
Mo doping and Se vacancy engineering for boosting electrocatalytic water oxidation by regulating the electronic structure of self-supported Co9Se8@NiSe†
Lin
Tian
 *abc,
Zhenyang
Chen
a,
Tingjian
Wang
a,
Ming
Cao
a,
Xinhua
Lu
a,
Wenjing
Cheng
bc,
Changchun
He
a,
Ju
Wang
a and
Zhao
Li
*a
*abc,
Zhenyang
Chen
a,
Tingjian
Wang
a,
Ming
Cao
a,
Xinhua
Lu
a,
Wenjing
Cheng
bc,
Changchun
He
a,
Ju
Wang
a and
Zhao
Li
*a
aSchool of Materials and Chemical Engineering, Xuzhou University of Technology, Xuzhou 221018, PR China. E-mail: xzittl@xzit.edu.cn; xzitlz@xzit.edu.cn
bUniversity and College Key Lab of Natural Product Chemistry and Application in Xinjiang, School of Chemistry and Environmental Science, Yili Normal University, Yili 835000, China
cKey Laboratory of Pollutant Chemistry and Environmental Treatment, School of Chemistry and Environmental Science, Yili Normal University, Yili 835000, China
First published on 28th November 2022
Abstract
Oxygen evolution reactions (OERs) are regarded as the rate-determining step of electrocatalytic overall water splitting, which endow OER electrocatalysts with the advantages of high activity, low cost, good conductivity, and excellent stability. Herein, a facile H2O2-assisted etching method is proposed for the fabrication of Mo-doped ultrathin Co9Se8@NiSe/NF-X heterojunctions with rich Se vacancies to boost electrocatalytic water oxidation. After step-by-step electronic structure modulation by Mo doping and Se vacancy engineering, the self-standing Mo-Co9Se8@NiSe/NF-60 heterojunctions deliver a current density of 50 mA cm−2 with an overpotential of 343 mV and a cell voltage of only 1.87 V at 50 mA cm−2 for overall water splitting in 1.0 M KOH. Our study opens up the possibility of realizing step-by-step electronic structure modulation of nonprecious OER electrocatalysts via heteroatom doping and vacancy engineering.
1. Introduction
Developing an effective green hydrogen production strategy for solving the enormous energy crisis and accompanied environmental pollution has become increasingly important.1,2 Water electrolysis involving anodic oxygen evolution reactions (OERs) and cathodic hydrogen evolution reactions (HERs) is regarded as a promising technology for large-scale hydrogen production.3,4 However, the efficiency of overall water splitting is seriously hampered by the anodic half-reaction, which involves a four-electron process.5,6 Currently, the state-of-the-art OER electrocatalysts are Ru/Ir and their oxides, mainly due to their suitable electronic structure and good durability.7,8 Unfortunately, their scarcity and high cost have seriously impeded their industrial applications.9,10 Therefore, it is imperative to design and prepare OER electrocatalysts with high activity, low cost, good conductivity, and excellent stability.Nowadays, several strategies including morphology engineering,11,12 composition tuning,13–15 metal/non-metal doping,15,16 and surface/interface engineering17,18 are applied for designing and preparing high-performance OER electrocatalysts. Defective engineering, a recently proposed strategy, is regarded as an effective method for improving the intrinsic OER activity of metal oxides, metal phosphides, metal sulfides, and selenides. Nanocatalysts with rich P vacancies, S vacancies, O vacancies are reported to achieve outstanding OER performance, as vacancies can significantly affect the electronic structure of electrocatalysts, thus providing an ideal binding energy for the intermediates, which is a crucial requirement for developing highly efficient OER electrocatalysts.19,20 Among these anion vacancies, Se vacancies have been reported to be favourable for the modulation of the oxidation state of metal ions. In addition, Se vacancies can also optimize the Gibbs free energy and partial density of states, thereby contributing to the outstanding OER performance. However, due to the complexity of preparation, there are few reports on Se-defective electrocatalysts.21 Apart from defect engineering, doping is an established strategy for improving the electrocatalytic OER performance due to its versatile capability for simultaneously tailoring the electronic structure and introducing extra active sites.22,23 In view of this, integrating a nonprecious electrocatalyst with rich Se vacancies and doped heteroatom will greatly enhance electrocatalytic water oxidation.
ZIF-67, a typical metal–organic framework material has been widely applied to synthesize Co-based selenides via high-temperature calcination and using Se powder as a precursor, which consumes enormous energy, and the shape is also hard to control.24 In our previous work, we have successfully fabricated CoSe2 using ZIF-67 as a precursor and SeO2 as a Se source.25 Supports such as nickel foams, carbon cloth, and graphenes were widely used in the synthesis process of catalysts to improve the conductivity and stability.26,27 Given this, a self-standing nanomaterial with a modulated electronic structure and rich Se vacancies may be a potential outstanding anodic catalyst for electrocatalytic water splitting.
Considering these points, in this work, Mo-doped Co9Se8@NiSe/NF with rich Se vacancies and heterointerfaces was facilely prepared by a wet-chemical method followed by H2O2 etching. The as-prepared electrocatalysts, which possessed ultrathin nanosheets, strong interfacial interaction, and tuned electronic structure, exhibited enviable electrocatalytic performance. In particular, the optimized Mo-Co9Se8@NiSe/NF-60 heterojunction needed an overpotential of only 343 mV to drive a current density of 50 mA cm−2, and the Tafel slope is only 61.86 mV dec−1. More importantly, the two-electrode coupled Co9Se8@NiSe/NF-60 heterojunction with Pt/C achieved a current density of 50 mA cm−2 for overall water electrolysis with a cell voltage of 1.87 V.
