shown bioactive properties, such as plant root growth promotion (hexylitaconic acid) and anti-fungal (hexylaconitic acid anhydride) bioactivity.225,226 Their BGC and the transcriptional factor (akcR) have been identified recently in A. niger by examining the functional annotation of BGCs predicted from genomic data (Fig. 30). The transcriptional factor akcR was overexpressed, leading to the production of several alkylcitric acids. In addition, an unlinked tailoring gene (hexylaconitic acid decarboxylase gene, hadA) and akcR gene were overexpressed together in A. niger, leading to the overproduction of alkylcitric acids and several previously unreported alkylcitric acids.154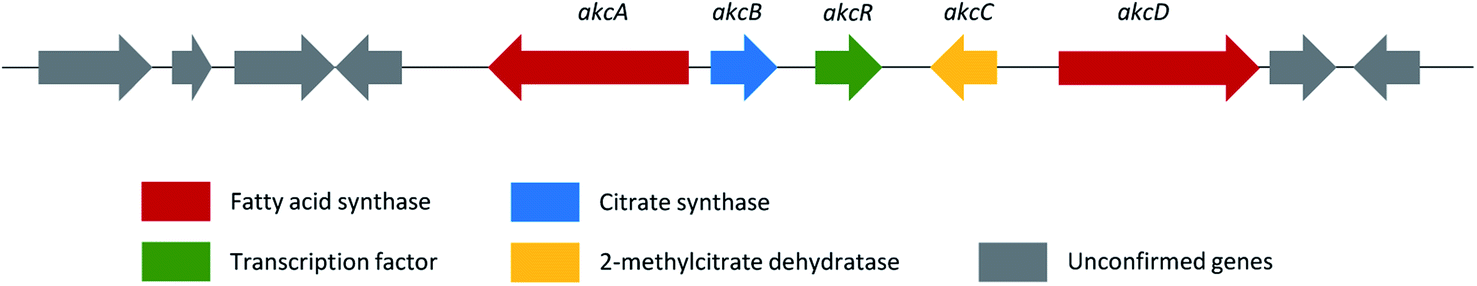 | ||
| Fig. 30 The gene cluster akc in A. niger. The boundaries of the gene cluster has not been defined.154 | ||
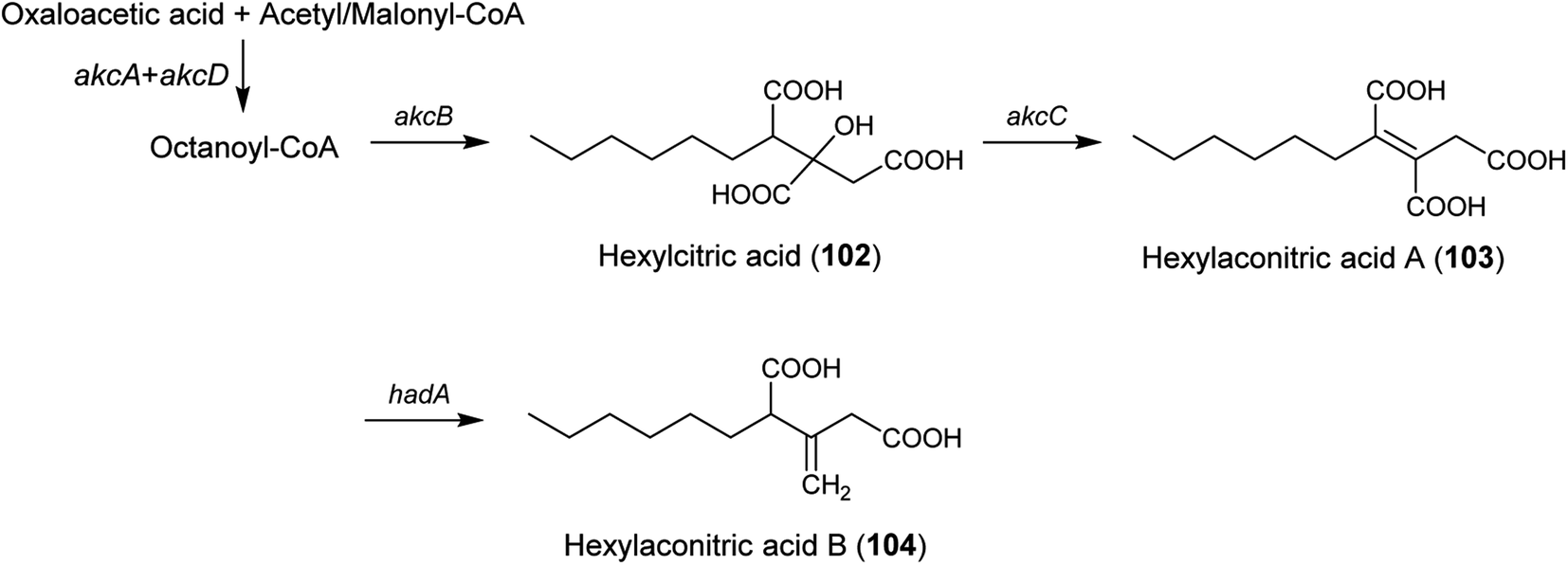 | ||
| Fig. 31 Predicted biosynthetic pathway of alkylcitric acids.154 | ||
The proposed alkylcitrate pathway starts with the production of hexylcitric acid (102) catalyzed by a citrate synthase (akcB) and a fatty acid synthase (akcA and akcD). Next, the dihydroxylation of hexylcitric is catalyzed by 2-methylcitrate dehydratase (akcC) to produce hexylaconitic acid A (103). Finally, a decarboxylation reaction is catalyzed by hexylaconitic acid decarboxylase (hadA), yielding hexylitaconic acid B (104) (Fig. 31).154
8 Conclusion and future perspectives
In this review we have highlighted and described the nineteen biosynthetic pathways that have so far been characterized from species belonging to the group of black aspergilli, linking the secondary metabolites to their corresponding BGCs. Considering that Vesth et al. recently identified 455 BGC families, across 37 genomes in section Nigri,6 this is a surprisingly small number taking the importance of black aspergilli as food contaminants and industrial workhorses for enzyme production into account. First, this means that many of the 176 secondary metabolites from which structures have already been reported from black aspergilli need to be linked to their respective BGCs. Secondly, the many unknown secondary metabolites predicted from these fungi need to be discovered and characterized, including the approximately 8.75 unique clusters found among both uni- and bi-seriate black aspergilli.2 With the continued rapid collection of high quality genomic information, the development of new integrated omics technologies and more effective strategies for manipulation and mining of fungal genomes, the number of fully characterized fungal BGCs is expected to increase substantially in the coming years.2279 Conflicts of interest
There are no conflicts to declare.10 Acknowledgements
This work was supported by the Danish National Research Foundation (DNRF137) for the Center for Microbial Secondary Metabolites (CeMiSt), as well as a grant from the Novo Nordic Foundation (NNF18OC0034952) and from the China Scholarship Council (No. 201806250032) and the Department of Biotechnology and Biomedicine at the Technical University of Denmark.11 References
- T. O. Larsen, J. Smedsgaard, K. F. Nielsen, M. E. Hansen and J. C. Frisvad, Nat. Prod. Rep., 2005, 22, 672–695 RSC
.
