
New horizons of MBenes: highly active catalysts for the CO oxidation reaction†
Bikun
Zhang
ab,
Jian
Zhou
 *ab and
Zhimei
Sun
*ab
*ab and
Zhimei
Sun
*ab
aSchool of Materials Science and Engineering, Beihang University, Beijing 100191, China. E-mail: jzhou@buaa.edu.cn; zmsun@buaa.edu.cn
bCenter for Integrated Computational Materials Engineering, International Research Institute for Multidisciplinary Science, Beihang University, Beijing 100191, China
First published on 8th December 2022
Abstract
The search for materials with high intrinsic carbon monoxide oxidation reaction (COOR) catalytic activity is critical for enhancing the efficiency of reducing CO contamination. COOR catalysts, however, have long relied heavily on noble metals and CeO2. Herein, in order to search for non-noble COOR catalysts that are more active than CeO2, 18 oxygen-functionalized MBenes with orthorhombic and hexagonal crystal structures, denoted as orth-M2B2O2 and hex-M2B2O2 (M = Ti, V, Cr, Zr, Nb, Mo, Hf, Ta and W), were investigated in terms of their COOR catalytic activity by high-throughput first-principles calculations. Hex-Mo2B2O2, orth-Mo2B2O2, hex-V2B2O2 and hex-Cr2B2O2 were found to be more active than CeO2 and possess structural stability below 1000 K, showing the potential to replace CeO2 as the substrates of COOR catalysts. Moreover, orth-Mo2B2O2, hex-V2B2O2 and hex-Cr2B2O2 exhibit even higher COOR catalytic activity than Pt–CeO2 and Au–CeO2, and are expected to be applied as COOR catalysts directly. Further investigations showed that the formation energy of oxygen vacancies could be used as the descriptor of COOR catalytic activity, which would help to reduce the amount of calculations significantly during the catalyst screening process. This work not only reports a series of 2D materials with high COOR catalytic activity and opens up a new application area for MBenes, but also provides a reliable strategy for highly efficient screening for COOR catalysts.
Introduction
Carbon monoxide (CO) is one of the major air pollutants and is not only toxic to human beings, but also results in the poisoning of Pt-based catalysts in proton-exchange membrane fuel cells (PEMFC).1,2 CO originates mostly from the exhaust gas of internal combustion engines,3 and one of the most common methods for CO treatment is the carbon monoxide oxidation reaction (COOR), in which CO is oxidized to carbon dioxide (CO2) at low temperature in an O2 atmosphere without combustion.4–7 There exist three typical mechanisms for COOR catalysis: the Langmuir–Hinshelwood (L–H) mechanism, the Eley–Rideal (E–R) mechanism and the Mars–van Krevelen (M–vK) mechanism.3 In the L–H and E–R mechanisms, all the CO molecules are oxidized by O2 molecules and the lattices of catalysts remain intact during the whole catalytic process. As illustrated in Scheme 1, in the L–H mechanism, the catalysts adsorb both CO and O2 molecules, while in the E–R mechanism, the catalysts only adsorb O2 molecules. However, in the M–vK mechanism, the CO molecules are oxidized by both O2 molecules and the lattice oxygen atoms on the surfaces of the catalysts, which means that the catalysts are involved in the elementary reactions directly. During the catalytic process, oxygen vacancies (VaO) are generated when the lattice oxygen atoms are used to oxidize CO molecules, and they will be filled with O2 molecules in the next elementary step.8,9 In general, on catalysts with metal active sites (including single atoms, clusters and nanoparticles) and inert substrates (such as graphene7), the COOR occurs via the L–H and E–R mechanisms, while only on catalysts containing accessible lattice oxygen atoms could the COOR occur via the M–vK mechanism.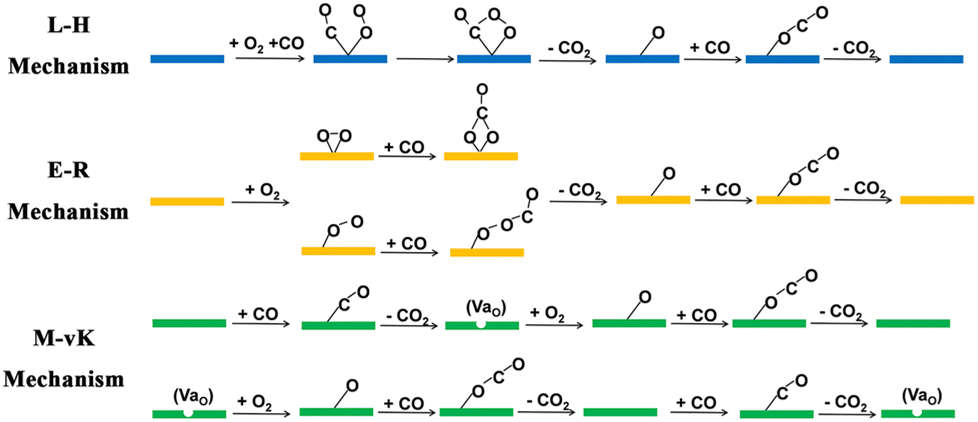 | ||
| Scheme 1 Three different mechanisms of COOR catalysis. | ||
So far, noble metals, such as Pt, Pd, and Au, are indispensable for most COOR catalysts. To reduce the use of noble metals and control the cost, one popular strategy is to load single atoms or nanoparticles of noble metals or their oxides on non-noble substrates, such as Pt/CeO2, Pd/CeO2 and Au/CeO2,5,8–23 which have been universally adopted in three-way catalytic converters (TWCs) in the exhaust systems of vehicles to remove CO, which is one of the major harmful gases. The most commonly used COOR catalysts is CeO2-supported noble metals because the lattice oxygen atoms in CeO2 could be easily utilized, making the COOR occur via the M–vK mechanism with high activity.8,11,14,15,19,24–26 The intrinsic catalytic activity of substrates contributes a lot to the overall performance of COOR catalysts since the substrates are involved in the oxidation of CO directly via the M–vK mechanism. However, the intrinsic catalytic activity of CeO2 is relatively poor.3 Therefore, the search for materials with higher intrinsic COOR catalytic activity to replace CeO2 will be beneficial for enhancing the COOR catalytic performance of combined catalysts.
