
Cyanophycin and its biosynthesis: not hot but very cool
Itai
Sharon
a,
Donald
Hilvert
 b and
T. Martin
Schmeing
*a
b and
T. Martin
Schmeing
*a
aDepartment of Biochemistry and Centre de Recherche en Biologie Structurale, McGill University, Montréal, QC, Canada H3G 0B1. E-mail: martin.schmeing@mcgill.ca
bLaboratory of Organic Chemistry, ETH Zürich, CH-8093 Zürich, Switzerland
First published on 26th May 2023
Abstract
Covering: 1878 to early 2023
Cyanophycin is a biopolymer consisting of a poly-aspartate backbone with arginines linked to each Asp sidechain through isopeptide bonds. Cyanophycin is made by cyanophycin synthetase 1 or 2 through ATP-dependent polymerization of Asp and Arg, or β-Asp-Arg, respectively. It is degraded into dipeptides by exo-cyanophycinases, and these dipeptides are hydrolyzed into free amino acids by general or dedicated isodipeptidase enzymes. When synthesized, chains of cyanophycin coalesce into large, inert, membrane-less granules. Although discovered in cyanobacteria, cyanophycin is made by species throughout the bacterial kingdom, and cyanophycin metabolism provides advantages for toxic bloom forming algae and some human pathogens. Some bacteria have developed dedicated schemes for cyanophycin accumulation and use, which include fine temporal and spatial regulation. Cyanophycin has also been heterologously produced in a variety of host organisms to a remarkable level, over 50% of the host's dry mass, and has potential for a variety of green industrial applications. In this review, we summarize the progression of cyanophycin research, with an emphasis on recent structural studies of enzymes in the cyanophycin biosynthetic pathway. These include several unexpected revelations that show cyanophycin synthetase to be a very cool, multi-functional macromolecular machine.
 Itai Sharon | Itai Sharon performed undergraduate studies in biology and chemistry at Tel-Aviv University, Israel, and will soon receive his Ph.D. in Biochemistry from McGill University, Canada. His Ph.D. research has focused on the structural and functional characterization of all steps of cyanophycin metabolism, using cryo-electron microscopy, X-ray crystallography, biochemistry and microbiology. |
 Donald Hilvert | Donald Hilvert received a PhD in Chemistry from Columbia University, New York. After postdoctoral studies at Rockefeller University, New York, he joined the faculty of the Scripps Research Institute at La Jolla, California, where he pursued his interests in chemical biology. In 1997 he moved to his current position as Professor of Chemistry at the ETH Zurich. His research program aims to understand the origins of enzyme catalysis, mimic and expand upon these properties, and engineer protein compartments for diverse applications. |
 T. Martin Schmeing | Martin Schmeing performed graduate research with Tom Steitz at Yale University, studying the architecture and mechanism of the large ribosomal subunit. He then undertook postdoctoral training at the LMB, Cambridge, with Venki Ramakrishnan, using cryo-EM and X-ray crystallography to investigate initiation and elongation of translation. Martin established his laboratory at McGill University in 2010, where he studies nonribosomal peptide synthetases and other macromolecular biosynthetic machines. Martin is currently a James McGill Professor in the Department of Biochemistry and the Director of the McGill Centre de recherche en biologie structurale. |
1 Introduction
Cyanophycin is a natural biopolymer consisting of a long poly-L-Asp backbone with L-Arg residues attached to each of the β-carboxylate side chains through isopeptide bonds1 (Fig. 1). First observed in 1878 as granules within cyanobacterial cells,2,3 cyanophycin is produced by a wide range of bacteria and can be degraded by many more.4–6 The nitrogen content of cyanophycin, 24% by mass, is higher than that of other biopolymers.7 It is insoluble at physiological pH, and so spontaneously forms large, inert granules (Fig. 2).8,9 These properties make cyanophycin an ideal molecule to store fixed nitrogen in cells. Cyanobacteria have developed several dedicated modes of accumulation and subsequent mobilization of nitrogen stored in cyanophycin.10–12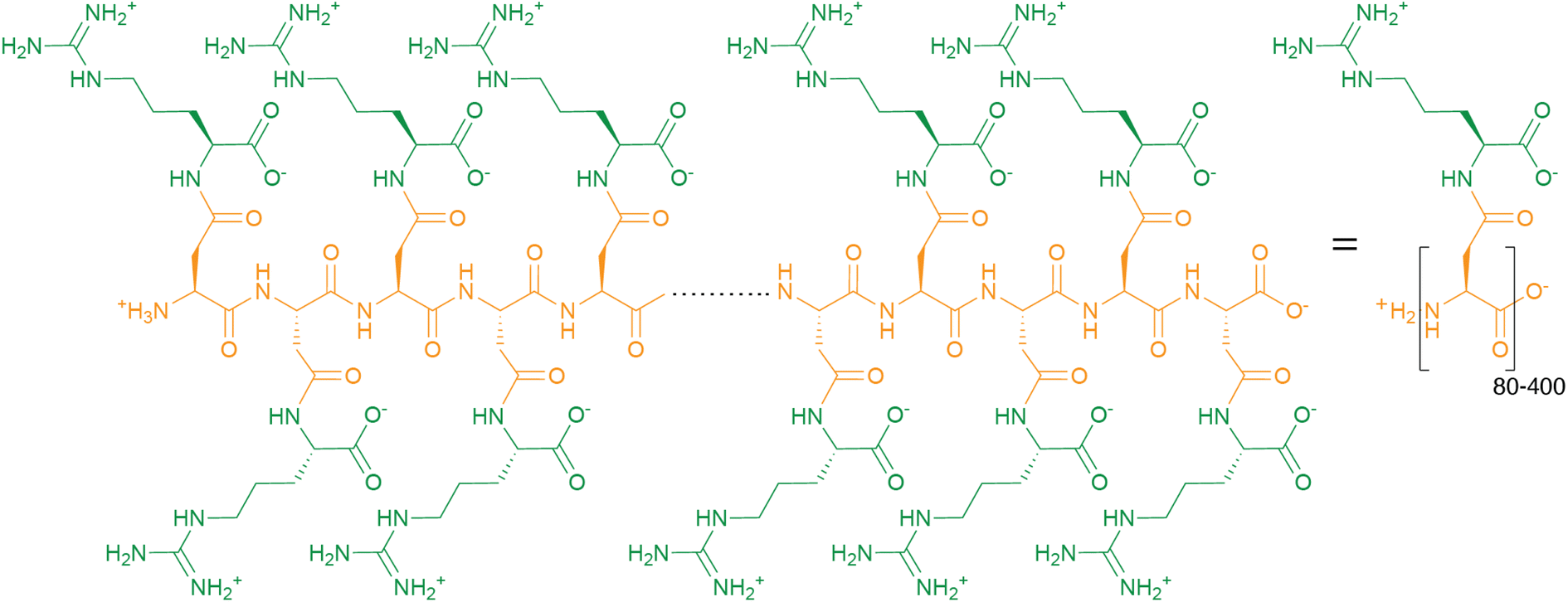 | ||
| Fig. 1 The chemical structure of cyanophycin. | ||
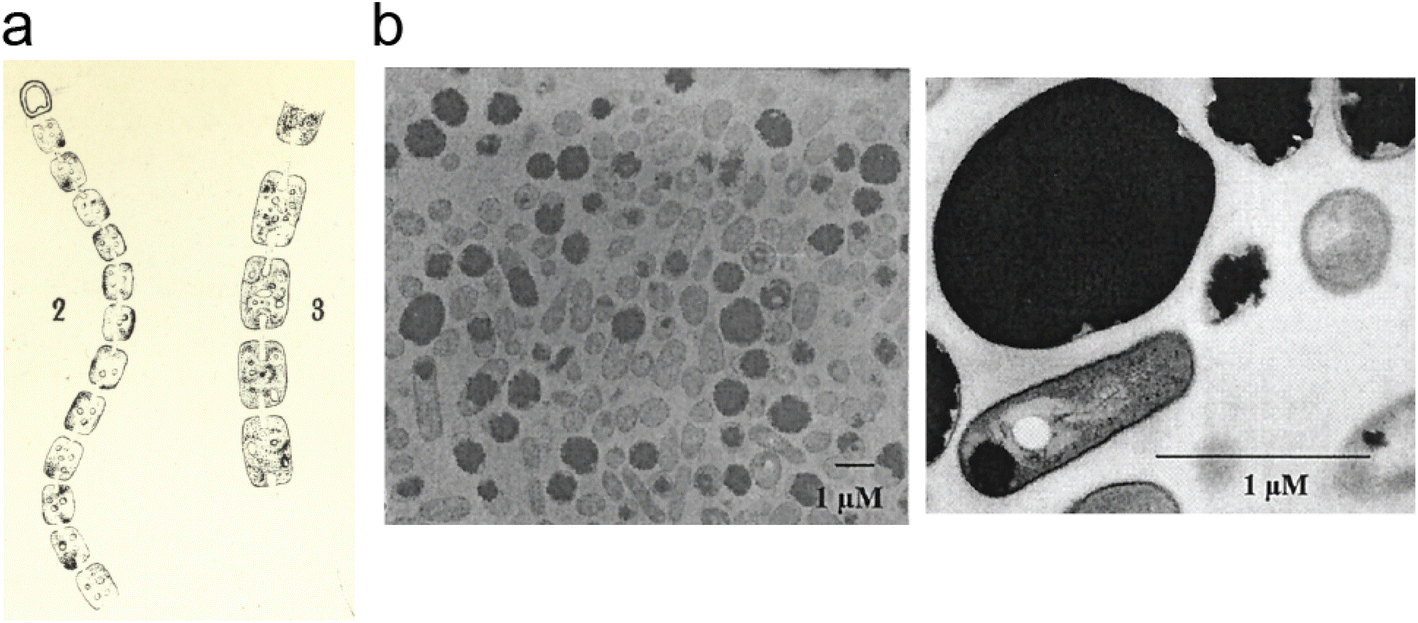 | ||
| Fig. 2 Cyanophycin granules in cells. (a) An image, hand-drawn by Antonino Borzì in ∼1886 showing cyanophycin in cyanobacterial cells. Reproduced from ref. 3. The image is in the public domain. (b) Electron micrographs of A. calcoaceticus grown under optimal conditions for cyanophycin production, showing cyanophycin granules which take up most of the cell.41 Reproduced from Fig. 3 of ref. 41 with permission from the American Society for Microbiology. | ||
The chemistry of cyanophycin metabolism is simple. It is biosynthesized in an ATP-dependent manner by one of two related enzymes. Cyanophycin synthetase 1 (CphA1), a common bacterial enzyme, iteratively incorporates Asp and Arg by alternating reactions at two different active sites.13 Cyanophycin synthetase 2 (CphA2), a cyanobacterial enzyme, polymerizes β-Asp-Arg dipeptides at a single catalytic site.14 Cyanophycin synthetases are more efficient when supplied with primers, i.e., short segments of cyanophycin.15,16 To use the stored nutrients, the polymer is degraded into Asp and Arg in two steps. First, cyanophycinase hydrolyzes cyanophycin into β-Asp-Arg dipeptides.17 Then, isodipeptidases degrade these dipeptides into Asp and Arg.18 The free amino acids can then feed into primary metabolism to provide the cell with fixed nitrogen, carbon and energy.
In addition to its biological significance, cyanophycin has a variety of potential industrial and biomedical applications. It has promising properties for self-assembling nano-vesicles19 and as a wound-healing bandage material.20 Cyanophycin can be used as a precursor for β-Asp-Arg, a nutritional supplement,21 and for poly-Asp, a biodegradable water softener, super-swelling material and useful biodegradable polymer.22–24 This has led to efforts by many groups endeavoring to increase the production levels of cyanophycin by assessing cyanophycin synthetases in vitro and in a variety of native and heterologous hosts,25,26 and to modify the material properties of cyanophycin and its derivatives.27,28
In this review, we endeavor to provide a holistic view of the advances in cyanophycin research since its discovery over a century ago. Although many readers will not have heard of cyanophycin (“not hot”), it is increasingly recognized as an important, widespread polymer. It is found in many different environments and microbiomes,29 and producing or scavenging it provides advantages to bacteria that are impactful on society. We particularly highlight the leap in molecular understanding gained by the recent flurry of crystal and electron microscopy structures covering enzymes that catalyze every step of cyanophycin metabolism. These include a series of structures of the star of the cyanophycin show – cyanophycin synthetase 1, a remarkable multi-domain, multi-functional biosynthetic machine, inside which we can finally look.15,30,31
2 Cyanophycin and the bacteria that produce it
2.1 Discovery and characterization of the cyanophycin polymer
Around 140 years ago, the eminent Italian botanist Antonino Borzì observed that cyanobacterial cells can contain large light-refracting granules (Fig. 2a).2,3 Because the analytical tools at his disposal were a light microscope, stains and basic chemical treatments, Borzì could not ascertain the nature of the material forming these granules, but named it “cianoficina”3 after the cyanobacteria (then called Cyanophyceæ) in which it was discovered. Although a role as a nutrient store and a “proteid” character was suggested early on,3,32–34 its nature and purpose was hotly debated (see Macallum,32 Fritsch35 and references therein), and almost 100 years passed before Robert Simon ascertained that the granules consist of long chains of poly-aspartate with arginine residues attached to the sidechain of each aspartate residue9,36,37 (Fig. 1), formally called “multi-L-arginyl-poly(L-aspartic acid)”. Simon9,37 and later researchers determined that individual chains of cyanophycin ranged from around 80 to 400 dipeptide residues, meaning that an average cyanophycin chain is more massive than the average protein.38,39 Another seminal contributor to cyanophycin research, Alexander Steinbüchel, later found that lysine can substitute for arginine, typically at low levels.40The chemical structure of cyanophycin endows it with unique properties. While its backbone is peptidic, it is resistant to proteolytic degradation by a variety of proteases36 because of the Arg decoration on all sidechains. The 24% nitrogen content by mass makes it the most nitrogenous common biopolymer, above typical proteins (∼13–19%),7 nucleic acids (∼16%),7 fat (0%) and glycogen (0%). Cyanophycin also has interesting solubility properties, which depend on the amount of lysine incorporated. Canonical (β-Asp-Arg)n cyanophycin is soluble in acidic or basic aqueous solutions,1,36 but very insoluble at physiological pH,36 causing spontaneous aggregation into the membrane-less granules Borzì observed in cells.41 Its net neutral charge and tendency to segregate into granules renders it inert, preventing it from affecting osmotic pressure or interfering with cellular processes. Increased lysine content leads to higher solubility at neutral pH, but this does not appear to adversely affect cell growth.
