
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing dynamic covalent chemistry in a 2D boroxine framework by in situ near-ambient pressure X-ray photoelectron spectroscopy†
Paul
Leidinger
 a,
Mirco
Panighel
b,
Virginia
Pérez Dieste
c,
Ignacio J.
Villar-Garcia
c,
Pablo
Vezzoni
d,
Felix
Haag
d,
Johannes V.
Barth
d,
Francesco
Allegretti
d,
Sebastian
Günther
a and
Laerte L.
Patera
*ae
a,
Mirco
Panighel
b,
Virginia
Pérez Dieste
c,
Ignacio J.
Villar-Garcia
c,
Pablo
Vezzoni
d,
Felix
Haag
d,
Johannes V.
Barth
d,
Francesco
Allegretti
d,
Sebastian
Günther
a and
Laerte L.
Patera
*ae
aDepartment of Chemistry and Catalysis Research Center, Technical University of Munich, 85748 Garching, Germany
bCNR-IOM, Laboratorio TASC, 34149 Trieste, Italy
cCELLS-ALBA Synchrotron Radiation Facility, 08290 Cerdanyola del Valles, Spain
dPhysics Department E20, Technical University of Munich, 85748 Garching, Germany
eInstitute of Physical Chemistry, University of Innsbruck, 6020 Innsbruck, Austria. E-mail: laerte.patera@uibk.ac.at
First published on 1st December 2022
Abstract
Dynamic covalent chemistry is a powerful approach to design covalent organic frameworks, where high crystallinity is achieved through reversible bond formation. Here, we exploit near-ambient pressure X-ray photoelectron spectroscopy to elucidate the reversible formation of a two-dimensional boroxine framework. By in situ mapping the pressure–temperature parameter space, we identify the regions where the rates of the condensation and hydrolysis reactions become dominant, being the key to enable the thermodynamically controlled growth of crystalline frameworks.
 Laerte L. Patera | Dr. Laerte L. Patera is an Assistant Professor at the University of Innsbruck (Austria). He received his PhD in Nanotechnology from the University of Trieste (Italy) in 2016 and worked as a postdoctoral researcher at the University of Regensburg (Germany) from 2016 to 2019. In 2020, he started his independent career at the Technical University of Munich (Germany). He was awarded the Gustav Hertz Prize of the German Physical Society (2019) and an ERC Starting Grant (2022). His research is focused on the synthesis and imaging of two-dimensional covalent organic frameworks for energy conversion. |
Introduction
Covalent organic frameworks (COFs) are materials formed by the covalent linkage of organic building blocks to create periodic structures with predefined topologies.1 COFs feature reticular structures with an ordered organic backbone, whose flexible choice allows tuning the structural and optoelectronic properties.2,3 While 3D-COFs have covalent bonds extended through the entire network,4 2D-COFs are restrained to in-plane bonds, placing them in the field of 2D materials. Among the variety of possible coupling schemes,5 covalent bonding is typically achieved through condensation reactions, yielding water as a by-product.6,7 Recently, the formation of extended 2D-COFs has been demonstrated by on-surface synthesis, which exploits atomically flat surfaces as templates to restrict and guide reaction pathways in two dimensions.7–11 However, on-surface synthesis of atomically thin COFs based on condensation reactions under ultra-high vacuum (UHV) conditions typically results in a low degree of crystallinity.12–18 This issue is related to the irreversibility of the covalent bond formation, as the water by-product from the condensation reaction is promptly pumped away. In contrast, applying the dynamic covalent chemistry strategy, the addition of water enables a certain rate of backreaction, providing a mechanism for error correction and, thus, facilitating the formation of a thermodynamically most stable crystalline structure.1,6,19–23 For the case of boronic acid condensation, where three boronic acid groups form a six-membered boroxine ring (B3O3),24,25 thermodynamically controlled crystallization has been reported by introducing water vapor during the reaction.19,20 However, the water partial pressure (pH2O) required for the crystallization could not be determined.21 A recent study exploited in situ scanning tunnelling microscopy (STM) at the solid–liquid interface to quantitatively extract information on crystallization parameters, such as critical nucleus size, nucleation and growth rate.26 Despite the unprecedented fundamental understanding provided by such a microscopic approach, the experiments were performed at room temperature only and in a limited humidity range. As pH2O is a key parameter to determine the chemical equilibrium of the system, systematic studies under different experimental conditions (i.e. pH2O and temperature T) are required to identify the fundamental thermodynamic reaction values and the parameter space where highly crystalline 2D-COFs can be achieved.27Recent developments in surface-science techniques capable of working at elevated pressures offer an appealing approach for novel in situ investigations of chemical reactions occurring at surfaces.28–33 In particular, differentially pumped electron optics allowed pushing X-ray Photoelectron Spectroscopy (XPS) up to ambient pressures. In this way, surface species can be monitored with elemental and chemical specificity under conditions closer to those present in industrial chemical reactors.28,34–40 This approach has successfully been exploited to probe atomic scale mechanisms governing the growth of 2D materials in realistic environments.41–45
Here, we studied the polymerization and hydrolysis of an atomically thin boroxine (B3O3) framework on Au(111). By controlling pH2O and T, the chemical equilibrium could be tuned, leading to reversible formation or dissolution of the framework. By exploiting near-ambient pressure X-ray photoelectron spectroscopy (NAP-XPS), we monitored in situ the evolution of the surface species and shed light on the parameter space governing the thermodynamically controlled bond formation in boroxine-based 2D-COFs.