Lattice oxygen atoms are indispensable to the intrinsic COOR catalytic activity of non-noble catalysts.3,27 In the past decade, two-dimensional transition metal carbides/nitrides/carbonitrides, MXenes, have been widely studied as catalysts for various reactions. The exposed transition metal atoms on the surfaces of MXenes are easily functionalized by oxygen atoms which can tune their properties.28–30 According to the previous research by Tahini et al.,27 the interaction between the transition metal atoms and the surface oxygen atoms in Cr3C2O2, V3C2O2 and Sc3C2O2 MXenes is relatively weak, and thus the surface oxygen atoms could be easily utilized to oxidize CO molecules. By studying the COOR catalytic activity, it was found that these O-functionalized MXenes are more active than CeO2via the M–vK mechanism. In 2017, MBenes, a family of two-dimensional transition metal borides, were reported,31 and they possess similar structures to MXenes.32 MBenes are widely studied as catalysts or substrates of various reactions, including the hydrogen evolution reaction (HER),31,33–36 the oxygen evolution reaction (OER)/oxygen reduction reaction (ORR),34,37 the nitrogen reduction reaction (NRR)38–40 and the CO2 reduction reaction (CO2RR).41,42 Similar to the situation for MXenes, the exposed transition metal surfaces of pristine MBenes can be easily functionalized by O atoms.35,37,43,44 Therefore, it is anticipated that the surface oxygen atoms on O-functionalized MBenes could also be utilized to oxidize CO molecules leading to high COOR catalytic activity via the M–vK mechanism.
In this work, 18 O-functionalized MBenes, denoted as orth-M2B2O2 and hex-M2B2O2, where M represents Ti, V, Cr, Zr, Nb, Mo, Hf, Ta, and W, and “orth/hex” indicates orthorhombic/hexagonal crystal systems, were investigated to explore their intrinsic COOR catalytic activity and mechanism by means of the first-principles method. Our calculated results show that five M2B2O2 compounds demonstrate higher COOR catalytic activity than CeO2 at room temperature and good thermal stability at a high temperature of 1000 K, among which, orth-Mo2B2O2, orth-W2B2O2, hex-V2B2O2 and hex-Cr2B2O2 are eligible to serve as independent COOR catalysts. Moreover, it has been found that oxygen vacancy formation energy could reflect the maximum reaction energy for the COOR directly. Therefore, by taking oxygen vacancy formation energy as the descriptor for COOR catalytic activity, the screening efficiency for COOR catalysts could be highly improved.
Computational methods
All first-principles calculations were performed based on density functional theory (DFT) using the Vienna ab initio Simulation Package (VASP)45,46 with the projector-augmented wave (PAW) method.47 The interaction between ion cores and valence electrons was described using the Perdew–Burke–Ernzerhof (PBE) functional.48,49 All the atomic structures were fully relaxed under the energy and force convergence criteria of 10−4 eV per atom and 0.02 eV per angstrom, respectively. For the calculation of energy, 3 × 3 × 1 supercells were used, while for ab initio molecular dynamics (AIMD) simulation, 4 × 4 × 1 supercells were applied, during which a canonical NVT ensemble was used, and the temperature was controlled with the algorithm of Nosé.50In this work, COOR catalysis on 18 orth/hex-M2B2O2via the M–vK mechanism was studied, which involves three phases: the adsorption of O2 (I:* + O2 → *OO), the formation of the first CO2 (II: *OO + CO → *OOCO and III: *OOCO → CO2 + *O) and the formation of the second CO2 (IV: *O + CO → *CO2 and V: *CO2 → * + CO2). Here * represents M2B2O2 with O vacancies. The Gibbs free energy of steps I∼V is denoted as ΔG1 ∼ ΔG5. The computational details of the energy terms are included in the ESI.†