- T. C. Vesth, J. L. Nybo, S. Theobald, J. C. Frisvad, T. O. Larsen, K. F. Nielsen, J. B. Hoof, J. Brandl, A. Salamov, R. Riley, J. M. Gladden, P. Phatale, M. T. Nielsen, E. K. Lyhne, M. E. Kogle, K. Strasser, E. McDonnell, K. Barry, A. Clum, C. Chen, K. LaButti, S. Haridas, M. Nolan, L. Sandor, A. Kuo, A. Lipzen, M. Hainaut, E. Drula, A. Tsang, J. K. Magnuson, B. Henrissat, A. Wiebenga, B. A. Simmons, M. R. Mäkelä, R. P. de Vries, I. V. Grigoriev, U. H. Mortensen, S. E. Baker and M. R. Andersen, Nat. Genet., 2018, 50, 1688–1695 CrossRef CAS PubMed
- J. Houbraken, S. Kocsubé, C. M. Visagie, N. Yilmaz, X. C. Wang, M. Meijer, B. Kraak, V. Hubka, K. Bensch, R. A. Samson and J. C. Frisvad, Stud. Mycol., 2020, 95, 5–169 CrossRef CAS PubMed
- I. V. Grigoriev, R. Nikitin, S. Haridas, A. Kuo, R. Ohm, R. Otillar, R. Riley, A. Salamov, X. Zhao, F. Korzeniewski, T. Smirnova, H. Nordberg, I. Dubchak and I. Shabalov, Nucleic Acids Res., 2014, 42, 699–704 CrossRef PubMed
- I. V. Grigoriev, D. A. Martinez and A. A. Salamov, Appl. Mycol. Biotechnol., 2006, 6, 123–142 CAS
- T. C. Vesth, J. L. Nybo, S. Theobald, J. C. Frisvad, T. O. Larsen, K. F. Nielsen, J. B. Hoof, J. Brandl, A. Salamov, R. Riley, J. M. Gladden, P. Phatale, M. T. Nielsen, E. K. Lyhne, M. E. Kogle, K. Strasser, E. McDonnell, K. Barry, A. Clum, C. Chen, K. LaButti, S. Haridas, M. Nolan, L. Sandor, A. Kuo, A. Lipzen, M. Hainaut, E. Drula, A. Tsang, J. K. Magnuson, B. Henrissat, A. Wiebenga, B. A. Simmons, M. R. Mäkelä, R. P. de Vries, I. V. Grigoriev, U. H. Mortensen, S. E. Baker and M. R. Andersen, Nat. Genet., 2018, 50, 1688–1695 CrossRef CAS PubMed
- S. A. Kautsar, K. Blin, S. Shaw, T. Weber and M. H. Medema, Nucleic Acids Res., 2021, 49, D490–D497 CrossRef CAS PubMed
- N. Ziemert, A. Lechner, M. Wietz, N. Millań-Aguiñaga, K. L. Chavarria and P. R. Jensen, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E1130–E1139 CrossRef CAS PubMed
- C. Rajendran and B. N. Muthappa, Proc. - Indian Acad. Sci., Plant Sci., 1980, 185–191 CrossRef
- J. C. Frisvad, L. M. Petersen, E. K. Lyhne and T. O. Larsen, PLoS One, 2014, 9, e94857 CrossRef PubMed
- V. Ellena, D. Bucchieri, E. Arcalis, M. Sauer and M. G. Steiger, Fungal Biol., 2021, 125, 485–494 CrossRef CAS PubMed
- A. Mageswari, J. S. Kim, K. H. Cheon, S. W. Kwon, O. Yamada and S. B. Hong, Mycobiology, 2016, 44, 269–276 CrossRef PubMed
- J. Varga, S. Kocsubé, B. Tóth, J. C. Frisvad, G. Perrone, A. Susca, M. Meijer and R. A. Samson, Int. J. Syst. Evol. Microbiol., 2007, 57, 1925–1932 CrossRef CAS PubMed
- A. Al-Musallam, Antonie van Leeuwenhoek, 1980, 46, 407–411 CrossRef CAS PubMed
- P. Noonim, W. Mahakarnchanakul, J. Varga, J. C. Frisvad and R. A. Samson, Int. J. Syst. Evol. Microbiol., 2008, 58, 1727–1734 CrossRef CAS PubMed
- J. Varga, J. C. Frisvad, S. Kocsubé, B. Brankovics, B. Tóth, G. Szigeti and R. A. Samson, Stud. Mycol., 2011, 69, 1–17 CrossRef CAS PubMed
- J. J. da Silva, B. T. Iamanaka, L. S. Ferranti, F. P. Massi, M. H. Taniwaki, O. Puel, S. Lorber, J. C. Frisvad and M. H. P. Fungaro, J. Fungi, 2020, 6, 1–20 Search PubMed
- S. Khuna, N. Suwannarach, J. Kumla, J. C. Frisvad, K. Matsui, W. Nuangmek and S. Lumyong, Front. Microbiol., 2021, 12, 1–20 Search PubMed
- K. Barrett, K. Jensen, A. S. Meyer, J. C. Frisvad and L. Lange, Sci. Rep., 2020, 10, 1–12 CrossRef PubMed
- R. A. Samson, C. M. Visagie, J. Houbraken, S. B. Hong, V. Hubka, C. H. W. Klaassen, G. Perrone, K. A. Seifert, A. Susca, J. B. Tanney, J. Varga, S. Kocsubé, G. Szigeti, T. Yaguchi and J. C. Frisvad, Stud. Mycol., 2014, 78, 141–173 CrossRef CAS PubMed
- J. C. Frisvad and T. O. Larsen, Appl. Microbiol. Biotechnol., 2015, 99, 7859–7877 CrossRef CAS PubMed
- J. L. Steenwyk, X. X. Shen, A. L. Lind, G. H. Goldman and A. Rokas, mBio, 2019, 10, e00925-19 CrossRef PubMed
- C. M. Visagie, Y. Hirooka, J. B. Tanney, E. Whitfield, K. Mwange, M. Meijer, A. S. Amend, K. A. Seifert and R. A. Samson, Stud. Mycol., 2014, 78, 63–139 CrossRef CAS PubMed
- R. Mosseray, Cellule, 1934, 43, 203–285 Search PubMed
-
K. B. Raper, and D. I. Fennell, The Genus Aspergillus, Williams Wilkins, Co., Baltimore, Maryland, 1965, p. 686 Search PubMed
- H. Murakami, J. Soc. Brew., Jpn., 1976, 71, 952–956 CrossRef
- H. Murakami, Soc. Brew., Jpn., 1976, 71, 956–959 CrossRef
- F. Noro, H. Murakami, K. Yoshida and K. Yoshida, J. Soc. Brew., Jpn., 1979, 74, 466–470 CrossRef
- F. Howard and H. Ferris, J. Soc. Brew., Jpn., 1976, 71, 952–955 CrossRef
- H. Murakami, J. Soc. Brew., Jpn., 1979, 74, 323–327 CrossRef
- H. Murakami, J. Soc. Brew., Jpn., 1979, 74, 842–848 CrossRef
- H. Murakami, J. Soc. Brew., Jpn., 1979, 74, 849–853 CrossRef
- H. Murakami, J. Soc. Brew., Jpn., 1979, 854–858 Search PubMed
