
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorinated dendritic amphiphiles, their stomatosome aggregates and application in enzyme encapsulation†
Tiffany
Guitton-Spassky
 a,
Florian
Junge
a,
Abhishek Kumar
Singh
a,
Boris
Schade
b,
Katharina
Achazi
a,
Marta
Maglione
ac,
Stephan
Sigrist
c,
Rashmi
Rashmi
a and
Rainer
Haag
*a
a,
Florian
Junge
a,
Abhishek Kumar
Singh
a,
Boris
Schade
b,
Katharina
Achazi
a,
Marta
Maglione
ac,
Stephan
Sigrist
c,
Rashmi
Rashmi
a and
Rainer
Haag
*a
aInstitut für Chemie und Biochemie, Organische Chemie, Freie Universität Berlin, Takustraße 3, Berlin, 14195 Germany. E-mail: haag@chemie.fu-berlin.de
bForschungszentrum für Elektronenmikroskopie, Institut für Chemie und Biochemie, Freie Universität Berlin, Fabeckstraße 36a, Berlin, 14195 Germany
cInstitute for Biology, Freie Universität Berlin, Takustraße 6, Berlin, 14195 Germany
First published on 31st March 2023
Abstract
Enzymes are more selective and efficient than synthetic catalysts but are limited by difficult recycling. This is overcome by immobilisation, namely through encapsulation, with the main drawback of this method being slow diffusion of products and reactants, resulting in effectively lowered enzyme activity. Fluorinated dendritic amphiphiles were reported to self-assemble into regularly perforated bilayer vesicles, so-called “stomatosomes”. It was proposed that they could be promising novel reaction vessels due to their increased porosity while retaining larger biomolecules at the same time. Amphiphiles were synthesised and their aggregation was analysed by cryogenic transmission electron microscopy (cryo-TEM) and dynamic light scattering (DLS) in buffered conditions necessary for enzyme encapsulation. Urease and albumin were encapsulated using the thin-film hydration method and investigated by confocal and time-gated stimulated emission depletion microscopy (gSTED). Their release was then used to probe the selective retention of cargo by stomatosomes. Free and encapsulated enzyme activity were compared and their capacity to be reused was evaluated using the Berthelot method. Urease was successfully encapsulated, did not leak out at room temperature, and showed better activity in perforated vesicles than in closed vesicles without perforations. Encapsulated enzyme could be reused with retained activity over 8 cycles using centrifugation, while free enzyme had to be filtrated. These results show that stomatosomes may be used in enzyme immobilisation applications and present advantages over closed vesicles or free enzyme.
1 Introduction
Enzymes are biological catalysts produced by living organisms to accelerate biochemical reactions, which can then be extracted from cells to catalyse synthetic processes. Due to the multitude of compounds present in cellular environments, high selectivity and efficiency are necessary for these catalysts.1 Enzymes are therefore important components of green chemistry and are widely used in industry, food processing and development of pharmaceutics.2–4 They are biocompatible, biodegradable and renewable while surpassing synthetic catalysts in their selectivity, remaining efficient in mild aqueous conditions.However, this advantage in biological systems becomes a disadvantage in synthetic ones: enzymes are highly water soluble homogeneous catalysts and difficult to recover without expensive filtration systems. Recuperating desired products and reusing enzymes for further reaction cycles is made difficult. Therefore, new methods for recovering enzymes are currently an important research topic.5 A major method to this end is immobilisation, which may be done by trapping the enzyme within an insoluble carrier or attaching it through covalent bonds to an insoluble support.6 Enzymes may also be immobilised by encapsulation, where the enzyme remains dissolved in the reaction solution but is confined in a particulate nano-/microstructure surrounded by a barrier. This can be done with shell-forming proteins, DNA, silica or polymers.7 Alternatively, amphiphiles can be used to form vesicles: polymersomes in the case of amphiphilic polymers or liposomes for lipids.8,9 Using encapsulation, only little enzyme is denatured as it does not interact with the carrier. The effective increase in size and subsequent change in physical properties after encapsulation means the enzymes can be separated by either centrifugation or filtration. However, this method suffers from mass transfer limitations and leakage of enzyme into solution.10 Products and reactants move more slowly to and from the enzyme, which becomes effectively less active. Enzyme can leak into solution, contaminating the product and being lost for further reactions. Pore size should be limited such that products and reactants can pass through unhindered but without enzyme leaking out, i.e. they should behave as a sieve or filter. Such selectivity is key in enzyme encapsulation applications.
Due to their special properties, namely stability and tendency to form lower curvature structures, fluorinated systems have been a subject of interest for applications and to investigate the effect of fluorine on aggregation.11–15 The stability of these structures can be explained by the perfluorination of the amphiphile tails, due to highly electronegative fluorine,16 and the stable C–F bond. Perfluorinated moieties have extremely low polarisability, as dipole moments between C and electronegative F cancel each other in perfluorinated chains,17 making them both hydrophobic and lipophobic and leading to the formation of fluorous domains.18,19 Due to this strong phase separation, self-assembled layers are less permeable. They can be combined with hydrophilic dendritic oligoglycerol head groups, which imparts solubility and biocompatibility. Because of the advantageous properties described, the assembly of amphiphiles containing oligoglycerols head groups and fluorinated tails has been systematically evaluated.20,21 Recently, branched fluorinated amphiphiles have been of interest due to their potentially less hazardous nature.22–24
Stomatosomes, or perforated vesicles,25 were reported to form from single-component fluorinated dendritic amphiphiles.20 They are stable with regularly sized and spaced perforations and are formed from one component, unlike the majority of existing stomatosome systems. These usually include amphiphiles, with sizes ranging from small molecules to polymers, and additives such as surfactants.26–30 Tuning of salt concentration is often necessary in the case of charged compounds.31 The systems used in this work are therefore advantageous due to their simple formulation.