Results and discussion
The 18 orth/hex-M2B2 compounds studied in this work have been proved to be stable dynamically according to the previous research studies.31–33,37,51–55 Experimentally, oxygen functional groups have been detected on the surface of hex-Mo4/3B2 MBenes.56 Meanwhile, theoretical studies of O-functionalized MBenes have been carried out in many studies.31,35,36,43,54,57,58 First of all, we studied the atomic structures of 18 M2B2O2 and the corresponding formation energy of oxygen functional groups. There exist four and two sites with different chemical environments for O atoms binding on the surfaces of orth-M2B2 and hex-M2B2, respectively (Fig. S1(a) and S2(a)†). Considering the symmetry of orth-M2B2 and hex-M2B2, we investigated four types of atomic structures for orth-M2B2, including types I–III shown in Fig. 1(a–c) and type IV shown in Fig. S1(b),† and three types of atomic structures for hex-M2B2, including types A and B shown in Fig. 1(d and e) and type C shown in Fig. S2(b).† After studying the total energy of all the structures of the 18 M2B2O2, the most stable structures for each are shown in Fig. 1. There are three types of atomic structures for orth-M2B2O2: type-I for M = Ti, Zr, Hf, Mo; type-II for M = V, Nb, Ta, W; and type-III for M = Cr, while there are two different atomic structures for hex-M2B2O2: type-A for M = Ti, Zr, Hf, Mo, W and type-B for M = V, Nb, Ta, Cr. For the most stable structure for each M2B2O2, the formation energy of the oxygen functional group (ΔGOf) was calculated with eqn (S4),† and the results are listed in Table S2.† For all 18 M2B2O2, the values of ΔGOf are negative, indicating the spontaneity of the formation of oxygen functional groups under an O2 atmosphere thermodynamically. These surface oxygen atoms could serve as the oxidants of CO molecules in COOR catalysis.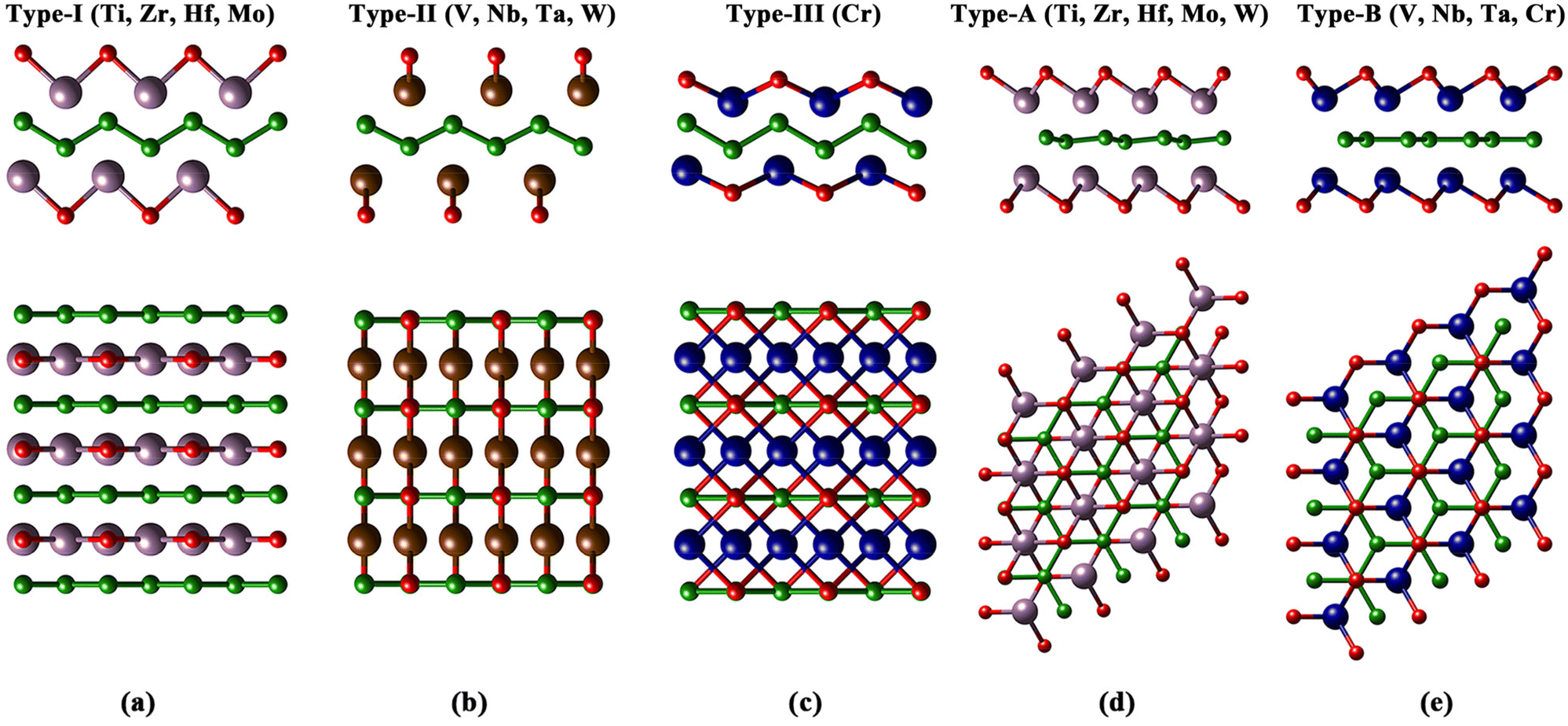 | ||
| Fig. 1 The optimized atomic structures of (a) type-I, (b) type-II, (c) type-III for orth-M2B2O2 and (d) type-A, (e) type-B for hex-M2B2O2 in side (up) and top (down) views. Green and red balls represent B and O atoms, respectively, and the balls with other colors represent transition metal atoms. | ||
In general, the surface vacancies on O-functionalized MXenes are inevitable.28,59 Therefore, we considered the existence of oxygen vacancies on the surface of M2B2O2 in this work. Here the concentration of O vacancies is considered to be 5.56%, corresponding to one O vacancy in a 3 × 3 × 1 supercell of M2B2O2. The formation energy of oxygen vacancies, ΔGVaf, was calculated with eqn (S5)† and the results are listed in Table S2.† The results show that the values of ΔGVaf for the 18 M2B2O2 range from 2.55 eV to 5.59 eV. In addition, the values of ΔGVaf for two O-functionalized MXenes, Ti2CO2 and Ti3C2O2, were calculated to be 4.5 eV and 5.0 eV (the concentration of O vacancies = 5.56%), respectively, which are higher than those of most of the M2B2O2, as displayed in Fig. 2. Since O vacancies on Ti2CO2 and Ti3C2O2 are very common in experiments,28,60 the M2B2O2 studied here can also easily become defective with O vacancies, which could capture CO molecules.