2. Experimental section
2.1 Reagents and chemicals
Zinc nitrate hexahydrate (Zn(NO3)2·6H2O), 2-methylimidazole (2-MIM), sodium molybdate dihydrate (Na2MoO4·2H2O), selenium dioxide (SeO2), hydrogen peroxide (H2O2, 30%), and ethanol (C2H5OH) were purchased from Aladdin (Shanghai, China).2.2 Apparatus and measurements
Transmission electron microscopy (TEM), high-resolution TEM (HRTEM), and high-angle aberration-corrected dark-field scan transmission electron microscopy (HAADF-STEM) were performed using a JEM 2100F transmission electron microscope at a voltage of 200 kV. Field emission scanning electron microscopy (FESEM) studies were carried out using a JEOL S4800 scan electron microscopy operating at 15 kV for obtaining FESEM images and elemental mapping. X-ray photoelectron spectroscopy (XPS) characterizations were carried out using a VG Scientific ESCALAB 210 electron spectrometer equipped with two ultrahigh vacuum chambers. Powder X-ray diffraction (PXRD) patterns were recorded using a Rigaku diffractometer with Cu-Kα radiation (λ = 0.15418 nm).2.3 Preparation of ZIF-67/NF
A nickel foam (NF, 2 cm × 3 cm) was sonicated in acetone and ethanol for 15 min. Then, 0.821 g of 2-MIM was dissolved in 20 mL of deionized water to obtain solution A. Solution B was obtained by dissolving 0.728 g of Co(NO3)2·2H2O in 20 mL of water. Then, solution B was injected into solution A and sonicated for 5 minutes. The pre-treated NF was immersed in the solution. After 4 h, the samples were obtained by washing with DI water and dried at 60 °C.2.4 Preparation of Mo-ZIF-67/NF
First, 100 mg of NaMo2O4·2H2O was dissolved in 50 mL of DI water and sonicated for complete dissolution. The as-obtained ZIF-67/NF was immersed in the solution to react for 25 min. Subsequently, Mo-doped ZIF-67/NF was obtained by washing several times with deionized water and dried at 60 °C.2.5 Preparation of Mo-Co9Se8@NiSe/NF
First, 10 mg of SeO2 and 0.1 mL of N2H4·H2O were dissolved in 20 mL DI water. After that, the obtained Mo-ZIF-67/NF (2 cm × 1.5 cm) was placed into this solution within a 100 mL Teflon-lined stainless autoclave for continuous reaction for 8 h at 160 °C. After cooling to room temperature, Mo-Co9Se8@NiSe/NF was washed several times with DI water and dried at 60 °C.2.6 Preparation of Mo-Co9Se8@NiSe/NF
To obtain Mo-Co9Se8@NiSe/NF with different numbers of Se vacancies, Mo-Co9Se8@NiSe/NF was etched for 40 s, 60 s and 80 s with 5 mol L−1 H2O2 at room temperature. Then, they were washed with DI water and dried at 60 °C. The samples were labeled as Mo-Co9Se8@NiSe/NF-40, Mo-Co9Se8@NiSe/NF-60, and Mo-Co9Se8@NiSe/NF-80, respectively.2.7 Electrochemical measurement
Electrochemical measurements were done using an electrochemical workstation in a three-electrode system, where a glassy carbon electrode covered with catalysts, a Ag/AgCl electrode, and a graphene rod were employed as the working, reference and counter electrodes, respectively. Linear sweep voltammetry at a scan rate of 5 mV s−1 was conducted at room temperature. For Tafel plots, the linear portion is fitted to the Tafel equation. All the potentials reported in the present study were converted to a reversible hydrogen electrode (RHE) scale. The long-term stability was also tested at a current density of 50 mA cm−2. EIS was performed in the frequency range of 0.01 to 100 000 Hz and an amplitude of 10 mV.3. Results and discussion
The scheme presents the wet-chemical synthesis of Mo-Co9Se8@NiSe/NF-60 (Fig. 1a). As shown in Fig. S1a,† the pretreated nickel foam has an obvious three-dimensional network structure.28 After in situ growth of ZIF-67 on the surface of the nickel foam by a hydrothermal method, is clearly observed that intensive triangular column-like ZIF-67 is anchored on the surface of the nickel foam (Fig. 1b). When the obtained ZIF-67/NF was immersed in a sodium molybdate solution for 25 min, an ion-exchange process will happen, which will lead to the formation of Mo-ZIF-67/NF ultrathin nanosheets (Fig. 1c).29 Mo-Co9Se8@NiSe/NF was obtained by selenizing Mo-ZIF-67/NF employing SeO2 as a selenium source under a mild hydrothermal condition. Fig. 1d and e show that Mo-Co9Se8@NiSe/NF could well inherit the morphology of ultrafine Mo-ZIF-67/NF. After immersion of Mo-Co9Se8@NiSe/NF in 5 M H2O2 for 60 s, a small amount of selenium ions can be oxidized into selenium by H2O2 and fall off the compound to form vacancies, and the obtained product was noted as Mo-Co9Se8@NiSe/NF-60.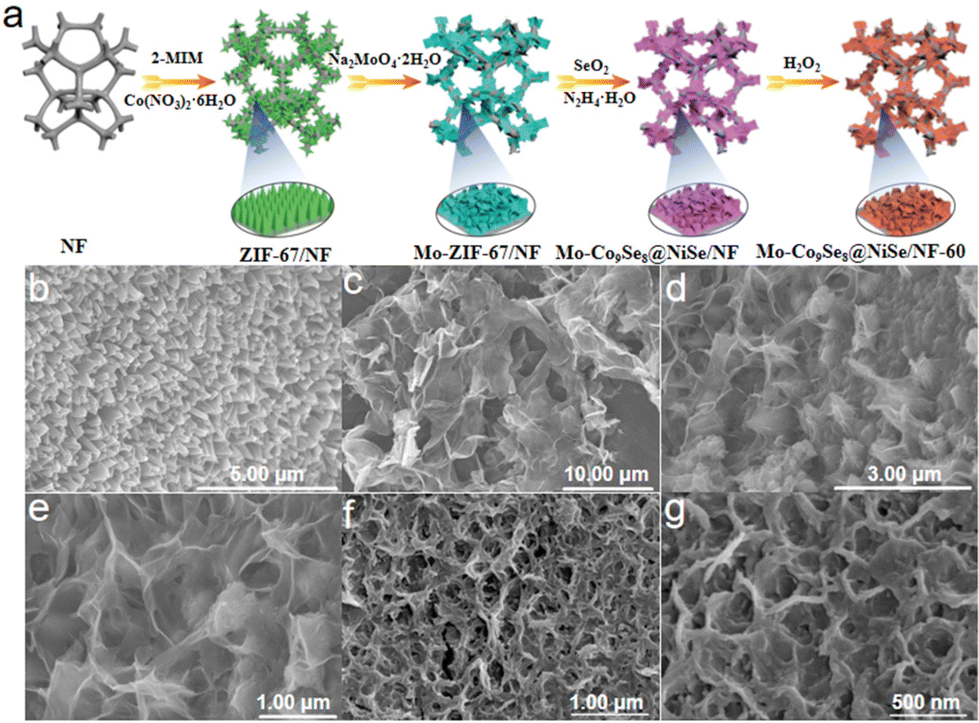 | ||