Results and discussion
As a model system to study the reversible condensation of boronic acid, we focused on a 2D boroxine framework resulting from the cyclocondensation of tetrahydroxydiboron (THDB) on Au(111),46 as shown in Scheme 1. This reaction has been previously studied, showing that polycondensation leads to an atomically thin amorphous film composed of covalently connected boroxine rings.46 The condensation reaction of boronic acid is exothermic:| 3THDBad → 2B3O3 + 6H2O↑ |
| 3THDBad ⇌ 2B3O3 + 6H2O↑↓ |
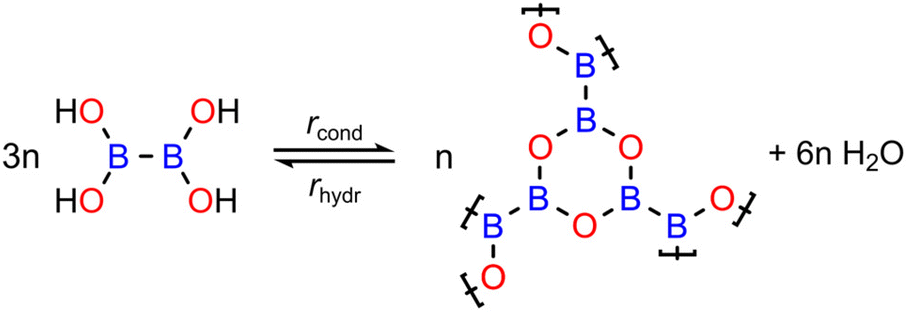 | ||
| Scheme 1 Reversible condensation reaction of tetrahydroxydiboron (THDB) leading to a 2-dimensional boroxine (B3O3) framework. The rates of condensation (rcond) and hydrolysis (rhydr) are indicated. | ||
Considering also the possible THDB desorption from the surface, the overall reaction can be written as:
| 3THDBg↑ ← 3 THDBad ⇌ 2B3O3 + 6H2O↑↓ |
In this work we show that by tuning pH2O and T, the rate of the backreaction (rhydr) can be controlled, enabling reversible covalent bond formation. In this way it is possible to identify the conditions which enable dynamic covalent chemistry to occur in a 2D boroxine framework.
THDB molecules were sublimated on the Au(111) surface kept at different Ts, ranging from 200 K to 393 K. The temperature stability of the THDB precursor has been determined through temperature-programmed desorption (TPD) experiments in UHV. Fig. 1a shows a TPD spectrum for the m/z value of 89 amu corresponding to the intact THDB molecule. A multilayer film was obtained by evaporating THDB on Au(111) kept at 200 K. Multilayer desorption sets in at ≈260 K, peaking at ≈290 K, while the peak at ≈300 K is attributed to the desorption of the second adsorbed layer.47 Further desorption is observed at ≈330 K, indicating partial desorption of the monolayer film.
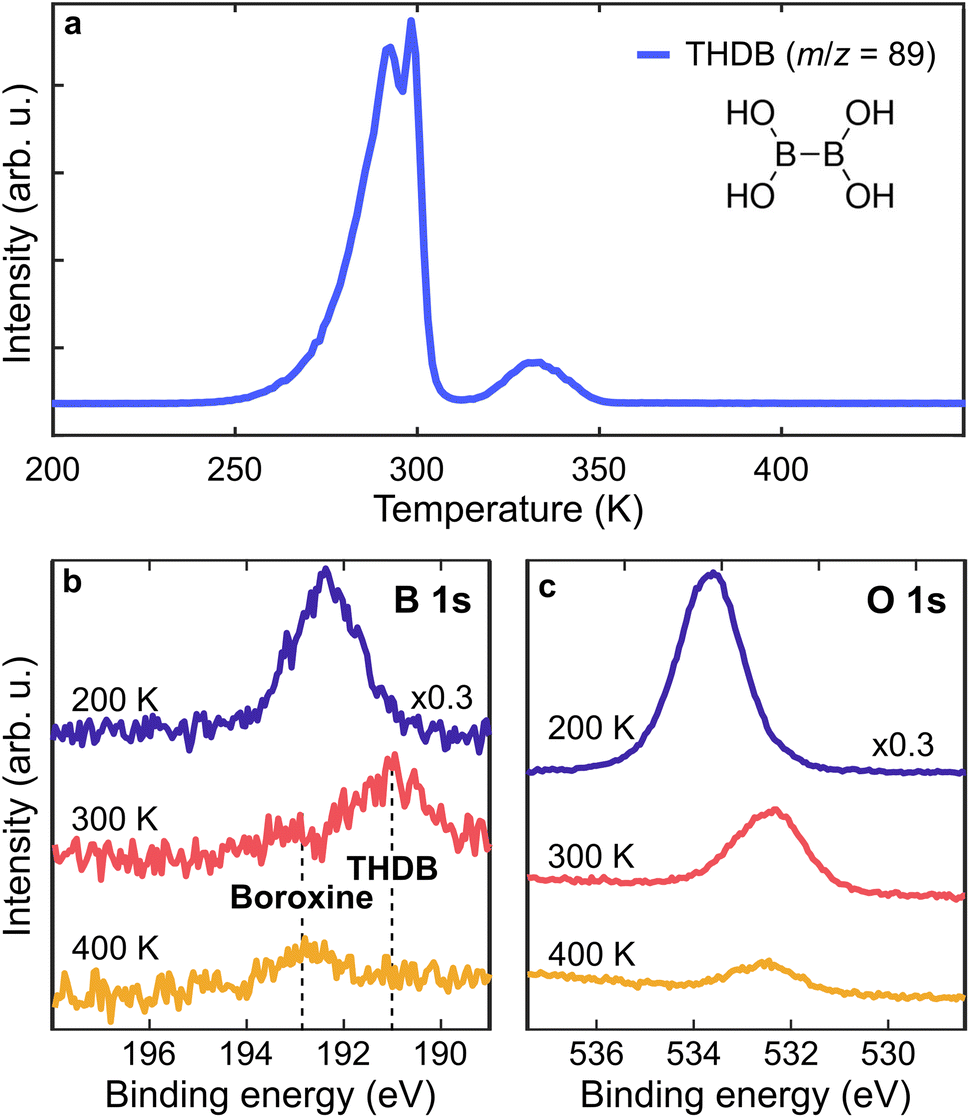 | ||
| Fig. 1 On-surface formation of the boroxine framework in UHV. (a) TPD measurement for the intact THDB molecule (m/z = 89 amu, linear heating rate of 1 K s−1). (b and c) B 1s and O 1s spectra of THDB/Au(111) after initial multilayer deposition at 200 K (blue), and after annealing to 300 K (red) and 400 K (orange), respectively. Photon energy = 1253.6 eV. | ||
To identify the surface species we acquired X-ray Photoelectron (XP) spectra of the B 1s and O 1s core levels after annealing a multilayer film to 200, 300 and 400 K (Fig. 1b and c). The B 1s and O 1s spectra acquired at 200 K are attributed to the multilayer THDB regime. The pronounced intensity reduction upon annealing to 300 K indicates the desorption of the multilayer. The B 1s spectrum exhibits a shift from ≈192.3 eV to ≈191.2 eV (due to the formation of a molecular monolayer), together with the formation of a small component at ≈192.8 eV. Stredansky et al. attributed the latter species to the boroxine framework,46 while a larger peak at ≈191.2 eV is here assigned to the adsorbed, unreacted THDB monomers. Upon annealing to 400 K, a complete vanishing of the monomer species is accompanied by enhanced growth of the boroxine component. Overall, the combined TPD and XPS results show that the cyclocondensation of the THDB monolayer to the boroxine framework in UHV already (slowly) occurs at 300 K and is completed at 400 K, accompanied by the desorption of the THDB monomers. While the O 1s spectra exhibit a clear energy shift once annealed from 200 K to 300 K (multilayer to monolayer transition), further annealing to 400 K does not lead to a pronounced change in the binding energy (≈200 meV), making the O 1s core level not sensitive enough to probe the transition from the THDB monolayer to the boroxine framework. Therefore, in the following we focus mainly on the changes in the B 1s spectra.