Amino acid polymers of simple composition are quite rare in nature. ε-Poly-lysine42 and the related polymers δ-poly-diaminobutanoic acid (δ-poly-DAB)43 and γ-poly-diaminopropionic acid (γ-poly-DAP)44 are made by some strains of Streptomyces, with ε-poly-Lys in wide use in Asia as a food preservative.42 γ-Poly-glutamate is an edible, water soluble polymer produced in Bacillus that has multiple industrial applications.45 γ-Poly-Glu is also synthesized in mammals, not as a free molecule but as a post-translational modification (PTM) on brain tubulin.46 However, these are all very different in mechanism of biosynthesis (see Section 4.1.3) and in nature from cyanophycin, being homopolymers and often of shorter length (e.g., ∼30 residues is typical for ε-poly-lysine47), making cyanophycin a truly unique molecule.
2.2 Modes of cyanophycin accumulation discovered
For decades, cyanophycin was known to exist only in cyanobacteria, so most studies have been performed with strains in this phylum. In pioneering work, Simon48 showed that exposing Anabaena cylindrica to the ribosome inhibitor chloramphenicol led to high accumulation of cyanophycin, and that upon chloramphenicol removal protein synthesis resumed and cyanophycin was degraded. This demonstrated that cyanophycin is made in a ribosome-independent manner. Moreover, Simon showed cyanophycin synthesis to be energy consuming and correctly posited that following its degradation, nitrogen from the cyanophycin was used for protein synthesis,10,48 giving the first clue to its primary role as a nitrogen reservoir.Following these first manipulations of cellular cyanophycin levels came detailed studies on conditions that influence cyanophycin accumulation. Perhaps predictably, cyanophycin accumulation was found to depend on the availability of sufficient amounts of carbon and fixed nitrogen (as nitrate or arginine) in Aphanocapsa sp. PCC 6308.8 Several sub-optimal cell growth conditions also increased accumulation of cyanophycin: Low levels of light, phosphorus and sulfur all lead to reduced cell growth, but more relative cyanophycin, measured as percentage of dry weight.8,49 That cyanobacteria can accumulate cyanophycin when the steady supply of nutrients is compromised was observed again in Agmenellum,50Synechocystis sp. PCC 630811 and Anabaena cylindrica.11 Other stressors seen to increase cyanophycin accumulation included high salinity (in Scytonema51), dehydration (in Nostoc elipsosporum),3 low temperature (in Aphanocapsa PCC 63088), and various antibiotics (in Agmenellum quadruplicatum52 and Fremyella diplosiphon53).
2.3 Use for dynamic nitrogen storage
The suggestion that cyanophycin is a dynamic nitrogen store for nitrogen-fixing cyanobacteria54 was logical, because that would be very useful to address a fundamental challenge these organisms face: Many cyanobacteria are both photosynthetic, oxidizing water to O2, and diazotrophic, fixing atmospheric N2 to ammonia.55 Diazotrophic bacteria are less dependent on the availability of fixed nitrogen in the environment, and so have a clear advantage under nitrogen-limited conditions. However, the key enzyme required for N2 fixation, nitrogenase, contains an iron-sulfur cluster which becomes oxidized in the presence of O2, leading to irreversible inactivation of the enzyme.56 Thus, nitrogen fixation is incompatible with photosynthesis.57Cyanobacteria that exist as a single cell type often separate nitrogen fixation and photosynthesis temporally.57,58 Diazotrophic, unicellular Cyanothece sp. ATCC 51142 possesses a day/night metabolic cycle, where photosynthesis occurs in daylight and nitrogenase activity is elevated at night. Sherman and coworkers observed that cyanophycin accumulation follows the same schedule: synthesis during dark periods and degradation in the light.58 This pattern was also observed in diazotrophic, colony-forming Trichodesmium,59 but not in the non-diazotrophic strain Synechocystis 680358 or in the heterocyst-forming Gloeothece and Anabaena cylindrica,60 consistent with cyanophycin serving as a dynamic reservoir of fixed nitrogen. The low solubility and reactivity of cyanophycin make it much better suited for this role than NH4+ or arginine, which at very high concentrations would interfere with cellular processes.
Bacteria that differentiate into specialized cell types can separate nitrogen fixation and photosynthesis spatially. Anabaena sp. PCC 7120 have vegetative cells, which perform photosynthesis and have high levels of cytosolic oxygen, as well as heterocysts, which perform nitrogen fixation and maintain low levels of cytosolic oxygen.55 Heterocysts can accumulate cyanophycin near their connection to vegetative cells.61 For the vegetative cells to access this nitrogen store, cyanophycin is first degraded in heterocysts by cyanophycinase, making β-Asp-Arg dipeptides,62 which are shuttled to vegetative cells. There, high levels of isoaspartyl dipeptidase enzyme degrade the dipeptides into free Asp and Arg, allowing rapid funneling of cyanophycin-derived material into other metabolic processes.61,63
Heterocyst-forming cyanobacterial species can also use cyanophycin for nitrogen storage.64 These species form akinetes when environmental conditions are unfavorable. Akinetes are similar to spores, having thick walls and slow metabolism to allow survival at elevated temperature, high salinity or low nutrient availability and germinating once favorable conditions return.64 Under akinete-inducing conditions, Aphanizomenon ovalisporum and Anabaena variabilis ATCC 29413 use a multi-step process to transiently accumulate cyanophycin (and glycogen) in cells that differentiate into akinetes.3,65,66 Large amounts of cyanophycin are observed in akinetes during differentiation, although the amount is much lower following their maturation,67 and germination of akinetes is not dependent on cyanophycin metabolism.66
Interestingly, the ability to make cyanophycin appears to provide a fitness advantage to Synechocystis sp. PCC 6308 cells under conditions of limited nitrogen despite only low levels of cyanophycin accumulation.68 This suggests that cyanophycin's function as a transient nitrogen sink allows the cells to assimilate nitrogen more efficiently.68
2.4 Cyanophycin and cyanobacterial blooms
Many species of cyanobacteria form harmful algal blooms, a condition in which cells multiply to vast quantities and dominate the phytoplanktonic community.69,70 These blooms are often accompanied by the release of toxins,71 leading to extensive ecological harm, economical damage and health risks to humans.72 Nitrogen availability is a major factor in cyanobacterial blooms,70 and cyanophycin facilitates these blooms: Planktothrix agardhii changes the expression levels of cyanophycin-metabolizing genes in response to seasonal variations in nitrogen availability,73 with anabolic genes upregulated during high nitrogen availability and catabolic genes expressed when nitrogen is scarce. Similarly, Raphidiopsis raciborskii accumulates cyanophycin during periods of nitrogen fluctuation and degrades it during low nitrogen availability,12 including in nitrogen-deficient blooms. Indeed, Lu et al. conclude that nitrogen derived from cyanophycin, rather than from de novo fixation, is what supports persistent R. raciborskii blooms, which presents an unexpected challenge to mitigating these devastating events.122.5 Cyanophycin in non-cyanobacterial species
Cyanophycin is well studied in cyanobacteria, but its existence and roles in other bacteria are severely underappreciated and understudied. Indeed, publications continue to refer to it erroneously as “unique to cyanobacteria”.74 Füser and Steinbüchel75 had already reported cyanophycin-metabolising genes from non-cyanobacteria strains in 2007, and of the non-redundant protein sequences currently available in databases, only ∼16% are cyanobacterial (Fig. 3). To date, only one study has investigated the role of cyanophycin in non-cyanobacterial species. In the firmicute Clostridium perfringens SM 101, cyanophycin was shown to be involved in spore formation:76 Cyanophycinase was detected in a set of membrane-associated proteins of germinated C. perfringens spores, and mutants deficient in cyanophycin production produced fewer and smaller spores. Cyanophycin use in spores is reminiscent of that in cyanobacterial akinetes, but only a small fraction of the bacteria that encode cyanophycin synthetase are cyanobacterial or spore-forming strains, so additional unknown cellular roles for cyanophycin almost certainly exist.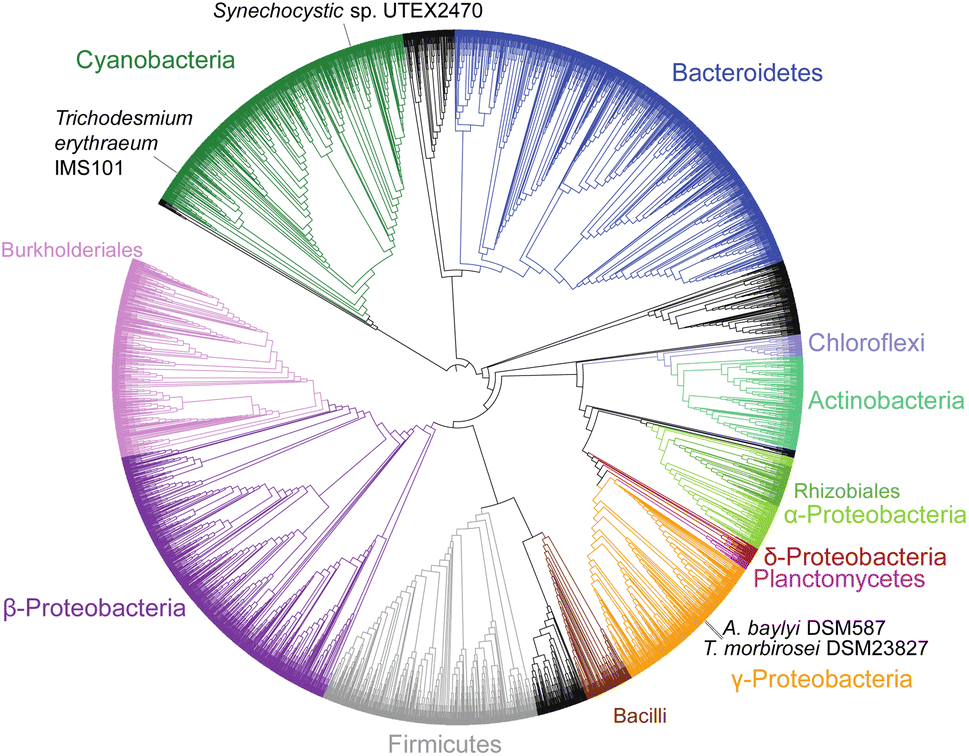 | ||
| Fig. 3 Phylogenetic tree of 4229 CphA1 sequences. The positions of the four CphA1 enzymes with determined structures, SuCphA1, TeCphA1, TmCphA1 and AbCphA1, marked. | ||
2.6 Cyanophycin-scavenging microorganisms
Cyanophycin-producing organisms exist in many environments, so it should not be surprising that non-producers have also evolved the ability to use nutrients stored within cyanophycin. Steinbüchel and coworkers screened samples from forest soil,77 aerobic78 and anaerobic79 pond sediments, and the gut flora of many different animals80 and found cyanophycin degradation activity from each environment. Remarkably, bacterial strains or consortia capable of using cyanophycin both as a sole nitrogen and carbon source were isolated from all environments.81 The isolates included likely non-producers79 that express an exported version of cyanophycinase (CphE) which degrades cyanophycin extracellularly. The cphE gene can be found in fungi82 and in bacteria, either distal from other cyanophycin genes or within dedicated operons that encode both cyanophycinase and isoaspartyl dipeptidase.4 Since no mechanism for extruding cyanophycin polymer from live cells is known, it is likely that extracellular cyanophycin comes from lysed cells.We recently discovered that the human pathogen Pseudomonas aeruginosa and many other Pseudomonas species encode the ability to import and survive on cyanophycin-derived material.6 Their aot operon encodes a multi-subunit arginine transporter (AotJQMP), an arginine-dependent transcription activator (ArgR), and a previously uncharacterized enzyme, AotO.83,84 We found AotO to be a member of a new family of cyanophycin dipeptide hydrolase enzymes (see Section 4.4.1) specific for β-Asp-Arg/Lys, and that AotJQMP can transport β-Asp-Arg in addition to Arg. This machinery allows P. aeruginosa to use β-Asp-Arg as a sole, but rather poor carbon source, and as a sole nitrogen source as effective as NH4+. Many AotO homologs exist, suggesting cyanophycin scavenging is quite common.
