
Deciphering chemical logic of fungal natural product biosynthesis through heterologous expression and genome mining
Chen-Yu
Chiang
a,
Masao
Ohashi
 *a and
Yi
Tang
*ab
*a and
Yi
Tang
*ab
aDept. of Chemical and Biomolecular Engineering, 5531 Boelter Hall, 420 Westwood Plaza, Los Angeles, CA 90095, USA. E-mail: yitang@ucla.edu; gph422001@ucla.edu
bDept. of Chemistry and Biochemistry, 5531 Boelter Hall, 420 Westwood Plaza, Los Angeles, CA 90095, USA
First published on 20th September 2022
Abstract
Covering: 2010 to 2022
Heterologous expression of natural product biosynthetic gene clusters (BGCs) has become a widely used tool for genome mining of cryptic pathways, bottom-up investigation of biosynthetic enzymes, and engineered biosynthesis of new natural product variants. In the field of fungal natural products, heterologous expression of a complete pathway was first demonstrated in the biosynthesis of tenellin in Aspergillus oryzae in 2010. Since then, advances in genome sequencing, DNA synthesis, synthetic biology, etc. have led to mining, assignment, and characterization of many fungal BGCs using various heterologous hosts. In this review, we will highlight key examples in the last decade in integrating heterologous expression into genome mining and biosynthetic investigations. The review will cover the choice of heterologous hosts, prioritization of BGCs for structural novelty, and how shunt products from heterologous expression can reveal important insights into the chemical logic of biosynthesis. The review is not meant to be exhaustive but is rather a collection of examples from researchers in the field, including ours, that demonstrates the usefulness and pitfalls of heterologous biosynthesis in fungal natural product discovery.
 Chen-Yu Chiang | Chen-Yu Chiang received his B.S. in Chemistry from National Taiwan University in 2019. He is currently working toward his PhD under the guidance of Prof. Yi Tang at the University of California, Los Angeles. His research interests focus on discovering new enzymes in the biosynthetic pathway. |
 Masao Ohashi | Masao Ohashi received his PhD in 2015 in Medicinal Chemistry from Okayama University, Japan. In 2015, he joined the Department of Pharmaceutical Sciences at the University of Shizuoka as a designated assistant professor. In 2016, he joined Prof. Yi Tang's lab as a postdoctoral scholar at the University of California, Los Angeles. He is currently an assistant project scientist in the Tang lab, and his current research interests focus on the identification of new enzymes that catalyze unusual reactions in nature. |
 Yi Tang | Yi Tang received his undergraduate degree in Chemical Engineering and Material Science from Penn State University. He received his PhD in Chemical Engineering from California Institute of Technology in 2002. After NIH postdoctoral training in Chemical Biology at Stanford University, he started his independent career at the University of California Los Angeles in 2004. He is currently the Ralph M. Parsons Foundation Chair Professor in Department of Chemical and Biomolecular Engineering; and Professor in the Department of Chemistry and Biochemistry. His lab is interested in natural product biosynthesis, biocatalysis and protein engineering. |
1. Introduction
Genome mining, a term first mentioned in the context of natural products in 2005,1 has brought a renaissance to the research fields of natural product discovery, biosynthesis and chemical biology. More recently, the marriage of biosynthesis and synthetic biology has further broadened interests in natural products. Advances in next-generation DNA sequencing, DNA synthesis and gene editing technologies have rapidly enhanced our abilities to identify, construct and characterize biosynthetic gene clusters (BGCs). Among the different approaches to mine new natural products or rediscover biological activities of known compounds, heterologous expression of BGCs in model organisms is a key strategy.2,3 Heterologous expression of BGCs, coupled with host engineering, has been successfully demonstrated for nearly all major families of natural products from bacterial, fungal, plant and animal pathways.4–7 Our lab has been involved in the genome mining of natural products from filamentous fungi in the last decade. Genome sequencing of Ascomycota, such as Penicillium and Aspergillus species, have revealed a significant amount of BGCs are unexplored and have no associated metabolites.8,9 It is estimated that over 97% of fungal BGCs are not associated with known natural products, a number that is in line with that estimated for bacterial BGCs.10,11In this review, we highlight a number of examples from ours and other researchers in genome mining of fungal BGCs. In Section 2, we describe the general workflow of BGC refactoring and expression in different heterologous hosts. Several commonly used hosts, such as Aspergillus nidulans, Aspergillus oryzae, and Saccharomyces cerevisiae are discussed with representative examples. Considerations to increase successes of reconstitution are discussed. In Section 3, we focus on strategies to expand natural product chemical space through mining of BGCs with unique features. In Section 4, we recount several case studies in which intermediates and shunt products from heterologous reconstitution led to new discoveries of biosynthetic logic. Collectively, this review illustrates the successes and failures of genome mining in fungi, and underscores the vast unexplored biosynthetic potential in this kingdom of eukaryotic organisms.
2. Overview of heterologous expression of fungal natural products
2.1. Why heterologous expression?
While the focus of this review is on the use of heterologous hosts for fungal BGC expression, the importance of working with native producing strains cannot be overstated. If the original producing host can be sourced from the many different collections around the world, working directly in a genome-sequenced strain has unmatched advantages. First, the strain can be grown under a plethora of media conditions to produce different metabolic profiles. This is the “one strain many compounds” (OSMAC) strategy that can activate a subset of BGCs.12–14 Second, the compounds produced in a native host are highly likely to be the final natural product of the pathway; and may be produced at sufficient titers for full structural characterization. This is especially important in cases where the BGC is too large to be refactored using heterologous host/vector systems, or has ambiguous definitions of cluster boundaries.15 Lastly, if the fungal strain can be genetically modified, many tools can be directly applied to the native host including BGC-specific transcriptional factor (TF) overexpression,16–19 global epigenetic modifications,20,21 CRISPR-Cas9 mediated pathway activation,22 and top-down genetic inactivation to assign functions to individual pathway genes (Fig. 1).23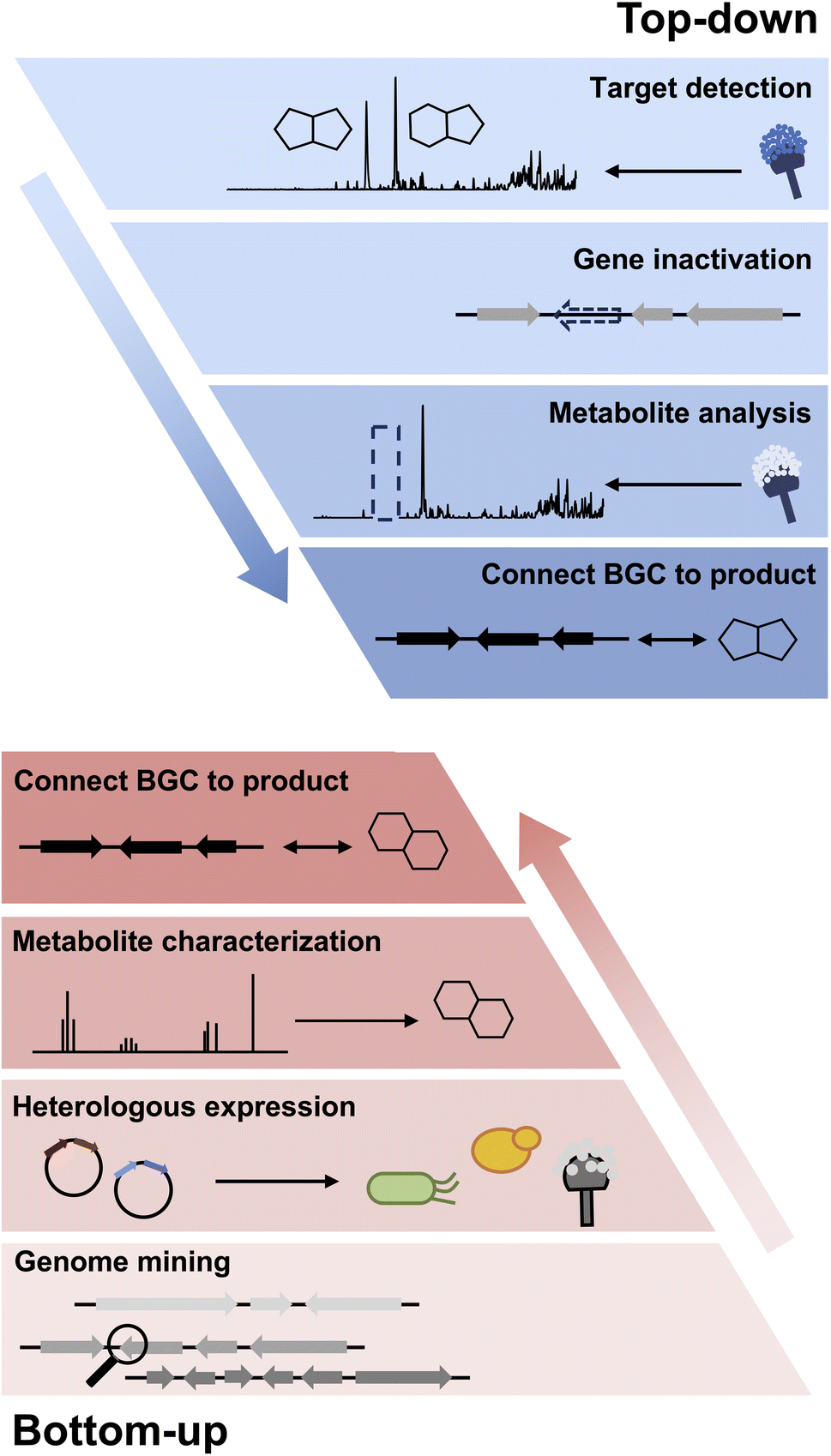 | ||
| Fig. 1 Top-down and bottom-up approaches in studying biosynthesis. | ||
Clearly, if the native strain cannot be sourced or is genetically intractable, transplanting the BGC of interest into a chassis organism for heterologous expression is the only alternative. Heterologous hosts are genetically well-characterized with robust molecular biology tools. An ideal heterologous host is also fast growing and has a minimal metabolic background. For bacterial BGCs, E. coli and different Streptomyces organisms are used frequently.24–27 For plant BGCs, Saccharomyces cerevisiae (Baker's yeast, also referred to as yeast throughout the review) is the go-to microbial chassis,28 while the tobacco plant Nicotiana benthamiana is the most used plant heterologous host.29 For fungi, a number of strains are available, including Saccharomyces cerevisiae and well-characterized Aspergillus sp. that are discussed in Section 2.2. Several other fungal species have also been reported to be useful heterologous hosts.30,31
Even when the native fungal host can be genetically manipulated, heterologous expression offers several enabling advantages in analysis of BGCs. First, with a reduced metabolic background and rewired precursor fluxes dedicated to heterologously expressed pathways, the titers of the target metabolite may be engineered to be significantly higher than from the native host. However, metabolite titers in a heterologous host can be highly variable and depend on the BGC of interest. Titers from less than 1 mg L−1 to well over 1 g L−1 have been reported under similar growth conditions for different BGCs. The exact reasons for such variation are not well-understood; and empirical methods are used to optimize the titers. For the purpose of this review, titers are generally not discussed, and readers should refer to the primary literature for such information. Second, many BGCs in native hosts cannot be transcriptionally activated despite the myriad of tools mentioned above. Through complete refactoring of BGCs for heterologous expression, such cryptic regulation can be bypassed. Third, if the native strain is difficult to obtain due to logistical reasons, reconstitution of homologous BGC from accessible organisms can overcome such hurdles. For example, in our effort to study sambutoxin (1) biosynthesis, the producing strain Fusarium sambucinum was not obtainable.32 Comparison of candidate BGCs revealed the wide-spread presence of the candidate BGC in other species, including Fusarium oxysporum of which the genomic DNA can be obtained. Heterologous reconstitution of the six genes in the BGC in Aspergillus nidulans confirmed the cluster indeed produced sambutoxin. Similarly, the biosynthesis of the potent oxidative phosphorylation inhibitor ilicicolin H (2) was reconstituted in A. nidulans by expressing a homologous BGC from Penicillium variabile, which was not known to be a producer prior to the study.33 Lastly, with a modular refactoring approach, mix-and-matching of pathway genes between target BGC and homologous BGCs can be readily performed in heterologous hosts. This can overcome expression or pathway flux bottleneck attributed to one particular enzyme from a BGC, as well as to generate structural variants of natural products (Scheme 1).34,35
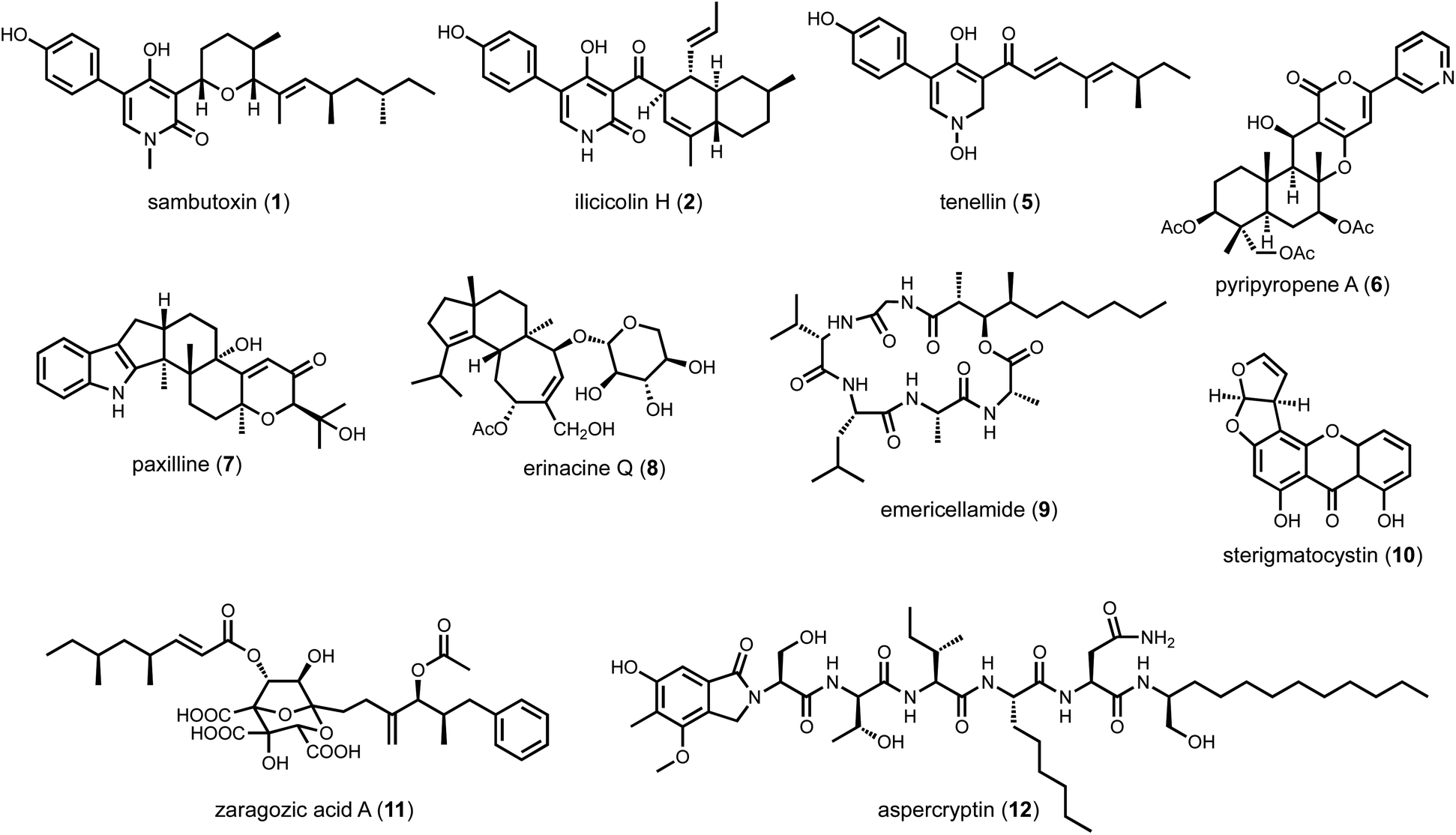 | ||
| Scheme 1 Metabolites discussed in Section 2. | ||
Heterologous reconstitution enables the gene-by-gene, bottom-up approach to examine individual steps of biosynthesis and allows functional association between BGCs and natural products in an unambiguous way (Fig. 1). While the genetic inactivation-based, top-down approach in native hosts can be highly informative, several examples of misinterpretation of enzyme function have been noted in the literature due to pleotropic effects of gene inactivation. We note two examples here, in which subsequent heterologous reconstitution studies led to revision of initial BGC assignments. In the first example, Chankhamjon et al. identified AoiQ as the halogenase responsible for biosynthesis of 8-methyldichlorodiaporthin (3) from Aspergillus oryzae. RIB40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 363 is a coumarin-containing polyketide with a gem-dichlorinated sp3 carbon α to a secondary alcohol. Based on genetic knockout in the native host, the authors concluded an unclustered, nonreducing polyketide synthase (NRPKS) AoiG is involved in biosynthesis of the unchlorinated substrate. This led to the speculation that AoiQ, a flavin-dependent halogenase, can chlorinate an unactivated sp3 carbon via an unprecedented radical mechanism. Subsequent heterologous expression work, however, showed a different NRPKS DiaA, which is clustered with AoiQ, is involved in synthesizing the coumarin core.37 DiaA synthesizes a 1,3-diketone containing substrate that can be halogenated by AoiQ with canonical flavin-dependent halogenation mechanism via an enolate intermediate. Nonenzymatic deacetylation and short-chain dehydrogenase/reductase (SDR)-catalyzed ketoreduction afforded dichlorodiaporthin 3 (Fig. 2A).
363 is a coumarin-containing polyketide with a gem-dichlorinated sp3 carbon α to a secondary alcohol. Based on genetic knockout in the native host, the authors concluded an unclustered, nonreducing polyketide synthase (NRPKS) AoiG is involved in biosynthesis of the unchlorinated substrate. This led to the speculation that AoiQ, a flavin-dependent halogenase, can chlorinate an unactivated sp3 carbon via an unprecedented radical mechanism. Subsequent heterologous expression work, however, showed a different NRPKS DiaA, which is clustered with AoiQ, is involved in synthesizing the coumarin core.37 DiaA synthesizes a 1,3-diketone containing substrate that can be halogenated by AoiQ with canonical flavin-dependent halogenation mechanism via an enolate intermediate. Nonenzymatic deacetylation and short-chain dehydrogenase/reductase (SDR)-catalyzed ketoreduction afforded dichlorodiaporthin 3 (Fig. 2A).
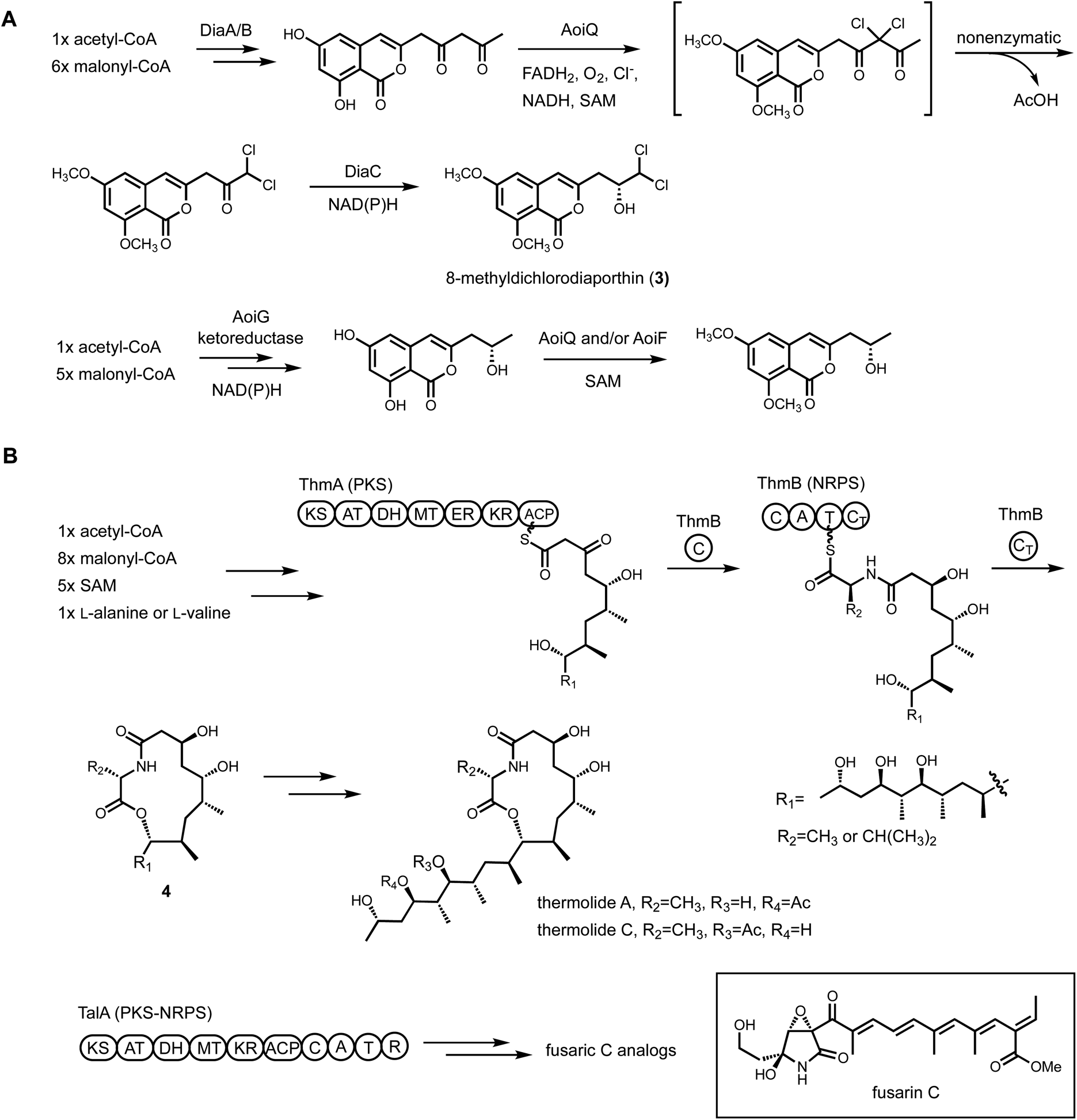 | ||
| Fig. 2 Cluster reassignment using heterologous expression of BGCs. (A) Revised biosynthetic pathway of dichlorodiaporthins. (B) Revised biosynthetic pathway of thermolides. | ||
The second example of misinterpretation of gene deletion results is that of thermolide biosynthesis.38 Thermolides are a class of nematocidal natural products isolated from the thermophilic fungus Talaromyces thermophilus NRRL2155, featuring an uncommon macrolactone linkage between the polyketide and nonribosomal peptide portions. Inactivation of the polyketide synthase-nonribosomal peptide synthetase (PKS-NRPS) TalA in the native producing organism abolished the production of thermolides. This led to the putative assignment of TalA as the scaffold-generating enzyme, although the predicted functions of remaining enzymes in the tal BGC were not consistent with thermolide structural features. The Zou lab subsequently performed heterologous expression of all candidate BGCs in A. nidulans to functionally characterize the PKS-NRPS. They demonstrated that the tal BGC was in fact responsible for biosynthesis of analogs of fusarin C. The bona fide thermolide BGC encodes a PKS (ThmA) and an NRPS (ThmB) as two separate proteins, with a terminal C domain (CT) in the NRPS enzyme catalyzing the macrolactonization step (Fig. 2B). Coexpression of ThmA and ThmB led to the production of the macrolactone 4. The earlier gene inactivation of talA may have caused pleotropic silencing of the thermolides BGC.
2.2. Host/vector systems for heterologous expression of fungal BGCs
To date, several well-developed organisms have been used as microbial chassis for heterologous expression of fungal BGCs. Useful heterologous hosts share many advantageous features: fast growth under laboratory culturing conditions; genetical tractability; and not posing biosafety hazards when grown in large volumes. Additional considerations in choosing the model organism include (i) the number of heterologous genes that can be expressed from episomal vectors or chromosomal integrations; (ii) crosstalk with endogenous metabolism (both primary and secondary) or detoxification pathways; (iii) endogenous secondary metabolite levels that can interfere with compound identification or purification; and (iv) most importantly, abilities to correctly splice introns during mRNA maturation and perform essential post-translational modifications (PTMs) of foreign biosynthetic enzymes.High-throughput and scalable genome mining of fungal BGCs using yeast as the heterologous host was reported using the HEx platform.58 In this study, the host JHY702 was a heavily modified strain to combine the above features, including integration of npgA and atCPR, as well as modifications of genes involved in sporulation, respiratory growth, and protein expression, to improve strain robustness. Furthermore, the study examined the use of different ADH2-like promoters, which are inducible promoters activated upon glucose depletion and ethanol accumulation, from various yeast species. The sequentially divergent but functionally equivalent promoters allowed rapid pathway refactoring through yeast homologous recombination. Using HEx platform, 22 out of 41 tested BGCs from diverse fungal species produced detectable compounds, including polyketides, peptides and terpenes.
Despite the ease of use of yeast as a model host, several deficiencies have severely limited its application in genome mining. The most glaring is its inability to splice fungal introns. Although yeast has a spliceosome machinery,59,60 it is not able to recognize nor process fungal introns. This deficiency requires fungal genes with introns to be manually spliced to give uninterrupted open reading frame (ORFs) for yeast expression. As a result, accurate prediction of start codons and introns are indispensable to successful reconstitution. Often times, this would require isolation of mRNA from the native host, followed by RT-PCR and sequencing; or by first expressing the gene in a fungal heterologous host followed by mRNA isolation if the BGC of interest is silent. The limitation of intron splicing was attributed as a major reason for the failed reconstitution of several BGCs using the Hex platform.58 In addition, we and others have noted foreign P450 expression in yeast, especially multiple P450s, can be highly unpredictable and problematic.61 As a result of these limitations, the engineered yeast strains are limited to reconstitution of core biosynthetic enzymes or shorter pathways, while leaving genome mining of more complex BGCs to the filamentous fungi discussed below.
Cox and coworkers reported the first successful reconstitution a complete fungal BGC, that of tenellin (5),64 in A. oryzae M-2-3 using argB auxotroph as well as glufosinate and bleomycin resistant markers. Refactoring the cluster and placing the genes under control of inducible amyB promoters, the titer of 5 (243 mg L−1) was five times higher than that of the native host.64 In the same year, heterologous reconstitution using the same strain to study biosynthesis of fungal meroterpenoid pyripyropene A (6) was published by Abe and coworkers.65 Five biosynthetic genes were individually cloned into pTAex3 and pPTRI. Coexpression of different combinations of gene with feeding of precursors led to the discovery of early biosynthetic steps to pyripyropene A. In subsequent years, the Abe group has used A. oryzae extensively for meroterpenoid biosynthesis reconstitution.66 Stepwise reconstitution, followed by isolation and characterization of biosynthetic intermediates and shunt products enabled functional assignment of many meroterpenoid pathways. The Oikawa group has used A. oryzae to examine the biosynthesis of indole-diterpene natural products such as paxilline (7).67 One feature of the A. oryzae system is that the plasmids designed for protein expression cannot be autonomously replicated, therefore requiring all genes to be integrated into the genome for expression. As a result, colony-to-colony gene expression variation is an issue since the integrated sites can vary in each transformant. To have more control over gene integration and expression levels, Liu et al. identified high expression loci in A. oryzae, and developed a CRISPR-Cas9-based technique for targeted chromosomal integration.68 This new strategy successfully led to the reconstitution of erinacine Q (8) and its intermediates in higher titer (4.7 mg L−1 for 8).
Although Aspergillus heterologous hosts can splice most fungal introns correctly, one must remain cautious that incorrect splicing, which is detrimental to reconstitution, can occur. One published example of incorrect intron processing in A. oryzae was reported by Song et al. during the expression of a Magnaporthe oryzae PKS-NRPS encoded by ACE1.69 RT-PCR of ACE1 expression revealed that one of three introns was not spliced correctly, resulting in no production of any metabolites from the A. oryzae transformant. Further heterologous expression attempts using intron-free DNA led to detection of new metabolites, confirming incomplete splicing was the obstacle. Intron splicing is also an issue when expressing Basidiomycota genes in Aspergillus strains since genes from Basidiomycota can contain many introns. Thus, intron-free cDNAs are typically required for refactoring the corresponding pathways.70 Recently, A. oryzae was examined as a suitable host for expressing Basidiomycota diterpene genes directly using genomic DNA sequences.71 Nagamine et al. screened thirty terpene synthases from two Basidiomycota strains to show that A. oryzae can splice most of those genes correctly. In cases where partial splicing is observed, the errors were analyzed and were mostly predictable according to the authors.