- F. Noro and H. Murakami, J. Soc. Brew., Jpn., 1979, 74, 462–465 CrossRef
- K. Yoshida and H. Murakami, J. Soc. Brew., Jpn., 1979, 74, 328–331 CrossRef
- K. Yoshida and H. Murakami, J. Soc. Brew., Jpn., 1979, 74, 459–461 CrossRef
- L. Pařenicová, J. A. E. Benen, R. A. Samson and J. Visser, Mycol. Res., 1997, 101, 810–814 CrossRef
- L. Pařenicová, P. Skouboe, J. Frisvad, R. A. Samson, L. Rossen, M. Ten Hoor-Suykerbuyk and J. Visser, Appl. Environ. Microbiol., 2001, 67, 521–527 CrossRef PubMed
- K. Yokoyama, L. Wang, M. Miyaji and K. Nishimura, FEMS Microbiol. Lett., 2001, 200, 241–246 CrossRef CAS PubMed
- M. L. Abarca, F. Accensi, J. Cano and F. Cabañes, Antonie van Leeuwenhoek, 2004, 86, 33–49 CrossRef CAS PubMed
- R. P. de Vries, J. C. Frisvad, P. J. I. van de Vondervoort, K. Burgers, A. F. A. Kuijpers, R. A. Samson and J. Visser, Antonie van Leeuwenhoek, 2005, 87, 195–203 CrossRef CAS PubMed
- A. González-Salgado, B. Patiño, C. Vázquez and M. T. González-Jaén, FEMS Microbiol. Lett., 2005, 245, 353–361 CrossRef PubMed
- M. Bau, G. Castellá, M. R. Bragulat and F. J. Cabañes, Int. J. Food Microbiol., 2006, 111, 18–21 CrossRef PubMed
- R. Serra, F. J. Cabañes, G. Perrone, G. Castellá, A. Venâncio, G. Mulè and Z. Kozakiewicz, Mycologia, 2006, 98, 295–306 CrossRef PubMed
- D. M. Geiser, M. A. Klich, J. C. Frisvad, S. W. Peterson, J. Varga and R. A. Samson, Stud. Mycol., 2007, 59, 1–10 CrossRef CAS PubMed
- J. C. Frisvad, T. O. Larsen, R. De Vries, M. Meijer, J. Houbraken, F. J. Cabañes, K. Ehrlich and R. A. Samson, Stud. Mycol., 2007, 59, 31–37 CrossRef CAS PubMed
- G. Perrone, G. Mulè, A. Susca, P. Battilani, A. Pietri and A. Logrieco, Appl. Environ. Microbiol., 2006, 72, 680–685 CrossRef CAS PubMed
- G. Perrone, A. Susca, G. Cozzi, K. Ehrlich, J. Varga, J. C. Frisvad, M. Meijer, P. Noonim, W. Mahakamchanakul and R. A. Samson, Stud. Mycol., 2007, 59, 53–66 CrossRef CAS PubMed
- M. R. Bragulat and F. J. Cabañes, J. Microbiol. Methods, 2008, 75, 81–85 CrossRef CAS PubMed
- G. Perrone, J. Varga, A. Susca, J. C. Frisvad, G. Stea, S. Kocsubé, B. Tóth, Z. Kozakiewicz and R. A. Samson, Int. J. Syst. Evol. Microbiol., 2008, 58, 1032–1039 CrossRef PubMed
- S. W. Peterson, Mycologia, 2008, 100, 205–226 CrossRef CAS PubMed
- L. M. Ferracin, J. C. Frisvad, M. H. Taniwaki, B. T. Iamanaka, D. Sartori, M. E. Schapovaloff and M. H. P. Fungaro, Braz. Arch. Biol. Technol., 2009, 52, 241–248 CrossRef
- M. L. Chiotta, M. L. Ponsone, M. Combina, A. M. Torres and S. N. Chulze, Int. J. Food Microbiol., 2009, 136, 137–141 CrossRef CAS PubMed
- G. Perrone, G. Stea, F. Epifani, J. Varga, J. C. Frisvad and R. A. Samson, Fungal Biol., 2011, 115, 1138–1150 CrossRef CAS PubMed
- J. D. Palumbo and T. L. O'Keeffe, Lett. Appl. Microbiol., 2015, 60, 188–195 CrossRef CAS PubMed
- D. M. Silva, L. R. Batista, E. F. Rezende, M. H. P. Fungaro, D. Sartori and E. Alves, Braz. J. Microbiol., 2011, 42, 761–773 CrossRef PubMed
- A. Sørensen, P. S. Lübeck, M. Lübeck, K. F. Nielsen, B. K. Ahring, P. J. Teller and J. C. Frisvad, Int. J. Syst. Evol. Microbiol., 2011, 61, 3077–3083 CrossRef PubMed
- M. L. Castrillo, M. I. Fonseca, G. A. Bich, G. Jerke, M. A. Horianski and P. D. Zapata, BAG, J. Basic Appl. Genet., 2012, 23, 19–27 Search PubMed
- V. Hubka and M. Kolarik, Persoonia, 2012, 1–10 CAS
- Ž. Jurjevic, S. W. Peterson, G. Stea, M. Solfrizzo, J. Varga, V. Hubka and G. Perrone, IMA Fungus, 2012, 3, 159–173 CrossRef PubMed
- S. B. Hong, M. Lee, D. H. Kim, J. Varga, J. C. Frisvad, G. Perrone, K. Gomi, O. Yamada, M. Machida, J. Houbraken and R. A. Samson, PLoS One, 2013, 8, e63769 CrossRef CAS PubMed
- S. B. Hong, O. Yamada and R. A. Samson, Appl. Microbiol. Biotechnol., 2014, 98, 555–561 CrossRef CAS PubMed
- A. Susca, A. Moretti, G. Stea, A. Villani, M. Haidukowski, A. Logrieco and G. Munkvold, Int. J. Food Microbiol., 2014, 188, 75–82 CrossRef CAS PubMed
- G. Garmendia and S. Vero, Int. J. Food Microbiol., 2016, 216, 31–39 CrossRef CAS PubMed
- L. de S. Ferranti, M. H. P. Fungaro, F. P. Massi, J. J. da Silva, R. E. S. Penha, J. C. Frisvad, M. H. Taniwaki and B. T. Iamanaka, Int. J. Food Microbiol., 2018, 268, 53–60 CrossRef CAS PubMed
- E. A. A. Duarte, C. L. Damasceno, T. A. S. de Oliveira, L. de O. Barbosa, F. M. Martins, J. R. Q. de Silva, T. E. F. de Lima, R. M. da Silva, R. B. Kato, D. E. Bortolini, V. Azevedo, A. Góes-Neto and A. C. F. Soares, Front. Microbiol., 2018, 9, 1227 CrossRef PubMed
- E. D’hooge, P. Becker, D. Stubbe, A. C. Normand, R. Piarroux and M. Hendrickx, Med. Mycol., 2019, 57, 773–780 CrossRef PubMed
- J. Gil-Serna, M. García-Díaz, C. Vázquez, M. T. González-Jaén and B. Patiño, Food Microbiol., 2019, 82, 240–248 CrossRef CAS PubMed