After having clarified the temperature evolution and stability of the THDB precursors and the conditions required for the formation of the boroxine framework on Au(111) under UHV conditions, we focus on the effect of pH2O on the presence of the boroxine framework. For this purpose, we performed near-ambient pressure XPS (NAP-XPS). Fig. 2a and b show the B 1s and O 1s XP spectra acquired at T = 323 K for a boroxine framework under different pH2O values (the corresponding survey spectra are shown in Fig. SI1†). By increasing pH2O up to 6 × 10−3 mbar no change can be detected in the XP spectra. A slight decrease in peak intensity can be observed at pH2O = 0.1 mbar (see the time-resolved B 1s spectra in Fig. SI2†), while at 1 mbar the boron and most of the oxygen signals vanish. This is confirmed also by the spectra acquired after evacuation of the cell (pH2O < 1 × 10−6 mbar). Here, the residual oxygen signal is attributed to surface contamination due to the water-induced displacement of oxygen-containing carbonaceous species from the internal chamber walls,48,49 as suggested also by the C 1s spectra shown in Fig. SI3.† As hydrolysis of boroxine can already occur at low temperatures,50 the behaviour shown in Fig. 2a and b can be rationalized as a water-induced dissolution of the boroxine framework (rhydr > rcond, where rcond and rhydr are the rates of condensation and hydrolysis reactions), for pH2O as low as 0.1 mbar. In particular, given that the THDB monomers are not stable on the surface above about 320 K (see Fig. 1a), the hydrolysis of the framework leads to instantaneous desorption of the hydrolysed THDB molecules (rdes ≫ rhydr − rcond, where rdes is the rate of THDB desorption).
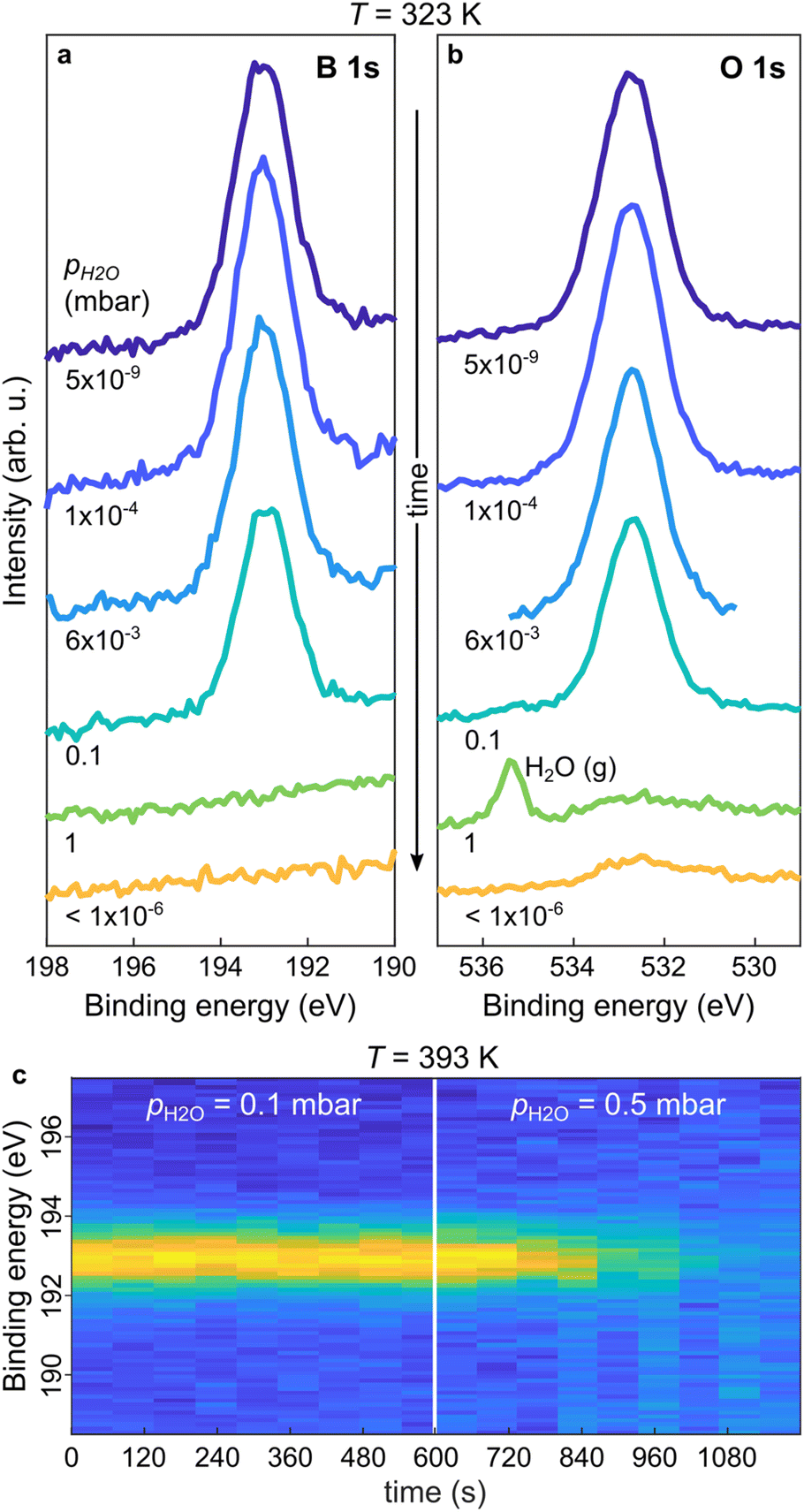 | ||
| Fig. 2 In situ NAP-XPS characterization of hydrolysis in the boroxine framework upon water vapor exposure. (a and b) B 1s and O 1s spectra of the boroxine framework on Au(111) upon sequential exposure to increasing pH2O (shown in shades of blue and green) and after cell evacuation (indicated in orange) at T = 323 K. The water gas-phase peak, H2O(g), is indicated. (c) Contour plot of B 1s spectra acquired during exposure of the boroxine framework to water vapor pressures at T = 393 K, with pH2O increased from 0.1 to 0.5 mbar after 600 s. Photon energy = 650 eV. | ||
To understand the role of pH2O under conditions typically used for the crystallization of boroxine-based COFs, we monitored the time evolution of the boron species at higher temperatures.20,21,51Fig. 2c shows a time series of B 1s spectra acquired during water exposure at T = 393 K. At pH2O = 0.1 mbar the boroxine XPS intensity is constant. In contrast, rising pH2O to 0.5 mbar results in a rapid decrease of the signal, completely vanishing after 300 s. This suggests that at T = 393 K and pH2O = 0.1 mbar, the boroxine hydrolysis rate is still smaller than the one for THDB condensation (rhydr < rcond), but increasing pH2O to 0.5 mbar initiates this reaction path (rhydr > rcond), combined with the immediate desorption of the formed monomer THDB species (rdes ≫ rhydr − rcond).
As the hydrolysed molecules were not detected at T = 393 K, the spectra in Fig. 2c do not represent direct proof of the reversible condensation reaction of the boroxine framework. Therefore, we carried out further in situ studies at T = 300 K, at which the THDB monomers are stable on the gold surface and should coexist with the condensed species (see Fig. 1). Fig. 3a and b show the B 1s and O 1s spectra acquired at 300 K. Given that at very low pH2O values the condensation reaction of boronic acid is exothermic (rcond > 0, rhydr ≈ 0),18,26 a fraction of the THDB molecules forms boroxine species (rhydr < rcond) upon deposition at 300 K. This is evident from the lineshape of the B 1s spectrum acquired at pH2O = 5 × 10−9 mbar (Fig. 3a), exhibiting two components, corresponding to the intact THDB molecules (red) and COF boroxine framework (blue). At pH2O = 7 × 10−3 mbar, partial elimination of THDB is indicated by the decrease in the absolute intensity of the XPS component (see Fig. 3c). This is accompanied by a reduction of the boroxine peak intensity, revealing a significant rate of hydrolysis (rhydr > rcond). As the THDB to boroxine ratio decreases (see Fig. 3c) the THDB desorption rate (rdes) is larger than that of boroxine dissolution, revealing that at T = 300 K and pH2O = 7 × 10−3 mbar, rdes > rhydr − rcond. The adsorption of water due to the polar nature of the boronic acid groups and the presence of oxygen-containing contaminations (see Fig. SI3†), in addition to the close components corresponding to the THDB and boroxine frameworks, complicate the quantitative interpretation of the O 1s spectra. At pH2O values of 0.1 and 0.5 mbar, the ratio of THDB to boroxine rises, due to a higher hydrolysis rate. Indeed, time-resolved spectra (see Fig. SI4 and 5†) acquired at pH2O = 0.5 mbar show that boroxine hydrolysis proceeds faster than THDB desorption (rhydr − rcond > rdes). Instead, upon subsequent evacuation of the reaction cell (pH2O < 1 × 10−6 mbar), the THDB to boroxine ratio decreases (rhydr < rcond), due to the suppression of the water-induced hydrolysis of boroxine (rhydr ≈ 0), and the absolute intensity of the boroxine component rises, indicating full control over the hydrolysis process through the adjustment of pH2O. Notably, this represents the first in situ direct observation of dynamic covalent chemistry in 2D-COFs.
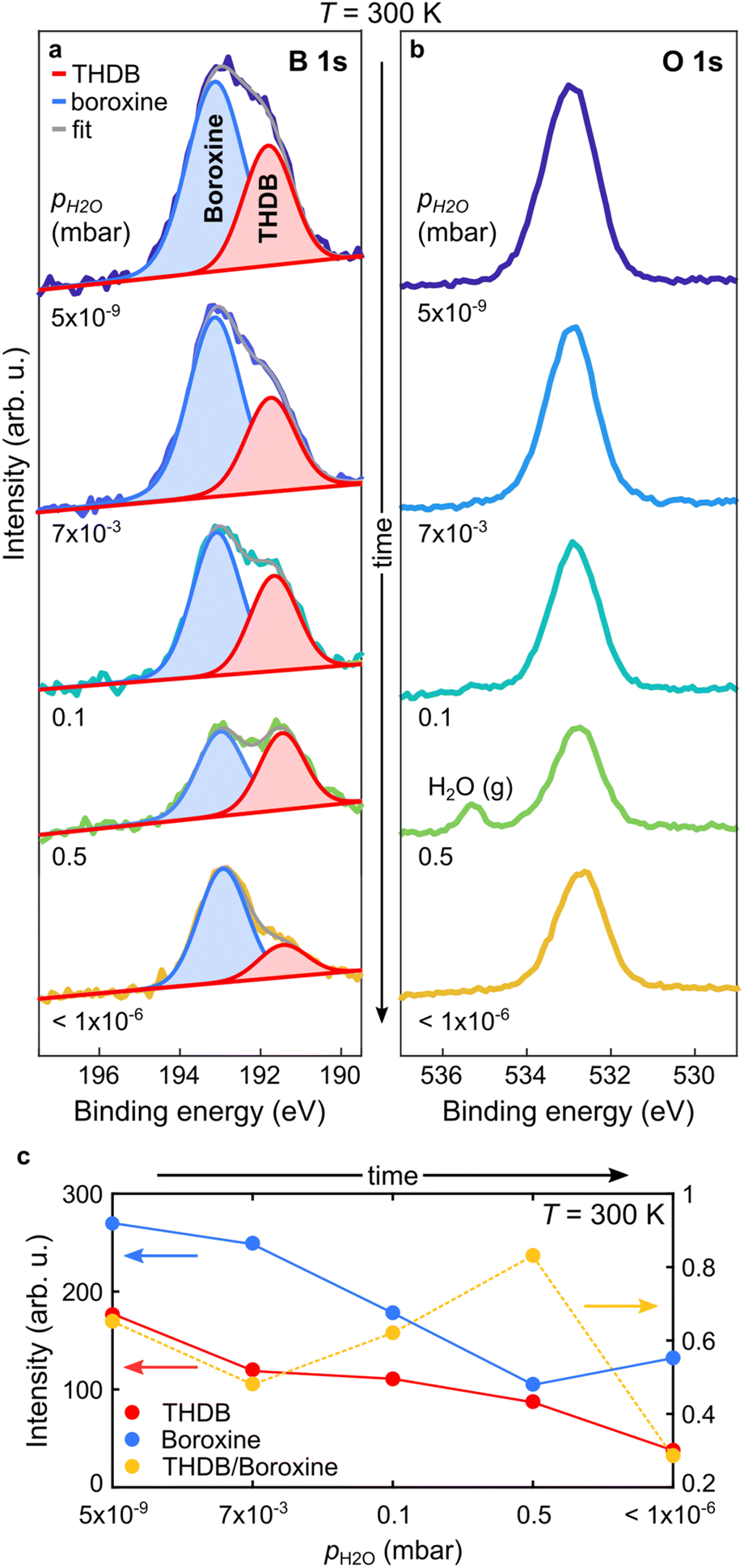 | ||
| Fig. 3 In situ NAP-XPS characterization of a mixed boroxine and THDB monolayer upon water vapor exposure. (a and b) B 1s and O 1s spectra acquired at T = 300 K upon a sequential increase of pH2O and after cell evacuation. Photon energy = 650 eV. (a) Individual components obtained by the fitting procedure are superimposed as solid peaks. (c) Absolute intensities of the two B 1s components of panel (a) and their intensity ratio are plotted versus pH2O. | ||
Aiming at achieving crystalline COFs, it is pivotal to identify the parameter space where the rates of the reaction and backreaction (almost) equalize. Fig. 4 shows a pressure–temperature diagram of the reversible condensation reaction obtained from the presented datasets (Fig. 2 and 3). While at a given T, it is difficult to determine the exact pH2O value where rhydr and rcond are equal (given the limited measurement time), we defined upper and lower boundaries, where significant and no hydrolysis, respectively, is observed, as marked in Fig. 4 (see Methods). Exploiting the diagram, a crystalline framework could be, in principle, obtained by using a condition where the rate of condensation is slightly larger than the rate of hydrolysis. However, in this specific case, the loss of reactant THDB molecules hinders the formation of a crystalline 2D boroxine framework. In fact, the conditions favouring bond reversibility coincide with the desorption of the hydrolysed molecules.
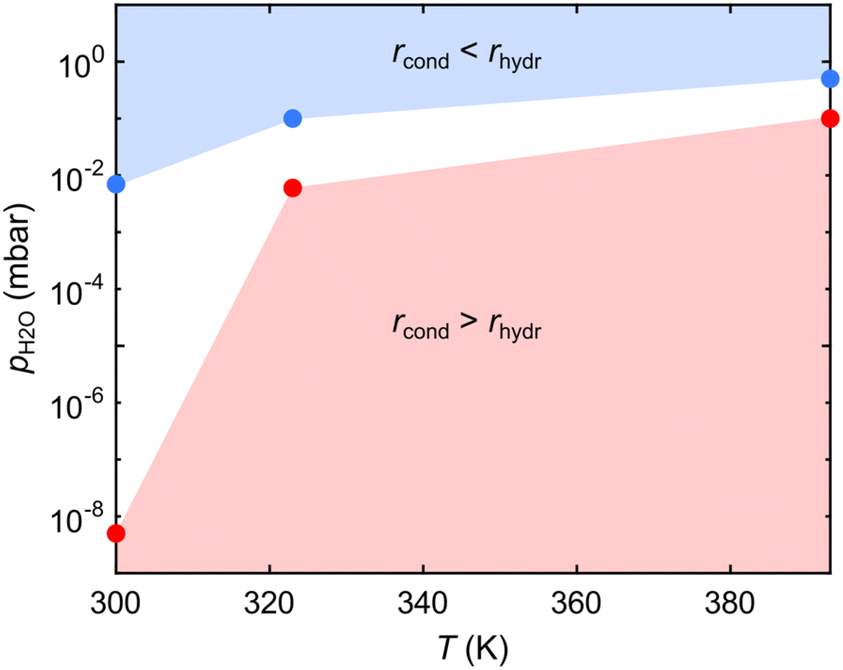 | ||
| Fig. 4 Pressure–temperature diagram of the reversible condensation reaction of tetrahydroxydiboron (THDB) leading to a 2D boroxine framework. The regions where the rates of the condensation (rcond) and hydrolysis (rhydr) reactions become dominant are indicated in red and blue, respectively. | ||
The effect of even higher pH2O (>1 mbar) was investigated ex situ by means of STM imaging (Fig. 5). The region imaged in Fig. 5a displays four ascending Au(111) terraces (from left to right), separated by monoatomic steps. Each terrace is partly covered by a network phase which resembles the boroxine phase and which appears slightly higher than the supporting Au terrace in the STM image. While the incomplete filling is readily observed in the large overview, the zoomed area shown in the inset of Fig. 5a proves the porous and amorphous nature of the network structure, which has been previously reported.46 Exposure of the boroxine framework to 20 mbar of water vapor at 300 K for 15 minutes results in the complete hydrolysis of the boroxine framework and in the desorption of the entire THDB monolayer (Fig. 5b). As a result, flat Au(111) terraces are observed when imaging the surface after water exposure by STM as shown in Fig. 5b, where the characteristic herringbone reconstruction of the Au(111) surface is clearly resolved.52
 | ||
| Fig. 5 STM images acquired at T = 300 K after (a) on-surface synthesis of the boroxine framework on Au(111) at T = 393 K and (b) after exposure to water vapor at 20 mbar at T = 300 K for 15 minutes. (a) Inset: a molecular-resolved image of the disordered boroxine framework. (b) Inset: zoom on a flat terrace, showing the herringbone reconstruction of the Au(111) surface. (a) V = 0.55 V; I = 0.8 nA. (b) V = 0.28 V; I = 2 nA. | ||
Conclusions
We exploited NAP-XPS to enable in situ observations of dynamic covalent chemistry in 2D-COFs, in particular for the case of the polymerization and hydrolysis of an atomically thin boroxine (B3O3) framework on Au(111). By controlling the sample temperature and the water partial pressure in the reaction chamber, the shift of the prevalent surface species can be tracked in real time. While at T = 300 K, hydrolysis of the boroxine network is observed already at pH2O = 7 × 10−3 mbar, under typical conditions for 2D-COF growth (T = 393 K), a pH2O of ≈0.5 mbar is needed to induce a shift in equilibrium towards the reactant monomers. By mapping the pressure–temperature parameter space, we identified the regions where the rates of the condensation and hydrolysis reactions become dominant. The synthesis of a crystalline boroxine framework has been hindered by the competing desorption of the THDB precursors occurring upon hydrolysis of the network, as confirmed by STM. Importantly, we have shown that reversible bond formation in 2D boroxine frameworks occurs in the (sub-)mbar pressure regime, enabling the use of current NAP-XPS analysers36,53 for the in situ investigation of dynamic covalent chemistry. The presented approach can be extended to a large variety of 2D-COFs. Furthermore, in combination with rigorous analysis,27 it opens the possibility of precisely determining relevant parameters, such as the kinetic barriers for reversible reactions, as well as the Gibbs free energy ruling the dynamic covalent bond formation.Methods
THDB molecules (purity: 95%) were purchased from Sigma Aldrich and sublimated on the Au(111) surface from a Knudsen cell kept at a temperature of about 350 K.UHV-XPS and TPD experiments were performed in a home-built UHV chamber. The XPS measurements were carried out using the non-monochromatized Mg Kα line (hν = 1253.6 eV) from a standard X-ray tube and a SPECS Phoibos 100 CCD hemispherical analyser combined with a PCO pixelfly camera, with an overall energy resolution of ≈900 meV. The binding energies of all XP spectra were calibrated against the Au 4f7/2 core level of the Au(111) substrate at a binding energy of 84.00 eV. The TPD measurements were performed with a quadrupole mass spectrometer mounted behind a Feulner cap.54 As the poor signal to noise ratio of the B 1s spectra hinders a quantitative estimation of the surface coverage, no fit is presented for the B 1s spectra shown in Fig. 1b.
The NAP-XPS experiments were performed at the NAPP station of the CIRCE beamline of the ALBA synchrotron (Barcelona, Spain).55 Au/mica films have been used as a substrate. Several sputtering (1 keV, Ar+) and annealing (623–673 K) cycles were performed in the preparation chamber. THDB molecules were evaporated in a dedicated chamber. After transfer to the analysis chamber, the sample cleanliness was checked at a sample temperature of 300 K. B 1s, C 1s, Au 4f and O 1s core level spectra were recorded at a photon energy of 650 eV, with an overall energy resolution of ≈200 meV. Carbon contamination was detected in the XP spectra acquired immediately after THDB evaporation, as a C 1s component at 284.0 eV, and attributed to the incomplete outgassing of the organic powder. The binding energies of all XP spectra were calibrated against the Au 4f7/2 core level of the Au(111) substrate at a binding energy of 84.00 eV. The analyser axis is at 54.7° with respect to the incident X-ray beam. The pressure in the cell was measured with a VACOM BARION passive hot cathode ionization gauge for pressures up to 10−3 mbar and with a Type 121A MKS Baratron Absolute Pressure Transducer for higher pressures. Since hot ion-gauge filaments have been reported to facilitate the formation of radicals due to water cracking,49 most of the experiments were performed with the hot-ion gauge filament turned off. Instead, for the spectra acquired at pH2O = 1 × 10−4 mbar (see Fig. 2a and b) the hot ion-gauge was used. However, no increase in the oxygen/boron peak ratio was observed compared to the one obtained for pH2O = 5 × 10−9 mbar, indicating a negligible contamination effect. We observed that the X-ray beam increases the rates of polymerization and hydrolysis (depending on the experimental conditions).56 For this reason, the X-ray beam intensity was reduced by about a factor of 100 with respect to the standard operating conditions of the beamline, by detuning the undulator. As a result, a beam size of 50 × 100 μm2 was used. This resulted in a negligible beam effect over the acquisition time of a spectrum (about 120 s). Measurements have been performed by raster-scanning the sample surface with a step size of 300 μm, allowing for the acquisition of each spectrum on a fresh sample spot. The B 1s core-level spectra were fitted with symmetric Voigt functions and a linear background. The Lorentzian width was constrained to 500 meV during the fit, while the Gaussian width was not constrained, resulting in an overall full width half maximum (FWHM) of 1.4–1.6 eV. The B 1s binding energies were not strictly constrained to account for small shifts due to the interaction with water molecules. Errors arising from the fitting procedure are negligible. The pressure–temperature diagram in Fig. 4 has been constructed from a series of three experiments performed at T = 300, 323 and 393 K, where pH2O has been stepwise increased. For each experiment, we marked the highest pH2O value at which the boroxine intensity in the B 1s spectra was constant for at least 600 s (red points) and the lowest pH2O value at which a time-dependent decrease of the signal could be detected (blue points).
STM experiments were performed at 300 K in a UHV chamber (p = 5 × 10−10 mbar) that hosts a high-temperature scanning tunnelling microscope (SPECS Aarhus 150 HT-NAP). Au(111) single-crystal samples were cleaned by several sputtering (Ar+, 1.5 keV) and annealing (673 K) cycles. Images were acquired in the constant current mode, with the bias voltage applied to the sample and the tip at the ground. Pt/Ir tips were used for imaging. Water vapor exposure for the STM experiments was done in the STM load lock at room temperature. The sample was then directly transfered to the STM chamber without breaking the vacuum.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), through the Project ID PA3628/1, the TUM International Graduate School of Science and Engineering (IGSSE, GSC81), and the Germany's Excellence Strategy (e-conversion Cluster of Excellence EXC2089/1-390776260). This project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 101007417, having benefited from the access provided by by ALBA in Barcelona (Spain) and CNR-IOM in Trieste (Italy) within the framework of the NFFA-Europe Pilot Transnational Access Activity, proposal ID075. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 101039746) funded by the European Union. Views and opinions expressed are however those of the authors only and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the granting authority can be held responsible for them.References
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- M. Lu, J. Liu, Q. Li, M. Zhang, M. Liu, J.-L. Wang, D.-Q. Yuan and Y.-Q. Lan, Angew. Chem., 2019, 131, 12522–12527 CrossRef.