| Fig. 1 (a) Schematic illustration of the preparation of Mo-Co9Se8@NiSe/NF-60. SEM images of (b) ZIF-67/NF, (c) Mo-ZIF-67/NF, (d and e) Mo-Co9Se8@NiSe/NF, and (f and g) Mo-Co9Se8@NiSe/NF-60. | ||
As shown in Fig. 1f and g, the same morphology as that of Mo-Co9Se8@NiSe/NF was also observed, implying that H2O2 etching would not damage the ultrathin structure. For comparison, the samples with different immersion durations were also prepared, and both Mo-Co9Se8@NiSe/NF-40 and Mo-Co9Se8@NiSe/NF-80 possessed a similar morphology to that of Mo-Co9Se8@NiSe/NF-60 (Fig. S1b and c†), which suggested that this method had good versatility. Moreover, the samples without H2O2 etching and Mo doping were also prepared by a simple method, which also exhibited an analogous morphology (Fig. S2†). Fig. S3† presents the XRD patterns of samples at different stages and their locally enlarged images. As shown in Fig. S3,† ZIF-67/NF and Mo-ZIF-67/NF showed similar XRD patterns to that of pre-treated NF, but the locally enlarged images of ZIF-67/NF showed faint peaks around 7° and 10°, which were assigned to the characterized peaks of ZIF-67, implying that ZIF-67 successfully grew on the surface of the NF.30,31 After Mo doping, the appearance of a broad and large peak located 24.3° indicated the amorphous structure of Mo-ZIF-67/NF, suggesting that Mo doping not only destroyed the original morphology, but also altered its crystal structure. After further selenization and H2O2 etching, the XRD patterns of Mo-Co9Se8@NiSe/NF with different etching durations have similar peaks. After careful observation, it was clearly found that the peaks at 28.4° and 29.6° were pointed to the (222) plane of Co9Se8 (PDF # 09-0233). Meanwhile, the peaks around 30.8°, 33.9°, 38.5°, 46.5°, and 56.2° were ascribed to the (300), (021), (211), (131), and (401) crystal planes of NiSe (PDF # 18-0887), respectively, implying that Mo-Co9Se8@NiSe/NF-X was successfully fabricated.32
In order to obtain detailed information of Mo-Co9Se8@NiSe/NF-60, the ultrathin nanosheet morphology was further established and explored. As shown in Fig. 2a, the folded ultrathin morphology of Mo-Co9Se8@NiSe/NF-60 is clearly observed. High-resolution TEM, which is assigned to the olive square area of Fig. 2b, shows two different lattice fringes at 0.291 nm and 0.240 nm, corresponding to the (222) lattice plane of Co9Se8 and the (211) lattice plane of NiSe, respectively.33,34 The corresponding selected area electron diffraction (SAED) pattern (Fig. 2c, the purple circle area in Fig. 2a) obviously exhibited two sets of diffraction rings, which could be indexed to the (222) plane of Co9Se8 and the (211) plane of NiSe, which is consistent with the results of HRTEM. High-angle annular dark-field scanning TEM (HAADF-STEM; Fig. 2d and e) and EDS were also employed to explore the distribution of surface elements, which presented the uniform distribution of Co, Ni, Se, and Mo elements on the surface of Mo-Co9Se8@NiSe/NF-60. The line-scan profiles (corresponding the orange arrow line in Fig. 2d) and EDS results showed the coexistence of Ni, Co, Ni, Se, and Mo throughout Mo-Co9Se8@NiSe/NF-60, and the atomic number ratio of Co, Ni, Se, and Mo was measured to be about 68/148/73/1 (Fig. S4 and S5†). Fig. 2f presented the locally enlarged images and the Se vacancies can be clearly observed in the orange rectangle area, suggesting that H2O2 etching also generates Se vacancies. Electron paramagnetic resonance (EPR) was used to further study the relationship of etching time with numbers of vacancies (Fig. 2g). Mo-Co9Se8@NiSe/NF has a faint intensity, indicating that samples without H2O2 etching nearly have no Se vacancies. When Mo-Co9Se8@NiSe/NF was etched in a H2O2 solution for 40 s, the EPR intensity becomes stronger, implying the appearance of more defects and unpaired electrons.35,36 The results also indicate that H2O2 etching is an effective method for generating Se vacancies.
