
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling the orientation and dynamics of enzymes in unstructured artificial compartments of metal–organic frameworks (MOFs)†
Yanxiong
Pan
 *a,
Qiaobin
Li
b,
Wei
Liu
a,
Zoe
Armstrong
b,
Austin
MacRae
b,
Li
Feng
b,
Charles
McNeff
b,
Pinjing
Zhao
b,
Hui
Li
*c and
Zhongyu
Yang
*b
*a,
Qiaobin
Li
b,
Wei
Liu
a,
Zoe
Armstrong
b,
Austin
MacRae
b,
Li
Feng
b,
Charles
McNeff
b,
Pinjing
Zhao
b,
Hui
Li
*c and
Zhongyu
Yang
*b
aState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, China. E-mail: yxpan@ciac.ac.cn
bDepartment of Chemistry and Biochemistry, North Dakota State University, Fargo, ND 58108, USA
cDepartment of Plant Sciences, North Dakota State University, Fargo, ND 58108, USA. E-mail: hui.li.3@ndsu.edu
First published on 12th January 2023
Abstract
Confining enzymes in well-shaped MOF compartments is a promising approach to mimic the cellular environment of enzymes and determine enzyme structure–function relationship therein. Under the cellular crowding, however, enzymes can also be confined in unstructured spaces that are close to the shapes/outlines of the enzyme. Therefore, for a better understanding of enzymes in their physiological environments, it is necessary to study enzymes in these unstructured spaces. However, practically it is challenging to create compartments that are close to the outline of an enzyme and probe enzyme structural information therein. Here, for proof-of-principle, we confined a model enzyme, lysozyme, in the crystal defects of a MOF via co-crystallization, where lysozyme served as the nuclei for MOF crystal scaffolds to grow on so that unstructured spaces close to the outline of lysozyme are created, and determined enzyme relative orientation and dynamics. This effort is important for understanding enzymes in near-native environments and guiding the rational design of biocatalysts that mimic how nature confines enzymes.
Confining enzymes in nanoscale compartments has improved biocatalysis and become a promising approach to mimic the confined environment that enzymes experience in cells.1–4 Upon confinement, enzyme activity depends on substrate recognition and access, which further rely on the dynamics and relative orientation of the enzyme.5–8 Thus far, MOFs and Covalent–Organic Frameworks (COFs) are the most advanced enzyme confinement compartments due to their controllable size, shape, and hydrophobicity,8–15 altering/fine-tuning which has led to changes in the conformational dynamics of a confined enzyme and correspondingly, the catalytic efficiency.16
While most of these studies are focused on structured compartments, in cells, enzymes can also be (partially) confined in unstructured spaces (slightly larger than themselves due to crowding or partially embedded in membranes) and still display the optimal catalytic activity (likely optimized by nature during evolution).17–20 Therefore, it becomes necessary to study enzymes in these unstructured compartments and probe their backbone dynamics and relative orientation therein. This effort will complement the current knowledge of enzymes confined in well-shaped compartments16 and provide a thorough understanding of enzymes confined in spaces with unstructured shapes that occur in cells, shedding light on nature-inspired biocatalyst design.
It is challenging to create unstructured compartments for enzymes using most available porous materials. A potential solution is co-crystallizing enzymes in the crystal defects of MOFs,21–23 where enzymes serve as the nuclei for MOF crystal scaffolds to grow upon. The resultant composites appear to have enzymes entrapped in the “defects” of the MOF scaffolds. However, the popular Zeolitic Imidazolate Framework prepared in methanol (ZIF-8)21,24 creates high restrictions on enzyme backbone dynamics due to the well-packed crystals and does not serve as a good platform to mimic the unstructured enzyme spaces in cells. It is also challenging to probe the dynamics and orientation of enzymes under the interference of the materials of compartments using most experimental techniques.
