
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Co3−xFexO4 inverse opals with tunable catalytic activity for high-performance overall water splitting†
Thi Hong Trang
Nguyen
 ab,
Zviadi
Zarkua
a,
Chinnabathini Vana
Chinnappa
a,
Wenjian
Hu
ac,
Sreeprasanth Pulinthanathu
Sree
de,
Didier
Grandjean
a,
Deepak
Pant
c and
Ewald
Janssens
*a
ab,
Zviadi
Zarkua
a,
Chinnabathini Vana
Chinnappa
a,
Wenjian
Hu
ac,
Sreeprasanth Pulinthanathu
Sree
de,
Didier
Grandjean
a,
Deepak
Pant
c and
Ewald
Janssens
*a
aQuantum Solid-State Physics (QSP), Department of Physics and Astronomy, KU Leuven, Celestijnenlaan 200 D, 3001 Leuven, Belgium. E-mail: ewald.janssens@kuleuven.be
bDepartment of Physics and Materials Science, Faculty of Natural Sciences, Quy Nhon University, 170 An Duong Vuong, Quy Nhon, Binh Dinh, Vietnam
cSeparation and Conversion Technology, Flemish Institute for Technological Research (VITO), Boeretang 200, 2400 Mol, Belgium
dCenter for Surface Chemistry and Catalysis, KU Leuven, B-3001, Leuven, Belgium
eDepartment of Materials Engineering, KU Leuven, 3001 Leuven, Belgium
First published on 30th May 2023
Abstract
The development of earth-abundant and high-performance bifunctional catalysts for both the oxygen evolution reaction (OER) and the hydrogen evolution reaction (HER) in alkaline electrolytes is required to efficiently produce hydrogen by electrochemical water splitting, but remains a challenge. We have fabricated mesoporous cobalt iron oxide inverse opals (m-CFO IO) with different mole ratios of cobalt and iron by a wet chemical method using polystyrene beads as a hard template, followed by calcination in air. The performance of the m-CFO IO as OER and HER electrocatalysts was investigated. The as-prepared catalyst with equal concentrations of Fe and Co exhibits remarkable OER and HER performances with low overpotentials of 261 and 157 mV to attain 10 mA cm−2 and small Tafel slopes of 63 and 56 mV dec−1, respectively. An alkaline water electrolyzer with a two-electrode configuration achieves 10 mA cm−2 at 1.55 V with excellent long-term stability, outperforming the combination of noble metal IrO2 and Pt/C benchmark catalysts. The superior catalytic performance is ascribed to the synergistic effects of particle size, crystallinity, oxygen efficiency, a large number of active sites, and the large specific surface area of the porous inverse opal structure.
Introduction
Several alternative sources of renewable energy are being developed the last decades to address the world's growing energy demands and environmental threats caused by the depletion of natural fossil resources.1,2 Catalytic splitting of water is a valuable path to produce clean hydrogen and oxygen that can be used in fuel cells and metal–air batteries. However, large overpotentials are typically required to facilitate hydrogen and oxygen evolution reactions (HER and OER, respectively), which reduces the overall water splitting efficiency.3,4 While platinum (Pt) is the most effective catalyst for HER,5 ruthenium oxide (RuO2) and iridium oxide (IrO2) has been proven to be highly efficient OER catalysts.6,7 However the high cost and scarcity of the noble metals impede their widespread use.8–11 Cheap earth-abundant catalysts such as transition metal oxides are a possible alternative. Another promising option to contribute to the sustainability of the overall water splitting reaction are bi-functional catalysts that allow coupling the HER and OER reactions in the same electrolyte. However this remains a challenge as HER catalysts perform best in acidic condition, whereas alkaline solutions are more suited for OER catalysts.12,13Due to its unique electronic structure and high corrosion stability, the spinel-phase Co3O4 electrocatalyst has attracted considerable attention for OER.14 Co possesses multiple oxidation states in the spinel structure, with high-spin Co2+ occupying tetrahedral sites (Co2+Td) and Co3+ octahedral sites (Co3+Oct). These octahedral and tetrahedral sites can be substituted with a range of other first-row transition metals, providing new physicochemical properties and adaptive catalytic centers.15–17 It was recently reported that Fe substitution serves as a supportive core element in spinel, contributing to the long-term durability.18 Theoretical calculations and magnetic measurements revealed that the introduction of Fe3+ ions into the Co3O4 network causes delocalization of the Co 3d electrons and a spin-state transition. Fe3+ ions can effectively activate adjacent Co3+ ions, providing the binary spinel with an enhanced intrinsic oxygen catalytic activity.19,20
Particular efforts have been devoted to the development and further optimization of the OER and HER activity of spinel-type cobalt oxide-based materials, either by tuning the morphology14,21 and defect engineering22,23 or by doping with other transition metals.14,24,25 Waag et al. reported dose-controlled pulsed laser fragmentation in a liquid to generate structural defects on the surface of CoFe2O4.22 Moon et al. studied in situ formation of active cobalt species and KIT-6 silica nanocasted cobalt iron oxide systems showed that the OER activity could be boosted by the incorporation of a small amount of iron.14 Jiang et al. developed a low Tafel slope hydrogen evolution material of Co3O4 supported on nitrogen-doped carbon nanotubes.26 Although the role of iron in OER has been widely studied in cobalt spinel systems, there is limited information about the correlations between the stoichiometry, inversion degree, morphology, and electrocatalytic activity. Moreover, the role of iron in cobalt spinel systems for HER is less understood.
Despite numerous studies, the development of high-performance bifunctional electrocatalysts through a rational design is still desired. An efficient electrocatalyst is expected to: (i) exhibit a high catalytic activity with a large number of active sites for OER and HER processes; (ii) possess sufficient mass transfer pathways for fast electrode kinetics; and (iii) be chemically stable with robust material and/or electrode architecture for high durability. In general, the activity of the electrocatalyst can be enhanced by increasing the number and the intrinsic activity of the active sites. Substantial work dealt with the development of new synthetic approaches to realize this.27–29
Shaping electrocatalyst in an ordered porous morphology brings advantageous features, e.g., facile charge transport via an interconnected nano framework, appropriate oxygen and hydrogen transport via the porous structure, and a high surface area to accomodate the active sites and increase contact between the catalyst and the electrolyte. Template methods have been considered as convenient and effective methods to prepare mesoporous inverse opal (IO) structure, the negative replica of the opal structure, in which the solid spheres are replaced by air phase forming pores and the space between the spheres is filled with a new material. Olivares-Marín et al. proposed binder-free carbon IO with controlled macropore size as Li–O2 cathode in which the dual porosity enabled the best performance.30 Li et al. demonstrated the development of pomegranate-like bifunctional electrocatalyst incorporating Co3O4 nanocrystals in a nitrogen-doped partially graphitized carbon framework for Zn–air batteries.31 Benefiting from the unique pomegranate-like architecture, the composite catalysts possess abundant active sites, strong synergetic coupling, enhanced electron transfer, and high efficiencies in the oxygen reduction and evolution reactions.
In this work, we report on the design of a range of ordered mesoporous metal oxides IO materials. The IO grown on nickel foam (NF) substrates include mesoporous cobalt oxide Co3O4, iron oxide Fe2O3, and cobalt–iron oxide (m-CFO) m-Co3−xFexO4 IOs. The work focuses on the influence of iron on the phase transformation and electrocatalytic performance of the designed oxides. Varying the iron concentration during the synthesis changes the size of the crystallites and the oxidation state of cobalt. The OER and HER activities of cobalt oxide are significantly affected by iron incorporation. An optimal amount of iron decreases the charge transfer resistance and enhances the water splitting performance of the electrocatalyst.
Experimental methods
Substrate preparation
Nickel foam (NF, TMAX-China, L × W × T 1.0 mm × 1.0 mm × 1.0 mm, porosity: 75–98%, density: 0.1–0.8 g cm−3) substrates were ultrasonically cleaned for 5 min in 0.5 M hydrochloric acid (HCl) and for 30 min in ethanol (EtOH) and distilled (DI) water to remove any contaminants and amorphous surface oxides. Fluorine-doped tin oxide coated glass slides (FTO, Merck-Belgium, L × W × T 1.0 mm × 1.0 mm × 2.2 mm, surface resistivity ∼75 Ω m−2) were ultrasonically cleaned for 30 min in EtOH and DI. Thereafter, they were dried for 12 h in air in a box furnace at 60 °C.Preparation of polystyrene (PS) nanobeads
PS spheres were prepared by emulsion polymerization using sodium dodecyl sulfate (SDS, Sigma Aldrich), potassium persulfate (PPS, Sigma Aldrich), and DI water as an emulsifier, initiator, and dispersion medium, respectively. A mixture of 8 mg SDS surfactants, 100 mg PPS initiators, and 30 ml DI water was kept 30 min at 70 °C under N2 flow in a three-necked reaction flask. 6 ml of styrene monomers, filtered with alumina, were injected into the solution. After polymerization at 70 °C for 4 h, the obtained PS dispersion was dialysis filtered in deionized water for 3 days to remove unreacted reagents. The obtained PS beads have an average diameter of 280 nm.Fabrication of ordered mesoporous cobalt iron oxide IO on substrates (m-CFO IO)
A PS solution was prepared by diluting 1 ml of as-prepared PS nanobead solution with 35 ml of DI and 14 ml of anhydrous EtOH (Merck-Belgium, 200 proof, ≥99.5%). NF or FTO substrates were vertically immersed into 50 mL of PS solution. By keeping the substrates 48 h in the solution at 65 °C, which is until the solution was completely evaporated, the PS beads uniformly coat the substrates. The precursors cobalt nitrate hexahydrate (Co(NO3)2·6H2O, Sigma Aldrich) and iron nitrate nonahydrate (Fe(NO3)2·9H2O, Sigma Aldrich) were slowly added into anhydrous EtOH and stirred at ambient conditions for 1 h. Catalysts with different Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe ratios were grown by varying the precursor ratio, while the summed concentrations of the metal salts was always 0.2 M. After stirring, the solution was dropped on the PS coated substrate (20 μl cm−2) by a micropipette. The electrodes were dried overnight in ambient conditions, followed by thermal annealing in air at 200 °C for 3 h. Finally, the precursor solution-treated PS coated substrates were transferred to a box furnace and annealed at 500 °C for 3 h (ramping rate of 5 °C min−1) in the air to remove the PS template and form the m-CFO. The synthesis procedure is summarized in Scheme 1.
:Fe ratios were grown by varying the precursor ratio, while the summed concentrations of the metal salts was always 0.2 M. After stirring, the solution was dropped on the PS coated substrate (20 μl cm−2) by a micropipette. The electrodes were dried overnight in ambient conditions, followed by thermal annealing in air at 200 °C for 3 h. Finally, the precursor solution-treated PS coated substrates were transferred to a box furnace and annealed at 500 °C for 3 h (ramping rate of 5 °C min−1) in the air to remove the PS template and form the m-CFO. The synthesis procedure is summarized in Scheme 1.