Only recently have stomatosomes been investigated for applications. Unlike other self-assembled structures like cubosomes and hexosomes32 or vesicles,33 studies testing for applications have been limited to a few papers34–38 or focused on polymeric capsules.39
It was expected that stomatosome pores would increase mass transfer, i.e. reactants and products would be able to diffuse in and out of vesicles with faster kinetics. Additionally, the presence of fluorine leads to higher stability and regularity of pore size, potentially leading to reduced leakage and a selective porosity. Being one-component systems, formulation is straight-forward and less likely to interfere with the reaction that is catalysed. For these reasons, we decided to evaluate these systems as carriers for enzyme encapsulation applications.
Jack bean urease is a relevant enzyme in research and industry with many applications.40,41 It is a large enzyme (MW: 590 kDa),42 which exceeds in size the pores of stomatosomes formed from the synthesised fluorinated amphiphiles (13 nm diameter compared to 10 nm for holes)20,43 and so was not expected to leak out significantly. It was encapsulated using the thin-film hydration method then encapsulation was confirmed by fluorescence microscopy and quantified through bicinchoninic acid (BCA) assays.44 A smaller enzyme, bovine serum albumin, was also encapsulated to test the theory that stomatosomes can act as sieves and retain cargo selectively.
Urease activity was measured using the Berthelot method45 and was compared between free and encapsulated enzyme. Closed vesicles were used as controls and activity was compared within closed and perforated vesicles. Enzyme was recycled over 8 reuse cycles. The stability of these systems was evaluated through leakage studies.
2 Introduction
2.1 Amphiphile structures
The synthesis was carried out following already reported procedures from our group.20,23 Amphiphiles were selected based on their ability to form stomatosomes reliably (Fig. 1). Two different chain lengths were used to investigate further the effect of the structure change on the assembly behaviour and capacity to encapsulate. The branched amphiphile was chosen as it was shown to form closed vesicles, and so its aggregates were used as a control system.23 The syntheses involve click-coupling as well as enzyme-catalysed reactions, thus improving the efficiency and ecological impact of certain steps.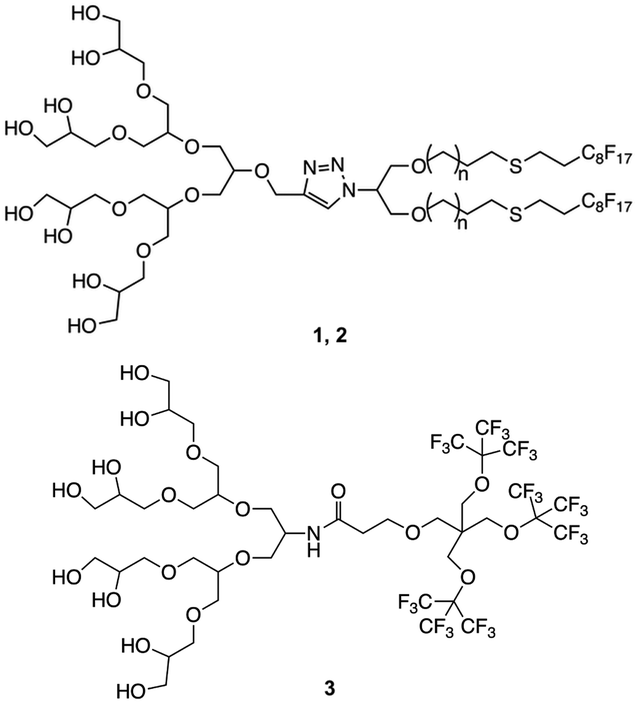 | ||
| Fig. 1 Structure of amphiphiles forming stomatosomes (n = 1 for 1 and n = 4 for 2) and amphiphile forming closed vesicles 3. | ||
2.2 Comparison of amphiphile aggregation in water and buffer
The branched amphiphile has a CAC of 83 μM.23 The shorter fluorous moiety here is associated with a higher CAC, as a decrease in the fluorination leads to a weaker fluorophobicity. Although this is higher than for the two linear amphiphiles, it still remains very low compared to most alkylated amphiphilic compounds.
The CACs of 1 and 2 were also determined in PBS solution, as it has to be used as a medium in enzyme encapsulation. It was not expected to see a large change in value as the amphiphiles are non-ionic. Nevertheless, the CACs were found to be lower with 1.4 μM for the shorter chain amphiphile 1 and 1.6 μM for the longer chain amphiphile 2. This may be due to the higher ionic strength of the medium.
 | ||
| Fig. 2 Cryo-TEM micrographs of self-assemblies formed from amphiphiles 1 (left) and 2 (right) proving the formation of stomatosomes in PBS. Spherical stomatosomes (black arrows) can be clearly distinguished from planar bilayers (white arrows) due to their dark edges, which result from the view along the boundaries of the hollow structures, whereas planar bilayers do not feature these edges. While the shorter compound 1 mainly forms the spherical stomatosomes we found conspicuously more planar bilayers and even some micellar threads (gray arrows) from compound 2. Concentration used was 2 mM. | ||
DLS was used to compare aggregation in PBS and ultrapure water (Table 1). An increase was observed in the z-average diameter going from water to PBS while the PDIs remained within error range of each other. It seems that the additional salinity increases the size of the aggregates. It has been shown previously that addition of salt resulted in enhanced ordering of nonionic surfactant bilayers,52 which could result in a change in aggregation.
