
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of surface and intermolecular interactions on the properties of supported polyoxometalates†
Oliva M.
Primera-Pedrozo
 a,
Shuai
Tan
a,
Difan
Zhang
a,
Brian T.
O'Callahan
b,
Wenjin
Cao
a,
Eric T.
Baxter
a,
Xue-Bin
Wang
a,
Patrick Z.
El-Khoury
a,
Venkateshkumar
Prabhakaran
a,
Vassiliki-Alexandra
Glezakou
c and
Grant E.
Johnson
*a
a,
Shuai
Tan
a,
Difan
Zhang
a,
Brian T.
O'Callahan
b,
Wenjin
Cao
a,
Eric T.
Baxter
a,
Xue-Bin
Wang
a,
Patrick Z.
El-Khoury
a,
Venkateshkumar
Prabhakaran
a,
Vassiliki-Alexandra
Glezakou
c and
Grant E.
Johnson
*a
aPacific Northwest National Laboratory, Physical Sciences Division, P.O. Box 999, MSIN J7-10, Richland, Washington 99352, USA. E-mail: grant.johnson@pnnl.gov
bPacific Northwest National Laboratory, Earth and Biological Sciences Division, P.O. Box 999, MSIN K8-88, Richland, Washington 99352, USA
cOak Ridge National Laboratory, 1 Bethel Valley Road, Oak Ridge, Tennessee 37830, USA
First published on 27th February 2023
Abstract
Polyoxometalates (POMs) with localized radical or open-shell metal sites have the potential to be used as transformative electronic spin based molecular qubits (MQs) for quantum computing (QC). For practical applications, MQs have to be immobilized in electronically or optically addressable arrays which introduces interactions with supports as well as neighboring POMs. Herein, we synthesized Keggin POMs with both tungsten (W) and vanadium (V) addenda atoms. Ion soft landing, a highly-controlled surface modification technique, was used to deliver mass-selected V-doped POMs to different self-assembled monolayer surfaces on gold (SAMs) without the solvent, counterions, and contaminants that normally accompany deposition from solution. Alkylthiol, perfluorinated, and carboxylic-acid terminated monolayers were employed as representative model supports on which different POM-surface and POM–POM interactions were characterized. We obtained insights into the vibrational properties of supported V-doped POMs and how they are perturbed by interactions with specific surface functional groups using infrared reflection absorption and scattering-type scanning near-field optical microscopy, as well as tip enhanced Raman spectroscopy. Different functional groups on SAMs and nanoscale heterogeneity are both shown to modulate the observed spectroscopic signatures. Spectral shifts are also found to be dependent on POM–POM interactions. The electronic structure of the V-doped POMs was determined in the gas phase using negative ion photoelectron spectroscopy and on surfaces with scanning Kelvin probe microscopy. The chemical functionality and charge transfer properties of the SAMs are demonstrated to exert an influence on the charge state and electronic configuration of supported V-doped POMs. The geometric and electronic structure of the POMs were also calculated using density functional theory. Our joint experimental and theoretical findings provide insight into how V substitution as well as POM-surface and POM–POM interactions influence the vibrational properties of POMs.
Introduction
Several envisioned quantum computing (QC) architectures take full advantage of coherent electronic spins in open shell metal containing species and molecular radicals.1 In practice, such spin centers have to be arranged into optically or electrically accessible quantum gates (QGs) capable of performing algorithmic operations.2 Indeed, large scale QC will only be achieved if an appropriate and addressable set of QGs are arranged in a controlled and scalable manner.3 Coordination complexes and magnetic molecules may be used to assemble two-qubit QGs that provide notable advantages over solid-state systems.4 In particular, all molecules synthesized following a given chemical reaction are identical and their structures may be tailored extensively using established chemistry.2 Coordination chemistry also provides a way of arranging multiple paramagnetic ions or assemblies of strongly coupled open shell metals within a molecule.5 These species may be connected across multiple length scales through convenient and potentially switchable interactions.6 Molecular metal oxides, known as polyoxometalates (POMs), have additional advantages for QC such as being robust species that maintain their structural integrity in solution, in the solid state, and potentially in 2-dimensional (2D) arrays.7 POMs may host single magnetic ions or groups of ions at defined sites in their rigid structure, leading to large magnetic molecules and clusters with defined topologies and symmetry.2 Due to their multielectron reduction–oxidation (redox) activity, POMs may also accommodate different numbers of electrons while maintaining their geometric structure, making them valuable model systems with which to study the interplay between electron delocalization and magnetic exchange in high nuclearity clusters.7–9Despite their advantages, increasing the spin coherence times of MQs supported in arrays remains a challenge that is hindering their use in QC.3 Ensembles of identical weakly interacting spin centers are favored in this regard because there is a fine line between optimum coherent signal generation and onset of decoherence from spin centers interacting with one another and their environment.10 Knowledge gaps remain regarding how the geometric and electronic structures of POMs and supports may be tailored to minimize different decoherence mechanisms (i.e., nuclear spin diffusion, electron spin coupling, methyl rotation, spin–lattice relaxation, and spin diffusion barriers) and design qubit arrays with sufficiently long coherence times (T > 100 μs) for practical QC applications.11 In particular, spins in supported molecules relax through spin–lattice interactions, making the surface-qubit interface an influential parameter determining the performance of MQ arrays.12 Innovative approaches are needed to prepare and characterize well-defined arrays of supported MQs to achieve transformational advances in the understanding of spin decoherence mechanisms. Synthetic methods currently used to prepare magnetically doped POMs in solution usually result in a broad and poorly-defined distribution of products. The undefined composition of doped MQs, combined with the presence of solvent and counterions, often results in uncontrolled formation of aggregates and crystallites following drop casting onto surfaces from solution. Collectively, these confounding factors make it challenging to establish the structure–property relationships needed to inform the predictive design of MQ arrays with extended coherence times.
Ion soft landing (ISL) is a versatile technique that facilitates the preparation and modification of homogeneous films of mass-selected polyatomic ions in predetermined charge states through controlled deposition of gas-phase ions onto surfaces.13–21 This distinguishing capability allows supported ions to be investigated in the absence of interference from counterions, solvent molecules, and other impurities that are present at interfaces prepared using solution based deposition.22,23 In addition, soft landed ions are deposited without aggregation and crystallite formation.24,25 Correspondingly, ISL is able to prepare the highest achievable coverage of discrete polyatomic ions and clusters dispersed homogeneously on surfaces. In particular, ISL has recently demonstrated itself to be a powerful technique for the controlled fabrication of well-defined POM-based interfaces for energy storage applications.8,24–28
Retention of charge by polyatomic ions soft landed onto self-assembled monolayers (SAMs) on gold was initially observed by Cooks and co-workers.29 In subsequent investigations of inherently charged cations, we found that the 3+ charge state of Au113+ clusters was retained on fluorinated monolayers (FSAMs) up to a threshold surface coverage, while SAMs terminated with carboxyl (COOH–SAM) and methyl groups (HSAM) stabilized soft landed Au113+ clusters predominantly in the 2+ and 1+ charge states, respectively.30,31 On HSAMs, cationic clusters underwent rapid charge reduction and neutralization through electron transfer from the underlying gold surface, while on FSAMs cationic clusters exhibited much slower charge reduction.30 Retention of positive charge by soft landed cations on FSAMs is attributed to an energy barrier for electron transfer resulting from the electronegativity of the fluorine atoms in the SAM which induce a dipole at the Au–S interface.32 In comparison, the charge retention and neutralization of negatively charged anions soft landed onto SAMs is remarkably different to that of cations. Specifically, triply charged POM anions were previously found to retain charge on all three SAM surfaces.26,27 The unexpected charge retention was attributed to the large electron binding energies of the POM anions.33
POMs are known to undergo rapid and reversible multielectron redox processes while maintaining their geometric structure,34 making them promising systems for a wide range of applications including electrochemical energy storage,34 photochromic films,35 and microelectronics.36 In the context of MQs for potential QC, we focus herein on vanadium-doped tungsten Keggin POMs [PVxW12−xO40](3+x)− because they are amenable to solution synthesis with different numbers of V atoms, each of which introduces a spin-unpaired electron to the cluster. Alkylthiol (HSAM), hydrophobic perfluorinated alkylthiol (FSAM), and hydrophilic carboxylic acid (COOH–SAM) terminated monolayers on gold were selected as well-defined representative model supports on which to characterize POM-surface and POM–POM interactions relevant to potential QC applications.
In this paper, we report the electronic structure of PVW11O404− (VPOM4−), which was synthesized in solution and measured in isolation in the gas phase using electrospray ionization mass spectrometry (ESI-MS) and negative ion photoelectron spectroscopy (NIPES). The electronic properties of VPOMs soft landed on different SAMs was revealed using scanning Kelvin probe microscopy (KPFM). We investigated the vibrational properties of supported VPOMs and how they are influenced by interactions with different SAM surfaces using infrared reflection absorption spectroscopy (IRRAS) and nanoindentation enhanced tip enhanced Raman spectroscopy (TERS). The effect of POM surface coverage, and thereby POM–POM interactions on the vibrational modes of VPOMs, was also characterized using scattering-type scanning near-field optical microscopy (s-SNOM). The electronic structure and vibrational spectra of the POMs were modeled using density functional theory (DFT) calculations. Our joint experimental and theoretical investigation offers insight into how intramolecular interactions between the substituted V atom and other atoms (W and O), as well as POM-surface and POM–POM intermolecular interactions influence the electronic and vibrational properties of supported POMs.
Results and discussion
The experimental and theoretical results are separated into two sections. The first section describes the electronic properties of VPOM4− in the gas phase and supported on different SAMs. The second section presents the vibrational properties of SAM-supported VPOM4−.Electronic properties of VPOM4− in the gas phase and supported on SAMs
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NaVO3 salts in a molar ratio of 11:1 produced several V-doped POM anions with VPOM4− exhibiting the highest relative abundance in the mass spectrum. The mass spectrum of WPOM3− is provided in Fig. 1b for comparison. Notably, the ESI mass spectrum in Fig. 1a includes both the quadruply (VPOM4−) and triply charged [H+] [VPOM]4− anions with the latter species resulting from association of a proton [H+] from water onto VPOM4− which helps to Coulombically stabilize the highly-charged 4- anion. To be confident of the molecular formula assignments, we obtained high mass resolution spectra for each of the anions observed in Fig. 1a. We compared the m/z accuracy and relative abundance of each of the experimentally measured isotopes in the overall isotopic envelopes of each POM anion against those calculated using the freely available mMass software (https://www.mmass.org/). Comparisons of the m/z accuracy of the experimentally measured isotopes with the corresponding theoretically calculated values for VPOM4− are presented in Fig. S1 and Table S1 of the ESI.† The results confirm the accurate formula assignment of VPOM4− (685.79 m/z) which matches the corresponding calculated theoretical m/z values with an error Δm/z of −0.003.
:NaVO3 salts in a molar ratio of 11:1 produced several V-doped POM anions with VPOM4− exhibiting the highest relative abundance in the mass spectrum. The mass spectrum of WPOM3− is provided in Fig. 1b for comparison. Notably, the ESI mass spectrum in Fig. 1a includes both the quadruply (VPOM4−) and triply charged [H+] [VPOM]4− anions with the latter species resulting from association of a proton [H+] from water onto VPOM4− which helps to Coulombically stabilize the highly-charged 4- anion. To be confident of the molecular formula assignments, we obtained high mass resolution spectra for each of the anions observed in Fig. 1a. We compared the m/z accuracy and relative abundance of each of the experimentally measured isotopes in the overall isotopic envelopes of each POM anion against those calculated using the freely available mMass software (https://www.mmass.org/). Comparisons of the m/z accuracy of the experimentally measured isotopes with the corresponding theoretically calculated values for VPOM4− are presented in Fig. S1 and Table S1 of the ESI.† The results confirm the accurate formula assignment of VPOM4− (685.79 m/z) which matches the corresponding calculated theoretical m/z values with an error Δm/z of −0.003.
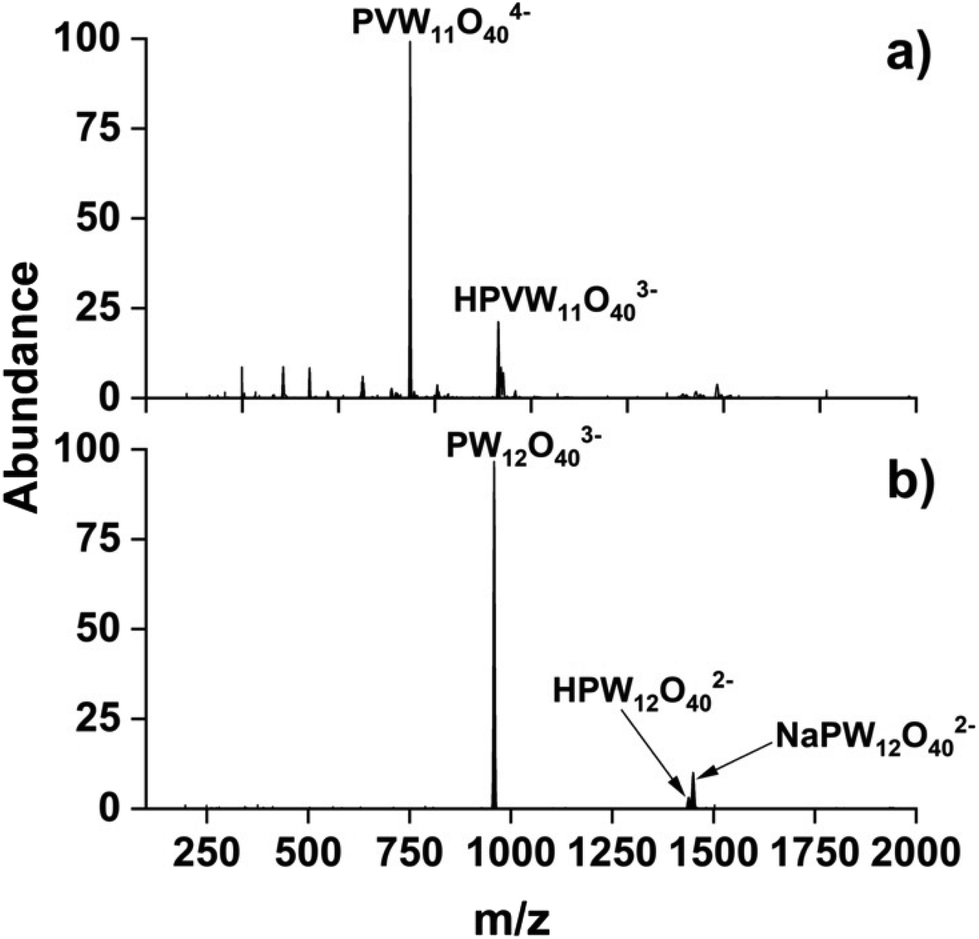 | ||
| Fig. 1 Representative negative mode electrospray ionization mass spectra of (a) 100 μM VPOM4− solution in water prepared using an 11:1 molar ratio of Na3PO4·12WO3·xH2O:NaVO3 salts, and (b) 100 μM WPOM3− solution in methanol prepared using Na3[PW12O40]·xH2O. | ||
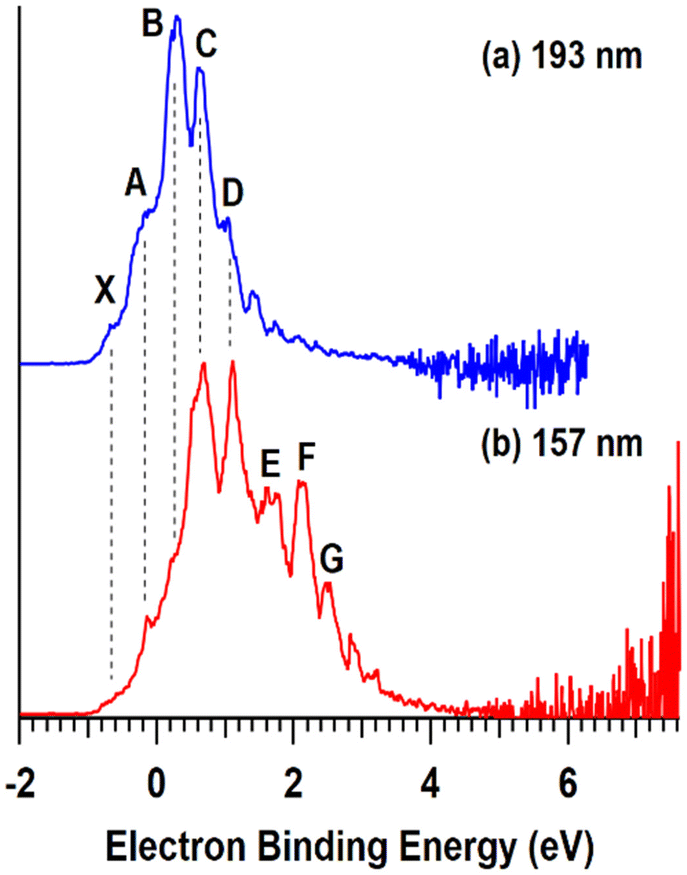 | ||
| Fig. 2 T = 20 K negative ion photoelectron spectra of VPOM4− obtained at photodetachment wavelengths of (a) 193 and (b) 157 nm. | ||
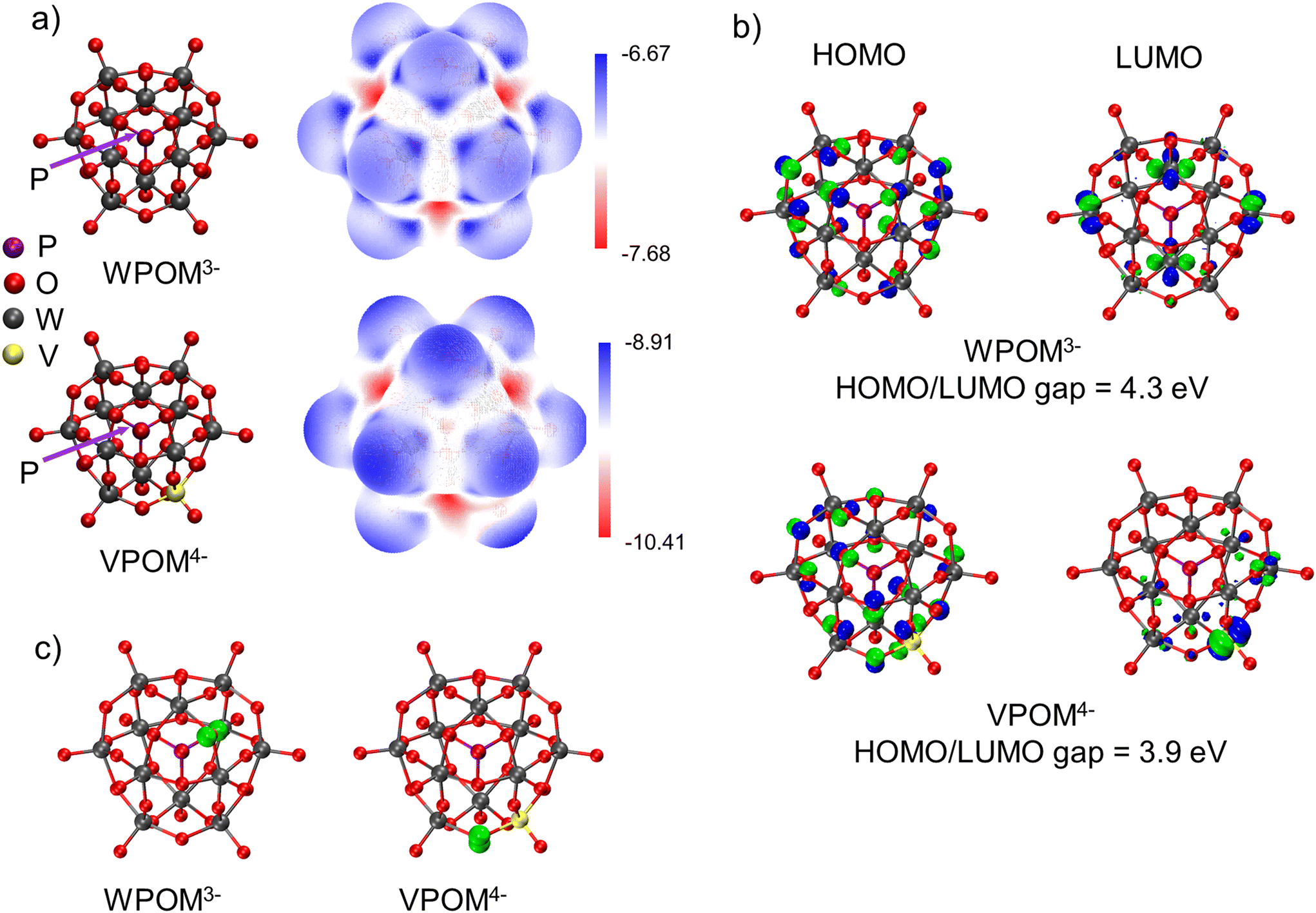 | ||
| Fig. 3 (a) Calculated structures and electrostatic potential maps of WPOM3− and VPOM4− (P atom is located behind central O atom), (b) the highest occupied/lowest unoccupied molecular orbitals (HOMO/LUMO) of WPOM3− and VPOM4−. Atoms follow the color scheme in (a), (c) calculated electron spin density of WPOM3− and VPOM4−, highlighted in green. Atoms follow the color scheme in (a). | ||
As shown in Fig. 4a, the measured WFs of the bare SAMs increase in the order HSAM (5.03 eV) < COOH-SAM (5.09 eV) < FSAM (5.52 eV) (Table 1), revealing a trend that is consistent with previous studies.44,46 Due to charge reduction and ion neutralization processes that occur between supported ions and SAM substrates, soft landed species may become partially neutralized following deposition.13,14 Previous studies from our group demonstrated an approach combining experimental IRRAS measurements with DFT calculations to determine the distribution of anionic charge states of POMs supported on different SAMs. This method relies on correlations between the wavenumbers and shifts of characteristic vibrational modes [terminal metal–oxygen band (M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ot), bridging metal–oxygen bands (M–Ob1,2–M)] and the ionic charge state of supported POMs. In our previous study, WPOM3− was found to partially retain its 3- charge on FSAMs while partially oxidized WPOM2− was observed to be the most abundant species on HSAMs. In general, the charge retention of soft landed anions on SAMs follows the order qHSAM < qCOOH-SAM < qFSAM. Herein, we used the measured WFs to better understand the ionic charge states and electronic structure of supported POMs. As shown in Fig. 4b, following soft landing of WPOM3− the WFs of all three SAMs were reduced and found to increase in the order HSAM (4.77 eV) < COOH-SAM (4.84 eV) < FSAM (4.94 eV). The global reduction in the WFs following deposition of WPOM3− on all three SAMs results from accumulation of negative charge at the SAM interface. In this case, electrons are emitted from the WPOM3− anions at the SAM interface where increased negative Coulombic repulsion is present, rather than from the underlying Au surface. As shown in Fig. 4c, the measured WFs of soft landed VPOM4− were found to be closer to those of the bare SAMs than WPOM3−. Interestingly, the WF was observed to be larger (5.36 eV) on the HSAM than on the COOH-SAM (5.17 eV) or FSAM (5.11 eV). This finding contrasts with the results for the bare SAMs and SAMs with soft landed WPOM3− where the relative WF increased in the order HSAM < COOH-SAM < FSAM. For VPOM4−, the largest negative charge is expected to accumulate on the FSAM, resulting in increased Coulombic repulsion that reduces the WF (assuming electron emission from VPOM4− rather than the Au surface). This repulsion builds up during deposition because ISL delivers only anions to the surface without any solvent or counter cations. In comparison, less negative charge from VPOM4− is expected to be retained on the HSAM, resulting in less Coulombic repulsion and a higher WF. Similar to the results for WPOM3−, in this case electrons are emitted from VPOM4− anions at the vacuum interface rather than from the underlying Au surface. Based on the gas phase NIPES results presented in Fig. 2, the ADE and VDE of WPOM3− are much higher than those of VPOM4−, due to increased Coulombic repulsion in quadruply charged VPOM4− compared to triply charged WPOM3−. In comparison, the consistently larger WFs measured for VPOM4− compared to WPOM3− on all three SAMs suggests that the surface plays an important role in stabilizing POMs toward electron detachment, possibly through an image potential interaction with the underlying polarizable gold surface.47 It is also possible that the larger charge accumulation resulting from soft landing of VPOM4− compared to WPOM3− was sufficient to promote spontaneous electron dissociation or transfer to the underlying gold surface.31 In either case, this would result in formation of partially oxidized VPOM3−,2−,1− species with reduced overall negative charge and Coulombic repulsion and, therefore, increased WFs (assuming electron emission from VPOM4−). These findings illustrate how substitution of a W atom with V influences the electronic structure and stability of VPOM4− supported on surfaces with different chemical functionality and electron transfer properties. Finding the right balance of substitution and surface functionality will be critical to preserving the electronic and vibrational properties of POMs for potential use in MQ arrays.
Ot), bridging metal–oxygen bands (M–Ob1,2–M)] and the ionic charge state of supported POMs. In our previous study, WPOM3− was found to partially retain its 3- charge on FSAMs while partially oxidized WPOM2− was observed to be the most abundant species on HSAMs. In general, the charge retention of soft landed anions on SAMs follows the order qHSAM < qCOOH-SAM < qFSAM. Herein, we used the measured WFs to better understand the ionic charge states and electronic structure of supported POMs. As shown in Fig. 4b, following soft landing of WPOM3− the WFs of all three SAMs were reduced and found to increase in the order HSAM (4.77 eV) < COOH-SAM (4.84 eV) < FSAM (4.94 eV). The global reduction in the WFs following deposition of WPOM3− on all three SAMs results from accumulation of negative charge at the SAM interface. In this case, electrons are emitted from the WPOM3− anions at the SAM interface where increased negative Coulombic repulsion is present, rather than from the underlying Au surface. As shown in Fig. 4c, the measured WFs of soft landed VPOM4− were found to be closer to those of the bare SAMs than WPOM3−. Interestingly, the WF was observed to be larger (5.36 eV) on the HSAM than on the COOH-SAM (5.17 eV) or FSAM (5.11 eV). This finding contrasts with the results for the bare SAMs and SAMs with soft landed WPOM3− where the relative WF increased in the order HSAM < COOH-SAM < FSAM. For VPOM4−, the largest negative charge is expected to accumulate on the FSAM, resulting in increased Coulombic repulsion that reduces the WF (assuming electron emission from VPOM4− rather than the Au surface). This repulsion builds up during deposition because ISL delivers only anions to the surface without any solvent or counter cations. In comparison, less negative charge from VPOM4− is expected to be retained on the HSAM, resulting in less Coulombic repulsion and a higher WF. Similar to the results for WPOM3−, in this case electrons are emitted from VPOM4− anions at the vacuum interface rather than from the underlying Au surface. Based on the gas phase NIPES results presented in Fig. 2, the ADE and VDE of WPOM3− are much higher than those of VPOM4−, due to increased Coulombic repulsion in quadruply charged VPOM4− compared to triply charged WPOM3−. In comparison, the consistently larger WFs measured for VPOM4− compared to WPOM3− on all three SAMs suggests that the surface plays an important role in stabilizing POMs toward electron detachment, possibly through an image potential interaction with the underlying polarizable gold surface.47 It is also possible that the larger charge accumulation resulting from soft landing of VPOM4− compared to WPOM3− was sufficient to promote spontaneous electron dissociation or transfer to the underlying gold surface.31 In either case, this would result in formation of partially oxidized VPOM3−,2−,1− species with reduced overall negative charge and Coulombic repulsion and, therefore, increased WFs (assuming electron emission from VPOM4−). These findings illustrate how substitution of a W atom with V influences the electronic structure and stability of VPOM4− supported on surfaces with different chemical functionality and electron transfer properties. Finding the right balance of substitution and surface functionality will be critical to preserving the electronic and vibrational properties of POMs for potential use in MQ arrays.
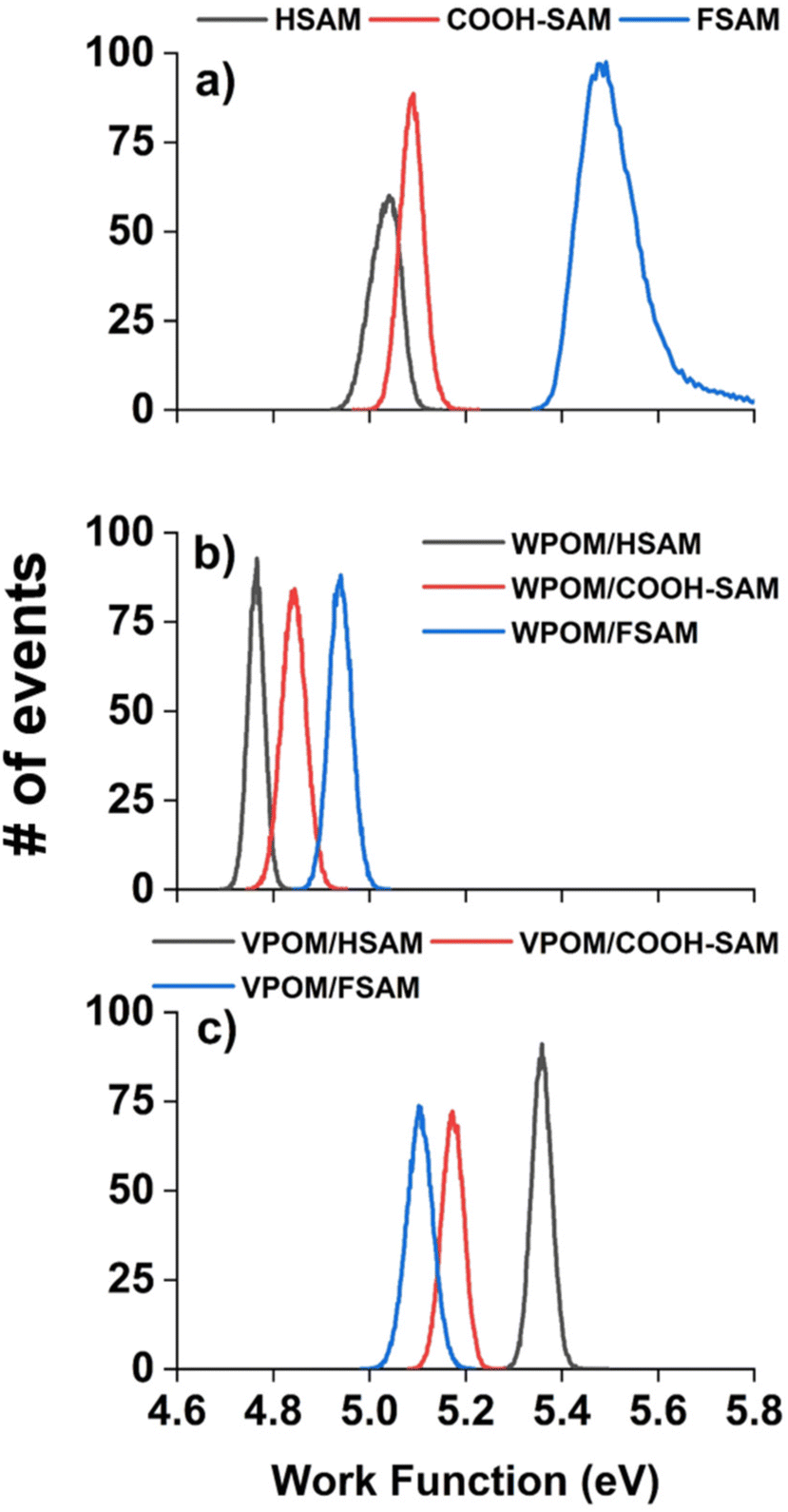 | ||
| Fig. 4 Distribution of work function values measured over a 4 μm2 area of (a) bare HSAM, COOH-SAM, and FSAM, (b) 1 × 1014 WPOM3− soft landed on HSAM, COOH-SAM, and FSAM, and (c) 1 × 1014 VPOM4− soft landed on HSAM, COOH-SAM, and FSAM. | ||
| Material | Work function (eV) | Material | Work function (eV) | Material | Work function (eV) |
|---|---|---|---|---|---|
| Bare HSAM | 5.03 | WPOM3−/HSAM | 4.77 | VPOM4−/HSAM | 5.36 |
| Bare COOH-SAM | 5.09 | WPOM3−/COOH-SAM | 4.84 | VPOM4−/COOH-SAM | 5.17 |
| Bare FSAM | 5.52 | WPOM3−/FSAM | 4.94 | VPOM4−/FSAM | 5.11 |
Vibrational properties of supported VPOM4−
Ot stretch appears between 950–970 cm−1.26,27,49–51 Two different types of bridging tungsten–oxygen bands also contribute vibrational features observed at ∼860–880 cm−1 (W–Ob2–W) and ∼805–820 cm−1 (W–Ob1–W), respectively. Oxygen sites are identified as Oi (internal), Ot (terminal), Ob1, (bridging 1), and Ob2 (bridging 2). All of these characteristic IR bands of POMs have been reported previously in the literature and are observed in the IRRAS spectra of WPOM3− or VPOM4− presented in Fig. 5a. The presence of these peaks confirms the intact deposition of WPOM3− and VPOM4− onto the SAM surfaces. Additional deposition experiments performed at higher kinetic energies which induce fragmentation of POMs did not reveal these characteristic peaks.
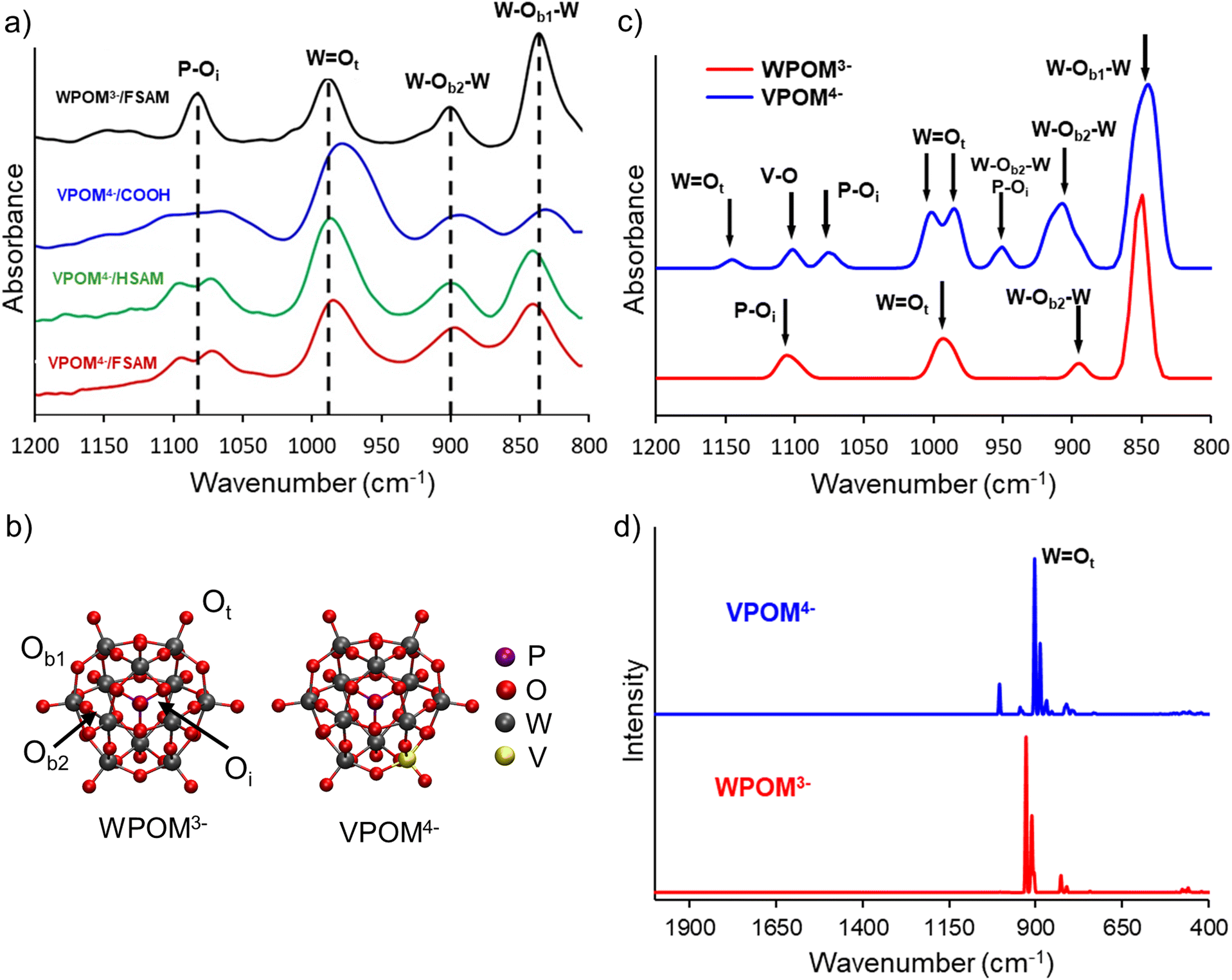 | ||
| Fig. 5 (a) Experimental infrared reflection absorption spectroscopy spectra obtained following deposition of ∼1 × 1014 VPOM4− ions on HSAM, COOH-SAM, and FSAM surfaces on Au. WPOM3− on FSAM is also shown for comparison, (b) theoretically calculated ground state structures of WPOM3− and VPOM4−, four types of oxygen sites are identified as Oi (internal), Ot (terminal), Ob1, and Ob2 (bridging), (c) theoretically calculated IR spectra of gas phase WPOM3− and VPOM4−, (d) theoretically calculated Raman spectrum of gas phase VPOM4− and WPOM3−. | ||
To better understand and interpret the experimental IR features presented in Fig. 5a and the implications of their shifts on different SAMs, we used DFT calculations to determine the gas phase IR spectra of isolated WPOM3− and VPOM4−. Snapshots of the structures used in these calculations are illustrated in Fig. 5b, and the theoretically predicted IR/Raman vibrational spectra are presented in Fig. 5c and d, respectively. In addition to the IR modes reported previously, the simulated vibrational spectra validate the peak assignments in the experimental spectrum (Fig. 5a). Specifically, WPOM3− exhibits two W–Ob1,2–W peaks, one WOt peak, and one P–Oi peak in the range of 800–1200 cm−1. Furthermore, the simulations identify additional peaks in the vibrational spectra of VPOM4− resulting from substitution of one W atom with V (1093–1107 cm−1). For example, an additional peak is found at ∼950 cm−1 in Fig. 5c which is not attributed to a single bond, but rather a combination band with contributions from both the P–Oi and W–Ob–W stretching vibrations. The WOt mode in VPOM4− is comprised of two equally intense peaks located close to each other instead of the single peak observed for WPOM3−. This difference in the IR spectra is consistent with the loss of molecular symmetry resulting from substitution of V for a W atom. In addition, the P–Oi peak of VPOM4− is shifted to lower wavenumber compared to WPOM3− and a new V–O peak is also observed at ∼1100 cm−1. Another weak peak associated with WOt is observed at higher wavenumber ∼1150 cm−1 for VPOM4− that is not present in WPOM3−. Previous reports in the literature used variations in this spectral feature to investigate vibrational signatures of V-substituted POMs.52,53 Our results agree with this previous work on mono-substituted ∝-(TBA)4[PVW11O40] where two vibrational bands were found to be present. However, V2POM5− substituted with two V atoms, showed triplet combinations of vibrational bands for P–Oi, VOt, and WOt.
Comparison of the different IR spectra presented in Fig. 5 and Table 2 provides insight into how V substitution for W and interaction of POMs with the different terminal functional groups of SAMs influence their vibrational properties. For instance, the P–O mode, which appears as a single peak at 1084 cm−1 for WPOM3− on FSAM, is observed as two peaks spread across a broader wavenumber range for VPOM4− on all three SAMs. This difference is consistent with the reduction in molecular symmetry that accompanies the substitution of W with a V atom in the external metal–oxygen cage of VPOM4−. In addition, the WOt peak, which appears at 988 cm−1 for WPOM3− on FSAM, is shifted to lower wavenumber for VPOM4− on all three SAMs. This shift is most pronounced for VPOM4− on the COOH-SAM surface where the peak appears at 978 cm−1. A substantial shift in the WOt mode is reasonable considering that these bonds are located on the outside of the POM and, therefore, interact intimately with the SAM surface and adjacent POMs. Compared to WPOM3−, shifts to lower wavenumber are also observed for the W–Ob2–W band of VPOM4− where the effect is again most prominent on the COOH-SAM. Compared to WPOM3−, the W–Ob1–W bands of VPOM4− exhibit shifts to lower wavenumber on COOH-SAM and higher wavenumber on HSAM and FSAM. This difference may be attributed to the polar anionic character of carboxylate (COO−) groups that may form through partial deprotonation of COOH. In comparison, the terminal CH3 and CF3 groups of the HSAM and FSAM are both nonpolar and comparatively inert.
| IR assignment | WPOM3− (cm−1) | VPOM4− (cm−1) | |||
|---|---|---|---|---|---|
| FSAM (exp.) | FSAM (exp.) | HSAM (exp.) | COOH-SAM (exp.) | Gas-phase (calc.) | |
| VOt |
1093 | 1093 | 1095 | 1107 | |
| P–Oa | 1084 | 1074 (10) | 1071 (13) | 1067 (17) | 1097 |
| WOt |
988 | 988 (0) | 987 (1) | 978 (10) | 974 |
| W–Ob2–W | 900 | 901 (−1) | 899 (1) | 892 (8) | 887 |
| W–Ob1–W | 836 | 845 (−9) | 842 (−6) | 835 (1) | 845 |
In a previous study, we calculated how the different vibrational modes of WPOM3− shift as a function of the charge state of the supported POM.27 Shifts in vibrational frequencies are also related to the interaction of Mo or W with the rest of the Keggin structure. For example, the IR peak positions of WPOM3− anions soft-landed on an FSAM surface shifted toward higher wavenumber when compared with other soft-landed anions such as MoPOM.3− This finding may be related to the weaker bonding in MoPOM3− compared to WPOM.3− The WOt band, in particular, exhibited a pronounced shift to higher wavenumber as the charge state of WPOM decreased from 3- to 2- to 1- to 0 due to oxidation. The W–Ob1,2–W bands also exhibited modest shifts to higher wavenumber with decreasing anionic charge of the WPOM. However, as shown in Fig. 5 and Table 2, most of the shifts observed for VPOM4− on the three SAMs were in the opposite direction toward lower wavenumber. Based on these findings, it appears that only a small amount of charge reduction or neutralization of VPOM4− occurs on the SAMs following soft landing, which is beneficial for maintaining the native electronic configuration of VPOM4−.
Ot band (∼988 cm−1) is found to shift depending on the position (and surface coverage) as well as the type of SAM. For example, on the FSAM, the peak shifted from ∼980 cm−1 at the edge of the deposited spot to ∼987 cm−1 at the center. On the HSAM, we observed similar but less pronounced behavior where the WOt peak shifted from 978 cm−1 to 981 cm−1 across the deposited spot. In contrast, on the COOH-SAM a consistent peak position at 974 cm−1 was observed that did not change with the position or coverage of VPOM4−. The shift of the WOt band to higher wavenumber near the center of the FSAM and HSAM may be explained by an increase in the surface coverage of VPOM4−. Specifically, near the edges of the spot where the surface coverage is low, the soft landed VPOM4− interact primarily with the SAM surfaces because there is a large space between neighboring POMs. However, as the surface coverage increases near the center of the spot, the deposited POMs interact more with each other and less with the underlying support. Indeed, at sufficiently high coverage multiple layers of POMs may form that are not in direct contact with the SAM but rather with previously deposited POMs. Interestingly, POMs supported on the COOH-SAM do not exhibit the same shift in wavenumber as those deposited on HSAM and FSAM. The WOt band is also much broader on the COOH-SAM compared to the HSAM and FSAM, where this band appears sharper. The lack of a shift on the COOH-SAM may result from the presence of a fraction of deprotonated carboxylate anions (COO−) on the surface that give the COOH-SAM a partial negative charge. A mixture of COOH and COO− surface sites would also explain the broader WOt peak observed on the COOH-SAM. In comparison, no negative charge is present on the bare HSAM and FSAM so the change in charge at the surface following soft landing of VPOM4− may be more substantial than on the COOH-SAM. These results show that in addition to POM-surface interactions, POM–POM interactions play an important role in determining the vibrational properties of supported POMs.
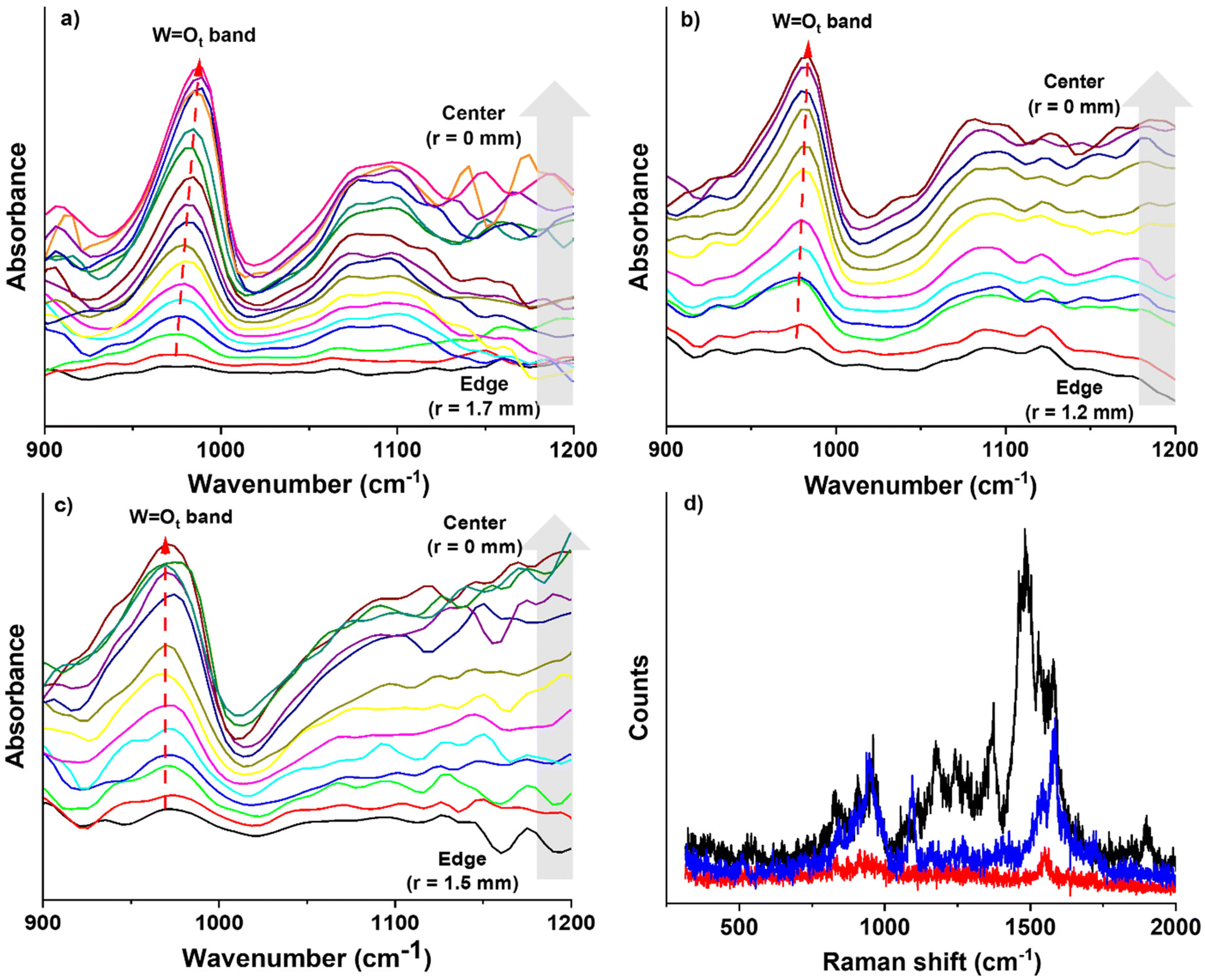 | ||
| Fig. 6 Spatially-resolved infrared s-SNOM spectra obtained following deposition of 1 × 1014 VPOM4− ions on (a) FSAM, (b) HSAM, and (c) COOH-SAM surfaces on Au. The feature appearing at ∼980 cm−1 corresponds to the terminal WOt vibrational band. Arrows from the bottom to the top indicate data collected from the edge (r = 1.2–1.7 mm) of the soft-landed sample to the center (r = 0 mm), (d) TERS spectra of VPOM4− obtained at three different positions in the same nanoindentation on FSAM/Au. The peaks observed beyond 1150 cm−1 may result from inter-cluster vibrations and the underlying SAM on Au. | ||
Ot vibrational modes reported previously in the literature for WPOM3−.55,56 The simulated Raman vibrational spectra from DFT calculations also reproduces such WOt peaks between 900–1000 cm−1, as illustrated in Fig. 5d. Furthermore, unlike the experimental Raman spectra, the DFT simulated spectra do not show any peaks beyond 1150 cm−1. Since the simulated Raman results correspond to the vibrations of POM clusters in the gas phase, the additional peaks observed in the experiments beyond 1150 cm−1 may result from inter-cluster vibrations and the underlying SAM on Au. The varying intensity of the three Raman spectra indicate different levels of enhancement depending on the position in a single nanoindentation. Differences in the relative intensity of the peaks between spectra suggest that even when using a highly-controlled deposition technique such as ISL, there is still substantial heterogeneity between different regions of supported POMs. These differences may result from varying orientations of the reduced symmetry VPOM4− on the surface as well as from disordered regions of the SAMs. These findings emphasize the importance of spatially-resolved spectroscopic probes in understanding the role of nanoscale heterogeneity in determining the properties of materials for potential QC applications.
Conclusions
POMs have the potential to be used as transformative electronic spin-based MQs for QC applications. In practical devices, MQs have to be immobilized in addressable arrays to form QGs that are capable of performing algorithmic operations. The array environment introduces unavoidable interactions of MQs with supports as well as with neighboring POMs. Therefore, understanding POM-surface and POM–POM interactions and how they may be harnessed to maximize spin coherence times is central to developing addressable MQ arrays. In this contribution, ISL was used to deposit mass-selected VPOM4− onto three different SAM surfaces on gold which were used as representative model systems on which to characterize support and intermolecular interactions. ISL avoids the complications inherent to deposition from solution and provides well-defined samples that are amenable to detailed spectroscopic characterization and high-level theoretical modeling. The electronic structure of VPOM4− synthesized in solution was determined in the gas phase using NIPES and on three different SAMs using scanning KPFM. The terminal chemical functionality and charge transfer properties of the SAMs were shown to exert an influence on the charge state and electronic configuration of supported VPOM4−. The vibrational modes of VPOM4− and their perturbation through interactions with surfaces were also explored using IRRAS and TERS revealing that different functional groups on SAMs and nanoscale heterogeneity both modulate the observed spectroscopic signatures. The effect of surface coverage and intermolecular interactions on the vibrational properties of VPOM4− was also investigated using spatially-resolved s-SNOM revealing spectral shifts that are dependent on POM-support and POM–POM interactions. The geometric and electronic structure of the POMs, along with their vibrational spectra, were also calculated using DFT to provide molecular-level insight into the stability and local spin density in VPOM4−. Collectively, our findings provide insight into how V substitution for W as well as different POM-surface and POM–POM interactions influence the electronic and vibrational properties of supported POMs. The vibrational properties of POMs modulate spin–lattice decoherence rates which determine the suitability of MQ arrays for potential QC applications.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
O. M. P., S. T., D. Z., B. T. O., and E. T. B acknowledge support from the Laboratory Directed Research and Development Program at PNNL. G. E. J., V. P., and V.-A. G. were partially supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division, project 72353 (Interfacial Structure and Dynamics in Separations). P. Z. E., X.-B.W., and W. C. were partially supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences (Chemical Kinetics and Dynamics at Interfaces, FWP 16248). V. A. G. was partially supported by the laboratory directed research and development program (LDRD) at Oak Ridge National Laboratory. Part of this work was performed using EMSL, a National Scientific User Facility sponsored by the DOE's Office of Biological and Environmental Research and located at the Pacific Northwest National Laboratory (PNNL). PNNL is a multiprogram national laboratory operated by Battelle for the U.S. Department of Energy under contract DE-AC05-76RL01830. Computer resources were provided by Research Computing at PNNL and the National Energy Research Scientific Computing Center (NERSC), a DOE Office of Science User Facility operated under contract DE-AC02-05CH11231.References
- M. R. Wasielewski, et al., Exploiting chemistry and molecular systems for quantum information science, Nat. Rev. Chem., 2020, 4, 490–504 CrossRef CAS.
- G. Aromi, D. Aguila, P. Gamez, F. Luis and O. Roubeau, Design of magnetic coordination complexes for quantum computing, Chem. Soc. Rev., 2012, 41, 537–546 RSC.
- M. J. Graham, J. M. Zadrozny, M. S. Fataftah and D. E. Freedman, Forging Solid-State Qubit Design Principles in a Molecular Furnace, Chem. Mater., 2017, 29, 1885–1897 CrossRef CAS.
- J. Lehmann, A. Gaita-Arino, E. Coronado and D. Loss, Quantum computing with molecular spin systems, J. Mater. Chem., 2009, 19, 1672–1677 RSC.
- A. Gaita-Arino, F. Luis, S. Hill and E. Coronado, Molecular spins for quantum computation, Nat. Chem., 2019, 11, 301–309 CrossRef CAS PubMed.
- R. E. P. Winpenny, Quantum Information Processing Using Molecular Nanomagnets As Qubits, Angew. Chem., Int. Ed., 2008, 47, 7992–7994 CrossRef CAS PubMed.
- J. M. Clemente-Juan, E. Coronado and A. Gaita-Arino, Magnetic polyoxometalates: from molecular magnetism to molecular spintronics and quantum computing, Chem. Soc. Rev., 2012, 41, 7464–7478 RSC.
- V. Prabhakaran, G. E. Johnson, B. B. Wang and J. Laskin, In situ solid-state electrochemistry of mass-selected ions at well-defined electrode-electrolyte interfaces, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 13324–13329 CrossRef CAS PubMed.
- T. Ueda, Electrochemistry of Polyoxometalates: From Fundamental Aspects to Applications, ChemElectroChem, 2018, 5, 823–838 CrossRef CAS.
- G. A. Timco, et al., Engineering the coupling between molecular spin qubits by coordination chemistry, Nat. Nanotechnol., 2009, 4, 173–178 CrossRef CAS PubMed.
- C. J. Yu, et al., Spin and Phonon Design in Modular Arrays of Molecular Qubits, Chem. Mater., 2020, 32, 10200–10206 CrossRef CAS.
- M. J. Amdur, et al., Chemical control of spin-lattice relaxation to discover a room temperature molecular qubit, Chem. Sci., 2022, 13, 7034–7045 RSC.
- J. Laskin, G. E. Johnson, J. Warneke and V. Prabhakaran, From Isolated Ions to Multilayer Functional Materials Using Ion Soft Landing, Angew. Chem., Int. Ed., 2018, 57, 16270–16284 CrossRef CAS PubMed.
- G. E. Johnson, D. Gunaratne and J. Laskin, Soft- and reactive landing of ions onto surfaces: Concepts and applications, Mass Spectrom. Rev., 2016, 35, 439–479 CrossRef CAS PubMed.
- J. Cyriac, T. Pradeep, H. Kang, R. Souda and R. G. Cooks, Low-Energy Ionic Collisions at Molecular Solids, Chem. Rev., 2012, 112, 5356–5411 CrossRef CAS PubMed.
- S. Rauschenbach, M. Ternes, L. Harnau and K. Kern, Mass Spectrometry as a Preparative Tool for the Surface Science of Large Molecules, Annu. Rev. Anal. Chem., 2016, 9, 473–498 CrossRef CAS PubMed.
- S. Rauschenbach, et al., Electrospray ion beam deposition of clusters and biomolecules, Small, 2006, 2, 540–547 CrossRef CAS PubMed.
- A. Tata, C. Salvitti and F. Pepi, From vacuum to atmospheric pressure: A review of ambient ion soft landing, Int. J. Mass Spectrom., 2020, 450, 116309 CrossRef CAS.
- A. Walz, K. Stoiber, A. Huettig, H. Schlichting and J. V. Barth, Navigate Flying Molecular Elephants Safely to the Ground: Mass- Selective Soft Landing up to the Mega-Dalton Range by Electrospray Controlled Ion-Beam Deposition, Anal. Chem., 2022, 94, 7767–7778 CrossRef CAS PubMed.
- S. Kahle, et al., The Quantum Magnetism of Individual Manganese-12-Acetate Molecular Magnets Anchored at Surfaces, Nano Lett., 2012, 12, 518–521 CrossRef CAS PubMed.
- A. Saywell, et al., Self-assembled aggregates formed by single-molecule magnets on a gold surface, Nat. Commun., 2010, 1, 4392 Search PubMed.
- G. E. Johnson, Q. C. Hu and J. Laskin, Soft Landing of Complex Molecules on Surfaces, Annu. Rev. Anal. Chem., 2011, 4, 83–104 CrossRef CAS PubMed.
- N. Vats, et al., Electron microscopy of polyoxometalate ions on graphene by electrospray ion beam deposition, Nanoscale, 2018, 10, 4952–4961 RSC.
- V. Prabhakaran, et al., Rational design of efficient electrode-electrolyte interfaces for solid-state energy storage using ion soft landing, Nat. Commun., 2016, 7, 11399 CrossRef CAS PubMed.
- G. E. Johnson, et al., DRILL Interface Makes Ion Soft Landing Broadly Accessible for Energy Science and Applications, Batteries Supercaps, 2018, 1, 97–101 CrossRef PubMed.
- K. D. D. Gunaratne, et al., Controlling the Charge State and Redox Properties of Supported Polyoxometalates via Soft Landing of Mass-Selected Ions, J. Phys. Chem. C, 2014, 118, 27611–27622 CrossRef CAS.
- K. D. D. Gunaratne, V. Prabhakaran, A. Andersen, G. E. Johnson and J. Laskin, Charge retention of soft-landed phosphotungstate Keggin anions on self-assembled monolayers, Phys. Chem. Chem. Phys., 2016, 18, 9021–9028 RSC.
- V. Prabhakaran, et al., Controlling the Activity and Stability of Electrochemical Interfaces Using Atom-by-Atom Metal Substitution of Redox Species, ACS Nano, 2019, 13, 458–466 CrossRef CAS PubMed.
- S. A. Miller, H. Luo, S. J. Pachuta and R. G. Cooks, Soft-landing of polyatomic ions at fluorinated self-assembled monolayer surfaces, Science, 1997, 275, 1447–1450 CrossRef CAS.
- G. E. Johnson, T. Priest and J. Laskin, Charge Retention by Gold Clusters on Surfaces Prepared Using Soft Landing of Mass Selected Ions, ACS Nano, 2012, 6, 573–582 CrossRef CAS PubMed.
- G. E. Johnson, T. Priest and J. Laskin, Coverage-Dependent Charge Reduction of Cationic Gold Clusters on Surfaces Prepared Using Soft Landing of Mass-Selected Ions, J. Phys. Chem. C, 2012, 116, 24977–24986 CrossRef CAS.
- D. M. Alloway, et al., Interface dipoles arising from self-assembled monolayers on gold: UV-photoemission studies of alkanethiols and partially fluorinated alkanethiols, J. Phys. Chem. B, 2003, 107, 11690–11699 CrossRef CAS.
- T. Waters, et al., Photoelectron Spectroscopy of free multiply charged keggin anions alpha-[PM12O40]3− (M = Mo, W) in the gas phase, J. Phys. Chem. A, 2006, 110, 10737–10741 CrossRef CAS PubMed.
- H. Wang, et al., In Operand X-ray Absorption Fine Structure Studies of Polyoxometalate Molecular Cluster Batteries: Polyoxometalates as Electron Sponges, J. Am. Chem. Soc., 2012, 134, 4918–4924 CrossRef CAS PubMed.
- S. Q. Liu, H. Mohwald, D. Volkmer and D. G. Kurth, Polyoxometalate-based electro- and photochromic dual-mode devices, Langmuir, 2006, 22, 1949–1951 CrossRef CAS PubMed.
- D. Velessiotis, et al., Molecular junctions made of tungsten-polyoxometalate self-assembled monolayers: Towards polyoxometalate-based molecular electronics devices, Microelectron. Eng., 2011, 88, 2775–2777 CrossRef CAS.
- M. T. Ma, et al., Gas-Phase Fragmentation of Polyoxotungstate Anions, Inorg. Chem., 2009, 48, 598–606 CrossRef CAS PubMed.
- X.-B. Wang and L.-S. Wang, Photoelectron Spectroscopy of Multiply Charged Anions, Annu. Rev. Phys. Chem., 2009, 60, 105–126 CrossRef CAS PubMed.
- L.-S. Wang and X.-B. Wang, Probing Free Multiply Charged Anions Using Photodetachment Photoelectron Spectroscopy, J. Phys. Chem. A, 2000, 104, 1978–1990 CrossRef CAS.
- X.-B. Wang and L.-S. Wang, Observation of negative electron-binding energy in a molecule, Nature, 1999, 400, 245–248 CrossRef CAS.
- J. Warneke, et al., Electronic Structure and Stability of [B12X12]2− (X = F–At): A Combined Photoelectron Spectroscopic and Theoretical Study, J. Am. Chem. Soc., 2017, 139, 14749–14756 CrossRef CAS PubMed.
- X. Yang, et al., Photoelectron spectroscopy of free polyoxoanions Mo6O192− and W6O192− in the gas phase, J. Phys. Chem. A, 2004, 108, 10089–10093 CrossRef CAS.
- F. Rissner, et al., Understanding the Electronic Structure of Metal/SAM/Organic−Semiconductor Heterojunctions, ACS Nano, 2009, 3, 3513–3520 CrossRef CAS PubMed.
- H. Fukushima, et al., Microstructure, Wettability, and Thermal Stability of Semifluorinated Self-Assembled Monolayers (SAMs) on Gold, J. Phys. Chem. B, 2000, 104, 7417–7423 CrossRef CAS.
- H. Wang, S. Chen, L. Li and S. Jiang, Improved Method for the Preparation of Carboxylic Acid and Amine Terminated Self-Assembled Monolayers of Alkanethiolates, Langmuir, 2005, 21, 2633–2636 CrossRef CAS PubMed.
- H. Ron, S. Matlis and I. Rubinstein, Self-assembled monolayers on oxidized metals. 2. Gold surface oxidative pretreatment, monolayer properties, and depression formation, Langmuir, 1998, 14, 1116–1121 CrossRef CAS.
- N. D. Lang and W. Kohn, Theory of Metal-Surfaces - Induced Surface Charge and Image Potential, Phys. Rev. B, 1973, 7, 3541–3550 CrossRef CAS.
- J. C. Orozco, D. T. Shuaib, C. L. Marshall and M. I. Khan, Divanadium substituted keggin [PV2W10O40] on non-reducible supports-Al2O3 and SiO2: synthesis, characterization, and catalytic properties for oxidative dehydrogenation of propane, React. Kinet., Mech. Catal., 2020, 131, 753–768 CrossRef CAS.
- R. Thouvenot, M. Fournier, R. Franck and C. Rocchicciolideltcheff, Vibrational Investigations of Polyoxometalates .3. Isomerism in Molybdenum(VI) and Tungsten(VI) Compounds Related to the Keggin Structure, Inorg. Chem., 1984, 23, 598–605 CrossRef CAS.
- P. Su, V. Prabhakaran, G. E. Johnson and J. Laskin, In Situ Infrared Spectroelectrochemistry for Understanding Structural Transformations of Precisely Defined Ions at Electrochemical Interfaces, Anal. Chem., 2018, 90, 10935–10942 CrossRef CAS PubMed.
- X. Lopez, J. M. Maestre, C. Bo and J. M. Poblet, Electronic properties of polyoxometalates: A DFT study of alpha/beta-[XM12O40]n− relative stability (M=W, Mo and X a main group element), J. Am. Chem. Soc., 2001, 123, 9571–9576 CrossRef CAS PubMed.
- M. J. Watras and A. V. Teplyakov, Infrared and computational investigation of vanadium-substituted Keggin [PVnW12−-nO40]n+3− polyoxometallic anions, J. Phys. Chem. B, 2005, 109, 8928–8934 CrossRef CAS PubMed.
- O. Dmitrenko, et al., Effect of cations in infrared and computational analysis of vanadium-containing six-coordinate oxotungstates, J. Phys. Chem. B, 2003, 107, 7747–7752 CrossRef CAS.
- C. F. Wang, B. T. O'Callahan, A. Krayev and P. Z. El-Khoury, Nanoindentation-enhanced tip-enhanced Raman spectroscopy, J. Chem. Phys., 2021, 154, 241101 CrossRef CAS PubMed.
- C. Rocchicciolideltcheff, M. Fournier, R. Franck and R. Thouvenot, Vibrational Investigations of Polyoxometalates .2. Evidence for Anion Anion Interactions in Molybdenum(VI) and Tungsten(VI) Compounds Related to the Keggin Structure, Inorg. Chem., 1983, 22, 207–216 CrossRef CAS.
- A. J. Bridgeman, Density functional study of the vibrational frequencies of alpha-Keggin heteropolyanions, Chem. Phys., 2003, 287, 55–69 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and theoretical methods. Spectrum and table comparison of the m/z agreement between the experimentally measured and theoretically calculated isotope values for VPOM4− (PDF). See DOI: https://doi.org/10.1039/d2nr06148a |
| This journal is © The Royal Society of Chemistry 2023 |