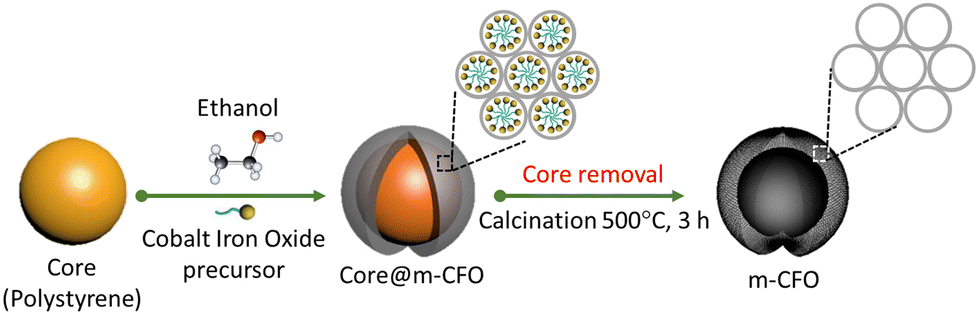 | ||
| Scheme 1 Schematic diagram showing the synthesis procedure of the mesoporous inverse opal m-CFO structure. | ||
Catalyst characterization
The crystal structure of the synthesized materials was characterized by X-ray diffraction (X'Pert PRO MRD system, using Cu Kα radiations of 1.5405 Å with a scanning rate of 2° per min and the 2θ angle ranging from 20° to 75° at current 40 mA and voltage 50 kV). Scanning electron microscopy (SEM) measurements were performed using a Raith EBL-SEM system. The chemical stoichiometry and oxidation states of Fe, Co, and O in the composite were studied by energy dispersive X-ray spectroscopy (EDX) and X-ray photoelectron spectroscopy (XPS). The EDX spectra were recorded using an INCA x-act Oxford instrument (Model 51-ADD0007). XPS measurements were performed at room temperature using Mg KαX-rays (XR4 twin anode X-ray source, non-monochromatic) and an Alpha 110 hemispherical analyser. The spectra were acquired in ultra-high vacuum (UHV) (base pressure ∼10−8 mbar) with an energy step of 0.2 eV, an energy resolution of ≈1 eV, and a beam diameter of 5 mm. The specific surface area and pore size distribution of as-prepared catalysts were derived via the Brunauer–Emmett–Teller (BET) method by using a Tristar 3000 micromeritics system.Electrocatalytic experiments
All electrochemical measurements were carried out in a three-electrode system at room temperature using a potentiostat (CS2350 BiPotentiostat, Corrtest Instruments, China), with a Pt plate (1 × 1 cm2), a Hg|HgO electrode, and 1.0 M KOH as counter electrode, reference electrode, and electrolyte, respectively. The nickel foam covered with m-CFO was directly used as the working electrode with a catalyst loading of about 0.85 mg cm−2. Linear sweep voltammetry (LSV) at a scan rate of 5 mV s−1 and electrochemical impedance spectroscopy (EIS) analysis using a 10 mV amplitude AC signal over a frequency range from 100 kHz to 10 mHz were performed on the electrochemical workstation. Commercial IrO2 and Pt/C were used for comparison as state-of-the-art OER and HER catalysts, respectively.The potential against Hg|HgO (EHg|HgO) electrode can be converted to potential vs. RHE (ERHE):
 | (1) |
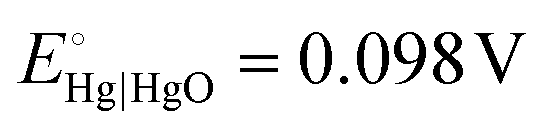 and pH = 14 for 1.0 M KOH.
and pH = 14 for 1.0 M KOH.
The overpotential ηj for OER can be obtained as:
| ηj = (EHg|HgO + 0.924) − 1.23 V, | (2) |
In the case of HER, the thermodynamic potential is zero therefore, for HER:
| ηj = (EHg|HgO + 0.924) V. | (3) |
The two-electrode water electrolysis was performed by using m-CFO both as anode and cathode in 1.0 M KOH.
Analysis of the gaseous products
The gas products (H2, O2) were identified and quantified by a gas chromatography instrument (Compact GC4.0, Interscience) equipped with two thermal conductivity detectors via injection of known amounts of pure hydrogen and oxygen. The detection of H2 and O2 was performed using a Biologic VMP3 potentiostat in a two-electrode cell with a 1Co1Fe–1Co1Fe electrode-pair in a 1.0 M KOH aqueous solution. Before the detection of the gaseous products, the cell was firmly sealed and purged with nitrogen for 30 min. Upon conducting the electrolysis at a current density of 100 mA cm−2, the gaseous products were analyzed to determine the faradaic efficiencies (FEs). For each tested condition, the experiment was repeated at least three times to establish the statistical significance for the obtained data. FEs were calculated according to the equation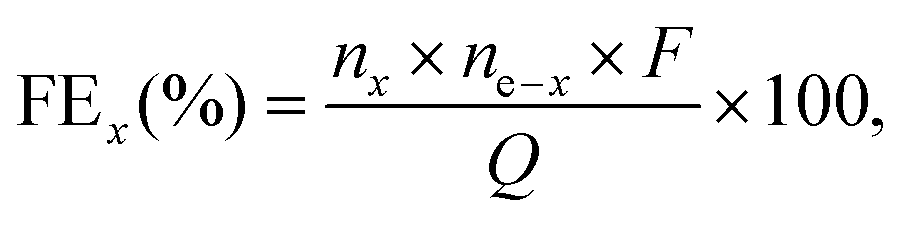 | (4) |
685 C mol−1), and Q the charge passed to produce nx.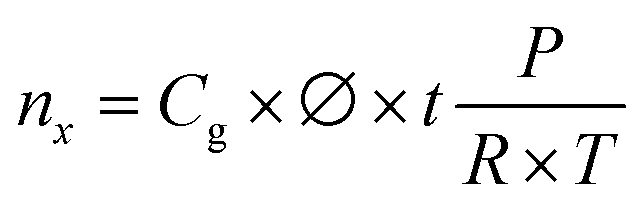 | (5) |
Results and discussion
Morphology analysis
A series of m-CFO IO materials with varying Co:Fe precursor ratios of 1.0:0, 2.0:1.0, 1.0:1.0, 1.0:2.0 and 0:1.0 were grown on NF using PS beads as template. They are denoted hereafter as Co, 2Co1Fe, 1Co1Fe, 1Co2Fe, and Fe, respectively.
The m-CFO IO materials were imaged by SEM to investigate the morphology of their nanostructures at different steps of the synthesis procedure in Scheme 1. The low-magnification SEM images show a regular coating layer of PS beads on the NF substrate (Fig. 1a) featuring a relatively uniform average diameter of ca. 280 nm, a smooth surface, and a rather ordered arrangement. The SEM image (Fig. 1b) of the precursor solution-treated PS-coated NF, after natural drying at room temperature followed by thermal annealing at 200 °C for 3 h in air, reveals that the honeycomb structure is incompletely formed because the PS spheres are only partially burned. The mesoporosity of the material is still low, as clearly visible in Fig. 1e. Fig. 1c shows the morphology of the final mesoporous 1Co1Fe IO structure, evidencing the stability of the nanostructures following the high temperature calcination step. Homogeneously distributed ≈280 nm wide voids, corresponding to the former PS spheres, are organized in a regular 3D honeycomb of ca. 2.5 μm thickness (Fig. S2†) with interconnecting pores corresponding the contact points between the former PS spheres. These pores may act as diffusion channels, and hence, facilitate charge exchange processes, possibly contributing to the catalytic activity. The voids are delimited by mesoporous walls consisting of nanoparticles with sizes of 10 to 15 nm (Fig. 1f). Although similar ordered IO structures are formed for the other Fe/Co precursor ratios (Co, 2Co1Fe, 1Co2Fe, and Fe materials shown in Fig. S3†), a significant reduction in the particle size corresponding to an increase of the porosity is observed upon Fe concentration increase. A specific surface area of 67 m2 g−1 and a pore size distribution of 5 to 20 nm for the prepared 1Co1Fe sample were determined by the Brunauer–Emmett–Teller (BET) method using nitrogen isothermal adsorption–desorption. BET krypton adsorption isotherms for 2Co1Fe and 1Co2Fe reveal that these samples have surface areas of approximately 50 m2 g−1 and 63 m2 g−1, respectively (see Fig. S4 in the ESI†). Thus the m-CFO IOs all have relatively high surface areas and pore sizes that may significantly contribute to their electrochemical performance.
 | ||
| Fig. 1 SEM images illustrating the different steps of the fabrication process of the 1Co1Fe mesoporous inverse opal structure: (a) PS beads as hard template, (b) the precursor solution-treated PS-coated substrates after drying and annealing at 200 °C for 3 h, (c) 1Co1Fe IO obtained after calcination at 500 °C for 2 h. Scale bars are 1 μm. The respective high-resolution enlarged SEM images in the right column (d–f) have scale bars of 100 nm. | ||
The crystal structures of the Co, 2Co1Fe, 1Co1Fe, 1Co2Fe, and Fe samples grown on NF and FTO substrates were examined by X-ray diffraction (XRD). As the diffraction peaks corresponding to the NF substrate overlap with those of the IO materials and dominate the diffractograms (Fig. S5, ESI†), we focus here on the samples grown on FTO, whose XRD patterns are shown in Fig. 2.
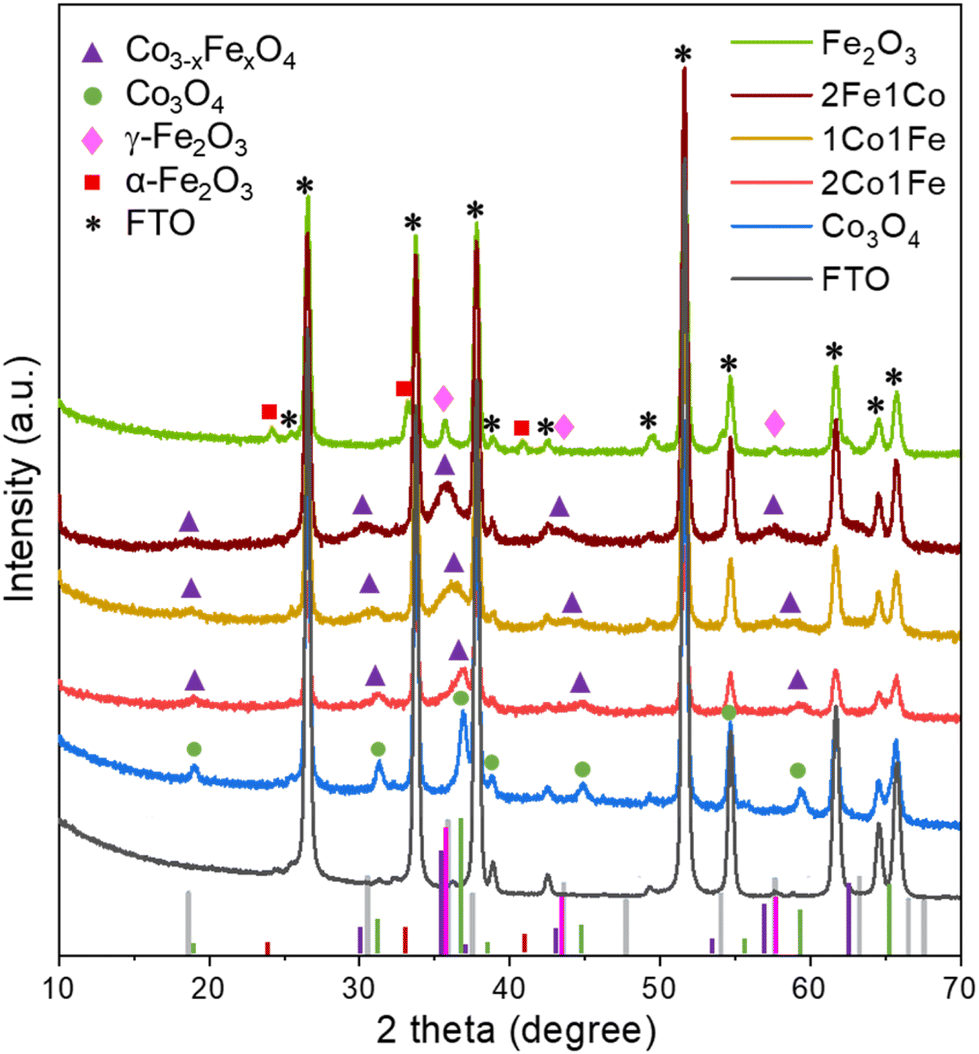 | ||
| Fig. 2 XRD patterns of the Co3−xFexO4 IO structures on FTO. The asterisk, square, rhombus, circle, and triangle marks represent the XRD peaks from FTO, α-Fe2O3, γ-Fe2O3, Co3O4, and Co3−xFexO4, respectively. The vertical bars at the bottom are the XRD profiles of α-Fe2O3 (red, JCPDS card No. 89-8104), γ-Fe2O3 (pink, JCPDS card No. 39-1346), Co3O4 (green, JCPDS card No. 42-1467), CoFe2O4 (gray, JCPDS card No. 04-018-8948) and Co3−xFexO4 (purple, JCPDS card No. 743417). | ||
The XRD pattern of the pure Co sample can be assigned to the spinel cobalt oxide Co3O4 (JCPDS No. 42-1467)28 that has diffraction peaks at 2θ equal to 18.95° (111), 31.20° (220), 36.96° (311), 38.43° (222), 44.83° (400), 55.60° (422), and 59.17° (511). Peak positions and relative intensities of the pure Fe sample with diffraction peaks at 30.36°, 35.76°, 43.49°, and 57.29° are indexed to (220), (311), (400), and (511) crystalline planes of cubic spinel γ-Fe2O3 maghemite (JCPDS No. 39-1346),32 confirming that the main crystalline phase in Fe IO sample is γ-Fe2O3. Additional diffraction peaks at 2θ equal to 24.2°, 33.0° and 41.2° coincide with the characteristic peaks of hematite α-Fe2O3 (JCPDS No. 89-8104), which suggests that some crystallites of α-Fe2O3 are present in the Fe IO sample as well.