Here, for proof-of-principle, we show a possible approach to create unstructured compartments for enzymes and determine enzyme backbone dynamics and orientation therein. In doing so, we co-crystallized a model enzyme, lysozyme (lys; 2.5 × 3 × 4.5 nm), with Zn2+ and imidazolate in the aqueous phase (designated as aq-ZIF), which resulted in less intensely packed composites with enhanced lys freedom as compared in ZIF-8 (aperture 0.6–0.7 nm). We then probed the site-specific backbone dynamics of lys confined in the crystal defects of these composites at the residue level using site-directed spin labeling (SDSL) Electron Paramagnetic Resonance (EPR) spectroscopy, which is immune of the complexities of the background25–29 and can reveal enzyme dynamics and orientation.16,21,30 We found that the enzyme was confined both on the crystal surface (partially embedded) and within crystal defects, a condition also discovered in our recent works.21,30–36 We then distinguished the latter from the former using a recently reported urea-perturbation strategy,21 followed by revealing the backbone dynamics and contact residues of lys in the co-crystal defects. To our best knowledge, this is the first report on experimental unveiling of enzyme dynamics in unstructured artificial compartments. The findings are important for guiding the design of novel biocatalysts and compartments that can better mimic the cellular environment of enzymes.37–39
Lys was selected as the model enzyme in this study because of its capability of forming enzyme@ZIF composites and its extensively studied dynamics using SDSL-EPR.21,40 The one-pot co-crystallization of lys with Zn2+ and imidazolate was carried out in water (details see the ESI†). Transmission Electron Microscopy (TEM) images of the co-crystals (designated as aq-ZIF/lys) show morphology (Fig. 1B) similar to that without lys (Fig. 1A). The shape of crystals is different from the hexagon-like ZIF-8 formed in MeOH,21 likely due to the less intensely packed crystal scaffolds.
 | ||
| Fig. 1 TEM images of aq-ZIF in the absence (A) and presence of lys (B). | ||
Powder X-ray Diffraction (PXRD; Fig. 2A) shows that the incorporation of lys did not cause major changes in the crystal scaffold. Thermal gravimetric analysis (TGA) shows a ∼2.9% weight loss (Fig. 2B, 200 to 400 °C), which is close to the loading capacity estimated by disassembling the co-crystals and measuring lys concentration. The loading capacity is lower as compared to lys@ZIF-8 prepared in MeOH,21,23 which is reasonable due to the reduced packing intensity in water. Degradation of aq-ZIF and aq-ZIF/lys is also present (Fig. 2B, 581.7 and 584.8 °C, respectively). N2 absorption experiments indicate a decrease in the porosity of the crystal from 1628.4 m2 g−1 without lys to 1306.6 m2 g−1 upon lys trapping (Fig. 2C). Lys activity was assessed by monitoring the optical density of bacterial cell walls at 450 nm (OD450). Upon confirming no change in OD450 was caused by aq-ZIF alone (Fig. 2D blue), we observed a decay of OD450 (Fig. 2D red) from aq-ZIF/lys. The decay rate is slower than that of lys in buffer (Fig. 2D black), indicating lys trapped on aq-ZIF is only partially active. This is consistent with our recent finding.21 Because the substrates of lys is bacterial cell walls (μm scale), which is much larger than aq-ZIF apertures (0.6–0.7 nm), the presence of active lys indicates that lys must be partially exposed above the aq-ZIF crystal surface, in order to contact the substrates.