 | ||
| Fig. 2 (a) TEM image, (b) HRTEM images (inset I-1 and II-2 correspond to the area of red square I and orange square II), (c) SAED patterns, (d) HAADF-STEM image, (e) EDX images, and (f) local HRTEM images of Mo-Co9Se8@NiSe/NF-60. (g) EPR spectra of etched Mo-Co9Se8@NiSe/NF with different etching durations compared to Mo-Co9Se8@NiSe/NF. | ||
Considering its ultrathin nanosheet configuration, heterointerfaces, polycrystalline structure, and rich Se vacancies, the electrocatalytic OER performance of Mo-Co9Se8@NiSe/NF was confidently evaluated. Fig. 3a shows that optimized Mo-Co9Se8@NiSe/NF-60 exhibited outstanding OER activity, which outperformed the referenced RuO2, Co9Se8@NiSe/NF, and Mo-Co9Se8@NiSe/NF. The large oxidation peaks in the range of 1.3 to 1.5 V were ascribed to the oxidation of Ni species.37 Moreover, other samples also showed better OER activity than that of RuO2, Co9Se8@NiSe/NF, and Mo-Co9Se8@NiSe/NF, indicating that the prepared Mo-Co9Se8@NiSe/NF was a series of promising OER electrocatalysts.38 For intuitively comparing the OER activity of different electrocatalysts, the overpotentials of different samples at 50 mA cm−2 are shown in Fig. 3b, where Mo-Co9Se8@NiSe/NF-60 needed an overpotential of only 343 mV to achieve 50 mA cm−2, which was 31 mV, 7 mV, 61 mV, 56 mV and 111 mV lower than those of Mo-Co9Se8@NiSe/NF-40 (374 mV), Mo-Co9Se8@NiSe/NF-80 (350 mV), Mo-Co9Se8@NiSe/NF (404 mV), Co9Se8@NiSe/NF-60 (399) and RuO2 (454 mV), respectively, implying that Mo doping and Se vacancies synergistically improve the electrocatalytic OER performance.
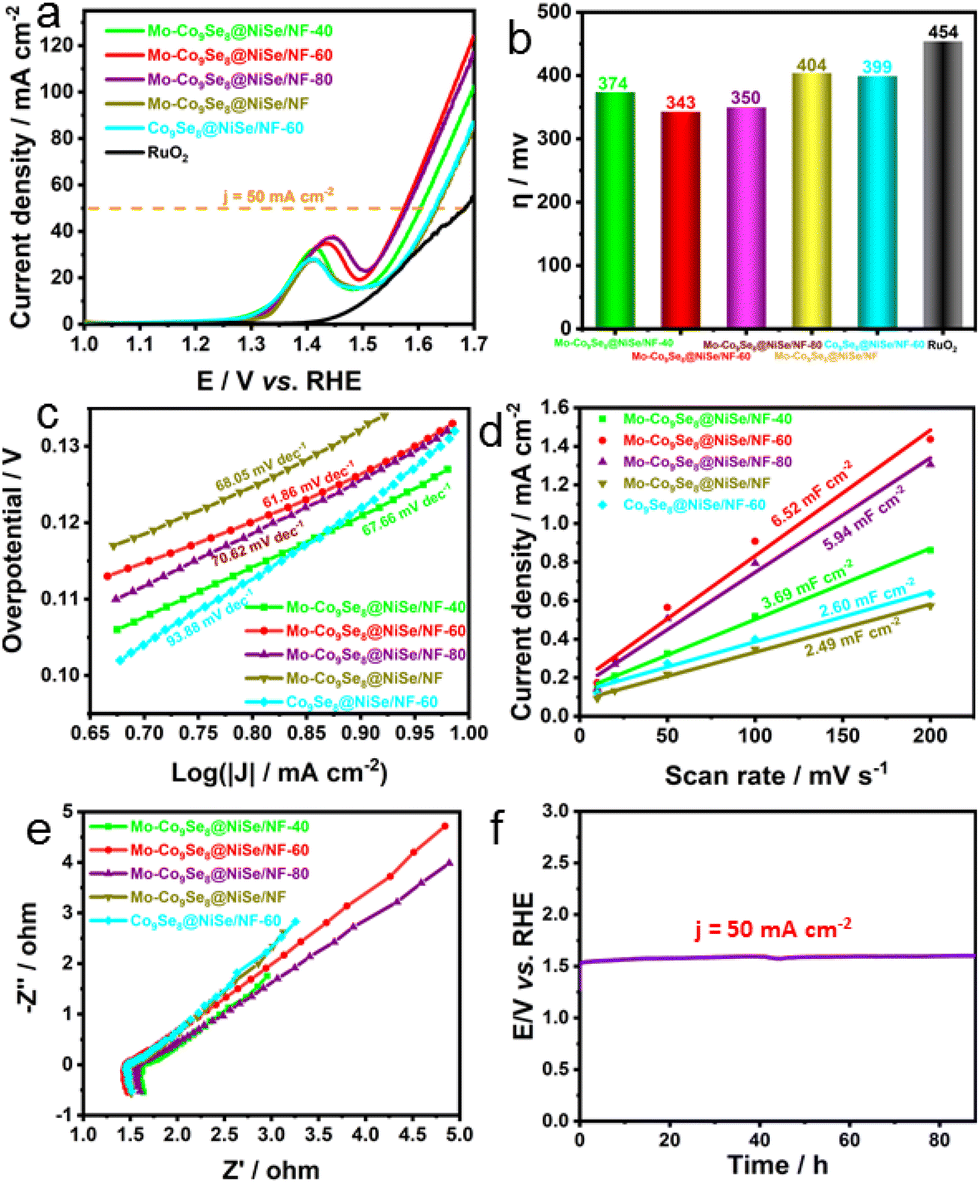 | ||
| Fig. 3 (a) LSV polarization curves of Mo-Co9Se8@NiSe/NF-40, Mo-Co9Se8@NiSe/NF-60, Mo-Co9Se8@NiSe/NF-80, Mo-Co9Se8@NiSe/NF, Co9Se8@NiSe/NF-60 and RuO2. (b) Corresponding histograms of overpotentials of different products at 50 mA cm−2. (c) Tafel plots of various OER electrocatalysts. (d) Current density as a function of the scan rate for different electrodes. (e) Nyquist plots of different products at 0.4 V. (f) Prolonged CP of Mo-Co9Se8@NiSe/NF-60 at a current density of 50 mA cm−2. | ||
Subsequently, the Tafel plots were employed to evaluate the OER kinetics of different products. Fig. 3c shows that Mo-Co9Se8@NiSe/NF-60 possessed the smallest Tafel slope of 61.86 mV dec−1. Compared with Mo-Co9Se8@NiSe/NF-40 (67.66 mV dec−1), Mo-Co9Se8@NiSe/NF-80 (70.62 mV dec−1), Mo-Co9Se8@NiSe/NF (68.05 mV dec−1), and Co9Se8@NiSe/NF-40 (93.88 mV dec−1), it was clearly found that Mo-Co9Se8@NiSe/NF-60 owned the optimal OER kinetics in all the samples. Fig. 3d represents the Cdl values of various samples, and Co9Se8@NiSe/NF-60 could obtain a value of 6.52 mF cm−2, which was much higher than those of Mo-Co9Se8@NiSe/NF-40 (3.69 mF cm−2), Mo-Co9Se8@NiSe/NF-80 (5.94 mF cm−2), Mo-Co9Se8@NiSe/NF (2.49 mF cm−2), and Co9Se8@NiSe/NF-40 (2.60 mF cm−2), manifesting that the appropriate etching time was crucial for improving the electrochemical active surface area (ECSA). The Nyquist plots displayed in Fig. 3e show that all the samples had similar charge transfer resistance (Rct) values, implying that the introduction of supports can effectively improve the conductivity of the materials. The long-term durability evaluated by performing chronopotentiometry is another important criterion for evaluating potential applications. Fig. 3f shows that the electrode modified by the Mo-Co9Se8@NiSe/NF-60 electrode can maintain an unchanged voltage for undergoing at least 88-hour CP tests at a current density of 50 mA cm−2, strongly implying that Mo-Co9Se8@NiSe/NF-60 has excellent stability.