| 1 | 2 | |||
|---|---|---|---|---|
| PDI | z-Average/nm | PDI | z-Average/nm | |
| Ultrapure water | 0.242 ± 0.009 | 62 ± 3 | 0.32 ± 0.05 | 92 ± 5 |
| PBS | 0.27 ± 0.05 | 134 ± 9 | 0.31 ± 0.04 | 115 ± 8 |
Planar bilayers can be observed in the cryo-TEM micrographs. They are perforated like the stomatosomes but do not have darker regions at the periphery, hence do not curve into vesicles. The planar bilayers are found more often for the long-chain amphiphile 2, potentially due to its lower spontaneous curvature, which discourages bending into vesicles.53–55 This could be because of the longer alkyl chain linking the fluorinated moiety with the polar head. Firstly, this extends the hydrophobic tail, thus increasing its volume relative to the head and making the molecule more symmetrical. The alkyl chain is also more flexible than the fluorinated chain. This increases the number of possible conformations, i.e. the entropy, which can increase the volume occupied by the chain. Again, this would decrease the spontaneous curvature. The effect of structure on aggregate curvature was also observed in other fluorinated amphiphiles synthesised by this research group, where an increased symmetry from a smaller head group volume lead to a decrease in curvature.21
The salinity had a greater effect on the aggregation behaviour of the shorter-chain amphiphile 1. Cryo-TEM experiments pointed to an enhanced formation of vesicles rather than perforated planar bilayers by this compound. Vesicles may be more sensitive to the salinity than planar bilayers and show a larger increase in size when the salinity of the solution is changed.
Lastly, some cylindrical micelles are also present, which sometimes are joined at both ends like doughnuts. These are higher curvature structures than stomatosomes or planar bilayers.
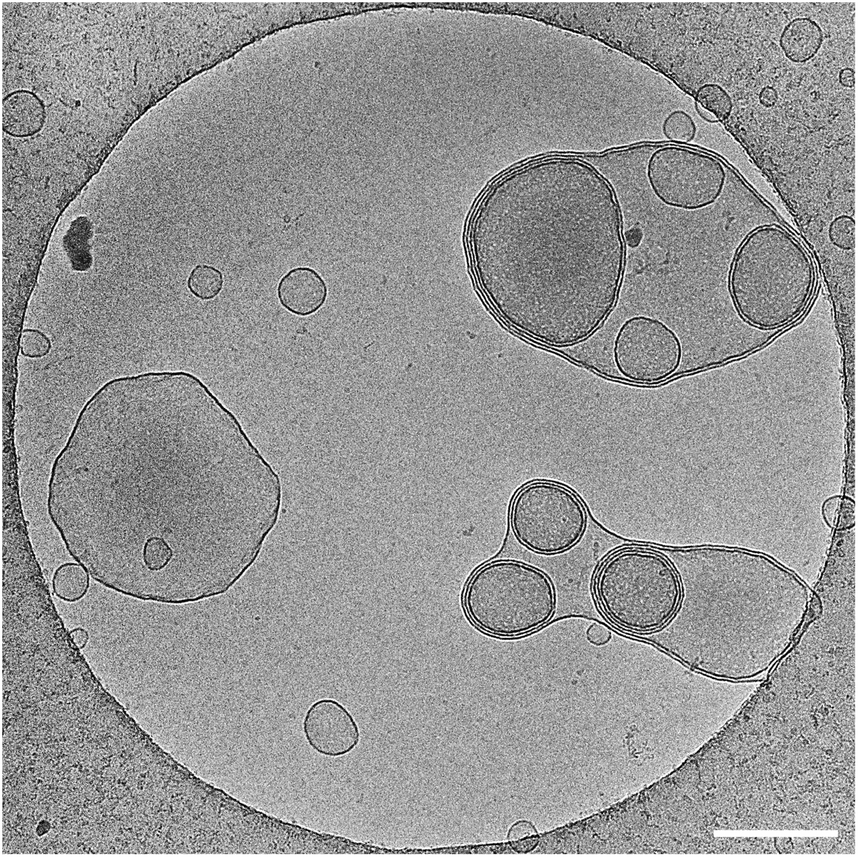 | ||
| Fig. 3 Cryo-TEM micrograph of amphiphiles 3 showing the formation of multilayered closed vesicles. Concentration used was 3.5 mM, scale bar is 200 nm. | ||
2.3 Enzyme encapsulation in stomatosomes
 | ||
| Fig. 4 Schematic representation of the encapsulation of urease within stomatosomes, Cy5 and amphiphiles are represented in red and ATTO 565-urease in cyan. | ||
The hydrophobic dye Cy5 was dissolved with amphiphile 1 in step 1 in Fig. 4 and then evaporated to form a thin film inside a round-bottom flask. A solution of urease dissolved in PBS buffer was added in step 2, the amphiphile layer was dissolved in step 3 and free enzyme was removed by centrifugation in step 4. Encapsulated enzyme is larger and heavier than free enzyme and so the latter can be removed with the supernatant. Centrifuging and removing the supernatant three times was enough to remove all free enzyme. This was verified using BCA assays, which were used to determine the protein concentration in the supernatant. No more enzyme could be detected in the supernatant after 3 centrifugation cycles, even after extensive washing. The amount of enzyme removed was lower than the initial amount added, thus we concluded that some was retained by the stomatosomes.
The stomatosomes and its encapsulated enzyme were then visualised using fluorescence microscopy in order to confirm the procedure had been successful, both using 2 channels 2D time-gated STED (or gSTED) and confocal microscopy (Fig. 5). Urease was labelled with ATTO 565 as it can be imaged well using gSTED fluorescence microscopy. Simply by using confocal microscopy, distinct vesicles could be observed and then imaged by 3D reconstruction (Fig. 6). Cy5 was used as a hydrophobic dye. Pyrene was initially used to this end, as its fluorescence spectrum changes when it is in a polar or apolar environment. Comparing spectra allowed us to verify that it could be incorporated in the hydrophobic layer. Cy5 had better efficiency and did not overlap with the spectra of the other dyes, thus avoiding cross-talk, and so was used in following experiments instead of pyrene.