3 Biotechnological production and uses of cyanophycin
3.1 Industrial and biomedical uses
Cyanophycin and its derivatives have promising industrial and medical uses. For example, Tseng et al. showed that polyethylene glycol-conjugated cyanophycin can form self-assembling nanovesicles which reversibly encapsulate small molecules in a temperature and pH-dependent manner.19 These could have possible uses in drug delivery, as Grogg et al. found that intravenous injection of cyanophycin had no adverse effects in mice.85,86 Cyanophycin has also been proposed as a wound dressing, as layers of cyanophycin and hyaluronic acid or γ-polyglutamic acid increased cell migration in cultures, which should potentiate healing.20 In addition, cyanophycin is a candidate for adsorption of anionic pollutants in wastewater.29Cyanophycin has also been processed to materials with commercial applications. It can be enzymatically hydrolyzed to dipeptides,17 or chemically hydrolyzed to dipeptides or poly-Asp depending on the conditions.87 β-Asp-Arg/Lys dipeptides can serve as a nutritional amino acid source, since dipeptides are thought to have higher bioavailability than free amino acids or protein.88 Dipeptides have also been proposed as tyrosinase inhibitors.89 Polyaspartate is currently synthesized chemically and is a biodegradable, biocompatible polymer with multiple biomedical24 and industrial23 applications, for example as a green antiscalant or water softener.90
3.2 Biotechnological production of cyanophycin in vivo
To realize its full commercial potential, cyanophycin must be produced in large amounts and at low cost. The most promising approach is in vivo production, and many studies have been performed with different native8,38,48,50,91,92 and heterologous25,26,87,93–100 hosts, with various CphA1 enzymes15,16,91,96,100–106 and growth conditions. An excellent review by Frommeyer, Wiefel and Steinbüchel summarizes this field comprehensively.28 Briefly, for cyanophycin production in native hosts, various bioengineering approaches and optimized growth conditions have been explored.8,38,48,50,91,92,107 Currently, an engineered Synechocystis sp. PCC 6803 holds the record for native-source yield at a remarkable 57% (w/w) of cell dry mass,108 with R. eutropha107 and Acinetobacter baylyi strain ADP1 (ref. 41) not far behind at 48% and 46%, respectively (Fig. 2b). A key feature of these high-producing strains is alterations of primary metabolism, for example with mutations to increase the flux through the arginine anabolism pathway, to provide cyanophycin synthetase with higher levels of substrate.41,107,108 Heterologous production organisms can be relatively simple to construct (“Just add cphA1”), and heterologous hosts assayed include bacteria (Escherichia coli,93,109Corynebacterium glutamicum,94,95Bacillus megaterium,96Ralstonia eutropha,96Sinorhizobium meliloti,97Pseudomonas putida96), fungi (Saccharomyces cerevisiae,87,110Pichia pastoris,26Rhizopus oryzae98) and plants111 (tobacco,25,99,112 potato100). Generally, bacterial hosts have been the most successful with yields up to 44% of cell dry mass in S. meliloti97 and P. putida,113 but plants are useful too. A recent analysis concluded that large-scale production of cyanophycin in tobacco is already commercially viable.25 Remarkably, dedicated cyanophycin synthesis may not even be necessary: bacterial sludge in wastewater treatment plants contains relatively high levels of cphA1, and large amounts of cyanophycin could be isolated from sludge samples, suggesting it could be an essentially free source of the polymer.293.3 In vivo production of cyanophycin variants
Various cyanophycin production systems vary the characteristics of the resulting polymer. The backbone is essentially always polyAsp,28 and Arg is typically the β-linked amino acid, with low levels of Lys often observed.28 By varying the CphA1, host, and growth conditions, cyanophycin-like polymers with high levels of other amino acids in place of Arg can be obtained. In one notable study, Steinle et al. expressed CphA1 from Synechocystis sp. PCC 6308 in yeast strains harboring mutations that inactivate arginine metabolism.87 Deletion of argininosuccinate synthetase produced cyanophycin with citrulline present as up to 40% of the β-linked residues, and deletion of ornithine carbamoyltransferase led to 16% ornithine at these positions. Similarly, citrulline is incorporated in 18% of β-linked positions when Synechocystis sp. PCC 6308 CphA1 is expressed in P. putida ATCC 4359.113 These results highlight the potential promiscuity of CphA1 under certain metabolic conditions.The composition of cyanophycin is important because it affects the polymer's properties. Cyanophycin is often purified in an “insoluble” form, which (more precisely) is insoluble at neutral pH, and highly soluble at high or low pH. A “soluble” form, which dissolves in water regardless of pH, is sometimes also produced. Frommeyer et al. first reported that the major difference between soluble and insoluble forms of cyanophycin is the lysine content.40 Cyanophycin from heterologous expression of various CphA1 enzymes had variable lysine content, and the soluble form of the polymer cyanophycin can have Lys in up to 50% of the β-linked positions, whereas the insoluble form had ∼10%. A later study found a similar trend and reported that higher temperature and Lys content increases solubility,114 which allows for easy separation of polymer fractions with different characteristics.
Ambitious attempts to alter the products of cyanophycin synthetase more markedly, including the direct synthesis of poly-Asp, have been undertaken by us and others, with breakthroughs yet to come. It is not clear that cyanophycin synthetase is a better starting point than poly-Lys or poly-Glu producing enzymes to bioengineer into a poly-Asp polymerase.42–46
4 Biochemistry and structural biology of cyanophycin metabolizing enzymes
As mentioned in the introduction, cyanophycin can be synthesized from the proteinogenic amino acids Asp and Arg by a single enzyme, and broken down to Asp and Arg by two enzymes (Fig. 4), making its metabolic pathway conceptually simple. However, the biochemistry and structural biology of these enzymes show the enzymes, especially cyanophycin synthetase 1, to be remarkable and elegant.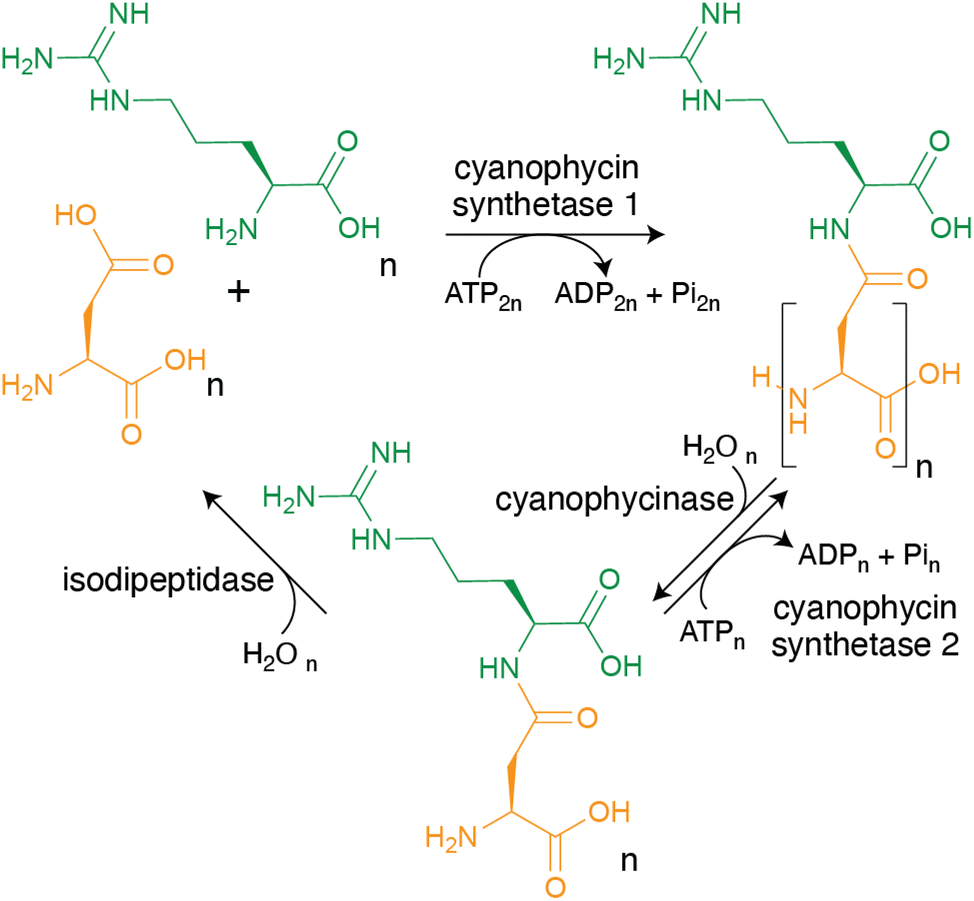 | ||
| Fig. 4 The steps of cyanophycin biosynthesis and biodegradation. CphA1 polymerizes Asp and Arg into cyanophycin. Cyanophycinase degrades cyanophycin to β-Asp-Arg dipeptides. These dipeptides can either be re-polymerized by CphA2, or hydrolyzed into Asp and Arg by isodipeptidases (isoaspartyl dipeptidase, isoaspartyl aminopeptidase or cyanophycin dipeptide hydrolase). | ||
4.1 Cyanophycin synthetase 1 (CphA1)
Following the discovery that cyanophycin was synthesized in a ribosome-independent manner,48 Simon used ammonium sulfate fractionation and ion exchange chromatography to prepare a sample with 92-fold enrichment in cyanophycin synthesis activity.37 The enriched enzyme(s), which he named multi-L-arginyl-poly(L-aspartic acid) synthetase, required Asp, Arg, ATP, MgCl2 and KCl. Twenty-two years later, Lockau and coworkers13 proved that cyanophycin synthetase (first called CphA, later CphA1 (ref. 75)) was a single (multimeric) enzyme by cloning a cphA1 gene and showing it was sufficient to heterologously produce cyanophycin.Identification of cphA1 and the ever-increasing availability of gene and genome sequences has allowed progressively better characterization of CphA1 enzymes, in silico, in vivo28 and in vitro (Table 1). Amino acid sequences revealed CphA1 to be ∼100 kDa in mass, with three regions that we30 later named the N domain (N terminal domain; residues 1–160 in Synechocystis sp. UTEX2470 CphA1 (SuCphA1)), the G domain (glutathione synthetase-like domain; residues 161–470), and the M domain (Mur ligase like domain; residues 471–873) (Fig. 6). Mutagenesis proved that the G domain ligates Asp to the growing cyanophycin chain and thus that the M domain likely ligates Arg115 (Fig. 5). These two synthetic active sites were shown to act iteratively13,101,116–118 with chemical mechanisms likely analogous to their ATP-grasp and Mur ligase relatives, respectively.13,101,117,118KM values for Asp (240–500 μM) and for Arg (15–50 μM) were measured, and the two KM values for ATP (38 and 210 μM) supported the two active site model.119 CphA1 requires Mg2+ for substrate phosphorylation by ATP, but as with many other enzymes,120,121 a definitive explanation for its K+ requirement is not clear. CphA1 was reported to form dimers13,16 or tetramers122 in solution and tends to associate with cyanophycin polymer/granules,68 a behavior promoted by Mg2+ ions119 and decreased during cyanophycin catabolism.68 In addition, most characterized CphA1s were described as primer dependent,13 meaning they can only extend existing chains of cyanophycin, not start polymerization de novo. Some molecules other than cyanophycin, such as N-acetylglucosamine, are thought to also serve as primers, albeit with low efficiency.123
| Organism | Purification | Oligomerization | Year |
|---|---|---|---|
| Anabaena cylindrica | Yes | — | Simon, 1976 (ref. 37) |
| Anabaena variabilis | Yes | Dimer | Ziegler et al., 1998 (ref. 13) |
| Synechococcus sp. MA19 | Yes | — | Hai et al., 1999 (ref. 38) |
| Synechocystis sp. PCC 6803 | Partial | — | Aboulmagd et al., 2000 (ref. 154) |
| Anabaena variabilis ATCC 29413 | Yes | — | Berg et al., 2000 (ref. 116) |
| Synechocystis sp. PCC 6308 | Yes | Dimer | Aboulmagd et al., 2000 (ref. 16) |
| Synechococcus sp. MA19 | Yes | — | Hai et al., 2002 (ref. 123) |
| Acinetobacter baylyi ADP1 | No | — | Krehenbrink et al., 2002 (ref. 93) |
| Desulfitobacterium hafniense | No | — | Ziegler et al., (ref. 2002 155) |
| Acinetobacter baylyi ADP1 | Yes | — | Krehenbrink et al., 2004 (ref. 119) |
| Anabaena sp. PCC 7120 | No | — | Voss et al., 2004 (ref. 96) |
| Nostoc ellipsosporum | No | — | Hai et al., 2006 (ref. 101) |
| Thermosynechococcus elongatus BP-1 | Yes | Tetramer | Arai et al., 2008 (ref. 122) |
| Nostoc ellipsosporum | Yes | Dimer | Hai et al., 2008 (ref. 132) |
| Unknown cyanobacterium 49 | Yes | — | Du et al., 2013 (ref. 106) |
| Acinetobacter baylyi DSM 587 | Yes | Tetramer | Sharon et al., 2021 (ref. 30) |
| Tatumella morbirosei DSM 23827 | Yes | Tetramer | Sharon et al., 2021 (ref. 30) |
| Synechocystis sp. UTEX 2470 | Yes | Tetramer | Sharon et al., 2021 (ref. 30) |
| Trichodesmium erythraeum IMS 101 | Yes | Tetramer | Miyakawa et al., 2022 (ref. 31) |
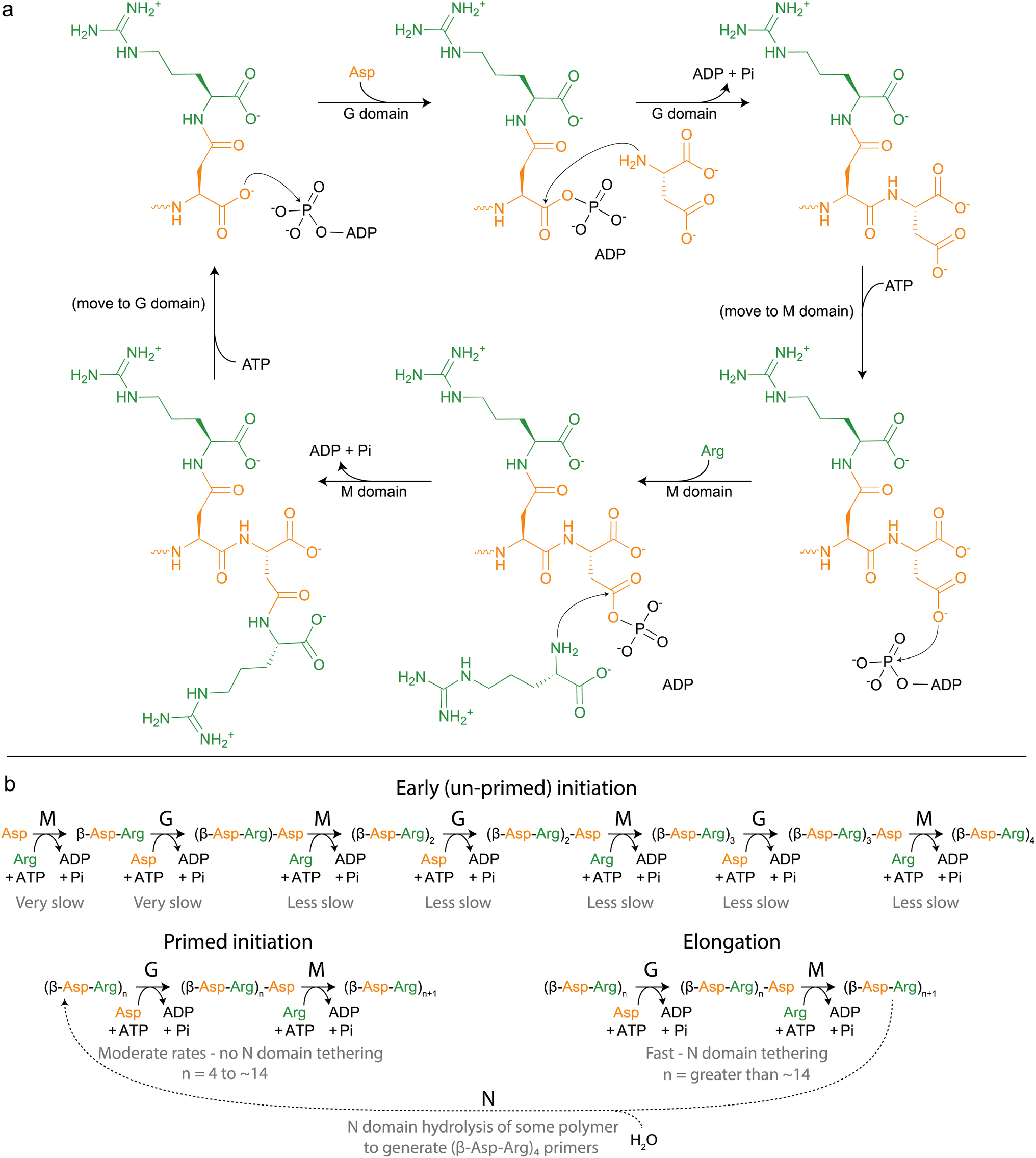 | ||
| Fig. 5 Cyanophycin biosynthesis by CphA1. (a) Schematic diagram of the reactions catalyzed by CphA1, initially proposed by Berg et al., 2000.116 (b) More complete model of cyanophycin biosynthesis, grouped into phases and including action of the N domain. Relative rates of each reaction have not been independently measured, but are inferred by rates of overall cyanophycin biosynthesis from various starting materials. | ||
With that foundation of knowledge, recent structural and functional studies on several CphA1 enzymes by us15,30 and Miyakawa et al.31 finally provided an exciting look inside this multi-functional biosynthetic machine. Overall, CphA1 has an elegant dimer-of-dimers architecture (Fig. 6). In cyanobacterial SuCphA1 (ref. 15 and 30) and Trichodesmium erythraeum IMS 101 (Te)CphA1,31 tri-lobed protomers make extensive interactions through their G domains to form a constituent dimer. The contacts that build the tetramer from constituent dimers, conversely, are strikingly small: SuCphA1 W672 residues of each M domain pack into shallow pockets in the G domains of the adjacent dimers, burying only ∼450 Å2 of surface area each. The resulting tetramer architecture resembles a spikey, hollow ball. Proteobacterial Tatumella morbirosei DSM 23827 (Tm)CphA1 and Acinetobacter baylyi DSM 587 (Ab)CphA1 display the same protomer and constituent dimer configuration, but a very different tetramer form. An extensive ∼1800 Å2 interface results in a spikey ring shape with a large central cavity. The equivalent of W672 is found in ∼30% of unique CphA1 sequences, suggesting they probably all adopt the spherical tetramer architecture, but it is not clear if all CphA1s that lack this residue adopt the ring architecture.