One disadvantage of A. nidulans is its robust endogenous secondary metabolism. With well over fifty identified BGCs, the host can produce a significant background of compounds under laboratory cultivating conditions. Furthermore, upon introduction of the replicative-competent vectors, additional metabolic pathways can be activated which lead to emergence of new compounds that can complicate target BGC analysis. To alleviate this problem, multiple labs have engineered the host to abolish some of the most abundant endogenous metabolites. For example, our lab used CRISPR-Cas9 facilitated homologous recombination to delete easA and stcA, responsible for emericellamide (9) and sterigmatocystin (10) biosynthesis, respectively, to arrive at A. nidulans A1145ΔSTΔEM.76 These knockouts enabled the analysis of the zaragozic acid (11) pathway, of which the LC-MS features of a key intermediate were masked by the presence of sterigmatocystin prior to stcA deletion. The Wang lab reported a highly engineered strain LO8030 with deletion of eight endogenous BGCs,77 which facilitated the discovery of aspercryptin (12) as an additional endogenous metabolite in A. nidulans due to a cleaner metabolic profile. The Keller lab developed A. nidulans RJW256, which is an auxotrophic strain of pyrG89 and pyroA4 with the deletion of sterigmatocystin gene cluster, for applications in working with fungal artificial chromosomes (FACs).78,79
In addition to the three model hosts discussed above, a number of other fungal species have been evaluated and engineered to be a chassis for fungal BGC expression. These strains include Aspergillus niger,40Penicillium rubens,42Fusarium heterosporum,39Fusarium graminearum,30Trichoderma reesei31etc. While each of these strains offers some unique advantages (titer, splicing, etc.) over the Aspergillus strains for specific BGCs, they are not as widely adopted for natural product genome mining applications.
3. Examples of genome mining guided by biosynthetic gene cluster features
Most researchers performing natural product genome mining are interested in discovering one or more of the following: (1) new chemical structures that have not been previously reported; (2) natural products with new or more potent biological activities; and (3) new enzymes catalyzing synthetically challenging reactions. With an estimated more than one million BGCs from microbial genome sequences, a key step is prioritization of the clusters based on individual research objectives. The approach for finding new biological activity is certainly different from finding novel chemical scaffolds, although one would expect new activities should reside in new chemical space.With a chemocentric focus for this review, we will not elaborate on genome mining for new biological activities, except a brief detour on the topic of self-resistance gene guided search. This has been extensively reviewed by us and others recently.80,81 Briefly, the premise of this approach is that for a producing host to survive the (potent) activity of a natural product, proteins or enzymes that can confer self-resistance must be coexpressed with the pathway enzymes in the BGC. In the case of a self-resistance enzyme (SRE), it is a homolog of the housekeeping target of the natural product, but sufficiently mutated to confer resistance and keep the producing host alive. The colocalization of SRE with biosynthetic enzymes in a BGC provides a predicative window of the biological activity of the molecule encoded in the BGC; and can be leveraged to query database of BGCs for the desired biological activity. This colocalizations phenomenon is widely observed in both bacteria and fungi, and has been used to find BGCs of compounds with known activities, such as fumagillin (13),82 fellutamide (14)83 (Scheme 2A); or to assign functions to known natural product following identification of BGCs.84 One application of SRE-guide genome mining is the successful identification of a natural product inhibitor for fungal and plant dihydroxyacid dehydratase (DHAD) involved in branched chain amino acid biosynthesis (Fig. 3).57 Using yeast as the heterologous host, a terpene biosynthetic pathway encoding one terpene cyclase, two P450s and a putative self-resistant DHAD was shown to produce the submicromolar inhibitor aspterric acid (15). 15 is a promising herbicide lead and represents the first reported natural product inhibitor of DHAD. Following reconstitution in A. nidulans and identification of a SRE, the previously known fungal natural product harzianic acid (16) was shown to be an inhibitor of acetolactate synthase (ALS),85 another enzyme in the branched chain amino acid pathway. 16 binds to ALS in a different mode compared to all synthetic inhibitors, which can explain how it can evade the widely found resistance mutations. Overall, while only a small fraction of BGCs contain SREs and some putative SREs turn out to be biosynthetic rather than self-resistant, this approach offers the promise to rapidly associate biological activity to BGC-driven genome mining.
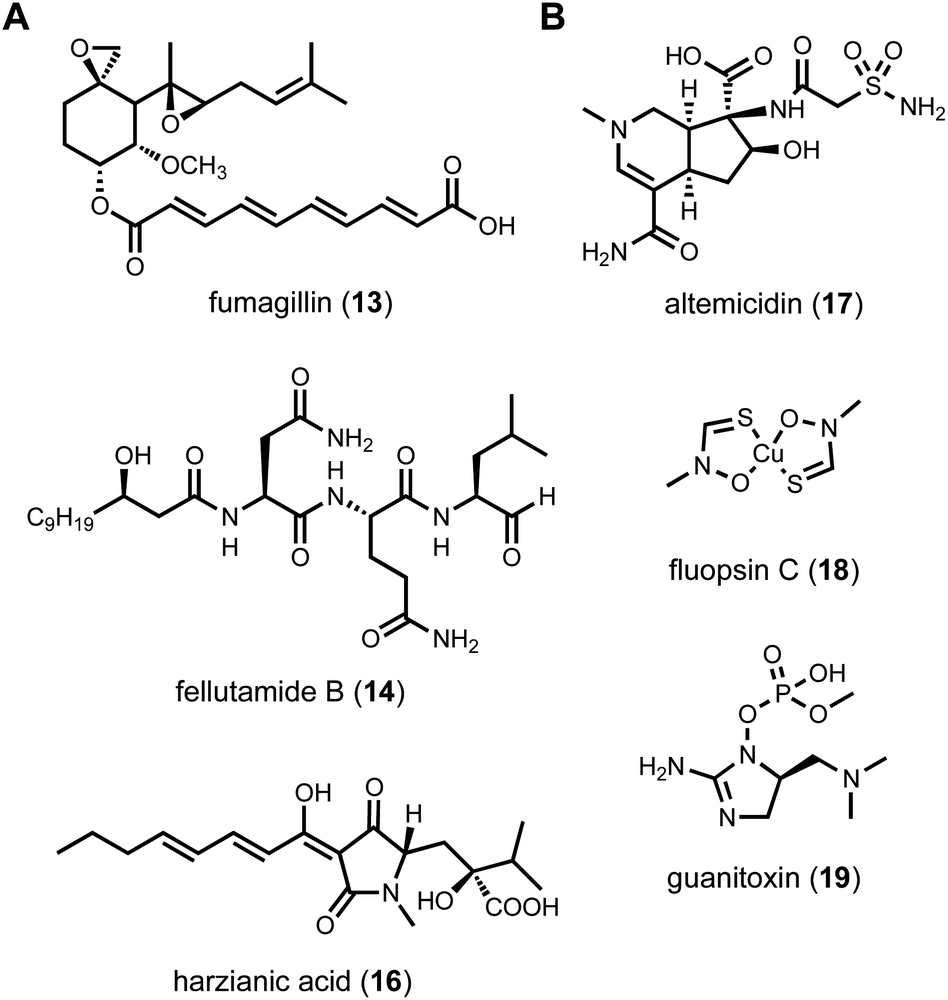 | ||
| Scheme 2 (A) Examples of metabolites found by SRE-guided genome mining. (B) Discoveries of natural product can be classified into unknown-known. | ||
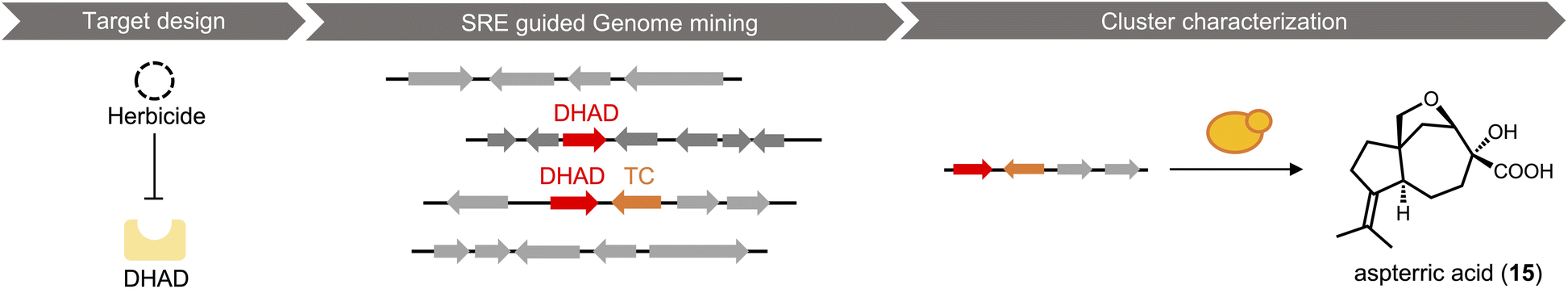 | ||
| Fig. 3 Example of Self-Resistance Enzyme (SRE)-guided genome mining. With DHAD as a target, the cluster of aspterric acid was identified through SRE-guided genome mining. The product of the cluster was revealed by heterologous expression in yeast. | ||
To prioritize genome mining towards new natural product structures and enzymes, we can consider the classification officially coined by Biermann and Helfrich,86 in which a quadrant system (2 × 2) relating natural product structures to BGCs is used (Fig. 4). Here both the BGC and natural products can be classified as either known or unknown. A known BGC refers to one that can be predicted to make a certain compound class based on the presence of a canonical core biosynthetic enzyme, such as polyketide synthase (PKS), nonribosomal peptide synthetase (NRPS), terpene synthase/terpene cyclase (TS/TC), prenyltransferase (PT), etc. These core enzymes are the basis of bioinformatics algorithms that catalog BGCs from sequenced genomes.87 An unknown BGC refers to one that has no identifiable core enzyme, and the compound type cannot be readily predicted. With regard to the natural product, known vs. unknown simply refers to whether the compound has been identified and structurally characterized. One can place most of the genome mining efforts into one of the four quadrants. Once the BGC-natural product association has been confirmed using native or heterologous host, that pair is placed in the known (BGC)-known (metabolite) category (I). If the natural product structure does not readily suggest a biosynthetic origin and the cluster is unknown, then that pair is placed into quadrant of unknown (BGC)-known (metabolite) (III). These compounds can be exciting targets to pursue new biosynthetic chemistry, as represented by recent discoveries of BGCs for altemicidin (17),88 fluopsin C (18),89 and guanitoxin (19)90 (Scheme 2B). The known (BGC)-unknown (metabolite) (II) is where most of the BGC-driven genome mining activities originate and are discussed in detail below. The last quadrant (IV), which represents the true biosynthetic dark matter, is the unknown (BGC)-unknown (metabolite) category, in which BGCs of unknown functions are predicted to produce new natural product structures. To focus on efforts in unknown-unknowns, researchers must deemphasize or deprioritize known core enzymes. While this represents the most challenging and nascent area of genome mining, it is likely that truly novel chemical structures and biological activities will arise from BGCs currently classified in this quadrant.
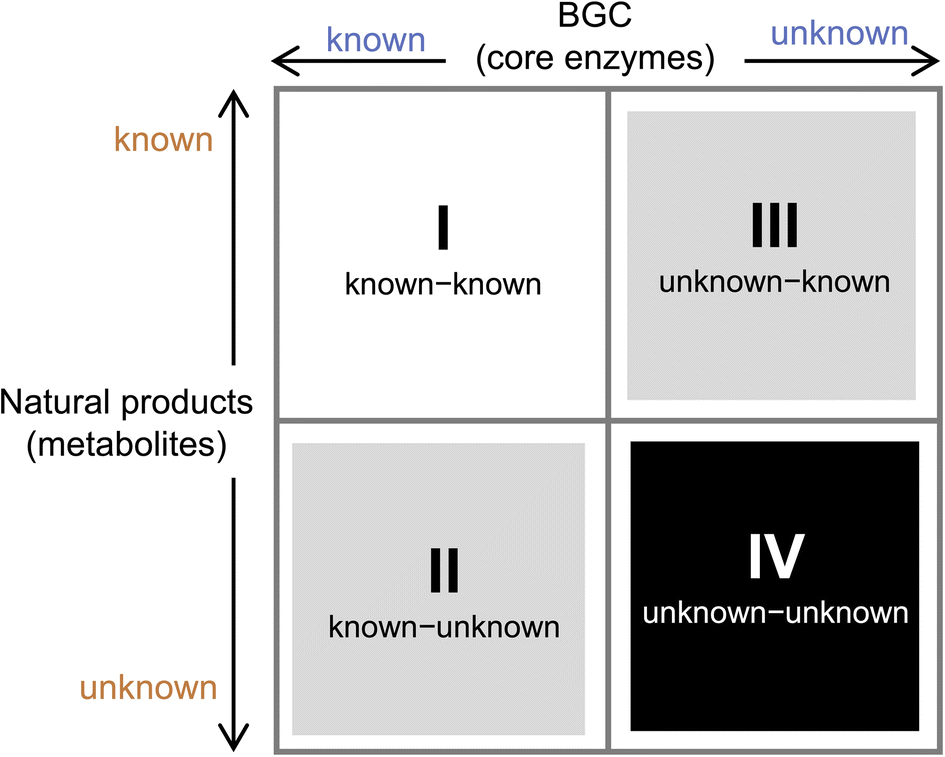 | ||
| Fig. 4 Natural product classification using a quadrant system. X-axis refers to known or unknown gene clusters based on core enzyme prediction. Y-axis refers to known or unknown metabolites. This review mainly focuses on the second quadrant. | ||
The known–unknowns (II in Fig. 4) represent most of the ongoing genome mining efforts in both bacteria and fungi. Using the known core enzymes as queries, a catalog of BGCs can be rapidly generated from each genome. In fungi that are prolific producers of natural products, it is typical to find more than 50 BGCs per genome using PKS, NRPS, TS/TC as leads. Enzymes that are frequently found in natural product BGCs, such as prenyltransferases (PTs) and RiPPs maturation enzymes can also populate the bioinformatics query. Compared to bacteria, other families of natural products such as aminoglycosides, phosphonates, etc. are not typically produced by fungi. To prioritize BGCs that may produce new chemical structures, several search criteria have been employed by many labs including ours. These include search for: (1) multidomain core enzymes, such as PKS and NRPS, that have unusual domains or domain arrangements; (2) BGCs that contain more than one core enzymes, which indicate potential convergent assembly of a more elaborate natural product scaffold; (3) BGCs that contain a large number of “tailoring” enzymes, such as oxygenases (P450s, flavin-dependent, nonheme iron-dependent, etc.), transferases (acyl, methyl glycosyl), PLP-dependent (transaminases, racemases, β- and γ-replacement enzymes), etc. In addition, clustering of predicted protein products that are hypothetic proteins (HPs) or contain domains of unknown function (DUF) are also strong indicators of potential new enzymology and chemical modifications; and (4) a combination of the above features. In this section, we will summarize a few recent examples of genome mining using these prioritization strategies.
3.1. Unusual core enzyme domain arrangement
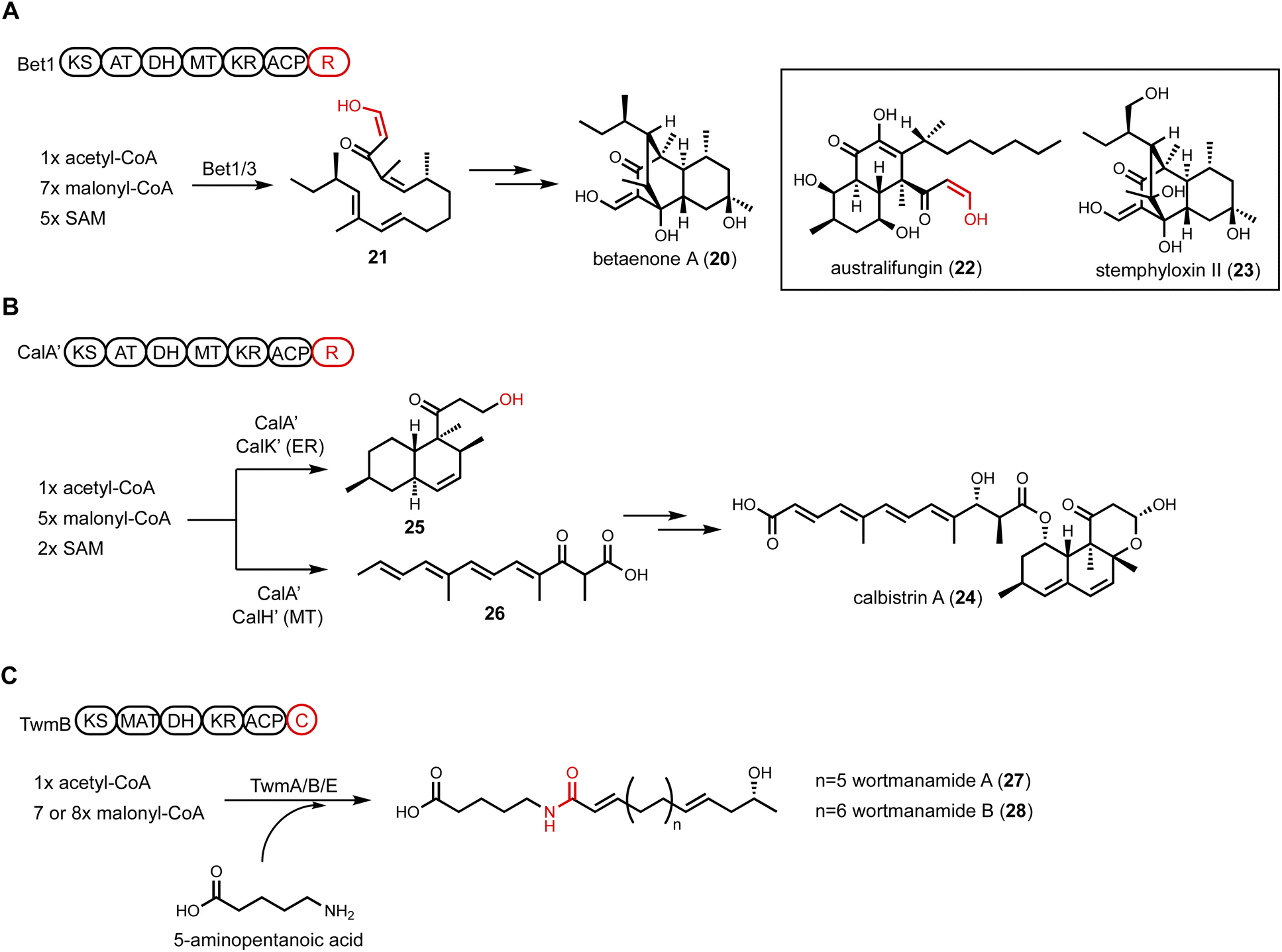 | ||
| Fig. 5 Unusual HRPKS domain arrangement. (A) PKS-R in betaenone biosynthesis. Terminal R domain catalyzes reductive product release. The enol form of aldehyde is highlighted in red. (B) PKS-R in calbistrin A biosynthesis. CalA′ can collaborate with different tailoring enzymes to generate two different polyketide scaffolds. Terminal R domain catalyzes consecutive two-electron reduction to release the decalin product as an alcohol, which is highlighted in red. (C) PKS-C in wortmanamide biosynthesis. The C domain fused with PKS is able to catalyze long chain N-acyl amide formation. | ||
An additional HRPKS-R example was found in calbistrin A (24) biosynthesis published by Tao et al.101 The revised structure of 24 consists of a decalin and a polyene that are esterified together. Retrobiosynthetic analysis would have predicted the involvement of two PKSs in the pathway to synthesize the two distinctive polyketide-derived fragments, as seen in the lovastatin biosynthesis.102 However, comparative genome analysis focused on HRPKSs in three producing fungi led to a candidate cal BGC that encodes a single HRPKS.103 Heterologous expression of a homologous cluster, cal′, was performed in A. oryzae to investigate the biosynthesis (Fig. 5B). Unexpectedly, the coexpression of CalA′(HRPKS) and CalK′(ER) led to the production of decalin product 25, while the coexpression of CalA′ and CalH′(MT) produced the polyene fragment 26. This result demonstrates that a single HRPKS is engaged in producing two distinct products with the aid of different tailoring enzymes. From the structure of 25, chain release was proposed to be catalyzed by R domain in CalA′ via consecutive two-electron reduction to generate an alcohol product. The polyene 26 isolated from CalA′/CalH′ coexpression transformant was release as a carboxylic acid, indicating the R domain can only reductively release the decalin product 25. The use of a single HRPKS in biosynthesis of two different portions of the final polyketide product is reminiscent of the HRPKS in chaetoviridine biosynthesis, in which two triketides of different degrees of reduction are synthesized and intercepted by separate releasing enzymes to build the final product.104
A single-module NRPS can be fused at the C-terminal of a HRPKS to form a PKS-NRPS megasynthetase. The NRPS module incorporates a nitrogen-containing functional group such as an amino acid, into the natural product. Such fusion of PKS and NRPS can generate a variety of new scaffolds as will be discussed below. A few PKS-NRPS enzymes only contain the condensation (C) module of an NRPS, while the adenylation (A) and thiolation (T) domains are absent. Furthermore, in this PKS-C subgroup, the characteristic HHxxxDG motif of C domain active site is mutated, suggesting noncanonical functions of the C domain. One well-known PKS-C example is lovastatin nonaketide synthase LovB, in which the second histidine in the C domain active site is mutated to an arginine. Based on in vitro and yeast reconstitution studies, the proposed function of the C domain is to catalyze the endo [4 + 2] cycloaddition of the hexaketide trienyl intermediate to form the trans-decalin.52 However, the exact function is still not confirmed, despite an available crystal structure of the standalone C domain,105 as well as the cryo-EM structure of the entire LovB.106 Bioinformatics analysis of fungal LovB-like enzymes revealed a clade of PKS-C in which the second histidine residue in the C domain active site is mutated to a proline. To investigate the product of this PKS-C enzyme, Hai et al. expressed one candidate cluster from Talaromyces wortmanii in A. nidulans.107 The expression of TwmB (PKS-C), TwmE (ER), and TwmA (TE) resulted in the biosynthesis of the N-acylamide compounds wortmanamide A and B (27 and 28). Both compounds incorporate the rare ω-amino acid, 5-aminopentanoic acid, that is amidated with a long chain polyketide product (Fig. 5C). The function of C domain was further studied by expressing TwmB-ΔC, which is a truncated version of TwmB with only the PKS portion. While no product was detected from TwmB-ΔC alone, coexpression with the standalone C domain restored the production of 27 and 28. This result implicates the C domain in TwmB is involved in the release of polyketide acyl chain through amide bond formation with ω-amino acid.
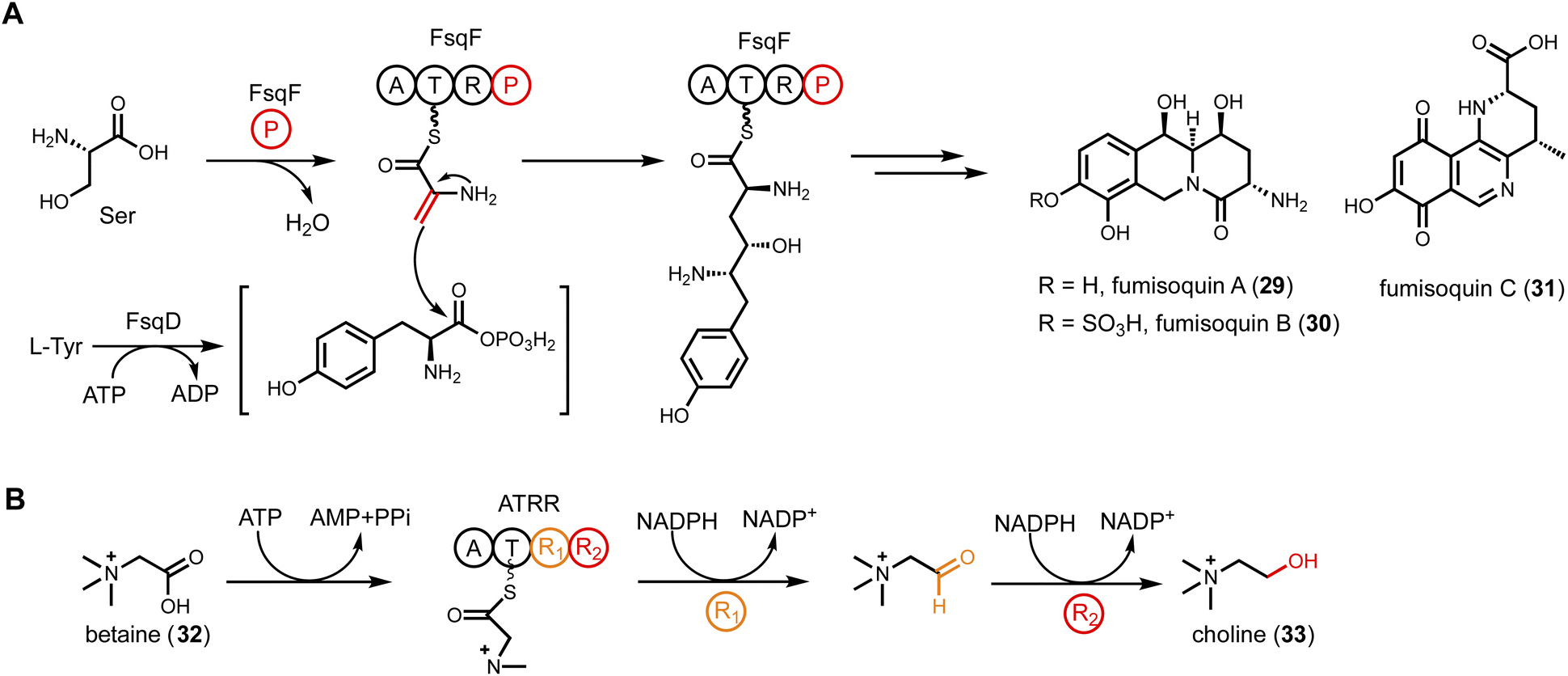 | ||
| Fig. 6 Noncanonical single module NRPS-like enzyme. (A) FsqF (A-T-R-PLP) in fumisoquin biosynthesis. PLP domain is fused with NRPS, catalyzing dehydration of serine and forming a new C–C bond with FsqD-activated tyrosine. (B) ATRR (A–T–R1–R2) in choline biosynthesis. Two consecutive R domains catalyze sequential reduction to convert betaine to choline. | ||
Another example of a CAR containing an additional domain is the ATRR enzyme widely conserved in fungi.110 The enzyme group is named ATRR because an additional C-terminal YdfG-like short chain dehydrogenase/reductase (SDR) domain is fused to a CAR, giving the domain architecture A-T-R1-R2. This enzyme, initially thought as a candidate to produce a new natural product, is involved in primary metabolism and catalyzes sequential, ATP- and NADPH-dependent reduction of betaine (32) to choline (33) (Fig. 6B). While the oxidation of choline to betaine is known in fungi, the reverse reaction to reduce betaine to choline was not biochemically characterized. Initial inspection of ATRR domain arrangement pointed to a sequential two-electron reductions of a carboxylic acid to an alcohol, with the A domain specifying the identity of the carboxylic acid substrate. Bioinformatics analysis of A domain active site showed mutation of a highly conserved aspartate residue that electrostatically interacts with the α-amino group of amino acids, hence excluding amino acids as substrates. Structural-based prediction using a homology model based on A domain of TycA showed that the A domain in ATRR positions three aromatic amino acids at the end of the active site pocket into an aromatic cage that may interact with a quaternary ammonium group through cation-π interactions. Based on this observation, screening of ATRR activity towards betaine substrates was performed using purified enzyme. Rapid consumption of NADPH was observed in the presence of glycine betaine and ATP. The stepwise reduction was supported by trapping the intermediate glycine betaine aldehyde with phenylhydrazine. In addition, site-directed mutagenesis was performed to generate single R domain mutants. Mutation of R1 abolished the carboxylic acid reductase activity, while mutation of R2 abolished the aldehyde reductase activity. Fusion of the second R domain was postulated to enhance substrate channeling in the tandem reduction reaction and to prevent dissociation of the aldehyde that can be readily hydrated. While the genome mining efforts aimed at ATRR did not lead to discovery of a new natural product, the findings provided insights into choline metabolism and betaine homeostasis in fungal organisms.