All the XRD peaks of the different m-CFOs can be indexed to the face-centered cubic phase of a spinel Co3−xFexO4 (JCPDS No. 74-3417)33 and CoFe2O4 (JCPDS card No. 04-018-8948). XRD patterns of the mixed m-CFO materials show clear similarities with that of the pure Co sample. Considering a spinel cubic phase of general formula AB2O4, where A and B correspond to tetrahedral and octahedral cationic sites, Fe3+ may substitute cobalt at the different sites to result in a Co3−xFexO4 spinel phase. The lattice parameters of such Fe doped Co3O4 cubic Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m spinel structure is expected to increase34 due to the larger radius of Fe3+ (0.65 Å) compared to that of Co3+ (0.61 Å).35 This conjecture agrees with the observed shift of the diffraction peaks towards smaller angles with increasing iron concentration (i.e., from Co over 2Co1Fe and 1Co1Fe to 1Co2Fe). The lattice parameter increase is a good indication of the successful incorporation of iron into the cobalt oxide lattice. The intensity decrease and the broadening of the diffraction peaks upon incorporation of Fe indicate the formation of smaller crystalline particles with possibly a lower crystallinity. The average m-CFO crystallite size calculated using the Debye–Scherrer equation and listed in Table 1 together with the expected molecular formula based on XRD and EDX. These results are consistent with previous reports showing that the partial replacement of Co3+ by larger Fe3+ expands the unit cell.36 The increase in cell volume and a lower crystallinity of m-CFO may have beneficial effects on the catalytic activity by increasing the electrolyte permeability.37,38 The crystallite size ranging from 9 in pure Fe to 13.5 nm in pure Co samples is in good agreement with apparent particle size range observed in the SEM images.
m spinel structure is expected to increase34 due to the larger radius of Fe3+ (0.65 Å) compared to that of Co3+ (0.61 Å).35 This conjecture agrees with the observed shift of the diffraction peaks towards smaller angles with increasing iron concentration (i.e., from Co over 2Co1Fe and 1Co1Fe to 1Co2Fe). The lattice parameter increase is a good indication of the successful incorporation of iron into the cobalt oxide lattice. The intensity decrease and the broadening of the diffraction peaks upon incorporation of Fe indicate the formation of smaller crystalline particles with possibly a lower crystallinity. The average m-CFO crystallite size calculated using the Debye–Scherrer equation and listed in Table 1 together with the expected molecular formula based on XRD and EDX. These results are consistent with previous reports showing that the partial replacement of Co3+ by larger Fe3+ expands the unit cell.36 The increase in cell volume and a lower crystallinity of m-CFO may have beneficial effects on the catalytic activity by increasing the electrolyte permeability.37,38 The crystallite size ranging from 9 in pure Fe to 13.5 nm in pure Co samples is in good agreement with apparent particle size range observed in the SEM images.
| Sample | Co:Fe precursor ratio |
Average crystallite size determined from XRD (nm) | Expected molecular formula based on XRD/EDX |
|---|---|---|---|
| Co | 1:0 |
13.5 | Co3O4 |
| 2Co1Fe | 2:1 |
12.2 | Co1.9Fe1.1O4 |
| 1Co1Fe | 1:1 |
11.7 | Co1.5Fe1.5O4 |
| 1Co2Fe | 1:2 |
11.1 | Co0.7Fe2.3O4 |
| Fe | 0:1 |
9.0 | Fe2O3 |
Chemical structure analysis
XPS analyses were performed to verify the surface chemical composition and the valence states of m-CFO IO catalysts on NF substrates. XPS survey spectra (see Fig. S7 of the ESI†) confirm the presence of Fe, Co, O, and Ni. Trace amounts of carbon suggest low carbonation level of the metal oxides when exposed to air, a phenomenon which may progress into the bulk to a different extent depending on the material and the aging time.The XPS data was analysed by fitting peaks with Voigt functions and assuming a nonlinear background. Two major peaks observed in the Co 2p binding energy spectrum (Fig. 3a–c) around 780 eV and 795 eV can be ascribed to Co 2p3/2 and Co 2p1/2, respectively. The separation within this 2p doublet (15.1 eV) is slightly smaller than the previously reported value of 15.8 eV for Co3O4,39 which reflect the changed Co electronic properties due to the Fe incorporation. The Co 2p3/2 and Co 2p1/2 peaks are asymmetric and can both be fitted by two Voigt functions to account for the contributions of Co2+ and Co3+ sites. The Co3+/Co2+ ratio is found to be 2.0 ± 0.3, 2.4 ± 0.4, and 1.5 ± 0.3 in 2Co1Fe, 1Co1Fe, and 1Co2Fe, respectively. The results are listed in Table 2. The change in the average Co valence state can be attributed to the amount of Fe that is incorporated in the cobalt oxide spinel structure to form cobalt ferrite spinel.
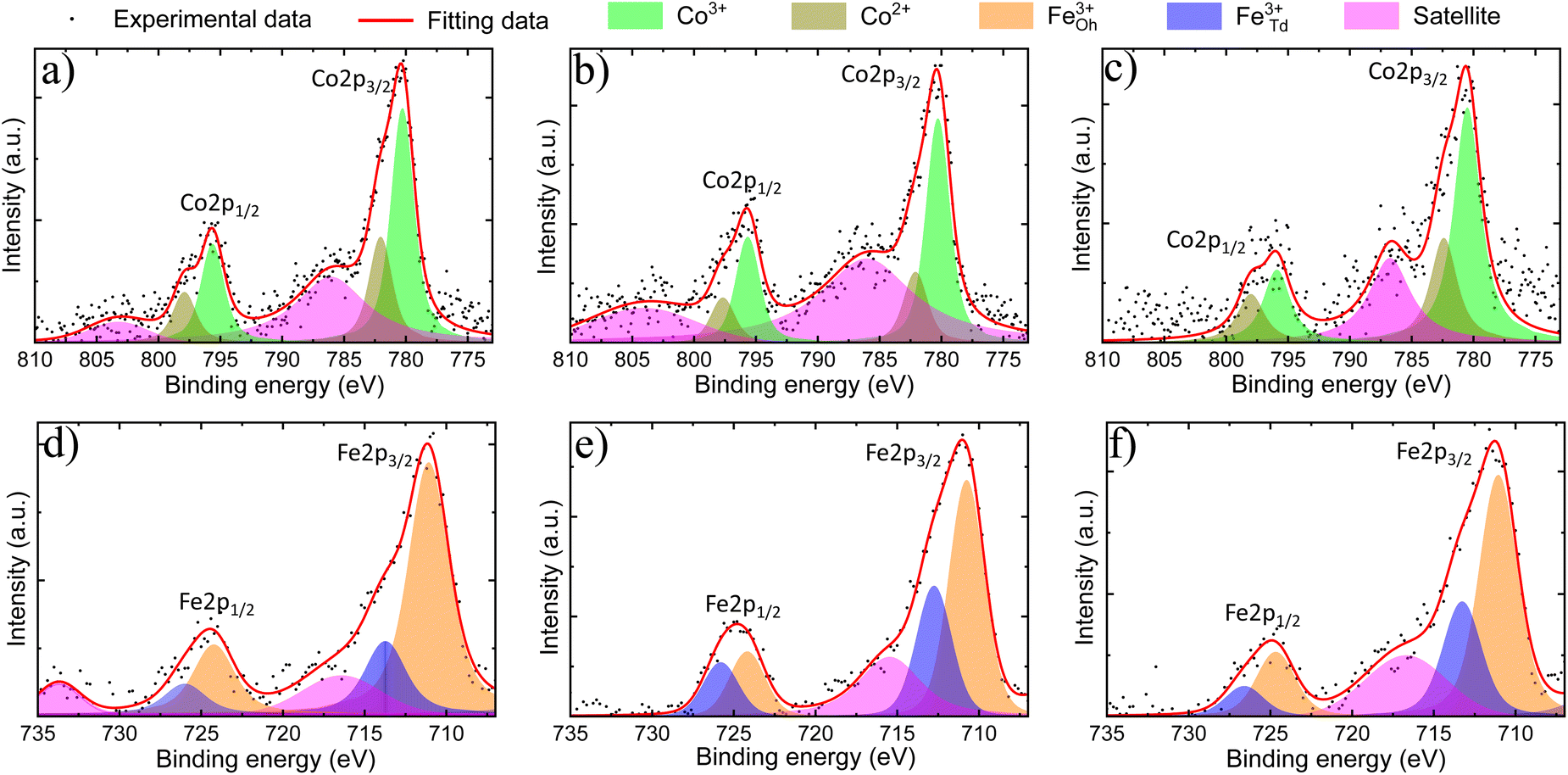 | ||
| Fig. 3 Co 2p (a, b and c) and Fe 2p (d, e and f) XPS spectra of 2Co1Fe (a and d), 1Co1Fe (b and e) and 1Co2Fe (c and f). The black points are the experimental data and the red lines are the corresponding fitting curves. The shaded areas underneath the fitting curves are Voigt functions, representing the individual contributions. | ||
| Samples | Fe3+Td/Fe3+Oh | Co3+/Co2+ | Fex+ | Coy+ | O needed for crystalline phases | O deficiency % |
|---|---|---|---|---|---|---|
| 2Co1Fe | 0.36 ± 0.10 | 2.0 ± 0.3 | 3.0 | 2.66 | 4.18 | 4.2 |
| 1Co1Fe | 0.7 ± 0.2 | 2.4 ± 0.4 | 3.0 | 2.70 | 4.28 | 6.4 |
| 1Co2Fe | 0.47 ± 0.15 | 1.5 ± 0.4 | 3.0 | 2.60 | 4.36 | 8.3 |
The Fe 2p core-level binding energy spectrum of the catalysts (Fig. 3d–f) shows the presence of the two characteristic peaks corresponding to Fe 2p doublet Fe 2p3/2 and Fe 2p1/2 at 711 eV and 725 eV, respectively. The absence of characteristic peaks related to Fe0 at 706 eV (ref. 40) and to Fe2+ around 708 eV and 709 eV,41 indicates that all iron is in a Fe3+ state. This implies that iron mainly replaces the Co3+ in octahedral sites, which agrees with a decreased proportion of Co3+ in the material with the highest iron content.19 The Fe 2p spectra were deconvoluted in Oh and Td peaks following the procedure developed in previous studies, where the Td peak was identified as a shoulder on the high binding energy side of the Oh peak.42–44 The Fe3+Td/Fe2+Oh ratio is found to be 0.36 ± 0.10, 0.7 ± 0.20, and 0.47 ± 0.15 in 2Co1Fe, 1Co1Fe, and 1Co2Fe, respectively.