To further analyze the reasons for the performance improvement, XPS was performed for Mo-Co9Se8@NiSe/NF-60, Mo-Co9Se8@NiSe/NF, and Co9Se8@NiSe/NF-60. Fig. S6† manifests the coexistence of Co, Ni, Se, and Mo elements on the surface of Mo-Co9Se8@NiSe/NF-60, indicating that the final products were successfully prepared. As shown in Fig. 4a, the Co 2p spectrum of Mo-Co9Se8@NiSe/NF-60 can be split into six characteristic peaks. The two peaks located at 781.70 and 775.92 eV could be assigned to Co3+ 2p3/2 and Co2+ 2p3/2, respectively.39 Meanwhile, the peaks at 797.70 and 803.43 eV could be ascribed to Co3+ 2p1/2 and Co2+ 2p1/2, respectively.40 Moreover, two accompanying satellite peaks were approximately located at 787.00 and 808.02 eV, respectively.41 For the Mo 3d spectrum of Mo-Co9Se8@NiSe/NF-60 (Fig. 4b), the two main peaks at binding energies of 229.24 and 233.73 eV were assigned to Mo 3d5/2 and Mo 3d3/2, respectively.42 In the high-resolution spectrum of Ni 2p (Fig. 4c), the two peaks at 852.90 and 856.22 eV were attributed to Ni3+ 2p3/2 and Ni2+ 2p3/2, respectively,43 the peaks located at 873.96 eV were ascribed to Ni 2p1/2,44 while the two accompanying peaks at 861.89 and 879.86 eV belonged to satellite peaks.45 As for the Se 3d spectra displayed in Fig. 4d, the peaks at binding energies of 54.11 and 59.09 eV were ascribed to Mo 3d5/2 and Mo 3d3/2, respectively.46 As for Mo-Co9Se8@NiSe/NF-60, the intensity of Se 3d was obviously reduced, indicating that H2O2 etching can effectively induce Se defects in Mo-Co9Se8@NiSe/NF-60.47 Notably, the binding energies of Co 2p for Mo-Co9Se8@NiSe/NF-60 showed a step-by-step positive shift in comparison with Mo-Co9Se8@NiSe/NF without H2O2 etching and Co9Se8@NiSe/NF-60 without Mo doping. Meanwhile, Ni 2p also showed a similar change when compared with Co 2p. To further establish the stepwise electronic structure regulation, the surface valence band spectra and the calculated d-band centers of different electrocatalysts were obtained (Fig. S7†). Compared with Co9Se8@NiSe/NF-60, the calculated Ed (energy of the d-band center) of Mo-Co9Se8@NiSe/NF-60 was found to be closer to the Fermi level (0 eV). According to the reports in the literature, this phenomenon indicated stronger adsorption between electrochemically active sites and OER intermediates (OH*, OOH*, and O*). In addition, numerous experiments have witnessed that a higher Ed value was positively associated with the OER activity. Meanwhile, from the XPS results, the incorporation of Mo has changed the electronic structure of Mo-Co9Se8@NiSe/NF-60, leading to a higher binding energy. More interestingly, compared with Co9Se8@NiSe/NF, the calculated Ed value of Co9Se8@NiSe/NF-60 was also closer to the Fermi level, indicating that Se vacancies can elevate the value of Ed by regulating the electronic structure, thereby contributing to the substantial improvement in electrocatalytic OER activity. These results indicated that Mo doping and Se vacancies can regulate the electronic structure of Co9Se8@NiSe/NF step by step, which optimized the adsorption energy for oxygenated intermediates and thus promoted the catalytic activity.48–52
 | ||
| Fig. 4 High-resolution XPS spectra of (a) Co 2p, (b) Mo 3d, (c) Ni 2p, and (d) Se 3d in Mo-Co9Se8@NiSe/NF-60, Mo-Co9Se8@NiSe/NF, and Co9Se8@NiSe/NF-60. | ||
Considering the promising OER performance of Mo-Co9Se8@NiSe/NF-60, its overall water splitting (OWS) performance was further explored. The equipment was assembled by coupling Mo-Co9Se8@NiSe/NF-60 with Pt/C to achieve overall water splitting (Fig. 5a). Fig. 5b shows that the Mo-Co9Se8@NiSe/NF-60||Pt/C couple exhibited good catalytic activity towards overall water splitting, which required a cell voltage of only 1.87 V to achieve 50 mA cm−2, remarkably surpassing the state-of-the-art RuO2||Pt/C coupling. Moreover, LSV showed that the Mo-Co9Se8@NiSe/NF-60||Pt/C couple retained