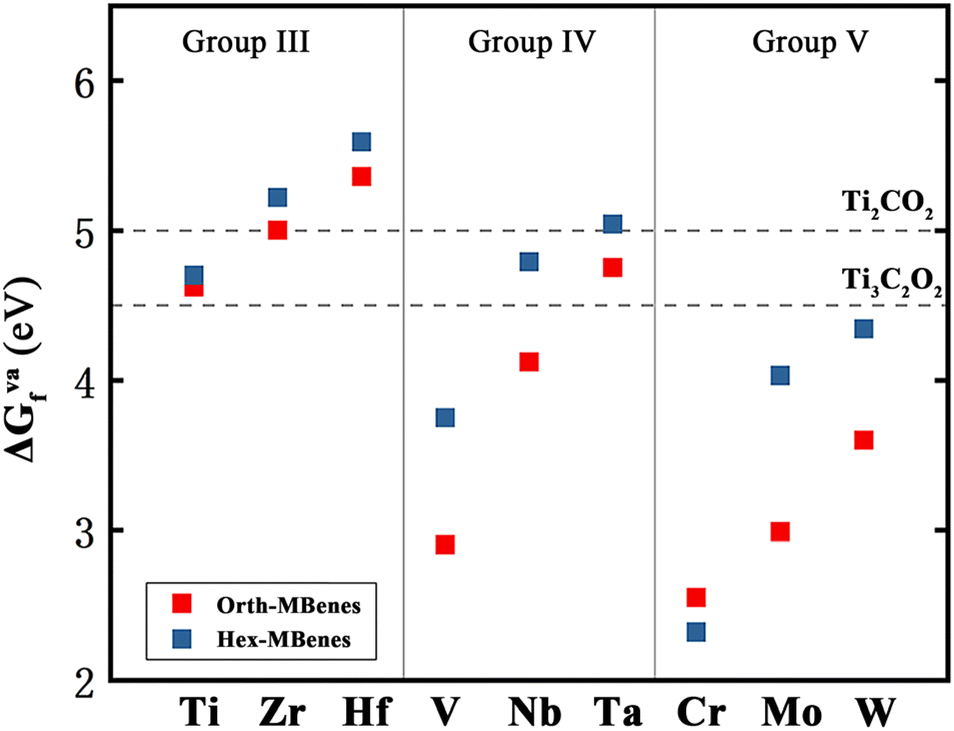 | ||
| Fig. 2 The formation energy of oxygen vacancies of 18 M2B2O2, in comparison with those of Ti2CO2 and Ti3C2O2. | ||
From Fig. 2, it is found that for orth/hex-M2B2O2 with M in the same group, the ΔGVaf values increase with increasing atomic number (Z), while for M2B2O2 with M in the same period, the ΔGVaf values decrease with increasing electron layer number (n). Therefore, orth/hex-Cr2B2O2 has the lowest ΔGVaf value, while orth/hex-Hf2B2O2 has the highest ΔGVaf value. Apparently, the variation of ΔGVaf is in accordance with the periodic law of elements: the larger Z and the lower n M has, the lower ΔGVaf value M2B2O2 has.
The electrons on the surfaces of defective M2B2O2 are not evenly distributed due to the existence of O vacancies, which are easy to fill with gaseous molecules. According to the results of Bader charge calculations, the transition metal atoms nearest to the O vacancies have lower coordination number than the other transition metal atoms, and thus they possess more electrons. The extra electrons could transfer to the 2p orbitals of the O atoms in O2 or CO molecules to drive the electron distribution of the transition metal layers equally. Theoretically, both O2 and CO molecules could adsorb at the O vacancy sites. However, if CO molecules fill the O vacancies, the C atoms would be left on the surface, which could not be removed by combining with O2 to form CO2. This would result in the blockage of O2 adsorption sites and deactivation of the catalysts. Therefore, we investigated the adsorption selectivity of O vacancies by calculating the adsorption energy of O2 and CO, as shown in Fig. 3(a). Apparently, the adsorption of O2 on all 18 defective M2B2O2 is highly spontaneous with the O2 adsorption energy being lower than −1 eV, while the values of CO adsorption energy are all higher than 0. Taking orth-Cr2B2O2 and hex-Cr2B2O2 as examples, after adsorbing CO, the O atoms of CO could not fill the O vacancies and the CO molecules would stay above the surfaces, as displayed in Fig. S3(a and b).† Therefore, all 18 defective M2B2O2 compounds have higher adsorption selectivity towards O2 than towards CO, avoiding the deactivation of catalysts.
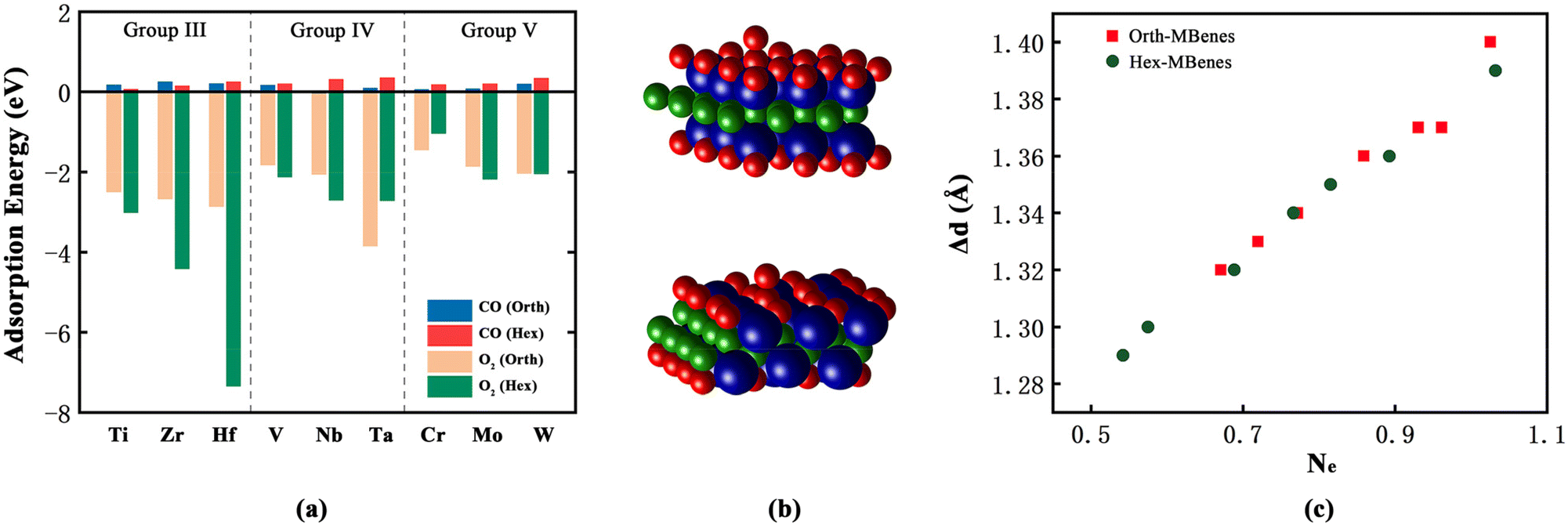 | ||
| Fig. 3 (a) The adsorption energy of O2 and CO on 18 defective M2B2O2. (b)The structures after O2 adsorption on hex-Cr2B2O2 (up) and orth-Cr2B2O2 (down). Blue, green and red balls represent transition metal, B and O atoms, respectively. (c) The relationship between the O–O bond length (Δd) and the numbers of electrons that the O2 molecules obtains from the substrates (Ne). | ||
As shown in Fig. 3(b), an O2 molecule adsorbs on the O vacancy site perpendicularly to the surfaces of most M2B2O2. However, for orth-Ta2B2O2 and hex-Zr2B2O2, O2 molecules are parallel with the surfaces (Fig. S3(c and d)†), while for hex-Hf2B2O2, after the vacancy is filled with one O atom of O2, the other O atom adsorbs on the top of the Hf atom (Fig. S3(e)†). To study the mechanism, we further calculated the change in the O–O bond length (Δd) and the number of electrons that the O2 molecules obtain from the substrates (Ne), which are listed in Table S3.† The O–O bond length of O2 increases on all 18 M2B2O2 after adsorption, indicating that the O2 molecules are activated by M2B2O2 and the O–O bonds become weaker. By analyzing the relationship between Δd and Ne, it is found from Fig. 3(c) that Δd is approximately proportional to Ne, implying that the more electrons the O2 molecules obtain, the stronger the O2 activation degree is, contributing to a longer O–O bond.