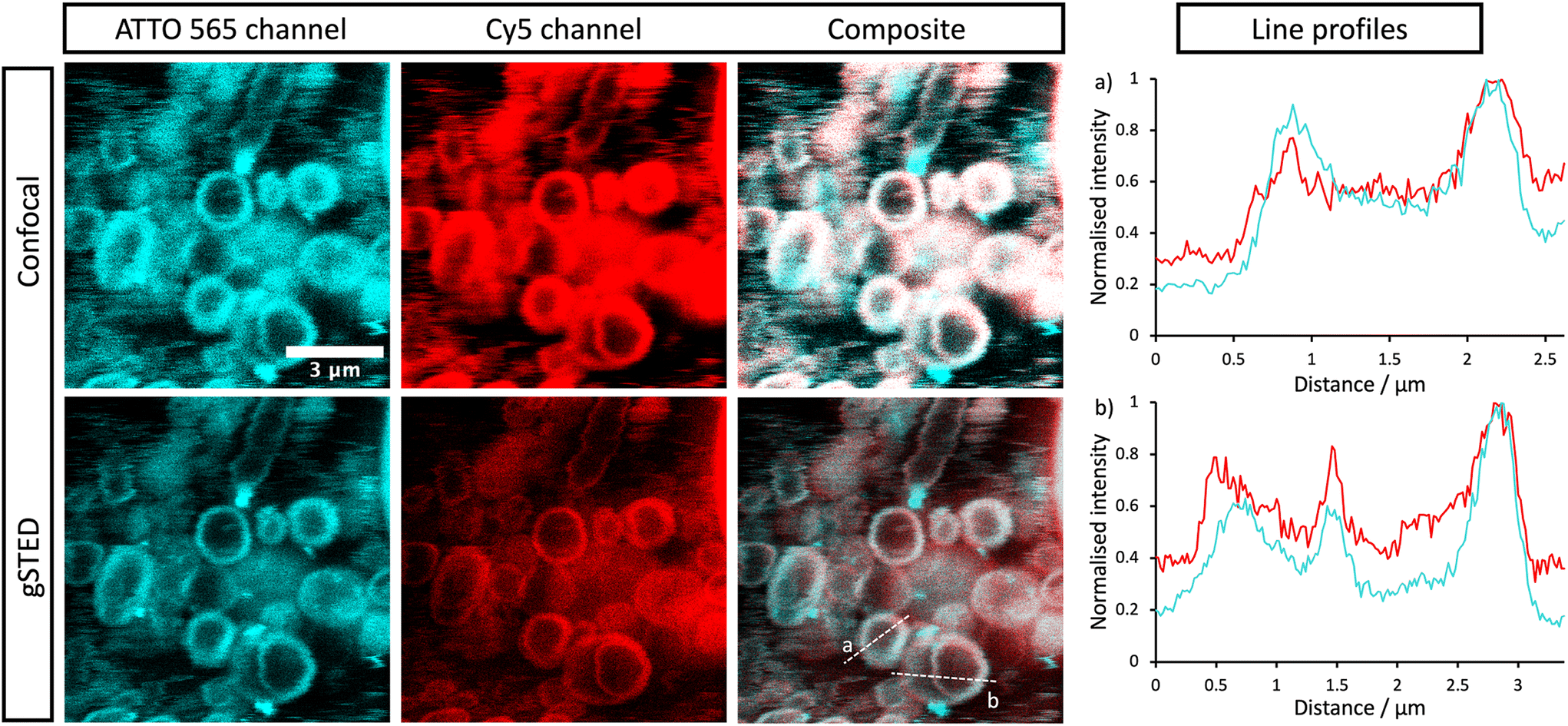 | ||
| Fig. 5 Confocal and gSTED micrographs of encapsulated labelled urease within stomatosomes. ATTO 565 (cyan) is bound to urease and Cy5 (red) resides in the stomatosome bilayer formed from 1. Composite images show the overlap (white) and colocalisation of the two dyes. Concentration of amphiphile used was initially 12 mM, which may have been reduced after encapsulation. | ||
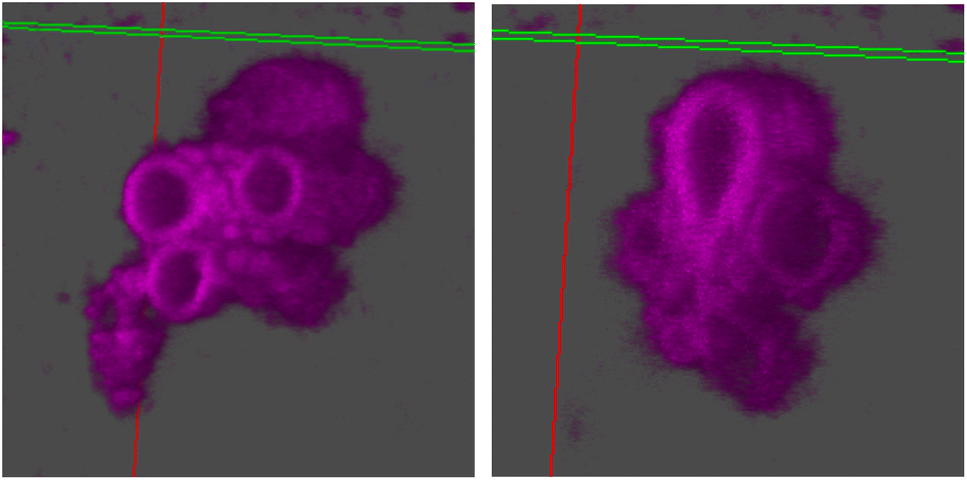 | ||
| Fig. 6 3D reconstruction using z-stacks taken in the Cy5 channel in confocal microscopy. The line perpendicular to the plane cutting through the vesicle is the z-axis. The hollow vesicular structure of stomatosomes is shown, going down the z-axis from the right image to the left. | ||
Both enzyme and stomatosomes were detected in the same regions, with overlap shown as white in the composite images. This showed that the enzyme was successfully trapped by the aggregates. Due to the presence of perforations, it was possible that some leakage would have occurred, thus making encapsulation inefficient and transient. This was not the case here, and encapsulated enzyme as well as the carriers could be visualised.