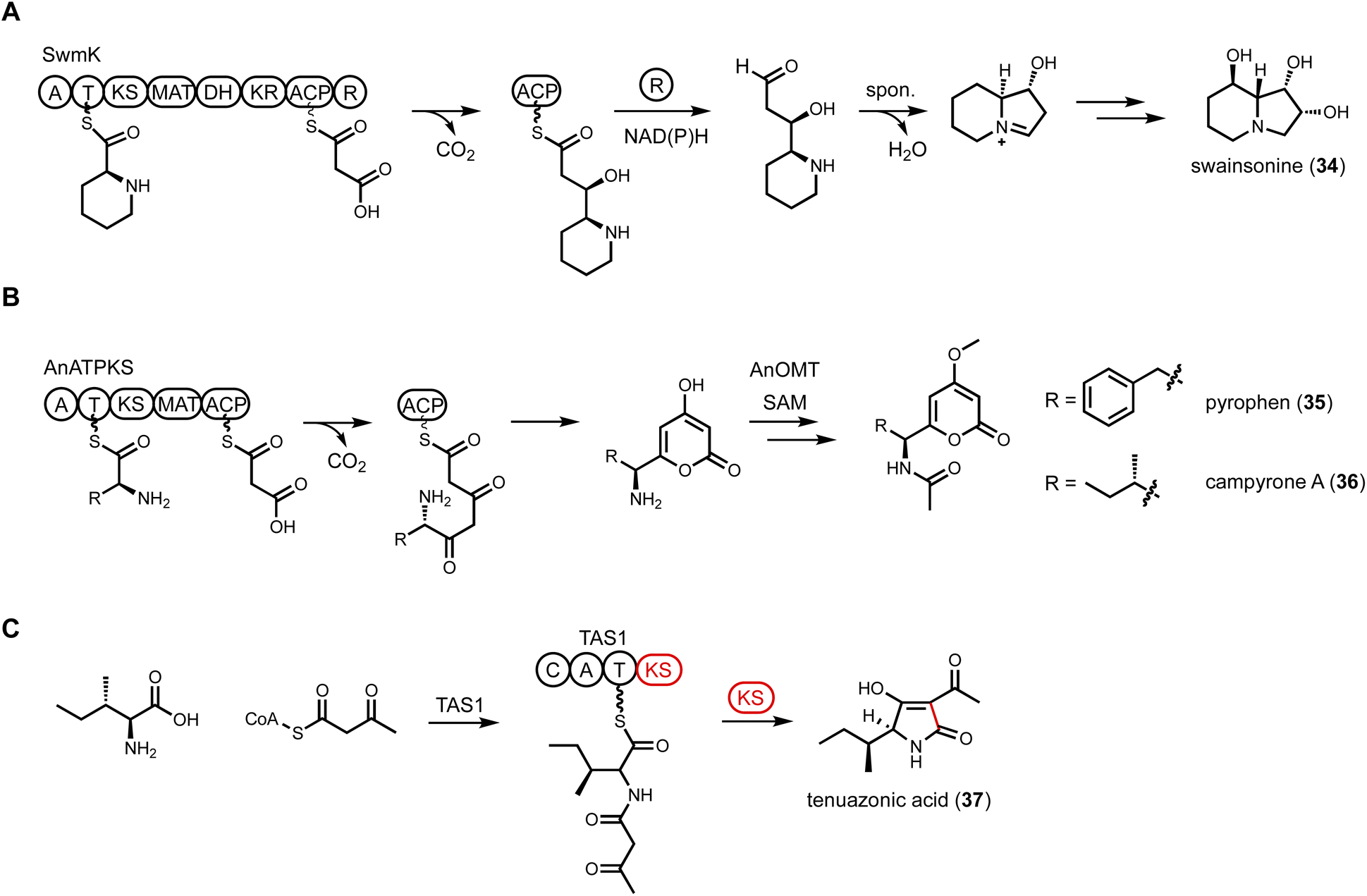 | ||
| Fig. 7 NRPS-PKS hybrid enzymes. (A) Proposed swainsonine biosynthetic pathway. SwmK catalyzes the formation of indolizidine core. (B) Proposed pyrophen and campyrone biosynthetic pathway. AnATPKS is involved in the formation of amino acid-containing α-pyrone natural products. (C) Proposed tenuazonic acid biosynthetic pathway. A KS domain is fused with NRPS to catalyze cyclization and product release. | ||
A recent example of NRPS-PKS discovered through genome mining was shown to be involved in formation of pyrophen (35) and campyrone A (36),112 which are amino acid-containing α-pyrone compounds. The BGC encoding an NRPS-PKS was mined from Aspergillus niger. In addition to NRPS-PKS (AnATPKS), an O-methyltransferase (AnOMT) is colocalized in the cluster, matching the methoxy moiety observed in pyrophen and campyrone A. Heterologous expression of AnATPKS and AnOMT in A. nidulans confirmed the cluster is responsible for the formation of 35 and 36. The NRPS domain activates L-phenylalanine which is used as starter unit of the PKS module. Two decarboxylative Claisen elongation steps followed by a cyclization release step generate the α-pyrones (Fig. 7B). Feeding experiment with nonproteinogenic amino acid showed that AnATPKS A domain has relaxed substrate specificity and can accept different aromatic amino acids to arrive at pyrophen analogs.
Tetramate or pyrrolidine-containing natural products from fungi are biosynthesized by PKS-NRPS megasynthetases. After aminoacylation of the β-ketoacyl PKS intermediate, a Dieckmann cyclization catalyzed by an R* domain, or a reductase release by an R domain followed by Knoevenagel condensation, yields either a tetramate or a pyrrolidine, respectively. However, the discovery of tenuazonic acid (37) biosynthesis revealed a different strategy to form this core structure from one isoleucine and two acetate units (diketide).113 BGC for the eukaryotic translation inhibitor 37 was identified from Magnaporthe oryzae using comparative RNA sequencing under producing and non-producing conditions. While none of the PKS-NRPS encoding gene displayed change in transcriptional levels, one gene encoding a NRPS-PKS homolog (TAS1) was found to be upregulated under 37 producing conditions. The involvement of TAS1 in 37 biosynthesis was confirmed by knockout. 37 has an N-terminal NRPS domain and a single KS domain at the C-terminus. In vitro assays treating TAS1 with different CoA substrates revealed that acetoacetyl-CoA is directly incorporated, hence no decarboxylative Claisen condensation is required. Using N-acetoacetyl-L-Ile-SNAC to mimic the linear substrate attached to the T domain in NRPS, the authors showed that the standalone KS domain can cyclize the substrate into 37 (Fig. 7C). The likely mechanism is KS-catalyzed enolization of the diketo functionality to initiate tetramate formation.
3.2. Multiple core enzyme combinations
To mine compounds with structural complexity, focusing on BGCs containing multiple core enzymes is an attractive option. Molecules derived from such pathways display combined structural features accessible through individual core enzymes. Several modes of core enzyme collaboration have been characterized: (1) one core enzyme is responsible for biosynthesis of a building block for the other core enzyme. For example, in the biosynthesis of the immunosuppressant drug cyclosporine A (38),114 the unnatural amino acid (4R)-4-[(E)-2-butenyl]-4-methyl-L-threonine (Bmt) incorporated by the cyclosporine NRPS is biosynthesized by a HRPKS in the BGC (Scheme 3A). Release of the polyketide product is subjected to α-oxidation to a ketone followed by transamination to yield Bmt. Another example is in the biosynthesis of ergotamine alkaloids (39),115 which involves first the generation of lysergic acid by the actions of numerous enzymes including 4-dimethylallyl tryptophan synthase (4-DMATS), followed by a NRPS that uses lysergic acid as a starter unit to arrive at the final product; (2) sequential biosynthesis of chemically distinct portions of a natural product by different core enzymes. This division of labor is most well-characterized in the collaboration between NRPKS and HRPKSs in the biosynthesis of resorcylic acid lactone (RAL) natural products,116 such as hypomycetin (40)117 and zearalenone (41).118 In these pathways, the HRPKS generates the highly reduced polyketide chain that is transferred to the NRPKS for elongation and cyclization into the 2,4-dihydroxybenzoic acid moiety; and (3) convergent synthesis of the final product by core enzymes, as exemplified in the biosynthesis of lovastatin (42). The lovastatin nonaketide synthase LovB synthesizes the decalin product dihydromonacolin L (DML), while the lovastatin diketide synthase (LovF) synthesizes the α-methylbutyrate side chain that is transferred to the oxidized DML (monacolin J).52 In this section, we will highlight some recent examples of genome mining prioritized by the presence of two of more core enzymes in the same BGC.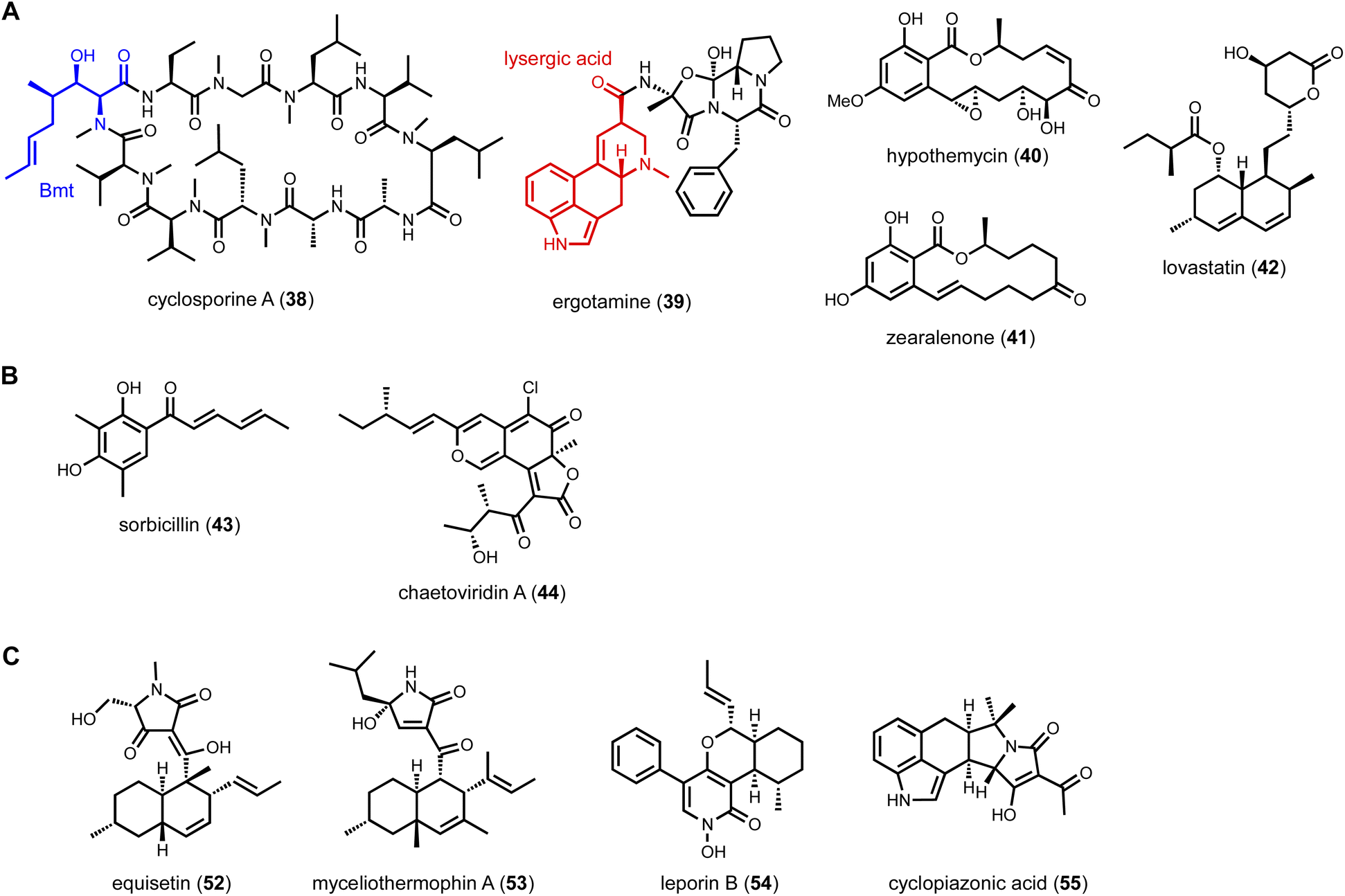 | ||
| Scheme 3 (A) Selected example of metabolites required collaboration of multiple core enzymes. (B) Representative natural products synthesized by dual PKSs. (C) Selected PKS-NRPS metabolites using proteinogenic amino acid as building block. | ||
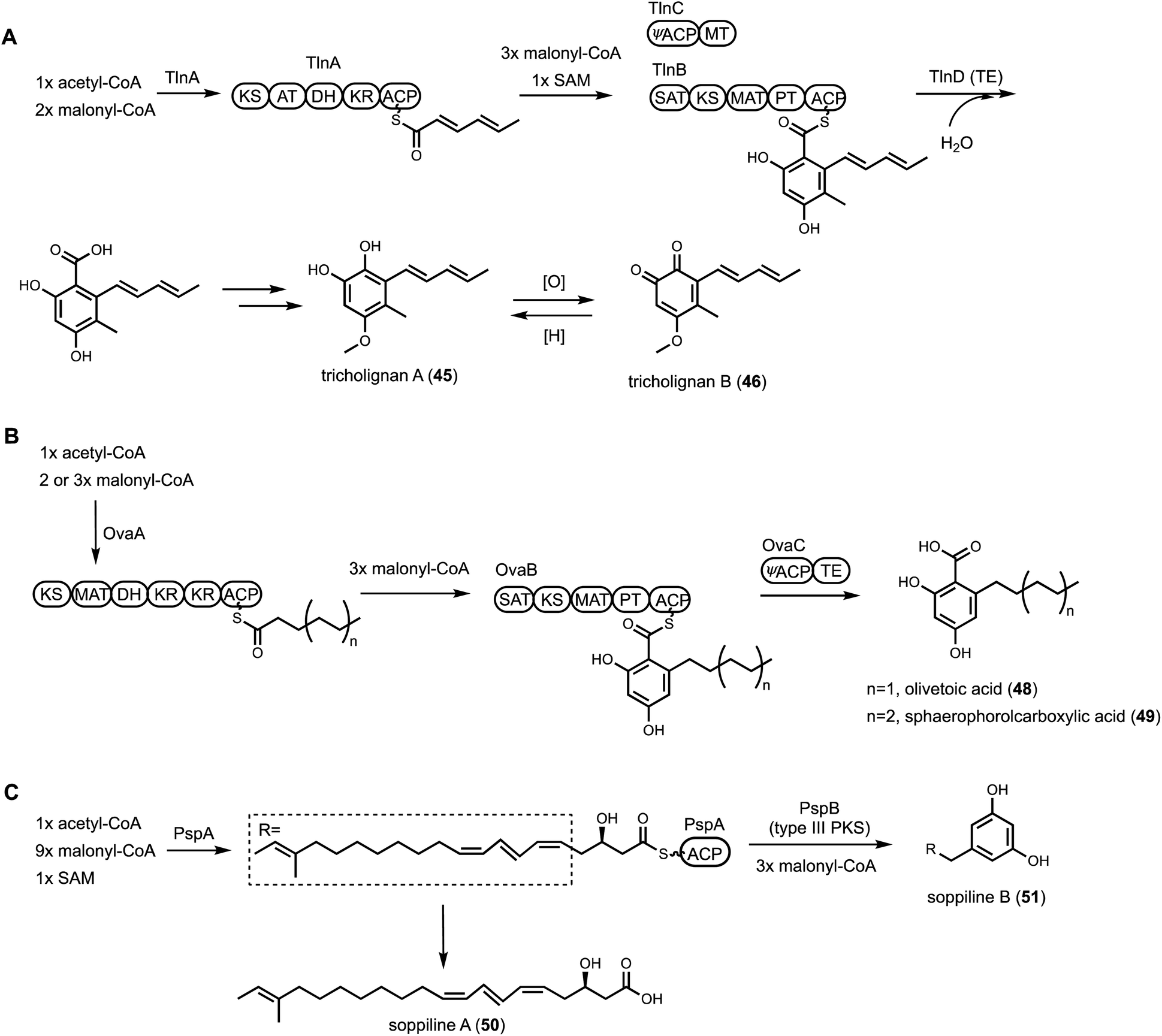 | ||
| Fig. 8 Pathways in which multiple PKSs collaborate. (A) Biosynthetic pathway of tricholigan A and B. The pathway requires TlnC, which is a didomain protein with a ψACP fused with MT. (B) A fungal biosynthetic pathway to olivetolic acid. The cluster contains a didomain enzyme with an ψACP fused with TE. (C) Proposed pathway of soppiline. A type III PKS collaborates with HRPKS in chain elongation and product cyclization. | ||
The role of the ψACP-MT (TlnC) was examined in detail using purified HPRKS, NRPKS and TE. It was determined that the ψACP domain of TlnC is involved in recruiting the MT domain to NRPKS for a single Cα-methylation step during chain elongation cycle of the NRPKS. Interestingly, when the active site residue in ψACP was mutated back to serine, the ACP domain regained ability to be phosphopantetheinylated, but the MT domain lost the function to perform the programmed methylation. This was attributed to the gain-of-function pPant arm that can enter the cis-fused MT active site and block the in trans C-methylation of the polyketide intermediate attached to TlnB pPant arm. Taken together, this indicated ψACP is important for protein–protein interactions between the megasynthase and trans-acting MT domain. This finding was used to mine other BGCs encoding ψACP fusion proteins. One such example is a tandem HRPKS and NRPKS-encoding BGC found in Metarhizium anisopliae, which encodes a ψACP-TE fusion didomain protein.121 The ψACP active site is also mutated from the conserved amino acid sequence DSL to NQI, indicating loss of phosphopantetheinylation. Reconstitution of the two PKSs and the ψACP-TE fusion in A. nidulans led to biosynthesis of both olivetolic acid (48) and the longer sphaerophorolcarboxylic acid (49) (Fig. 8B). Both compounds are precursors to plant-derived cannabinoids: 48 can be geranylated to cannabigerolic acid (CBGA) and further cyclized into tetrahydrocannabinolic acid in the plant pathways. The A. nidulans host, after minimal optimization, was able to produce >1 g L−1 of 49. Mining of additional homologous BGCs led to pathway that can exclusively produce 48. It is speculated here that ψACP recruits the fused TE partner to NRPKS to hydrolytically releases the free carboxylic acid products.
Kaneko et al. found a new HRPKS releasing mechanism through collaboration with a type III PKS.122 Genome mining of Penicillium soppi revealed a simple but unique BGC, which consists of a HRPKS (PspA), a type III PKS (PspB), and a P450 (PspC). Type III PKSs are ACP-independent KS dimers that can catalyze one or more cycles of decarboxylative Claisen condensation with malonyl-CoA. Fungal type III PKSs are functionally less diverse compared to plant type III PKS.123 Heterologous expression of PspA, or PspA together with PspB in A. oryzae, led to the production of two new metabolites, soppiline A (50) and B (51), respectively (Fig. 8C). The structural difference between 50 and 51 suggests that PspA synthesizes an unsaturated polyketide molecules, which is transferred to PspB as a starter unit, followed by three cycles of chain extension and cyclization to complete the biosynthesis of alkylresorcinol soppiline B. This is the first example of a biosynthetic collaboration between a HRPKS and a type III PKS.
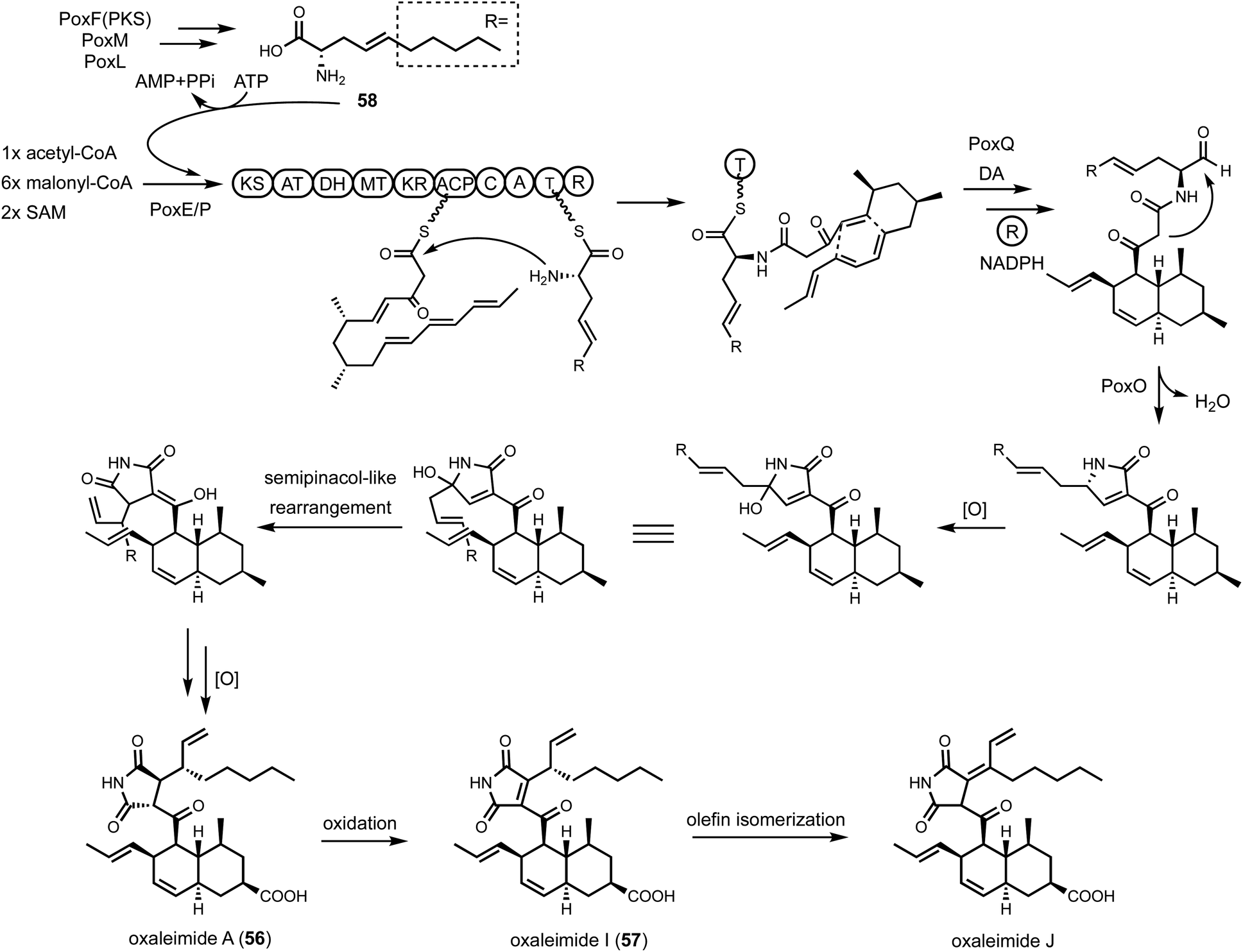 | ||
| Fig. 9 Proposed biosynthetic pathway of oxaleimides. The biosynthesis requires HRPKS (PoxF) with PoxM and PoxL to generate a nonproteinogenic amino acid 58, which is incorporated by PKS-NRPKS (PoxE) assembly line. Knoevenagel condensation followed by hydroxylation and a semipinacol-like rearrangement complete the formation of the substituted succinimide. | ||
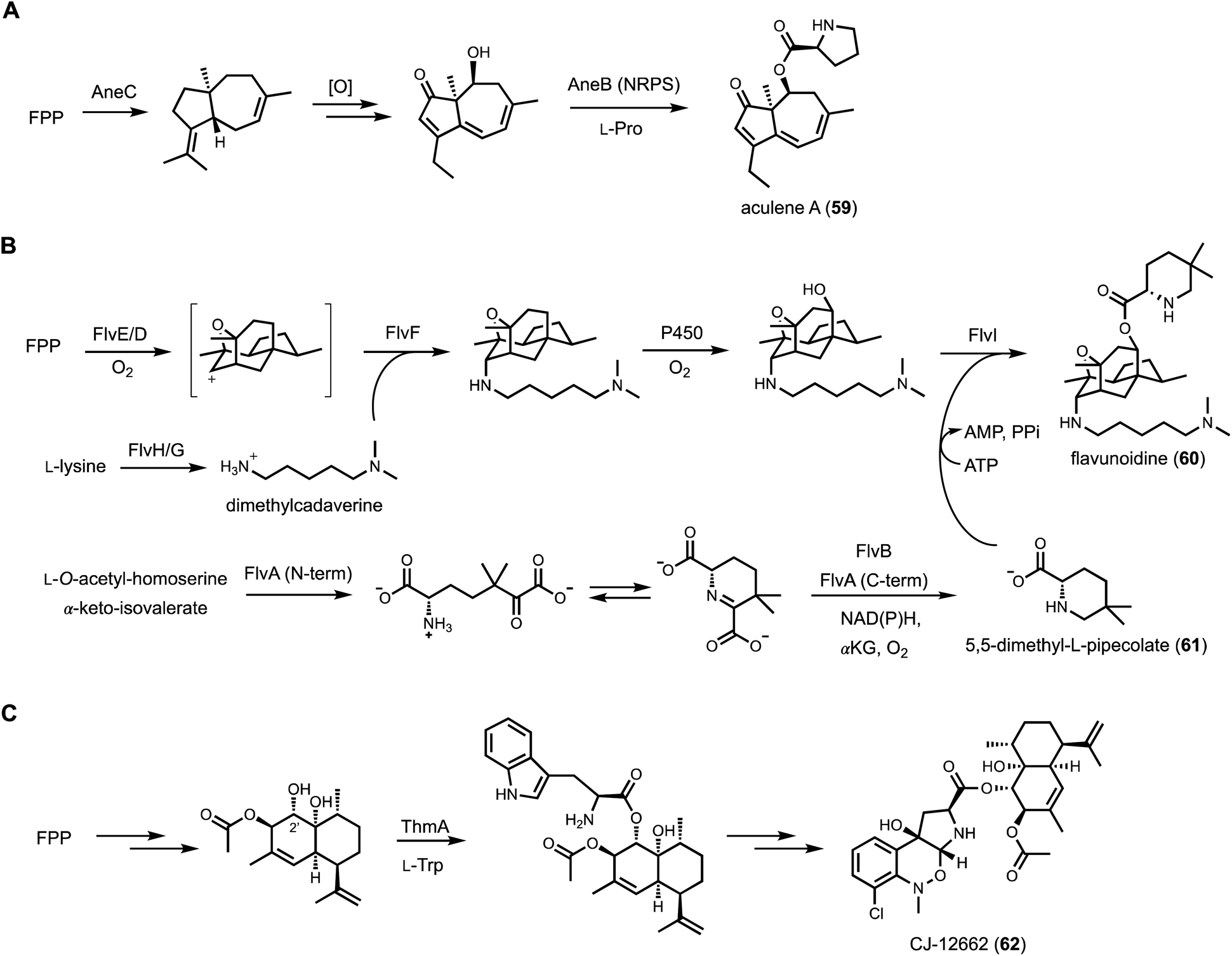 | ||
| Fig. 10 Examples of natural products produced from BGCs that encode both terpene cyclase and NRPS. FPP: farnesyl diphosphate. (A) Simplified aculene biosynthesis pathway. The proline moiety is incorporated into terpene scaffold via a single module NRPS AneB. (B) Proposed flavunoidine biosynthetic pathway. Two unique amino acid building blocks are highlighted in the pathway. N,N-Dimethylcadaverine is synthesized from L-lysine, which then connected to the tetracyclic core via an axial C–N bond. The other substituent, 5,5-dimethyl-L-pipecolate is formed by PLP-dependent enzyme FlvA. The connection of 5,5-dimethyl- L-pipecolate to the tetracyclic structure is catalyzed by a single module NRPS FlvI. (C) Simplified CJ12662 biosynthesis pathway. L-Tryptophan is esterified with the terpene scaffold in an ATP-dependent step catalyzed by ThmA. | ||
To explore the biosynthetic diversity of TC-NRPS hybrid pathways, Yee and Kakule performed genome mining and showed such hybrid clusters are in fact wide-spread in fungi. Focusing on the flv cluster from Aspergillus flavus, a heterologous expression approach using A. nidulans was performed. The flv cluster is particularly interesting in that it contains two predicted TCs (FlvE and FlvF) and one single-module NRPS (FlvI).132 In addition, numerous accessory enzymes are encoded in the BGC, including two P450s, an SDR (FlvB), an ornithine decarboxylase (FlvG), and a didomain enzyme (FlvA) with a non-heme iron-dependent (NHI) oxygenase fused to a PLP-dependent enzyme. Heterologous expression of the entire BGC led to biosynthesis of a new natural product flavunoidine A (60), a tripartite molecule (Fig. 10B). The heavily oxidized terpenoid portion is synthesized by FlvE and oxidized by one of the P450 (FlvD). Surprisingly, a dimethylcadaverine substituent is connected to the terpene core via an axial C–N bond. Systematic reconstitution efforts showed that dimethylcadaverine is synthesized from L-lysine by FlvG and a cryptic methyltransferase (FlvH) initially annotated as an HP. The second TC in the BGC, FlvF, is responsible for formation of the C–N bond, only in the presence of the P450 enzyme FlvD. In the absence of FlvF, the terpenoid core is instead connected via C–N bond to ethanolamine in both axial and equatorial configurations, suggesting nonenzymatic quenching of a possible cation intermediate. The proposed mechanism is the P450 FlvD oxidizes the terpenoid core in sequential one-electron steps to generate a secondary carbocation, which is intercepted by FlvF with dimethylcadaverine. Therefore, FlvF does not function as a bona fide TC, but may be responsible for stereoselective C–N bond formation that is not seen in terpenoid maturation.133 The third part of flavunoidine is a new-to-nature 5,5-dimethyl-L-pipecolate (61) esterified to a hydroxyl group introduced by the P450 FlvE in the terpenoid core. This unusual amino acid is synthesized from the tandem actions of FlvA and FlvB, of which a key step is proposed to be the γ-replacement reaction using O-acetyl-L-homoserine as the latent vinyl glycine donor (see Section 3.3.2). Finally, the NRPS FlvI adenylates and esterifies 61 to the terpenoid to generate flavunoidine. The unexpected structural features of flavunoidine showcases how TC-NRPS hybrid pathways can afford new and complex natural products.