- R. Zhu, J. Ding, L. Jin and H. Pang, Coord. Chem. Rev., 2019, 389, 119–140 CrossRef CAS.
- L. Grill and S. Hecht, Nat. Chem., 2020, 12, 115–130 CrossRef CAS PubMed.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- D. Cui, D. F. Perepichka, J. M. MacLeod and F. Rosei, Chem. Soc. Rev., 2020, 49, 2020–2038 RSC.
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470–473 CrossRef CAS PubMed.
- Y.-C. Chen, T. Cao, C. Chen, Z. Pedramrazi, D. Haberer, D. G. De Oteyza, F. R. Fischer, S. G. Louie and M. F. Crommie, Nat. Nanotechnol., 2015, 10, 156–160 CrossRef CAS PubMed.
- M. Bieri, M. T. Nguyen, O. Gröning, J. Cai, M. Treier, K. Aït-Mansour, P. Ruffieux, C. A. Pignedoli, D. Passerone, M. Kastler, K. Müllen and R. Fasel, J. Am. Chem. Soc., 2010, 132, 16669–16676 CrossRef CAS PubMed.
- S. Clair and D. G. de Oteyza, Chem. Rev., 2019, 119, 4717–4776 CrossRef CAS PubMed.
- T. Joshi, C. Chen, H. Li, C. S. Diercks, G. Wang, P. J. Waller, H. Li, J. L. Bredas, O. M. Yaghi and M. F. Crommie, Adv. Mater., 2019, 31, 6–11 Search PubMed.
- C. Chen, T. Joshi, H. Li, A. D. Chavez, Z. Pedramrazi, P. N. Liu, H. Li, W. R. Dichtel, J. L. Bredas and M. F. Crommie, ACS Nano, 2018, 12, 385–391 CrossRef CAS PubMed.
- D. J. Rizzo, Q. Dai, C. Bronner, G. Veber, B. J. Smith, M. Matsumoto, S. Thomas, G. D. Nguyen, P. R. Forrester, W. Zhao, J. H. Jørgensen, W. R. Dichtel, F. R. Fischer, H. Li, J.-L. Bredas and M. F. Crommie, Nano Lett., 2020, 20, 963–970 CrossRef CAS PubMed.
- M. Treier, R. Fasel, N. R. Champness, S. Argent and N. V. Richardson, Phys. Chem. Chem. Phys., 2009, 11, 1209–1214 RSC.
- F. Klappenberger, Y.-Q. Zhang, J. Björk, S. Klyatskaya, M. Ruben and J. V. Barth, Acc. Chem. Res., 2015, 48, 2140–2150 CrossRef CAS PubMed.
- M. Treier, N. V. Richardson and R. Fasel, J. Am. Chem. Soc., 2008, 130, 14054–14055 CrossRef CAS.
- N. A. A. Zwaneveld, R. Pawlak, M. Abel, D. Catalin, D. Gigmes, D. Bertin and L. Porte, J. Am. Chem. Soc., 2008, 130, 6678–6679 CrossRef CAS PubMed.
- J. F. Dienstmaier, A. M. Gigler, A. J. Goetz, P. Knochel, T. Bein, A. Lyapin, S. Reichlmaier, W. M. Heckl and M. Lackinger, ACS Nano, 2011, 5, 9737–9745 CrossRef CAS PubMed.
- C.-Z. Guan, D. Wang and L.-J. Wan, Chem. Commun., 2012, 48, 2943–2945 RSC.
- J. F. Dienstmaier, D. D. Medina, M. Dogru, P. Knochel, T. Bein, W. M. Heckl and M. Lackinger, ACS Nano, 2012, 6, 7234–7242 CrossRef CAS PubMed.
- X.-H. Liu, C.-Z. Guan, D. Wang and L.-J. Wan, Adv. Mater., 2014, 26, 6912–6920 CrossRef CAS PubMed.
- R. Tanoue, R. Higuchi, N. Enoki, Y. Miyasato, S. Uemura, N. Kimizuka, A. Z. Stieg, J. K. Gimzewski and M. Kunitake, ACS Nano, 2011, 5, 3923–3929 CrossRef CAS PubMed.
- S. Lin, J. Gu, H. Zhang, Y. Wang and Z. Chen, FlatChem, 2018, 9, 27–32 CrossRef CAS.
- D. Toffoli, M. Stredansky, Z. Feng, G. Balducci, S. Furlan, M. Stener, H. Ustunel, D. Cvetko, G. Kladnik, A. Morgante, A. Verdini, C. Dri, G. Comelli, G. Fronzoni and A. Cossaro, Chem. Sci., 2017, 8, 3789–3798 RSC.
- G. Zhan, Z.-F. Cai, K. Strutyński, L. Yu, N. Herrmann, M. Martínez-Abadía, M. Melle-Franco, A. Mateo-Alonso and S. D. Feyter, Nature, 2022, 603, 835–840 CrossRef CAS PubMed.
- P. Leidinger and S. Günther, J. Phys. Chem. C, 2021, 125, 12663–12671 CrossRef CAS.
- M. Salmeron and R. Schlögl, Surf. Sci. Rep., 2008, 63, 169–199 CrossRef CAS.
- M. Rößler, P. Geng and J. Wintterlin, Rev. Sci. Instrum., 2005, 76, 023705 CrossRef.
- C. T. Herbschleb, P. C. Van Der Tuijn, S. B. Roobol, V. Navarro, J. W. Bakker, Q. Liu, D. Stoltz, M. E. Cañas-Ventura, G. Verdoes, M. A. Van Spronsen, M. Bergman, L. Crama, I. Taminiau, A. Ofitserov, G. J. C. Van Baarle and J. W. M. Frenken, Rev. Sci. Instrum., 2014, 85, 083703 CrossRef CAS PubMed.
- B. L. M. Hendriksen, S. C. Bobaru and J. W. M. Frenken, Top. Catal., 2005, 36, 43–54 CrossRef CAS.
- B. Hendriksen, S. Bobaru and J. Frenken, Catal. Today, 2005, 105, 234–243 CrossRef CAS.
- B. Hendriksen and J. Frenken, Phys. Rev. Lett., 2002, 89, 046101 CrossRef CAS PubMed.