The additional resolution imparted by gSTED microscopy gave interesting results. The enzyme seemed to be concentrated in the same regions as the hydrophobic dye, i.e., the stomatosome membrane. This could be explained by the labelling of the enzyme, which could have increased their hydrophobicity. ATTO 565 has 6 hydrocarbon rings, including 2 piperidines and 4 aromatics. This makes it more hydrophobic than the enzyme and prone to interact with the membrane, this in the same way as Cy5, which also has a structure based on an extended aromatic nitrogen-containing system. The degree of labelling with the hydrophobic dye ATTO 565 used to visualise the urease was 0.340, so lower than one. This means not all enzyme present has been labelled with dye. It is therefore possible that the more hydrophobic labelled enzyme interacts with the hydrophobic bilayer while the non-labelled enzyme remains in the aqueous solution in the interior of the stomatosomes. Only the labelled enzyme is visible in fluorescence microscopy, which could explain the ring patterns obtained (Fig. 5, gSTED images). Minor interaction between protein and membrane has previously been suggested to explain similar distributions in previous works.58 Another factor mentioned was the scattering and reflection of fluorescent light between the membrane–cavity interface. This could also be taking place in our case and may in part explain the distribution obtained.
Another possibility is that the permeability from the perforations lead to a high concentration of enzyme in the same region as the membrane. Similar distributions have been observed in cases where the membrane is at least partially porous (either smooth vesicle or disrupted membrane).59,60 In those cases, a darker outline can be seen. When polymers or gold is used instead of lipids, the porosity is greatly reduced and a more uniform distribution is observed.61 When the membrane is disrupted, a similar disk can again be seen.62
Bright cyan spots of fluorescence can be observed. These are likely to be due to agglomerated urease, where enzymes aggregate with other enzyme in solution.
From gSTED and confocal microscopy, we can note the surprisingly large size of the aggregates, which exceeds what we would expect from previous cryo-TEM and DLS results. This could be due to the large polydispersity in vesicle sizes. Samples were not filtered on membranes to narrow the size distribution, unlike in DLS measurements, this to avoid changes in the aggregation and potential loss of stomatosome structures. In cryo-TEM, smaller aggregates are observed as larger ones are destroyed in the freezing process due to being too large for the thin layer of ice formed. In gSTED, the image is produced sequentially by scanning pixel by pixel, and so smaller aggregates diffusing faster appear blurry and can not be imaged. This can be seen in the background of gSTED and confocal images (Fig. 5). For these reasons, vesicle sizes vary a lot when measured with different methods, and when taken together, this reveals large dispersity. The exact assembly process and whether there is hierarchical self-assembly taking place is not known.
Interestingly, little fluctuation was observed between topologies and vesicles remained stable as they did not convert into other structures. It was also verified that stomatosomes were still present after encapsulation (Fig. 7). They were still present and remained stable after the encapsulation procedure. Urease could not be visualised by this method due to the high contrast from the fluorinated chains. Due to this, it was not possible to explain the distribution observed in fluorescence microscopy from cryo-TEM images and determine the exact localisation of the encapsulated enzyme. In any case, enzyme was successfully retained by stomatosome carriers.
 | ||
| Fig. 7 Cryo-TEM micrograph of amphiphile 1 after encapsulation of urease and separation of free enzyme. Concentration of amphiphile used was initially 12 mM, which may have been reduced after encapsulation. | ||
We know from gSTED and confocal microscopy that enzyme is in part concentrated in close proximity to the membrane. From extensive washing of the aggregates and lack of background fluorescence, the enzyme is probably not in the external solution. The retained enzyme is either within the vesicle (interacting or not with the membrane) or attached on the outside of the vesicle. This could not be observed by cryo-TEM due to the low contrast of unlabelled urease compared to the fluorinated amphiphiles. To further understand the distribution of encapsulated urease, we measured zeta potentials of different samples (Fig. 8) following conditions taken from previous work.63 We investigated the surface charge of both empty vesicles, vesicles carrying enzyme and free enzyme by studying their electrophoretic mobility. Urease has a negative charge at buffer pH (isoelectric point of urease is 5.1).64 These results confirmed that urease was not stuck on the outside of the vesicles, both in the case of stomatosomes and closed vesicles. If it was the case, vesicles would have had a negative surface charge,59 but instead they retained their slightly positive charge after encapsulation of urease.
 | ||
| Fig. 8 Surface charge evaluation given by electrophoretic mobility measurements. We measured 5 samples: free jack bean urease (JBU), empty stomatosomes (stom.), stomatosomes encapsulating urease (stom. + JBU), empty closed vesicles and closed vesicles encapsulating urease (closed + JBU). All vesicles had slightly positive surface charge while urease had a highly negative surface charge. Amphiphile concentration was initially 12 mM then diluted 100× before measurement, free urease had a concentration of 0.4 mg ml−1. | ||
The encapsulation efficiency (EE%, eqn (1)) was determined using BCA assays. The supernatant was removed and assayed to find the concentration of urease. The amount lost after centrifugation was subtracted from the initial amount in solution.
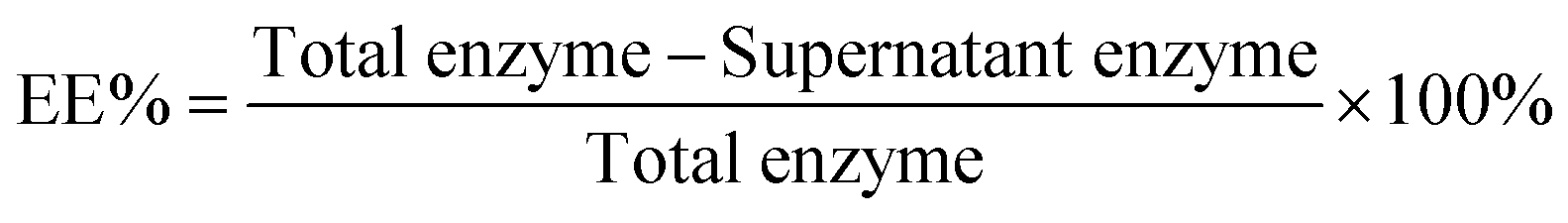 | (1) |
Due to the presence of fluorous moieties, the aggregates tend to preform before they encapsulate, thus the EE% is lowered overall.65 However, free enzyme left after encapsulation can be recovered by centrifugation in the supernatant and used again.