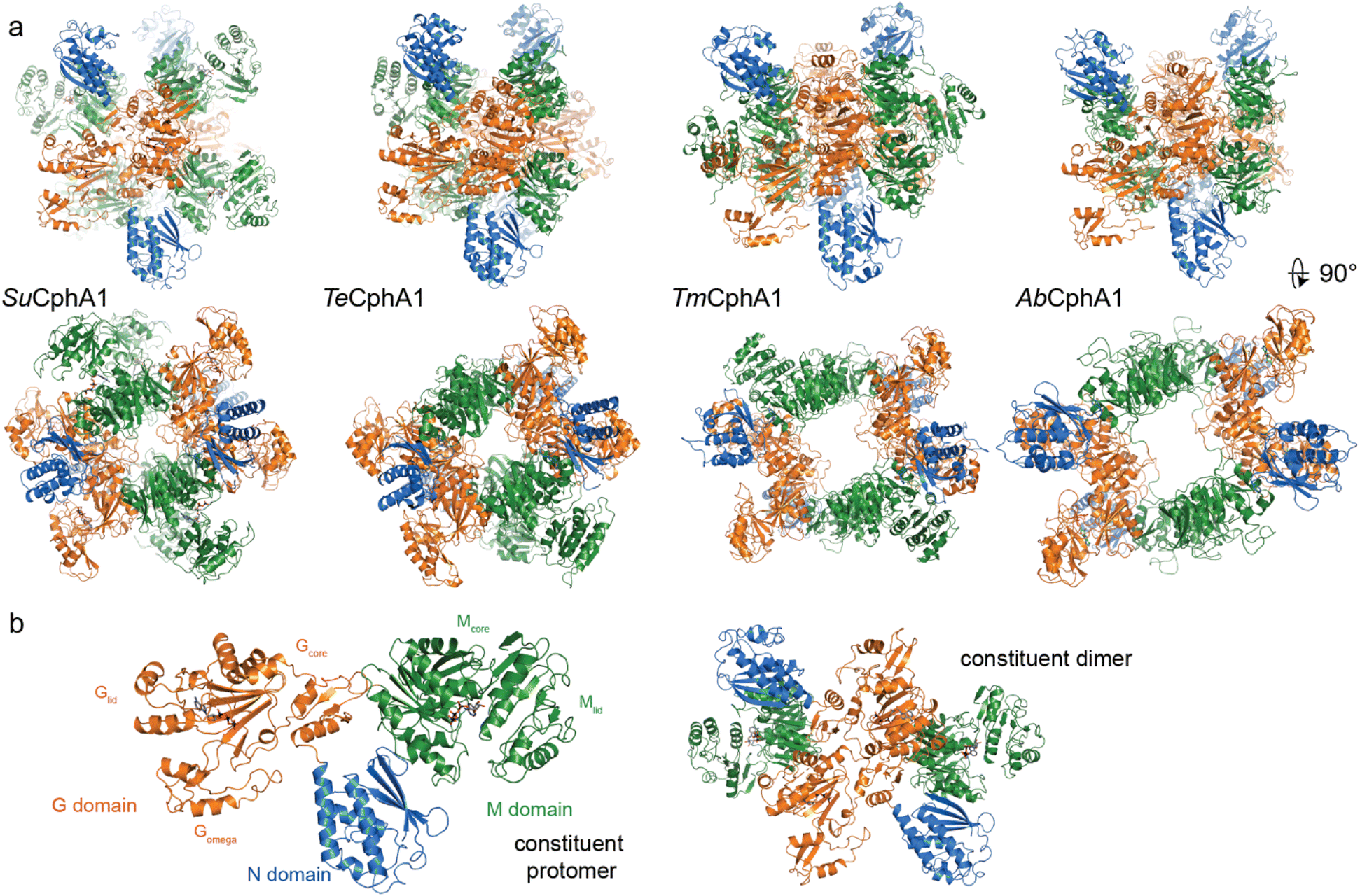 | ||
| Fig. 6 Structures of cyanophycin synthetase 1 (a) Architectures of the four CphA1 enzymes with determined structures.15,30,31Synechocystis sp. UTEX 2470 SuCphA1,15,30Trichodesmium erythraeum IMS101 TeCphA1 (ref. 31) and Tatumella morbirosei DSM 23827 TmCphA1 (ref. 30) were solved as tetramers, while Acinetobacter baylyi DSM 587 AbCphA1 (ref. 30) is a tetramer in solution but mainly dissociated to constituent dimers on the EM grid. The tetrameric AbCphA1 shown here was re-assembled using TmCphA1 as a guide. Cyanobacterial SuCphA1 and TeCphA1 share 70% identity, gammaproteobacterial TmCphA1 and AbCphA1 share 75% identity, while SuCphA1 and TmCphA1 share 41% identity. (b) The monomer and dimer architectures of SuCphA1, which are very similar amongst the 4 enzymes. | ||
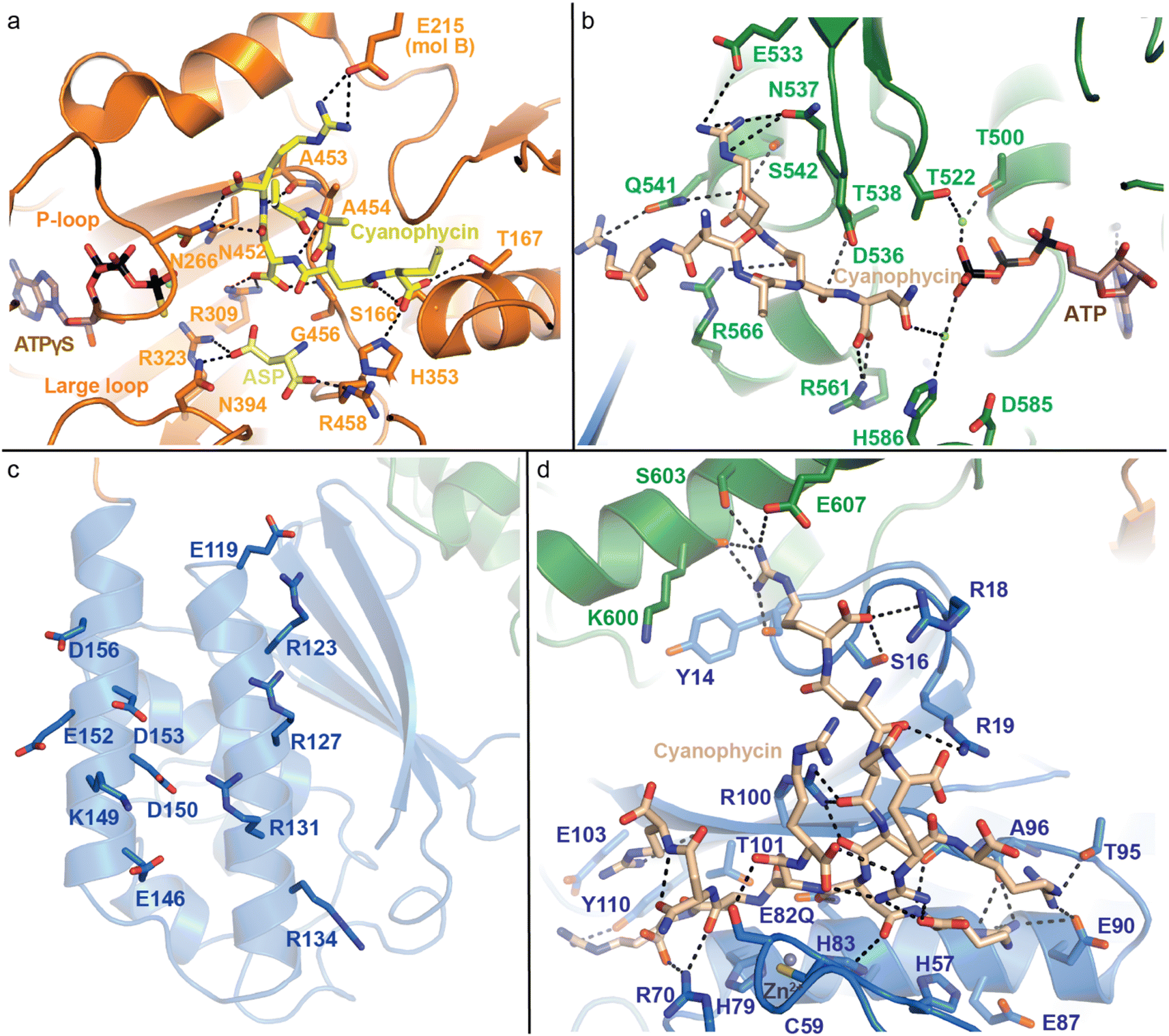 | ||
| Fig. 7 Active domains of CphA1 with bound substrates. (a) The G domain of TeCphA1 bound to cyanophycin, Asp and an ATP analog (PDB 7WAE).31 (b) The M domain of SuCphA1 with cyanophycin and ATP (PDB 7LGQ).30 (c) The charged residues on the N domain helices of SuCphA1 (PDB 7LGQ)30 loosely bind cyanophycin polymer. (d) The hydrolytic active site in the N domain SuCphA1(E82Q) bound to cyanophycin (PDB 7TXV).15 | ||
The M domain likewise performs two-step amide bond formation by an analogous pathway to the G domain, and to the Mur ligases to which the M domain is related.131 Like Mur ligases, the M domain has core and lid subdomains, although it lacks the Mur ligase N-terminal domain. Note that Mlid and Mcore have different folds from Glid and Gcore and the domains are not evolutionarily related, despite catalyzing analogous reactions. Mcore provides an extensive binding site for the cyanophycin intermediate (β-Asp-Arg)n-Asp, including a key SuCphA1 R561 interaction with the terminal Asp residue's α-carboxylate (Fig. 7b). This interaction positions the reactive β-carboxylate near ATP, allowing CphA1 to differentiate between these two very similar, proximal moieties.30 This ATP is bound to the Mlid, which is known to be mobile in Mur ligases and shows positional variability in CphA1 cryo-EM and crystal structures. Movement of Mlid (like that of Glid) is important for phosphorylation and nucleotide exchange, and its truncation inactivates CphA1.101,132 The incoming Arg substrate likely binds in the crevice between Mcore and Mlid, but, like in structural studies of many Mur ligases, the substrate was not observed.
SuCphA1 structures also helped reveal a completely unexpected catalytic role for the N domain. We had observed that omitting primer from cyanophycin synthesis reactions delayed SuCphA1 activity by only ∼15 minutes, while TmCphA1 made no cyanophycin at all in the absence of exogenous primer.15 After extensive mutagenesis and residue swapping experiments on G and M domains, we found that the N domain of SuCphA1, provided as an extruded domain or a chimera, allowed TmCphA1 to synthesize cyanophycin in the absence of exogenous primer. A re-examination of the SuCphA1 N domain revealed a hitherto unrecognized Cx19HxxEH motif on the “back” side of the N domain, reminiscent of the inverted zinc metallopeptidase HxxEH motif. The cryptic active site residues are all conserved in over 80% of CphA1 enzymes, including SuCphA1 and TeCphA, but hydrophobic residues take their place in a minority of CphA1s, including TmCphA1 and AbCphA1. New structures of SuCphA1 with cyanophycin polymer caught the pre- and post-hydrolysis states: SuCphA1E82Q showed that the N domain binds a stretch of 7 cyanophycin dipeptide residues, with the scissile peptide bond positioned directly over the Zn2+ ion (Fig. 7d), while wildtype SuCphA1 showed a (β-Asp-Arg)4 cleavage product. Mass spectrometry confirmed the N domain to be an endo-cyanophycinase with a preference for cleaving the polymer to (β-Asp-Arg)4. Notably, systematic assessment of primer activity of progressively longer cyanophycin segments showed (β-Asp-Arg)4 to be an excellent primer, in agreement with the four dipeptide residues observed ordered at the G domain active site. Furthermore, tetramerization of SuCphA1 contributes to the efficacy of primer-independent activity, possibly by increasing the local concentration of N domain active sites for nascent polymer chains.15 Thus, most CphA1s neatly solve the problem of primer independence by encoding a hydrolytic site that can produce primers of optimal length.