The pyrrolobenzoxazine terpenoid CJ-12662 (62) from Aspergillus fischeri var. thermomutatus ATCC 18618 is another example of such fusion between terpene and amino acid. A known compound with potent anthelmintic activity, CJ-12662 contains two distinct substructures that are esterified together, indicating NRPS involvement. The terpenoid portion is a heavily hydroxylated amorpha-4,11-diene, while the pyrrolobenzoxazine portion is clearly derived from oxidative rearrangement of L-tryptophan. Genome scanning identified the candidate thm BGC, which was reconstituted using A. nidulans to confirm its role in CJ-12662 biosynthesis (Fig. 10C).134 Amorpha-4,11-diene, a famous plant terpene that is the precursor to artemisinin, is synthesized by the TC ThmB and triply hydroxylated by three P450 enzymes in the BGC. The resulting triol is esterified regioselectively at the C2′ alcohol with L-tryptophan by the NRPS ThmA. The indole portion is then oxidatively modified to pyrroloindole via the action of a flavin-dependent epoxidase, followed by N-methylation, chlorination, and N-oxidation to trigger a [1,2]-Meisenheimer rearrangement and give CJ-12662. A related pathway involved in biosynthesis of an epoxidized version of CJ-12662 was independently reconstituted in A. nidulans by Hu and coworkers.135 The authors in this study suggested a possible role of the N-oxygenase in facilitating the Meisenheimer rearrangement.
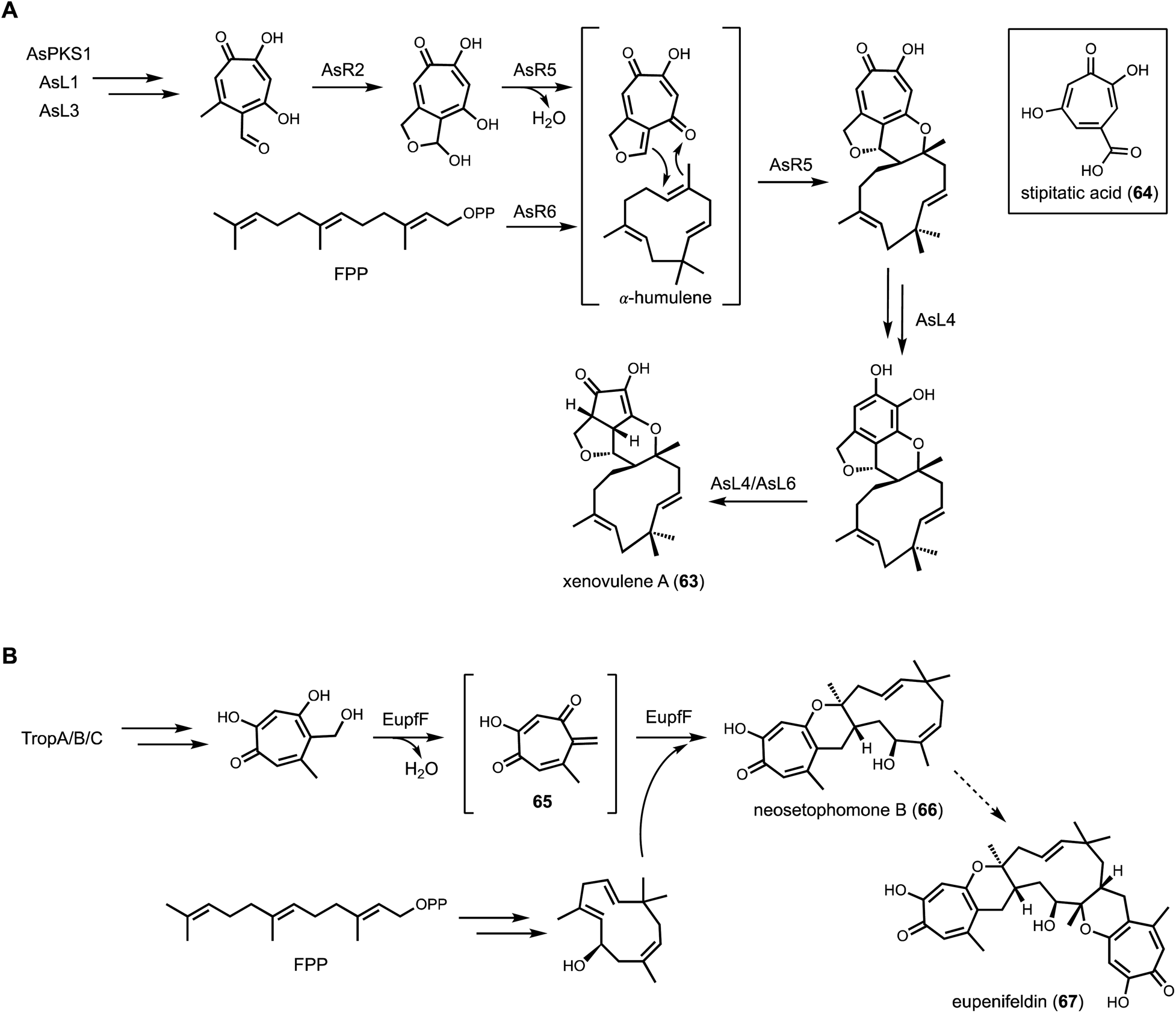 | ||
| Fig. 11 Cryptic terpene cyclase functions with NRPKS. (A) Proposed biosynthetic pathway of xenovulene A. AsR6 was confirmed as a new class of terpene cyclase, catalyzing the formation of humulene. (B) Proposed biosynthetic pathway of eupenifeldin. EupfF was characterized as a hetero Diels–Alderase, catalyzing the cycloaddition between the quinone methide 65 and sesquiterpene. | ||
To investigate the function of AsR5 and AsR6, recombinant proteins were obtained from E. coli. Incubation of AsR6 with farnesyl pyrophosphate (FPP) and Mg2+ led to the production of α-humulene, indicating the function of AsR6 is indeed a TC. Although sequence alignment with type I terpene cyclase indicated AsR6 lacks well-conserved magnesium binding residues, the enzyme is Mg2+-dependent as confirmed through biochemical assays.138 The fusion between tropolone and α-humulene was proposed to occur through an intermolecular hetero-Diels–Alder reaction. In a follow-up study, Hu and coworkers functionally characterized the homolog of AsR6, EupfF, in the BGC of related compound eupenifeldin (67).140 Dehydration of the tropolone to form a reactive o-quinine methide (65) followed by hetero-Diels–Alder reaction with hydroxyl-humulene led to the product neosetophomone B (66) that can be further processed into eupenifeldin (Fig. 11B). Therefore, the BGCs of 63 and 67 contain two cryptic enzymes: a sesquiterpene cyclase and a pericyclase, that were functionally reconstituted in heterologous host. Cox and coworkers subsequently reported the combinatorial biosynthesis using A. oryzae as heterologous host.141 Coexpression of genes from three arpks1 homologous clusters led to the generation of unnatural tropolone sesquiterpenoids. Recently, the total synthesis of the related pycnodione and DFT calculations led to the conclusion that the second hetero-Diels–Alder reaction observed in such bistropolone sesquiterpenes must also be enzyme-catalyzed.142 The responsible enzyme and substrates have not been identified to date.
3.3. Combinations of accessory enzymes
One strategy used by the community to mine known-unknown BGCs is focusing on those that encode a multitude of accessory enzymes in addition to a core enzyme. Clustering of oxidative enzymes such as P450s, NHI oxygenases, and/or flavin-containing monooxygenases (FMOs), etc. is usually correlated with extensive structural modifications. Examples of such modifications have been discussed in some of the previous examples, and additional examples will be presented in 3.3.1. Other accessory enzymes such as transferases, hydrolases are also scaffold modifying, and often function in series with the oxidative enzymes. In recent years two additional classes of enzymes have emerged to be strong indicators of chemical complexity. The PLP-dependent enzymes will be discussed in 3.3.2, while the pericyclases family of enzymes will be discussed in Section 4.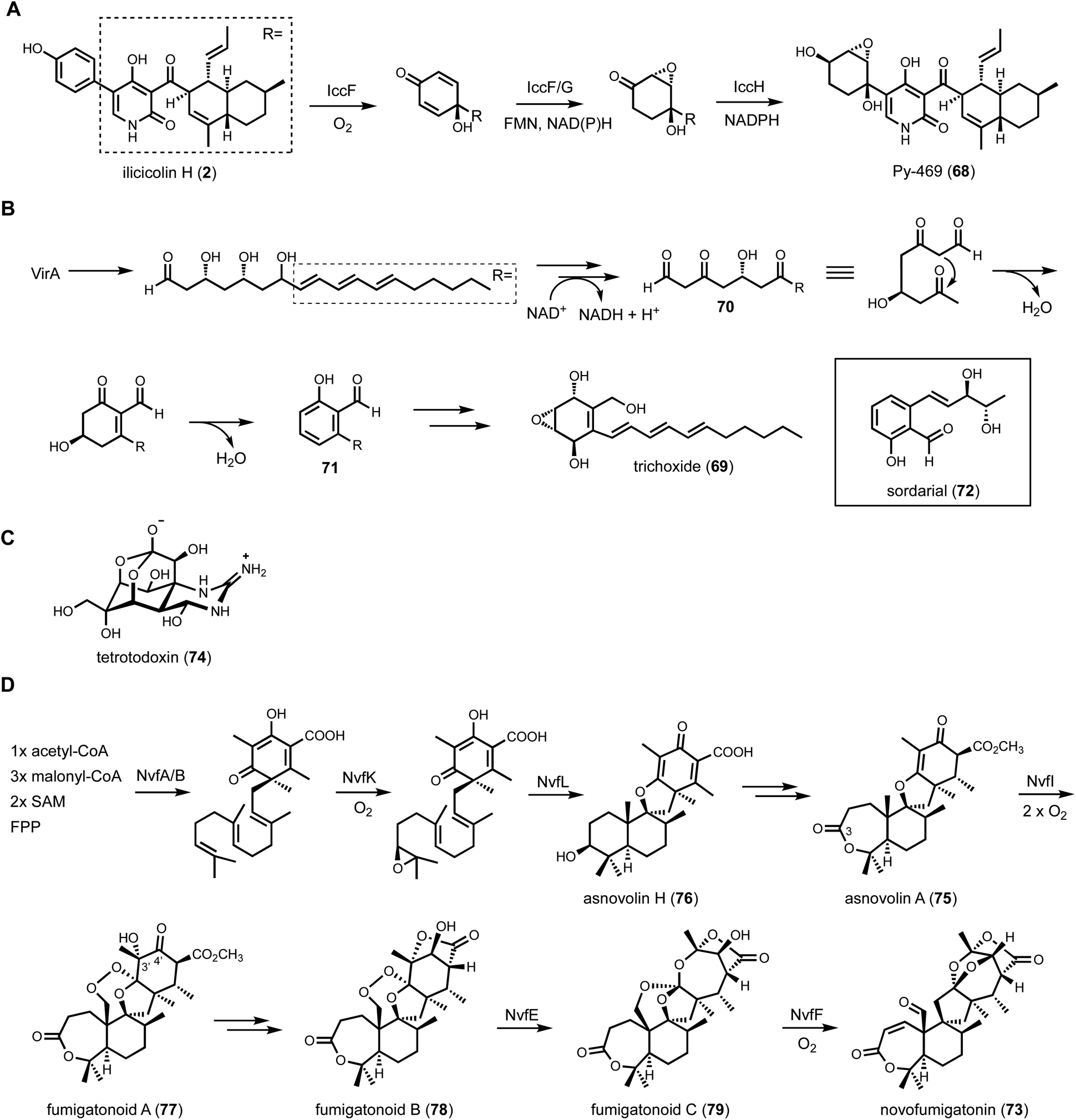 | ||
| Fig. 12 Post-core enzyme modifications by redox enzymes. (A) Biosynthetic pathway of Py-469 highlights three consecutive redox enzymes in modifying benzene ring to 2,3-epoxy-1,4-cyclohexane diol. The relative stereochemistry was determined by MicroED. (B) Multitude of redox enzymes in the biosynthesis of trichoxide reveals a unique strategy to synthesize aromatic polyketide structure using HRPKS. A similar pathway was also found in sodarial biosynthesis. (C) Structure of tetrotodoxin, which is an orthoester containing natural product. (D) Simplified biosynthetic pathway of novofumigatonin. The structure is highly oxygenated with a series of oxygen-handling enzymes, in particular the formation of orthoester functional group by two non-heme iron enzymes, NvfE and NvfF. | ||
Another genome mining example involving a BGC with a multitude of redox enzymes is that of trichoxide from Trichoderma virens.146 Tabulation of the HRPKS-encoding BGCs from this well-studied fungal host revealed the vir cluster that is particularly interesting. In addition to the HRPKS VirA, the BGC encodes potentially nine redox enzymes, including four SDRs, one P450, two flavin-dependent oxidoreductase and two cupid-domain containing oxidoreductases. Heterologous expression of the entire cluster in A. nidulans led to the discovery of the new metabolic trichoxide (69), which is a new member of the epoxycyclohexenol containing natural products. Bottom-up reconstitution starting with the HRPKS showed the biosynthetic pathway can be divided into two parts, the formation of an aromatic salicylaldehyde, and dearomatization to the epoxycyclohexanol (Fig. 12B). Formation of an aromatic intermediate in a HRPKS-containing BGC was surprising, considering HRPKSs generate highly reduced compounds. Analysis of shunt products and intermediate, however, revealed the biosynthetic strategy. VirA synthesizes a reduced polyketide in which the last three ketides are reduced to β-hydroxyl groups. The triol is released reductively to an aldehyde, which can cyclize into a cyclic hemiacetal. Stepwise oxidations of two of three β-alcohols yield a poly-β-ketone intermediate (70) that more resembles a NRPKS product. Intramolecular aldol cyclization followed by dehydration and aromatization give the salicylaldehyde intermediate (71). This unexpected logic of using an HPRKS in combination with redox enzyme to generate salicylaldehyde was also seen in the biosynthesis of the mycotoxin sordarial (72) from Neurospora crassa, using A. nidulans as a heterologous host.147 From the salicylaldehyde, the remaining redox enzymes dearomatize the aromatic ring through a series of hydroxylation, epoxidation and reduction steps, a sequence of reactions similar to that take place during biosynthesis of 68 (Fig. 12A).
One additional notable example of how redox enzymes can heavily modify a core scaffold is in the biosynthesis of novofunigatonin (73), which is isolated and characterized from Aspergillus novofumigatus IBT 16806.148 Novofumigatonin is a dramatically oxygenated meroterpenoid containing an orthoester group, two lactone rings, and an aldehyde group. Orthoester is a unique functional group that contains three alkoxyl groups attached to a single carbon atom. The classic example of an orthoester natural product is tetrodotoxin (74) (Fig. 12C).149 The precursor of novofumigatonin was proposed to be derived from asnovolin A (75), which is synthesized from the aromatic polyketide 3,5-dimethylorsellinic acid and the C15 isoprenoid farnesyl-diphosphate (Fig. 12D). To solve the biosynthetic pathway of 73, Matsuda et al. employed CRISPR-Cas9 based gene deletion, heterologous expression in A. oryzae, and in vitro analysis. The oxidative decoration begins from the NvfK-catalyzed epoxidation of farnesyl-dimethylorsellinate to initiate cyclization of the tetracyclic asnovolin H (76). The C3-OH is then oxidized to a ketone by NvfC and expanded to the seven-member lactone by a Baeyer–Villiger type of oxygenase NvfH. Formation of the hydroxy endoperoxide in fumigatonoid A (77) is catalyzed by a nonheme, Fe(II)/α-ketoglutarate-dependent enzyme, NvfI, consuming two molecules of oxygen: the first oxygen is incorporated intact as the bridging endoperoxide, while an oxygen atom from the second molecular oxygen is incorporated as a hydroxyl group at C3’ position via a radical rebound step. Formation of the lactone and reduction of C4′ ketone affords fumigatonoid B (78). Two additional NHI enzymes, NvfE and NvfF, complete the biosynthesis of novofumigatonin with the orthoester moiety. NvfE is responsible for the formation of orthoester moiety in fumigatonoid C (79) with no change in oxidation state, indicating that it functions as an isomerase instead of an oxygenase. Further in vitro analysis showed that NvfE is a cofactor- and cosubstrate-free enzyme with mutation of the highly conserved glutamate required for α-ketoglutarate binding. The last enzyme NvfF is a bona fide Fe(II)/αKG-dependent dioxygenase that catalyzes orthoester exchange and oxidation of the released alcohol to aldehyde. Overall, three out of eight oxygen atoms in novofumigatonin are from the orsellinic acid building block. The other five oxygen atoms are incorporated from four molecular oxygens by a series oxygen-handling enzymes, showcasing how nature generates a remarkable set of oxygen functional groups in a compact framework.
The first example of a PLP-dependent enzyme catalyzing γ-replacement, C–C bond forming reaction is CndF found in the citrinadin BGC from Penicillium citrinum.152 Citrinadin A (80) and B (81) (Fig. 13A) are prenylated indole alkaloids derived from a tryptophanyl-6-methyl-pipecolate ketopiperazine biosynthesized by a bimodular NRPS. The nonproteinogenic (2S,6S)-6-methyl-pipecolate (82) is not a previously known metabolite and must therefore be biosynthesized from dedicated enzymes encoded in the cnd BGC. Comparative analysis with other BGCs of indole alkaloids that do not incorporate 82 in the scaffold suggested three cnd enzymes, CndE (SDR), CndF (PLP-dependent enzyme with 36% sequence identity to cystathionine-γ-synthase), and CndG (HMG-CoA lyase), may be involved. Heterologous expression of the three enzymes in A. nidulans indeed led to the accumulation of 82 (Fig. 13B). Exclusion of CndG in the heterologous host did not abolish the biosynthesis of 82, albeit led to decreased levels. Since CndG is predicted as an HMG-CoA lyase that can cleave HMG-CoA into acetyl-CoA and acetoacetate, it was hypothesized that acetoacetate could be the three-carbon nucleophile used by CndF in the PLP-dependent γ-replacement reaction. Feeding experiment using [2,4-13C2] ethyl acetoacetate, which can be hydrolyzed by endogenous esterase in A. nidulans expressing CndE and CndF, resulted in C5 and C6-methyl doubly labeled 82. Based on additional biochemical evidences, CndF is proposed to catalyze C–C bond formation between acetoacetate and enzyme-bound ketimine form of vinyl glycine to give (S)-2-amino-6-oxoheptanoate (83), which upon release from the enzyme can cyclize to form the Schiff base (84). The SDR CndE then catalyzes stereospecific imine reduction to afford 82. Encouraged by the finding that β-keto carboxylates such as acetoacetate can be generated in cellulo by endogenous esterase from ethyl esters, A. nidulans expressing CndE and CndF was used as a biotransformation host to convert β-keto ethyl esters to 6-alkyl pipecolate derivatives. These studies showed CndF has considerable promiscuity towards α- and γ- substituted β-keto carboxylate compounds, including bulky and cyclic substrates. Thus, CndF may be further repurposed as a biocatalyst to construct 6-alkyl pipecolate derivatives.
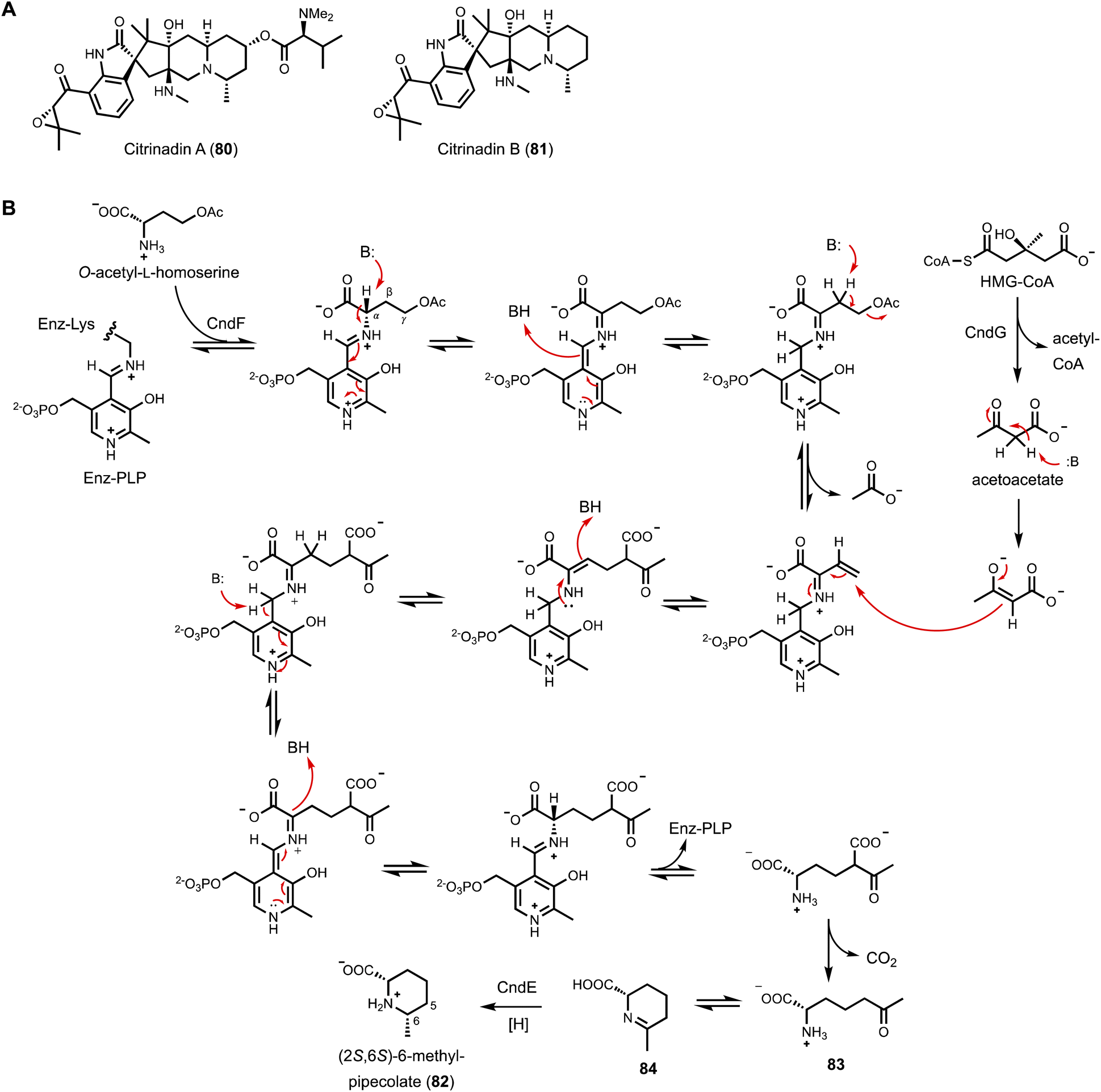 | ||
| Fig. 13 (A) Structure of citrinadins. (B) Biosynthetic pathway of forming (2S,3S)-6-methylpipecolate. The mechanism of γ-substitution catalyzed by CndF was proposed. O-Acetyl-L-homoserine was first bound to PLP as the external aldimine. After eliminating acetate, acetoacetate generated from HMG-CoA by HMG-CoA lyase then attacks the γ position to form new C–C bond. | ||
In a parallel study, a two-enzyme combination, including a PLP-dependent enzyme (Fub7) and an FMN-dependent oxidase (Fub9), was discovered in fusaric acid (85) biosynthetic pathway to construct 5-alkyl substituted picolinic acid.153 Fusaric acid is an inhibitor of dopamine β-hydroxylase produced by Fusarium species, and BGC was previously identified through genetic knockout experiments.154 A 6π electrocyclization step was proposed to generate the picolinic acid moiety. Interestingly, the BGC contains a HRPKS (Fub1) and a NRPS-like CAR (Fub8), as well as a few other enzymes including Fub7 and Fub9. Heterologous expression in A. nidulans was performed to clarify the biosynthetic route as shown in Fig. 14A. Coexpression Fub1, Fub4 (α/β hydrolase), Fub6 (reductase), Fub7, Fub8 and Fub9 led to biosynthesis of 85, establishing the minimal set of enzymes needed. In vitro reconstitution showed that Fub1, 4, 6 and 8 collectively synthesizes n-hexanal, a rather simple aldehyde substrate that requires the collaborative functions of an HRPKS (carbon backbone generation) and a CAR (reductive release as aldehyde). The α-carbon of n-hexanal turned out to be the carbon nucleophile in the Fub7-catalyzed γ-replacement C–C bond formation using O-acetyl-L-homoserine as the latent electrophile. The resulting (2S)-2-amino-5-formylnonanoic acid (86), upon release from the PLP enzyme, can cyclized into the Schiff base tetrahydrofusaric acid (87). Fub9 was confirmed to catalyze the four-electron oxidation to generate 85. Similar to CndF, Fub7 showed substantial promiscuity towards aldehyde substrates with different carbon length and substituents. Combining Fub7 and Fub9 activities, a panel of 5-alkyl, 5,5-dialkyl, and 5,5,6-trialkyl-pipcolic acids were prepared. PLP-enzymes with sequence homology to CndF and Fub9 are widely found in fungal BGC and can serve as new leads to in genome mining efforts.