- D. O. A. Vanzela, R. A. dos Santos, T. M. M. Nunes, J. P. S. Monteiro, M. M. Ribeiro, M. I. Rezende and D. Sartori, Braz. J. Anim. Environ. Res., 2020, 3, 3856–3866 CrossRef
- M. Doilom, J. W. Guo, R. Phookamsak, P. E. Mortimer, S. C. Karunarathna, W. Dong, C. F. Liao, K. Yan, D. Pem, N. Suwannarach, I. Promputtha, S. Lumyong and J. C. Xu, Front. Microbiol., 2020, 11, 1–24 CrossRef PubMed
- M. A. Kusters-van Someren, R. A. Samson and J. Visser, Curr. Genet., 1991, 19, 21–26 CrossRef CAS
- F. Accensi, J. Cano, L. Figuera, M. L. Abarca and F. J. Cabañes, FEMS Microbiol. Lett., 1999, 180, 191–196 CrossRef CAS PubMed
- A. Z. Kozakiewicz, J. C. Frisvad, D. L. Hawksworth, J. I. Pitt, R. A. Samson and C. Stolk, Taxon, 1992, 41, 109–113 CrossRef
- H. J. Pel, J. H. De Winde, D. B. Archer, P. S. Dyer, G. Hofmann, P. J. Schaap, G. Turner, R. P. De Vries, R. Albang, K. Albermann, M. R. Andersen, J. D. Bendtsen, J. A. E. Benen, M. Van Den Berg, S. Breestraat, M. X. Caddick, R. Contreras, M. Cornell, P. M. Coutinho, E. G. J. Danchin, A. J. M. Debets, P. Dekker, P. W. M. Van Dijck, A. Van Dijk, L. Dijkhuizen, A. J. M. Driessen, C. D'Enfert, S. Geysens, C. Goosen, G. S. P. Groot, P. W. J. De Groot, T. Guillemette, B. Henrissat, M. Herweijer, J. P. T. W. Van Den Hombergh, C. A. M. J. J. Van Den Hondel, R. T. J. M. Van Der Heijden, R. M. Van Der Kaaij, F. M. Klis, H. J. Kools, C. P. Kubicek, P. A. Van Kuyk, J. Lauber, X. Lu, M. J. E. C. Van Der Maarel, R. Meulenberg, H. Menke, M. A. Mortimer, J. Nielsen, S. G. Oliver, M. Olsthoorn, K. Pal, N. N. M. E. Van Peij, A. F. J. Ram, U. Rinas, J. A. Roubos, C. M. J. Sagt, M. Schmoll, J. Sun, D. Ussery, J. Varga, W. Vervecken, P. J. J. Van De Vondervoort, H. Wedler, H. A. B. Wösten, A. P. Zeng, A. J. J. Van Ooyen, J. Visser and H. Stam, Nat. Biotechnol., 2007, 25, 221–231 CrossRef PubMed
- O. Yamada, R. Takara, R. Hamada, R. Hayashi, M. Tsukahara and S. Mikami, J. Biosci. Bioeng., 2011, 112, 233–237 CrossRef CAS PubMed
- W. Gong, Z. Cheng, H. Zhang, L. Liu, P. Gao and L. Wang, Genome Announc., 2016, 4, 1 Search PubMed
- R. P. de Vries, R. Riley, A. Wiebenga, G. Aguilar-Osorio, S. Amillis, C. A. Uchima, G. Anderluh, M. Asadollahi, M. Askin, K. Barry, E. Battaglia, Ö. Bayram, T. Benocci, S. A. Braus-Stromeyer, C. Caldana, D. Cánovas, G. C. Cerqueira, F. Chen, W. Chen, C. Choi, A. Clum, R. A. C. dos Santos, A. R. de Lima Damásio, G. Diallinas, T. Emri, E. Fekete, M. Flipphi, S. Freyberg, A. Gallo, C. Gournas, R. Habgood, M. Hainaut, M. L. Harispe, B. Henrissat, K. S. Hildén, R. Hope, A. Hossain, E. Karabika, L. Karaffa, Z. Karányi, N. Kraševec, A. Kuo, H. Kusch, K. LaButti, E. L. Lagendijk, A. Lapidus, A. Levasseur, E. Lindquist, A. Lipzen, A. F. Logrieco, A. MacCabe, M. R. Mäkelä, I. Malavazi, P. Melin, V. Meyer, N. Mielnichuk, M. Miskei, Á. P. Molnár, G. Mulé, C. Y. Ngan, M. Orejas, E. Orosz, J. P. Ouedraogo, K. M. Overkamp, H. S. Park, G. Perrone, F. Piumi, P. J. Punt, A. F. J. Ram, A. Ramón, S. Rauscher, E. Record, D. M. Riaño-Pachón, V. Robert, J. Röhrig, R. Ruller, A. Salamov, N. S. Salih, R. A. Samson, E. Sándor, M. Sanguinetti, T. Schütze, K. Sepčić, E. Shelest, G. Sherlock, V. Sophianopoulou, F. M. Squina, H. Sun, A. Susca, R. B. Todd, A. Tsang, S. E. Unkles, N. van de Wiele, D. van Rossen-Uffink, J. V. de Castro Oliveira, T. C. Vesth, J. Visser, J. H. Yu, M. Zhou, M. R. Andersen, D. B. Archer, S. E. Baker, I. Benoit, A. A. Brakhage, G. H. Braus, R. Fischer, J. C. Frisvad, G. H. Goldman, J. Houbraken, B. Oakley, I. Pócsi, C. Scazzocchio, B. Seiboth, P. A. VanKuyk, J. Wortman, P. S. Dyer and I. V. Grigoriev, Genome Biol., 2017, 18, 1–45 CrossRef PubMed
- S. Paul, Y. Ludeña, G. K. Villena, F. Yu, D. H. Sherman and M. Gutiérrez-Correa, Stand. Genomic Sci., 2017, 12, 1–8 CrossRef PubMed
- W. Wang, J. Gong, X. Liu, C. Dai, Y. Wang, X. N. Li, J. Wang, Z. Luo, Y. Zhou, Y. Xue, H. Zhu, C. Chen and Y. Zhang, J. Nat. Prod., 2018, 81, 1578–1587 CrossRef CAS PubMed
- W. Sun, L. Liu, Y. Yu, B. Yu, C. Liang, H. Ying, D. Liu and Y. Chen, ACS Omega, 2020, 5, 19737–19746 CrossRef CAS PubMed
- T. Futagami, K. Mori, A. Yamashita, S. Wada, Y. Kajiwara, H. Takashita, T. Omori, K. Takegawa, K. Tashiro, S. Kuhara and M. Goto, Eukaryotic Cell, 2011, 10, 1586–1587 CrossRef PubMed
- O. Yamada, M. Machida, A. Hosoyama, M. Goto, T. Takahashi, T. Futagami, Y. Yamagata, M. Takeuchi, T. Kobayashi, H. Koike, K. Abe, K. Asai, M. Arita, N. Fujita, K. Fukuda, K. I. Higa, H. Horikawa, T. Ishikawa, K. Jinno, Y. Kato, K. Kirimura, O. Mizutani, K. Nakasone, M. Sano, Y. Shiraishi, M. Tsukahara and K. Gomi, DNA Res., 2016, 23, 507–515 CrossRef CAS PubMed
- M. Shimizu, Y. Kusuya, Y. Alimu, C. Bian, H. Takahashi and T. Yaguchi, Microbiol. Resour. Announce., 2019, 8, 24–25 Search PubMed