After the adsorption of O2, the outer O atom combines with the gaseous CO molecule, thus forming the OOCO intermediates (step II). This step is highly spontaneous for all 18 M2B2O2 compounds, with ΔG2 ranging from −4 eV to −6 eV. It is notable that the OOCO intermediates exist as separate adsorbed O and isolated OCO from the M2B2O2 substrates, as illustrated in Fig. 4(a). The isolated OCO is located at a distance of over 2.8 Å above the surfaces of M2B2O2, which suggests that the binding strength between the M2B2O2 substrates and the outer O atom is extremely weak. Consequently, it is very easy for the outer O atoms to detach from the substrates and combine with the CO molecules, which is attributed to the O2 activation by substrates. Further study of Bader charge indicated that there is barely electron transfer between the substrates and OCO. In other words, no chemisorption exists between the two moieties and the binding strength is relatively low. Therefore, step III is also exothermic, generating gaseous CO2 molecules with ΔG3 ranging from 0 to −0.5 eV. The values of Gibbs free energy change for the formation of the first CO2 (ΔG2 + ΔG3) are listed in Table S4.†
 | ||
| Fig. 4 (a) Gibbs free energy profile for steps II and III. (b) ΔGmax for COOR of orth-V2B2O2, orth-Cr2B2O2, orth-Nb2B2O2, orth-Mo2B2O2, orth-W2B2O2, hex-V2B2O2, hex-Cr2B2O2, hex-Mo2B2O2, compared with CeO2, Pt–CeO2 and Au–CeO2. | ||
After desorption of the first CO2, the surfaces of substrates become complete and flat, and all the O atoms could serve as the adsorption sites for CO molecules. During this process, the values of ΔG4 are negative for orth-V2B2O2 (−0.59 eV), orth-Cr2B2O2 (−0.56 eV), orth-Mo2B2O2 (−0.60 eV) and hex-Cr2B2O2 (−0.87 eV). For these materials, the subsequent CO2 desorption process requires more energy input with ΔG5 ranging from −0.28 eV to 0.1 eV, and thus ΔG5 is the maximum Gibbs free energy change (ΔGmax) for the whole COOR. For the other M2B2O2 compounds, the adsorption of the second CO molecules is endothermic and the most energy-consuming step, among which orth-Nb2B2O2, orth-W2B2O2, hex-V2B2O2 and hex-Mo2B2O2 have ΔGmax lower than 1 eV. The values of ΔG4 and ΔG5 for all 18 M2B2O2 are listed in Table S5.† Besides, the COOR catalytic activity of CeO2 was also studied. The supercell of CeO2 contains 18 surface O atoms, which is the same as for the M2B2O2 studied in this work, and the ΔGmax for CeO2 is calculated to be 0.93 eV. It was found that the features in terms of reaction energy are similar for CeO2 and M2B2O2: easy adsorption of O2, easy formation of the first CO2, but hard formation of the second CO2. As shown in Fig. 4(b), the eight M2B2O2 mentioned above are more active than CeO2 for COOR catalysis. Moreover, except hex-Mo2B2O2, these M2B2O2 compounds possess COOR catalytic activities even higher than those of CeO2-based single atom catalysts, Pt–CeO2 and Au–CeO2 (ΔGmax = 0.60 eV and 0.45 eV, respectively), which have been considered as highly active COOR catalysts experimentally according to the previous studies.11,26 Since the COOR is widely used for CO removal in exhaust gas, the catalysts should maintain the stability of their structures at high temperature. By ab initio molecular dynamics (AIMD) simulation at 1000 K for 10 ps, hex-Mo2B2O2, orth-Mo2B2O2, hex-V2B2O2 and hex-Cr2B2O2 were found to be stable with tiny energy fluctuation and structural distortion, as seen in Fig. S4.† Therefore, we conclude that hex-Mo2B2O2, orth-Mo2B2O2, hex-V2B2O2 and hex-Cr2B2O2 could all serve as the substitutes for CeO2 as the substrates of COOR catalysts. Furthermore, orth-Mo2B2O2, hex-V2B2O2 and hex-Cr2B2O2 are expected to be used as COOR catalysts directly.