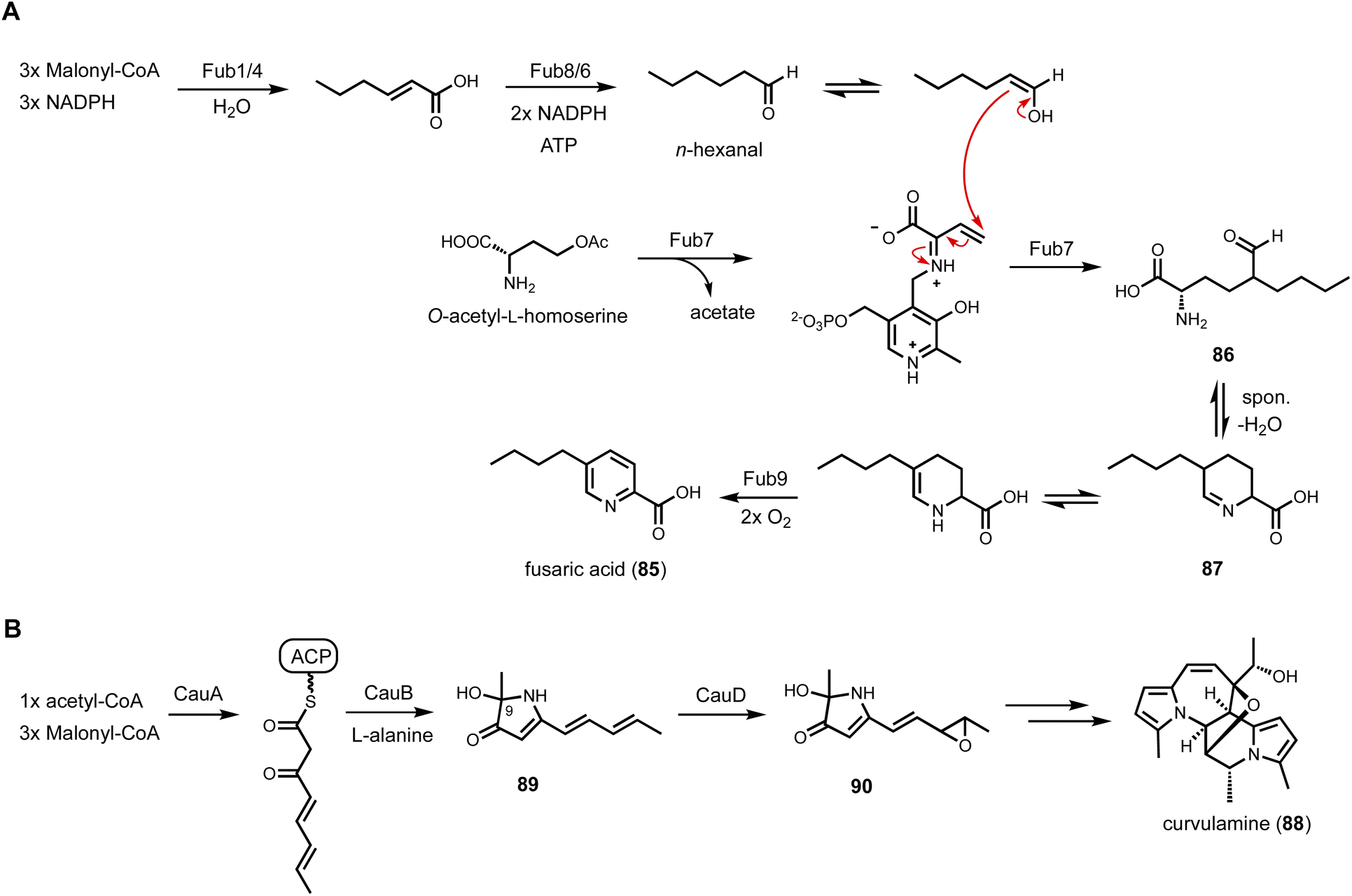 | ||
| Fig. 14 (A) Proposed biosynthetic pathway of fusaric acid. Fub7 is a PLP-dependent enzyme that catalyzes γ-replacement C–C bond formation. The mechanism is similar to CndF. (B) Simplified biosynthetic pathway of curvulamine, highlighting a bifunctional PLP-dependent enzyme, CauB, that can catalyze Claisen condensation to release PKS product as well as hydroxylation on the substrate. | ||
A bifunctional PLP-dependent enzyme was discovered to work together with an HRPKS, leading to the discovery of new biosynthetic logic to form the indolizidine scaffold.155 The indolizidine structure is commonly found in various plant alkaloids, including vinblastine and vincristine. An early study of indolizidine formation in fungi was that of swainsonine (34) biosynthesis.111 It was reported that the formation of indolizidine requires the reductive release of polyketide chain from a NRPS-PKS assembly line (Fig. 7A). However, the same biosynthetic logic cannot rationalize the biosynthesis of curvulamine (88) (Fig. 14B). Dai et al., discovered a new indolizidine forming pathway through a collaboration between a HRPKS and a PLP-dependent enzyme. The putative BGC was identified from analyzing RNA expression levels and verified with gene inactivation. The BGC encodes a HRPKS (CuaA), a PLP-dependent aminotransferase (CuaB), a SDR (CuaC) and a FMO (CuaD). Heterologous expression of CuaA and CuaB in both A. oryzae and yeast led to accumulation of 89, which contains a 3H-pyrrol-3-one structure with hydroxylation at C9 position. Further coexpression of CuaD led to epoxidation of 89 to 90. Biochemical characterization showed that CuaB alone can catalyze formation of 89 when using a SNAC-tethered polyketide substrate mimic. The origin of the C9 hydroxyl group was confirmed to derive from molecular oxygen, leading to a proposed mechanism in which a resonance stabilized carbanion activates molecular oxygen to perform substrate oxidation, which has been noted for PLP-dependent enzymes in natural product biosynthesis.156 CuaB is therefore a bifunctional PLP-dependent enzyme in biosynthesis of curvulamine. It catalyzes a Claisen condensation to release the polyketide chain from a HRPKS, followed by substrate-hydroxylation in a paracatalytic reaction.
3.4. Other considerations
The above examples rely on the clustered nature of biosynthetic genes, in which a BGC encodes all the necessary proteins and enzymes for biosynthesis of target compound. In fungi, this appears to be overwhelmingly the case, which has greatly facilitated biosynthetic reconstitution and genome mining. However, given the intrinsic difficulty in finding unclustered genes, the occurrence of unclustered BGCs is surely underestimated. One example is that of the meroterpenoid austinol (91) discovered by the Wang group.157 The BGC of 91 was initially searched for by using PKS and prenyltransferase as targets. However, no such enzyme combination was found to be colocalized in A. nidulans genome. An intensive gene inactivation experiment that afforded 20 different mutant strains was carried out to identify gene candidates involved in biosynthesis of 91, which were located in two different BGCs on the A. nidulans genome.In the case where a single enzyme is unclustered with the BGC, heterologous biosynthesis can serve as a facile approach to screen potential candidates. Such a case study can be found in the identification of unclustered terpene cyclases in the biosynthesis of fungal indole diterpenes, such as aflavinine (92) and anominine (93) (Scheme 4).55 These compounds are antiinsectants and are produced in the sclerotia of several Aspergillus species. All biosynthetic genes for related compounds such as paxilline (5) and aflatram (94) are clustered, which include a geranylgeranyl diphosphate synthase (GGPPS), a GGPP transferase that forms geranylgeranyl indole, multiple epoxidases and an indole diterpene cyclase (IDTC). However, no gene encoding an IDTC was clustered with the other biosynthetic genes in the hosts that produce 92, 93 and tubingensin A (95). A phylogenetic analysis of all the IDTC homologues found in the genomes of the producing strains led to the identification of numerous candidates. Using yeast as a heterologous host, the IDTC candidates were individually coexpressed with the other biosynthetic enzymes followed by metabolite analysis. This led to the verification of AfB and AtS2B as unclustered IDTCs in the pathways of anominine and aflavinine, respectively. The evolutionary reason for such unclustered feature is unknown. One possible explanation is that these IDTCs are co-regulated with sclerotia-specific genes and are not co-regulated with the upstream enzymes in the BGCs that synthesize the uncyclized indole diterpene. Regardless of the biological intentions, one should keep in mind the possibility of unclustered biosynthetic genes in genome mining efforts.
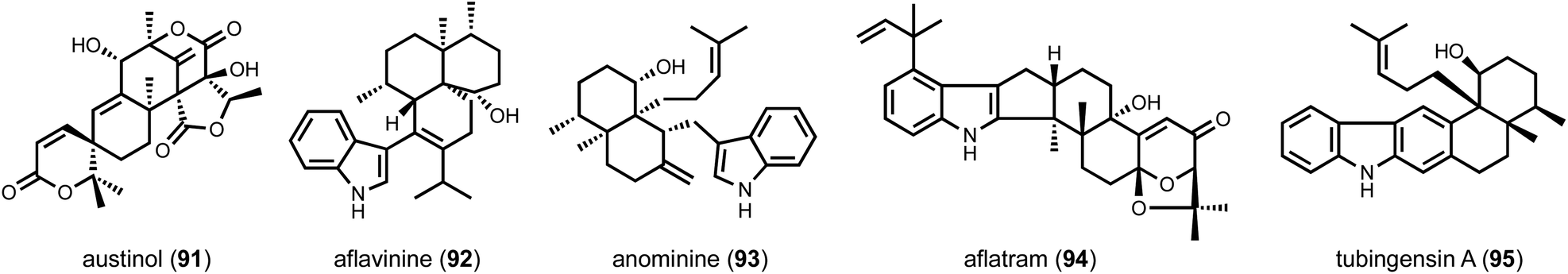 | ||
| Scheme 4 Examples of fungal indole diterpenes. | ||
On the other hand, duplication of biosynthetic genes into multiple BGCs in the same host has also been noted and should be considered in genome mining. One such example is in the biosynthesis of the heptacyclic duclauxin (96) from Penicillium duclauxi (Fig. 15).158 Duclauxin belongs to fungal aromatic polyketides that has a characteristic 6/6/6/5/6/6/6 ring system derived from the dimerization of oxaphenalenones. Structural variations among duclauxin derivatives arise from different oxaphenalenone monomers and dimerization regiospecificities. The precursor of oxaphenalenone monomers, phenalenone (97) is synthesized by an NRPKS and oxidatively rearranged by a FMO.159 In the producing strain Talaromyces stipitatus ATCC 10500, the dux1 cluster was identified using NRPKS and FMO as BLAST queries. Gene deletion of NRPKS (duxI), completely abolished the production of 96. Remaining redox enzymes encoded in the dux1 cluster include two P450s (DuxD and DuxL), oxidoreductase (DuxB), NAD(P)H-dependent reductase (DuxA, DuxG, and DuxJ), and a cupin family oxygenase (DuxM), which were proposed to be essential in transforming phenalenone to oxaphenalenone. While inactivation of some of these, such as ΔduxH and ΔduxM led to the complete abolishment of 96, other knockouts such as ΔduxA and ΔduxL had no effects. Detailed BLAST analysis revealed a separate cluster, dux2, encoding DuxA′, DuxB′, DuxC′, DuxD′, DuxG′, DuxH′, DuxL′, and DuxM′, all of which showed over 70% sequence identity to the corresponding proteins in dux1 cluster. RT-PCR analysis further confirmed that both clusters were expressed when the production of 96 was detected in T. stipitatus. Because of such gene duplication of the dux clusters, heterologous expression turned out to be the more informative approach to understand individual dux gene functions. Stepwise reconstitution in both yeast and A. nidulans solved the pathway of 96, highlighting DuxM is the key enzyme for diversifying the structures of oxaphenalenone monomers. DuxM initiates the redox reaction sequence by an oxidative cleavage of phenalenone through a transient hemiketal-oxaphenalenone intermediate (98). DuxM can further catalyze decarboxylation of 98 to give the anhydride 100, while three other enzymes (DuxJ, DuxD, and DuxG) in the biosynthetic pathway can morph 98 into 99. Lastly, P450 DuxL catalyzes the oxidative heterocoupling of 99 and 100 to form cryptoclauxin (101), or homocoupling of 99 to duclauxin (96) (Fig. 15).
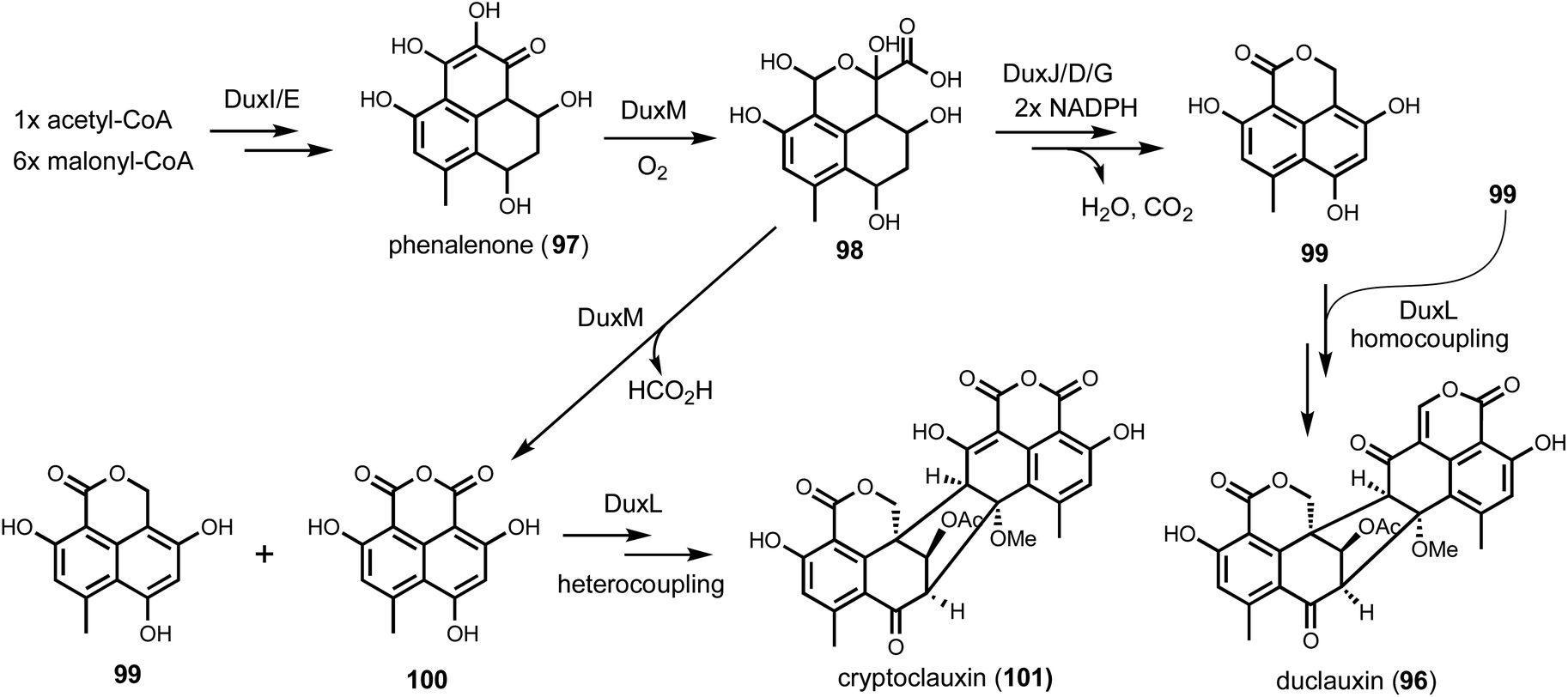 | ||
| Fig. 15 Proposed pathway of duclauxins. DuxM is the key enzyme to start the redox modifications, leading to the production of two different monomers 99 and 100. DuxL then catalyzes coupling reaction to give duclauxin or cryptoclauxin. | ||
4. Shunt product formation during heterologous reconstitution
Shunt product or intermediate? That is the question asked by nearly all researchers working on biosynthesis of natural products. Regardless of using native hosts or heterologous hosts, a change in metabolite profile following either a gene knockout or gene introduction is an exciting result. Identifying an on-pathway intermediate is a desired outcome in reconstruction of the biosynthetic pathways. However, off-pathway shunt products are frequently formed due to chemical instability of intermediates or crosstalk between targeted pathway and endogenous metabolism. In most cases, formation of shunt products presents an obstacle in pathway reconstitution because downstream enzymes cannot recognize the compound to advance the pathway. Incorrect assignment of a shunt product as a biosynthetic intermediate can significantly derail the investigation and lead to misinterpretations of the chemical logic. On the other hand, careful analysis of shunt product structures can provide revealing chemical insights into biosynthetic pathways. One such example is from studying the iterative HPRKSs from fungi, typified by LovB from the lovastatin pathway. Shunt product characterization from the bottom-up reconstitution in heterologous hosts, as well as from interruption of the HRPKS domain functions, revealed highly complex programming rules in chain length control, permutative reductive tailoring and methylations.52,160 HRPKS programming will not be elaborated further here, and the readers can refer to the excellent review by Cox that is also in this volume.161 In this section, we will discuss recent representative case studies of how identification of shunt products has impacted the biosynthetic mining and investigation of fungal natural products.4.1. Shunt products formed from cellular crosstalk
Shunt products can form from crosstalk of target pathway with endogenous enzymes, especially those involved in detoxification of reactive compounds such as aldehydes and α,β-unsaturated Michael-acceptors, etc. Members of the alcohol dehydrogenases (ADHs), aldehyde dehydrogenases and short-chain dehydrogenase/reductases (SDRs) families from fungi have promiscuous substrate specificities and can rapidly reduce aldehydes into the corresponding primary alcohol. In fungal biosynthetic pathways, one route to generate an aldehyde intermediate is through the reductive release of thioesters by R domains appended at the end of assembly lines (see Section 3). In PKS-NRPS assembly lines terminated with R domains, the released aldehyde is immediately cyclized via Knoevenagel condensation with the α-carbon of the 1,3-diketo portion of the polyketide chain to yield a pyrrolidine ring. Although this reaction can occur spontaneously in some pathways, a dedicated hydrolase-like enzyme is required in most studied pathways.128,162 In the absence of the hydrolase, the aldehyde is readily reduced to the shunt product alcohol. This was observed in cytochalasin reconstitution in A. oryzae and in mining of oxaleimides in A. nidulans.69,128,163 In the oxaleimide example, which was discussed in detail above, structure of the alcohol shunt product led to clarification of the biosynthetic origin of the unusual succinimide group. One strategy found in fungal biosynthesis to counter the propensity of aldehydes to be reduced to alcohol shunt products is the presence a flavin-dependent oxidase that oxidizes the alcohol to the aldehyde. This strategy was uncovered in the biosynthesis of pyriculol (102) which is a devastating mycotoxin. Genome mining of a HRPKS-containing pathway in Neurospora crassa led to the biosynthesis of sordarial (72), a related salicylaldehyde.147 In analogous steps to the trichoxide example in Fig. 11B, the salicylaldehyde is synthesized by the HRPKS and a cadre of redox enzymes. However, the aldehyde (103) is readily reduced to yield the salicylic alcohol (104) in A. nidulans (Fig. 16A). Introduction of a flavin-dependent oxidase, however, restored the formation of the salicylaldehyde that is further processed into sordarial. This is also the role of a homologous oxidase in the pyriculol pathway in Magnaporthe oryzae, as knockout of this enzyme led only to biosynthesis of the nonvirulent dihydroxypyriculol (105).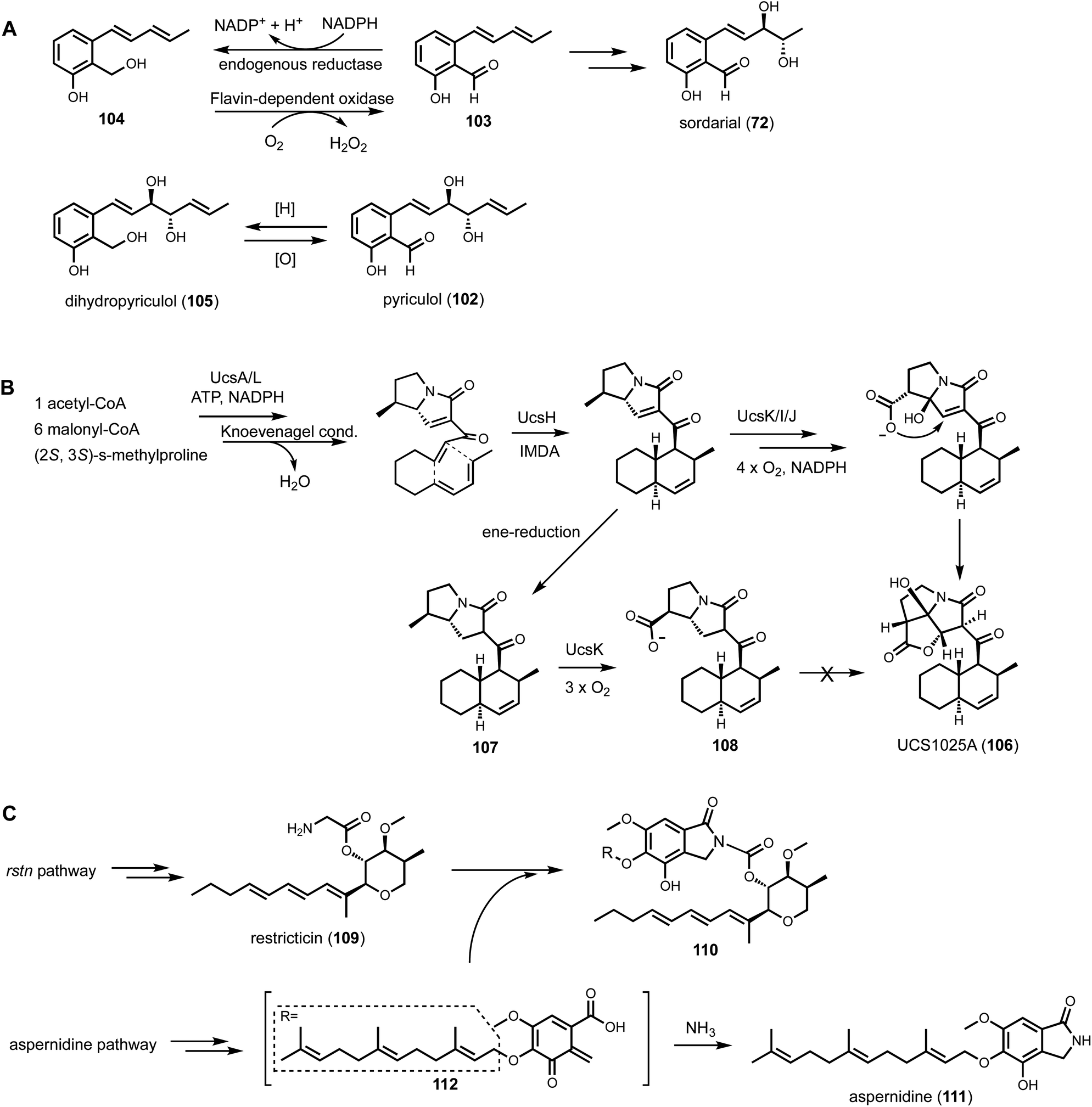 | ||
| Fig. 16 Shunt products formed by cellular crosstalk. (A) Two examples of endogenous NADPH-dependent reduction convert aldehydes to alcohols. The shunt products can be reoxidized by flavin-dependent oxidase to be further modified in the biosynthetic pathway of salicylaldehyde products. (B) Biosynthesis of UCS1025A as an example of ene reduction hampering heterologous reconstitution. (C) Proposed cellular crosstalk between restricticin and A. nidulans endogenous metabolite aspernidine. | ||
The reduction of α,β-unsaturated moieties by endogenous ene-reductases is another common occurrence that can hamper heterologous reconstitution efforts. Ene-reductases are flavin-dependent enzymes that can perform a 1,4-addition with a hydride to yield a saturated shunt product. This was most clearly demonstrated in the attempted reconstitution of UCS1025A (106) in A. nidulans.164 UCS1025A is a pyrrolizidinone (azabicyclo [3.3.0] octanone)-containing natural product and is a strong telomerase inhibitor.165 The pyrrolizidinone is fused with a γ-lactone to give a furopyrrolizidine that is further connected to a polyketide derived trans-decalin. While UCS1025A was initially isolated from Acremonium sp. KY4917 and a putative gene cluster was located, difficulties involved in growing Acremonium sp. and subsequent molecular biology experiments, led to the mining of a homologous cluster found in Myceliophthora thermophila. Transcriptional factor overexpression led to production of UCS1025A in this host and confirmed the metabolite of the ucs cluster. The pyrrolizidinone ring is generated by the function of a PKS-NRPS, which aminoacylates the polyketide with (4S, 5S)-4-methylproline. Reductive release and Knoevenagel condensation afford the pyrrolizidinone ring system. However, attempts to characterize the oxidative transformations to furopyrrolizidine using heterologous hosts such as yeast was not successful. This was primarily due to the facile reduction of the ene moiety in pyrrolizidinone to the saturated product. Although these shunt products cannot be elaborated into UCS1025A, the 6S-methyl group in an early shunt product 107 can be oxidized by a P450 enzyme UcsK into the corresponding carboxylic acid (108), which confirmed the role of UcsK in the pathway. Characterization of the shunt products also led to an improved biosynthetic pathway proposal in which an oxa-Michael cyclization involving the carboxylate group forms the final product (Fig. 16B).