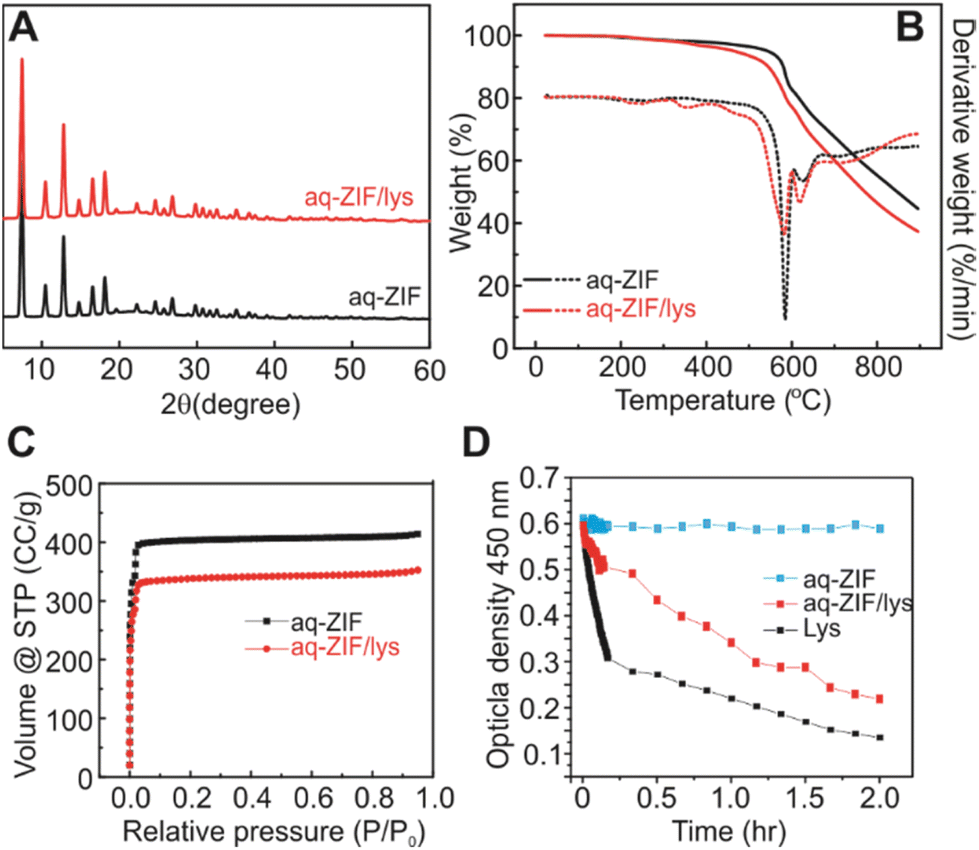 | ||
| Fig. 2 PXRD (A), TGA (B), and nitrogen absorption experiments (C) of aq-ZIF (black) and aq-ZIF/lys composite (red). (D) Activity assay of lys (black), aq-ZIF (blue), and aq-ZIF/lys composite (red). | ||
To probe the site-specific backbone dynamics of lys confined in the crystal defects of aq-ZIF, SDSL were conducted by creating 6 cysteine mutants of the recombinant T4 phage lysozyme (T4L), one at a time, scanning most regions of the enzyme, followed by spin-labeling (forming the R1 sidechain; Fig. 3A).25 Continuous wave (CW) EPR was used to probe the dynamics of each labeled site (Fig. 3B–G). For all sites, CW EPR results in first derivative spectra with the low-, mid-, and high-field regions due to the hyperfine splitting.25 Within each region, an immobile component and a mobile one are resolved (marked as “im” and “m” in the low-field; Fig. 3B–G grey shades). The spectrum of each component is decided by three motions, protein rotational tumbling, backbone dynamics, and R1 intrinsic motion.25,41,42 When the rotational tumbling is restricted, the resultant spectra have the typical line shapes shown in Fig. 3B–G (dotted curves). If the labeled site is in contact with some species (ca. aq-ZIF scaffolds), the backbone dynamics and R1 intrinsic motion are restricted, resulting in broader spectra with the “im” component populated (blue curves in Fig. 3B–G).
 | ||
| Fig. 3 (A) Schematic illustration of SDSL and generation of the R1 sidechain. (B) to (G) CW EPR spectra of the six labelled mutants in buffer (black), upon immobilized on a sepharose surface (dots), trapped in aq-ZIF (blue), and upon treatment of urea (orange). (H) Illustration of the origins of the “im” and “m” spectral components and upon treatment of urea (I). | ||
We observed both “m” and “im” components for all sites. As discussed above, the “im” component is caused by R1 in contact with a defect (Fig. 3H blue arrow); scanning multiple enzyme surface regions can reveal which lys regions contact the cavity and lys orientation. The “m” component can originate from two sources, the labelled residue being exposed to the solvent (Fig. 3H yellow arrow21) or facing a space within a defect (but not contact the scaffold; Fig. 3H orange arrow). Revealing the “m” component in the defect (Fig. 3H orange arrow) will lead to site-specific backbone dynamics of lys. The contribution of “m” component from the enzyme portion exposed to the solvent can be distinguished from that within the defects using a urea-perturbation method (Fig. 3I), wherein the former shows enhanced dynamics (sharpened “m” peak) while the latter is unchanged (Fig. 3I yellow vs. orange arrows). When all cases are present, spectral analysis can be used to deconvolute each case's contribution.
To quantitatively understand the data, we carried out spectral simulation by varying the rate and order parameters of R1 motion until the best fit was reached (details see the ESI†).43 The resultant parameters (Tables S2†) confirmed that the “im” component is from a highly ordered, slow motion caused by R1 in contact with the defects. The “m” component shows relatively low order and fast motion, consistent with R1 having certain freedom. The relative population of the “m” component varies between 25–56% depending on labeled sites (Table 1 column 2). To determine the “m” component in the defects, we introduced 6 M urea to unfold the solvent-exposed portion of lys (but not the buried ones44). The resultant “m” component population is shown in column 3 of Table 1.
| Mutant | Mobile%a | Urea%b | Δmobile%c |
|---|---|---|---|
| a Population percentage of the mobile component in aq-ZIF/lys. b Population percentage of the mobile component upon treatment of urea. c Population difference between the former two cases. | |||
| 44R1 | 42 | 32 | 10 |
| 65R1 | 53 | 29 | 24 |
| 72R1 | 56 | 21 | 25 |
| 118R1 | 25 | 18 | 7 |
| 131R1 | 46 | 17 | 29 |
| 151R1 | 38 | 13 | 25 |
The difference between columns 2 and 3 of Table 1 is the “m” component of the labelled sites in the defects. As indicate by column 4 of Table 1, two sites, 44R1 and 118R1, have a lower chance to have such freedom thus a higher chance to contact the inner walls of the cavities as compared to other sites. Based on this observation, we propose the relative arrangement of enzyme in cavities as shown in Fig. 4B, wherein 65, 72, 131, and 151 regions have less spatial restriction, or, more dynamics, than other protein regions.
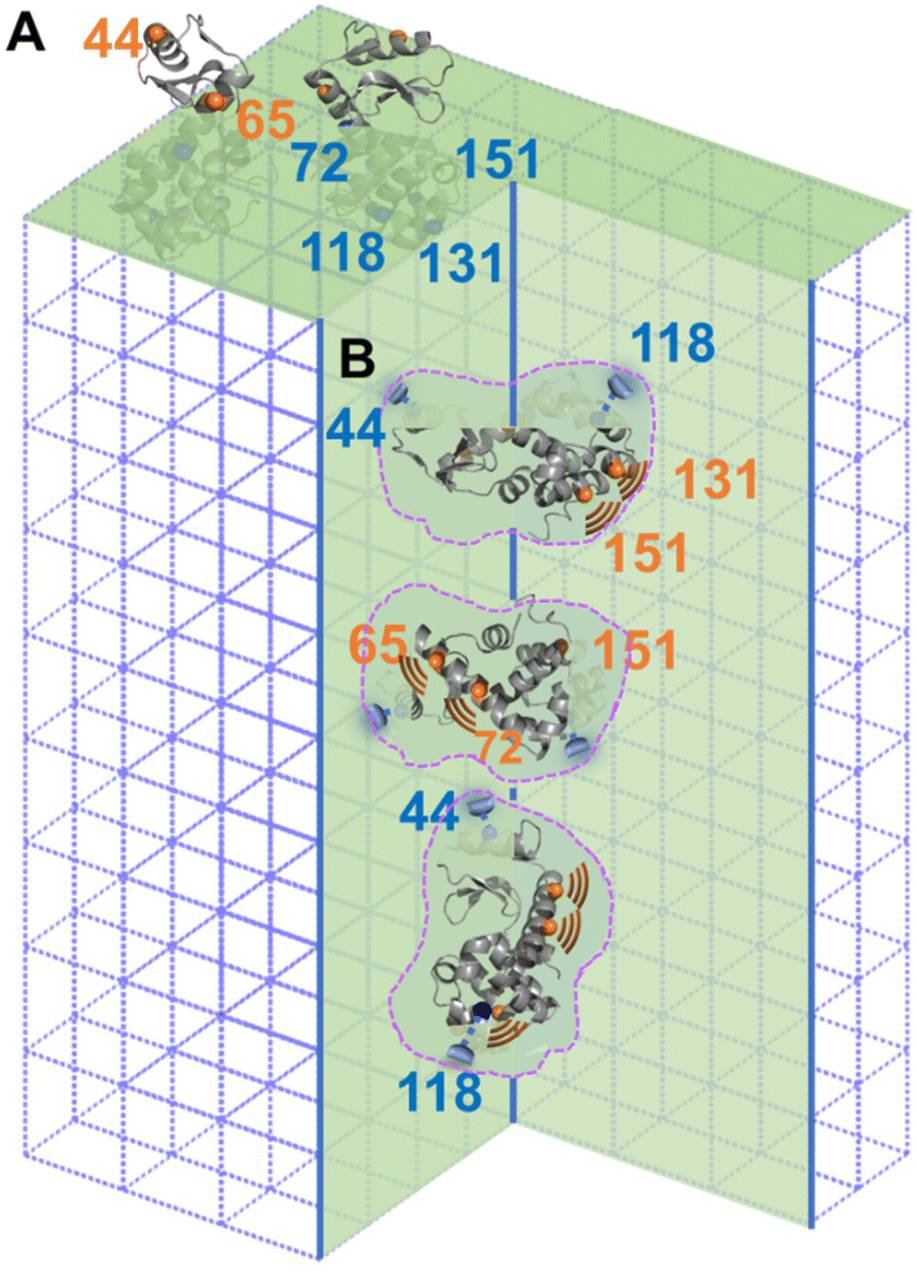 | ||
| Fig. 4 Schematic illustration of lys trapped on the surface (with two views; A) and inside of the crystal defects (with three views; B). Sites forming stronger interaction with aq-ZIF are shown in blue while those tend to have less interactions and, therefore, a higher DOF, are shown in orange. Dotted areas indicate crystal defects in the aq-ZIF scaffold. | ||
A careful look at column 3 of Table 1 indicates that 44R1 and 65R1 have a higher chance to be exposed to solvents compared to other labelled sites, indicating that the N-terminus is more likely to be exposed above the crystal surface. This led to a preferred enzyme orientation on the crystal surface as modelled in Fig. 4A. The overall picture of how lys being encapsulated both on the surface and in the cavities of aq-ZIF is shown in Fig. 4.
Co-crystallization of enzymes with certain metal ions and ligands is a unique way to immobilize enzymes so that some enzymes are “implanted” on MOFs (like a tree where the “root” is buried under the crystal surface and the “leaves and branches” are exposed above; Fig. 4A) while others completely buried under MOF surfaces (Fig. 4B). Such a unique enzyme immobilization approach has been observed and confirmed in several of our recent works.21,30–36 In this communication, we report the case of aq-ZIF. Because urea can only unfold the exposed portion of enzymes (the buried portion is protected by the MOF scaffolds),21,44 column 3 of Table 1 reports the chance for different lys regions to be exposed (13–32% depending on labelled regions). Thus, the majority of the enzyme is buried under aq-ZIF surface. It has to be noted that the crystallinity and porosity of aq-ZOF were not changed significantly, because only 2.9% w/w of aq-ZIF is occupied by lys. In other words, the majority of the aq-ZIF shows the expected crystal and porous structure. These also confirmed that lys entrapment did not collapse the aq-ZIF skeleton. The “defects” were created by lys which serves as the crystallization nuclei (instead of loading lys to big defects formed after crystallization). Note that although aq-ZIF is a crystal, the lys enzyme trapped in aq-ZIF is not crystallized. Instead, these enzymes are active and dynamic, as shown in our EPR-based dynamics measurements.
Conclusions
We created unstructured spatial confinement on a model enzyme by co-crystallizing the enzyme with Zn2+ and imidazolate in the aqueous phase. Using SDSL-EPR and a urea-perturbation test, we determined the regions of the enzyme that prefer to contact the unstructured compartment and those possess more backbone dynamics, which revealed the orientation and dynamics of the model enzyme in the unstructured compartments. This effort is important for the rational design of biocatalysts based on enzymes confined in unstructured compartments. Knowing this information will also shed light on the general understanding of enzymes under the physiological conditions.45Author Contributions
Y. P., H. L., and Z. Y. designed the research. Y. P., Q. L., W. L., and H. L. carried out the data acquisition. Z. A., A. M. L. F., C. M., and P. Z. assisted in data analysis. All authors contributed to manuscript drafting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Science Foundation (MCB 1942596) to Z.Y. We sincerely appreciate Prof. Wayne Hubbell for providing the software for spectral simulation.References
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS.
- N. Carlsson, H. Gustafsson, C. Thörn, L. Olsson, K. Holmberg and B. Åkerman, Adv. Colloid Interface Sci., 2014, 205, 339–360 CrossRef CAS PubMed.
- Z. Zhou and M. Hartmann, Chem. Soc. Rev., 2013, 42, 3894–3912 RSC.
- C.-H. Lee, T.-S. Lin and C.-Y. Mou, Nano Today, 2009, 4, 165–179 CrossRef CAS.
- U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453–468 RSC.
- K. N. Houk, A. G. Leach, S. P. Kim and X. Zhang, Angew. Chem., Int. Ed., 2003, 42, 4872–4897 CrossRef CAS PubMed.
- V. Van Meervelt, M. Soskine, S. Singh, G. K. Schuurman-Wolters, H. J. Wijma, B. Poolman and G. Maglia, J. Am. Chem. Soc., 2017, 139, 18640–18646 CrossRef CAS PubMed.
- Q. Sun, C.-W. Fu, B. Aguila, J. Perman, S. Wang, H.-Y. Huang, F.-S. Xiao and S. Ma, J. Am. Chem. Soc., 2018, 140, 984–992 CrossRef CAS PubMed.
- X. Lian, Y. Fang, E. Joseph, Q. Wang, J. Li, S. Banerjee, C. Lollar, X. Wang and H.-C. Zhou, Chem. Soc. Rev., 2017, 46, 3386–3401 RSC.
- R. J. Drout, L. Robison and O. K. Farha, Coord. Chem. Rev., 2019, 381, 151–160 CrossRef CAS.
- Q. Sun, B. Aguila, P. C. Lan and S. Ma, Adv. Mater., 2019, 31, 1900008 CrossRef PubMed.
- Y. Song, Q. Sun, B. Aguila and S. Ma, Adv. Sci., 2019, 6, 1801410 CrossRef PubMed.
- M. B. Majewski, A. J. Howarth, P. Li, M. R. Wasielewski, J. T. Hupp and O. K. Farha, CrystEngComm, 2017, 19, 4082–4091 RSC.
- E. Gkaniatsou, C. M. Sicard, R. M. Ricoux, J.-P. Mahy, N. Steunou and C. Serre, Mater. Horiz., 2017, 4, 55–63 RSC.
- C. Doonan, R. Riccò, K. Liang, D. Bradshaw and P. Falcaro, Acc. Chem. Res., 2017, 50, 1423–1432 CrossRef CAS PubMed.
- Q. Sun, Y. Pan, X. Wang, H. Li, J. Farmakes, B. Aguila, Z. Yang and S. Ma, Chem, 2019, 5, 3184–3195 CAS.
- B. Ma and R. Nussinov, Dynamics in Enzyme Catalysis, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 123–137, DOI:10.1007/128_2012_316.
- H.-X. Zhou, G. N. Rivas and A. P. Minton, Ann. Rev. Biophys., 2008, 37, 375–397 CrossRef CAS PubMed.
- A. Zotter, F. Bäuerle, D. Dey, V. Kiss and G. Schreiber, J. Biol. Chem., 2017, 292, 15838–15848 CrossRef CAS PubMed.
- R. J. Ellis, Trends Biochem. Sci., 2001, 26, 597–604 CrossRef CAS PubMed.
- Y. Pan, H. Li, J. Farmakes, F. Xiao, B. Chen, S. Ma and Z. Yang, J. Am. Chem. Soc., 2018, 140, 16032–16036 CrossRef CAS PubMed.
- W. Liang, H. Xu, F. Carraro, N. K. Maddigan, Q. Li, S. G. Bell, D. M. Huang, A. Tarzia, M. B. Solomon, H. Amenitsch, L. Vaccari, C. J. Sumby, P. Falcaro and C. J. Doonan, J. Am. Chem. Soc., 2019, 141, 2348–2355 CrossRef CAS PubMed.
- C. Hu, Y. Bai, M. Hou, Y. Wang, L. Wang, X. Cao, C.-W. Chan, H. Sun, W. Li, J. Ge and K. Ren, Sci. Adv., 2020, 6, eaax5785 CrossRef CAS PubMed.
- F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761–5765 CrossRef CAS PubMed.
- W. L. Hubbell, C. J. López, C. Altenbach and Z. Yang, Curr. Opin. Struct. Biol., 2013, 23, 725–733 CrossRef CAS PubMed.
- Y. Pan, S. Neupane, J. Farmakes, M. Bridges, J. Froberg, J. Rao, S. Y. Qian, G. Liu, Y. Choi and Z. Yang, Nanoscale, 2017, 9, 3512–3523 RSC.
- D. S. Cafiso, Acc. Chem. Res., 2014, 47, 3102–3109 CrossRef CAS PubMed.
- H. S. McHaourab, P. R. Steed and K. Kazmier, Structure, 2011, 19, 1549–1561 CrossRef CAS PubMed.
- G. E. Fanucci and D. S. Cafiso, Curr. Opin. Struct. Biol., 2006, 16, 644–653 CrossRef CAS PubMed.
- S. Neupane, K. Patnode, H. Li, K. Baryeh, G. Liu, J. Hu, B. Chen, Y. Pan and Z. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 12133–12141 CrossRef CAS PubMed.
- D. Jordahl, Z. Armstrong, Q. Li, R. Gao, W. Liu, K. Johnson, W. Brown, A. Scheiwiller, L. Feng, A. Ugrinov, H. Mao, B. Chen, M. Quadir, Y. Pan, H. Li and Z. Yang, ACS Appl. Mater. Interfaces, 2022, 14, 51619–51629 CrossRef CAS PubMed.
- Y. Pan, Q. Li, H. Li, J. Farmakes, A. Ugrinov, X. Zhu, Z. Lai, B. Chen and Z. Yang, Chem. Catal., 2021, 1, 146–161 CrossRef.
- Y. Pan, H. Li, Q. Li, M. Lenertz, X. Zhu, B. Chen and Z. Yang, Chem. Catal., 2021, 1, 207–231 CrossRef.
- Y. Pan, H. Li, Q. Li, M. Lenertz, I. Schuster, D. Jordahl, X. Zhu, B. Chen and Z. Yang, STAR Protoc., 2021, 2, 100676 CrossRef CAS PubMed.
- Y. Pan, H. Li, M. Lenertz, Y. Han, A. Ugrinov, D. Kilin, B. Chen and Z. Yang, Green Chem., 2021, 33, 4466–4476 RSC.
- J. Farmakes, I. Schuster, A. Overby, L. Alhalhooly, M. Lenertz, Q. Li, A. Ugrinov, Y. Choi, Y. Pan and Z. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 23119–23126 CrossRef CAS PubMed.
- L. Yang, A. Gomez-Casado, J. F. Young, H. D. Nguyen, J. Cabanas-Danés, J. Huskens, L. Brunsveld and P. Jonkheijm, J. Am. Chem. Soc., 2012, 134, 19199–19206 CrossRef CAS PubMed.
- S. Ding, A. A. Cargill, I. L. Medintz and J. C. Claussen, Curr. Opin. Biotechnol., 2015, 34, 242–250 CrossRef CAS PubMed.
- R. C. Rodrigues, C. Ortiz, Á. Berenguer-Murcia, R. Torres and R. Fernández-Lafuente, Chem. Soc. Rev., 2013, 42, 6290–6307 RSC.
- C. J. López, M. R. Fleissner, Z. Guo, A. K. Kusnetzow and W. L. Hubbell, Prot. Sci., 2009, 18, 1637–1652 CrossRef PubMed.
- W. L. Hubbell, A. Gross, R. Langen and M. A. Lietzow, Curr. Opin. Struct. Biol., 1998, 8, 649–656 CrossRef CAS PubMed.
- W. L. Hubbell, D. S. Cafiso and C. Altenbach, Nat. Struct. Biol., 2000, 7, 735–739 CrossRef CAS PubMed.
- D. E. Budil, S. Lee, S. Saxena and J. H. Freed, J. Magn. Reson., Ser. A, 1996, 120, 155–189 CrossRef CAS.
- F. S. Liao, W. S. Lo, Y. S. Hsu, C. C. Wu, S. C. Wang, F. K. Shieh, J. V. Morabito, L. Y. Chou, K. C. W. Wu and C. K. Tsung, J. Am. Chem. Soc., 2017, 139, 6530–6533 CrossRef CAS PubMed.
- P. Li, Q. Chen, T. C. Wang, N. A. Vermeulen, B. L. Mehdi, A. Dohnalkova, N. D. Browning, D. Shen, R. Anderson, D. A. Gómez-Gualdrón, F. M. Cetin, J. Jagiello, A. M. Asiri, J. F. Stoddart and O. K. Farha, Chem, 2018, 4, 1022–1034 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr06659a |
| This journal is © The Royal Society of Chemistry 2023 |