Oxygen vacancies have been shown to play an important role in improving the catalytic activities for the OER and ORR45 The amount of oxygen needed to form well-defined crystalline phases can be determined by assuming a charge neutrality. The oxygen deficiency is subsequently calculated from the difference between the amount of oxygen required for charge neutrality and the amount of oxygen from the molecular formula which was obtained from EDX and XRD data (column 4, Table 1). Details of the calculation are given in the ESI.† The results are listed in Table 2. The oxygen deficiencies in 2Co1Fe, 1Co1Fe, and 1Co2Fe catalysts are 4.2%, 6.4%, and 8.3%, respectively. Information about the presence of oxygen vacancies can also be obtained from the O 1s XPS spectra. The O 1s spectra (see ESI Fig. S8†) can be deconvoluted into three peaks located around 530.0, 531.0, and 532.2 eV, attributable to lattice oxygen (O–M), oxygen vacancy (Ov), and adsorbed water (H–O–H), respectively.46,47 The increased proportion of Ov at the surface of the samples with respect to the lattice oxygen O–M (Ov/O–M ratio) from 0.20 for 2Co1Fe, over 0.25 for 1Co1Fe, to 0.34 for 1Co2Fe, is consistent with the data in Table 2, i.e. 1Co2Fe possesses more oxygen vacancies than 1Co1Fe and 2Co1Fe.48,49 It is our conjecture that the oxygen deficiency is optimal in the 1Co1Fe sample, serving for additional charge-transfer pathways.50
Based on these results, it can be concluded that a spinel-type binary iron–cobalt oxide was grown on the NF substrate. The m-CFO IO morphology with tunable particle size, crystallinity, and cell volume should enable efficient transportation of the electrolyte. Additionally, the tunable ratios of Fe3+Td/Fe3+Oct and Co3+/Co2+ could vary the number of active sites available for catalytic reactions. Whilst the intimate contact between the active catalyst and current collector (NF), coupled with the absence of any external binders, can minimize the resistance associated with the charge transfer process.
Electrocatalytic experiments
Catalysts’ activities towards OER and HER are typically compared by two key descriptors, namely the overpotential (η10), i.e. the minimum potential needed to attain a current density of 10 mA cm−2, and the Tafel slope which represents the sensitivity of reaction rate towards applied potential.51Electrocatalytic oxygen evolution reaction (OER)
The influence of the m-CFO IO's composition on the OER activity was investigated in 1 M KOH for the different Co:Fe ratios with linear sweep voltammetry (LSV), Tafel analysis, electrochemical impedance spectroscopy (EIS), and chronopotentiometry (CP). The results are summarized in Fig. 4.
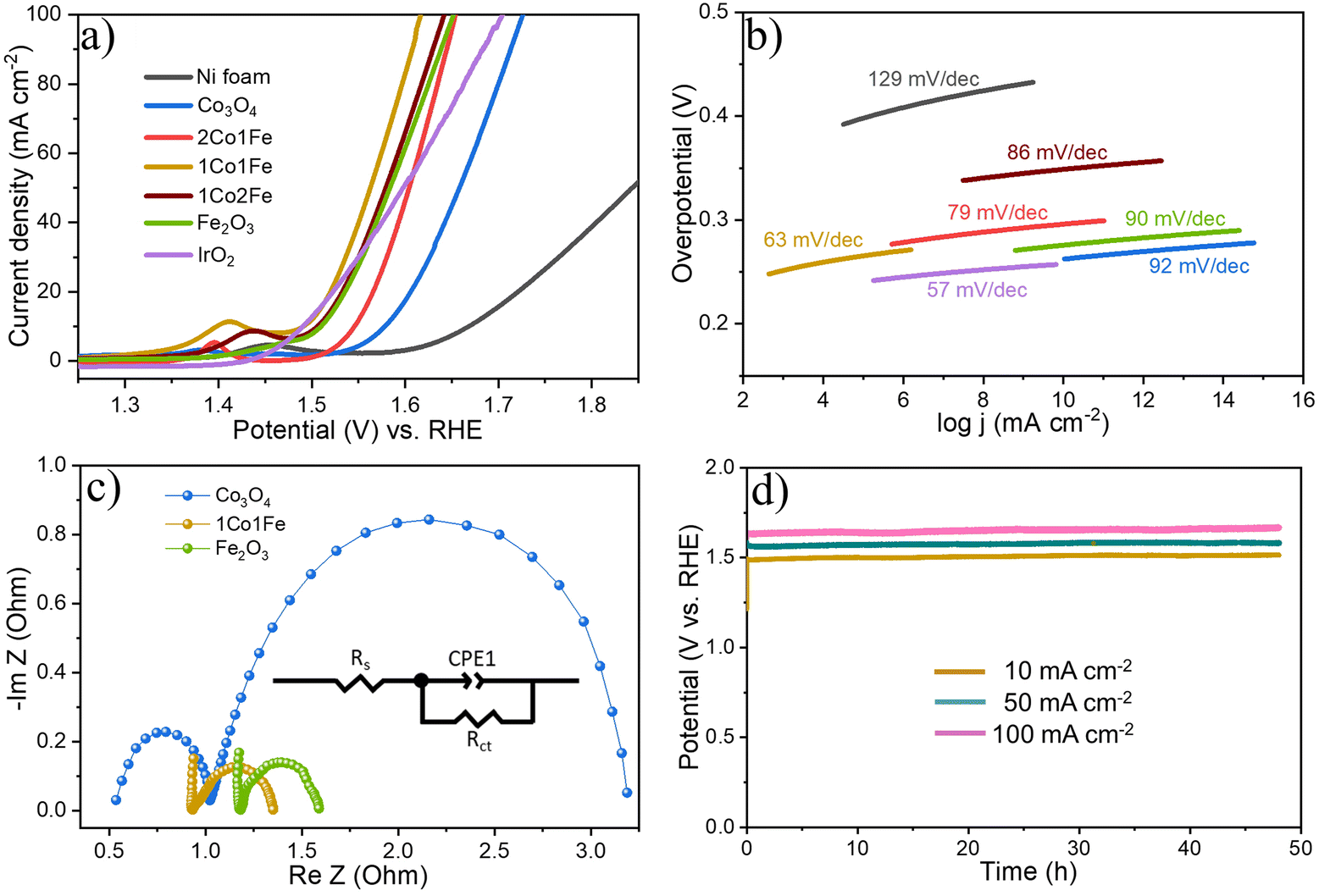 | ||
| Fig. 4 Evaluation of the electrocatalytic activity of the m-CFO IO catalysts, bare Ni foam substrate, and benchmark IrO2 for oxygen evolution reaction. (a) Polarization curves of different catalysts collected at a scan rate of 5 mV sec−1 in 1 M KOH. (b) Corresponding Tafel plots. (c) Nyquist plots at an overpotential of 350 mV for pure m-Co3O4, m-Fe2O3, and 1Co1Fe electrodes. The inset is the best fitted equivalent circuit. (d) Chronopotentiometric curves of the 1Co1Fe catalyst, obtained at different current densities of 10, 50, and 100 mA cm−2. | ||
Fig. 4a shows representative LSV polarization curves of the different m-CFO IOs as well as that of the bare Ni foam substrate and a benchmark IrO2 catalyst. 1Co1Fe possesses the lowest overpotentials of 261 mV and 386 mV at current densities of 10 mA cm−2 and 100 mA cm−2, respectively, to be compared with those of pure Co (η10 = 349 mV, η100 = 496 mV), 2Co1Fe (η10 = 296 mV, η100 = 427 mV), 1Co2Fe (η10 = 276 mV, η100 = 411 mV), and Fe (η10 = 293 mV, η100 = 422 mV) (ESI Fig. S12†). Using Tafel plots, the OER kinetics is further analyzed. The Tafel slope b was obtained by fitting the data in Fig. 4b with Tafel equation η = blog(j), where η is the overpotential and j the current density. The 63 mV dec−1 Tafel slope of 1Co1Fe is much lower than those of m-Co3O4 (92 mV dec−1), 2Co1Fe (79 mV dec−1), 1Co2Fe (86 mV dec−1), and m-Fe2O3 (90 mV dec−1), implying that the reaction kinetics is the fastest in the mesoporous 1Co1Fe IO.
Table 3 provides a comparison of recently reported cobalt-based OER catalysts52–58 with our 1Co1Fe catalyst, demonstrating its excellent low overpotential for O2 production, concomitant with a quite small Tafel slope.
| OER catalyst | Electrolyte | η 10 (mV) | Tafel slope (mV dec−1) | Ref. |
|---|---|---|---|---|
| CoOx | 1 M KOH | 309 | 28 | 52 |
| CoFe2O4 | 1 M NaOH | 378 | 73 | 53 |
| Co3O4 | 1 M NaOH | 500 | 61 | 54 |
| Co3O4–CuCo2O4 | 0.1 M KOH | 498 | — | 55 |
| Co3O4−x-C@ | ||||
| Fe2−yCoyO3 | 1 M KOH | 350 | 38 | 56 |
| Janus Co/CoP | 1 M KOH | 340 | 80 | 57 |
| Au@Co3O4 | 0.1 M KOH | 387 | 60 | 58 |
| m-Co1.5Fe1.5O4 | 1 M KOH | 261 | 63 | This work |
EIS was performed to reveal the electronic conductivity and charge transfer process of the OER catalysts. Nyquist impedance spectra are shown in Fig. 4c for impedance measurements carried out at a potential of 1.565 V vs. RHE. The recorded data was fitted with an equivalent circuit (inset of Fig. 4c) composed of a solution resistance Rs, a charge transfer resistance Rct, and a constant phase element CPE1. Based on this equivalent electrical scheme we found Rs values of 0.8, 1.0, and 1.15 Ω for 1Co1Fe, m-Co3O4, and m-Fe2O3, respectively. Also the Rct of 1Co1Fe (1.35 Ω) was found to be lower than those of m-Co3O4 (1.6 Ω) and m-Fe2O3 (3.1 Ω). The lower charge transfer and solution resistances reflect a higher electrocatalytic activity of 1Co1Fe. Also, the LSV polarization curves confirm that 1Co1Fe is catalytically more active than m-Co3O4 and m-Fe2O3. The low film resistance of the 1Co1Fe electrode may originate from the good electrical conductivity of the Co-based oxide IO, where the incorporation of Fe3+ may further contribute to the low Rct.