the initial voltage even after 1000 potential cycles, implying that the assembled electrode couple possessed excellent durability for overall water splitting. Furthermore, a chronoamperometry (CP) test was also employed to further establish its durability. Fig. 5c shows the ignorable voltage degradation maintaining 50 mA cm−2 for at least 25 h continuously. In order to analyze the structural stability of Mo-Co9Se8@NiSe/NF-60, the electronic structure, phase, and morphology of spent catalysts were characterized. As shown in Fig. S8,† the XPS spectra of the catalyst before and after CP tests were obtained to depict the electronic structure. Co 2p and Ni 2p showed a significant negative shift compared with the initial one, which suggested that the electronic structure was noticeably altered. As displayed in Fig. S9,† the spent catalysts showed a similar pattern with the initial one (Fig. S9a†), and the locally enlarged area also showed the (222) plane of Co9Se8 (PDF # 09-0233) and the (300), (021), (211), (131) and (401) planes of NiSe (PDF # 18-0887) (Fig. S9b†). Fig. S9c and d† further prove the unchanged ultrathin morphology after 25-hour CP tests.
 | ||
| Fig. 5 (a) Schematic illustration of the electrocatalytic overall water splitting based on a Mo-Co9Se8@NiSe/NF-60∥Pt/C couple. (b) LSV polarization curves of RuO2∥Pt/C, initial Mo-Co9Se8@NiSe/NF-60∥Pt/C, and Mo-Co9Se8@NiSe/NF-60∥Pt/C after 1000 circles toward water electrolysis. (c) Long-term CP curve of the Mo-Co9Se8@NiSe/NF-60∥Pt/C couple at 50 mA cm−2. | ||
Combining the results of electrochemical tests and characterizations, we can reach the conclusion that the excellent electrocatalytic OER performance of Mo-Co9Se8@NiSe/NF-60 is primarily generated from the electronic structure modification originating from Mo doping and Se vacancies (Fig. 6). For one thing, Mo doping can modify the electronic configuration and optimize the adsorption energy of reaction intermediates, resulting in enhanced activities. For another, Se vacancies can also effectively modify the electronic or surface structures to boost the numbers and intrinsic activities of electroactive sites, which further promotes the catalytic OER activity. Moreover, the ultrathin and self-supported structure also contributes to the substantial improvement in its OER electrocatalytic activity and stability.
 | ||
| Fig. 6 Schematic illustration of the mechanism of OER activity based on Mo-Co9Se8@NiSe/NF-60. | ||
4. Conclusions
In summary, a dual modulation strategy was presented for tuning the electronic structure of Co9Se8@NiSe/NF ultrathin nanosheet arrays with Mo doping and Se vacancies. It is demonstrated that a trace amount of Mo doping and H2O2 etching can modulate the electronic structure of Co9Se8@NiSe/NF step by step, which can optimize the binding strength with oxygenated intermediates. Moreover, the self-supported ultrathin structure can also expose more accessible active sites for intermediates. Benefitting from the modulated electronic structure and self-supported ultrathin architecture, such Mo-Co9Se8@NiSe/NF-60 can exhibit superb electrocatalytic OER performance with an overpotential of 343 mV at 50 mA cm−2, and a Tafel slope of 61.86 mV dec−1 in 1.0 M KOH. This work demonstrated the power of step by step electronic structure modification in designing outstanding OER electrocatalysts by doping and defect engineering. It is highly anticipated that more works on the electronic structure and morphology engineering of other potential materials may be stimulated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financed by the National Natural Science Foundation of China (22202169).References
- H. Xu, J. Yuan, G. He and H. Chen, Coord. Chem. Rev., 2023, 475, 214869 CrossRef CAS.
- Y. Ding, K.-W. Cao, J.-W. He, F.-M. Li, H. Huang, P. Chen and Y. Chen, Chin. J. Catal., 2022, 43, 1535 CrossRef CAS.
- Q. Xue, X.-Y. Bai, Y. Zhao, Y.-N. Li, T.-J. Wang, H.-Y. Sun, F.-M. Li, P. Chen, P.J. Jin, S.-B. Yin and Y. Chen, J. Energy Chem., 2022, 65, 102 CrossRef.
- W. Shen, J. Yin, J. Jin, Y. Hu, Y. C. Hou, J. T. Xiao, Y.-Q. Zhao and P. X. Xi, Adv. Energy Sustain. Res., 2022, 3, 2200036 CrossRef.
- Z. Li, D. Liu, X. Lu, M. Du, Z. Chen, J. Teng, R. Sha and L. Tian, Dalton Trans., 2022, 51, 1527–1532 RSC.
- H. Xu, Y. Zhao, Q. Wang, G. He and H. Chen, Coord. Chem. Rev., 2022, 451, 214261 CrossRef CAS.
- J. Yin, J. Jin, M. Lu, B. L. Huang, H. Zhong, Y. Peng, P. X. Xi and C.-H. Yan, J. Am. Chem. Soc., 2020, 142, 18378–18386 CrossRef CAS PubMed.
- H. Xu, Y. Zhao, G. He and H. Chen, Int. J. Hydrogen Energy, 2022, 47, 14257–14279 CrossRef CAS.
- L. Tian, Z. Li, X. Xu and C. Zhang, J. Mater. Chem. A, 2021, 9, 13459–13470 RSC.
- T.-J. Wang, H.-Y. Sun, Q. Xue, M.-J. Zhong, F.-M. Li, X.L. Tian, P. Chen, S.-B. Yin and Y. Chen, Sci. Bull., 2021, 66, 2079 CrossRef CAS.
- Z. Li, X. Xu, X. Lu, C. He, J. Huang, W. Sun and L. Tian, J. Colloid Interface Sci., 2022, 615, 273–281 CrossRef CAS PubMed.
- L. Tian, X. Zhai, X. Wang, X. Pang, J. Li and Z. Li, Electrochim. Acta, 2020, 337, 135823 CrossRef CAS.
- H. Xu, B. Huang, Y. Zhao, G. He and H. Chen, Inorg. Chem., 2022, 61, 4533–4540 CrossRef CAS.
- Y. Wang, S. Wang, S. L. Zhang and X. W. D. Lou, Angew. Chem., Int. Ed., 2020, 59, 11918–11922 CrossRef CAS.
- Y. Wang, X. Li, M. Zhang, J. Zhang, Z. Chen, X. Zheng, Z. Tian, N. Zhao, X. Han, K. Zaghib, Y. Wang, Y. Deng and W. Hu, Adv. Mater., 2022, 34, e2107053 CrossRef.
- H. Xu, H. Shang, C. Wang and Y. Du, Adv. Funct. Mater., 2020, 30, 2000793 CrossRef CAS.
- M. M. Alsabban, M. K. Eswaran, K. Peramaiah, W. Wahyudi, X. Yang, V. Ramalingam, M. N. Hedhili, X. Miao, U. Schwingenschlogl, L. J. Li, V. Tung and K. W. Huang, ACS Nano, 2022, 16, 3906–3916 CrossRef CAS PubMed.
- L. Tian, H. Chen, X. Lu, D. Liu, W. Cheng, Y. Liu, J. Li and Z. Li, J. Colloid Interface Sci., 2022, 628, 663–672 CrossRef CAS PubMed.
- J. Yin, Y. X . Li, F. Lv, M. Lu, K. Sun, W. Wang, L. Wang, F. Y. Cheng, Y. F. Li, P.X. Xi and S.J. Guo, Adv. Mater., 2017, 29, 1704681 CrossRef.
- J. Wang, X. Zheng, G. Wang, Y. Cao, W. Ding, J. Zhang, H. Wu, J. Ding, H. Hu, X. Han, T. Ma, Y. Deng and W. Hu, Adv. Mater., 2022, 34, e2106354 CrossRef.
- M. Wang, Z. Sun, H. Ci, Z. Shi, L. Shen, C. Wei, Y. Ding, X. Yang and J. Sun, Angew. Chem., Int. Ed., 2021, 60, 24558–24565 CrossRef CAS PubMed.
- H. Xu, C. Wang, G. He, H. Chen and Y. Du, Inorg. Chem., 2022, 61, 14224–14232 CrossRef CAS.
- H. Liu, M. Zha, Z. Liu, J. Tian, G. Hu and L. Feng, Chem. Commun., 2020, 56, 7889–7892 RSC.
- S. Tang, Y. Zhou, X. Lu, Z. Chen, Z. Huang, Z. Li and L. Tian, J. Alloys Compd., 2022, 924, 166415 CrossRef CAS.
- Z. Li, Z. Jiang, W. Zhu, C. He, P. Wang, X. Wang, T. Li and L. Tian, Appl. Surf. Sci., 2020, 504, 144368 CrossRef CAS.