- K. Mori, C. Kadooka, A. Nishitani, K. Okutsu, Y. Yoshizaki, K. Takamine, K. Tashiro, M. Goto, H. Tamaki and T. Futagami, Microbiol. Resour. Announce., 2021, 10, 1–3 Search PubMed
- N. Yamamoto, N. Watarai, H. Koyano, K. Sawada, A. Toyoda, K. Kurokawa and T. Yamada, Fungal Genet. Biol., 2021, 155, 103601 CrossRef CAS PubMed
- E. Choque, C. Klopp, S. Valiere, J. Raynal and F. Mathieu, BMC Genomics, 2018, 19, 1–16 CrossRef PubMed
- K. K. I. Yoshika, H. Takahashi, Y. Kusuya and T. Yaguchi, Microbiol. Resour. Announce., 2020, 9, e00702-20 CrossRef PubMed
- J. Wang, Z. Gao, Y. Qian, X. Hu, G. Li, F. Fu, J. Guo and Y. Shan, Front. Microbiol., 2021, 12, 1–15 CAS
- S. Zhao, D. Koffi, J.-P. Latge, K. Sylla and J. G. Gibbons, Microbiol. Resour. Announce., 2021, 10, 99–101 Search PubMed
- F. J. C. M. L. Abarca and M. R. Bragulat, Appl. Environ. Microbiol., 1993, 60, 2650–2652 CrossRef PubMed
- G. Perrone, A. Susca, F. Epifani and G. Mulè, Int. J. Food Microbiol., 2006, 111, 22–27 CrossRef PubMed
- J. C. Frisvad, T. O. Larsen, U. Thrane, M. Meijer, J. Varga, R. A. Samson and K. F. Nielsen, PLoS One, 2011, 6, 2–7 CrossRef PubMed
- A. Medina, R. Mateo, L. López-Ocaña, F. M. Valle-Algarra and M. Jiménez, Appl. Environ. Microbiol., 2005, 71, 4696–4702 CrossRef CAS PubMed
- M. Storari, L. Bigler, C. Gessler and G. A. L. Broggini, Food Addit. Contam., Part A: Chem., Anal., Control, Exposure Risk Assess., 2012, 29, 1450–1454 CrossRef CAS PubMed
- Y. Gherbawy, H. Elhariry, S. Kocsubé, A. Bahobial, B. El Deeb, A. Altalhi, J. Varga and C. Vágvölgyi, Foodborne Pathog. Dis., 2015, 12, 414–423 CrossRef CAS PubMed
- F. P. Massi, D. Sartori, L. de Souza Ferranti, B. T. Iamanaka, M. H. Taniwaki, M. L. C. Vieira and M. H. P. Fungaro, Int. J. Food Microbiol., 2016, 221, 19–28 CrossRef CAS PubMed
- N. Khaldi and K. H. Wolfe, Int. J. Evol. Biol., 2011, 2011, 1–7 CrossRef PubMed
- J. D. Palumbo, T. L. O'Keeffe and L. Gorski, Mycologia, 2013, 105, 277–284 CrossRef CAS PubMed
- K. F. Nielsen, J. C. Frisvad and A. Logrieco, J. Food Prot., 2015, 78, 6–8 CrossRef CAS PubMed
- A. Susca, R. H. Proctor, M. Morelli, M. Haidukowski, A. Gallo, A. F. Logrieco and A. Moretti, Front. Microbiol., 2016, 7, 1412 Search PubMed
-
R. A. Samson, J. Houbraken, U. Thrane, J. C. Frisvad and B. Andersen, Food indoor fungi, Westerdijk Fungal Biodivers Institute, Utrecht, 2nd edn, 2019, p. 481 Search PubMed
- J. C. Frisvad, L. L. H. Møller, T. O. Larsen, R. Kumar and J. Arnau, Appl. Microbiol. Biotechnol., 2018, 102, 9481–9515 CrossRef CAS PubMed
- L. Poulsen, M. R. Andersen, A. E. Lantz and J. Thykaer, PLoS One, 2012, 7, e50596 CrossRef CAS PubMed
- J. Niu, M. Arentshorst, P. D. S. Nair, Z. Dai, S. E. Baker, J. C. Frisvad, K. F. Nielsen, P. J. Punt and A. F. J. Ram, G3: Genes, Genomes, Genet., 2016, 6, 193–204 CrossRef CAS PubMed
- T. Linde, M. Zoglowek, M. Lübeck, J. C. Frisvad and P. S. Lübeck, J. Ind. Microbiol. Biotechnol., 2016, 43, 1139–1147 CrossRef CAS PubMed
- A. Klitgaard, R. J. N. Frandsen, D. K. Holm, P. B. Knudsen, J. C. Frisvad and K. F. Nielsen, J. Nat. Prod., 2015, 78, 1518–1525 CrossRef CAS PubMed
- S. Theobald, T. C. Vesth, J. K. Rendsvig, K. F. Nielsen, R. Riley, L. M. de Abreu, A. Salamov, J. C. Frisvad, T. O. Larsen, M. R. Andersen and J. B. Hoof, Sci. Rep., 2018, 8, 1–12 Search PubMed
- M. A. S. Lima., M. da C. F. de. Oliveira, A. T. Á. Pimenta and P. K. S. Uchôa, J. Braz. Chem. Soc., 2019, 30, 2029–2259 CAS
- K. F. Nielsen, J. M. Mogensen, M. Johansen, T. O. Larsen and J. C. Frisvad, Anal. Bioanal. Chem., 2009, 395, 1225–1242 CrossRef CAS PubMed
- M. R. Andersen, M. L. Nielsen and J. Nielsen, Mol. Syst. Biol., 2008, 4, 178 CrossRef PubMed
- J. Brandl, M. V. Aguilar-pontes, P. Schäpe, A. Noerregaard, M. Arvas, A. F. J. Ram, V. Meyer, A. Tsang, R. P. de Vries and M. R. Andersen, Fungal Biol. Biotechnol., 2018, 5, 1–12 CrossRef PubMed
- K. M. N. Burgess, J. B. Renaud, T. McDowell and M. W. Sumarah, ACS Chem. Biol., 2016, 11, 2618–2625 CrossRef CAS PubMed
- S. Yu, Y. X. Zhu, C. Peng and J. Li, Nat. Prod. Res., 2019, 1–8 Search PubMed
- L. M. Petersen, J. C. Frisvad, P. B. Knudsen, M. Rohlfs, C. H. Gotfredsen and T. O. Larsen, J. Antibiot., 2015, 68, 603–608 CrossRef CAS PubMed
- L. M. Petersen, C. Hoeck, J. C. Frisvad, C. H. Gotfredsen and T. O. Larsen, Molecules, 2014, 19, 10898–10921 CrossRef PubMed
- J. Rabenstein, G. Davis and D. D. Baker, J. Antibiot., 2000, 53, 110–113 CrossRef PubMed
- S. P. B. Ovenden, G. Sberna, R. M. Tait, H. G. Wildman, R. Patel, B. Li, K. Steffy, N. Nguyen and B. M. Meurer-Grimes, J. Nat. Prod., 2004, 67, 2093–2095 CrossRef CAS PubMed