With the identification of the N domain active site, it is simple to predict which enzymes can self-provide primers. Interestingly, while installing the N domain active site into a CphA1 that lacks it can improve heterologous cyanophycin yields in vivo, CphA1 enzymes lacking the N domain active site can still produce large amounts of cyanophycin in heterologous hosts,91 perhaps using non-cyanophycin primers.16,116 The presence of a metalloprotease site in the large majority of CphA1s indicates that it provides an advantage to the producing organisms, perhaps by increasing the speed with which they can switch from cyanophycin degradation to cyanophycin accumulation modes.
This scheme of biosynthesis is unique among nature's polypeptide makers. A comprehensive discussion comparing each strategy is beyond the scope of this review, but key features of seven polypeptide polymerases are summarized in Table 2.
| System | Polypeptide product | Typical length of product | Linkage | Building block substrate range | Substrate selection | Energy source per bond | Mode of activation | Bond formation catalytic strategy |
|---|---|---|---|---|---|---|---|---|
| Ribosome (and translation machinery)156 | Proteins and peptides; also RiPPs157 | ∼Tens – thousands of residues | Peptide | Proteinogenic amino acids | Each residue dictated by the order of codons in mRNA, read by aa-tRNA | 1 ATP plus 2 GTP (used by EF-Tu, EF-G) | Adenylation by aminoacyl-tRNA synthetase | Repeated use of single active site (peptidyl transferase center) |
| Nonribosomal peptide synthetases (canonical, modular)158 | Nonribosomal peptides | ∼2–20 residues | Peptide | Amino acids and ∼500 other carboxylic acids | Each residue dictated by direct binding to each module's adenylation domain | 1 ATP | Adenylation by the adenylation domain | Single use of dedicated condensation domains in each elongation module |
| Nonribosomal peptide synthetases (membrane bound poly-acyl synthetases)42–44 | γ-poly-DAP, γ-poly-DAB, ε-poly-Lys | ∼5–35 residues | Isopeptide | DAP, DAB, Lys | All residues dictated by repeated action of single adenylation domain | 1 ATP | Adenylation by the adenylation domain | Repeated use of single active site in integral membrane condensation domain |
| γ-PGA biosynthetase159 | γ-poly-Glu | ∼80–8000 residues | Isopeptide | D/L-Glu | Dictated as Glu by direct binding to PGA complex | 1 ATP | Phosphorylation by an M domain – like subunit | Repeated use of single active site in the M domain-like subunit |
| Tubulin polyglutamylase (TTLL)46 | γ-poly-Glu PTM on tubulin | ∼1–20 residues | Isopeptide | L-Glu | Dictated as Glu by direct binding to TTLL | 1 ATP | Phosphorylation by TTLL | Repeated use of single active site in TTLL |
| Cyanophycin synthetase 1 | Cyanophycin | ∼80–400 dipeptide residues | Peptide and isopeptide | Backbone: Asp; decoration: Arg, some Lys, rarely Orn, Cit | Backbone dictated as Asp by direct binding to G domain; decoration dictated by direct binding to M domain | 1 ATP for each peptide, isopeptide bond | Phosphorylation by G domain and M domains | Iterative use of active sites in G domain and M domain |
| Cyanophycin synthetase 2 | Cyanophycin | ∼80–400 dipeptide residues | Isopeptide | β-Asp-Arg dipeptide | Dictated as β-Asp-Arg by direct binding to G domain | 1 ATP | Phosphorylation by G domain | Repeated use of active site in G domain |
4.2 Cyanophycin synthetase 2
For some bacteria, one flavour of cyanophycin synthetase isn't enough. Herrero and coworkers133 showed that Anabaena sp. PCC 7120 has a canonical cphA1 gene as well as a gene encoding a related enzyme, cyanophycin synthetase 2 (CphA2). CphA2 sequences are shorter than those of CphA1, because they lack the M domain ATP binding site. Although their N domains share low sequence identity with CphA1 N domains, CphA2 clearly evolved from CphA1.14 In 2016, Lockau, Volkmer and colleagues14 showed that CphA2 catalyzes a single kind of ligation – the ATP-dependent polymerization of β-Asp-Arg dipeptides to form cyanophycin.14In vitro, CphA2 is robustly active with primer, but primer independent activity is only observed at high β-Asp-Arg concentrations (e.g. 100 mM).14 Like CphA1, CphA2 requires Mg2+ and K+ ions for its activity. The only known source of β-Asp-Arg dipeptides is cyanophycin degradation by cyanophycinase, so CphA2 is a “re-polymerase” (Fig. 4).CphA2 is found in unicellular and multicellular diazotrophic cyanobacteria that also encode CphA1, suggesting that the two enzymes play complementary roles.14,134 Knockout experiments showed that under N2-fixing conditions ΔcphA2 cells accumulated 10–20% less cyanophycin14,133 and displayed impaired growth.14 In Anabaena sp. PCC 7120, cphA1 and cphA2 are each found in a cluster with a copy of cphB,133 and their expression pattern differs somewhat: CphA1 is expressed in the presence of ammonium, nitrate or N2, but at higher levels in the absence of all exogenous nitrogen, while CphA2 is expressed in the presence of ammonium, nitrate or N2, but at higher levels in the absence of ammonium. A recent metatranscriptomic study showed that in the toxic-blooming cyanobacteria Planktothrix, cphA2 mRNA is more prevalent during seasons with low nitrogen availability, and cphA1 mRNA more prevalent when ammonium is abundant.73
The ability to separately control the relative CphA1 and CphA2 expression within a single cell, as well as differentially between cell types or based on position in a filament, provide cyanobacteria with mechanisms to control the balance between cyanophycin production and degradation.14,134 This could allow an advantageous fine-tuning of their arginine and aspartate budget and the amount of nitrogen that flows into primary metabolic processes.134
Our recent structures135,136 revealed two distinct architectures for CphA2. Of 9 enzymes we characterized biochemically, most exist as dimers,14,135 but one is hexameric (Figs. 8a and b, Table 3). The crystal structure of dimeric Gloeothece citriformis PCC 7424 (Gc)CphA2 and the EM structure of hexameric Stanieria sp. NIES 3757 (St)CphA2 show that CphA2 has the same domain structure as CphA1 (other than the absent Mlid) and that CphA2 protomers form dimers in much the same way as those in CphA1. StCphA2 further assembles these dimers into an esthetically pleasing open-ring, 2-fold symmetric hexamer.136 The CphA2 G domain binds cyanophycin much like CphA1 does30,136 (Figs. 8c and d). Its incoming substrate is β-Asp-Arg14 (not Asp as in CphA1), and although we were not able to visualize β-Asp-Arg bound to CphA2, the substrate specificity difference between CphA1 and CphA2 is likely dictated by differences in the sequence of Gomega. Mutational analysis and the substrate recognition role of the large loop of ATP-grasp enzymes support this proposed binding site.30,127,135
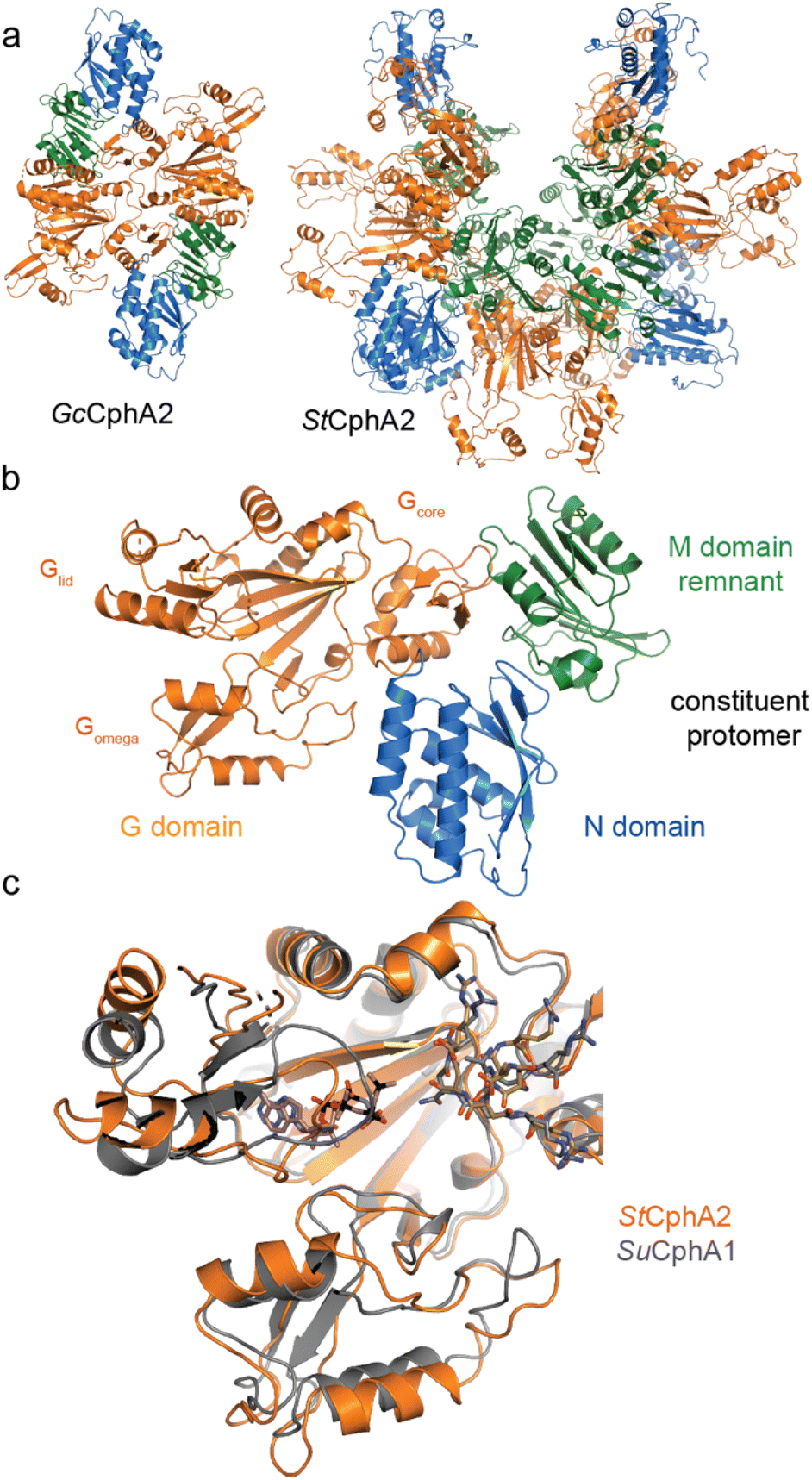 | ||
| Fig. 8 Structures of CphA2. (a) Architectures of GcCphA2 (PDB 7TA5)135 and StCphA2 (PDB 8FXH),136 which are a dimer and hexamer, respectively. (b) Protomer architecture of GcCphA2. (c) Overlay of the G domains of StCphA2 and SuCphA1 showing similar binding of cyanophycin. | ||
| Organism | Purification | Oligomerization | Year |
|---|---|---|---|
| Anabaena variabilis ATCC 29413 | Yes | Trimer/tetramer | Friederike et al., 2016 (ref. 14) |
| Cyanothecesp. PCC 7425 | Yes | — | Friederike et al., 2016 (ref. 14) |
| Gloeothece citriformis PCC 7424 | Yes | Dimer | Sharon et al., 2022 (ref. 135) |
| Anabaena variabilis PCC 7120 | Yes | Dimer | Sharon et al., 2022 (ref. 135) |
| Anabaena sp. UTEX 2576 | Yes | Dimer | Sharon et al., 2022 (ref. 135) |
| Calothrix elsteri CCALA 953 | Yes | Trimer/hexamer | Sharon et al., 2022 (ref. 135) |
| Leptolyngbya boryana NIES 2135 | Yes | Dimer | Sharon et al., 2022 (ref. 135) |
| Stenomitos frigidus ULC 18 | Yes | Dimer | Sharon et al., 2022 (ref. 135) |
| Mastigocladus laminosus UU 774 | Yes | Dimer | Sharon et al., 2022 (ref. 135) |
| Stanieria sp. NIES 3757 | Yes | Hexamer | Sharon et al., 2022 (ref. 135 and 136) |
| Tolypothrix sp. NIES 4075 | Yes | Dimer | Sharon et al., 2022 (ref. 135) |
The N domain does not appear to be as important or as interesting in CphA2. In CphA2, it does not have the same charged patches used by CphA1 to bind nascent cyanophycin chains and does not contain the primer-generating hydrolytic active site. The primer-independent activity observed at exceedingly high β-Asp-Arg concentrations presumably reflects the binding of the dipeptide to both cyanophycin and dipeptide binding sites. GcCphA2 was the only CphA2 we studied that could not synthesize cyanophycin from 100 mM β-Asp-Arg in the absence of primer, but we could impart this activity with a single point mutation in its G domain. The unremarkableness of the CphA2 N domain is easily explained by its physiological context. CphA2 has only one active site, so does not need to efficiently transfer the growing chain between G and M domains, obviating the need for charged patches in the N domain. Also, primer availability is less likely to be an issue for CphA2, because CphA2 is typically found in bacteria with CphA1 that possesses N domain hydrolytic sites,14,15 so the CphA1 can make primer. In addition, as a re-polymerase, CphA2 must be active only following cyanophycin degradation into β-Asp-Arg. This degradation would need to be extremely thorough so as not to leave any (β-Asp-Arg)≥3, which is an excellent primer.135
The reduced complexity of CphA2 compared to CphA1 may make it an easier template for bioengineering experiments to construct desirable homopolymers. However, because of the extensive contacts between the Arg residues of cyanophycin and the CphA2 G domain, as well as the elusiveness of complexes with all substrates bound, such bioengineering will not be easy.
4.3 Cyanophycinase
Cyanophycinases are C-terminal exo-peptidase enzymes which hydrolyze cyanophycin to β-Asp-Arg dipeptides.17,81 A dedicated and specific cyanophycinase is crucial for cyanophycin catabolism because cellular proteases and peptidases are unable to digest this biopolymer.17,81 First isolated in 1999 from Synechocystis sp. PCC 6803, cyanophycinase displays sequence similarity to nonclassical serine proteases like peptidase E.17 Cyanophycinases have been sub-classified into CphB (dimeric, intracellular enzymes of ∼30 kDa protomers17), CphI (∼80 kDa pseudodimeric enzymes in which only one active site is maintained75), and CphE (∼45 kDa enzymes that are exported from the cell for cyanophycin scavenging81).Bacteria normally have one cyanophycinase gene, for example either cphB or cphI, which are often found in genomes adjacent to cphA1, forming a minimal cyanophycin metabolism cluster.5,75 As expected from their respective catabolic and anabolic roles, expression of CphA1 and cyanophycinase can be differentially regulated, though under some conditions both are expressed.8,50,133,137 The cphE gene that encodes secreted cyanophycinase is found in bacteria (and some fungi) that do not make cyanophycin. Little is known about the mechanism controlling its expression, but it is notable that extracellular cyanophycinase activity has been detected in many bacterial isolates from a variety of environments.77,80,81
Kimber and colleagues138 determined the structure of Synechocystis sp. PCC 6803 (Sy)CphB in the absence of substrate, and we used a genetic code expansion approach139,140 to observe it in complex with an acyl-enzyme intermediate (Fig. 9). CphB displays a Ser-His-Glu catalytic triad,17 similar to the classic Ser-His-Asp triad, and the structures and accompanying mutagenesis experiments show how CphB possesses modified substrate binding regions specialized for cyanophycin. Proteases often use deep binding pockets to accommodate sidechains, but Arg-decorated Asp sidechains of cyanophycin are much bulkier than those of canonical protein residues. Cyanophycinase instead has very shallow binding pockets at the active site, allowing cyanophycin to make specific contacts with both the Asp and the Arg portions of P1 and P1′ β-Asp-Arg peptide residues and position the scissile peptide bond above the catalytic serine.
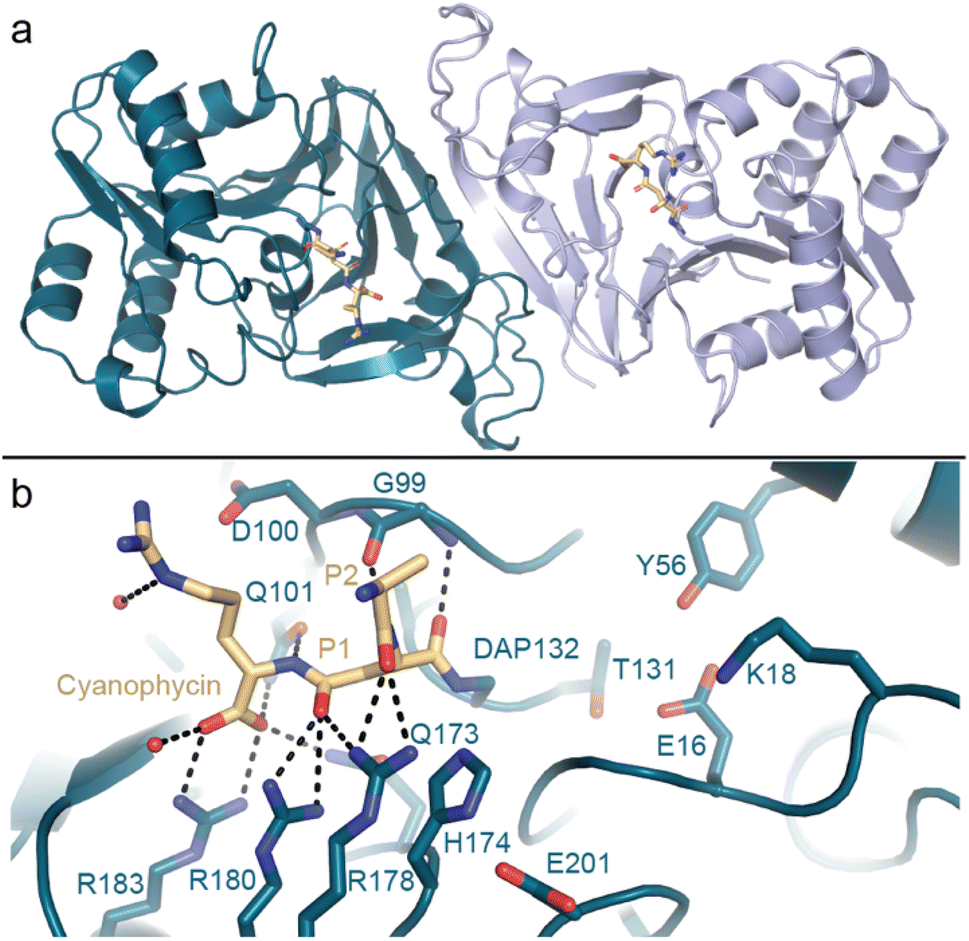 | ||
| Fig. 9 The crystal structure of cyanophycinase. (a) Structure of the SyCphBDAP dimer (PDB 7UQW).139 (b) The active site of SyCphBDAP with a covalently-bound cyanophycin fragment. | ||
4.4 General and specific cyanophycin dipeptide hydrolases
An enzyme that hydrolyzes β-Asp-Arg is required for cyanophycin metabolism, because the only known degradation pathway (Fig. 4) goes through β-Asp-Arg as an intermediate. Lockau et al.18 first showed that IaaA enzymes from Synechocystis and Anabaena can degrade β-Asp-Arg/Lys dipeptides. IadA was assumed to be involved in cyanophycin metabolism as well, but this had not been experimentally confirmed.17,75
Recently, we structurally and functionally characterized an IaaA and an IadA enzyme whose genes cluster with cphA1 and cphB in “complete” cyanophycin metabolism clusters5 (Fig. 10a). IadA from Leucothrix mucor DSM 2157 (LmIadA) and IaaA from Roseivivax halodurans DSM 15395 (RhIaaA) could each hydrolyze β-Asp-Arg and β-Asp-Lys, but were not specific to Arg/Lys as the β-linked amino acid. Their structures revealed their distinct structures and confirmed their mechanisms of substrate promiscuity: LmIadA forms an octameric assembly with binuclear Zn2+ active sites, whereas RhIaaA is a α2β2 tetramer with a catalytic Thr nucleophile. The architecture and dipeptide binding sites of both LmIadA and RhIaaA are very similar to previously characterized homologs from E. coli which are not involved in cyanophycin metabolism.148,149 Both binding sites provide specific interactions only with the Asp moiety, while the β-linked amino acid points towards solution. Since their source organisms evolved to cluster genes for LmIadA and RhIaaA with cyanophycin metabolism genes, their expression may be regulated for function in cyanophycin degradation, but they have not developed specificity for it.
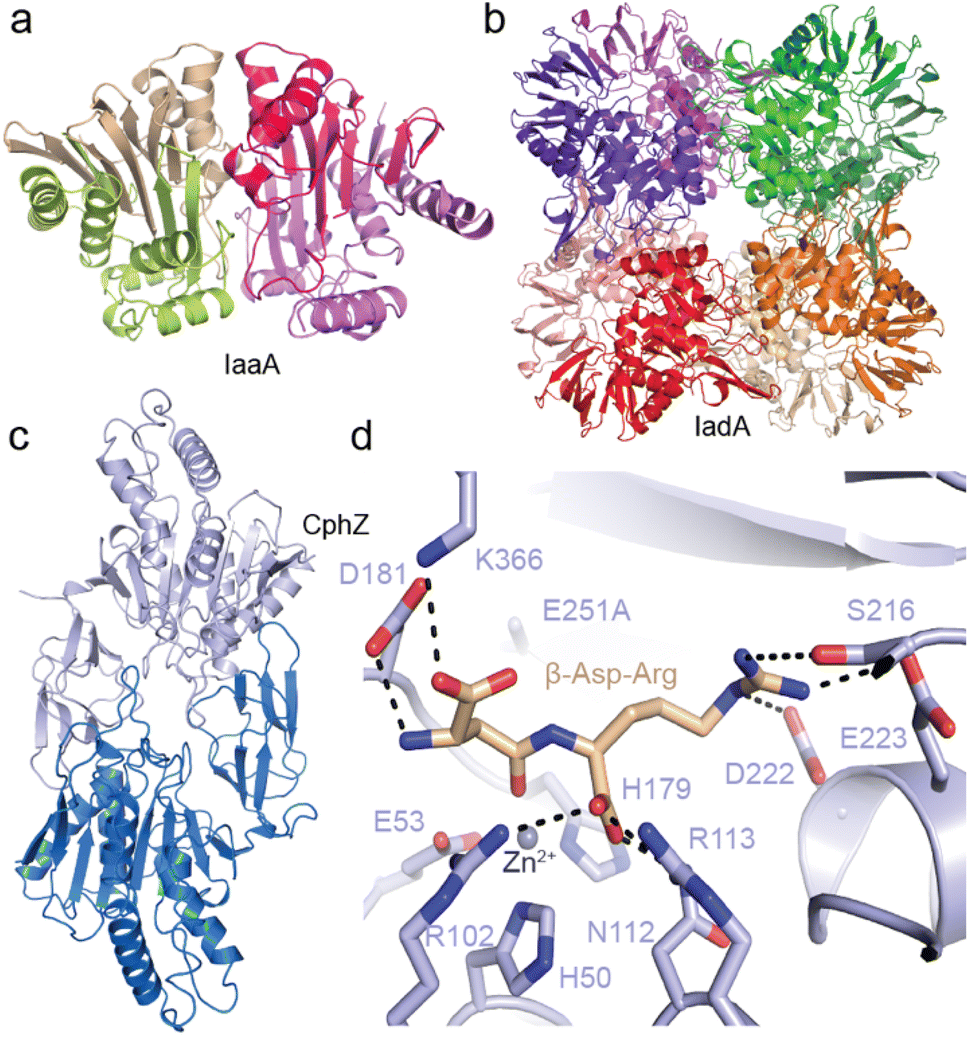 | ||
| Fig. 10 Structures of isoaspartyl dipeptidases and cyanophycin dipeptide hydrolase. (a) The crystal structure of LmIadA (PDB 8DQN).5 (b) The crystal structure of RhIaaA (PDB 8DQM).5 (c) The crystal structure of AbCphZ (PDB 8EIN).6 (d) The active site of AbCphZ E251A complexed with β-Asp-Arg (PDB 8EIP).6 | ||
We also showed that Pseudomonas aeruginosa AotO,6,83,84 the enzyme mentioned in Section 2.6, allows P. aeruginosa to use β-Asp-Arg as a sole carbon source and as a remarkably good sole nitrogen source, is a bone fide CphZ. There are nearly 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 more CphZ sequences in nonredundant protein databases, highlighting the broad utility of this newly-described enzyme.
000 more CphZ sequences in nonredundant protein databases, highlighting the broad utility of this newly-described enzyme.
5 Outlook – the final frontiers for cyanophycin research
Cyanophycin metabolism genes are much more common than might be expected: Around 11% of complete bacterial genomes in the NCBI RefSeq database encode at least one gene for cyanophycin metabolism.6 For comparison, ∼38% of these genomes encode at least one gene for glycogen metabolism. These numbers highlight how common it is for bacteria to be cyanophycin producers or scavengers, much more so than currently recognized. It is likely that a greater appreciation of cyanophycin's pervasiveness will lead to new research avenues and practical applications. For example, since the human pathogen P. aeruginosa seems to gain an advantage from cyanophycin dipeptides in the environment, it may be worth exploring whether inhibition of its uptake or hydrolysis could limit proliferation of pathogenic strains in complex microbiomes. Likewise, inhibiting cyanophycin synthesis or degradation could be a promising strategy to combat toxic algal blooms.Bioinformatic analyses indicate that more surprises are in store:5,6,75 Some genomes encode CphA1 but not a cyanophycinase, or encode a cyanophycinase but neither CphZ, IaaA nor IadA. Some incomplete sets of cyanophycin-metabolizing genes might be explained by the existence of enzymes too distantly related to their characterized homologs to be recognized by gene sequence or task sharing in the microbial community for extracellular cyanophycin scavenging.78 We believe that new families of cyanophycinases, cyanophycin dipeptide hydrolases and cyanophycin dipeptide importers are waiting to be found.75 The discovery of new enzymes, for example a “cyanophycin isopeptidase” enzyme that prunes Arg directly from the long chains of cyanophycin polymer, or an enzyme that harvests nitrogen from β-Asp-Arg without breaking the isopeptide bond, would reveal new pathways for cyanophycin degradation.
Likewise, some already identified enzymes have yet to be characterized, and are likely to broaden known cyanophycin metabolism. For example, Füser & Steinbüchel noted that many betaproteobacteria encode two adjacent genes with high similarity to CphA1,75 and named them CphA3 and CphA3′. Steinbüchel's experiments153 suggest that CphA3 is a typical primer-dependent cyanophycin synthetase, and our sequence analysis indicates that CphA3′ contains inactive G and M domains, with an intact N domain hydrolytic site. Conceivably, CphA3′ evolved into a dedicated primer-making enzyme, encoded separately from cyanophycin synthetase to allow for better control of polymerization vs. hydrolysis. This conjecture remains to be verified experimentally.
Bioengineering and bioproduction of cyanophycin and its derivatives will continue to be an important focus of activity.95,101 Studies showing that cyanophycin can already constitute as much as one-half of all the dry weight of a cell91,96,97 and that tobacco-expressed cyanophycin is already commercially viable25 suggest bioproduction of cyanophycin is more a business than a scientific challenge. Altering the substate specificity of cyanophycin synthetase to produce different polymers seems more daunting.95 The structures of CphA1 and CphA2 provide a good resource for rational bioengineering, but they reveal extensive interactions between the synthetase and the growing cyanophycin chain, highlighting the challenge of bioengineering production of anything substantially different from cyanophycin than (β-Asp-Lys)n or (β-Asp-Orn)n.28 Mutation of CphA1 to enable incorporation of non-basic amino acids in place of Arg may require extensive modifications of G, M and N domains. Likewise, bioengineering a poly-Asp synthetase from cyanophycin synthetase will require a tour-de-force; it may be easier to find or design a cyanophycin isopeptidase enzyme to express alongside CphA1 to produce poly-Asp. Nonetheless, bioengineering and selection approaches have become so powerful that they seem sure to bring exciting breakthroughs in manipulation of cyanophycin biosynthesis in the near future.
6 Conflicts of interest
There are no conflicts to declare.7 Acknowledgements
Thanks to members of the Schmeing and Hilvert labs for important discussions, Michael Tarry and Nancy Rogerson for proofreading drafts, our co-authors on studies of cyanophycin metabolism (Marcel Grogg, Asfarul Haque, Indrajit Lahiri, Andres Leschziner, Linda Markus, Geoff McKay, Nic Moitessier, Kim Munro, Dao Nguyen, Sharon Pinus, Dieter Seebach, Mike Strauss), and staff at synchrotrons (Canadian Light Source, Advanced Light Source, Advanced Photon Source) and the McGill Facility for EM Research for facilitating our studies of cyanophycin metabolism. We thank Yong Xiong, and staff at McGill University interlibrary loans and the libraries of the University of Michigan and Yale University for reproductions of nineteenth century images, and Prof. Dr Yasser Elbahloul, Prof. Dr Alexander Steinbüchel, and the American Society for Microbiology for allowing us to reprint their striking micrographs in Fig. 2b. TMS is funded by CIHR Project Grant 178084 and McGill University, and DH is supported by ETH Zurich.8 References
- R. D. Simon, N. H. Lawry and G. L. McLendon, Biochim. Biophys. Acta, 1980, 626, 277–281 CrossRef CAS PubMed.
- A. Borzi, Nuovo G. Bot. Ital., 1878, 1, 236–288 Search PubMed.
- A. Borzi, Malpighia – Rassegna mensuale di botanica, 1887, vol. 1, pp. 1886–1887 Search PubMed.
- W. Ben Hania, M. Joseph, B. Bunk, C. Spröer, H.-P. Klenk, M.-L. Fardeau and S. Spring, Environ. Microbiol., 2017, 19, 1134–1148 CrossRef CAS PubMed.
- I. Sharon and T. M. Schmeing, Sci. Rep., 2023, 13, 8314 CrossRef PubMed.
- I. Sharon, G. A. McKay, D. Nguyen and T. M. Schmeing, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2216547120 CrossRef CAS PubMed.
- F. Mariotti, D. Tome and P. P. Mirand, Crit. Rev. Food Sci. Nutr., 2008, 48, 177–184 CrossRef CAS PubMed.
- M. M. Allen, F. Hutchison and P. J. Weathers, J. Bacteriol., 1980, 141, 687–693 CrossRef CAS PubMed.
- R. D. Simon, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 265–267 CrossRef CAS PubMed.
- N. H. Kolodny, D. Bauer, K. Bryce, K. Klucevsek, A. Lane, L. Medeiros, W. Mercer, S. Moin, D. Park, J. Petersen, J. Wright, C. Yuen, A. J. Wolfson and M. M. Allen, J. Bacteriol., 2006, 188, 934–940 CrossRef CAS PubMed.
- A. H. Mackerras, N. M. de Chazal and G. D. Smith, Microbiology, 1990, 136, 2057–2065 CrossRef CAS.
- Z. Lu, J. Ye, Z. Chen, L. Xiao, L. Lei, B. P. Han and H. W. Paerl, Water Res., 2022, 214, 118215 CrossRef CAS PubMed.
- K. Ziegler, A. Diener, C. Herpin, R. Richter, R. Deutzmann and W. Lockau, Eur. J. Biochem., 1998, 254, 154–159 CrossRef CAS PubMed.
- F. Klemke, D. J. Nurnberg, K. Ziegler, G. Beyer, U. Kahmann, W. Lockau and T. Volkmer, Microbiology, 2016, 162, 526–536 CrossRef CAS PubMed.
- I. Sharon, S. Pinus, M. Grogg, N. Moitessier, D. Hilvert and T. M. Schmeing, Nat. Commun., 2022, 13, 3923 CrossRef CAS PubMed.
- E. Aboulmagd, F. B. Oppermann-Sanio and A. Steinbuchel, Appl. Environ. Microbiol., 2001, 67, 2176–2182 CrossRef CAS PubMed.
- R. Richter, M. Hejazi, R. Kraft, K. Ziegler and W. Lockau, Eur. J. Biochem., 1999, 263, 163–169 CrossRef CAS PubMed.
- M. Hejazi, K. Piotukh, J. Mattow, R. Deutzmann, R. Volkmer-Engert and W. Lockau, Biochem. J., 2002, 364, 129–136 CrossRef CAS PubMed.
- W. C. Tseng, T. Y. Fang, Y. C. Lin, S. J. Huang and Y. H. Huang, Biomacromolecules, 2018, 19, 4585–4592 CrossRef CAS PubMed.
- Z. Uddin, T. Y. Fang, J. Y. Siao and W. C. Tseng, Macromol. Biosci., 2020, 20, e2000132 CrossRef PubMed.
- H. Nausch, M. Dorn, A. Frolov, S. Hoedtke, P. Wolf and I. Broer, Front. Plant Sci., 2020, 11, 842 CrossRef PubMed.
- U. Conrad, Trends Plant Sci., 2005, 10, 511–512 CrossRef CAS PubMed.
- H. Adelnia, I. Blakey, P. J. Little and H. T. Ta, Front. Chem., 2019, 7, 755 CrossRef CAS PubMed.
- H. Adelnia, H. D. N. Tran, P. J. Little, I. Blakey and H. T. Ta, ACS Biomater. Sci. Eng., 2021, 7, 2083–2105 CrossRef CAS PubMed.
- J. Huckauf, B. P. Brandt, C. Dezar, H. Nausch, A. Hauerwaas, U. Weisenfeld, O. Elshiewy, M. Rua, J. Hugenholtz, J. Wesseler, K. Cingiz and I. Broer, Front Bioeng Biotechnol, 2022, 10, 896863 CrossRef PubMed.
- A. Steinle, S. Witthoff, J. P. Krause and A. Steinbuchel, Appl. Environ. Microbiol., 2010, 76, 1062–1070 CrossRef CAS PubMed.
- M. Obst and A. Steinbüchel, in Inclusions in Prokaryotes, ed. J. M. Shively, Springer Berlin Heidelberg, Berlin, Heidelberg, 2006, pp. 167–193, DOI: DOI:10.1007/3-540-33774-1_7.
- M. Frommeyer, L. Wiefel and A. Steinbuchel, Crit. Rev. Biotechnol., 2016, 36, 153–164 CrossRef CAS PubMed.
- K. Zou, Y. Huang, B. Feng, T. Qing, P. Zhang and Y. P. Chen, Appl. Environ. Microbiol., 2022, e0074222, DOI:10.1128/aem.00742-22.
- I. Sharon, A. S. Haque, M. Grogg, I. Lahiri, D. Seebach, A. E. Leschziner, D. Hilvert and T. M. Schmeing, Nat. Chem. Biol., 2021, 17, 1101–1110 CrossRef CAS PubMed.
- T. Miyakawa, J. Yang, M. Kawasaki, N. Adachi, A. Fujii, Y. Miyauchi, T. Muramatsu, T. Moriya, T. Senda and M. Tanokura, Nat. Commun., 2022, 13, 5097 CrossRef CAS PubMed.
- A. B. Macallum, Trans. Can. Inst., 1898-99,(6), 439–506 Search PubMed.
- B. M. Davis, Am. Nat., 1905, 39, 695–740 CrossRef.
- A. Fischer, Bot. Ztg., 1905, 4, 51 Search PubMed.
- F. E. Fritsch, The structure and reproduction of the algae, Cambridge University Press, vol. 2, 1945 Search PubMed.
- R. D. Simon and P. Weathers, Biochim. Biophys. Acta, 1976, 420, 165–176 CrossRef CAS PubMed.
- R. D. Simon, Biochim. Biophys. Acta, 1976, 422, 407–418 CrossRef CAS PubMed.
- T. Hai, F. B. Oppermann-Sanio and A. Steinbuchel, FEMS Microbiol. Lett., 1999, 181, 229–236 CrossRef CAS PubMed.
- A. Tiessen, P. Pérez-Rodríguez and L. J. Delaye-Arredondo, BMC Res. Notes, 2012, 5, 85 CrossRef CAS PubMed.
- M. Frommeyer and A. Steinbuchel, Appl. Environ. Microbiol., 2013, 79, 4474–4483 CrossRef CAS PubMed.
- Y. Elbahloul, M. Krehenbrink, R. Reichelt and A. Steinbuchel, Appl. Environ. Microbiol., 2005, 71, 858–866 CrossRef CAS PubMed.
- M. Kunioka, Appl. Microbiol. Biotechnol., 1997, 47, 469–475 CrossRef CAS.
- M. Takehara, M. Saimura, H. Inaba and H. Hirohara, FEMS Microbiol. Lett., 2008, 286, 110–117 CrossRef CAS PubMed.
- Z. Xu, Z. Sun, S. Li, Z. Xu, C. Cao, Z. Xu, X. Feng and H. Xu, Sci. Rep., 2015, 5, 17400 CrossRef CAS PubMed.
- I. Bajaj and R. Singhal, Bioresour. Technol., 2011, 102, 5551–5561 CrossRef CAS PubMed.
- K. K. Mahalingan, E. Keith Keenan, M. Strickland, Y. Li, Y. Liu, H. L. Ball, M. E. Tanner, N. Tjandra and A. Roll-Mecak, Nat. Struct. Mol. Biol., 2020, 27, 802–813 CrossRef CAS PubMed.
- K. Yamanaka, C. Maruyama, H. Takagi and Y. Hamano, Nat. Chem. Biol., 2008, 4, 766–772 CrossRef CAS PubMed.
- R. D. Simon, Arch. Mikrobiol., 1973, 92, 115–122 CAS.
- G. Trentin, F. Piazza, M. Carletti, B. Zorin, I. Khozin-Goldberg, A. Bertucco and E. Sforza, Appl. Microbiol. Biotechnol., 2023, 107(1), 97–110 CrossRef CAS PubMed.
- S. E. Stevens and D. A. Paone, Plant Physiol., 1981, 67, 716–719 CrossRef CAS PubMed.
- M. Page-Sharp, C. A. Behm and G. D. Smith, FEMS Microbiol. Lett., 1998, 160, 11–15 CrossRef CAS.
- L. O. Ingram, E. L. Thurston and C. Van Baalen, Arch. Mikrobiol., 1972, 81, 1–12 CAS.
- Y. S. Yalcin, B. N. Aydin, M. Sayadujjhara and V. Sitther, Front. Aquat. Microbiol., 2022, 13, 930357 CrossRef PubMed.
- N. G. Carr, Proc. Phytochem. Soc. Eur., 1988, 28, 14–21 Search PubMed.
- B. A. Whitton and M. Potts, in Ecology of cyanobacteria II: Their diversity in space and time, ed. B. A. Whitton, Springer Netherlands, Dordrecht, 2012, pp. 1–13, DOI: DOI:10.1007/978-94-007-3855-3_1.
- O. Einsle and D. C. Rees, Chem. Rev., 2020, 120, 4969–5004 CrossRef CAS PubMed.
- J. R. Gallon, New Phytol., 1992, 122, 571–609 CrossRef CAS.
- H. Li, D. M. Sherman, S. Bao and L. A. Sherman, Arch. Microbiol., 2001, 176, 9–18 CrossRef CAS PubMed.
- J. A. Finzi-Hart, J. Pett-Ridge, P. K. Weber, R. Popa, S. J. Fallon, T. Gunderson, I. D. Hutcheon, K. H. Nealson and D. G. Capone, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6345–6350 CrossRef CAS PubMed.
- A. H. Mackerras, B. N. Youens, R. C. Weir and G. D. Smith, Microbiology, 1990, 136, 2049–2056 CrossRef CAS.
- D. M. Sherman, D. Tucker and L. A. Sherman, J. Phycol., 2000, 36, 932–941 CrossRef CAS.
- M. Burnat, A. Herrero and E. Flores, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 3823–3828 CrossRef CAS PubMed.
- R. Popa, P. K. Weber, J. Pett-Ridge, J. A. Finzi, S. J. Fallon, I. D. Hutcheon, K. H. Nealson and D. G. Capone, ISME J., 2007, 1, 354–360 CrossRef CAS PubMed.
- D. G. Adams and P. S. Duggan, New Phytol., 1999, 144, 3–33 CrossRef.
- A. Sukenik, I. Maldener, T. Delhaye, Y. Viner-Mozzini, D. Sela and M. Bormans, Front. Aquat. Microbiol., 2015, 6 DOI:10.3389/fmicb.2015.01067.
- R. Garg, M. Luckner, J. Berger, K. Hipp, G. Wanner, K. Forchhammer and I. Maldener, Life, 2022, 12, 429 CrossRef CAS PubMed.
- R. Perez, K. Forchhammer, G. Salerno and I. Maldener, Microbiology, 2016, 162, 214–223 CrossRef CAS PubMed.
- B. Watzer and K. Forchhammer, Appl. Environ. Microbiol., 2018, 84(20), e01298 CrossRef CAS PubMed.
- M. J. Harke, M. M. Steffen, C. J. Gobler, T. G. Otten, S. W. Wilhelm, S. A. Wood and H. W. Paerl, Harmful Algae, 2016, 54, 4–20 CrossRef PubMed.
- H. W. Paerl and T. G. Otten, Microb. Ecol., 2013, 65, 995–1010 CrossRef CAS PubMed.
- L. Bláha, P. Babica and B. Maršálek, Interdiscip. Toxicol., 2009, 2, 36–41 Search PubMed.
- M. M. Coffer, B. A. Schaeffer, K. Foreman, A. Porteous, K. A. Loftin, R. P. Stumpf, P. J. Werdell, E. Urquhart, R. J. Albert and J. A. Darling, Water Res., 2021, 201, 117377 CrossRef CAS PubMed.
- J. J. Hampel, M. J. McCarthy, M. Neudeck, G. S. Bullerjahn, R. M. L. McKay and S. E. Newell, Harmful Algae, 2019, 81, 42–52 CrossRef CAS PubMed.
- P. Singh, A. Khan and A. Srivastava, in Advances in Cyanobacterial Biology, ed. P. K. Singh, A. Kumar, V. K. Singh and A. K. Shrivastava, Academic Press, 2020, pp. 235–248 Search PubMed.
- G. Fuser and A. Steinbuchel, Macromol. Biosci., 2007, 7, 278–296 CrossRef PubMed.
- H. Liu, W. K. Ray, R. F. Helm, D. L. Popham and S. B. Melville, J. Bacteriol., 2016, 198, 1773–1782 CrossRef CAS PubMed.
- M. Obst, A. Sallam, H. Luftmann and A. Steinbuchel, Biomacromolecules, 2004, 5, 153–161 CrossRef CAS PubMed.
- M. Obst, A. Krug, H. Luftmann and A. Steinbuchel, Appl. Environ. Microbiol., 2005, 71, 3642–3652 CrossRef CAS PubMed.
- A. Sallam and A. Steinbuchel, Appl. Environ. Microbiol., 2008, 74, 3434–3443 CrossRef CAS PubMed.
- A. Sallam and A. Steinbuchel, J. Appl. Microbiol., 2009, 107, 474–484 CrossRef CAS PubMed.