- Y. Zhang, L. You, Q. Liu, Y. Li, T. Li, Z. Xue and G. Li, ACS Appl. Mater. Interfaces, 2021, 13, 2765–2771 CrossRef CAS.
- L. Tian, Z. Huang, W. Na, Y. Liu, S. Wang, Y. He, W. Cheng, T.-Z. Huang, Z. Li and T. Li, Nanoscale, 2022, 14, 15340–15347 RSC.
- Y. Ding, Q. Xue, Q. L. Hong, F. M. Li, Y. C. Jiang, S. N. Li and Y. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 4026–4033 CrossRef CAS PubMed.
- L. Tian, X. Pang, H. Xu, D. Liu, X. Lu, J. Li, J. Wang and Z. Li, Inorg. Chem., 2022, 61, 16944–16951 CrossRef CAS PubMed.
- B. Fang and L. Feng, Acta Phys. -Chim. Sin, 2020, 36, 1905023–1905020 Search PubMed.
- X. Wang, Y. Chen, Y. Fang, J. Zhang, S. Gao and X. W. D. Lou, Angew. Chem., Int. Ed., 2019, 58, 2675–2679 CrossRef CAS PubMed.
- A. M. Kwiecińska, D. Kutyła, K. Kołczyk-Siedlecka, K. Skibińska, P. Żabiński and R. Kowalik, J. Electroanal. Chem., 2019, 848, 113278 CrossRef.
- Z.-B. Zhai, K.-J. Huang and X. Wu, Nano Energy, 2018, 47, 89–95 CrossRef CAS.
- X. Zhai, X. Pang, X. Wang, H. Xu, C. Zhang, Q. Zhang, Y. Zhou and L. Tian, J. Saudi Chem. Soc., 2022, 26, 101469 CrossRef CAS.
- X. Wang, Y. Zhang, H. Si, Q. Zhang, J. Wu, L. Gao, X. Wei, Y. Sun, Q. Liao, Z. Zhang, K. Ammarah, L. Gu, Z. Kang and Y. Zhang, J. Am. Chem. Soc., 2020, 142, 4298–4308 CrossRef CAS PubMed.
- Z. F. Huang, S. Xi, J. Song, S. Dou, X. Li, Y. Du, C. Diao, Z. J. Xu and X. Wang, Nat. Commun., 2021, 12, 3992 CrossRef CAS.
- Q. Zhang, K. Wang, M. Zhang, T. Chen, L. Li, S. Shi and R. Jiang, CrystEngComm, 2022, 24, 5580–5587 RSC.
- X. Yu, J. Zhao and M. Johnsson, Adv. Funct. Mater., 2021, 31, 2101578 CrossRef CAS.
- W. Shen, J. Jin, Y. Hu, Y. C. Hou, J. Yin, Z. H. Ma, Y.-Q. Zhao and P.X. Xi, Chin. J. Catal., 2022, 43, 1485–1492 CrossRef CAS.
- H. Xu, H. Shang, C. Wang, L. Jin, C. Chen, C. Wang and Y. Du, Appl. Catal., B, 2020, 265, 118605 CrossRef CAS.
- Z. Li, C. Li, J. Huang, W. Sun, W. Cheng, C. He and L. Tian, Int. J. Hydrogen Energy, 2022, 47, 15189–15197 CrossRef CAS.
- H. Xu, H. Shang, L. Jin, C. Chen, C. Wang and Y. Du, J. Mater. Chem. A, 2019, 7, 26905–26910 RSC.
- J. Yin, J. Jin, H. Zhang, M. Lu, Y. Peng, B.L. Huang, P.X. Xi and C.-H. Yan, Angew. Chem., Int. Ed., 2019, 58, 18676–18682 CrossRef CAS PubMed.
- D. Yang, L. Yang, L. Zhong, X. Yu and L. Feng, Electrochim. Acta, 2019, 295, 524–531 CrossRef CAS.
- Z. Xue, X. Li, Q. Liu, M. Cai, K. Liu, M. Liu, Z. Ke, X. Liu and G. Li, Adv. Mater., 2019, 31, e1900430 CrossRef PubMed.
- G. Zhou, X. Wu, M. Zhao, H. Pang, L. Xu, J. Yang and Y. Tang, ChemSusChem, 2021, 14, 699–708 CrossRef CAS PubMed.
- L. Zhang, C. Lu, F. Ye, R. Pang, Y. Liu, Z. Wu, Z. Shao, Z. Sun and L. Hu, Adv. Mater., 2021, 33, e2007523 CrossRef PubMed.
- Y. Wang, S. Wang, Z. L. Ma, L. T. Yan, X. B. Zhao, Y. Y. Xue, J. M. Huo, X. Yuan, S. N. Li and Q. G. Zhai, Adv. Mater., 2022, 34, e2107488 CrossRef PubMed.
- Y. Sun, Z. Xue, Q. Liu, Y. Jia, Y. Li, K. Liu, Y. Lin, M. Liu, G. Li and C. Y. Su, Nat. Commun., 2021, 12, 1369 CrossRef CAS.
- H. Xu, H. Shang, C. Wang and Y. Du, Coord. Chem. Rev., 2020, 418, 213374 CrossRef CAS.
- H. Xu, H. Shang, C. Wang and Y. Du, Adv. Funct. Mater., 2020, 30, 2006317 CrossRef CAS.
- L. Tian, Y. Liu, C. He, S. Tang, J. Li and Z. Li, Chem. Rec., 2022, 22, e202200213 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr05410h |
| This journal is © The Royal Society of Chemistry 2023 |