Cellular crosstalk can also come from nonenzymatic reactions between the reactive warhead of the target natural product and endogenous metabolites, via a possible mechanism of cellular detoxification. One such example is in the heterologous biosynthesis of restricticin (109).166109 is a potent antifungal natural product that inhibits lanosterol 14α-demethylase (CYP51), which is a validated antifungal target. A putative gene cluster for restricticin was found in Aspergillus nominus, a strain not known to produce the compound, using the SRE-guided approach. The rstn cluster encodes a HPRKS that is responsible for synthesizing carbon skeleton of the polyene-tetrahydropyran unit, a single-module NRPS that esterifies the hydroxylated tetrahydropyran with glycine, a set of accessory enzymes and a self-resistance version of CYP51. Heterologous expression of the cluster in A. nidulans in CDST media led to formation of trace amounts of restricticin, with the major product being the N-acetylated restricticin. N-acetylated 109 is significantly weaker as an antifungal since the primary amine in 109 is involved in coordinating to the heme iron in CYP51. The amine, however, is modified in a more surprising fashion when the heterologous strain was grown in the minimal CD media in which no restricticin was observed in the extract. Instead, an adduct (110) between restricticin and the A. nidulans metabolite aspernidine (111) was formed, in which the free amine is incorporated as part of the isoindolone ring. The adduct is proposed to form from the attack of restricticin on the quinone methide precursor (112) of 111, followed by cyclization (Fig. 16C). In comparison, 111 is formed when 112 is attacked by ammonia. It is not clear if this is a cellular strategy to scavenge free amine-containing compounds. In this example, even though restricticin was not isolated as a pure compound from the heterologous host, formation of acetylated and modified versions of the compound confirmed the role of the BGC, which led to identification of additional CYP51 inhibitors such as lanomycin using genome mining approaches.166
4.2. Shunt products from pericyclic reactions in biosynthesis
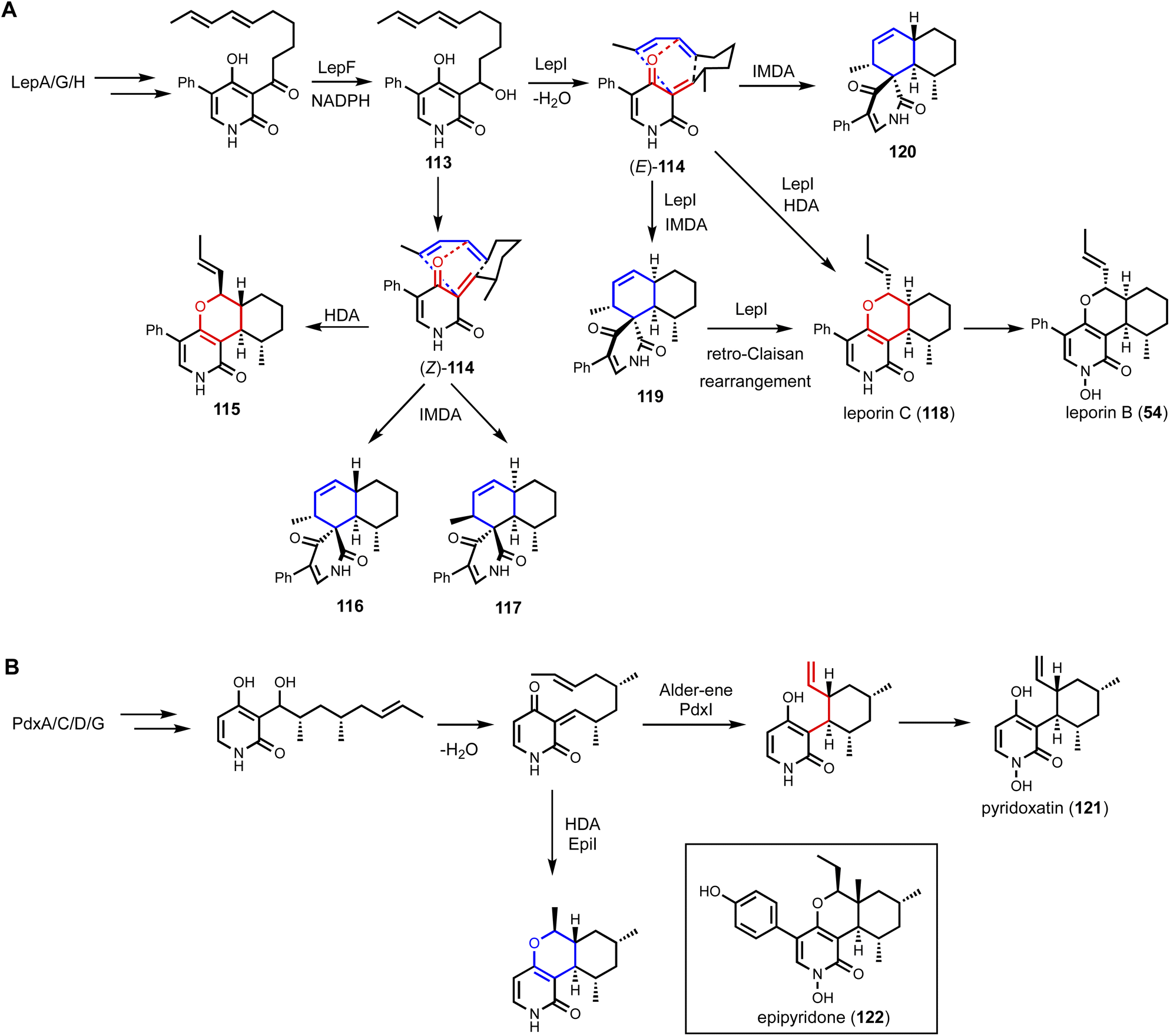 | ||
| Fig. 17 Pericyclases in 2-pyridones biosynthesis. (A) Biosynthetic pathway of leporin B. HDA reaction is highlighted in red, while IMDA reaction is labeled in blue. (B) Two O-MT-like pericyclases, PdxI and EpiI, catalyze Alder-ene reaction and a different HDA reaction, respectively. | ||
Interestingly, even though no O-methylation step is required for leporin B biosynthesis, the BGC contains a putative O-MT (LepI). Initial identification and gene knockout studies in the native producing strain showed lepI was required for leporin biosynthesis and unidentified shunt products were produced upon deletion.169 When LepI was coexpressed with other Lep enzymes in an A. nidulans strain that produced the aforementioned mixture of products 115–120, exclusive production of the desired HDA reaction product 118 was observed without any regioisomeric and stereoisomeric shunt products. Further biochemical characterization of LepI revealed that it is a multifunctional SAM-dependent pericyclase that catalyzes (i) stereoselective anti-dehydration of 113 to (E)-114; (ii) two pericyclic transformations: undesired IMDA and desired HDA reactions to form 119 and 118, respectively; and (iii) the first enzymatic retro-Claisen rearrangement of 119 to 118. Co-formation of 118 and 119 was calculated to be thermodynamically unavoidable, since the two compounds bifurcate from an ambimodal endo transition state from (E)-114. LepI therefore catalyzes the additional retro-Claisen rearrangement to convert the shunt product to 118. This represents a kinetic ‘by-product recycle’ process to overcome thermodynamic limitations and to arrive fully at the desired biosynthetic end product. Ensuing crystal structure analysis showed a modified O-MT-like active site, in which a histidine and an arginine were introduced to accelerate the reactions. The LepI-substrate and LepI-product structures also showed the presence of a hydrophilic wall in the active site to suppress formation of the undesired 120 from the exo IMDA reaction.170
This finding echoes Liu and coworkers' discovery of a bacterial MT-like enzyme, SpnF, in catalyzing the [4 + 2] cycloaddition reaction during spinosyn biosynthesis.171 Both studies showcased how Nature repurposes a common MT fold to afford precise control of regioselectivity, stereoselectivity and periselectivity of a pericyclic reaction. Heterologous reconstitution of leporin biosynthesis led to a genome mining effort using LepI as a lead to study other pericyclic reactions in 2-pyridone fungal natural products. Heterologous expression combined with biochemical and computational characterization of the enzymes were used in these studies to arrive at new pericyclases. Examples include the C-methyltransferase like enzyme IccD from ilicicolin H (2) biosynthetic pathway that catalyzes an inverse electron demand Diels–Alder reaction;33O-MT-like enzymes PdxI from the pyridoxatin (121) biosynthetic pathway that catalyzes an Alder-ene reaction; and a new HDAse EpiI from epipyridone (122) biosynthetic pathway (Fig. 17B).172
Although several decalin-forming Diels–Alderases have been discovered from bacterial pathways,171 fungal decalin-forming Diels–Alderases were only discovered recently. In 2015, Sato et al. reported the discovery of CghA from the biosynthetic pathway of Sch210972 (123) isolated from Chaetomium globossum.176 The structure of 123 contains a trans-fused decalin ring system connected to a tetramic acid moiety derived from γ-hydroxylmethyl-L-glutamic acid. The cgh BGC encodes a PKS-NRPS (CghG), a trans-ER (CghC), a lipocalin-like enzyme (CghA), an aldolase (CghB), and a transcription factor (CghD). The aldolase CghB was characterized to catalyze the aldol reaction of two pyruvate molecules to yield γ-hydroxylmethyl-L-glutamic acid.177 The only candidate enzyme remaining in the pathway to catalyze the IMDA reaction is CghA, which is a lipocalin-like protein with unknown function. CghA homologs are found in many fungal BGCs that are connected to products formed via IMDA reactions, including other pyrrolidine-2-one-bearing decalin compounds and cytochalasans such as cytochalasin E (124), in which the isoindolone ring system was proposed to be formed by the homologous CcsF.178 The role of CghA was tested through heterologous expression of CghG, CghC, and CghB with or without CghA in A. nidulans.176 Whereas heterologous expression of CghG, CghC, CghB, and CghA in A. nidulans gave the trans-decalin 123 exclusively, excluding CghA from the heterologous pathway led to the formation of not only 123 but also the cis-decalin diastereomer as the shunt product (125) (Scheme 5). 123 and 125 are formed through the endo and exo transition states, respectively, both of which were computationally predicted to be kinetically accessible. The heterologous expression results therefore indicated CghA is a pericyclases that catalyzes the endo-selective IMDA reaction while suppressing the exo-IMDA reaction. Recent structural and computational studies of CghA identified key residues in the active site that confer such stereoselectivity through steric interactions with the acyclic intermediate. Mutations to these residues led to inversion of stereoselectivity.179 At the time of CghA discovery, Osada and coworkers arrived at the same conclusion that Fsa2, a homolog of CghA from the equisetin (126) biosynthetic pathway, is a lipocalin-like Diels–Alderase that catalyzes the endo-selective decalin-forming Diels–Alder reaction.180 The pioneering discoveries of CghA and Fsa2 have led to the identification of other lipocalin-like pericyclases including Eqx3, PvhB,181 MycB,125 UcsH,164 and AspoB.182
 | ||
| Scheme 5 Metabolites discussed in Section 4.2.2. | ||
Depending on molecular orbitals (MO) involved in the reaction, DA reaction can be further classified as normal-electron demand Diels–Alder reactions (NEDDA) and inverse-electron demand Diels–Alder reactions (IEDDA). In the case of NEDDA, the DA reaction happens between an electron-rich dienophile and an electron-deficient diene, while in IEDAA, the reaction takes place between an electron-rich diene and an electron-poor dienophile. Among many known Diels–Alderases characterized so far, the biosynthesis of varicidin A (127) involves IEDDA to control the selectivity of IMDA reaction.181 The pvh BGC from Penicillium variabile was targeted for mining because the clustering of a PKS-NRPS (PvhA) and a lipocalin-like enzyme (PvhB). In addition to those two enzymes, the pvh cluster also encodes a trans-ER (PvhC) that functions collaboratively with PKS-NRPS (PvhA), N-methyltransferase (PvhD), and a P450 (PvhE). Coexpression of PvhA with PvhC in A. nidulans accumulated three metabolites 128–130 (Fig. 18A). 128 is an acyclic tetramate-containing compound that is the product of PKS-NRPS and trans-ER. The presence of the diene and dienophile pairs in 128 led to the initial proposal that this is the substrate of the Diels–Alderase. The pair of 129 and 130 were structurally characterized to be trans-decalin diastereomers, which are expected to derive from nonenzymatic IMDA of 128. This further solidified the hypothesis that PvhB may be required to control the stereochemistry to give either 129 or 130 as the desired biosynthetic intermediate. However, further coexpression of PvhB did not change the metabolic profile in A. nidulans transformant, suggesting 129 and 130 are in fact shunt products instead of intermediates. Coexpression of the P450 PvhE with PKS-NRPS and trans-ER led to the carboxylated compound 131, which does not undergo nonenzymatic IMDA reaction. Coexpression of PvhB in the above strain exclusively produced the cis-decalin 127, while no other diastereomer can be detected. Therefore, PvhB selectively catalyzes exo-cycloaddition of the carboxylated substrate in an IEDDA reaction to give the cis-decalin product. DFT calculations on either 128 or 131 showed the activation barrier for reaching the IEDDA reaction transition state is higher than that of the NEDDA reaction. However, upon carboxylation in 131, the exo transition state of IEDDA reaction required to form the cis-decalin becomes more kinetically accessible compared to the carboxylated endo transition state. Therefore, the biosynthesis of varicidin unveiled an interesting strategy for Nature to access the cis-decalin structure: carboxylative deactivation of the nonenzymatic NEDDA reaction followed by enzymatic IEDDA reaction to control the diastereoselectivity.
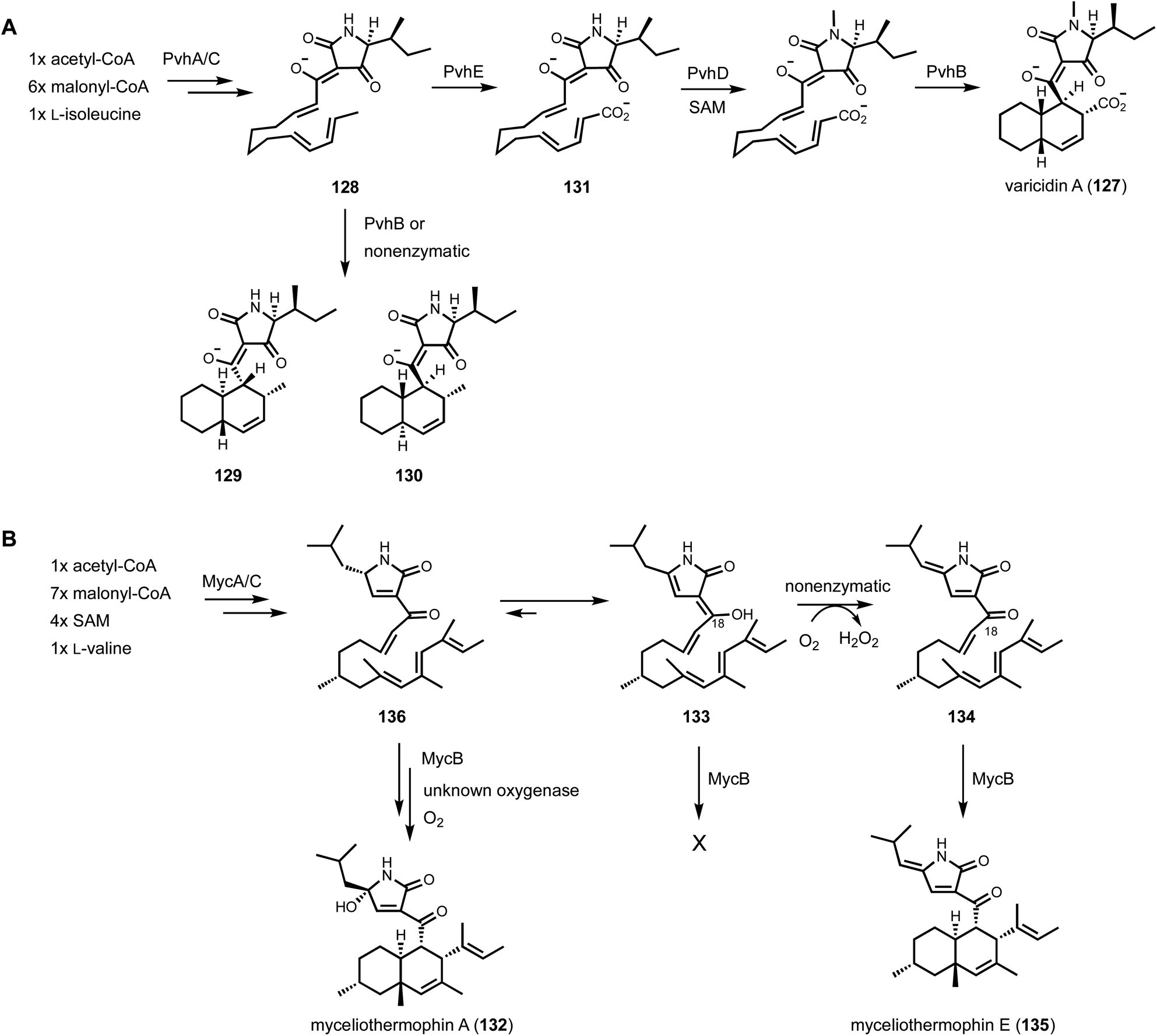 | ||
| Fig. 18 Pericyclases in biosynthesis of decalin-containing natural products. (A) Biosynthetic pathway of varicidin. Carboxylation catalyzed by PvhE is required for the formation of cis-decalin product via a IEDDA mechanism. (B) Biosynthetic pathway of myceliothermophin. The shunt product 134 isolated from heterologous host allowed in vitro reaction to verify the function of MycB. | ||
While shunt products derived from off-pathway oxidation or reduction are undesirable in heterologous reconstitution, in certain cases, such modification may slow down nonenzymatic reactions and can be useful in biochemical assays if the enzymatic recognition moiety in the structure remains unchanged. This was observed in the reconstitution of myceliothermophin A (132) biosynthesis using A. nidulans.125 The structure of 132 contains a trans-fused decalin ring system connected to a 3-pyrrolin-2-one moiety derived from reductive release from a PKS-NRPS. A compact three-gene myc BGC from Myceliophthora thermophila was identified and expressed in A. nidulans. Coexpression of PKS-NRPS (MycA) and trans-ER (MycC) gave two new metabolites 133 and 134, both with a 3-pyrrolin-2-one moiety. 133 is an acyclic polyolefin in the enol form at C18, while 134 is a further oxidized version of 133 with the keto form at C18 (Fig. 18B). The oxidation of 133 to 134 is nonenzymatic and involves the generation of hydrogen peroxide. Upon incubation with the lipocalin-like enzyme MycB, no reaction of the enol 133 can be observed, while complete transformation of 134 to the product myceliothermophin E (135) was seen. Based on this data, it is proposed that the ketone tautomer of 133, 136, is the true substrate of MycB and can undergo cycloaddition to the final product 132. In the absence of MycB, the inactive enol tautomer 133 can be formed and can undergo oxidation to 134.
Biosynthetically, pyrrocidines are derived from the 3-pyrrolin-2-one intermediate, which is synthesized by a PKS-NRPS in collaboration with a trans-ER as suggested by Oikawa.186 The intermediate is proposed to cyclize into the para-cyclophane and form ring C through either electrophilic cyclization or a radical mechanism, connecting the tyrosine phenol and the acyclic polyketide chain, as shown by Nay.187 A subsequent IMDA reaction is proposed to yield the decahydrofluorene core. Based on the total synthesis of hirsutellone B (139), formation of the cis-fused A/B ring system in pyrrocidines from the proposed IMDA reaction is highly challenging, which implicates the involvement of an exo-specific pericyclase in this pathway. Such retrobiosynthetic analysis led to identification of putative pyd cluster which encodes a PKS-NRPS (PydA), trans-ER partner (PydC), a lipocalin-like enzyme (PydB), a medium-chain dehydrogenase/reductase (MDR, PydE), an α/β hydrolase (PydG), and three small hypothetical proteins PydX, PydY, and PydZ. Heterologous expression of PydA, PydC, and PydG in A. nidulans led to the formation of three shunt products 140–142 (Fig. 19). Compound 140 was determined to be a polyolefinic shunt product that is converted from the enolization of the acyclic intermediate (143). The shunt product 141 is an air-oxidized shunt product of 140, while the shunt product 142 contains a cyclohexyl ring that corresponds to ring C in pyrrocidines. To advance the biosynthesis from the acyclic intermediate 143 to pyrrocidines, expression of additional enzymes in A. nidulans was performed. While coexpression with the MDR PydE and the putative Diels–Alderase PydB did not yield any new compounds, the additional coexpression of PydX and PydZ in this transformant led to the formation of pyrrocidine D (144) that is the trans diastereomer of 138. This suggested the four proteins (PydB, PydE, PydX, and PydZ) may work as a complex to cyclize 143 and form rings D and C in 139. It is proposed the acyclic chain in 143 is configured in an inverse S-shape (145) which enables the phenol to position near C13, and the fully saturated portion of the chain to form a chair-like conformation aided by the two equatorially substituted methyl groups at C9 and C11. Upon deprotonation, the phenolate attacks the C13 olefin, which drives the conjugated addition of C12 into the triene at C7 to form ring C. A hydride acceptor such as the NAD(P)+ cofactor in the MDR may be positioned near C1 to complete the reaction. In the absence of a downstream Diels–Alderase, the resultant product can undergo nonenzymatic cycloaddition through the kinetically accessible endo transition state to give the trans-fused adduct as the shunt product, which is further modified by a nonenzymatic hydroxylation of C2′ followed by the reduction catalyzed by an ene-reductase in A. nidulans to give pyrrocidine D (144).
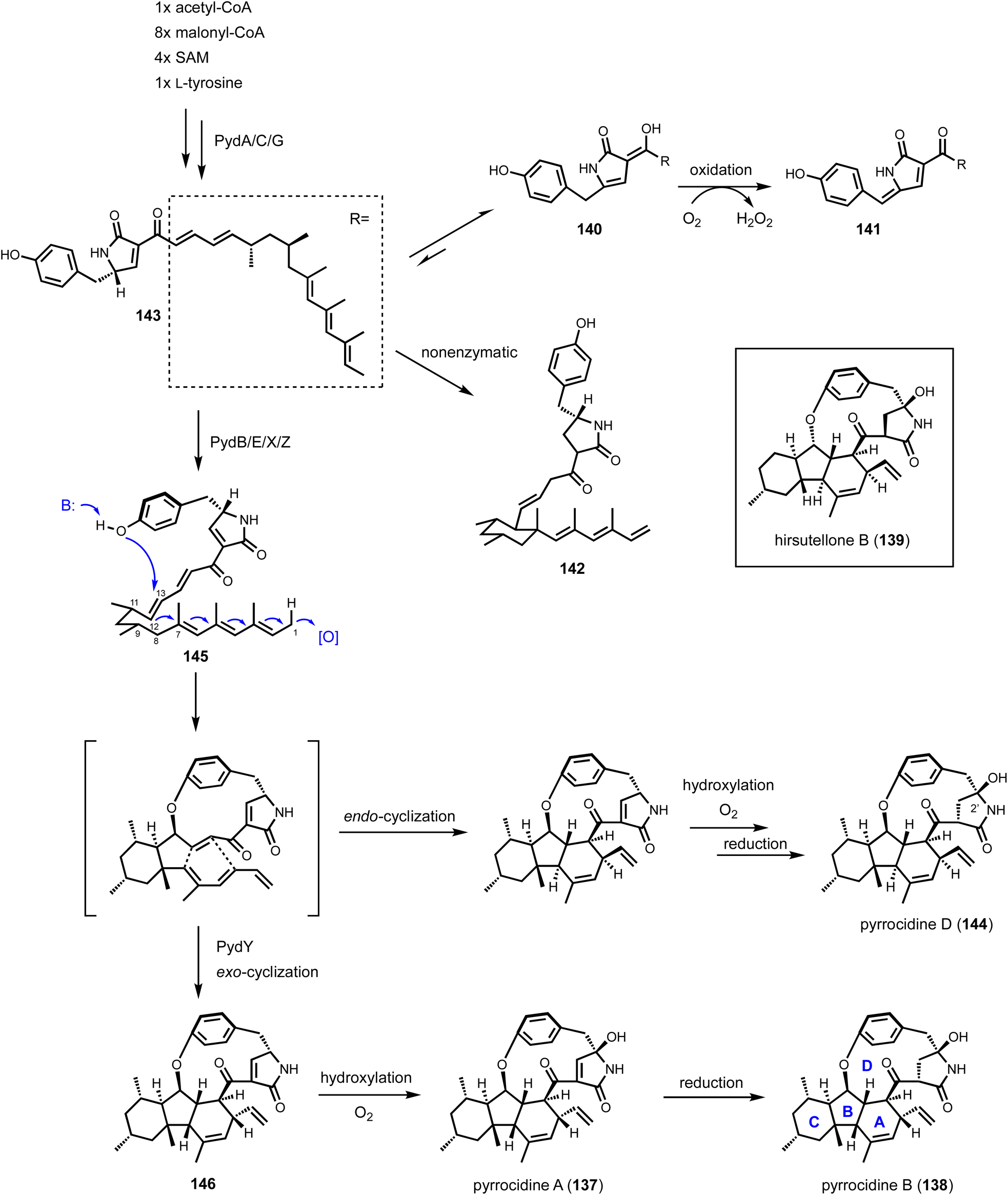 | ||
| Fig. 19 Biosynthetic pathway of pyrrocidine. Expression of the putative Diels–Alderase PydB led to the formation of shunt product, pyrrocidine D, through endo-cyclization. PydY, which is predicted as hypothetical protein, is the pericyclase that catalyzes exo-selective cycloaddition to form the ring structure of pyrrocidine A and B. | ||
Formation of shunt product 144, which was not isolated from the pyrrocidine-producing strain, suggested the lipocalin-like enzyme PydB is not the Diels–Alderase responsible for the exo-selective cycloaddition required to form 137. To test if the remaining hypothetical protein PydY is the Diels–Alderase, heterologously coexpression of this enzyme in the strain that produced 144 was performed. The resulting strain indeed produced a new compound that is structurally characterized to be the cis-fused decahydrofluorene 146. PydY, a small lipophilic protein but has no sequence homology to PydB, is therefore the responsible pericyclase in formation of the cis-decahydrofluorene. Genome mining of other BGCs in this family showed that homologs of PydY are absent in pathways that make trans-decahydrofluorene as seen in 144.
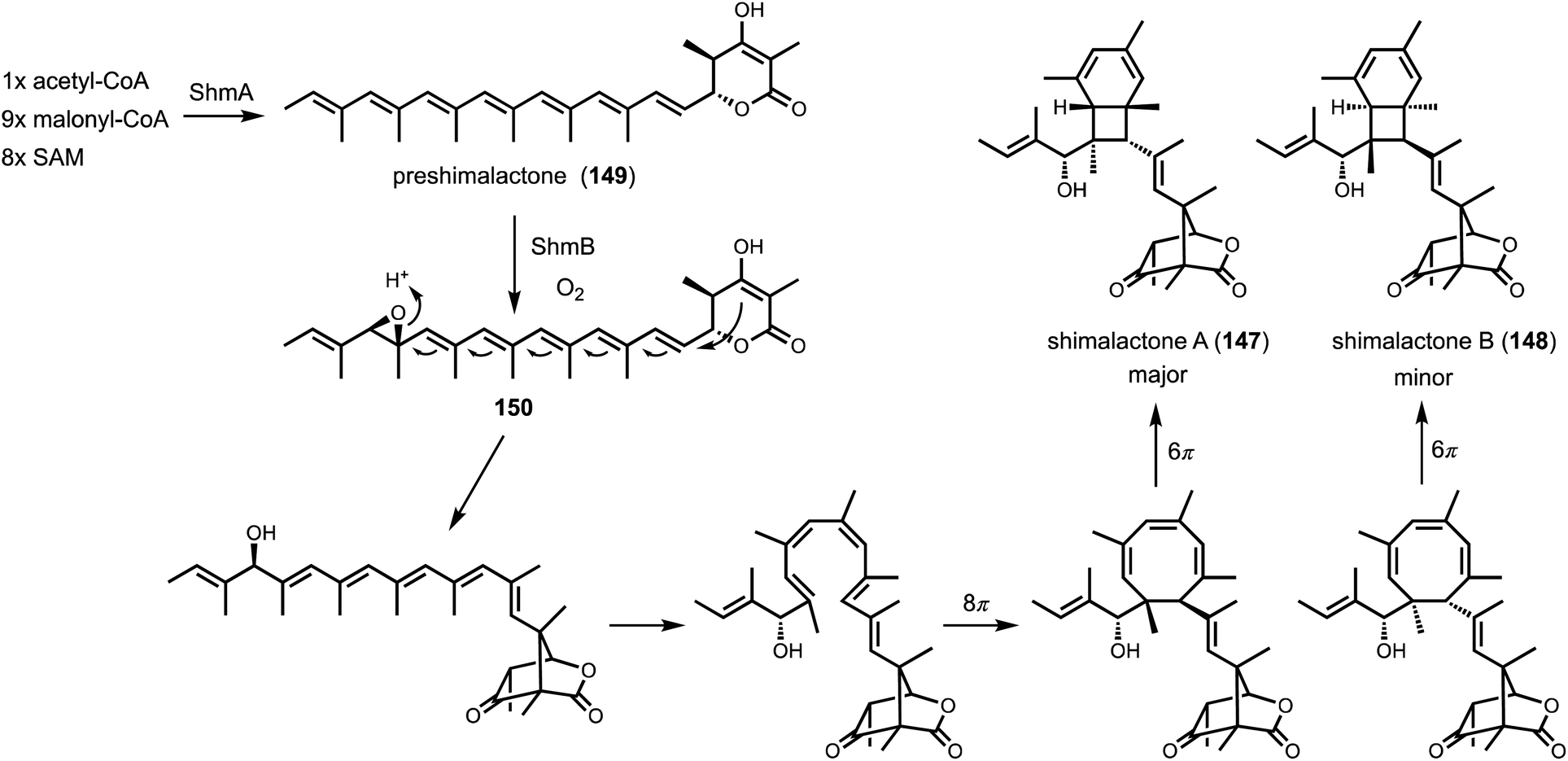 | ||
| Fig. 20 Biosynthetic pathway of Shimalactones involves nonenzymatic 8π-6π electrocyclization to generate a diastereomeric pair of products. | ||
One can conclude that a pericyclase is not involved in shimalactones biosynthesis since both 147 and 148, diastereomer of shimalactone, have been co-isolated from the same producing strain. 148 thus should not be considered as a shunt product in the biosynthesis of shimalactones. Therefore, discovering a pericyclase that catalyzes electrocyclic reactions remains an objective. It is worth noting that putative “electrocyclases” have been recently reported in bacterial biosynthetic pathways, although these electrocyclases would not be required to control the stereo- and regioselectivity based on the proposed products.189 We expect that new “electrocyclases” will be discovered from fungal or plant biosynthesis pathways in near future. As described before, such discovery will certainly involve identification of nonenzymatically formed shunt products, which are not formed in the presence of a dedicated “electrocyclase”.