- M. Obst, F. B. Oppermann-Sanio, H. Luftmann and A. Steinbuchel, J. Biol. Chem., 2002, 277, 25096–25105 CrossRef CAS PubMed.
- M. Johnson, I. Zaretskaya, Y. Raytselis, Y. Merezhuk, S. McGinnis and T. L. Madden, Nucleic Acids Res., 2008, 36, W5–W9 CrossRef CAS PubMed.
- T. Nishijyo, S. M. Park, C. D. Lu, Y. Itoh and A. T. Abdelal, J. Bacteriol., 1998, 180, 5559–5566 CrossRef CAS PubMed.
- C. D. Lu, Z. Yang and W. Li, J. Bacteriol., 2004, 186, 3855–3861 CrossRef CAS PubMed.
- M. Grogg, D. Hilvert, M. O. Ebert, A. K. Beck, D. Seebach, F. Kurth, P. S. Dittrich, C. Sparr, S. Wittlin, M. Rottmann and P. Maser, Helv. Chim. Acta, 2018, 101, e1800112 CrossRef PubMed.
- M. Grogg, D. Hilvert, A. K. Beck and D. Seebach, Synthesis, 2019, 51, 31–39 CrossRef CAS.
- A. Steinle, K. Bergander and A. Steinbuchel, Appl. Environ. Microbiol., 2009, 75, 3437–3446 CrossRef CAS PubMed.
- A. Sallam and A. Steinbuchel, Appl. Microbiol. Biotechnol., 2010, 87, 815–828 CrossRef CAS PubMed.
- A. Krzemińska, N. Kwiatos, F. Arenhart Soares and A. Steinbüchel, Int. J. Mol. Sci., 2022, 23, 3335 CrossRef PubMed.
- D. Hasson, H. Shemer and A. Sher, Ind. Eng. Chem. Res., 2011, 50, 7601–7607 CrossRef CAS.
- Y. Elbahloul and A. Steinbuchel, Appl. Environ. Microbiol., 2006, 72, 1410–1419 CrossRef CAS PubMed.
- M. M. Allen and P. J. Weathers, J. Bacteriol., 1980, 141, 959–962 CrossRef CAS PubMed.
- M. Krehenbrink, F.-B. Oppermann-Sanio and A. Steinbuchel, Arch. Microbiol., 2002, 177, 371–380 CrossRef CAS PubMed.
- E. Aboulmagd, I. Voss, F. B. Oppermann-Sanio and A. Steinbuchel, Biomacromolecules, 2001, 2, 1338–1342 CrossRef CAS PubMed.
- R. Wördemann, L. Wiefel, V. F. Wendisch and A. Steinbüchel, AMB Express, 2021, 11, 55 CrossRef PubMed.
- I. Voss, S. C. Diniz, E. Aboulmagd and A. Steinbuchel, Biomacromolecules, 2004, 5, 1588–1595 CrossRef CAS PubMed.
- Y. Abd-El-Karem, T. Elbers, R. Reichelt and A. Steinbuchel, Appl. Microbiol. Biotechnol., 2011, 89, 1177–1192 CrossRef CAS PubMed.
- B. J. Meussen, R. A. Weusthuis, J. P. M. Sanders and L. H. d. Graaff, Appl. Microbiol. Biotechnol., 2012, 93, 1167–1174 CrossRef CAS PubMed.
- H. Nausch, T. Hausmann, D. Ponndorf, M. Huhns, S. Hoedtke, P. Wolf, A. Zeyner and I. Broer, New Biotechnol., 2016, 33, 842–851 CrossRef CAS PubMed.
- M. Huhns, K. Neumann, T. Hausmann, F. Klemke, W. Lockau, U. Kahmann, L. Kopertekh, D. Staiger, E. K. Pistorius, J. Reuther, E. Waldvogel, W. Wohlleben, M. Effmert, H. Junghans, K. Neubauer, U. Kragl, K. Schmidt, J. Schmidtke and I. Broer, Plant Biotechnol. J., 2009, 7, 883–898 CrossRef PubMed.
- T. Hai, K. M. Frey and A. Steinbuchel, Appl. Environ. Microbiol., 2006, 72, 7652–7660 CrossRef CAS PubMed.
- Y. Elbahloul, K. Frey, J. Sanders and A. Steinbüchel, Appl. Environ. Microbiol., 2005, 71, 7759–7767 CrossRef CAS PubMed.
- T. Hai, H. Ahlers, V. Gorenflo and A. Steinbüchel, Appl. Microbiol. Biotechnol., 2000, 53, 383–389 CrossRef CAS PubMed.
- Y. Zhang, A. Kumar, P. V. Vadlani and S. Narayanan, J. Chem. Technol. Biotechnol., 2013, 88, 1321–1327 CrossRef CAS.
- M. Huhns, K. Neumann, T. Hausmann, K. Ziegler, F. Klemke, U. Kahmann, D. Staiger, W. Lockau, E. K. Pistorius and I. Broer, Plant Biotechnol. J., 2008, 6, 321–336 CrossRef PubMed.
- J. Du, L. Li, X. Ding, H. Hu, Y. Lu and S. Zhou, Appl. Microbiol. Biotechnol., 2013, 97, 8619–8628 CrossRef CAS PubMed.
- K. Lin, Y. Elbahloul and A. Steinbüchel, Appl. Microbiol. Biotechnol., 2012, 93, 1885–1894 CrossRef CAS PubMed.
- B. Watzer, A. Engelbrecht, W. Hauf, M. Stahl, I. Maldener and K. Forchhammer, Microb. Cell Fact., 2015, 14, 192 CrossRef PubMed.
- K. Swain, I. Sharon, W. Blackson, S. Parrish, S. Tekel, T. M. Schmeing, D. R. Nielsen and B. L. Nannenga, Biochem. Eng. J., 2023, 195, 108916 CrossRef CAS.
- A. Steinle, F. B. Oppermann-Sanio, R. Reichelt and A. Steinbuchel, Appl. Environ. Microbiol., 2008, 74, 3410–3418 CrossRef CAS PubMed.
- H. Nausch, J. Huckauf and I. Broer, Appl. Microbiol. Biotechnol., 2016, 100, 1559–1565 CrossRef CAS PubMed.
- K. Neubauer, M. Huhns, T. Hausmann, F. Klemke, W. Lockau, U. Kahmann, E. K. Pistorius, U. Kragl and I. Broer, J. Biotechnol., 2012, 158, 50–58 CrossRef CAS PubMed.
- L. Wiefel, A. Broker and A. Steinbuchel, Appl. Microbiol. Biotechnol., 2011, 90, 1755–1762 CrossRef CAS PubMed.
- L. Wiefel and A. Steinbuchel, Appl. Environ. Microbiol., 2014, 80, 1091–1096 CrossRef PubMed.
- A. Glieder, ChemBioChem, 2009, 10, 2111–2112 CrossRef CAS.
- H. Berg, K. Ziegler, K. Piotukh, K. Baier, W. Lockau and R. Volkmer-Engert, Eur. J. Biochem., 2000, 267, 5561–5570 CrossRef CAS PubMed.
- T. Hara, H. Kato, Y. Katsube and J. Oda, Biochemistry, 1996, 35, 11967–11974 CrossRef CAS PubMed.
- J. van Heijenoort, Nat. Prod. Rep., 2001, 18, 503–519 RSC.
- M. Krehenbrink and A. Steinbuchel, Microbiology, 2004, 150, 2599–2608 CrossRef CAS PubMed.
- M. J. Page and E. Di Cera, Physiol. Rev., 2006, 86, 1049–1092 CrossRef CAS PubMed.
- J. L. Pederick, A. P. Thompson, S. G. Bell and J. B. Bruning, J. Biol. Chem., 2020, 295, 7894–7904 CrossRef CAS PubMed.
- T. Arai and K. Kino, Appl. Microbiol. Biotechnol., 2008, 81, 69–78 CrossRef CAS PubMed.
- T. Hai, F. B. Oppermann-Sanio and A. Steinbuchel, Appl. Environ. Microbiol., 2002, 68, 93–101 CrossRef CAS PubMed.
- M. V. Fawaz, M. E. Topper and S. M. Firestine, Bioorg. Chem., 2011, 39, 185–191 CrossRef CAS PubMed.
- J. Stout, D. De Vos, B. Vergauwen and S. N. Savvides, J. Mol. Biol., 2012, 416, 486–494 CrossRef CAS PubMed.
- T. Hibi, T. Nishioka, H. Kato, K. Tanizawa, T. Fukui, Y. Katsube and J. Oda, Nat. Struct. Biol., 1996, 3, 16–18 CrossRef CAS PubMed.
- A. Galant, K. A. Arkus, C. Zubieta, R. E. Cahoon and J. M. Jez, Plant Cell, 2009, 21, 3450–3458 CrossRef CAS PubMed.
- W. Wang, T. J. Kappock, J. Stubbe and S. E. Ealick, Biochemistry, 1998, 37, 15647–15662 CrossRef CAS PubMed.
- H. Yamaguchi, H. Kato, Y. Hata, T. Nishioka, A. Kimura, J. Oda and Y. Katsube, J. Mol. Biol., 1993, 229, 1083–1100 CrossRef CAS PubMed.
- H. Li, W. Fast and S. J. Benkovic, Protein Sci., 2009, 18, 881–892 CrossRef CAS PubMed.
- C. A. Smith, J. Mol. Biol., 2006, 362, 640–655 CrossRef CAS PubMed.
- T. Hai, J.-S. Lee, T.-J. Kim and J.-W. Suh, Biochim. Biophys. Acta, 2009, 1794, 42–49 CrossRef CAS PubMed.
- S. Picossi, A. Valladares, E. Flores and A. Herrero, J. Biol. Chem., 2004, 279, 11582–11592 CrossRef CAS PubMed.
- K. Forchhammer and B. Watzer, Microbiology, 2016, 162, 727–729 CrossRef CAS PubMed.
- I. Sharon, M. Grogg, D. Hilvert and T. M. Schmeing, ACS Chem. Biol., 2022, 17(3), 680–700 CrossRef PubMed.
- L. M. D. Markus, I. Sharon, K. Munro, M. Grogg, D. Hilvert, M. Strauss and T. M. Schmeing, bioRxiv, 2023 DOI:10.1101/2023.04.15.537035v1.
- N. H. Lawry and R. D. Simon, J. Phycol., 1982, 18, 391–399 CrossRef CAS.
- A. M. Law, S. W. Lai, J. Tavares and M. S. Kimber, J. Mol. Biol., 2009, 392, 393–404 CrossRef CAS PubMed.
- I. Sharon, M. Grogg, D. Hilvert and T. M. Schmeing, Biochim. Biophys. Acta, Gen. Subj., 2022, 1866, 130217 CrossRef CAS PubMed.
- N. Huguenin-Dezot, D. A. Alonzo, G. W. Heberlig, M. Mahesh, D. P. Nguyen, M. H. Dornan, C. N. Boddy, T. M. Schmeing and J. W. Chin, Nature, 2019, 565, 112–117 CrossRef CAS PubMed.
- D. W. Aswad, M. V. Paranandi and B. T. Schurter, J. Pharm. Biomed. Anal., 2000, 21, 1129–1136 CrossRef CAS PubMed.
- E. Kim, J. D. Lowenson, D. C. MacLaren, S. Clarke and S. G. Young, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 6132–6137 CrossRef CAS PubMed.
- J. D. Lowenson, E. Kim, S. G. Young and S. Clarke, J. Biol. Chem., 2001, 276, 20695–20702 CrossRef CAS PubMed.
- J. D. Gary and S. Clarke, J. Biol. Chem., 1995, 270, 4076–4087 CrossRef CAS PubMed.
- J. B. Thoden, R. Marti-Arbona, F. M. Raushel and H. M. Holden, Biochemistry, 2003, 42, 4874–4882 CrossRef CAS PubMed.
- D. Borek, K. Michalska, K. Brzezinski, A. Kisiel, J. Podkowinski, D. T. Bonthron, D. Krowarsch, J. Otlewski and M. Jaskolski, Eur. J. Biochem., 2004, 271, 3215–3226 CrossRef CAS PubMed.
- A. Prahl, M. Pazgier, M. Hejazi, W. Lockau and J. Lubkowski, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1173–1176 CrossRef PubMed.
- R. Marti-Arbona, V. Fresquet, J. B. Thoden, M. L. Davis, H. M. Holden and F. M. Raushel, Biochemistry, 2005, 44, 7115–7124 CrossRef CAS PubMed.
- K. Michalska, K. Brzezinski and M. Jaskolski, J. Biol. Chem., 2005, 280, 28484–28491 CrossRef CAS PubMed.
- D. C. Rees, M. Lewis and W. N. Lipscomb, J. Mol. Biol., 1983, 168, 367–387 CrossRef CAS PubMed.
- J. H. Cho, D. H. Kim, S. J. Chung, N. C. Ha, B. H. Oh and K. Yong Choi, Bioorg. Med. Chem., 2002, 10, 2015–2022 CrossRef CAS PubMed.
- N. Cerda-Costa and F. X. Gomis-Ruth, Protein Sci., 2014, 23, 123–144 CrossRef CAS PubMed.
- K. Adames, K. Euting, A. Broker and A. Steinbuchel, Appl. Microbiol. Biotechnol., 2013, 97, 3579–3591 CrossRef CAS PubMed.
- E. Aboulmagd, F. B. Oppermann-Sanio and A. Steinbuchel, Arch. Microbiol., 2000, 174, 297–306 CrossRef CAS PubMed.
- K. Ziegler, R. Deutzmann and W. Lockau, Z. Naturforsch., C: J. Biosci., 2002, 57, 522–529 CrossRef CAS PubMed.
- T. M. Schmeing and V. Ramakrishnan, Nature, 2009, 461, 1234–1242 CrossRef CAS PubMed.
- S. Wang, S. Lin, Q. Fang, R. Gyampoh, Z. Lu, Y. Gao, D. J. Clarke, K. Wu, L. Trembleau, Y. Yu, K. Kyeremeh, B. F. Milne, J. Tabudravu and H. Deng, Nat. Commun., 2022, 13, 5044 CrossRef CAS PubMed.
- J. M. Reimer, A. S. Haque, M. J. Tarry and T. M. Schmeing, Curr. Opin. Struct. Biol., 2018, 49, 104–113 CrossRef CAS PubMed.
- Z. Luo, Y. Guo, J. Liu, H. Qiu, M. Zhao, W. Zou and S. Li, Biotechnol. Biofuels, 2016, 9, 134 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2023 |