- L. M. Petersen, T. T. Bladt, C. Dürr, M. Seiffert, J. C. Frisvad, C. H. Gotfredsen and T. O. Larsen, Molecules, 2014, 19, 9786–9797 CrossRef PubMed
- J. Zhan, G. M. K. B. Gunaherath, E. M. K. Wijeratne and A. A. L. Gunatilaka, Phytochemistry, 2007, 68, 368–372 CrossRef CAS PubMed
- X. He, B. Liu, G. Wang, X. Wang, L. Su, G. Qu and X. Yao, J. Steroid Biochem. Mol. Biol., 2006, 100, 87–94 CrossRef CAS PubMed
- D. H. Li, T. Han, L. P. Guan, J. Bai, N. Zhao, Z. L. Li, X. Wu and H. M. Hua, Nat. Prod. Res., 2016, 30, 1116–1122 CrossRef CAS PubMed
- R. Andersen, G. Büchi, B. Kobbe and A. L. Demain, J. Org. Chem., 1977, 42, 352–353 CrossRef CAS PubMed
- S. Ates, S. Ozenir and M. Gökdere, Appl. Biochem. Microbiol., 2006, 42, 500–501 CrossRef CAS
- L. M. Petersen, S. Kildgaard, M. Jaspars and T. O. Larsen, Tetrahedron Lett., 2015, 56, 1847–1850 CrossRef CAS
- M. E. Futyma, Y. Guo, C. Hoeck, J. B. Hoof, C. H. Gotfredsen, U. H. Mortensen and T. O. Larsen, Appl. Microbiol. Biotechnol., 2021, 105, 5113–5121 CrossRef CAS PubMed
- Y. M. Chiang, K. M. Meyer, M. Praseuth, S. E. Baker, K. S. Bruno and C. C. C. Wang, Fungal Genet. Biol., 2011, 48, 430–437 CrossRef CAS PubMed
- T. R. Jørgensen, J. Park, M. Arentshorst, A. M. van Welzen, G. Lamers, P. A. vanKuyk, R. A. Damveld, C. A. M. van den Hondel, K. F. Nielsen, J. C. Frisvad and A. F. J. Ram, Fungal Genet. Biol., 2011, 48, 544–553 CrossRef PubMed
- S. Obermaier and M. Müller, Biochemistry, 2019, 58, 2589–2593 CrossRef CAS PubMed
- C. Gil Girol, K. M. Fisch, T. Heinekamp, S. Günther, W. Hüttel, J. Piel, A. A. Brakhage and M. Müller, Angew. Chem., Int. Ed., 2012, 51, 9788–9791 CrossRef CAS PubMed
- Y. Li, Y. H. Chooi, Y. Sheng, J. S. Valentine and Y. Tang, J. Am. Chem. Soc., 2011, 133, 15773–15785 CrossRef CAS PubMed
- A. O. Zabala, W. Xu, Y. H. Chooi and Y. Tang, Chem. Biol., 2012, 19, 1049–1059 CrossRef CAS PubMed
- B. Wang, X. Li, D. Yu, X. Chen, J. Tabudravu, H. Deng and L. Pan, Microbiol. Res., 2018, 217, 101–107 CrossRef CAS PubMed
- H. H. Yeh, S. L. Chang, Y. M. Chiang, K. S. Bruno, B. R. Oakley, T. K. Wu and C. C. C. Wang, Org. Lett., 2013, 15, 756–759 CrossRef CAS PubMed
- S. S. Gao, A. Duan, W. Xu, P. Yu, L. Hang, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2016, 138, 4249–4259 CrossRef CAS PubMed
- L. M. Petersen, D. K. Holm, C. H. Gotfredsen, U. H. Mortensen and T. O. Larsen, ChemBioChem, 2015, 16, 2200–2204 CrossRef CAS PubMed
- A. Susca, R. H. Proctor, R. A. E. Butchko, M. Haidukowski, G. Stea, A. Logrieco and A. Moretti, Fungal Genet. Biol., 2014, 73, 39–52 CrossRef CAS PubMed
- J. M. Mogensen, J. C. Frisvad, U. Thrane and K. F. Nielsen, J. Agric. Food Chem., 2010, 58, 954–958 CrossRef CAS PubMed
- R. Gerber, L. Lou, J. Huffman, X. Zhu, T. Lin, L. Q. Li, I. Arreguin, R. A. E. Butchko, R. H. Proctor and L. Du, ACS Symp. Ser., 2009, 1031, 167–182 CrossRef CAS
- Q. Yue, L. Chen, X. Zhang, K. Li, J. Sun, X. Liu, Z. An and G. F. Bills, Eukaryotic Cell, 2015, 14, 698–718 CrossRef CAS PubMed
- W. Huttel, L. Youssar, B. A. Grüning, S. Günther and K. G. Hugentobler, BMC Genomics, 2016, 17, 1–8 CrossRef PubMed
- E. Geib, F. Baldeweg, M. Doerfer, M. Nett and M. Brock, Cell Chem. Biol., 2019, 26, 223–234 CrossRef CAS PubMed
- Y. Zhang, X. M. Li, P. Proksch and B. G. Wang, Steroids, 2007, 72, 723–727 CrossRef CAS PubMed
- W. Qiao, T. Tang and F. Ling, Sci. Rep., 2020, 10, 1–11 CrossRef PubMed
- T. Awakawa, X. L. Yang, T. Wakimoto and I. Abe, ChemBioChem, 2013, 14, 2095–2099 CrossRef CAS PubMed
- T. Yamamoto, Y. Tsunematsu, H. Noguchi, K. Hotta and K. Watanabe, Org. Lett., 2015, 17, 4992–4995 CrossRef CAS PubMed
- M. C. Tang, Y. Zou, D. Yee and Y. Tang, AIChE J., 2018, 64, 4182–4186 CrossRef CAS PubMed
- X. Wei, L. Chen, J. W. Tang and Y. Matsuda, Front. Microbiol., 2020, 11, 1–7 CrossRef PubMed
- A. Gallo, K. S. Bruno, M. Solfrizzo, G. Perrone, G. Mulè, A. Visconti and S. E. Baker, Appl. Environ. Microbiol., 2012, 78, 8208–8218 CrossRef CAS PubMed
- Y. Wang, L. Wang, F. Wu, F. Liu, Q. Wang, X. Zhang, N. Selvaraj, Y. Zhao, F. Xing, W-B. Yin and Y. Liu, Appl. Environ. Microbiol., 2018, 84, e01009-18 CrossRef PubMed
- P. B. Wolff, M. L. Nielsen, J. C. Slot, L. N. Andersen, L. M. Petersen, T. Isbrandt, D. K. Holm, U. H. Mortensen, C. S. Nødvig, T. O. Larsen and J. B. Hoof, Fungal Genet. Biol., 2020, 139, 103378 CrossRef CAS PubMed
- D. K. Holm, L. M. Petersen, A. Klitgaard, P. B. Knudsen, Z. D. Jarczynska, K. F. Nielsen, C. H. Gotfredsen, T. O. Larsen and U. H. Mortensen, Chem. Biol., 2014, 21, 519–529 CrossRef CAS PubMed
- T. S. Bugni, D. Abbanat, V. S. Bernan, W. M. Maiese, M. Greenstein, R. M. Van Wagoner and C. M. Ireland, J. Org. Chem., 2000, 65, 7195–7200 CrossRef CAS PubMed
- C. F. Lee, L. X. Chen, C. Y. Chiang, C. Y. Lai and H. C. Lin, Angew. Chem., Int. Ed., 2019, 58, 18414–18418 CrossRef CAS PubMed
- S. Palys, T. T. M. Pham and A. Tsang, Front. Microbiol., 2020, 11, 1378 CrossRef PubMed
- L. Liu, Z. Zhang, C. L. Shao and C. Y. Wang, Front. Microbiol., 2017, 8, 1–13 Search PubMed
- E. Choque, Y. El Rayess, J. Raynal and F. Mathieu, Appl. Microbiol. Biotechnol., 2015, 99, 1081–1096 CrossRef CAS PubMed
- A. Watanabe, I. Fujii, H. F. Tsai, Y. C. Chang, K. J. Kwon-Chung and Y. Ebizuka, FEMS Microbiol. Lett., 2000, 192, 39–44 CrossRef CAS PubMed
- I. Fujii, Y. Yasuoka, H. F. Tsai, Y. C. Chang, K. J. Kwon-Chung and Y. Ebizukal, J. Biol. Chem., 2004, 279, 44613–44620 CrossRef CAS PubMed
- S. E. Baker, Med. Mycol., 2006, 44, 17–21 CrossRef PubMed
- G. Büchi, D. H. Klaubert, S. M. Weinreb, K. C. Shank, G. N. Wogan, G. Büchi, D. H. Klaubert, S. M. Weinreb, K. C. Shank and G. N. Wogan, J. Org. Chem., 1971, 36, 1143–1147 CrossRef PubMed
- G. Büchi, K. C. Luk, B. Kobbe and J. M. Townsend, J. Org. Chem., 1977, 42, 244–246 CrossRef PubMed
- G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed
- L. S. Mazzaferro, W. Hüttel, A. Fries and M. Müller, J. Am. Chem. Soc., 2015, 137, 12289–12295 CrossRef CAS PubMed
- K. Kodukula, M. Arcuri, J. Q. Cutrone, R. M. Hugill, S. E. Lowe, D. M. Pirnik and Y. Shu, J. Antibiot., 1995, 48, 1055–1059 CrossRef CAS PubMed
- P. B. Fernandes, R. Seethala, J. Q. Cutrone, R. M. Hugill, S. E. Lowe, D. M. Pirnik and Y. Z. Shu, J. Antibiot., 1995, 48, 1055–1059 CrossRef PubMed
- J. F. Sanchez, A. D. Somoza, N. P. Keller and C. C. C. Wang, Nat. Prod. Rep., 2012, 29, 351–371 RSC
- H. Zhou, K. Qiao, Z. Gao, M. J. Meehan, J. W. H. Li, X. Zhao, P. C. Dorrestein, J. C. Vederas and Y. Tang, J. Am. Chem. Soc., 2010, 132, 4530–4531 CrossRef CAS PubMed
- Y. M. Chiang, E. Szewczyk, A. D. Davidson, N. Keller, B. R. Oakley and C. C. C. Wang, J. Am. Chem. Soc., 2009, 131, 2965–2970 CrossRef CAS PubMed
- S. M. Ma, J. W. H. Li, J. W. Choi, H. Zhou, K. K. M. Lee, V. A. Moorthie, X. Xie, J. T. Kealey, N. A. Da Silva, J. C. Vederas and Y. Tang, Science, 2009, 326, 589–592 CrossRef CAS PubMed
- N. Osmanova, W. Schultze and N. Ayoub, Phytochem. Rev., 2011, 10, 315–342 Search PubMed
- J. Romsdahl and C. C. C. Wang, MedChemComm, 2019, 10, 840–866 RSC
- B. Wang, X. Li, D. Yu, X. Chen, J. Tabudravu, H. Deng and L. Pan, Microbiol. Res., 2018, 217, 101–107 CrossRef CAS PubMed
- M. J. Koetsier, P. A. Jekel, M. A. van den Berg, R. A. L. Bovenberg and D. B. Janssen, Biochem. J., 2009, 417, 467–476 CrossRef CAS PubMed
-
R. J. Cox and E. J. Skellam, Fungal Non-Reducing Polyketide Synthases, Elsevier Ltd., 3rd edn, 2020, vol. 1, pp. 266–312 Search PubMed
- J. Inokoshi, K. Shiomi, R. Masuma, H. Tanaka, H. Yamada and S. Omura, J. Antibiot., 1999, 52, 1095–1100 CrossRef CAS PubMed
- Y. H. Chooi, C. Krill, R. A. Barrow, S. Chen, R. Trengove, R. P. Oliver and P. S. Solomon, Appl. Environ. Microbiol., 2015, 81, 177–186 CrossRef PubMed
- B. G. Hansen, B. Salomonsen, M. T. Nielsen, J. B. Nielsen, N. B. Hansen, K. F. Nielsen, T. B. Regueira, J. Nielsen, K. R. Patil and U. H. Mortensen, Appl. Environ. Microbiol., 2011, 77, 3044–3051 CrossRef CAS PubMed
- T. R. Jørgensen, K. F. Nielsen, M. Arentshorst, J. H. Park, C. A. van den Hondel, J. C. Frisvad and A. F. Ram, Appl. Environ. Microbiol., 2011, 77, 5270–5277 CrossRef PubMed
-
E. Bin Go and Y. Tang, Fungal Highly-Reducing Polyketide Synthases and Associated Natural Products, Elsevier Ltd., 3rd edn, 2020, vol. 1, pp. 333–364 Search PubMed
- W. C. A. Gelderblom, K. Jaskiewicz, W. F. O. Marasas, P. G. Thiel, R. M. Horak, R. Vleggaar and N. P. J. Kriek, Appl. Environ. Microbiol., 1988, 54, 1806–1811 CrossRef CAS PubMed
- A. Gallo, M. Ferrara and G. Perrone, Toxins, 2013, 5, 717–742 CrossRef CAS PubMed
- T. Bartók, Á. Szécsi, A. Szekeres, Á. Mesterházy and M. Bartók, Rapid Commun. Mass Spectrom., 2006, 20, 2447–2462 CrossRef PubMed
- M. Månsson, M. L. Klejnstrup, R. K. Phipps, K. F. Nielsen, J. C. Frisvad, C. H. Gotfredsen and T. O. Larsen, J. Agric. Food Chem., 2010, 58, 949–953 CrossRef PubMed
-
J. C. Frisvad, T. Isbrandt and T. O. Larsen, Fungal Partially Reducing Polyketides and Related Natural Products From Aspergillus, Penicillium, and Talaromyces, Elsevier Ltd., 3rd edn, 2020, vol. 1, pp. 313–332 Search PubMed
- A. E. Desjardins and R. H. Proctor, Int. J. Food Microbiol., 2007, 119, 47–50 CrossRef CAS PubMed
- K. Shimizu, H. Nakagawa, R. Hashimoto, D. Hagiwara, Y. Onji, K. Asano, S. Kawamoto, H. Takahashi and K. Yokoyama, Mycoscience, 2015, 56, 301–308 CrossRef CAS
- I. Abe and H. Morita, Nat. Prod. Rep., 2010, 27, 809–838 RSC
- G. Bills, Y. Li, L. Chen, Q. Yue, X. M. Niu and Z. An, Nat. Prod. Rep., 2014, 31, 1348–1375 RSC