To evaluate the intrinsic activity of the m-CFO IO catalysts, the redox peak method was used to calculate the relative electrochemical surface area (ECSA) by assuming that the number of Co3+ ions getting converted into Co4+ ions equals the number of active sites catalyzing the OER.59,60 The calculated areas for 2Co1Fe, 1Co1Fe, and 1Co2Fe are 1.75, 1.97, and 1.88 × 10−4 A V, respectively (ESI Fig. S10†), corresponding to 21.9, 24.6, and 23.5 × 1016 active sites involved in the OER reaction, respectively (see ESI† for details of the calculation).
Using the number of active sites, the relative ECSA was calculated by assuming that the number of active sites that are involved in OER with the 2Co1Fe sample exactly covers a geometrical area of 1 cm2. Hence, the relative ECSA of 1Co1Fe and 1Co2Fe to that of 2Co1Fe are 1.18, and 1.07, respectively. The relative ECSA-normalized LSVs are provided in Fig. 5a. From this, the calculated ηECSA10 for 2Co1Fe, 1Co1Fe, and 1Co2Fe are 315, 266, and 278 mV, respectively.
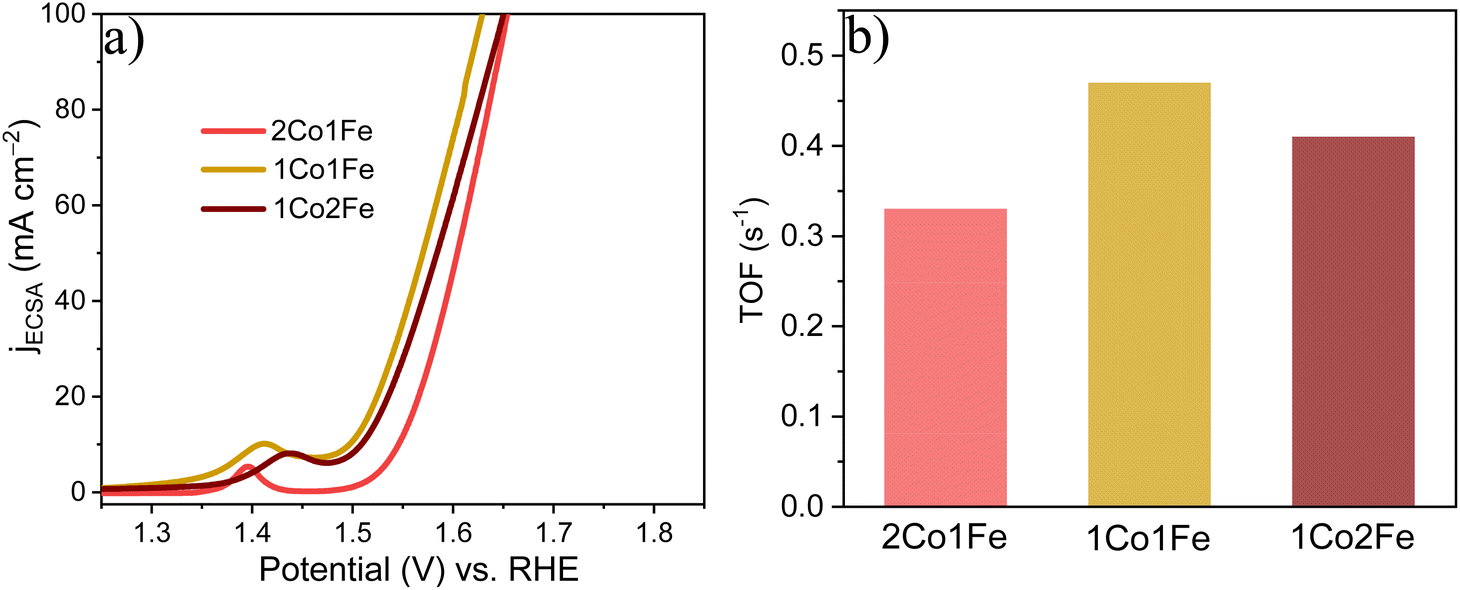 | ||
| Fig. 5 The intrinsic catalytic properties of m-CFO IOs in OER: (a) ECSA-normalized LSVs. (b) TOFs of the different catalysts. | ||
The turnover frequency (TOF) is an intrinsic activity parameter that can be derived from the current density at a fixed potential and the surface concentration of active sites. The TOFs for the three m-CFO IOs were calculated using the following equation:61
| TOF = jNA/nFΓ | (6) |
It is found that the OER performance is improved when Fe is incorporated and reaches a maximum if the Co:Fe precursor ratio is 1:1. It reduces greatly with excess Fe incorporation because high Fe concentration in Co3O4 decreases the amount of oxygen in the surface. The introduction of Fe as a modifying additive of the spinel structure of Co3O4 leads to Fe3+ ions, which appear to be a new active sites.62 In addition, during the oxidative decomposition of m-CFO, the redox reaction between Fe3+ and Co2+ leads to the formation of Co3+.63 Co3+ has been considered an active site for OER.64
The XPS result shows that the Co3+/Co2+ ratio among m-CFOs reaches a maximum of 0.83 and thus most OER active sites in the 1Co1Fe sample. Last, previous theoretical and experimental studies have shown that incorporation of Fe3+ causes polarization of cobalt cations, thereby leading to higher Co oxidation states. This increases the electrophilicity of the cobalt cations and accelerates the formation of OOH species by nucleophilic reaction with OH and other oxygen species, resulting in a better OER catalytic activity.39,65,66 Thus, the synergetic effect of the favorable electronic interaction, the larger number of active sites, the optimal oxygen deficiency, and the higher electrical conductivity leads to the favorable catalytic activity of 1Co1Fe as compared to other m-CFOs (2Co1Fe, 1Co2Fe) and many well-known OER catalysts.
Apart from the catalytic activity, the stability of the catalyst is also of paramount importance. We investigated the stability of the 1Co1Fe catalyst by performing CP tests at different constant current densities (Fig. 4d). The material retained most of its catalytic OER activity for more than 48 h. Ex situ XRD revealed that the catalysts are transformed into hydroxide phases after OER (see Fig. S5 in the ESI†). However, no visible changes in the morphology of the catalyst could be discerned even after conducting OER for 48 h (see Fig. S9 in the ESI†). The combination of exceptional activity and good stability make 1Co1Fe very suitable for practical OER catalysis applications.
Electrocatalytic hydrogen evolution reaction (HER)
The HER activity of the m-CFO was also evaluated using LSV, Tafel plots, EIS and CP. The results are summarized in Fig. 6. The LSV curves of different m-CFO IOs are compared in Fig. 6a with those of m-Co3O4, m-Fe2O3, platinum plate electrode, and bare NF, all measured under identical conditions. As expected, the Pt wire has the highest catalytic activity with a η10 of 84 mV. The bare NF displays a low HER activity (η10 = 277 mV), so the activity of the m-CFO IOs is not arising from the substrate. Among the different IO structures, m-CFO with Co:Fe ratio of 1:1 (1Co1Fe) displays by far the best performance with η10 = 157 mV, which is 106 mV, 46 mV, 10 mV and 80 mV less than the overpotential required to achieve 10 mA cm−2 using m-Co3O4, 1Co2Fe, 2Co1Fe, or m-Fe2O3, respectively. Although the Pt electrode has a better η10, the 1Co1Fe inverse opal structure exhibits a higher HER activity than the Pt/C electrode at large current densities (>140 mA cm−2), which can be seen clearly in Fig. 6a and Fig. S12 in the ESI.† The 1Co1Fe catalyst also proves to be highly competitive to many recently reported cobalt-based HER catalysts like Co3O4-NCTs/700 (η10 = 358 mV),26 NiCo2O4 (η10 = 104 mV),67 CeO2/Co(OH)2 (η10 = 317 mV),68 Ni/Co (η10 = 220 mV),69 Co0.85Se@N-doped graphene (η10 = 227 mV),70 Co–W/CeO2 (η10 = 166 mV),71 Co/Co3O4 core/shell (η10 = 90 mV)72 (see overview in Table 4).
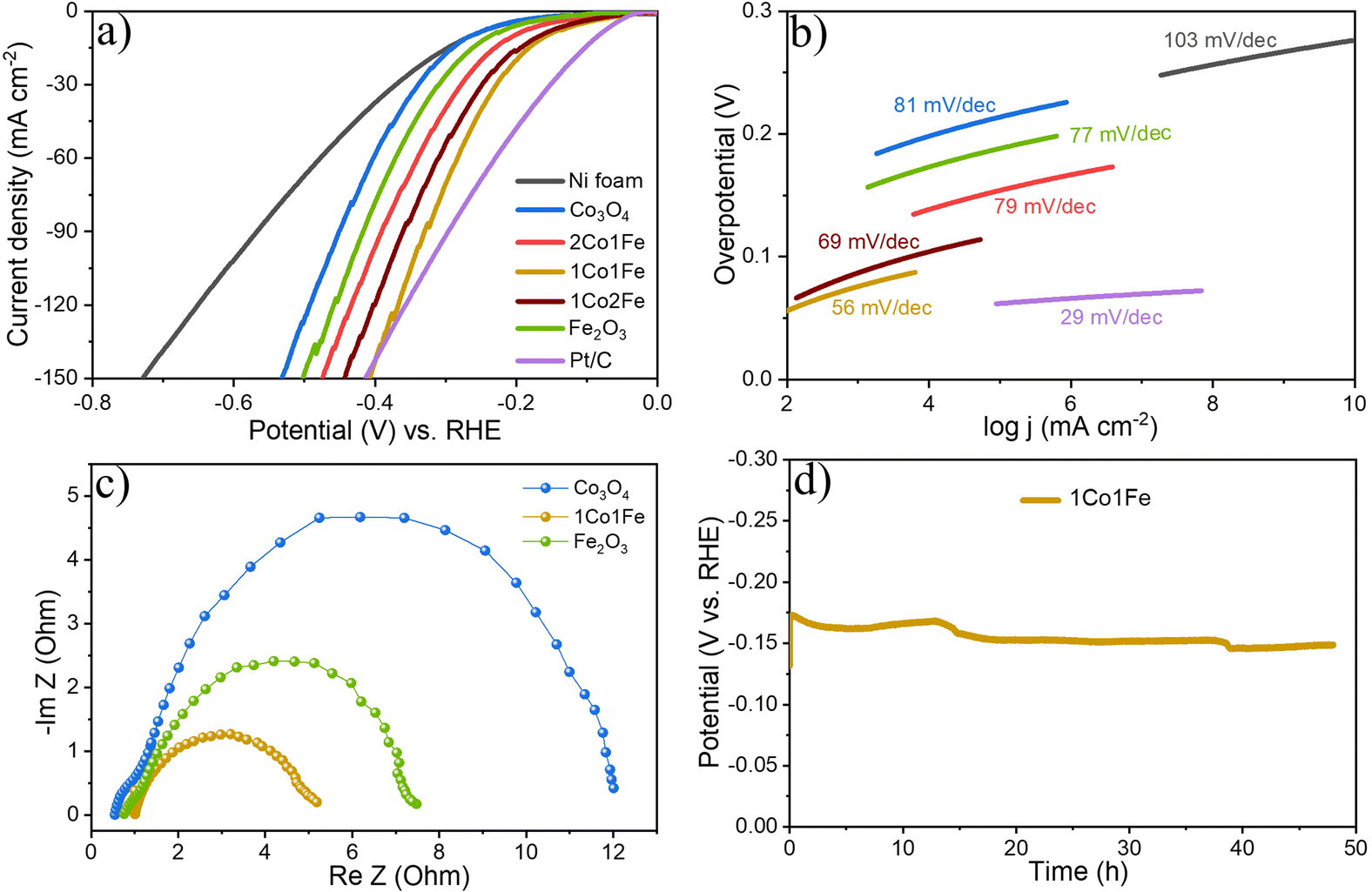 | ||
| Fig. 6 Evaluation of the electrocatalytic activity of different m-CFOs, the bare Ni foam substrate, and a Pt benchmark catalyst for the hydrogen evolution reaction. (a) Polarization curves of different catalysts collected at a scan rate of 5 mV sec−1 in 1 M KOH. (b) Corresponding Tafel plots. (c) Nyquist plots at the overpotential of 190 mV for pure Co3O4, Fe2O3, and 1Co1Fe electrodes. (d) Chronopotentiometric curves of 1Co1Fe catalyst obtained at a current density of 10 mA cm−2. | ||
| HER catalyst | Electro-lyte | η 10(mV) | Tafel slope (mV dec−1) | Ref. |
|---|---|---|---|---|
| Co3O4-NCTs/700 | 1 M KOH | 358 | 32.3 | 26 |
| NiCo2O4 | 1 M KOH | 104 | 112 | 67 |
| CeO2/Co(OH)2 | 1 M KOH | 317 | 144 | 68 |
| Ni/Co | 1 M KOH | 220 | 127 | 69 |
| Co0.85Se@NG | 1 M KOH | 227 | 76 | 70 |
| Co/β-Mo2C@ | ||||
| N-CNTs | 1 M KOH | 170 | 92 | 71 |
| Co/Co3O4 core/shell | 1 M KOH | 90 | 44 | 72 |
| m-Co1.5Fe1.5O4 | 1 M KOH | 157 | 56 | This work |
The Tafel slope (Fig. 6b) also highlights the excellent catalytic kinetics of 1Co1Fe. The Tafel slope of 1Co1Fe (56 mV dec−1) is smaller than that of the pure m-Co3O4 (81 mV dec−1), 2Co1Fe (79 mV dec−1), 1Co2Fe (69 mV dec−1), and m-Fe2O3 (77 mV dec−1). The relative ECSA-normalized LSVs of different m-CFO IO catalysts for HER are provided in the ESI (Fig. S11†). The calculated ηECSA10 for 2Co1Fe, 1Co1Fe, and 1Co2Fe are 203, 164, and 171 mV, respectively.
These results confirm that the electrocatalytic properties of Co3O4 can be enhanced by a suitable amount of Fe incorporation. The superior catalytic activity of 1Co1Fe could originate from the facile charge transfer between the catalyst and water molecules. Indeed, recent first principle calculations by Sun et al. suggested that the adsorption of H2O on Fe3+ is energetically more favorable than adsorption on Co2+ sites.73 Hence, the incorporation of Fe can promote the adsorption of water on the catalyst surface, which could facilitate the transfer of electrons to water molecules, thereby accelerating the formation of the intermediate active species of the HER.
The EIS response of the catalysts at a constant applied potential of 190 mV (vs. RHE) was recorded to study the charge transfer kinetics of the HER on catalysts. The Nyquist plots of different electrodes displayed in Fig. 6c, appear in the form of a semicircle. Rs values are 0.98, 0.55, 0.76 Ω for 1Co1Fe, m-Co3O4 and m-Fe2O3 respectively. Rct for 1Co1Fe is 4.27 Ω, whereas m-Co3O4 and m-Fe2O3 have higher values of 11.46 Ω, and 6.78 Ω respectively. The composition-dependent trends observed from the EIS analysis during HER and OER are consistent and indicate that the charge transfer resistance decreases with the incorporation of Fe in Co3O4. Cobalt sites, acting as an active center, possibly donate electrons to H2O and the adjacent Fe sites, resulting in a good electrical connection and efficient HER kinetics. Hence, it can be concluded that the superior catalytic activity of 1Co1Fe could be attributed to the improved charge transfer process and an optimum concentration of Fe3+ and Co3+ actives sites, which can be tuned by varying the Co:Fe precursor ratio.
Finally, the stability of the 1Co1Fe catalyst was examined by performing CP analysis at a constant current density of 10 mA cm−2. As evident from Fig. 6d, the 1Co1Fe catalyst has a good stability and retained 92% of its initial activity even after 48 h of electrolysis.
Overall water splitting
Encouraged by the excellent activity of 1Co1Fe for both HER and OER in alkaline media, an alkaline electrolyzer was assembled with 1Co1Fe serving both as cathode and anode. A small voltage of 1.55 V is sufficient to deliver a current density of 10 mA cm−2 for overall water splitting (Fig. 7a). The η10 value of the 1Co1Fe||1Co1Fe couple remarkably is 50 mV lower than that of the precious metal Pt/C||IrO2 benchmark combination (1.60 V). The operating potential of pure m-Co3O4 and m-Fe2O3 electrolyzers are 1.65 V and 1.63 V, respectively. Meanwhile, the 1Co1Fe||1Co1Fe water electrolyzer achieved a current density of 50 mA cm−2 at an overpotential of only 1.73 V. This outperforms the pure m-Co3O4 and m-Fe2O3 as well as the commercially available Pt/C||IrO2 system, which require cell voltages of 1.85 V, 1.82 V, and 1.77 V, respectively, to reach the same current density.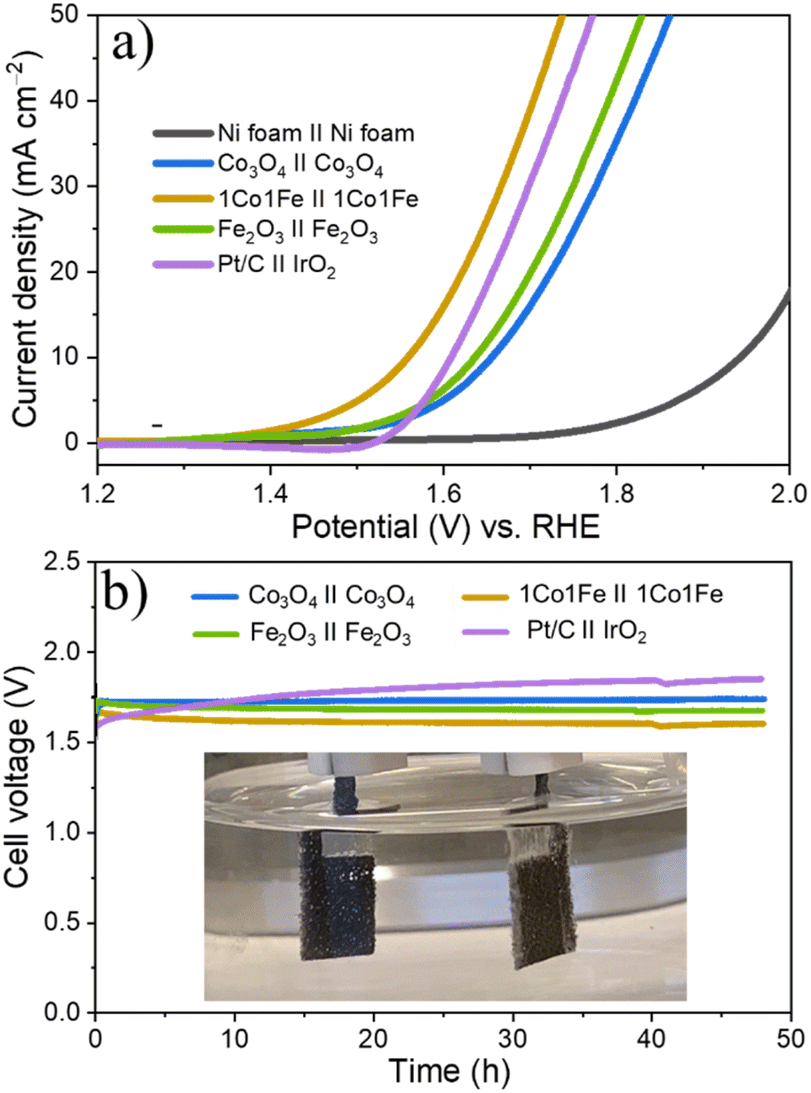 | ||
| Fig. 7 Overall water-splitting performance of different samples tested in 1 M KOH. (a) LSV curve obtained using a two-electrode set up with m-CFO serving as cathode and anode in 1 M KOH. (b) Chronopotentiometric curves of Co3O4||Co3O4, 1Co1Fe||1Co1Fe, Fe2O3||Fe2O3, and Pt/C||IrO2 as the benchmark at a current density of 10 mA cm−2. The optical photograph of the two-electrode configuration in the inset shows the generation of hydrogen and oxygen bubbles on the cathode and anode. | ||
As summarized in Fig. 874–94 the overall water-splitting performance of the 1Co1Fe||1Co1Fe couple competes with the best recently reported nonprecious metal-based catalysts. In addition, the 1Co1Fe catalyst features an excellent stability for full water splitting over 48 h at the current density of 30 mA cm−2 (Fig. 7b), with visible hydrogen and oxygen bubble formation on Ni foam (inset of Fig. 7b). Conversely, due to the poor stability of the IrO2 in alkaline conditions,95 the activity of the Pt/C||IrO2 electrolyzer was found to decrease more rapidly than that of 1Co1Fe||1Co1Fe. The combination of Pt/C and IrO2 is often chosen as the commercial noble metal catalyst benchmark for electrochemical water splitting,96,97 as Pt-group metals and Ir-based compounds are known to be among the most efficient precious metal-based electrocatalysts for HER and OER, respectively. These results clearly demonstrate that the activity and stability of 1Co1Fe||1Co1Fe is better than the noble metal catalyst, which in addition to its low cost and environmentally-friendly properties, make it a promising candidate for a widespread industrial use.
 | ||
| Fig. 8 Comparison of the overall water splitting activity of m-Co1.5Fe1.5O4 IO (1Co1Fe catalyst) with other non-noble metal-based electrocatalysts in alkaline electrolytes. | ||
Finally, gas chromatography (GC) measurements were conducted to detect H2 and O2 produced from overall water splitting. The faradaic efficiency of 1Co1Fe electrode for water splitting was calculated by measuring the experimentally generated amount of gas (Fig. 9a). Accordingly, the produced gas volume coincides with the calculated volume, and the molar ratio of the produced gases H2/O2 is 2/1.05, evidencing a faradaic efficiency of more than 91% after 9 h of electrolysis (Fig. 9b). All results indicate that 1Co1Fe is an excellent candidate for substituting the precious metal catalyst in commercial water electrolysis for H2 production.
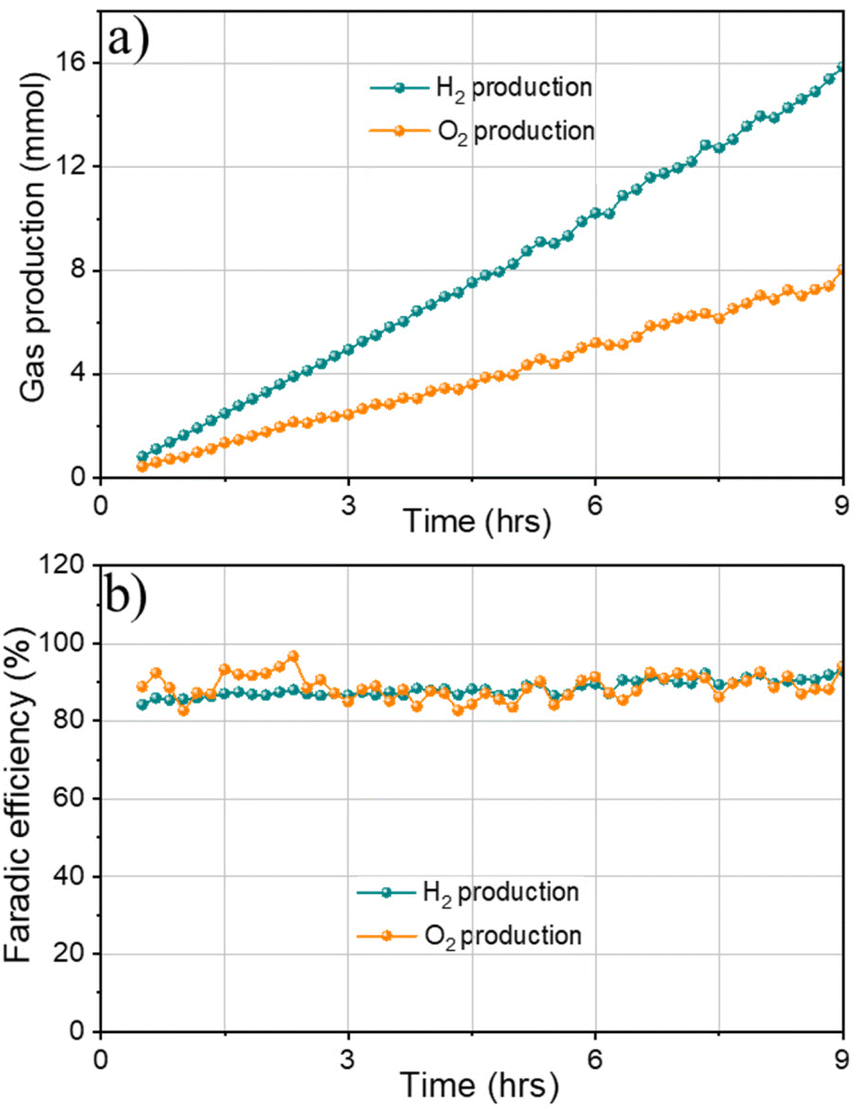 | ||
| Fig. 9 (a) Amounts of H2 and O2 gases and (b) faradaic efficiency obtained from 1Co1Fe electrode-pair setting (η = 540 mV) by GC measurement versus time for electrolysis with a current density of 100 mA cm−2 in 1 M KOH aqueous solution. | ||
Conclusions
In summary, mesoporous spinel iron cobalt oxide inverse opals were successfully grown on the surface of nickel foam. The resulting 1Co1Fe catalyst displayed an excellent catalytic activity for both OER and HER wherein a benchmark current density of 10 mA cm−2 could be achieved at a low overpotential of 266 mV and 164 mV respectively. In addition, an alkaline water electrolyzer constructed by using 1Co1Fe as both cathode and anode operated in a stable mode for more than 48 h. The remarkable catalytic activity and stability of 1Co1Fe could be attributed to the following factors: (i) A significant increase in the number of active sites and conductivity resulting from the incorporation of an optimum amount of Fe in the Co3O4 spinel structure the; (ii) An efficient transfer of the electrolyte and improved electrolyte permeability across the catalyst surface due to a good balance between the crystallite size, oxygen deficiency, and the large surface area of the mesoporous inverse opal structure. (iii) An intimate contact between the active catalyst, the electrolyte a and the substrate that is beneficial to reduce both the solution and charge transfer resistance and avoid the degradation of the catalyst during electrolysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the European Union's Horizon 2020 research and innovation program under grant agreement no. 955650 (CATCHY) and by Flanders Innovation & Entrepreneurship (VLAIO) under project no. HBC.2021.0586 (CLUE). T. H. T. Nguyen thanks the Research Foundation Flanders (FWO) for an FWO MSCA SoE postdoctoral fellowship with grant no. 12ZZI23N. We thank Margot F. K. Verstreken, cMACS, KU Leuven for conducting Kr physisorption measurements and providing BET data.References
- M. Gong, W. Zhou, M. C. Tsai, J. Zhou, M. Guan, M. C. Lin, B. Zhang, Y. Hu, D. Y. Wang, J. Yang, S. J. Pennycook, B. J. Hwang and H. Dai, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- M. Ledendecker, S. Krickcalderõn, C. Papp, H. P. Steinrück, M. Antonietti and M. Shalom, Angew. Chem., Int. Ed., 2015, 54, 12361–12365 CrossRef CAS PubMed.
- J. Duan, S. Chen and C. Zhao, Nat. Commun., 2017, 8, 1–7 CrossRef PubMed.
- J. N. Tiwari, S. Sultan, C. W. Myung, T. Yoon, N. Li, M. Ha, A. M. Harzandi, H. J. Park, D. Y. Kim, S. S. Chandrasekaran, W. G. Lee, V. Vij, H. Kang, T. J. Shin, H. S. Shin, G. Lee, Z. Lee and K. S. Kim, Nat. Energy, 2018, 3, 773–782 CrossRef CAS.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- X. Yu, M. Zhang, W. Yuan and G. Shi, J. Mater. Chem. A, 2015, 3, 6921–6928 RSC.
- B. K. Kang, M. H. Woo, J. Lee, Y. H. Song, Z. Wang, Y. Guo, Y. Yamauchi, J. H. Kim, B. Lim and D. H. Yoon, J. Mater. Chem. A, 2017, 5, 4320–4324 RSC.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267–9270 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, K. C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- X. Jia, Y. Zhao, G. Chen, L. Shang, R. Shi, X. Kang, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Energy Mater., 2016, 6, 1–6 Search PubMed.
- E. A. Hernández-Pagán, N. M. Vargas-Barbosa, T. Wang, Y. Zhao, E. S. Smotkin and T. E. Mallouk, Energy Environ. Sci., 2012, 5, 7582–7589 RSC.
- G. H. Moon, M. Yu, C. K. Chan and H. Tüysüz, Angew. Chem., Int. Ed., 2019, 58, 3491–3495 CrossRef CAS PubMed.
- T. Grewe, X. Deng and H. Tüysüz, Chem. Mater., 2014, 26, 3162–3168 CrossRef CAS.
- M. Yu, C. K. Chan and H. Tüysüz, ChemSusChem, 2018, 11, 605–611 CrossRef CAS PubMed.
- X. Deng, C. K. Chan and H. Tüysüz, ACS Appl. Mater. Interfaces, 2016, 8, 32488–32495 CrossRef CAS PubMed.
- H. Wang, R. Liu, Y. Li, X. Lü, Q. Wang, S. Zhao, K. Yuan, Z. Cui, X. Li, S. Xin, R. Zhang, M. Lei and Z. Lin, Joule, 2018, 2, 337–348 CrossRef CAS.
- X. T. Wang, T. Ouyang, L. Wang, J. H. Zhong, T. Ma and Z. Q. Liu, Angew. Chem., Int. Ed., 2019, 58, 13291–13296 CrossRef CAS PubMed.
- X. Gao, J. Liu, Y. Sun, X. Wang, Z. Geng, F. Shi, X. Wang, W. Zhang, S. Feng, Y. Wang and K. Huang, Inorg. Chem. Front., 2019, 6, 3295–3301 RSC.
- Y. Huang, W. Yang, Y. Yu and S. Hao, J. Electroanal. Chem., 2019, 840, 409–414 CrossRef CAS.
- F. Waag, B. Gökce, C. Kalapu, G. Bendt, S. Salamon, J. Landers, U. Hagemann, M. Heidelmann, S. Schulz, H. Wende, N. Hartmann, M. Behrens and S. Barcikowski, Sci. Rep., 2017, 7, 1–13 CrossRef CAS PubMed.
- G. Ou, F. Wu, K. Huang, N. Hussain, D. Zu, H. Wei, B. Ge, H. Yao, L. Liu, H. Li, Y. Shi and H. Wu, ACS Appl. Mater. Interfaces, 2019, 11, 3978–3983 CrossRef PubMed.
- K. Chakrapani, G. Bendt, H. Hajiyani, I. Schwarzrock, T. Lunkenbein, S. Salamon, J. Landers, H. Wende, R. Schlögl, R. Pentcheva, M. Behrens and S. Schulz, ChemCatChem, 2017, 9, 2988–2995 CrossRef CAS.
- Y. Liu, Y. Ying, L. Fei, Y. Liu, Q. Hu, G. Zhang, S. Y. Pang, W. Lu, C. L. Mak, X. Luo, L. Zhou, M. Wei and H. Huang, J. Am. Chem. Soc., 2020, 141, 8136–8145 CrossRef PubMed.
- J. Jiang, Y. Chen, H. Cong, J. Tang, Y. Sun, X. Hu, L. Wang, S. Han and H. Lin, Ionics, 2020, 26, 3437–3446 CrossRef CAS.
- X. Zou, J. Su, R. Silva, A. Goswami, B. R. Sathe and T. Asefa, Chem. Commun., 2013, 49, 7522–7524 RSC.
- S. A. Cho, Y. J. Jang, H.-D. Lim, J.-E. Lee, Y. H. Jang, T.-T. H. Nguyen, F. M. Mota, D. P. Fenning, K. Kang, Y. Shao-Horn and D. H. Kim, Adv. Energy Mater., 2017, 7, 1700391 CrossRef.
- E. Budiyanto, M. Yu, M. Chen, S. Debeer, O. Rüdiger and H. Tüysüz, ACS Appl. Energy Mater., 2020, 3, 8583–8594 CrossRef CAS.
- M. Olivares-Marín, P. Palomino, J. M. Amarilla, E. Enciso and D. Tonti, J. Mater. Chem. A, 2013, 1, 14270–14279 RSC.
- G. Li, X. Wang, J. Fu, J. Li, M. G. Park, Y. Zhang, G. Lui and Z. Chen, Angew. Chem., Int. Ed., 2016, 55, 4977–4982 CrossRef CAS PubMed.
- N. Husnain, E. Wang, S. Fareed and M. T. Anwar, Catalysts, 2019, 9, 1018–1034 CrossRef CAS.
- K. J. Kim, H. K. Kim, Y. R. Park, G. Y. Ahn, C. S. Kim and J. Y. Park, Hyperfine Interact., 2006, 169, 1363–1369 CrossRef CAS.
- N. Bahlawane, P. H. T. Ngamou, V. Vannier, T. Kottke, J. Heberle and K. Kohse-Höinghaus, Phys. Chem. Chem. Phys., 2009, 11, 9224–9232 RSC.
- G. Wu, J. Wang, W. Ding, Y. Nie, L. Li, X. Qi, S. Chen and Z. Wei, Angew. Chem., Int. Ed., 2016, 55, 1340–1344 CrossRef CAS PubMed.
- T. Baird, K. C. Campbell, P. J. Holliman, R. Hoyle, G. Noble, D. Stirling and B. P. Williams, J. Mater. Chem., 2003, 13, 2341–2347 RSC.
- M. Xing, L. B. Kong, M. C. Liu, L. Y. Liu, L. Kang and Y. C. Luo, J. Mater. Chem. A, 2014, 2, 18435–18443 RSC.
- T. Gao, Z. Jin, M. Liao, J. Xiao, H. Yuan and D. Xiao, J. Mater. Chem. A, 2015, 3, 17763–17770 RSC.
- Q. Qu, J.-H. Zhang, J. Wang, Q.-Y. Li, C.-W. Xu and X. Lu, Sci. Rep., 2017, 7, 41542 CrossRef CAS PubMed.
- J. Ahmed, B. Kumar, A. M. Mugweru, P. Trinh, K. V. Ramanujachary, S. E. Lofland, Govind and A. K. Ganguli, J. Phys. Chem. C, 2010, 114, 18779–18784 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- T. Aghavnian, J.-B. Moussy, D. Stanescu, R. Belkhou, N. Jedrecy, H. Magnan, P. Ohresser, M.-A. Arrio, P. Sainctavit and A. Barbier, J. Electron Spectrosc. Relat. Phenom., 2015, 202, 16–21 CrossRef CAS.
- Z. Zhou, Y. Zhang, Z. Wang, W. Wei, W. Tang, J. Shi and R. Xiong, Appl. Surf. Sci., 2008, 254, 6972–6975 CrossRef CAS.
- T. Fujii, F. M. F. de Groot, G. A. Sawatzky, F. C. Voogt, T. Hibma and K. Okada, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 3195–3202 CrossRef CAS.
- N.-I. Kim, Y. J. Sa, T. S. Yoo, S. R. Choi, R. A. Afzal, T. Choi, Y.-S. Seo, K.-S. Lee, J. Y. Hwang, W. S. Choi, S. H. Joo and J.-Y. Park, Sci. Adv., 2022, 4, 9360–9372 CrossRef PubMed.
- H. Ren, L. Zhang, J. Zhang, T. Miao, R. Yuan, W. Chen, Z. Wang, J. Yang and B. Zhao, Carbon, 2022, 198, 46–56 CrossRef CAS.
- G. Sun, H. Ren, Z. Shi, L. Zhang, Z. Wang, K. Zhan, Y. Yan, J. Yang and B. Zhao, J. Colloid Interface Sci., 2021, 588, 847–856 CrossRef CAS PubMed.
- J. Bao, X. Zhang, B. Fan, J. Zhang, M. Zhou, W. Yang, X. Hu, H. Wang, B. Pan and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 7399–7404 CrossRef CAS PubMed.
- G. Fang, C. Zhu, M. Chen, J. Zhou, B. Tang, X. Cao, X. Zheng, A. Pan and S. Liang, Adv. Funct. Mater., 2019, 29, 1808375 CrossRef.
- F. Cheng, J. Shen, B. Peng, Y. Pan, Z. Tao and J. Chen, Nat. Chem., 2011, 3, 79–84 CrossRef CAS PubMed.
- J. Li, P. Zhou, F. Li, J. Ma, Y. Liu, X. Zhang, H. Huo, J. Jin and J. Ma, J. Power Sources, 2016, 302, 343–351 CrossRef CAS.
- L. Gong, X. Y. E. Chng, Y. Du, S. Xi and B. S. Yeo, ACS Catal., 2018, 8, 807–814 CrossRef CAS.
- A. Kargar, S. Yavuz, T. K. Kim, C. H. Liu, C. Kuru, C. S. Rustomji, S. Jin and P. R. Bandaru, ACS Appl. Mater. Interfaces, 2015, 7, 17851–17856 CrossRef CAS PubMed.
- S. Jung, C. C. L. McCrory, I. M. Ferrer, J. C. Peters and T. F. Jaramillo, J. Mater. Chem. A, 2016, 4, 3068–3076 RSC.
- T. Grewe, X. Deng, C. Weidenthaler, F. Schüth and H. Tüysüz, Chem. Mater., 2013, 25, 4926–4935 CrossRef CAS.
- W. Xu, W. Xie and Y. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 28642–28649 CrossRef CAS PubMed.
- Z. H. Xue, H. Su, Q. Y. Yu, B. Zhang, H. H. Wang, X. H. Li and J. S. Chen, Adv. Energy Mater., 2017, 7, 1–7 CAS.
- Z. Zhuang, W. Sheng and Y. Yan, Adv. Mater., 2014, 26, 3950–3955 CrossRef CAS PubMed.
- M. E. G. Lyons and M. P. Brandon, Phys. Chem. Chem. Phys., 2009, 11, 2203–2217 RSC.
- S. Anantharaj and S. Kundu, ACS Energy Lett., 2019, 4, 1260–1264 CrossRef CAS.
- S. Anantharaj, S. R. Ede, K. Karthick, S. Sam Sankar, K. Sangeetha, P. E. Karthik and S. Kundu, Energy Environ. Sci., 2018, 11, 744–771 RSC.
- J. Sun, Y. Li, X. Liu, Q. Yang, J. Liu, X. Sun, D. G. Evans and X. Duan, Chem. Commun., 2012, 48, 3379–3381 RSC.
- R. Martínez-Garcia, M. Knobel and E. Reguera, J. Phys. Chem. B, 2006, 110, 7296–7303 CrossRef PubMed.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. García-Melchor, L. Han, J. Xu, M. Liu, L. Zheng, F. P. García de Arquer, C. T. Dinh, F. Fan, M. Yuan, E. Yassitepe, N. Chen, T. Regier, P. Liu, Y. Li, P. De Luna, A. Janmohamed, H. L. Xin, H. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- E. B. Castro and C. A. Gervasi, Int. J. Hydrogen Energy, 2000, 25, 1163–1170 CrossRef CAS.
- B. Wei, J. Wu, G. Mei, Z. Qi, W. Hu and Z. Wang, Int. J. Hydrogen Energy, 2019, 44, 6612–6617 CrossRef CAS.
- M. C. Sung, G. H. Lee and D. W. Kim, J. Alloys Compd., 2019, 800, 450–455 CrossRef CAS.
- Y. Li, X. Zhang, A. Hu and M. Li, Int. J. Hydrogen Energy, 2018, 3, 22012–22020 CrossRef.
- H. Wang, X. Wang, D. Yang, B. Zheng and Y. Chen, J. Power Sources, 2018, 400, 232–241 CrossRef CAS.
- T. Ouyang, Y. Q. Ye, C. Y. Wu, K. Xiao and Z. Q. Liu, Angew. Chem., Int. Ed., 2019, 58, 4923–4928 CrossRef CAS PubMed.
- X. Yan, L. Tian, M. He and X. Chen, Nano Lett., 2015, 15, 6015–6021 CrossRef CAS PubMed.
- F. Sun, L. Li, G. Wang and Y. Lin, J. Mater. Chem. A, 2017, 5, 6849–6859 RSC.
- Q. Liu, S. Gu and C. Li, J. Power Sources, 2015, 299, 342–346 CrossRef CAS.
- J. Luo, J. H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N. G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- H. Li, S. Chen, Y. Zhang, Q. Zhang, Q. Zhang, X. Jia, L. Gu, X. Sun, L. Song and X. Wang, Nat. Commun., 2018, 9, 1–12 CrossRef PubMed.
- Y. Pan, K. Sun, S. Liu, X. Cao, K. Wu, W. C. Cheong, Z. Chen, Y. Wang, Y. Li, Y. Liu, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 2610–2618 CrossRef CAS PubMed.
- B. Cao, Y. Cheng, M. Hu, P. Jing, Z. Ma, B. Liu, R. Gao and J. Zhang, Adv. Funct. Mater., 2019, 29, 1906316–1906333 CrossRef CAS.
- X. Yu, J. Zhao and M. Johnsson, Adv. Funct. Mater., 2021, 31, 2101578–2101588 CrossRef CAS.
- Z. Kang, H. Guo, J. Wu, X. Sun, Z. Zhang, Q. Liao, S. Zhang, H. Si, P. Wu, L. Wang and Y. Zhang, Adv. Funct. Mater., 2019, 29, 1807031–1807040 CrossRef.
- W. Fang, D. Liu, Q. Lu, X. Sun and A. M. Asiri, Electrochem. Commun., 2016, 63, 60–64 CrossRef CAS.
- J. Yu, Q. Li, Y. Li, C. Y. Xu, L. Zhen, V. P. Dravid and J. Wu, Adv. Funct. Mater., 2016, 26, 7644–7651 CrossRef CAS.
- Y. Sun, C. Wang, T. Ding, J. Zuo and Q. Yang, Nanoscale, 2016, 8, 18887–18892 RSC.
- R. Li, D. Zhou, J. Luo, W. Xu, J. Li, S. Li, P. Cheng and D. Yuan, J. Power Sources, 2017, 341, 250–256 CrossRef CAS.
- Q. Zhang, N. M. Bedford, J. Pan, X. Lu and R. Amal, Adv. Energy Mater., 2019, 9, 1–13 CrossRef.
- J. Jiang, P. Yan, Y. Zhou, Z. Cheng, X. Cui, Y. Ge and Q. Xu, Adv. Energy Mater., 2020, 10, 1–9 Search PubMed.
- T. L. L. Doan, D. C. Nguyen, S. Prabhakaran, D. H. Kim, D. T. Tran, N. H. Kim and J. H. Lee, Adv. Funct. Mater., 2021, 31, 2100233–2100247 CrossRef CAS.
- X. Gao, H. Zhang, Q. Li, X. Yu, Z. Hong, X. Zhang, C. Liang and Z. Lin, Angew. Chem., Int. Ed., 2016, 55, 6290–6294 CrossRef CAS PubMed.
- L. Yan, L. Cao, P. Dai, X. Gu, D. Liu, L. Li, Y. Wang and X. Zhao, Adv. Funct. Mater., 2017, 27, 1703455–1703464 CrossRef.
- X. Luo, P. Ji, P. Wang, R. Cheng, D. Chen, C. Lin, J. Zhang, J. He, Z. Shi, N. Li, S. Xiao and S. Mu, Adv. Energy Mater., 2020, 10, 1903891 CrossRef CAS.
- H. Ma, Z. Chen, Z. Wang, C. V. Singh and Q. Jiang, Adv. Sci., 2022, 9, 2105313 CrossRef CAS PubMed.
- B. Cao, Y. Cheng, M. Hu, P. Jing, Z. Ma, B. Liu, R. Gao and J. Zhang, Adv. Funct. Mater., 2019, 29, 1906316 CrossRef CAS.
- L. Hang, T. Zhang, Y. Sun, D. Men, X. Lyu, Q. Zhang, W. Cai and Y. Li, J. Mater. Chem. A, 2018, 6, 19555–19562 RSC.
- J. Wang, M. Zhang, G. Yang, W. Song, W. Zhong, X. Wang, M. Wang, T. Sun and Y. Tang, Adv. Funct. Mater., 2021, 31, 2101532 CrossRef CAS.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- Y. Xu, Y. Yan, T. He, K. Zhan, J. Yang, B. Zhao, K. Qi and B. Y. Xia, Carbon, 2019, 145, 201–208 CrossRef CAS.
- P. Wang, L. Zhang, Z. Wang, D. Bu, K. Zhan, Y. Yan, J. Yang and B. Zhao, J. Colloid Interface Sci., 2021, 597, 361–369 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr07300e |
| This journal is © The Royal Society of Chemistry 2023 |