As the COOR catalysis involves five elementary steps, it is very time-consuming to calculate the Gibbs free energy changes for all the elementary steps to screen the desired COOR catalysts from many candidates. Therefore, to simplify the screening process and reduce the amount of calculations, here we tried to find out a general descriptor for the COOR catalytic activity by investigating the relationship between ΔGmax of M2B2O2 and species of M. In Fig. 5(a and b) the ΔGmax values of orth-M2B2O2 and hex-M2B2O2 are shown in the form of a three-dimensional bar graph. It can be seen that the values of ΔGmax vary with the species of transition metal elements, in line with the periodic law of elements: for M in the same period, ΔGmax of M2B2O2 decreases with increasing atomic number (Z), while for M in the same group, ΔGmax of M2B2O2 becomes higher with increasing electron layer number (n). Therefore, orth/hex-Cr2B2O2 has the smallest ΔGmax while orth/hex-Hf2B2O2 has the largest ΔGmax.
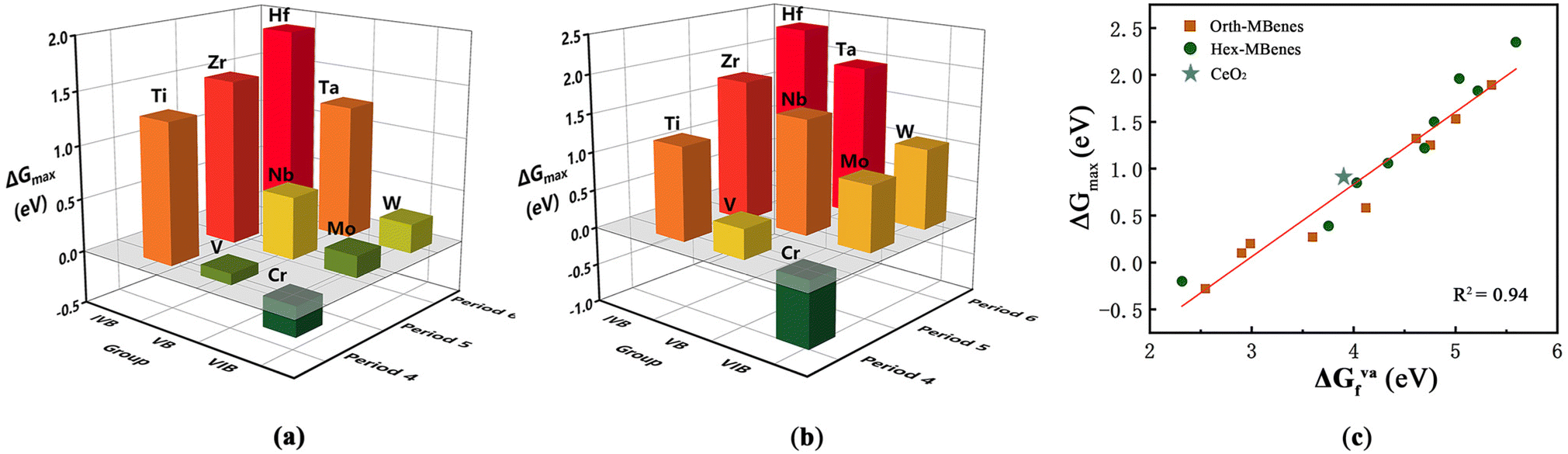 | ||
| Fig. 5 ΔGmax for COOR of (a) orth-M2B2O2 and (b) hex-M2B2O2 with M in different groups and periods. (c) Relationship between ΔGmax and ΔGVaf of orth/hex-M2B2O2. | ||
As discussed above, the periodic law of elements was also found in the variation of ΔGVaf with species of M. Therefore, we correlate ΔGmax with ΔGVaf, and the results are demonstrated in Fig. 5(c). The data dots in the figure indicate that there exists close correlation between ΔGmax and ΔGVaf. By linearly fitting the dot data, it is found that the determination coefficient, i.e., R2, reached 0.94, indicating the high degree of linear relationship between the two parameters. Meanwhile, the ΔGVaf value of CeO2 with an O vacancy density of 5.56% was calculated to be 3.94 eV. The coordinates (ΔGVaf, ΔGmax) for CeO2 are indicated with the blue star in Fig. 5(c), which is clearly very close to the fitted line. Therefore, the oxygen vacancy formation energy can directly reflect the COOR catalytic activity of materials. Generally, the lower the ΔGVaf, the lower the ΔGmax, which implies higher COOR catalytic activity.
By using ΔGVaf as the descriptor for the COOR catalytic activity, the screening of COOR catalysts with high activity will become much easier because only the values of ΔGVaf need to be calculated while the time-consuming calculations of ΔG for all the elementary steps are unnecessary. Subsequently, the investigation of the COOR catalytic activity of 9 M2CO2 MXenes was conducted, where M represents the same transition metal atoms as those of M2B2O2 in this work. Here, we calculated the ΔGVaf of the 9 M2CO2 first, which shows the periodic variation with M, the same as in the case of M2B2O2. Among the 9 M2CO2, only Cr2CO2 and V2CO2 have lower ΔGVaf than CeO2. Then, by calculating ΔGmax, it was found that Cr2CO2 and V2CO2 are the two most active catalysts with the values of ΔGmax being −0.2 eV (step II) and 0.26 eV (step I), respectively, as illustrated in Fig. S5.† Except Cr2CO2 and V2CO2, the other M2CO2 compounds have relatively low intrinsic COOR catalytic activity via the M–vK mechanism. For example, when used as a substrate of COOR catalysts, the surface oxygen atoms of Ti2CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6 are not involved in the elementary reactions and thus the COOR occurs on Ti2CO2-based catalysts via the L–H or E–R mechanism rather than the M–vK mechanism. Therefore, our findings are applicable to the case for M2CO2, which once again supports the conclusion that ΔGVaf can serve as the descriptor for COOR catalytic activity.
6 are not involved in the elementary reactions and thus the COOR occurs on Ti2CO2-based catalysts via the L–H or E–R mechanism rather than the M–vK mechanism. Therefore, our findings are applicable to the case for M2CO2, which once again supports the conclusion that ΔGVaf can serve as the descriptor for COOR catalytic activity.
Conclusions
By investigating the COOR catalytic activity of 18 M2B2O2 with first-principles methods, hex-Mo2B2O2, orth-Mo2B2O2, hex-V2B2O2, and hex-Cr2B2O2 were found to possess high COOR catalytic activity via the M–vK mechanism and high thermal stability under 1000 K. Hex-Mo2B2O2 is expected to serve as the substrate of COOR catalysts as an alternative to CeO2, and orth-Mo2B2O2, hex-V2B2O2, and hex-Cr2B2O2 are expected to be used as direct COOR catalysts with higher activity. Furthermore, it was found that the formation energy of O vacancies (ΔGVaf) can reflect the value of maximum Gibbs free energy change (ΔGmax) for COOR catalysis and serve as the descriptor for COOR catalytic activity. In general, materials with lower ΔGVaf have lower ΔGmax. Thus, during the process of COOR catalyst screening, only ΔGmax needs to be calculated, thereby saving large amounts of calculation cost and contributing to high screening efficiency.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 51871009 and 51872017).References
- S. Jiménez, J. Soler, R. X. Valenzuela and L. Daza, J. Power Sources, 2005, 151, 69–73 CrossRef.
- J. J. Baschuk and X. Li, Int. J. Energy Res., 2001, 25, 695–713 CrossRef CAS.
- H. J. Kim, M. G. Jang, D. Shin and J. W. Han, ChemCatChem, 2020, 12, 11–26 CrossRef CAS.
- B. K. Min and C. M. Friend, Chem. Rev., 2007, 107, 2709–2724 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- X. Zhang, J. Lei, D. Wu, X. Zhao, Y. Jing and Z. Zhou, J. Mater. Chem. A, 2016, 4, 4871–4876 RSC.
- Y. Li, Z. Zhou, G. Yu, W. Chen and Z. Chen, J. Phys. Chem. C, 2010, 114, 6250–6254 CrossRef CAS.
- G. Spezzati, A. D. Benavidez, A. T. DeLaRiva, Y. Su, J. P. Hofmann, S. Asahina, E. J. Olivier, J. H. Neethling, J. T. Miller, A. K. Datye and E. J. M. Hensen, Appl. Catal., B, 2019, 243, 36–46 CrossRef CAS.
- P. Schlexer, D. Widmann, R. J. Behm and G. Pacchioni, ACS Catal., 2018, 8, 6513–6525 CrossRef CAS.
- C. Cheng, X. Zhang, Z. Yang and Z. Zhou, ACS Appl. Mater. Interfaces, 2018, 10, 32903–32912 CrossRef CAS PubMed.
- Z. Jiang, P. Wang, X. Jiang and J. Zhao, Nanoscale Horiz., 2018, 3, 335–341 RSC.
- F. Li, Y. Li, X. C. Zeng and Z. Chen, ACS Catal., 2015, 5, 544–552 CrossRef CAS.
- Y. Lou, Y. Cai, W. Hu, L. Wang, Q. Dai, W. Zhan, Y. Guo, P. Hu, X.-M. Cao, J. Liu and Y. Guo, ACS Catal., 2020, 10, 6094–6101 CrossRef CAS.
- Y.-Q. Su, I. A. W. Filot, J.-X. Liu and E. J. M. Hensen, ACS Catal., 2018, 8, 75–80 CrossRef CAS PubMed.
- X.-m. Zhang, P. Tian, W. Tu, Z. Zhang, J. Xu and Y.-F. Han, ACS Catal., 2018, 8, 5261–5275 CrossRef CAS.
- C. Cheng, X. Zhang, Z. Yang and K. Hermansson, Adv. Theory Simul., 2019, 2, 1900006 CrossRef.
- S. Y. Hwang, E. Yurchekfrodl, C. Zhang and Z. Peng, ChemCatChem, 2016, 8, 97–101 CrossRef CAS.
- X. Zhang, C. Xu, Y. Zhang, C. Cheng, Z. Yang and K. Hermansson, Int. J. Hydrogen Energy, 2021, 46, 8477–8485 CrossRef CAS.
- J.-X. Liu, Y. Su, I. A. W. Filot and E. J. M. Hensen, J. Am. Chem. Soc., 2018, 140, 4580–4587 CrossRef CAS PubMed.
- M. Moses-DeBusk, M. Yoon, L. F. Allard, D. R. Mullins, Z. Wu, X. Yang, G. Veith, G. M. Stocks and C. K. Narula, J. Am. Chem. Soc., 2013, 135, 12634–12645 CrossRef CAS PubMed.
- A. J. Therrien, A. J. R. Hensley, M. D. Marcinkowski, R. Zhang, F. R. Lucci, B. Coughlin, A. C. Schilling, J.-S. McEwen and E. C. H. Sykes, Nat. Catal., 2018, 1, 192–198 CrossRef CAS.