4.3. Shunt products from enzymatic cyclization of polyethers
As seen in the previous example, epoxidation of polyene compounds is a useful strategy to generate complex ring structures. This strategy is most commonly seen in biosynthesis of polyethers derived from polyketides pathways.190 Baldwin's rule is widely accepted to explain the relative preference for ring forming reactions according to ring size, position of bond broken, and orbital geometry.191 An example of anti-Baldwin's rule from fungal biosynthesis was observed in aurovertin E (151),192 which has a unique 2,6-dioxa-bicyclo[3.2.1]octane (DBO) ring system. It is believed this complex bicyclic ring structure is derived from three epoxidation steps and a cascade of regioselective epoxide opening reactions. The gene cluster was found by using HRPKS as a lead. The cluster, aur, encodes an HRPKS (AurA), an O-MT (AurB), FMO (AurC), a predicted α/β hydrolase (AurD), a putative cyclase (AurE), and an acyltransferase (AurG). The function of each gene was confirmed by gene deletion as well as heterologous reconstitution in yeast. Coexpression of AurABC led to the formation of 152 and other minor metabolites including 153 (Fig. 21A). Feeding experiment confirmed that 152 is the only on-pathway intermediate, while other metabolites are shunt products that can be formed by nonenzymatic epoxide opening reactions. Additional expression of AurD with AurABC resulted in the formation of 151 together with a notable decrease in the accumulation of shunt products.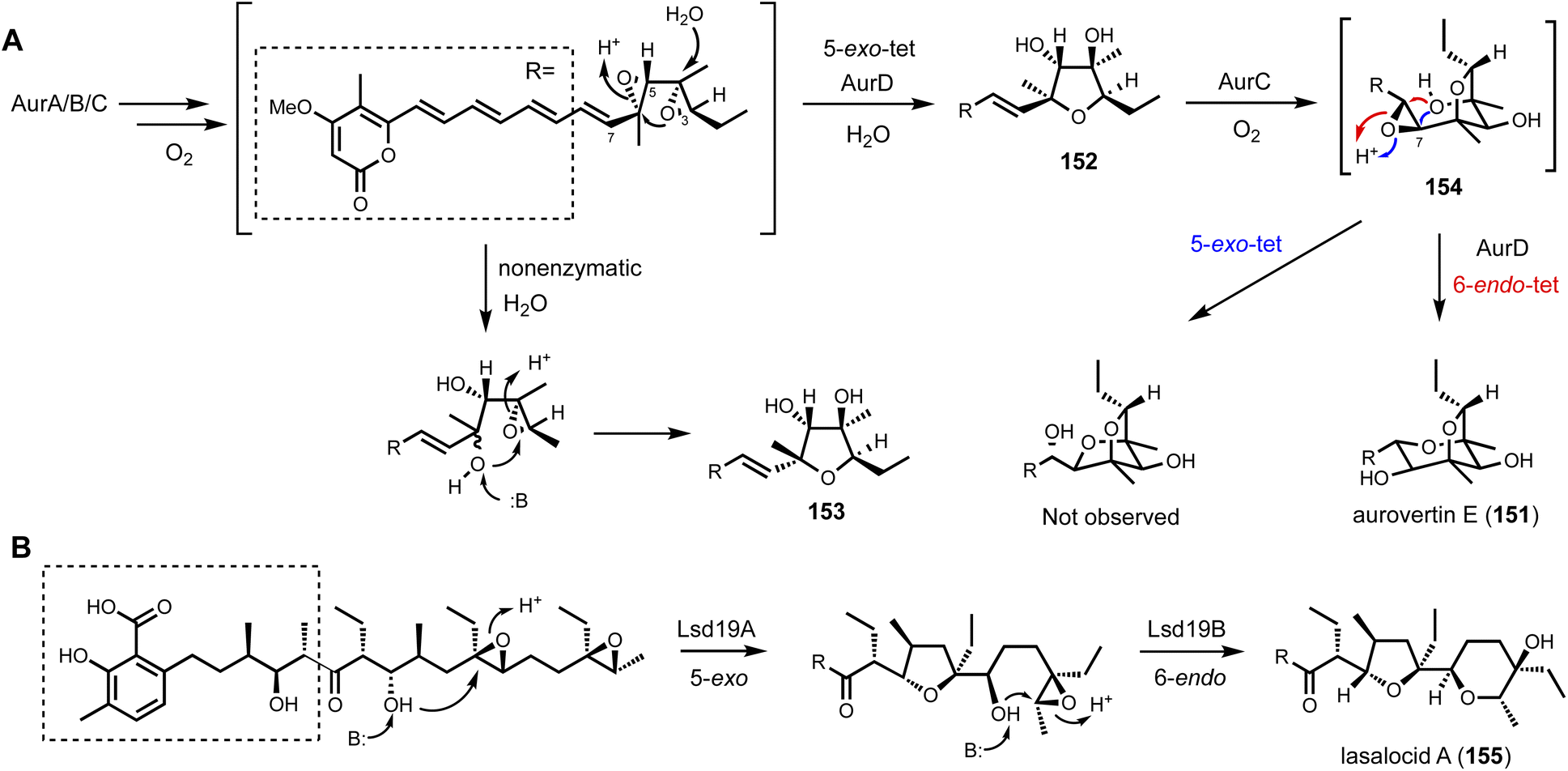 | ||
| Fig. 21 Epoxide opening reactions in polyether biosynthesis. (A) Sequential epoxide opening in biosynthesis of aurovertin. With the enzyme AurD, the final ring closure is via a 6-endo-tet reaction. (B) Sequential epoxide opening in biosynthesis of lasalocid A. The last 6-endo-tet reaction is classified as an anti-Baldwin's rule ring closure. | ||
Heterologous expression of the pathway with or without AurD led to the proposed iterative oxidation/cyclization pathway to form 151 from the polyene precursor. First, bis-epoxidation on C3–C4 and C5–C6 is catalyzed by AurC, which can nonenzymatically cyclize with no regioselective control. In the presence of AurD, regioselective 5-exo-tet epoxide ring opening leads to formation of 152. This is followed by a third epoxidation at C7–C8 catalyzed by AurC to form 154. The more favorable 5-exo-tet ring opening reaction as described by the Baldwin's rule does not take place from 154. Instead, AurD presumably catalyzes the unfavorable 6-endo-tet ring opening to give 151. It should be noted that Baldwin's rule is based on nucleophilic attack of the epoxide from an ideal angle. In the presence of an enzyme, the orientation of electrophile and nucleophile can be controlled in the active site leading to deviation of the rule. Structural insights of such deviation have been obtained from Lsd19, which is a polyether epoxide hydrolase that catalyzes disfavored 6-endo-tet epoxide ring opening during lasalocid (155) biosynthesis (Fig. 21B).193
4.4. Shunt products from dimeric radical coupling
Control of regio- and stereoselectivity during oxidative coupling reactions is essential in biosynthesis of natural products, especially those derived from dimerization of aryl monomers. In plant biosynthetic pathways, a special class of proteins known as dirigent proteins (from latin dirigere, to guide or align) controls the stereochemistry of radical-based dimerization of phenylpropanoids.194 For example, the radical-based coupling of p-coniferyl alcohol gives a 1:1 mixture of (+) and (−)-pinoresinol in the absence of the dirigent protein.194 However, in pea and Arabidopsis thaliana, dedicated dirigent protein controls stereoselectivity of the dimerization to give predominantly (+)- and (−)-pinoresinol, respectively.195,196 A similar mechanism of stereochemical control is exerted by a cotton dirigent protein in the biosynthesis of (+)-gossypol.197 Recently, a fungal version of dirigent protein that controls atroposelectivity was discovered by Chooi's group through heterologous reconstitution of bisnaphthopyrone viriditoxin biosynthesis.198 The vdt BGC was identified using naphtho-α-pyrone structure as a lead. Analysis of the metabolic profile of producing strain revealed that (M)-viriditoxin (156) is the major enantiomer and (P)-viriditoxin (157) is the minor product with an approximate 20:1 ratio. This observation suggested that the atroposelectivity of the coupling reaction is controlled by an enzyme. To investigate the pathway, coexpression of NRPKS (VdtA), O-methyltransferase (VdtC), Bayer–Villiger Monooxygease (VdtE), and SDR (VdtF) in A. nidulans led to formation of the monomer 158 (Fig. 22). Further coexpression VdtACEF with VdtB, which is a multicopper oxidase; and VdtD, a proposed hydrolase, led to the formation of 156 and 157 in 20:1 atropisomeric ratio, same as observed in the producing strain. Removing VdtD from the A. nidulans strain, however, the atropisomeric pair was produced at 1:2 ratio. In vitro assays using cell-free extracts from A. nidulans expressing VdtD and VdtB further supported that VdtD is crucial in controlling the stereoselectivity of biaryl coupling catalyzed by VdtB. This is the first example of a fungal dirigent protein, although the mechanism remains uncharacterized.
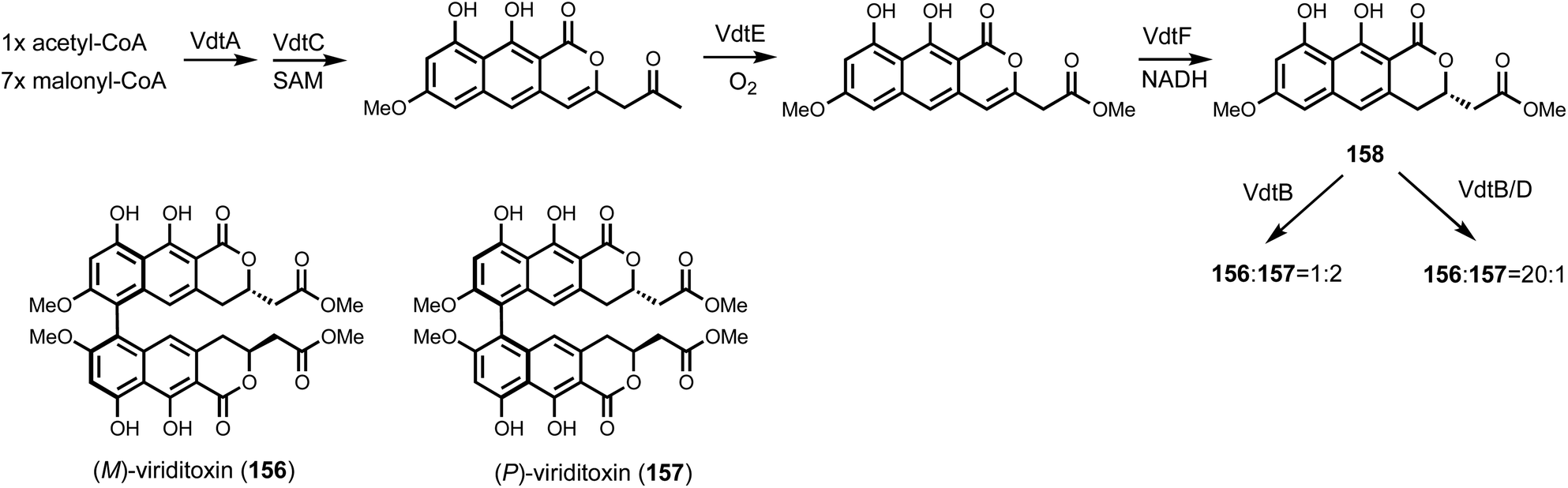 | ||
| Fig. 22 Biosynthetic pathway of virditoxin. Oxidative coupling reaction with the presence of VdtD led to high atroposelectivity for (M)-viriditoxin. | ||
5. Conclusion
Heterologous biosynthesis of natural products is a key tool for uncovering cryptic fungal BGCs and assigning functional roles to biosynthetic enzymes. S. cerevisiae, A. nidulans and A. oryzae are the most commonly used hosts, each offering unique advantages. However, it is evident that engineered Aspergillus hosts are most preferable since their abilities to correctly splice fungal introns. Genome mining of natural products in heterologous hosts can be driven by structural novelty, biological activity or both. Our review incorporated examples of how to prioritize gene clusters to find new-to-nature structures. Here, we classified these examples as known (types of BGCs) – unknowns (metabolites). However, it is anticipated that the true biosynthetic dark matter will come from the heterologous expression on unknown (no core enzyme)-unknowns (metabolites). With new advances in protein structural prediction (AlphaFold),199 high-throughput synthetic biology, and small molecular structural elucidation techniques such as MicroED,200,201 the goal of fully mapping the fungal secondary metabolome may be realized in the next decade.6. Abbreviations
| 4-DMATS | 4-Dimethylallyl tryptophan synthase |
| ACP | Acyl-carrier protein |
| ADH | Alcohol dehydrogenases |
| ALS | Acetolactate synthase |
| ATP | Adenosine 5′-triphosphate |
| BGC | Biosynthetic gene cluster |
| Bmt | (4R)-4-[(E)-2-Butenyl]-4-methyl-l-threonine |
| CAR | Carboxylic acid reductase |
| Cas9 | CRISPR-associated protein 9 |
| CBGA | Cannabigerolic acid |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| Cryo-EM | Cryogenic electron microscopy |
| CPR | Cytochrome P450 reductase |
| DBO | 2,6-Dioxa-bicyclo[3.2.1]octane |
| DFT | Density functional theory |
| DHAD | Dihydroxyacid dehydratase |
| DMAPP | Dimethylallyl pyrophosphate |
| DML | Dihydromonacolin L |
| DUF | Domain of unknown function |
| ER | Enoyl reductase |
| FAC | Fungal artificial chromosome |
| FAD | Flavin adenine dinucleotide |
| FMO | Flavin-containing monooxygenases |
| FPP | Farnesyl pyrophosphate |
| GRAS | Generally regarded as safe |
| HDA | Hetero-Diels–Alder |
| HMG-CoA | 3-Hydroxy-3-methylglutaryl coenzyme A |
| HP | Hypothetical protein |
| HRPKS | Highly-reducing polyketide synthase |
| IDTC | Indole diterpene cyclase |
| IEDDA | Inverse-electron demand Diels–Alder |
| IMDA | Intramolecular Diels–Alder |
| IPP | Isopentenyl pyrophosphate |
| LC-MS | Liquid chromatography-mass spectrometry |
| microED | Microcrystal electron diffraction |
| MO | Molecular orbital |
| MT | Methyltransferase |
| NADH | Nicotinamide adenine dinucleotide |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NEDDA | Normal-electron demand Diels–Alder |
| NMR | Nuclear magnetic resonance |
| NRPKS | Nonreducing polyketide synthase |
| NRPS | Nonribosomal peptide synthetase |
| OMT | O-Methyltransferase |
| ORF | Open reading frame |
| P450 | Cytochrome P450 |
| P450RE | Ring expansion cytochrome P450 |
| PCR | Polymerase chain reaction |
| PKS | Polyketide synthase |
| PLP | Pyridoxal 5′-phosphate |
| pPant | Phosphopantetheinyl |
| PRPKS | Partially-reducing polyketide synthase |
| PT | Prenyltransferase |
| PTS | Post-translational modification |
| RAL | Resorcylic acid lactone |
| RiPP | Ribosomally synthesized and post-translationally modified peptide |
| RT-PCR | Real-time polymerase chain reaction |
| SDR | Short-chain dehydrogenase/reductase |
| SRE | Self-resistance enzyme |
| TC | Terpene cyclase |
| TF | Transcription factor |
| TS | Terpene synthase |
6.1. Protein domain abbreviation
| KS | Ketosynthase |
| KR | Ketoreductase |
| DH | Dehydratase |
| ER | Enoyl reductase |
| ACP | Acyl-carrier protein |
| AT | Acyltransferase |
| MAT | Malonyl-CoA:ACP transacylase |
| SAT | Starter-unit:ACP transacylase |
| TE | Thioesterase |
| A | Adenylation |
| T | Thiolation |
| PCP | Peptidyl-carrier protein |
| C | Condensation |
| CT | Terminal condensation |
| R | Reduction |
7. Conflicts of interest
There are no conflicts to declare.8. References
- S. Lautru, R. J. Deeth, L. M. Bailey and G. L. Challis, Nat. Chem. Biol., 2005, 1, 265–269 CrossRef CAS PubMed.
- J. J. Zhang, X. Tang and B. S. Moore, Nat. Prod. Rep., 2019, 36, 1313–1332 RSC.
- X. Meng, Y. Fang, M. Ding, Y. Zhang, K. Jia, Z. Li, J. Collemare and W. Liu, Biotechnol. Adv., 2022, 54, 107866 CrossRef CAS.
- P. Brandt, M. García-Altares, M. Nett, C. Hertweck and D. Hoffmeister, Angew. Chem., Int. Ed., 2017, 56, 5937–5941 CrossRef CAS PubMed.
- K. L. Dunbar, H. Büttner, E. M. Molloy, M. Dell, J. Kumpfmüller and C. Hertweck, Angew. Chem., Int. Ed., 2018, 57, 14080–14084 CrossRef CAS PubMed.
- K. Scherlach and C. Hertweck, Nat. Commun., 2021, 12, 1–12 CrossRef PubMed.
- D. N. Chigumba, L. S. Mydy, F. de Waal, W. Li, K. Shafiq, J. W. Wotring, O. G. Mohamed, T. Mladenovic, A. Tripathi, J. Z. Sexton, S. Kautsar, M. H. Medema and R. D. Kersten, Nat. Chem. Biol., 2022, 18, 18–28 CrossRef CAS.
- A. A. Brakhage, Nat. Rev. Microbiol., 2013, 11, 21–32 CrossRef CAS.
- P. Wiemann and N. P. Keller, J. Ind. Microbiol. Biotechnol., 2014, 41, 301–313 CrossRef CAS.
- Y. F. Li, K. J. S. Tsai, C. J. B. Harvey, J. J. Li, B. E. Ary, E. E. Berlew, B. L. Boehman, D. M. Findley, A. G. Friant, C. A. Gardner, M. P. Gould, J. H. Ha, B. K. Lilley, E. L. McKinstry, S. Nawal, R. C. Parry, K. W. Rothchild, S. D. Silbert, M. D. Tentilucci, A. M. Thurston, R. B. Wai, Y. Yoon, R. S. Aiyar, M. H. Medema, M. E. Hillenmeyer and L. K. Charkoudian, Fungal Genet. Biol., 2016, 89, 18–28 CrossRef CAS.
- M. T. Robey, L. K. Caesar, M. T. Drott, N. P. Keller and N. L. Kelleher, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2020230118 CrossRef CAS.
- H. B. Bode, B. Bethe, R. Höfs and A. Zeeck, ChemBioChem, 2002, 3, 619–627 CrossRef CAS PubMed.
- R. T. Hewage, T. Aree, C. Mahidol, S. Ruchirawat and P. Kittakoop, Phytochemistry, 2014, 108, 87–94 CrossRef CAS.
- C. Yuan, Y.-H. Guo, H.-Y. Wang, X.-J. Ma, T. Jiang, J.-L. Zhao, Z.-M. Zou and G. Ding, Sci. Rep., 2016, 6, 19350 CrossRef CAS.
- P. Wiemann, C. J. Guo, J. M. Palmer, R. Sekonyela, C. C. C. Wang and N. P. Keller, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17065–17070 CrossRef CAS.
- S. Bergmann, J. Schümann, K. Scherlach, C. Lange, A. A. Brakhage and C. Hertweck, Nat. Chem. Biol., 2007, 3, 213–217 CrossRef CAS.
- M. Ahuja, Y. M. Chiang, S. L. Chang, M. B. Praseuth, R. Entwistle, J. F. Sanchez, H. C. Lo, H. H. Yeh, B. R. Oakley and C. C. C. Wang, J. Am. Chem. Soc., 2012, 134, 8212–8221 CrossRef CAS PubMed.
- Y. H. Chooi, J. Fang, H. Liu, S. G. Filler, P. Wang and Y. Tang, Org. Lett., 2013, 15, 780–783 CrossRef CAS.
- H. N. Lyu, H. W. Liu, N. P. Keller and W. B. Yin, Nat. Prod. Rep., 2020, 37, 6–16 RSC.
- C. E. Oakley, M. Ahuja, W. W. Sun, R. Entwistle, T. Akashi, J. Yaegashi, C. J. Guo, G. C. Cerqueira, J. Russo Wortman, C. C. C. Wang, Y. M. Chiang and B. R. Oakley, Mol. Microbiol., 2017, 103, 347–365 CrossRef CAS PubMed.
- M. F. Grau, R. Entwistle, C. E. Oakley, C. C. C. Wang and B. R. Oakley, ACS Chem. Biol., 2019, 14, 1643–1651 CrossRef CAS PubMed.
- I. Roux, C. Woodcraft, J. Hu, R. Wolters, C. L. M. Gilchrist and Y. H. Chooi, ACS Synth. Biol., 2020, 9, 1843–1854 CrossRef CAS.
- C. Wang, V. Hantke, R. J. Cox and E. Skellam, Org. Lett., 2019, 21, 4163–4167 CrossRef CAS PubMed.
- X. Bian, A. Plaza, Y. Zhang and R. Müller, J. Nat. Prod., 2012, 75, 1652–1655 CrossRef CAS.
- C. Olano, I. García, A. González, M. Rodriguez, D. Rozas, J. Rubio, M. Sánchez-Hidalgo, A. F. Braña, C. Méndez and J. A. Salas, Microb. Biotechnol., 2014, 7, 242–256 CrossRef CAS PubMed.
- M. K. Ahmadi and B. A. Pfeifer, Curr. Opin. Biotechnol., 2016, 42, 7–12 CrossRef CAS.
- M. Myronovskyi and A. Luzhetskyy, Nat. Prod. Rep., 2019, 36, 1281–1294 RSC.
- Y. Qu, M. L. A. E. Easson, J. Froese, R. Simionescu, T. Hudlicky and V. DeLuca, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6224–6229 CrossRef CAS.
- W. Lau and E. S. Sattely, Science, 2015, 349, 1224–1228 CrossRef CAS.
- M. R. Nielsen, R. D. Wollenberg, K. R. Westphal, T. E. Sondergaard, R. Wimmer, D. M. Gardiner and J. L. Sørensen, Fungal Genet. Biol., 2019, 132, 103248 CrossRef CAS.
- M. L. Shenouda, M. Ambilika, E. Skellam and R. J. Cox, J. Fungi, 2022, 8, 355 CrossRef CAS.
- E. Bin Go, L. J. Kim, H. M. Nelson, M. Ohashi and Y. Tang, Org. Lett., 2021, 23, 7819–7823 CrossRef.
- Z. Zhang, C. S. Jamieson, Y. L. Zhao, D. Li, M. Ohashi, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2019, 141, 5659–5663 CrossRef CAS.
- T. Asai, K. Tsukada, S. Ise, N. Shirata, M. Hashimoto, I. Fujii, K. Gomi, K. Nakagawara, E. N. Kodama and Y. Oshima, Nat. Chem., 2015, 7, 737–743 CrossRef CAS PubMed.
- K. Tsukada, S. Shinki, A. Kaneko, K. Murakami, K. Irie, M. Murai, H. Miyoshi, S. Dan, K. Kawaji, H. Hayashi, E. N. Kodama, A. Hori, E. Salim, T. Kuraishi, N. Hirata, Y. Kanda and T. Asai, Nat. Commun., 2020, 11, 1–12 CrossRef.
- P. Chankhamjon, Y. Tsunematsu, M. Ishida-Ito, Y. Sasa, F. Meyer, D. Boettger-Schmidt, B. Urbansky, K. D. Menzel, K. Scherlach, K. Watanabe and C. Hertweck, Angew. Chem., Int. Ed., 2016, 55, 11955–11959 CrossRef CAS PubMed.
- M. Liu, M. Ohashi, Y. S. Hung, K. Scherlach, K. Watanabe, C. Hertweck and Y. Tang, J. Am. Chem. Soc., 2021, 143, 7267–7271 CrossRef CAS.
- J. M. Zhang, H. H. Wang, X. Liu, C. H. Hu and Y. Zou, J. Am. Chem. Soc., 2020, 142, 1957–1965 CrossRef CAS PubMed.
- T. B. Kakule, R. C. Jadulco, M. Koch, J. E. Janso, L. R. Barrows and E. W. Schmidt, ACS Synth. Biol., 2015, 4, 625–633 CrossRef CAS PubMed.
- S. Boecker, S. Grätz, D. Kerwat, L. Adam, D. Schirmer, L. Richter, T. Schütze, D. Petras, R. D. Süssmuth and V. Meyer, Fungal Biol. Biotechnol., 2018, 5, 1–14 CrossRef.
- M. R. Nielsen, R. D. Wollenberg, K. R. Westphal, T. E. Sondergaard, R. Wimmer, D. M. Gardiner and J. L. Sørensen, Fungal Genet. Biol., 2019, 132, 103248 CrossRef CAS PubMed.
- C. Pohl, F. Polli, T. Schütze, A. Viggiano, L. Mózsik, S. Jung, M. de Vries, R. A. L. Bovenberg, V. Meyer and A. J. M. Driessen, Sci. Rep., 2020, 10, 1–16 CrossRef PubMed.
- D. K. Ro, E. M. Paradise, M. Quellet, K. J. Fisher, K. L. Newman, J. M. Ndungu, K. A. Ho, R. A. Eachus, T. S. Ham, J. Kirby, M. C. Y. Chang, S. T. Withers, Y. Shiba, R. Sarpong and J. D. Keasling, Nature, 2006, 440, 940–943 CrossRef CAS PubMed.
- S. Brown, M. Clastre, V. Courdavault and S. E. O'Connor, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3205–3210 CrossRef CAS.
- S. Galanie, K. Thodey, I. J. Trenchard, M. F. Interrante and C. D. Smolke, Science, 2015, 349, 1095–1100 CrossRef CAS.
- A. R. Awan, B. A. Blount, D. J. Bell, W. M. Shaw, J. C. H. Ho, R. M. McKiernan and T. Ellis, Nat. Commun., 2017, 8, 1–8 CrossRef.
- X. Luo, M. A. Reiter, L. d'Espaux, J. Wong, C. M. Denby, A. Lechner, Y. Zhang, A. T. Grzybowski, S. Harth, W. Lin, H. Lee, C. Yu, J. Shin, K. Deng, V. T. Benites, G. Wang, E. E. K. Baidoo, Y. Chen, I. Dev, C. J. Petzold and J. D. Keasling, Nature, 2019, 567, 123–126 CrossRef CAS.
- J. Misa, J. M. Billingsley, K. Niwa, R. K. Yu and Y. Tang, ACS Synth. Biol., 2022, 11, 1639–1649 CrossRef CAS.
- K. K. M. Lee, N. A. D. Silva and J. T. Kealey, Anal. Biochem., 2009, 394, 75–80 CrossRef CAS PubMed.
- H. D. Mootz, K. Schörgendorfer and M. A. Marahiel, FEMS Microbiol. Lett., 2002, 213, 51–57 CAS.
- B. A. Pfeifer, S. J. Admiraal, H. Gramajo, D. E. Cane and C. Khosla, Science, 2001, 291, 1790–1792 CrossRef CAS.
- S. M. Ma, J. W. H. Li, J. W. Choi, H. Zhou, K. K. M. Lee, V. A. Moorthie, X. Xie, J. T. Kealey, N. A. Da Silva, J. C. Vederas and Y. Tang, Science, 2009, 326, 589–592 CrossRef CAS PubMed.
- X. Gao, S. W. Haynes, B. D. Ames, P. Wang, L. P. Vien, C. T. Walsh and Y. Tang, Nat. Chem. Biol., 2012, 8, 823–830 CrossRef CAS.
- W. Xu, X. Cai, M. E. Jung and Y. Tang, J. Am. Chem. Soc., 2010, 132, 13604–13607 CrossRef CAS PubMed.
- M. C. Tang, H. C. Lin, D. Li, Y. Zou, J. Li, W. Xu, R. A. Cacho, M. E. Hillenmeyer, N. K. Garg and Y. Tang, J. Am. Chem. Soc., 2015, 137, 13724–13727 CrossRef CAS PubMed.
- W. Nam, Compr. Coord. Chem. II, 2004, 8, 281–307 CAS.
- Y. Yan, Q. Liu, X. Zang, S. Yuan, U. Bat-Erdene, C. Nguyen, J. Gan, J. Zhou, S. E. Jacobsen and Y. Tang, Nature, 2018, 559, 415–418 CrossRef CAS.
- C. J. B. Harvey, M. Tang, U. Schlecht, J. Horecka, C. R. Fischer, H.-C. Lin, J. Li, B. Naughton, J. Cherry, M. Miranda, Y. F. Li, A. M. Chu, J. R. Hennessy, G. A. Vandova, D. Inglis, R. S. Aiyar, L. M. Steinmetz, R. W. Davis, M. H. Medema, E. Sattely, C. Khosla, R. P. St. Onge, Y. Tang and M. E. Hillenmeyer, Sci. Adv., 2018, 4, eaar5459 CrossRef.
- D. A. Wassarman and J. A. Steitz, Science, 1992, 257, 1918–1925 CrossRef CAS PubMed.
- C. L. Will and R. Lührmann, Cold Spring Harbor Perspect. Biol., 2011, 3 DOI:10.1101/cshperspect.a003707.
- X. Zhang, J. Guo, F. Cheng and S. Li, Nat. Prod. Rep., 2021, 38, 1072–1099 RSC.
- K. Gomi, Y. Iimura and S. Hara, Agric. Biol. Chem., 1987, 51, 2549–2555 CAS.
- F. J. Jin, J. I. Maruyama, P. R. Juvvadi, M. Arioka and K. Kitamoto, FEMS Microbiol. Lett., 2004, 239, 79–85 CrossRef CAS.
- M. N. Heneghan, A. A. Yakasai, L. M. Halo, Z. Song, A. M. Bailey, T. J. Simpson, R. J. Cox and C. M. Lazarus, ChemBioChem, 2010, 11, 1508–1512 CrossRef CAS.
- T. Itoh, K. Tokunaga, Y. Matsuda, I. Fujii, I. Abe, Y. Ebizuka and T. Kushiro, Nat. Chem., 2010, 2, 858–864 CrossRef CAS.
- Y. Matsuda and I. Abe, Nat. Prod. Rep., 2016, 33, 26–53 RSC.
- K. Tagami, C. Liu, A. Minami, M. Noike, T. Isaka, S. Fueki, Y. Shichijo, H. Toshima, K. Gomi, T. Dairi and H. Oikawa, J. Am. Chem. Soc., 2013, 135, 1260–1263 CrossRef CAS.
- C. Liu, A. Minami, T. Ozaki, J. Wu, H. Kawagishi, J. I. Maruyama and H. Oikawa, J. Am. Chem. Soc., 2019, 141, 15519–15523 CrossRef CAS PubMed.
- Z. Song, W. Bakeer, J. W. Marshall, A. A. Yakasai, R. M. Khalid, J. Collemare, E. Skellam, D. Tharreau, M. H. Lebrun, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Chem. Sci., 2015, 6, 4837–4845 RSC.