- R. Arrigo, M. Hävecker, M. E. Schuster, C. Ranjan, E. Stotz, A. Knop-Gericke and R. Schlögl, Angew. Chem., Int. Ed., 2013, 52, 11660–11664 CrossRef CAS PubMed.
- S. Axnanda, E. J. Crumlin, B. Mao, S. Rani, R. Chang, P. G. Karlsson, M. O. M. Edwards, M. Lundqvist, R. Moberg, P. Ross, Z. Hussain and Z. Liu, Sci. Rep., 2015, 5, 9788 CrossRef CAS PubMed.
- D. E. Starr, Z. Liu, M. Hävecker, A. Knop-Gericke and H. Bluhm, Chem. Soc. Rev., 2013, 42, 5833–5857 RSC.
- F. Tao, M. E. Grass, Y. Zhang, D. R. Butcher, J. R. Renzas, Z. Liu, J. Y. Chung, B. S. Mun, M. Salmeron and G. A. Somorjai, Science, 2008, 322, 932–934 CrossRef CAS PubMed.
- P. Amann, D. Degerman, M.-T. Lee, J. D. Alexander, M. Shipilin, H.-Y. Wang, F. Cavalca, M. Weston, J. Gladh, M. Blom, M. Björkhage, P. Löfgren, C. Schlueter, P. Loemker, K. Ederer, W. Drube, H. Noei, J. Zehetner, H. Wentzel, J. Ahlund and A. Nilsson, Rev. Sci. Instrum., 2019, 90, 103102 CrossRef.
- P. Amann, B. Klötzer, D. Degerman, N. Köpfle, T. Götsch, P. Lömker, C. Rameshan, K. Ploner, D. Bikaljevic, H.-Y. Wang, M. Soldemo, M. Shipilin, C. M. Goodwin, J. Gladh, J. Halldin Stenlid, M Börner, C. Schlueter and A. Nilsson, Science, 2022, 376, 603–608 CrossRef CAS PubMed.
- J. Knudsen, T. Gallo, V. Boix, M. D. Strømsheim, G. D’Acunto, C. Goodwin, H. Wallander, S. Zhu, M. Soldemo, P. Lömker, F. Cavalca, M. Scardamaglia, D. Degerman, A. Nilsson, P. Amann, A. Shavorskiy and J. Schnadt, Nat. Commun., 2021, 12, 6117 CrossRef CAS PubMed.
- R. S. Weatherup, B. C. Bayer, R. Blume, C. Ducati, C. Baehtz, R. Schlögl and S. Hofmann, Nano Lett., 2011, 11, 4154–4160 CrossRef CAS PubMed.
- L. L. Patera, C. Africh, R. S. Weatherup, R. Blume, S. Bhardwaj, C. Castellarin-Cudia, A. Knop-Gericke, R. Schloegl, G. Comelli, S. Hofmann and C. Cepek, ACS Nano, 2013, 7, 7901–7912 CrossRef CAS PubMed.
- Y. Fan, K. Nakanishi, V. P. Veigang-Radulescu, R. Mizuta, J. C. Stewart, J. E. Swallow, A. E. Dearle, O. J. Burton, J. A. Alexander-Webber, P. Ferrer, G. Held, B. Brennan, A. J. Pollard, R. S. Weatherup and S. Hofmann, Nanoscale, 2020, 12, 22234–22244 RSC.
- Z.-J. Wang, J. Dong, L. Li, G. Dong, Y. Cui, Y. Yang, W. Wei, R. Blume, Q. Li, L. Wang, X. Xu, K. Liu, C. Barroo, J. W. M. Frenken, Q. Fu, X. Bao, R. Schlögl, F. Ding and M.-G. Willinger, ACS Nano, 2020, 14, 1902–1918 CrossRef CAS PubMed.
- P. R. Kidambi, B. C. Bayer, R. Blume, Z.-J. Wang, C. Baehtz, R. S. Weatherup, M.-G. Willinger, R. Schloegl and S. Hofmann, Nano Lett., 2013, 13, 4769–4778 CrossRef CAS PubMed.
- M. Stredansky, A. Sala, T. Fontanot, R. Costantini, C. Africh, G. Comelli, L. Floreano, A. Morgante and A. Cossaro, Chem. Commun., 2018, 54, 3971–3973 RSC.
- F. Haag, P. S. Deimel, P. Knecht, L. Niederegger, K. Seufert, M. G. Cuxart, Y. Bao, A. C. Papageorgiou, M. Muntwiler, W. Auwärter, C. R. Hess, J. V. Barth and F. Allegretti, J. Phys. Chem. C, 2021, 125, 23178–23191 CrossRef CAS.
- N. Comini, T. Huthwelker, J. T. Diulus, J. Osterwalder and Z. Novotny, J. Vac. Sci. Technol., A, 2021, 39, 043203 CrossRef CAS.
- L. Trotochaud, A. R. Head, S. Pletincx, O. Karslloǧlu, Y. Yu, A. Waldner, L. Kyhl, T. Hauffman, H. Terryn, B. Eichhorn and H. Bluhm, J. Phys. Chem. B, 2018, 122, 1000–1008 CrossRef CAS PubMed.
- K. Severin, Dalton Trans., 2009, 5254–5264 RSC.
- S. Spitzer, A. Rastgoo-Lahrood, K. Macknapp, V. Ritter, S. Sotier, W. M. Heckl and M. Lackinger, Chem. Commun., 2017, 53, 5147–5150 RSC.
- J. V. Barth, H. Brune, G. Ertl and R. Behm, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 9307 CrossRef CAS PubMed.
- D. F. Ogletree, H. Bluhm, G. Lebedev, C. S. Fadley, Z. Hussain and M. Salmeron, Rev. Sci. Instrum., 2002, 73, 3872–3877 CrossRef CAS.
- P. Feulner and D. Menzel, J. Vac. Sci. Technol., 1980, 17, 662–663 CrossRef CAS.
- V. Pérez-Dieste, L. Aballe, S. Ferrer, J. Nicolàs, C. Escudero, A. Milán and E. Pellegrin, J. Phys.: Conf. Ser., 2013, 425, 072023 CrossRef.
- S. Clair, O. Ourdjini, M. Abel and L. Porte, Chem. Commun., 2011, 47, 8028–8030 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr04949j |
| This journal is © The Royal Society of Chemistry 2023 |