- R. D. Süssmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770–3821 CrossRef PubMed
- X. Gao, S. W. Haynes, B. D. Ames, P. Wang, L. P. Vien, C. T. Walsh and Y. Tang, Nat. Chem. Biol., 2012, 8, 823–830 CrossRef CAS PubMed
- N. Kato, H. Suzuki, H. Takagi, Y. Asami, H. Kakeya, M. Uramoto, T. Usui, S. Takahashi, Y. Sugimoto and H. Osada, ChemBioChem, 2009, 10, 920–928 CrossRef CAS PubMed
- G. A. Payne, W. C. Nierman, J. R. Wortman, B. L. Pritchard, D. Brown, R. A. Dean, D. Bhatnagar, T. E. Cleveland, M. Machida and J. Yu, Med. Mycol., 2006, 44, 9–11 CrossRef PubMed
- K. Mizuno, A. Yagi, S. Satoi, M. Takada, T. Matsuda, M. Hayashi and K. Asano, J. Antibiot., 1977, 30, 297–302 CrossRef CAS PubMed
- R. A. Cacho, W. Jiang, Y. H. Chooi, C. T. Walsh and Y. Tang, J. Am. Chem. Soc., 2012, 134, 16781–16790 CrossRef CAS PubMed
- W. Jiang, R. A. Cacho, G. Chiou, N. K. Garg, Y. Tang and C. T. Walsh, J. Am. Chem. Soc., 2013, 135, 4457–4466 CrossRef CAS PubMed
- F. Zhang, H. Liu, T. Zhang, T. Pijning, L. Yu, W. Zhang, W. Liu and X. Meng, Appl. Microbiol. Biotechnol., 2018, 102, 7877–7890 CrossRef CAS PubMed
- W. Hüttel, Appl. Microbiol. Biotechnol., 2021, 105, 55–66 CrossRef PubMed
- M. Yukioka and T. Winnick, J. Bacteriol., 1966, 91, 2237–2244 CrossRef CAS PubMed
- Q. W. Tan, F. L. Gao, F. R. Wang and Q. J. Chen, Int. J. Mol. Sci., 2015, 16, 5750–5761 CrossRef CAS PubMed
- H. Wang, K. Sivonen and D. P. Fewer, Curr. Opin. Genet. Dev., 2015, 35, 79–85 CrossRef CAS PubMed
- E. Hühner, K. Öqvist and S. M. Li, Org. Lett., 2019, 21, 498–502 CrossRef PubMed
- C. Verrier, S. Moebs-Sanchez, Y. Queneau and F. Popowycz, Org. Biomol. Chem., 2018, 16, 676–687 RSC
- C. Piutti and F. Quartieri, Molecules, 2013, 18, 12290–12312 CrossRef CAS PubMed
-
C. Schmidt-Dannert, Biosynthesis of Terpenoid Natural Products in Fungi, Springer, 2014, pp. 19–61 Search PubMed
- M. B. Quin, C. M. Flynn and C. Schmidt-Dannert, Nat. Prod. Rep., 2014, 31, 1449–1473 RSC
- L. G. Bondarenko-gheorghiu, J. Serb. Chem. Soc., 2000, 65, 147–156 CrossRef CAS
- X. Zhou, H. Zhu, L. Liu, J. Lin and K. Tang, Appl. Microbiol. Biotechnol., 2010, 86, 1707–1717 CrossRef CAS PubMed
- W. Qiao, F. Ling, L. Yu, Y. Huang and T. Wang, Fungal Biol., 2017, 121, 1037–1044 CrossRef CAS PubMed
- R. Croteau, R. E. B. Ketchum, R. M. Long, R. Kaspera and M. R. Wildung, Phytochem. Rev., 2006, 5, 75–97 CrossRef CAS PubMed
- R. Riko, H. Nakamura and K. Shindo, J. Antibiot., 2014, 67, 179–181 CrossRef CAS PubMed
- J. Hiort, K. Maksimenka, M. Reichert, S. Perović-Ottstadt, W. H. Lin, V. Wray, K. Steube, K. Schaumann, H. Weber, P. Proksch, R. Ebel, W. E. G. Müller and G. Bringmann, J. Nat. Prod., 2005, 68, 1821 CrossRef CAS
- G. Schlingmann, T. Taniguchi, H. He, R. Bigelis, H. Y. Yang, F. E. Koehn, G. T. Carter and N. Berova, J. Nat. Prod., 2007, 70, 1180–1187 CrossRef CAS PubMed
- M. Johnson, I. Zaretskaya, Y. Raytselis, Y. Merezhuk, S. McGinnis and T. L. Madden, Nucleic Acids Res., 2008, 36, 5–9 CrossRef PubMed
- M. Yokoyama, Y. Hirayama, T. Yamamoto, S. Kishimoto, Y. Tsunematsu and K. Watanabe, Org. Lett., 2017, 19, 2002–2005 CrossRef CAS PubMed
- L. H. Meng, X. M. Li, Y. Liu and B. G. Wang, Chin. Chem. Lett., 2015, 26, 610–612 CrossRef CAS
- R. Geisen, Z. Mayer, A. Karolewiez and P. Färber, Syst. Appl. Microbiol., 2004, 507, 501–507 CrossRef PubMed
- A. Karolewiez and R. Geisen, Syst. Appl. Microbiol., 2005, 28, 588–595 CrossRef CAS PubMed
- A. Gallo, B. P. Knox, K. S. Bruno, M. Solfrizzo, S. E. Baker and G. Perrone, Int. J. Food Microbiol., 2014, 179, 10–17 CrossRef CAS PubMed
- J. O'Callaghan, A. Coghlan, A. Abbas, C. García-Estrada, J. F. Martín and A. D. W. Dobson, Int. J. Food Microbiol., 2013, 161, 172–181 CrossRef PubMed
- J. O'Callaghan, M. X. Caddick and A. D. W. Dobson, Microbiology, 2003, 149, 3485–3491 CrossRef PubMed
- A. Abbas, H. Valez and A. D. W. Dobson, Int. J. Food Microbiol., 2009, 135, 22–27 CrossRef CAS PubMed
- L. M. Petersen, D. K. Holm, P. B. Knudsen, K. F. Nielsen, C. H. Gotfredsen, U. H. Mortensen and T. O. Larsen, J. Antibiot., 2015, 68, 201–205 CrossRef CAS PubMed
- Y. Matsuda and I. Abe, Nat. Prod. Rep., 2016, 33, 26–53 RSC
- H. Nordberg, M. Cantor, S. Dusheyko, S. Hua, A. Poliakov, I. Shabalov, T. Smirnova, I. V. Grigoriev and I. Dubchak, Nucleic Acids Res., 2014, 42, 26–31 CrossRef PubMed
- G. Mondai, P. Dureja and B. Sen, Indian J. Exp. Biol., 2000, 38, 84–87 Search PubMed
- L. Koch, A. Lodin, I. Herold, M. Ilan, S. Carmeli and O. Yarden, Mar. Drugs, 2014, 12, 4713–4731 CrossRef CAS PubMed
- M. H. Medema, T. de Rond and B. S. Moore, Nat. Rev. Genet., 2021, 22, 553–571 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2023 |