- R. Nofiani, K. de Mattos-Shipley, K. E. Lebe, L.-C. Han, Z. Iqbal, A. M. Bailey, C. L. Willis, T. J. Simpson and R. J. Cox, Nat. Commun., 2018, 9, 3940 CrossRef.
- S. Nagamine, C. Liu, J. Nishishita, T. Kozaki, K. Sogahata, Y. Sato, A. Minami, T. Ozaki, C. Schmidt-Dannert, J. Maruyama and H. Oikawa, Appl. Environ. Microbiol., 2019, 85, e00409-19 CrossRef PubMed.
- T. Nayak, E. Szewczyk, C. E. Oakley, A. Osmani, L. Ukil, S. L. Murray, M. J. Hynes, S. A. Osmani and B. R. Oakley, Genetics, 2006, 172, 1557–1566 CrossRef CAS PubMed.
- D. A. Yee and Y. Tang, Engineering Natural Product Biosynthesis: Methods and Protocols, ed. E. Skellam, Springer US, New York, NY, 2022, pp. 41–52 Search PubMed.
- Y. M. Chiang, T. S. Lin, S. L. Chang, G. Ahn and C. C. C. Wang, ACS Synth. Biol., 2021, 10, 173–182 CrossRef CAS PubMed.
- A. D. Somoza, K.-H. Lee, Y.-M. Chiang, B. R. Oakley and C. C. C. Wang, Org. Lett., 2012, 14, 972–975 CrossRef CAS PubMed.
- N. Liu, Y. S. Hung, S. S. Gao, L. Hang, Y. Zou, Y. H. Chooi and Y. Tang, Org. Lett., 2017, 19, 3560–3563 CrossRef CAS.
- Y. M. Chiang, M. Ahuja, C. E. Oakley, R. Entwistle, A. Asokan, C. Zutz, C. C. C. Wang and B. R. Oakley, Angew. Chem., Int. Ed., 2016, 55, 1662–1665 CrossRef CAS PubMed.
- J. W. Bok, R. Ye, K. D. Clevenger, D. Mead, M. Wagner, A. Krerowicz, J. C. Albright, A. W. Goering, P. M. Thomas, N. L. Kelleher, N. P. Keller and C. C. Wu, BMC Genomics, 2015, 16, 1–10 CrossRef CAS.
- K. D. Clevenger, J. W. Bok, R. Ye, G. P. Miley, M. H. Verdan, T. Velk, C. Chen, K. H. Yang, M. T. Robey, P. Gao, M. Lamprecht, P. M. Thomas, M. N. Islam, J. M. Palmer, C. C. Wu, N. P. Keller and N. L. Kelleher, Nat. Chem. Biol., 2017, 13, 895–901 CrossRef CAS PubMed.
- Y. Yan, N. Liu and Y. Tang, Nat. Prod. Rep., 2020, 37, 879–892 RSC.
- K. H. Almabruk, L. K. Dinh and B. Philmus, ACS Chem. Biol., 2018, 13, 1426–1437 CrossRef CAS PubMed.
- H. C. Lin, Y. H. Chooi, S. Dhingra, W. Xu, A. M. Calvo and Y. Tang, J. Am. Chem. Soc., 2013, 135, 4616–4619 CrossRef CAS.
- H. H. Yeh, M. Ahuja, Y. M. Chiang, C. E. Oakley, S. Moore, O. Yoon, H. Hajovsky, J. W. Bok, N. P. Keller, C. C. C. Wang and B. R. Oakley, ACS Chem. Biol., 2016, 11, 2275–2284 CrossRef CAS PubMed.
- F. Panter, D. Krug, S. Baumann and R. Müller, Chem. Sci., 2018, 9, 4898–4908 RSC.
- L. Xie, X. Zang, W. Cheng, Z. Zhang, J. Zhou, M. Chen and Y. Tang, J. Am. Chem. Soc., 2021, 143, 9575–9584 CrossRef CAS PubMed.
- F. Biermann and E. J. N. Helfrich, mSystems, 2021, 6, 1–6 CrossRef.
- K. Blin, S. Shaw, A. M. Kloosterman, Z. Charlop-Powers, G. P. van Wezel, M. H. Medema and T. Weber, Nucleic Acids Res., 2021, 49, W29–W35 CrossRef CAS PubMed.
- Z. Hu, T. Awakawa, Z. Ma and I. Abe, Nat. Commun., 2019, 10, 1–10 CrossRef.
- J. B. Patteson, A. T. Putz, L. Tao, W. C. Simke, L. H. Bryant, R. D. Britt and B. Li, Science, 2021, 374, 1005–1009 CrossRef CAS PubMed.
- S. T. Lima, T. R. Fallon, J. L. Cordoza, J. R. Chekan, E. Delbaje, A. R. Hopiavuori, D. O. Alvarenga, S. M. Wood, H. Luhavaya, J. T. Baumgartner, F. A. Dörr, A. Etchegaray, E. Pinto, S. M. K. McKinnie, M. F. Fiore and B. S. Moore, J. Am. Chem. Soc., 2022, 144, 9372–9379 CrossRef CAS PubMed.
- R. J. Cox, Org. Biomol. Chem., 2007, 5, 2010–2026 RSC.
- K. Kasahara, T. Miyamoto, T. Fujimoto, H. Oguri, T. Tokiwano, H. Oikawa, Y. Ebizuka and I. Fujii, Chembiochem, 2010, 11, 1245–1252 CrossRef CAS PubMed.
- Y. H. Chooi and Y. Tang, J. Org. Chem., 2012, 77, 9933–9953 CrossRef CAS PubMed.
- J. M. Crawford, B. C. R. Dancy, E. A. Hill, D. W. Udwary and C. A. Townsend, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16728–16733 CrossRef CAS.
- X. Xie, M. J. Meehan, W. Xu, P. C. Dorrestein and Y. Tang, J. Am. Chem. Soc., 2009, 131, 8388–8389 CrossRef CAS.
- W. Xu, Y.-H. Chooi, J. W. Choi, S. Li, J. C. Vederas, N. A. Da Silva and Y. Tang, Angew. Chem., Int. Ed. Engl., 2013, 52, 6472–6475 CrossRef CAS.
- M. C. Tang, C. R. Fischer, J. V. Chari, D. Tan, S. Suresh, A. Chu, M. Miranda, J. Smith, Z. Zhang, N. K. Garg, R. P. S. Onge and Y. Tang, J. Am. Chem. Soc., 2020, 141, 8198–8206 CrossRef PubMed.
- T. Ugai, A. Minami, R. Fujii, M. Tanaka, H. Oguri, K. Gomi and H. Oikawa, Chem. Commun., 2015, 51, 1878–1881 RSC.
- I. Martín, F. Peláez, G. H. Harris, J. E. Curotto, W. Rozdilsky, M. B. Kurtz, R. A. Giacobbe, G. F. Bills and M. A. Cabello, J. Antibiot., 1995, 48, 349–356 CrossRef PubMed.
- H. Li, J. Hu, H. Wei, P. S. Solomon, K. A. Stubbs and Y. H. Chooi, Chem. - Eur. J., 2019, 25, 15062–15066 CrossRef CAS.
- H. Tao, T. Mori, X. Wei, Y. Matsuda and I. Abe, Angew. Chem., Int. Ed., 2021, 60, 8851–8858 CrossRef CAS.
- J. Kennedy, K. Auclair, S. G. Kendrew, C. Park, J. C. Vederas and C. R. Hutchinson, Science, 1999, 284, 1368–1372 CrossRef CAS PubMed.
- S. Grijseels, C. Pohl, J. C. Nielsen, Z. Wasil, Y. Nygård, J. Nielsen, J. C. Frisvad, K. F. Nielsen, M. Workman, T. O. Larsen, A. J. M. Driessen and R. J. N. Frandsen, Fungal Biol. Biotechnol., 2018, 5, 1–17 CrossRef.
- J. M. Winter, M. Sato, S. Sugimoto, G. Chiou, N. K. Garg, Y. Tang and K. Watanabe, J. Am. Chem. Soc., 2012, 134, 17900–17903 CrossRef CAS PubMed.
- L. Wang, M. Yuan and J. Zheng, Synth. Syst. Biotechnol., 2019, 4, 10–15 CrossRef PubMed.
- J. Wang, J. Liang, L. Chen, W. Zhang, L. Kong, C. Peng, C. Su, Y. Tang, Z. Deng and Z. Wang, Nat. Commun., 2021, 12, 1–10 CrossRef CAS.
- Y. Hai and Y. Tang, J. Am. Chem. Soc., 2018, 140, 1271–1274 CrossRef CAS PubMed.
- R. R. Forseth, S. Amaike, D. Schwenk, K. J. Affeldt, D. Hoffmeister, F. C. Schroeder and N. P. Keller, Angew. Chem., Int. Ed., 2013, 52, 1590–1594 CrossRef CAS.
- J. A. Baccile, J. E. Spraker, H. H. Le, E. Brandenburger, C. Gomez, J. W. Bok, J. MacHeleidt, A. A. Brakhage, D. Hoffmeister, N. P. Keller and F. C. Schroeder, Nat. Chem. Biol., 2016, 12, 419–424 CrossRef CAS.
- Y. Hai, A. M. Huang and Y. Tang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10348–10353 CrossRef CAS.
- D. Cook, B. G. G. Donzelli, R. Creamer, D. L. Baucom, D. R. Gardner, J. Pan, N. Moore, S. B. Krasnoff, J. W. Jaromczyk and C. L. Schardl, G3: Genes, Genomes, Genet., 2017, 7, 1791–1797 CrossRef CAS PubMed.
- Y. Hai, A. Huang and Y. Tang, J. Nat. Prod., 2020, 83, 593–600 CrossRef CAS.
- C. S. Yun, T. Motoyama and H. Osada, Nat. Commun., 2015, 6, 8758 CrossRef CAS PubMed.
- X. Yang, P. Feng, Y. Yin, K. Bushley, J. W. Spatafora and C. Wang, mBio, 2018, 9, e01211-18 CrossRef.
- N. Gerhards, L. Neubauer, P. Tudzynski and S. M. Li, Toxins, 2014, 6, 3281–3295 CrossRef CAS PubMed.
- N. Jana and S. Nanda, New J. Chem., 2018, 42, 17803–17873 RSC.
- H. Zhou, K. Qiao, Z. Gao, M. J. Meehan, J. W. H. Li, X. Zhao, P. C. Dorrestein, J. C. Vederas and Y. Tang, J. Am. Chem. Soc., 2010, 132, 4530–4531 CrossRef CAS PubMed.
- H. Zhou, J. Zhan, K. Watanabe, X. Xie and Y. Tang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6249–6254 CrossRef CAS.
- A. Al Fahad, A. Abood, K. M. Fisch, A. Osipow, J. Davison, M. Avramović, C. P. Butts, J. Piel, T. J. Simpson and R. J. Cox, Chem. Sci., 2014, 5, 523–527 RSC.
- M. Chen, Q. Liu, S. S. Gao, A. E. Young, S. E. Jacobsen and Y. Tang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 5499–5504 CrossRef CAS.
- I. C. Okorafor, M. Chen and Y. Tang, ACS Synth. Biol., 2021, 10, 2159–2166 CrossRef CAS PubMed.
- A. Kaneko, Y. Morishita, K. Tsukada, T. Taniguchi and T. Asai, Org. Biomol. Chem., 2019, 17, 5239–5243 RSC.
- M. Hashimoto, T. Nonaka and I. Fujii, Nat. Prod. Rep., 2014, 31, 1306–1317 RSC.
- J. W. Sims, J. P. Fillmore, D. D. Warner and E. W. Schmidt, Chem. Commun., 2005, 186–188 RSC.
- L. Li, P. Yu, M. C. Tang, Y. Zou, S. S. Gao, Y. S. Hung, M. Zhao, K. Watanabe, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2016, 138, 15837–15840 CrossRef CAS.
- M. Ohashi, F. Liu, Y. Hai, M. Chen, M. cheng Tang, Z. Yang, M. Sato, K. Watanabe, K. N. Houk and Y. Tang, Nature, 2017, 549, 502–506 CrossRef PubMed.
- M. Tokuoka, Y. Seshime, I. Fujii, K. Kitamoto, T. Takahashi and Y. Koyama, Fungal Genet. Biol., 2008, 45, 1608–1615 CrossRef CAS.
- M. Sato, J. E. Dander, C. Sato, Y. S. Hung, S. S. Gao, M. C. Tang, L. Hang, J. M. Winter, N. K. Garg, K. Watanabe and Y. Tang, J. Am. Chem. Soc., 2017, 139, 5317–5320 CrossRef CAS PubMed.
- D. W. Christianson, Chem. Rev., 2017, 117, 11570–11648 CrossRef CAS.
- Y. L. Du and K. S. Ryan, Nat. Prod. Rep., 2019, 36, 430–457 RSC.
- C. Lee, L. Chen, C. Chiang, C. Lai and H. Lin, Angew. Chem., 2019, 131, 18585–18589 CrossRef.
- D. A. Yee, T. B. Kakule, W. Cheng, M. Chen, C. T. Y. Chong, Y. Hai, L. F. Hang, Y. S. Hung, N. Liu, M. Ohashi, I. C. Okorafor, Y. Song, M. Tang, Z. Zhang and Y. Tang, J. Am. Chem. Soc., 2020, 142, 710–714 CrossRef CAS PubMed.
- M. A. Tararina, D. A. Yee, Y. Tang and D. W. Christianson, Biochemistry, 2022 DOI:10.1021/acs.biochem.2c00335.
- W. Cheng, M. Chen, M. Ohashi and Y. Tang, Angew. Chem., Int. Ed., 2022, 61, 1–5 Search PubMed.
- D. Yan, K. Wang, S. Bai, B. Liu, J. Bai, X. Qi and Y. Hu, J. Am. Chem. Soc., 2022, 144, 4269–4276 CrossRef CAS PubMed.
- P. Thomas, H. Sundaram, B. J. Krishek, P. Chazot, X. Xie, P. Bevan, S. J. Brocchini, C. J. Latham, P. Charlton, M. Moore, S. J. Lewis, D. M. Thornton, F. A. Stephenson and T. G. Smart, J. Pharmacol. Exp. Ther., 1997, 282, 513–520 CAS.
- J. Davison, A. al Fahad, M. Cai, Z. Song, S. Y. Yehia, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, Proc. Natl. Acad. Sci., 2012, 109, 7642–7647 CrossRef CAS.
- R. Schor, C. Schotte, D. Wibberg, J. Kalinowski and R. J. Cox, Nat. Commun., 2018, 9, 1693 CrossRef PubMed.
- F. Yu, S. Okamto, K. Nakasone, K. Adachi, S. Matsuda, H. Harada, N. Misawa and R. Utsumi, Planta, 2008, 227, 1291–1299 CrossRef CAS PubMed.
- Q. Chen, J. Gao, C. Jamieson, J. Liu, M. Ohashi, J. Bai, D. Yan, B. Liu, Y. Che, Y. Wang, K. N. Houk and Y. Hu, J. Am. Chem. Soc., 2019, 141, 14052–14056 CrossRef CAS PubMed.
- C. Schotte, L. Li, D. Wibberg, J. Kalinowski and R. J. Cox, Angew. Chem., Int. Ed., 2020, 59, 23870–23878 CrossRef CAS.
- C. Y. Bemis, C. N. Ungarean, A. S. Shved, C. S. Jamieson, T. Hwang, K. S. Lee, K. N. Houk and D. Sarlah, J. Am. Chem. Soc., 2021, 143, 6006–6017 CrossRef CAS PubMed.
- L. J. Kim, M. Ohashi, Z. Zhang, D. Tan, M. Asay, D. Cascio, J. A. Rodriguez, Y. Tang and H. M. Nelson, Nat. Chem. Biol., 2021, 17, 872–877 CrossRef CAS PubMed.
- S. B. Singh, W. Liu, X. Li, T. Chen, A. Shafiee, D. Card, G. Abruzzo, A. Flattery, C. Gill, J. R. Thompson, M. Rosenbach, S. Dreikorn, V. Hornak, M. Meinz, M. Kurtz, R. Kelly and J. C. Onishi, ACS Med. Chem. Lett., 2012, 3, 814–817 CrossRef CAS PubMed.
- S. B. Singh, W. Liu, X. Li, T. Chen, A. Shafiee, S. Dreikorn, V. Hornak, M. Meinz and J. C. Onishi, Bioorg. Med. Chem. Lett., 2013, 23, 3018–3022 CrossRef CAS PubMed.
- L. Liu, M. C. Tang and Y. Tang, J. Am. Chem. Soc., 2019, 141, 19538–19541 CrossRef CAS PubMed.
- Z. Zhao, Y. Ying, Y. S. Hung and Y. Tang, J. Nat. Prod., 2019, 82, 1029–1033 CrossRef CAS PubMed.
- C. Rank, R. K. Phipps, P. Harris, P. Fristrup, T. O. Larsen and C. H. Gotfredsen, Org. Lett., 2008, 10, 401–404 CrossRef CAS PubMed.
- M. Makarova, L. Rycek, J. Hajicek, D. Baidilov and T. Hudlicky, Angew. Chem., Int. Ed., 2019, 58, 18338–18387 CrossRef CAS PubMed.
- J. E. Schaffer, M. R. Reck, N. K. Prasad and T. A. Wencewicz, Nat. Chem. Biol., 2017, 13, 737–744 CrossRef CAS PubMed.
- Z. Cui, J. Overbay, X. Wang, X. Liu, Y. Zhang, M. Bhardwaj, A. Lemke, D. Wiegmann, G. Niro, J. S. Thorson, C. Ducho and S. G. Van Lanen, Nat. Chem. Biol., 2020, 16, 904–911 CrossRef CAS PubMed.
- M. Chen, C. T. Liu and Y. Tang, J. Am. Chem. Soc., 2020, 142, 10506–10515 CrossRef CAS PubMed.
- Y. Hai, M. Chen, A. Huang and Y. Tang, J. Am. Chem. Soc., 2020, 142, 19668–19677 CrossRef CAS PubMed.
- L. Studt, S. Janevska, E.-M. Niehaus, I. Burkhardt, B. Arndt, C. M. K. Sieber, H.-U. Humpf, J. S. Dickschat and B. Tudzynski, Environ. Microbiol., 2016, 18, 936–956 CrossRef CAS PubMed.
- G. Z. Dai, W. B. Han, Y. N. Mei, K. Xu, R. H. Jiao, H. M. Ge and R. X. Tan, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1174–1180 CrossRef CAS.
- Y. L. Du, R. Singh, L. M. Alkhalaf, E. Kuatsjah, H. Y. He, L. D. Eltis and K. S. Ryan, Nat. Chem. Biol., 2016, 12, 194–199 CrossRef CAS.
- H. C. Lo, R. Entwistle, C. J. Guo, M. Ahuja, E. Szewczyk, J. H. Hung, Y. M. Chiang, B. R. Oakley and C. C. C. Wang, J. Am. Chem. Soc., 2012, 134, 4709–4720 CrossRef CAS.
- S. S. Gao, T. Zhang, M. Garcia-Borràs, Y. S. Hung, J. M. Billingsley, K. N. Houk, Y. Hu and Y. Tang, J. Am. Chem. Soc., 2018, 140, 6991–6997 CrossRef CAS PubMed.
- S. S. Gao, A. Duan, W. Xu, P. Yu, L. Hang, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2016, 138, 4249–4259 CrossRef CAS PubMed.
- R. A. Cacho, J. Thuss, W. Xu, R. Sanichar, Z. Gao, A. Nguyen, J. C. Vederas and Y. Tang, J. Am. Chem. Soc., 2015, 137, 15688–15691 CrossRef CAS PubMed.
- R. J. Cox, Nat. Prod. Rep., 2022 10.1039/d2np00007e.
- M. C. Tang, Y. Zou, D. Yee and Y. Tang, AIChE J., 2018, 64, 4182–4186 CrossRef CAS PubMed.
- R. Fujii, A. Minami, K. Gomi and H. Oikawa, Tetrahedron Lett., 2013, 54, 2999–3002 CrossRef CAS.
- L. Li, M. C. Tang, S. Tang, S. Gao, S. Soliman, L. Hang, W. Xu, T. Ye, K. Watanabe and Y. Tang, J. Am. Chem. Soc., 2018, 140, 2067–2071 CrossRef CAS PubMed.
- T. Agatsuma, T. Akama, S. Nara, S. Matsumiya, R. Nakai, H. Ogawa, S. Otaki, S. Ikeda, Y. Saitoh and Y. Kanda, Org. Lett., 2002, 4, 4387–4390 CrossRef CAS PubMed.
- N. Liu, E. D. Abramyan, W. Cheng, B. Perlatti, C. J. B. Harvey, G. F. Bills and Y. Tang, J. Am. Chem. Soc., 2021, 143, 6043–6047 CrossRef CAS PubMed.
- K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed. Engl., 2002, 41, 1668–1698 CrossRef CAS.
- C. S. Jamieson, M. Ohashi, F. Liu, Y. Tang and K. N. Houk, Nat. Prod. Rep., 2019, 36, 698–713 RSC.
- J. W. Cary, V. Uka, Z. Han, D. Buyst, P. Y. Harris-Coward, K. C. Ehrlich, Q. Wei, D. Bhatnagar, P. F. Dowd, S. L. Martens, A. M. Calvo, J. C. Martins, L. Vanhaecke, T. Coenye, S. De Saeger and J. D. Di Mavungu, Fungal Genet. Biol., 2015, 81, 88–97 CrossRef CAS PubMed.
- Y. Cai, Y. Hai, M. Ohashi, C. S. Jamieson, M. Garcia-Borras, K. N. Houk, J. Zhou and Y. Tang, Nat. Chem., 2019, 11, 812–820 CrossRef CAS.
- H. J. Kim, M. W. Ruszczycky, S. H. Choi, Y. N. Liu and H. W. Liu, Nature, 2011, 473, 109–112 CrossRef CAS PubMed.
- M. Ohashi, C. S. Jamieson, Y. Cai, D. Tan, D. Kanayama, M. C. Tang, S. M. Anthony, J. V. Chari, J. S. Barber, E. Picazo, T. B. Kakule, S. Cao, N. K. Garg, J. Zhou, K. N. Houk and Y. Tang, Nature, 2020, 586, 64–69 CrossRef CAS PubMed.
- K. Klas, S. Tsukamoto, D. H. Sherman and R. M. Williams, J. Org. Chem., 2015, 80, 11672–11685 CrossRef CAS PubMed.
- N. Shionozaki, T. Yamaguchi, H. Kitano, M. Tomizawa, K. Makino and H. Uchiro, Tetrahedron Lett., 2012, 53, 5167–5170 CrossRef CAS.
- J. Xu, E. J. E. Caro-Diaz, L. Trzoss and E. A. Theodorakis, J. Am. Chem. Soc., 2012, 134, 5072–5075 CrossRef CAS.
- M. Sato, F. Yagishita, T. Mino, N. Uchiyama, A. Patel, Y. H. Chooi, Y. Goda, W. Xu, H. Noguchi, T. Yamamoto, K. Hotta, K. N. Houk, Y. Tang and K. Watanabe, ChemBioChem, 2015, 16, 2294–2298 CrossRef CAS.
- T. B. Kakule, S. Zhang, J. Zhan and E. W. Schmidt, Org. Lett., 2015, 17, 2295–2297 CrossRef CAS.
- K. Qiao, Y.-H. Chooi and Y. Tang, Metab. Eng., 2011, 13, 723–732 CrossRef CAS PubMed.
- M. Sato, S. Kishimoto, M. Yokoyama, C. S. Jamieson, K. Narita, N. Maeda, K. Hara, H. Hashimoto, Y. Tsunematsu, K. N. Houk, Y. Tang and K. Watanabe, Nat. Catal., 2021, 4, 223–232 CrossRef CAS.
- N. Kato, T. Nogawa, H. Hirota, J. H. Jang, S. Takahashi, J. S. Ahn and H. Osada, Biochem. Biophys. Res. Commun., 2015, 460, 210–215 CrossRef CAS PubMed.
- D. Tan, C. S. Jamieson, M. Ohashi, M. C. Tang, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2019, 141, 769–773 CrossRef CAS.
- J. M. Zhang, X. Liu, Q. Wei, C. Ma, D. Li and Y. Zou, Nat. Commun., 2022, 13, 225 CrossRef PubMed.
- M. Ohashi, T. B. Kakule, M. C. Tang, C. S. Jamieson, M. Liu, Y. L. Zhao, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2021, 143, 5605–5609 CrossRef CAS PubMed.
- H. Uchiro, R. Kato, Y. Arai, M. Hasegawa and Y. Kobayakawa, Org. Lett., 2011, 13, 6268–6271 CrossRef CAS PubMed.
- H. Sugata, K. Inagaki, T. Ode, T. Hayakawa, Y. Karoji, M. Baba, R. Kato, D. Hasegawa, T. Tsubogo and H. Uchiro, Chem.–Asian J., 2017, 12, 628–632 CrossRef CAS PubMed.
- H. Oikawa, J. Org. Chem., 2003, 68, 3552–3557 CrossRef CAS PubMed.
- A. Ear, S. Amand, F. Blanchard, A. Blond, L. Dubost, D. Buisson and B. Nay, Org. Biomol. Chem., 2015, 13, 3662–3666 RSC.
- I. Fujii, M. Hashimoto, K. Konishi, A. Unezawa, H. Sakuraba, K. Suzuki, H. Tsushima, M. Iwasaki, S. Yoshida, A. Kudo, R. Fujita, A. Hichiwa, K. Saito, T. Asano, J. Ishikawa, D. Wakana, Y. Goda, A. Watanabe, M. Watanabe, Y. Masumoto, J. Kanazawa, H. Sato and M. Uchiyama, Angew. Chem., Int. Ed., 2020, 59, 8464–8470 CrossRef CAS PubMed.
- J. Zhang, S. Yuzawa, W. L. Thong, T. Shinada, M. Nishiyama and T. Kuzuyama, J. Am. Chem. Soc., 2021, 143, 2962–2969 CrossRef CAS.
- A. R. Gallimore, Nat. Prod. Rep., 2009, 26, 266–280 RSC.
- B. J. E. Baldwin and J. C. S. Chem, Comm, 1976, 734–736 Search PubMed.
- X. M. Mao, Z. J. Zhan, M. N. Grayson, M. C. Tang, W. Xu, Y. Q. Li, W. B. Yin, H. C. Lin, Y. H. Chooi, K. N. Houk and Y. Tang, J. Am. Chem. Soc., 2015, 137, 11904–11907 CrossRef CAS PubMed.
- K. Hotta, X. Chen, R. S. Paton, A. Minami, H. Li, K. Swaminathan, I. I. Mathews, K. Watanabe, H. Oikawa, K. N. Houk and C. Y. Kim, Nature, 2012, 483, 355–358 CrossRef CAS PubMed.
- L. B. Davin, H. Bin Wang, A. L. Crowell, D. L. Bedgar, D. M. Martin, S. Sarkanen and N. G. Lewis, Science, 1997, 275, 362–366 CrossRef CAS.
- H. K. Seneviratne, D. S. Dalisay, K.-W. Kim, S. G. A. Moinuddin, H. Yang, C. M. Hartshorn, L. B. Davin and N. G. Lewis, Phytochemistry, 2015, 113, 140–148 CrossRef CAS.
- K.-W. Kim, S. G. A. Moinuddin, K. M. Atwell, M. A. Costa, L. B. Davin and N. G. Lewis, J. Biol. Chem., 2012, 287, 33957–33972 CrossRef CAS.
- J. Liu, R. D. Stipanovic, A. A. Bell, L. S. Puckhaber and C. W. Magill, Phytochemistry, 2008, 69, 3038–3042 CrossRef CAS.
- J. Hu, H. Li and Y. H. Chooi, J. Am. Chem. Soc., 2020, 141, 8068–8072 CrossRef PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- C. G. Jones, M. W. Martynowycz, J. Hattne, T. J. Fulton, B. M. Stoltz, J. A. Rodriguez, H. M. Nelson and T. Gonen, ACS Cent. Sci., 2018, 4, 1587–1592 CrossRef CAS PubMed.
- T. Gruene, J. T. C. Wennmacher, C. Zaubitzer, J. J. Holstein, J. Heidler, A. Fecteau-Lefebvre, S. De Carlo, E. Müller, K. N. Goldie, I. Regeni, T. Li, G. Santiso-Quinones, G. Steinfeld, S. Handschin, E. van Genderen, J. A. van Bokhoven, G. H. Clever and R. Pantelic, Angew. Chem., Int. Ed., 2018, 57, 16313–16317 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |