
Control of the stepwise self-assembly process of a pH-responsive amphiphilic 4-aminoquinoline-tetraphenylethene conjugate†
Yosuke
Hisamatsu
 *a,
Fangzhou
Cheng
b,
Katsuhiro
Yamamoto
c,
Hiroshi
Takase
d,
Naoki
Umezawa
a and
Tsunehiko
Higuchi
*a
*a,
Fangzhou
Cheng
b,
Katsuhiro
Yamamoto
c,
Hiroshi
Takase
d,
Naoki
Umezawa
a and
Tsunehiko
Higuchi
*a
aGraduate School of Pharmaceutical Sciences, Nagoya City University, 3-1 Tanabe-dori, Mizuho-ku, Nagoya 467-8603, Japan. E-mail: hisamatsu@phar.nagoya-cu.ac.jp; higuchi@phar.nagoya-cu.ac.jp
bFaculty of Pharmaceutical Sciences, Nagoya City University, 3-1 Tanabe-dori, Mizuho-ku, Nagoya 467-8603, Japan
cGraduate School of Engineering, Nagoya Institute of Technology, Gokiso-cho, Showa-ku, Nagoya 466-8555, Japan
dGraduate School of Medical Sciences, Nagoya City University, 1 Kawasumi, Mizuho-cho, Mizuho-ku, Nagoya 467-8601, Japan
First published on 19th January 2023
Abstract
Controlling the kinetic processes of self-assembly and switching their kinetic properties according to the changes in external environments are crucial concepts in the field of supramolecular polymers in water for biological and biomedical applications. Here we report a new self-assembling amphiphilic 4-aminoquinoline (4-AQ)-tetraphenylethene (TPE) conjugate that exhibits kinetically controllable stepwise self-assembly and has the ability of switching its kinetic nature in response to pH. The self-assembly process of the 4-AQ amphiphile comprises the formation of sphere-like nanoparticles, a transition to short nanofibers, and their growth to long nanofibers with ∼1 μm length scale at room temperature (RT). The timescale of the self-assembly process differs according to the pH-responsivity of the 4-AQ moiety in a weakly acidic to neutral pH range. Therefore, after aging for 24 h at RT, the 4-AQ amphiphile forms metastable short nanofibers at pH 5.5, while it forms thermodynamically favored long nanofibers at pH 7.4. Moreover, the modulation of nanofiber growth proceeding spontaneously at RT was achieved by switching the kinetic pathway through changing the pH between 7.4 and 5.5.
Introduction
Self-assembly is a promising strategy for the construction of supramolecular polymers with sophisticated structures through non-covalent interactions.1 Supramolecular polymers constructed in water2 have great potential for a broad range of biological and biomedical applications including drug delivery systems,3 tissue engineering,4 sensing systems,5 and contrast agents.6Responses to external stimuli (e.g., pH, temperature, light irradiation, ionic strength, chemical reactions) are fundamental attractive features of molecular assemblies.7 The shape of pH-sensitive supramolecular nanostructures is controlled by the protonation and deprotonation of building blocks.8 Slightly acidic extracellular microenvironments in tumors are targets for cancer therapy.9 Supramolecular nanocarriers that respond at acidic organelles such as endosomes (early endosomes: pH ∼6.3; late endosomes: pH ∼5.5) and lysosomes (pH ∼4.7) via endocytic pathways in cells are used as drug delivery systems.10 The pH-sensitive supramolecular polymers formed from monomer building blocks containing acidic and/or basic amino acid residues have been widely studied,11,12 and the self-assembly behavior of their charged building blocks can be regulated as a function of ionic strength.12,13 Such pH- and ionic strength-sensitive self-assembly systems are ubiquitous in nature, for instance the tobacco mosaic virus coat protein14 and amyloid fibrils,15 and through different strategies the fine-tuned design can functionalize the self-assembled nanostructure.
Traditionally, the construction of supramolecular polymers has focused on their thermodynamic equilibrium states.16 Recently, kinetically controlled self-assembly has received considerable attention.17 Kinetic control of the self-assembly can achieve greater control over the size and shape of supramolecular polymers17,18 and may thus be a promising strategy for developing supramolecular materials with desired properties. The kinetic processes of the self-assembly of various artificial monomer building blocks have been investigated in aqueous media.2i,19–23 Rybtchinski and co-workers reported a kinetically controlled self-assembly of a perylene diimide/peptide conjugate that can form three supramolecular polymers with different shapes in a stepwise manner in selected water/THF mixed solvent systems.20 Moreover, depending on the protocols via solvent processing, three self-assembled nanostructures were obtained under the same final conditions.
Self-assembling molecules that can readily control and access the kinetic processes of their self-assemblies through simple preparation protocols under biologically relevant conditions are promising candidates as supramolecular materials for biological and biomedical applications. Stupp and co-workers studied a peptide amphiphile having two distinct energy landscapes dependent on the ionic strength of aqueous solution.21 Besenius and co-workers reported a kinetically controlled stepwise self-assembly of Au(I)-metallopeptides in water.22 More recently, Fernández and co-workers reported a pH-responsive peptide amphiphile that exhibits kinetically controlled self-assembly at pH 7.4.23 In addition, modulations in pH, concentration and temperature allow the proceeding of diverse self-assembly pathways of the peptide amphiphile, resulting in the formation of six distinct nanostructures.23
Despite the recent achievements of kinetically controlled self-assembly systems in aqueous media,2i,19–23 the development of artificial monomer building blocks that exhibit kinetically controlled stepwise self-assembly and have the ability to switch their kinetic nature in response to the change in external environments under biologically relevant conditions is still a challenge.
In this study, we report tetraphenylethene (TPE)-conjugated 4-aminoquinoline (4-AQ) amphiphile 1 (Fig. 1) as a new self-assembling molecule exhibiting a kinetically controlled stepwise self-assembly process in water with 0.10 M NaCl (containing 0.5% (v/v) organic solvent) at pH 5.5 and 7.4. The self-assembly process of 1 comprises the formation of sphere-like nanoparticles (metastable product), an irreversible transition to short nanofibers (metastable product), and their growth to long nanofibers (thermodynamic product) at RT (25 ± 1 °C). The 4-AQ moiety of 1 shows the pH-responsivity in a weakly acidic to neutral pH range in the self-assembled states, and the timescale of the stepwise self-assembly process differs according to pH. Therefore, after aging for 24 h at RT, 1 formed metastable short nanofibers at pH 5.5 and formed thermodynamically favored long nanofibers with ∼1 μm length scale at pH 7.4. The outcome of the supramolecular polymerization suggests that the protonation and deprotonation of the 4-AQ moiety affect the timescale of each self-assembly step at RT. Also, the modulation (i.e., suppression and reacceleration) of the nanofiber growth of 1 proceeding spontaneously was achieved at RT by switching the kinetic pathway through changing the pH between 7.4 and 5.5. To the best of our knowledge, 4-AQ derivatives have not been used as a component of the self-assembling molecule for the creation of self-assembled nanofibers. Furthermore, the kinetically controlled stepwise self-assembly and switching its kinetic nature at RT in a biologically relevant pH range24 are unique properties of 1.
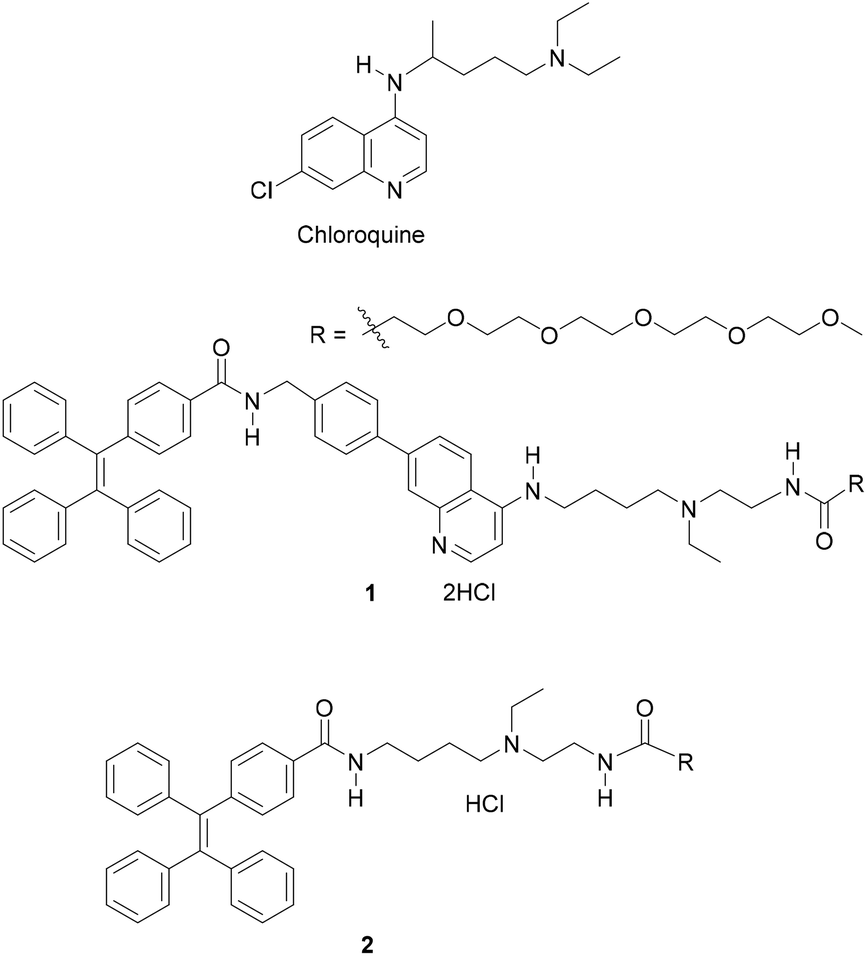 | ||
| Fig. 1 Structures of chloroquines 1 and 2. | ||
Results and discussion
Molecular design and synthesis
Our background to start this study is described. Previously, we have addressed the development of tweezer-type synthetic receptors for heme in aqueous media.25 Based on the knowledge that 4-AQ antimalarial agents,26 such as chloroquine (Fig. 1), exhibit the heme-binding ability,27 the synthetic receptors consist of two 4-AQ moieties bearing tertiary aliphatic amines at the terminal of their side chains (Fig. S1†).25 In that study, we found that a synthetic receptor bearing sulfo-Cy5 (Fig. S1b†) exhibits a change in the fluorescence intensity between pH 6 and 8 probably due to the self-assembly through the protonation and deprotonation of the 4-AQ moiety,25b although precise details were not determined. This finding motivated us to use 4-AQs as a component of self-assembling molecules to create supramolecular architectures with pH responsivity between weakly acidic to neutral pH conditions.9,10,24Compound 1 consists of tetraphenylethene (TPE), the 4-AQ moiety, a tertiary aliphatic amine moiety, and a tetraethylene glycol chain. The TPE and 4-AQ moieties were anticipated to contribute to self-assembly through non-covalent interactions including π–π stacking and hydrophobic interactions.25 Since 1 can exist in its neutral, monoprotonated, and diprotonated forms (Scheme 1), the pH-dependent self-assembly behavior of 1 was expected.
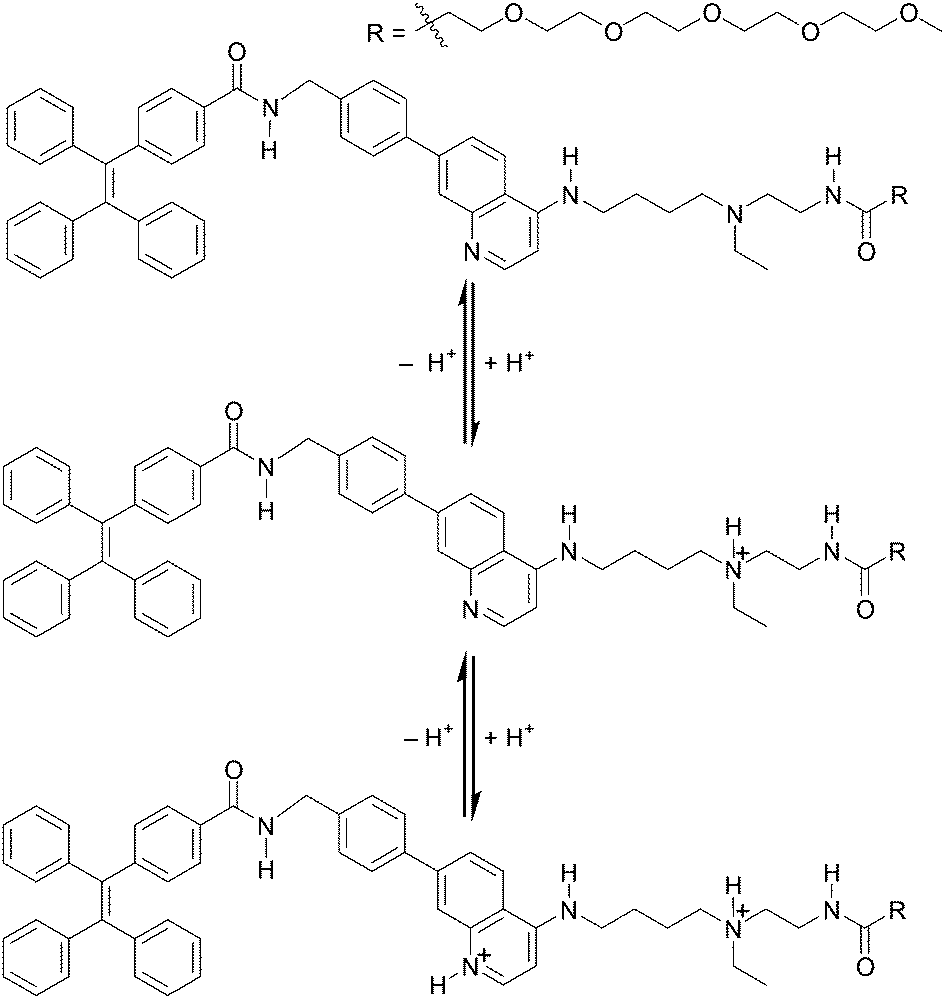 | ||
| Scheme 1 Structures of 1 in its neutral, monoprotonated, and diprotonated forms. | ||
The protonation of the 4-AQ moiety of 1 under acidic pH may prevent its self-assembly due to the electrostatic repulsion between diprotonated species. The pKa values of the protonated forms of quinoline and 4-AQ are reported to be 4.9 and 9.2, respectively.28 An increase in the pKa value is attributed to the effect of the amino group at the 4-position of the quinoline ring. The pKa values of chloroquine (Fig. 1) are reported to be 8.4 and 10.2 for the 4-AQ and tertiary amine moieties, respectively.29 Although the pKa value of the protonated 4-AQ appears to be too high for targeting the biologically relevant pH range,24 a shift in the apparent pKa value was expected due to the self-assembled nanostructure formation.25b On the other hand, salts such as NaCl can screen the repulsive force between charged species.12,13 Although the effect of salts on the protonated 4-AQ moiety is unknown, they may contribute to the self-assembly process of 1.
TPE, an aggregation induced emission (AIE) luminogen,30 was introduced at the hydrophobic part of 1. Conventional fluorescence dyes generally suffer from fluorescence quenching when exhibiting aggregated states. TPE exhibits strong fluorescence emission in aggregates due to the restriction of intramolecular rotation and is thus useful to detect self-assembly and future biological applications.11g,13b,19g,30
The synthesis of compound 1 was conducted according to Scheme S1.† Compound 2 without the 4-AQ moiety was also prepared as a reference compound (Scheme S2†).
Evaluation of the initial stage of self-assembly behavior based on AIE
Firstly, the initial stage of the self-assembly process of 1 was evaluated based on its AIE properties. The fluorescence of 1 (10 μM) corresponding to the AIE from TPE was observed at a water fraction (HEPES buffer, pH 7.4) of 80% (v/v) or greater (Fig. S2†).31 The result indicates that the self-assembly of 1 proceeded in high amounts of water fractions and suggests that its self-assembly is mainly driven by the hydrophobic interaction.The pH-dependent self-assembly of 1 (10 μM) was investigated using the fluorescence spectral measurement in 10 mM Good's buffer (Fig. 2a).‡ The fluorescence intensity of 1 at ca. 474 nm was very weak at pH 5.5; however, its fluorescence intensity increased dramatically at pH 6.0 and above due to self-assembly phenomena. Compound 1 exhibited a pH-dependent change in UV-Vis absorption spectra (Fig. S3a†), and the absorbance of 1 at 330 nm decreased when the pH value of solutions was increased (from pH 5.5 to 7.8). Changes in absorption spectra include the protonation and deprotonation of the 4-AQ moiety, and the apparent pKa value of the 4-AQ moiety of 1 was estimated to be approximately 6.3. This interpretation was supported by the pH-dependent change in the UV-Vis absorption spectra through the protonation of the 4-AQ moiety of chloroquine32 and our previous results.25 The shift in the apparent pKa value of the protonated 4-AQ28,29,32 of 1 (Fig. S3a†) is probably due to the self-assembly effects. Therefore, it is suggested that 1 exists mostly in its diprotonated form at pH 5.5, under which conditions electrostatic repulsion between the positively charged 4-AQ moieties disfavors self-assembly. At pH 6.0 or above, the self-assembly proceeded due to the decreasing repulsion force and increasing hydrophobicity as the amount of diprotonated species decreased. At pH 7.4, the major species of 1 should be the monoprotonated form based on the pH-dependent changes in the fluorescence and UV-Vis absorption spectra.
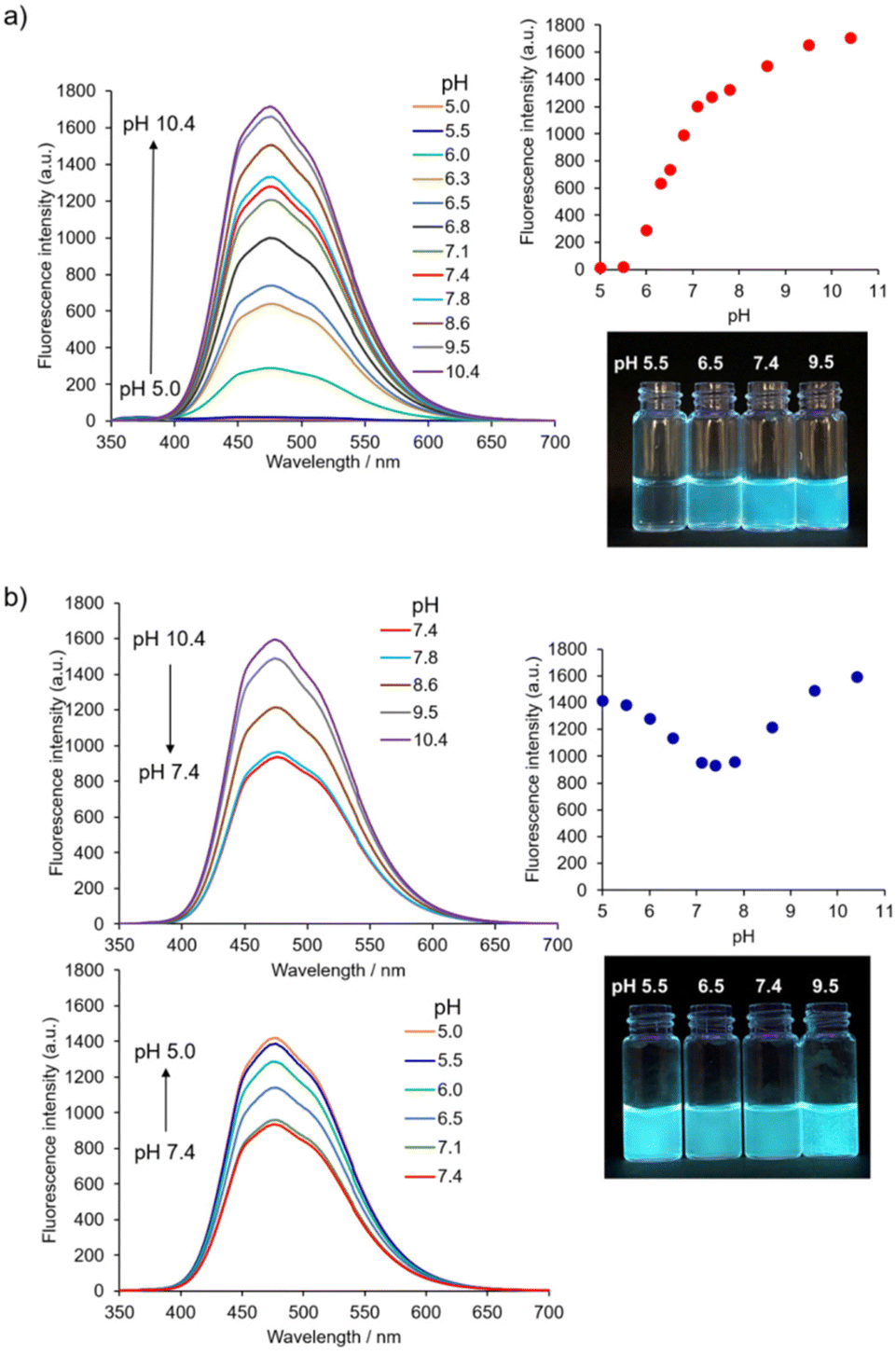 | ||
| Fig. 2 pH-dependent change in the fluorescence spectra of (a) 1 and (b) 1 with 0.10 M NaCl. Insets: plots of the pH-dependent change in the fluorescence intensity of 1 at 474 nm. Photographs show the fluorescence emission of 1 at various pH values. Ex. 365 nm. Conditions: [1] = 10 μM, 10 mM buffer (from pH 5.0 to 10.4) containing 0.5% (v/v) DMSO, 25 °C, Ex. 330 nm. | ||
The effect of ionic strength on self-assembly12,13 must be considered especially for biological and biomedical applications. The pH-dependent changes in the fluorescence spectra of 1 (10 μM) with 0.10 M NaCl are shown in Fig. 2b. The AIE of 1 was observed in all the measured pH range. Its fluorescence emission maximum was ca. 474 nm, i.e., the same as those in the absence of NaCl (Fig. 2a). When the pH value was reduced from pH 10.4 to 7.4, the fluorescence intensity of 1 also decreased. In contrast, as pH values decreased from pH 7.4 to 5.0, the fluorescence intensity of 1 increased, indicating the facilitation of self-assembly by NaCl at weakly acidic pH.
The pH-dependent change in the UV-Vis absorption spectra (Fig. S3b†) suggests that 1 mostly exists in its diprotonated form at pH 5.5,25,32 and that electrostatic repulsion was alleviated by the electrostatic screening effect imparted by 0.10 M NaCl.12,13 The absorbance of 1 at 330 nm continuously decreased when increasing the pH from 5.5 to 7.8. This change suggests that the major species of 1 at pH 7.4 was its monoprotonated species. However, it was not possible to estimate the apparent pKa value of the protonated 4-AQ of 1 because its absorption spectra were affected by scattering of the aggregates formed at alkaline pH.
As anticipated, these results imply that 1 shows different self-assembly behaviors between weakly acidic pH and neutral pH, which is a biologically relevant pH range.9,10,24 In this study, we have focused on pH 5.5 as a weakly acidic condition and pH 7.4 as a neutral condition. Based on the concentration-dependent change in the AIE of 1 (Fig. S4†), the critical aggregation concentration (CAC) values of the initial stage of the self-assembly process were estimated to be 18 ± 1 μM at pH 5.5 and 0.4 ± 0.1 μM at pH 7.4, respectively.33 Determination of the CAC values of 1 with 0.10 M NaCl at pH 5.5 and 7.4 by the AIE was unsuccessful because of the low values of CAC. Thus, CAC values were estimated at an upper limit of 0.4 μM which is more than 45-fold lower than that at pH 5.5 without NaCl (Table 1).34
The contribution of the 4-AQ moiety to the self-assembly was evaluated by comparison with the reference compound 2 without the 4-AQ moiety. Fig. 3 shows the fluorescence spectra of 2 (10 μM) with 0.10 M NaCl at various pH values.‡![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) § The fluorescence intensity of 2 at pH 5.5 remained very low and increased at pH 7.8 or higher. The apparent pKa value of the protonated tertiary aliphatic amine moiety of 2 was estimated to be approximately 8.2. Considering the apparent pKa value of 2, the deprotonation of the protonated tertiary aliphatic amine moiety of 1 occurs at alkaline pH, and an increase in the amount of the neutral form of 1 (Scheme 1) should facilitate further aggregation (Fig. 2 and Fig S3†). The CAC values of 2 with 0.10 M NaCl at pH 5.5 and 7.4 were more than 47-fold and 19-fold higher than those of 1, respectively (Table 1 and Fig. S7†). It clearly shows that the introduction of the 4-AQ moiety enhances the ability of self-assembly in comparison with 2.
§ The fluorescence intensity of 2 at pH 5.5 remained very low and increased at pH 7.8 or higher. The apparent pKa value of the protonated tertiary aliphatic amine moiety of 2 was estimated to be approximately 8.2. Considering the apparent pKa value of 2, the deprotonation of the protonated tertiary aliphatic amine moiety of 1 occurs at alkaline pH, and an increase in the amount of the neutral form of 1 (Scheme 1) should facilitate further aggregation (Fig. 2 and Fig S3†). The CAC values of 2 with 0.10 M NaCl at pH 5.5 and 7.4 were more than 47-fold and 19-fold higher than those of 1, respectively (Table 1 and Fig. S7†). It clearly shows that the introduction of the 4-AQ moiety enhances the ability of self-assembly in comparison with 2.
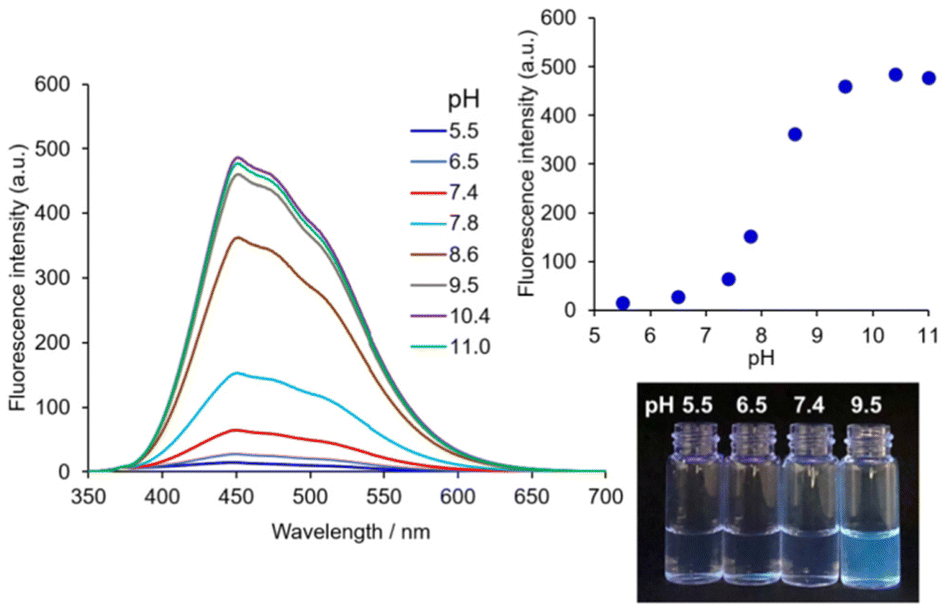 | ||
| Fig. 3 pH-dependent change in the fluorescence spectra of 2 with 0.10 M NaCl. Inset: a plot of the pH-dependent change in the fluorescence intensity of 2 at 450 nm. Conditions: [2] = 10 μM, 10 mM buffer (from pH 5.5 to 11) containing 0.5% (v/v) DMSO, 25 °C, Ex. 330 nm. A photograph shows the fluorescence emission of 2 at various pH values with 0.10 M NaCl. Ex. 365 nm. | ||
Although the electrostatic repulsion of 2 (monoprotonated species) at pH 5.5 appeared to be weaker than that of 1 (diprotonated species), the effect of NaCl on the self-assembly was greater for 1 than 2. When combined with the results of the stronger fluorescence intensity of 1 at pH 5.5 than at pH 7.4 (Fig. 2b), 0.10 M NaCl may further facilitate the self-assembly of 1 with the protonated 4-AQ moiety in addition to alleviating electrostatic repulsion.
Nanofiber formation and characterization of the self-assembly process
The initial stage of the self-assembly behavior of 1 evaluated by the AIE reveals that 1 is a more promising self-assembling molecule with responsivity to pH and NaCl than 2. Next, we focused on the characterization of supramolecular architectures created by 1 with 0.10 M NaCl at pH 5.5 and 7.4.The self-assembly behavior of 1 was investigated using transmission electron microscopy (TEM), dynamic light scattering (DLS), UV-Vis absorption spectroscopy, and small angle X-ray scattering (SAXS). The self-assembled nanostructures formed from 1 with 0.10 M NaCl at pH 5.5 were observed using negative-stain TEM (Fig. 4). The concentration of 1 used, 0.05 mM, is higher than the CAC values (Table 1). As shown in Fig. 4a, many short 1-dimensional (1D) nanofibers (worm-like micelles) of 1 were observed after 1 h at RT (25 ± 1 °C). The length distribution of these nanofibers was mostly less than 300 nm (Ln = 102 nm, Lw = 138 nm, PDI = 1.4) (Fig. 4f and Fig. S8†), and their widths were estimated to be approximately 8 nm (Fig. 4b). When the sample solution was aged for 24 h, nanofiber growth was very slow (Ln = 130 nm, Lw = 186 nm, PDI = 1.4) (Fig. 4c, g and Fig. S9†). Long nanofibers of around 1 μm in length were observed after aging for 7 days (Fig. 4d and Fig. S10†). Such nanofibers were observed, when the sample solution was heated at 80 °C for 30 min, followed by cooling to RT (Fig. S11†), while heating at 60 °C for 5 min was found to be insufficient (Fig. S12†). These results indicate that short nanofibers observed after aging for 1 h and 24 h are still in their metastable states and that the subsequent growth process toward long nanofibers is very slow probably due to the large energy barrier. We found that 1 initially formed sphere-like nanoparticles (2 min after sample preparation) (Fig. 4e and Fig. S13†), indicating that the transition of sphere-like nanoparticles to short nanofibers was rapid. The acceleration of the nanofiber formation of 1 by 0.10 M NaCl at pH 5.5 was confirmed by TEM observation of 1 in the absence of NaCl (Fig. S14†). In contrast to 1, the TEM images of 2 (0.05 mM) after aging for 1 h in the presence of 0.10 M NaCl show that the frequency of the nanostructures observed on the TEM grids was very low (Fig. S15†).
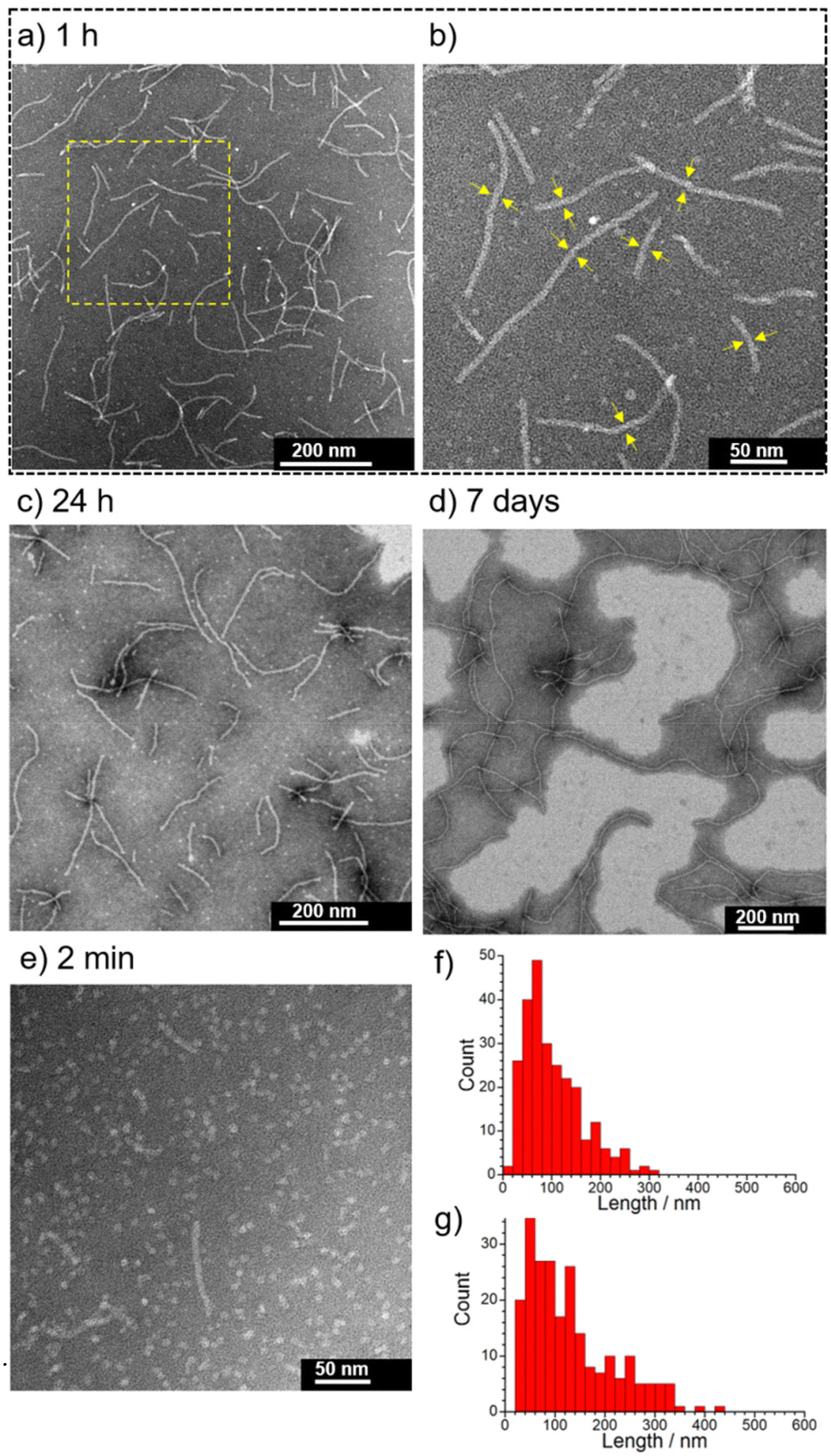 | ||
| Fig. 4 (a) Representative negative stain TEM images of 1 with 0.10 M NaCl at pH 5.5 after aging for 1 h and (b) high-magnification image showing the region of the yellow-dashed square in (a). The yellow arrows in (b) indicate the widths of the nanofibers (approximately 8 nm). Representative negative stain TEM images of 1 with 0.10 M NaCl at pH 5.5 after aging for (c) 24 h, (d) 7 days and (e) 2 min. The corresponding histogram of the TEM images after aging for (f) 1 h and (g) 24 h. Conditions: [1] = 0.05 mM, 10 mM MES buffer (pH 5.5, 0.10 M NaCl) containing 0.5% (v/v) MeOH. | ||
At pH 7.4 in the presence of 0.10 M NaCl, sphere-like nanoparticles were observed after aging for 30 min (Fig. 5a); the average hydrodynamic diameter was determined to be 9.4 ± 1.5 nm using DLS measurement (Fig. S16†). TEM observations after aging for 1, 4, 8 and 24 h revealed a transition from sphere-like nanoparticles to 1-D nanofibers (∼4 h), as well as their growth process (Fig. 5b–e and Fig. S17†). The nanofibers observed at 24 h were on the order of ∼1 μm in length (Fig. 5e). The width of the nanofibers was estimated to be approximately 8–9 nm (Fig. 5f), which is similar to that observed at pH 5.5 (Fig. 4b). In contrast to the case at pH 5.5 (Fig. S12†), long nanofibers readily formed at pH 7.4 by heating at 60 °C for 5 min (Fig. S18†).
 | ||
| Fig. 5 (a) Representative negative stain TEM images of 1 with 0.10 M NaCl at pH 7.4 after aging for (a) 30 min, (b) 1 h, (c) 4 h, (d) 8 h, (e) 24 h. (f) High-magnification image showing the region of the yellow-dashed square in (e). Yellow arrows indicate the widths of nanofibers (approximately 8–9 nm). Conditions: [1] = 0.05 mM, 10 mM HEPES buffer (pH 7.4, 0.10 M NaCl) containing 0.5% (v/v) MeOH. | ||
The self-assembly of 1 at pH 7.4 in the absence of NaCl was also investigated. Sphere-like nanoparticles were observed after aging for 1 h at RT, and the average hydrodynamic diameter was determined to be 9.5 ± 0.9 nm using DLS (Fig. S19†). Transitions from sphere-like nanoparticles to branched aggregates and then to short nanofibers were observed after 24 and 72 h, respectively (Fig. S20†), showing that the self-assembly of 1 at pH 7.4 was facilitated by 0.10 M NaCl.
Time-resolved UV-Vis absorption spectra (from 1.5 min to 24 h) of 1 (50 μM) in 10 mM MES buffer (pH 5.5, 0.10 M NaCl) were obtained (Fig. 6a, b and Fig. S21†). The change in the absorbance at 330 nm and 345 nm reached a plateau after aging for approximately 25 min, and the change in the absorption spectra of 1 was negligible after aging for 7 days (Fig. S21d†). On the other hand, the UV-Vis absorption spectra of 1 with 0.10 M NaCl at pH 7.4 (from 1.5 min to 24 h) showed that the change in the absorbance occurred after a lag time between 30 and 60 min, reaching a plateau after around 4 h (Fig. 6c, d and Fig. S22†). Using TEM, we confirmed that sphere-like nanoparticles had already formed within 2 min (Fig. S23†).
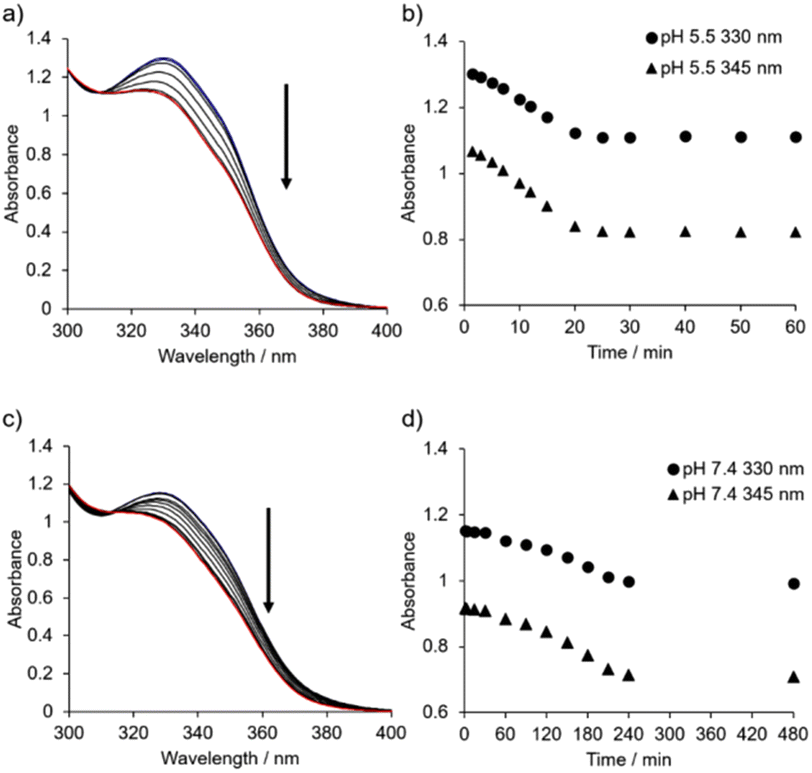 | ||
| Fig. 6 (a) Time-resolved UV-Vis absorption spectra of 1 with 0.10 M NaCl at pH 5.5 from 1.5 (blue line) to 60 min (red line). (b) Time-dependent change in the absorbance at 330 nm (closed circles) and 345 nm (closed triangles) of 1 from 1.5 to 60 min (c) Time-resolved UV-Vis absorption spectra of 1 with 0.10 M NaCl at pH 7.4 from 1.5 (blue line) to 480 min (red line). (d) Time-dependent change in the absorbance at 330 nm (closed circles) and 345 nm (closed triangles) of 1 from 1.5 to 480 min. Conditions: [1] = 50 μM, 10 mM buffer (pH5.5: MES; pH 7.4: HEPES) containing 0.10 M NaCl and 0.5% (v/v) MeOH at 25 °C. | ||
Combining the TEM results shown in Fig. 4, and 5, the timescale of the UV-Vis spectral change suggests a transition from sphere-like nanoparticles to short nanofibers at both pH 5.5 and 7.4, but the growth of nanofibers to reach thermodynamically favored products seems not to be reflected in the spectral change. Furthermore, the UV-Vis absorption spectra (molar absorptivity ε against the corresponding wavelength) of 1 (0.40 mM) at pH 5.5 and 7.4 after aging for 24 h were almost identical to those of 1 (50 μM) (Fig. S24†). These results imply that the products of molecular assemblies after sufficient aging time are biased toward nanofibers.
SAXS measurements of 1 were conducted to complement the TEM data with detailed insights into the shapes of self-assembled nanostructures in solution. A higher concentration (0.5 mM) of 1 was required in order to obtain sufficient scattering intensity. In the case of pH 7.4, the sample was prepared by heating to 60 °C and then cooling to RT to rapidly obtain long nanofibers. The formation of nanofibers from 1 under both pH conditions was confirmed using TEM (Fig. S25 and S26†). As shown in Fig. 7, the SAXS profiles show a power law regime of I ∝ q−1 which indicates the presence of cylinder structures, namely nanofibers. The SAXS profiles were fitted to a homogeneous cylinder model and the values of the core radius (Rcore), corresponding to the radius of nanofibers except for hydrophilic tetraethylene glycol,35 were determined to be 3.5 ± 0.2 nm for pH 5.5 and 3.8 ± 0.2 nm for pH 7.4.
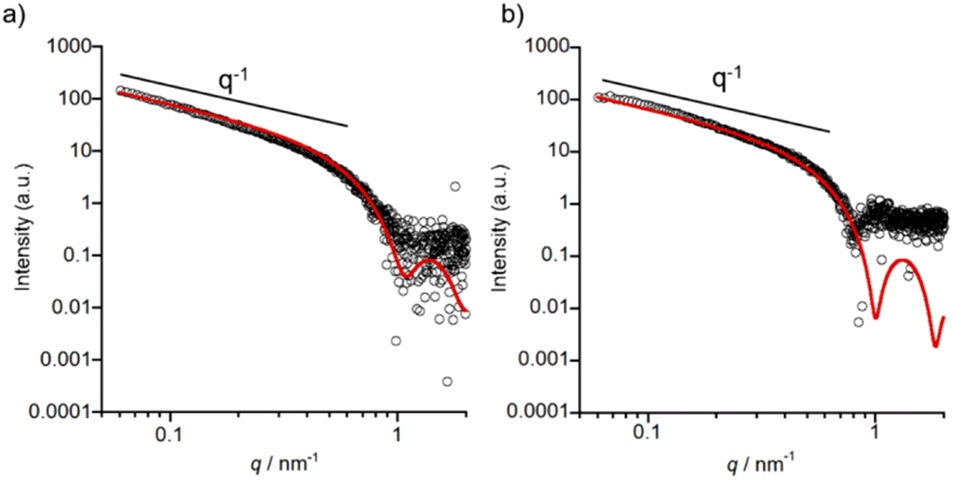 | ||
| Fig. 7 SAXS profile (black open circles) of 1 (a) at pH 5.5 and (b) at pH 7.4 (after heating at 60 °C for 5 min). Fitting curves are shown as a red solid line. Conditions: [1] = 0.5 mM, 10 mM buffer (pH5.5: MES, pH 7.4: HEPES) containing 0.10 M NaCl and 0.5% (v/v) MeOH. | ||
The optimized structures of the monoprotonated and diprotonated forms of 1 were calculated by the density functional theory (DFT) method (Fig. 8a and Fig. S27†). The length of 1 is ca. 5.1 nm, and the length of the core part is ca. 3.5 nm which almost agrees with the Rcore of nanofibers estimated from SAXS experiments. Moreover, the widths of nanofibers measured from the TEM images at pH 5.5 and 7.4 (Fig. 4b and 5f) were approximately 8–9 nm, roughly twice the molecular length of 1 (between 7 nm and 10 nm depending on the stretching of the tetraethylene glycol chain).35 Based on these TEM and SAXS results, a schematic representation of the proposed structure of the 1-D nanofiber is shown in Fig. 8 and Fig. S28.† It is suggested that the non-covalent interactions including the hydrophobic interaction and aromatic interactions36 of TPE and 4-AQ moieties play important roles in the self-assembly of 1.
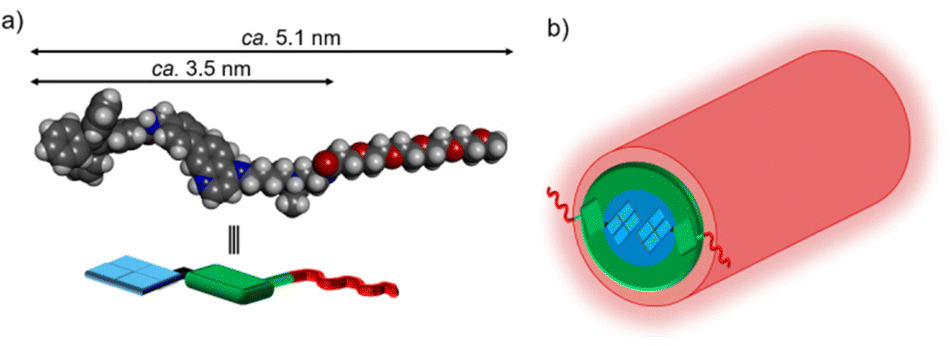 | ||
| Fig. 8 (a) The structure of the diprotonated form of 1 calculated by DFT (ωB97X-D/6-31+G*). (b) The proposed structure of the nanofiber formed from 1. | ||
Time-resolved SAXS measurements (from 10 min to 4 h) of 1 (0.4 mM) at pH 7.4 were conducted to understand the transition from sphere-like nanoparticles to nanofibers (Fig. 9a, b and Fig. S29a†).37 As shown in Fig. 9a, the SAXS profile of 1 at 10 min could be fitted with a homogeneous spherical model, and the core radius (Rcore) of sphere-like nanoparticles was determined to be 3.6 ± 0.7 nm. The scattering intensity increased over time (Fig. S29a†), and the homogeneous cylinder model (Rcore = 3.4 ± 0.7 nm) was found to give a good fitting result for the SAXS profile at 4 h (Fig. 9b). The self-assembly process of 1 (0.4 mM) was confirmed by TEM (Fig. S30†) and DLS (Fig. S31†).
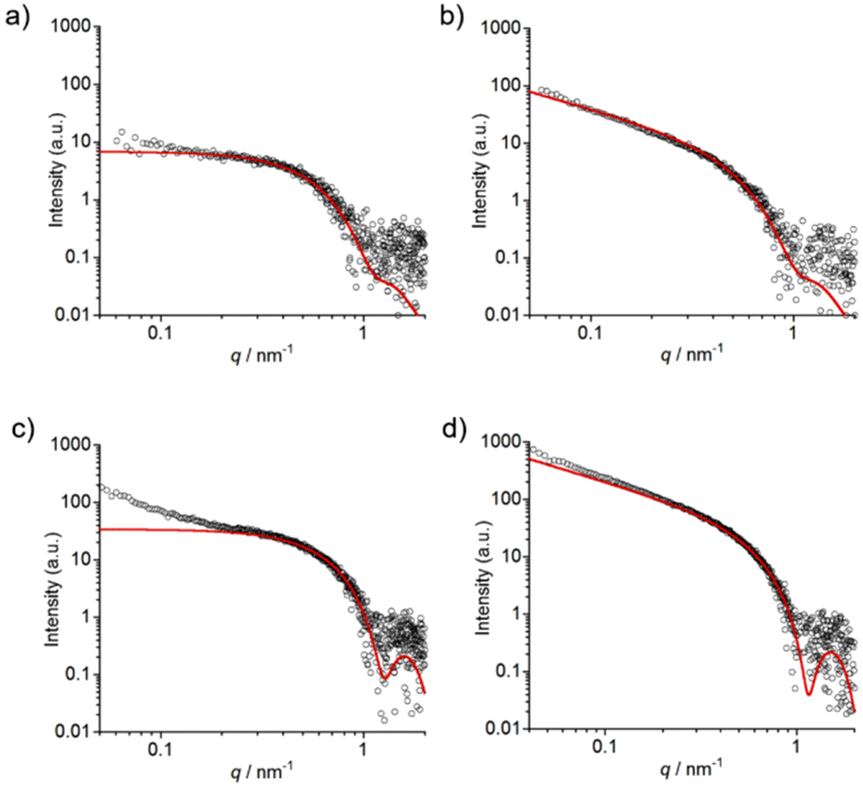 | ||
| Fig. 9 SAXS profiles (black open circles) of 1 with 0.10 M NaCl at pH 7.4 after aging for (a) 10 min and (b) 4 h. SAXS profiles (black open circles) of 1 with 0.10 M NaCl at pH 5.5 after aging for (c) 2 min and (d) 1 h. Fitting curves (red solid lines) using a homogeneous spherical model are shown in (a) and (c). Fitting curves (red solid lines) using a homogeneous cylinder model are shown in (b) and (d). Conditions: [1] = 0.4 mM, 10 mM buffer (pH 7.4: HEPES, pH5.5: MES) containing 0.10 M NaCl and 0.5% (v/v) MeOH. | ||
The time-resolved SAXS profiles of 1 with 0.10 M NaCl at pH 5.5 (from 2 min to 1 h) are shown in Fig. 9c, d and Fig. S29b.† A curve fitting using a homogeneous spherical model (Rcore = 3.5 ± 0.3 nm) shows that the SAXS profile at 2 min includes the profile of sphere-like nanoparticles (Fig. 9c). The upturn in the scattering intensity in the low q region of the profile at 2 min may be due to the production of larger aggregates (e.g., aggregates of sphere-like nanoparticles38 and/or nanofibers) as observed in the TEM images (Fig. S32†). As shown in Fig. 9d, the SAXS profile at 1 h could be fitted with a homogeneous cylinder model (Rcore = 3.3 ± 0.2 nm), and the nanofiber formation was confirmed by TEM (Fig. S32†).
The results of time-resolved SAXS measurements strongly support the transition from sphere-like nanoparticles to nanofibers. The Rcore values were in agreement with the proposed structures of sphere-like nanoparticles and nanofibers formed from 1, respectively (Fig. S27 and S28†).
Fig. 10 shows a schematic representation of the stepwise self-assembly process of 1. The timescale of the self-assembly process at pH 5.5 is different from that at pH 7.4. This result suggests that the protonation and deprotonation of the 4-AQ moiety play an important part in the selection of the self-assembly pathway. The transition of sphere-like nanoparticles to short nanofibers was faster at pH 5.5 (less than 1 h) than at pH 7.4 (∼4 h).¶ In addition to screening the electrostatic repulsion by NaCl,12,13 the chloride ion might contribute to the intermolecular interactions of the protonated 4-AQ moiety and therefore accelerate the self-assembly at pH 5.5.39 Although further studies should be conducted for understanding the mechanism of the transition from sphere-like nanoparticles to nanofibers, one of our hypotheses based on the TEM images (Fig. 5b and Fig. S34†) is that the sphere-like nanoparticles of 1 may join together and then transit to the nanofibers.37,40
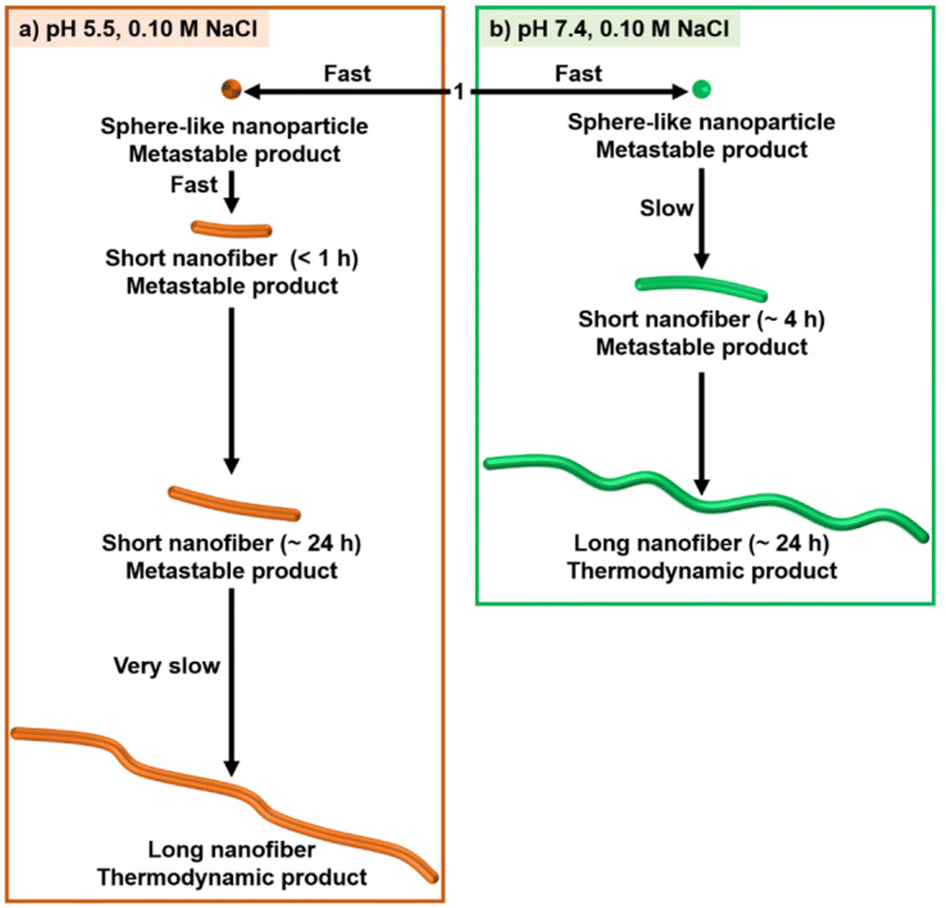 | ||
| Fig. 10 Schematic representation of the stepwise self-assembly process of 1 with 0.10 M NaCl at (a) pH 5.5 and (b) pH 7.4. Total aging times after the sample preparation shown in brackets were estimated based on the TEM results. | ||
On the other hand, the formation of long nanofibers, which was considered to be the thermodynamically favored product, at pH 7.4 was much faster (∼24 h) than that at pH 5.5 (∼7 days or more). At pH 5.5, the exquisite balance of the electrostatic repulsion and electrostatic screening effect by 0.10 M NaCl should be important for determining the length of nanofibers and their growth processes.13a Indeed, the lengths of nanofibers after aging for 1 h were partly elongated (Ln = 157 nm, Lw = 243 nm, PDI = 1.6) when the concentration of NaCl was increased to 0.20 M (Fig. S35†).
The outcome of the different self-assembly processes at RT suggests that the protonation and deprotonation of the 4-AQ moiety affect the timescale of each self-assembly step and do not affect the shape of the nanostructures created.
Switching of the self-assembly pathway through pH change
The metastable assemblies formed from 1 in a stepwise manner are readily available at RT. We anticipated that the self-assembly properties that affect the timescale of each self-assembly step can be applied for the modulation of the nanofiber growth proceeding spontaneously at RT by switching the self-assembly pathway through pH change.Therefore, switching the self-assembly pathway of 1 through pH changes was examined (Fig. 11A–D, Fig. S36 and S37†). Metastable sphere-like nanoparticles were primarily observed at pH 7.4 (in 1 h; see A in Fig. 11). When the pH was adjusted to 5.5, followed by aging for 1 h, short nanofibers were formed (B in Fig. 11). The growth of nanofibers remained to be suppressed after aging for an additional 23 h at pH 5.5 (C in Fig. 11). Subsequently, nanofiber growth was reaccelerated by the readjustment of pH at 7.4, and long nanofibers were formed after 24 h (D in Fig. 11). The pH-dependent self-assembly process was traced by changes in the UV-Vis absorption spectra (details in Fig. S38†). After the readjustment of pH from 5.5 (C in Fig. 11) to 7.4 and then aging for 30 min, short nanofibers were observed in the TEM images (Fig. S37†). This result suggests that the growth of short nanofibers restarted without returning to the formation of sphere-like nanoparticles (A in Fig. 11) and switching the self-assembly pathway occurred between the metastable short nanofibers at pH 5.5 and 7.4 (Fig. S37†). This interpretation was also supported by the change in the UV-Vis absorption spectra after the readjustment of pH from 5.5 to 7.4 (Fig. S38†). In contrast, the growth rate of nanofibers remained slow after aging for an additional 24 h from C in Fig. 11 without pH change (i.e., a total of 48 h at pH 5.5) (Fig. S39†). In addition, the transition process from sphere-like nanoparticles to nanofibers induced by a pH change (from A to B in Fig. 11) appeared to be faster than the self-assembly process at pH 7.4 after aging for 2 h without a pH change (Fig. S22 and S40†). These results demonstrate that spontaneous nanofiber growth in 1 can be controlled by switching the self-assembly pathway through pH change.
 | ||
| Fig. 11 Control of nanofiber growth by switching the self-assembly pathway of 1 through pH change. Conditions: [1] = 0.05 mM, 10 mM HEPES buffer (pH 7.4, 0.10 M NaCl) containing 0.5% (v/v) MeOH at 25 °C. | ||
Conclusions
In conclusion, we designed and synthesized the new amphiphilic 4-AQ-TPE conjugate 1 and evaluated its self-assembly properties in water using fluorescence and UV-Vis absorption spectroscopy, TEM, DLS, and SAXS. Our experimental results have revealed the kinetically controlled stepwise self-assembly of 1 with 0.10 M NaCl at RT, and its process comprises the formation of sphere-like nanoparticles, a transition to short nanofibers, and their growth to long nanofibers. The timescale of each self-assembly process of 1 differs according to pH (5.5 or 7.4). Thus, metastable short nanofibers were still formed after aging for 24 h at pH 5.5, while thermodynamically favored long nanofibers with ∼ 1 μm length scale were already formed after the same aging time at pH 7.4. The introduction of the 4-AQ moiety at the core of the self-assembling molecule contributes to the enhancement of the ability of self-assembly in comparison with that of 2.The outcome of the pH-responsive supramolecular polymerization of 1 with 0.10 M NaCl suggests that the protonation and deprotonation of the 4-AQ moiety affect the timescale of each self-assembly step at RT and do not affect the shape of the nanostructures of 1 formed. This self-assembly property of 1 can be applied for the modulation of nanofiber growth at RT through switching the self-assembly pathway in response to the pH change between 5.5 and 7.4. Further studies to deepen our understanding on self-assembly systems based on 4-AQ derivatives in water are now in progress.
The results reported herein may provide useful information for understanding kinetically controllable stepwise self-assembly and switching the kinetic nature under more biologically relevant conditions and may benefit the future design of self-assembling molecules to enable the creation of diverse supramolecular materials for various biological and biomedical applications.
Experimental section
Details of the experiments are described in the ESI.†Author contributions
Y. H. conceived the project. Y. H., F. C., K. Y. and H. T. performed the experiments and analyzed the data. Y. H. and T. H. supervised the project. Y. H., F. C., K. Y., N. U. and T. H. wrote and revised the manuscript. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Mr Go Toriyama (Faculty of Pharmaceutical Sciences, Nagoya City University) for the assistance of the synthesis of compounds. We are grateful for the assistance of the Research Equipment Sharing Center at Nagoya City University. This research was supported by JSPS KAKENHI (no. 21K06480) and the Research Foundation for Pharmaceutical Sciences. SAXS experiments were conducted at the Photon Factory of High Energy Accelerator Research Organization (approval 2020G071 and 2022G010).References
- (a) T. F. De Greef, M. M. Smulders, M. Wolffs, A. P. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS; (b) T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed; (c) P. K. Hashim, J. Bergueiro, E. W. Meijer and T. Aida, Prog. Polym. Sci., 2020, 105, 101250 CrossRef CAS.
- (a) J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS; (b) H. Fenniri, P. Mathivanan, K. L. Vidale, D. M. Sherman, K. Hallenga, K. V. Wood and J. G. Stowell, J. Am. Chem. Soc., 2001, 123, 3854–3855 CrossRef CAS PubMed; (c) S. Toledano, R. J. Williams, V. Jayawarna and R. V. Ulijn, J. Am. Chem. Soc., 2006, 128, 1070–1071 CrossRef CAS PubMed; (d) G. Zhang, W. Jin, T. Fukushima, A. Kosaka, N. Ishii and T. Aida, J. Am. Chem. Soc., 2007, 129, 719–722 CrossRef CAS PubMed; (e) P. Besenius, G. Portale, P. H. Bomans, H. M. Janssen, A. R. Palmans and E. W. Meijer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17888–17893 CrossRef CAS PubMed; (f) Z. Huang, H. Lee, E. Lee, S. K. Kang, J. M. Nam and M. Lee, Nat. Commun., 2011, 2, 459 CrossRef PubMed; (g) Z. Huang, S. K. Kang, M. Banno, T. Yamaguchi, D. Lee, C. Seok, E. Yashima and M. Lee, Science, 2012, 337, 1521–1526 CrossRef CAS; (h) W. Li, Y. Kim and M. Lee, Nanoscale, 2013, 5, 7711–7723 RSC; (i) E. Krieg, M. M. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS PubMed; (j) G. Yu, K. Jie and F. Huang, Chem. Rev., 2015, 115, 7240–7303 CrossRef CAS PubMed; (k) M. P. Hendricks, K. Sato, L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2017, 50, 2440–2448 CrossRef CAS PubMed; (l) J. Zhou, J. Li, X. Du and B. Xu, Biomaterials, 2017, 129, 1–27 CrossRef CAS PubMed; (m) P. Rajdev, S. Chakraborty, M. Schmutz, P. Mesini and S. Ghosh, Langmuir, 2017, 33, 4789–4795 CrossRef CAS; (n) T. Choisnet, D. Canevet, M. Salle, E. Nicol, F. Niepceron, J. Jestin and O. Colombani, Chem. Commun., 2019, 55, 9519–9522 RSC; (o) F. Aparicio, P. B. Chamorro, R. Chamorro, S. Casado and D. Gonzalez-Rodriguez, Angew. Chem., Int. Ed., 2020, 59, 17091–17096 CrossRef CAS; (p) R. Kubota, K. Nakamura, S. Torigoe and I. Hamachi, ChemistryOpen, 2020, 9, 67–79 CrossRef CAS PubMed.
- (a) J. B. Matson and S. I. Stupp, Chem. Commun., 2011, 47, 7962–7964 RSC; (b) M. H. Bakker, C. C. Lee, E. W. Meijer, P. Y. Dankers and L. Albertazzi, ACS Nano, 2016, 10, 1845–1852 CrossRef CAS; (c) H. Su, P. Zhang, A. G. Cheetham, J. M. Koo, R. Lin, A. Masood, P. Schiapparelli, A. Quinones-Hinojosa and H. Cui, Theranostics, 2016, 6, 1065–1074 CrossRef CAS PubMed; (d) M. J. Webber and R. Langer, Chem. Soc. Rev., 2017, 46, 6600–6620 RSC; (e) H. Su, F. Wang, Y. Wang, A. G. Cheetham and H. Cui, J. Am. Chem. Soc., 2019, 141, 17107–17111 CrossRef CAS PubMed.
- (a) G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352–1355 CrossRef CAS PubMed; (b) P. Y. Dankers, M. C. Harmsen, L. A. Brouwer, M. J. van Luyn and E. W. Meijer, Nat. Mater., 2005, 4, 568–574 CrossRef CAS PubMed; (c) P. Y. Dankers and E. W. Meijer, Bull. Chem. Soc. Jpn., 2007, 80, 2047–2073 CrossRef CAS; (d) J. B. Matson, R. H. Zha and S. I. Stupp, Curr. Opin. Solid State Mater. Sci., 2011, 15, 225–235 CrossRef CAS PubMed; (e) J. Boekhoven and S. I. Stupp, Adv. Mater., 2014, 26, 1642–1659 CrossRef CAS PubMed; (f) K. Sato, M. P. Hendricks, L. C. Palmer and S. I. Stupp, Chem. Soc. Rev., 2018, 47, 7539–7551 RSC.
- (a) S. Kiyonaka, K. Sada, I. Yoshimura, S. Shinkai, N. Kato and I. Hamachi, Nat. Mater., 2004, 3, 58–64 CrossRef CAS; (b) A. Wada, S. Tamaru, M. Ikeda and I. Hamachi, J. Am. Chem. Soc., 2009, 131, 5321–5330 CrossRef CAS PubMed; (c) M. Ikeda, T. Tanida, T. Yoshii, K. Kurotani, S. Onogi, K. Urayama and I. Hamachi, Nat. Chem., 2014, 6, 511–518 CrossRef CAS PubMed; (d) N. Mehwish, X. Dou, Y. Zhao and C.-L. Feng, Mater. Horiz., 2019, 6, 14–44 RSC.
- P. Besenius, J. L. Heynens, R. Straathof, M. M. Nieuwenhuizen, P. H. Bomans, E. Terreno, S. Aime, G. J. Strijkers, K. Nicolay and E. W. Meijer, Contrast Media Mol. Imaging, 2012, 7, 356–361 CrossRef CAS PubMed.
- (a) A. P. Blum, J. K. Kammeyer, A. M. Rush, C. E. Callmann, M. E. Hahn and N. C. Gianneschi, J. Am. Chem. Soc., 2015, 137, 2140–2154 CrossRef CAS PubMed; (b) W. Zhang and C. Gao, J. Mater. Chem. A, 2017, 5, 16059–16104 RSC; (c) M. Ikeda, Polym. J., 2019, 51, 371–380 CrossRef CAS.
- H. Frisch and P. Besenius, Macromol. Rapid Commun., 2015, 36, 346–363 CrossRef CAS PubMed.
- (a) L. Feng, Z. Dong, D. Tao, Y. Zhang and Z. Liu, Natl. Sci. Rev., 2018, 5, 269–286 CrossRef CAS; (b) S. Uthaman, K. M. Huh and I. K. Park, Biomater. Res., 2018, 22, 22 CrossRef PubMed.
- D. Pei and M. Buyanova, Bioconjugate Chem., 2019, 30, 273–283 CrossRef CAS PubMed.
- (a) K. J. van Bommel, C. van der Pol, I. Muizebelt, A. Friggeri, A. Heeres, A. Meetsma, B. L. Feringa and J. van Esch, Angew. Chem., Int. Ed., 2004, 43, 1663–1667 CrossRef CAS PubMed; (b) J. E. Goldberger, E. J. Berns, R. Bitton, C. J. Newcomb and S. I. Stupp, Angew. Chem., Int. Ed., 2011, 50, 6292–6295 CrossRef CAS PubMed; (c) A. Ghosh, M. Haverick, K. Stump, X. Yang, M. F. Tweedle and J. E. Goldberger, J. Am. Chem. Soc., 2012, 134, 3647–3650 CrossRef CAS PubMed; (d) H. Frisch, J. P. Unsleber, D. Ludeker, M. Peterlechner, G. Brunklaus, M. Waller and P. Besenius, Angew. Chem., Int. Ed., 2013, 52, 10097–10101 CrossRef CAS; (e) T. J. Moyer, J. A. Finbloom, F. Chen, D. J. Toft, V. L. Cryns and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 14746–14752 CrossRef CAS PubMed; (f) A. Uesaka, M. Ueda, A. Makino, T. Imai, J. Sugiyama and S. Kimura, Langmuir, 2014, 30, 1022–1028 CrossRef CAS PubMed; (g) C. Zhang, Y. Li, X. Xue, P. Chu, C. Liu, K. Yang, Y. Jiang, W. Q. Chen, G. Zou and X. J. Liang, Chem. Commun., 2015, 51, 4168–4171 RSC; (h) M. Pandeeswar and T. Govindaraju, Mol. Syst. Des. Eng., 2016, 1, 202–207 RSC; (i) A. Sarkar, J. C. Kolsch, C. M. Berac, A. Venugopal, R. Sasmal, R. Otter, P. Besenius and S. J. George, ChemistryOpen, 2020, 9, 346–350 CrossRef CAS PubMed.
- (a) H. Dong, S. E. Paramonov, L. Aulisa, E. L. Bakota and J. D. Hartgerink, J. Am. Chem. Soc., 2007, 129, 12468–12472 CrossRef CAS PubMed; (b) M. von Groning, I. de Feijter, M. C. A. Stuart, I. K. Voets and P. Besenius, J. Mater. Chem. B, 2013, 1, 2008–2012 RSC; (c) P. Ahlers, H. Frisch, D. Spitzer, Z. Vobecka, F. Vilela and P. Besenius, Chem. – Asian J., 2014, 9, 2052–2057 CrossRef CAS PubMed; (d) E. Fuentes, M. Gerth, J. A. Berrocal, C. Matera, P. Gorostiza, I. K. Voets, S. Pujals and L. Albertazzi, J. Am. Chem. Soc., 2020, 142, 10069–10078 CrossRef CAS PubMed.
- (a) I. de Feijter, P. Besenius, L. Albertazzi, E. W. Meijer, A. R. A. Palmans and I. K. Voets, Soft Matter, 2013, 9, 10025–10030 RSC; (b) C. Zhang, C. Liu, X. Xue, X. Zhang, S. Huo, Y. Jiang, W. Q. Chen, G. Zou and X. J. Liang, ACS Appl. Mater. Interfaces, 2014, 6, 757–762 CrossRef CAS PubMed; (c) R. Appel, S. Tacke, J. Klingauf and P. Besenius, Org. Biomol. Chem., 2015, 13, 1030–1039 RSC; (d) A. Rajbhandary, D. M. Raymond and B. L. Nilsson, Langmuir, 2017, 33, 5803–5813 CrossRef CAS PubMed.
- A. Klug, Philos. Trans. R. Soc. London, Ser. B, 1999, 354, 531–535 CrossRef CAS PubMed.
- (a) J. J. McManus, P. Charbonneau, E. Zaccarelli and N. Asherie, Curr. Opin. Colloid Interface Sci., 2016, 22, 73–79 CrossRef CAS; (b) A. Abelein, J. Jarvet, A. Barth, A. Graslund and J. Danielsson, J. Am. Chem. Soc., 2016, 138, 6893–6902 CrossRef CAS PubMed.
- M. M. Smulders, M. M. Nieuwenhuizen, T. F. de Greef, P. van der Schoot, A. P. Schenning and E. W. Meijer, Chem. – Eur. J., 2010, 16, 362–367 CrossRef CAS PubMed.
- (a) P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. Smulders, P. A. Hilbers, A. P. Schenning, T. F. De Greef and E. W. Meijer, Nature, 2012, 481, 492–496 CrossRef CAS PubMed; (b) A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476–5490 RSC; (c) J. Matern, Y. Dorca, L. Sanchez and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 16730–16740 CrossRef CAS PubMed; (d) M. Wehner and F. Würthner, Nat. Rev. Chem., 2020, 4, 38–53 CrossRef CAS; (e) G. Ghosh, P. Dey and S. Ghosh, Chem. Commun., 2020, 56, 6757–6769 RSC.
- (a) X. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644–647 CrossRef CAS PubMed; (b) J. B. Gilroy, T. Gadt, G. R. Whittell, L. Chabanne, J. M. Mitchels, R. M. Richardson, M. A. Winnik and I. Manners, Nat. Chem., 2010, 2, 566–570 CrossRef CAS PubMed; (c) S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188–195 CrossRef CAS PubMed; (d) J. Kang, D. Miyajima, T. Mori, Y. Inoue, Y. Itoh and T. Aida, Science, 2015, 347, 646–651 CrossRef CAS PubMed; (e) S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, J. Am. Chem. Soc., 2015, 137, 3300–3307 CrossRef CAS PubMed; (f) Y. Yan, J. Huang and B. Z. Tang, Chem. Commun., 2016, 52, 11870–11884 RSC; (g) T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2017, 9, 493–499 CrossRef CAS PubMed.
- (a) J. Baram, H. Weissman, Y. Tidhar, I. Pinkas and B. Rybtchinski, Angew. Chem., Int. Ed., 2014, 53, 4123–4126 CrossRef CAS PubMed; (b) P. A. Korevaar, C. J. Newcomb, E. W. Meijer and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 8540–8543 CrossRef CAS PubMed; (c) A. Levin, T. O. Mason, L. Adler-Abramovich, A. K. Buell, G. Meisl, C. Galvagnion, Y. Bram, S. A. Stratford, C. M. Dobson, T. P. Knowles and E. Gazit, Nat. Commun., 2014, 5, 5219 CrossRef CAS PubMed; (d) A. Pal, M. Malakoutikhah, G. Leonetti, M. Tezcan, M. Colomb-Delsuc, V. D. Nguyen, J. van der Gucht and S. Otto, Angew. Chem., Int. Ed., 2015, 54, 7852–7856 CrossRef CAS PubMed; (e) D. Gorl, X. Zhang, V. Stepanenko and F. Wurthner, Nat. Commun., 2015, 6, 7009 CrossRef PubMed; (f) A. Aliprandi, M. Mauro and L. De Cola, Nat. Chem., 2016, 8, 10–15 CrossRef CAS PubMed; (g) Y. Yan, J. Huang and B. Z. Tang, Chem. Commun., 2016, 52, 11870–11884 RSC; (h) M. P. Hendricks, K. Sato, L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2017, 50, 2440–2448 CrossRef CAS PubMed; (i) A. Singh, J. P. Joseph, D. Gupta, I. Sarkar and A. Pal, Chem. Commun., 2018, 54, 10730–10733 RSC; (j) A. Mishra, D. B. Korlepara, M. Kumar, A. Jain, N. Jonnalagadda, K. K. Bejagam, S. Balasubramanian and S. J. George, Nat. Commun., 2018, 9, 1295 CrossRef PubMed; (k) Q. Wan, X. S. Xiao, W. P. To, W. Lu, Y. Chen, K. H. Low and C. M. Che, Angew. Chem., Int. Ed., 2018, 57, 17189–17193 CrossRef CAS PubMed; (l) L. Zhao, S. Li, Y. Liu, R. Xing and X. Yan, CCS Chem., 2019, 1, 173–180 CAS; (m) C. Rest, D. S. Philips, T. Dunnebacke, P. Sutar, A. Sampedro, J. Droste, V. Stepanenko, M. R. Hansen, R. Q. Albuquerque and G. Fernández, Chem. – Eur. J., 2020, 26, 10005–10013 CrossRef CAS PubMed; (n) M. A. VandenBerg, J. K. Sahoo, L. Zou, W. McCarthy and M. J. Webber, ACS Nano, 2020, 14, 5491–5505 CrossRef CAS PubMed; (o) I. Helmers, G. Ghosh, R. Q. Albuquerque and G. Fernández, Angew. Chem., Int. Ed., 2021, 60, 4368–4376 CrossRef CAS PubMed; (p) G. Ghosh, T. Ghosh and G. Fernández, ChemPlusChem, 2020, 85, 1022–1033 CrossRef CAS PubMed; (q) H. Su, F. Wang, H. Wang, W. Zhang, C. F. Anderson and H. Cui, J. Am. Chem. Soc., 2021, 143, 18446–18453 CrossRef CAS PubMed; (r) R. Liu, R. Zhang, L. Li, Z. Kochovski, L. Yao, M. P. Nieh, Y. Lu, T. Shi and G. Chen, J. Am. Chem. Soc., 2021, 143, 6622–6633 CrossRef CAS PubMed; (s) D. Gupta, A. Bhatt, V. Gupta, C. Miglani, J. P. Joseph, J. Ralhan, D. Mandal, M. E. Ali and A. Pal, Chem. Mater., 2022, 34, 4456–4470 CrossRef CAS.
- Y. Tidhar, H. Weissman, S. G. Wolf, A. Gulino and B. Rybtchinski, Chem. – Eur. J., 2011, 17, 6068–6075 CrossRef CAS PubMed.
- F. Tantakitti, J. Boekhoven, X. Wang, R. V. Kazantsev, T. Yu, J. Li, E. Zhuang, R. Zandi, J. H. Ortony, C. J. Newcomb, L. C. Palmer, G. S. Shekhawat, M. O. de la Cruz, G. C. Schatz and S. I. Stupp, Nat. Mater., 2016, 15, 469–476 CrossRef CAS PubMed.
- B. Kemper, L. Zengerling, D. Spitzer, R. Otter, T. Bauer and P. Besenius, J. Am. Chem. Soc., 2018, 140, 534–537 CrossRef CAS PubMed.
- G. Ghosh, R. Barman, A. Mukherjee, U. Ghosh, S. Ghosh and G. Fernández, Angew. Chem., Int. Ed., 2022, 61, e202113403 CAS.
- (a) S. Harguindey, J. L. Arranz, J. D. Polo Orozco, C. Rauch, S. Fais, R. A. Cardone and S. J. Reshkin, J. Transl. Med., 2013, 11, 282 CrossRef PubMed; (b) J. Zhang, X. Jiang, X. Wen, Q. Xu, H. Zeng, Y. Zhao, M. Liu, Z. Wang, X. Hu and Y. Wang, J. Phys.: Mater., 2019, 2, 032004 CAS; (c) A. Jaworska, K. Malek and A. Kudelski, Spectrochim. Acta, Part A, 2021, 251, 119410 CrossRef CAS PubMed.
- (a) Y. Hisamatsu, N. Umezawa, H. Yagi, K. Kato and T. Higuchi, Chem. Sci., 2018, 9, 7455–7467 RSC; (b) Y. Hisamatsu, K. Otani, H. Takase, N. Umezawa and T. Higuchi, Chem. – Eur. J., 2021, 27, 6489–6499 CrossRef CAS PubMed.
- S. Deshpande and B. Kuppast, Med. Chem., 2016, 6, 001–011 Search PubMed.
- T. J. Egan, Mini-Rev. Med. Chem., 2001, 1, 113–123 CrossRef CAS PubMed.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC, Boca Raton, 83rd edn, 2002–2003 Search PubMed.
- D. C. Warhurst, J. C. Steele, I. S. Adagu, J. C. Craig and C. Cullander, J. Antimicrob. Chemother., 2003, 52, 188–193 CrossRef CAS PubMed.
- (a) H. Tong, Y. Hong, Y. Dong, M. Haussler, J. W. Lam, Z. Li, Z. Guo, Z. Guo and B. Z. Tang, Chem. Commun., 2006, 3705–3707 RSC; (b) J. Li, J. Wang, H. Li, N. Song, D. Wang and B. Z. Tang, Chem. Soc. Rev., 2020, 49, 1144–1172 RSC.
- L. N. Neupane, E. T. Oh, H. J. Park and K. H. Lee, Anal. Chem., 2016, 88, 3333–3340 CrossRef CAS PubMed.
- M. de Souza Santos, M. P. F. de Morais del Lama, A. Siuiti Ito and R. M. Zumstein Georgetto Naal, J. Lumin., 2014, 147, 49–58 CrossRef CAS.
- (a) W. Qin, Y. Wu, Y. Hu, Y. Dong, T. Hao and C. Zhang, ACS Appl. Bio Mater., 2021, 4, 1038–1044 CrossRef CAS; (b) W. Guan, T. Yang and C. Lu, Angew. Chem., Int. Ed., 2020, 59, 12800–12805 CrossRef CAS PubMed.
- H. Frisch, D. Spitzer, M. Haase, T. Basche, J. Voskuhl and P. Besenius, Org. Biomol. Chem., 2016, 14, 5574–5579 RSC.
- R. Appel, J. Fuchs, S. M. Tyrrell, P. A. Korevaar, M. C. Stuart, I. K. Voets, M. Schonhoff and P. Besenius, Chem. – Eur. J., 2015, 21, 19257–19264 CrossRef CAS PubMed.
- C. A. Hunter, K. R. Lawson, J. Perkins and C. J. Urch, J. Chem. Soc., Perkin Trans. 2, 2001, 651–669 RSC.
- G. V. Jensen, R. Lund, J. Gummel, T. Narayanan and J. S. Pedersen, Angew. Chem., Int. Ed., 2014, 53, 11524–11528 CrossRef CAS PubMed.
- D. Lombardo, G. Munao, P. Calandra, L. Pasqua and M. T. Caccamo, Phys. Chem. Chem. Phys., 2019, 21, 11983–11991 RSC.
- (a) A. Iscen and G. C. Schatz, J. Phys. Chem. B, 2019, 123, 7006–7013 CrossRef CAS PubMed; (b) J. C. Stendahl, M. S. Rao, M. O. Guler and S. I. Stupp, Adv. Funct. Mater., 2006, 16, 499–508 CrossRef CAS.
- X. Yan, Y. Cui, Q. He, K. Wang, J. Li, W. Mu, B. Wang and Z. C. Ou-Yang, Chem. – Eur. J., 2008, 14, 5974–5980 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of 1 and 2, experimental details, UV-Vis and fluorescence spectra, TEM images, and DLS and SAXS data. See DOI: https://doi.org/10.1039/d2nr05756e |
| ‡ In this study, MES buffer for pH 5.0, 5.5, 6.0, 6.3, and 6.5, HEPES buffer for pH 6.8, 7.1, 7.4, 7.8, and 8.2, CHES buffer for pH 8.6 and 9.5, and CAPS buffer for 10.4 and 11.0 were used. |
| § Fluorescence and UV-Vis absorption spectra of 2 without 0.10 M NaCl at various pH values are shown in Fig. S5 and S6.† |
| ¶ Similar short nanofibers formed from 1 were observed after 1 h when HEPES buffer (pH 5.5, 0.10 M NaCl) was used instead of MES buffer (Fig. S33†). |
| This journal is © The Royal Society of Chemistry 2023 |