- E. J. Peterson, A. T. DeLaRiva, S. Lin, R. S. Johnson, H. Guo, J. T. Miller, J. Hun Kwak, C. H. F. Peden, B. Kiefer, L. F. Allard, F. H. Ribeiro and A. K. Datye, Nat. Commun., 2014, 5, 4885 CrossRef CAS PubMed.
- C. Cheng, X. Zhang, M. Wang, S. Wang and Z. Yang, Phys. Chem. Chem. Phys., 2018, 20, 3504–3513 RSC.
- Y.-L. Song, L.-L. Yin, J. Zhang, P. Hu, X.-Q. Gong and G. Lu, Surf. Sci., 2013, 618, 140–147 CrossRef CAS.
- G. Glaspell, H. M. A. Hassan, A. Elzatahry, V. Abdalsayed and M. S. El-Shall, Top. Catal., 2008, 47, 22–31 CrossRef CAS.
- Y. Feng, Q. Wan, H. Xiong, S. Zhou, X. Chen, X. I. Pereira Hernandez, Y. Wang, S. Lin, A. K. Datye and H. Guo, J. Phys. Chem. C, 2018, 122, 22460–22468 CrossRef CAS.
- H. A. Tahini, X. Tan and S. C. Smith, ChemCatChem, 2020, 12, 1007–1012 CrossRef CAS.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS PubMed.
- Y. Wei, P. Zhang, R. A. Soomro, Q. Zhu and B. Xu, Adv. Mater., 2021, 33, 2103148 CrossRef CAS PubMed.
- Z. Guo, J. Zhou and Z. Sun, J. Mater. Chem. A, 2017, 5, 23530–23535 RSC.
- B. Zhang, J. Zhou and Z. Sun, J. Mater. Chem. A, 2022, 10, 15865–15880 RSC.
- B. Zhang, J. Zhou, Z. Guo, Q. Peng and Z. Sun, Appl. Surf. Sci., 2020, 500, 144248 CrossRef CAS.
- T. Zhang, B. Zhang, Q. Peng, J. Zhou and Z. Sun, J. Mater. Chem. A, 2021, 9, 433–441 RSC.
- B. Li, Y. Wu, N. Li, X. Chen, X. Zeng, Arramel, X. Zhao and J. Jiang, ACS Appl. Mater. Interfaces, 2020, 12, 9261–9267 CrossRef CAS PubMed.
- F. Li and Q. Tang, ACS Appl. Nano Mater., 2019, 2, 7220–7229 CrossRef CAS.
- E. Wang, B. Zhang, J. Zhou and Z. Sun, Appl. Surf. Sci., 2022, 604, 154522 CrossRef CAS.
- X. Guo, S. Lin, J. Gu, S. Zhang, Z. Chen and S. Huang, Adv. Funct. Mater., 2021, 31, 2008056 CrossRef CAS.
- Y. Xiao, C. Shen and T. Long, Chem. Mater., 2021, 33, 4023–4034 CrossRef CAS.
- X. Yang, C. Shang, S. Zhou and J. Zhao, Nanoscale Horiz., 2020, 5, 1106–1115 RSC.
- X. Liu, Z. Liu and H. Deng, J. Phys. Chem. C, 2021, 125, 19183–19189 CrossRef.
- Y. Xiao, C. Shen and N. Hadaeghi, J. Phys. Chem. Lett., 2021, 12, 6370–6382 CrossRef CAS PubMed.
- M. Yao, Z. Shi, P. Zhang, W.-J. Ong, J. Jiang, W.-Y. Ching and N. Li, ACS Appl. Nano Mater., 2020, 3, 9870–9879 CrossRef CAS.
- S. Xing, J. Zhou, B. Zhang and Z. Sun, J. Phys. Chem. C, 2022, 126, 14275–14282 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- S. Qi, Y. Fan, L. Zhao, W. Li and M. Zhao, Appl. Surf. Sci., 2021, 536, 147742 CrossRef CAS.
- T. Bo, P.-F. Liu, J. Xu, J. Zhang, Y. Chen, O. Eriksson, F. Wang and B.-T. Wang, Phys. Chem. Chem. Phys., 2018, 20, 22168–22178 RSC.
- N. Miao, J. Wang, Y. Gong, J. Wu, H. Niu, S. Wang, K. Li, A. R. Oganov, T. Tada and H. Hosono, Chem. Mater., 2020, 32, 6947–6957 CrossRef CAS.
- K. Rasoul, K. Mohammad, W. Vei, M. Nanxi, S. Chen, W. Jianfeng and W. Junjie, J. Phys.: Condens. Matter, 2020, 33, 155503 Search PubMed.
- M. Zafari, A. S. Nissimagoudar, M. Umer, G. Lee and K. S. Kim, J. Mater. Chem. A, 2021, 9, 9203–9213 RSC.
- J. Zhou, J. Palisaitis, J. Halim, M. Dahlqvist, Q. Tao, I. Persson, L. Hultman, P. O. Å. Persson and J. Rosen, Science, 2021, 373, 801–805 CrossRef CAS PubMed.
- J. Wang, T.-N. Ye, Y. Gong, J. Wu, N. Miao, T. Tada and H. Hosono, Nat. Commun., 2019, 10, 2284 CrossRef PubMed.
- T. Hu, M. Wang, X. Wang, Y. Zhou and C. Li, Comput. Mater. Sci., 2021, 199, 110810 CrossRef CAS.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
- W. Sun, S. A. Shah, Y. Chen, Z. Tan, H. Gao, T. Habib, M. Radovic and M. J. Green, J. Mater. Chem. A, 2017, 5, 21663–21668 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr05705k |
| This journal is © The Royal Society of Chemistry 2023 |