The EE% was found to be higher for the short-chain amphiphile 1 than for the long chain amphiphile 2 (27% ± 2 compared to 17% ± 2, Table 2). This is likely to be due to more vesicles being present for 1, which is the main difference in aggregation behaviour between the two compounds as was observed in cryo-TEM. Due to this difference in EE%, amphiphile 1 was used for following encapsulation experiments unless specified.
| Amphiphile | Enzyme | EE% |
|---|---|---|
| 1 | Urease | 27 ± 2 |
| 1 | BSA | 3 ± 0.4 |
| 2 | Urease | 17 ± 2 |
| 3 | Urease | 20 ± 2 |
Using heat and sonication, or increasing the time to hydrate the film by stirring at room temperature overnight, did not lead to significant improvements in the EE% and it remained within its margin of error. The EE% of urease within stomatosomes of 1 and 2 were comparable to past work relying on the thin-film hydration method.66 An EE% of approximately 10% was found for an equivalent amphiphile concentration. This shows that the preforming of vesicles that was expected did not lower the EE% significantly.
Other methods have previously been used to encapsulate proteins, some with higher expected EE% but also with additional considerations and drawbacks. These include, but are not limited to, freeze–thawing cycles (denaturation of enzyme and time-consuming) or extrusion (potential effect on aggregation) or microfluidics (expensive equipment and more complicated to set up).9,67,68 We therefore opted for the thin-film hydration method, as it is simple, low-cost, fast and should not denature the enzyme as much as other method, which was important for following activity assays. It nevertheless suffers from low EE% and high dispersity of vesicle sizes.
One can also evaluate the obtained EE% values by theoretical means by calculating the captured volume upon aggregation into vesicles.69,70 Using these methods and vesicle sizes from cryo-TEM and DLS (approximately 100 nm), the EE% of our systems may seem higher than expected, as we would predict between 5 and 10% captured volume for our concentrations used. Nevertheless, it is difficult in our case to make an accurate prediction due to multilamellarity and large size dispersity of vesicles. In addition to measuring by DLS the sizes of empty stomatosomes, we also did this for stomatosomes encapsulating enzyme (Table S1†). The major peak (84%) in the volume distribution had a size of approximately 1000 nm, while vesicles imaged in gSTED were as large as 2500 nm. Due to the large error on the DLS data (large PDI, non-spherical aggregates), these were not included here. Nevertheless, in combination with information from all other methods used, it shows us we have many large vesicles. This would increase the predicted EE% to values between 20 and 50%. Our experimental EE% falls within this, albeit large, range. In the future, obtaining a more narrow size distribution would be important for biomedical applications, but this is beyond the scope of this work. We can conclude from our comparison to theoretical predictions of captured volume that the presence of very large aggregates leads to an increased EE%. Using different characterisation methods reveals this dispersity.
Higher temperatures increase urease activity until 65 °C but also denature the enzyme faster.74 This is important to consider if the enzyme is to be reused over multiple reaction cycles and denaturation has to be limited. For this reason, we remained at room temperature to have enough heat energy available to react while not accelerating denaturation too much. At room temperature and in PBS buffer, no enzyme leaked out of perforated vesicles formed from fluorinated amphiphiles after a period of 2 days. This is necessary to establish for applications as enzyme leakage leads to mixing with the products and more difficult separation and down-stream processing. After ultrasonication for 5 min, 14.4 ± 0.7% of encapsulated enzyme was lost to solution. This was also reflected in cryo-TEM studies from past work,20 where ultrasonication resulted in fragments of vesicles being observed, which could explain the loss of enzyme here. Due to the stability of the system, it was concluded that activity assays could be carried out within a few days of encapsulation as long as the samples were not sonicated. In this way, measured enzyme activity would be solely reliant on the activity of encapsulated enzyme, whether within closed vesicles (smooth with no pores) or perforated vesicles. Activity would not be affected by leakage and varying concentration of free enzyme in solution.
2.4 Urease activity
Encapsulated enzyme (after separation from leftover free enzyme) was compared to free enzyme (Fig. 9) by diluting to the same concentrations and then assaying the enzyme solutions following procedures from the manufacturer. Closed vesicles encapsulating urease were prepared in exactly the same way as stomatosomes.
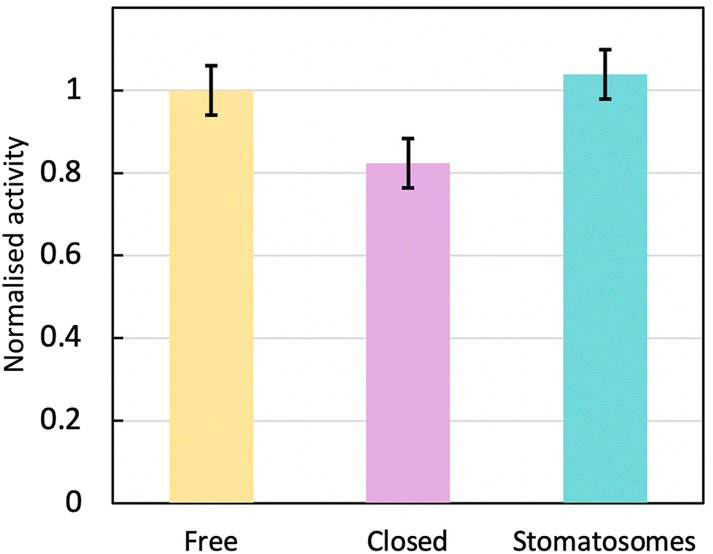 | ||
| Fig. 9 Normalised activity at room temperature for urease in different systems: free in solution, encapsulated within closed vesicles of 3 or encapsulated within perforated vesicles of 1, i.e. stomatosomes. The enzyme is the most active free or within stomatosomes, and less active within closed vesicles. | ||
The following factors were kept constant: substrate (urea) concentration, pH (same buffer used each time) and temperature. Everything is equivalent except the amphiphiles used, which form different types of vesicles – closed and perforated. The error on the activity values came mainly from the time taken for each step in the assay, which had to be kept constant. This may have varied between assays with varying number of samples, as time to for each step (take out of centrifuge, pipette into cuvette, add assay reagents) will depend on how many samples are being analysed. There is also a small error associated with the EE% (and so urease concentration) that is carried forward from the BCA assays into the activity assays.
The enzyme activity within stomatosomes was higher than in closed vesicles, probably because of their porous structure. Enzyme in closed vesicles exhibited on average 74 ± 7% of the activity of enzyme in perforated vesicle, only partially overcoming mass transfer limitations from encapsulation, i.e. with an activity reduction of 26 ± 7%. The fluorination makes bilayers more rigid and less fluid, which is offset by perforations increasing diffusion of smaller molecules to and from the enzyme. It is also likely that confinement effects increase activity of encapsulated enzyme in stomatosomes compared to free enzyme, which could explain why the latter does not have a significantly higher activity compared to the other systems.
When looking towards applications, this 26 ± 7% increase in activity could result in a substantial improvement of enzyme performance. For example, in the production of biosensors for the detection of ATP, one could get a faster response without needing additives that increase the membrane permeability. In one previous work, time to full response was usually around 6 minutes,76 which could be shortened to less than 5. Getting as close as possible to a real time response is important when analysing these systems in detail.77 With biosensors, a higher activity also results in a higher sensitivity, which is important for example when making biomedical devices that monitor physiological fluids.78 Furthermore, when developing cascade reaction models in cell mimicry, a semipermeable membrane is also needed. This cannot be achieved with closed vesicles as they are not permeable enough, so polymersomes are used instead.79 Instead of polymersomes, stomatosomes use amphiphiles, which form closer mimics to biological cells.80 Lastly, in the case of removal of dye toxins, these are longer processes that take around 6 h,81 where a 26% improvement would mean potentially saving more than 1 h. Here a much more efficient process would be enabled with additional porosity. Time is important for industrial applications and small gains in time can improve a process significantly over many reaction cycles.
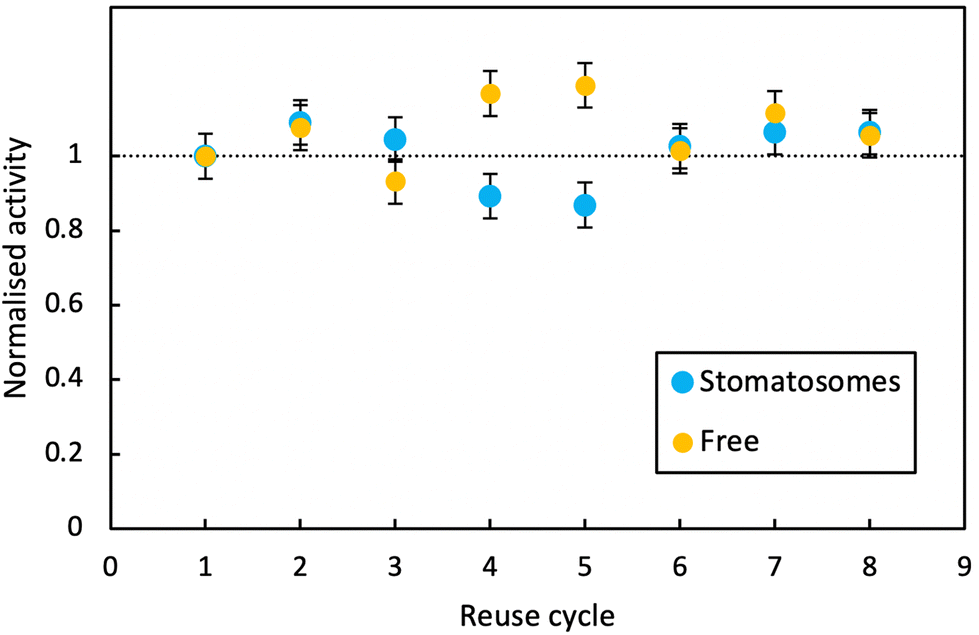 | ||
| Fig. 10 Activity at room temperature of free enzyme and enzyme encapsulated within stomatosomes formed from 1 after 8 consecutive reaction cycles. Activity of the enzyme is overall unchanged for both systems. | ||
In both cases, activity remained stable over 8 reaction cycles. Outliers were observed at reuse cycles 4 and 5, potentially due to clogging of filters and some product being released inconsistently after filtration (small increase in free enzyme activity)or irregular dissolution of the pellet after centrifugation (small decrease in activity for enzyme in stomatosomes). Here stomatosome encapsulated enzyme has a clear advantage over free enzyme with a fast, simple and cheap separation method. It showed similar results to other immobilisation methods like covalent grafting where activity was retained over reuse cycles.82 The loss in activity upon encapsulation being minimal, this is promising for applications.
3 Conclusions
The aim of this work was to evaluate the potential of stomatosomes as perforated reaction vessels for enzyme immobilisation and conservation. To this end, two fluorinated dendritic amphiphiles were synthesised and were found to reliably self-assemble into perforated bilayers under physiological buffer conditions. Due to their low water solubility, their critical aggregation concentration in water is as low as 5.5 μM and 1.4 μM in PBS, which renders their porous aggregates very stable at room temperature while leakage of enzymes is prevented for at least 2 days.By using fluorescent labels for both enzyme and stomatosomes, a detailed study of the localisation of encapsulated enzyme was performed using gSTED microscopy. This study revealed that the protein was concentrated in the stomatosome bilayer. Interestingly, an increase in encapsulation efficiency was observed for the short-chain amphiphile 1, which forms stomatosomes preferentially over planar bilayers. This points to enzyme at least partially residing in the stomatosome interior solution. Enzyme was successfully retained and it could be reused over 8 cycles with overall unaltered activity. Recycling was accomplished by just simple centrifugation, which can be used in batch reactions. Free enzyme had to be filtrated using expensive filtration systems, which tend to clog after reusing and so have to be replaced. Therefore, encapsulated enzyme in stomatosomes have a clear advantage over free enzyme.
Due to their porous structure, the application of stomatosomes improves the reaction kinetics compared to closed vesicles without leakage of enzymes. These results illustrate that stomatosomes could be promising reaction vessels for enzyme encapsulation. Tuning of the fluorinated amphiphile assemblies may enable the development of optimised carriers for various applications.
Author contributions
RH conceived and designed the study. TGS synthesised amphiphiles 1 and 2 with the support of FJ, RR and AKS. AKS synthesised amphiphile 3. TGS performed the aggregation, encapsulation and activity experiments with the support of FJ. TGS, KA, and MM performed confocal and gSTED microscopy and BS performed cryo-TEM. The evaluation and presentation of the data was done by TGS, FJ, AKS, BS, MM, SS and RH. All authors contributed to the writing of the manuscript and approved its final submitted version. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The study was financially supported by the Deutsche Forschungsgemeinschaft (SFB 1349 (Teilprojekt C6, Project-ID 387284271)). The authors would also like to acknowledge the assistance of the Core Facility BioSupraMol supported by the DFG and the research center SupraFAB. Lastly, Pierangelo Metrangolo, Marta Rosati, Valentina Dichiarante and Gabriella Cavallo from the Department of Chemistry, Materials and Chemical Engineering “Giulio Natta” at the Politecnico di Milano are gratefully acknowledged for their work in the development of the branched fluorinated amphiphile 3.References
- R. A. Sheldon, D. Brady and M. L. Bode, Chem. Sci., 2020, 11, 2587–2605 RSC.
- H. Sun, H. Zhang, E. L. Ang and H. Zhao, Bioorg. Med. Chem., 2018, 26, 1275–1284 CrossRef CAS PubMed.
- E. D. Yushkova, E. A. Nazarova, A. V. Matyuhina, A. O. Noskova, D. O. Shavronskaya, V. V. Vinogradov, N. N. Skvortsova and E. F. Krivoshapkina, J. Agric. Food Chem., 2019, 67, 11553–11567 CrossRef CAS PubMed.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed.
- R. A. Sheldon, A. Basso and D. Brady, Chem. Soc. Rev., 2021, 50, 5850–5862 RSC.
- Y. Wang, Q. Zhao, R. Haag and C. Wu, Angew. Chem., Int. Ed., 2022, 61, e202213974 CAS.
- F. Bialas, D. Reichinger and C. F. Becker, Enzyme Microb. Technol., 2021, 150, 109864–109884 CrossRef CAS PubMed.
- M. E. M. Cruz, M. L. Corvo, M. B. Martins, S. Simões and M. M. Gaspar, Pharmaceutics, 2022, 14, 531–547 CrossRef CAS PubMed.
- A. Mohammadi, S. M. Jafari, A. S. Mahoonak and M. Ghorbani, Food Bioprocess Technol., 2021, 14, 23–38 CrossRef CAS.
- N. R. Mohamad, N. H. C. Marzuki, N. A. Buang, F. Huyop and R. A. Wahab, Biotechnol. Biotechnol. Equip., 2015, 29, 205–220 CrossRef CAS PubMed.
- H. Omorodion, B. Twamley, J. A. Platts and R. J. Baker, Cryst. Growth Des., 2015, 15, 2835–2841 CrossRef CAS.
- O. Wagner, B. N. S. Thota, B. Schade, F. Neumann, J. L. Cuellar, C. Böttcher and R. Haag, Polym. Chem., 2016, 7, 2222–2229 RSC.
- O. Wagner, M. Zieringer, W. J. Duncanson, D. A. Weitz and R. Haag, Int. J. Mol. Sci., 2015, 16, 20183–20194 CrossRef CAS PubMed.
- M. P. Krafft, Adv. Drug Delivery Rev., 2001, 47, 209–228 CrossRef CAS PubMed.
- F. Junge, P.-W. Lee, A. K. Singh, J. Wasternack, M. P. Pachnicz, R. Haag and C. Schalley, Angew. Chem., Int. Ed., 2023, 62(12), e202213866 CrossRef CAS PubMed.
- J. E. Huheey, E. A. Keiter and R. L. Keiter, Inorganic chemistry, HarperCollins College Publishers, 1993 Search PubMed.
- A. Haupt, Organic and Inorganic Fluorine Chemistry, De Gruyter, Berlin, Germany, 2021, pp. 283–300 Search PubMed.
- J.-N. Marsat, M. Heydenreich, E. Kleinpeter, H. v. Berlepsch, C. Böttcher and A. Laschewsky, Macromolecules, 2011, 44, 2092–2105 CrossRef CAS.
- M. Cametti, B. Crousse, P. Metrangolo, R. Milani and G. Resnati, Chem. Soc. Rev., 2012, 41, 31–42 RSC.
- H. v. Berlepsch, B. N. S. Thota, M. Wyszogrodzka, S. de Carlo, R. Haag and C. Böttcher, Soft Matter, 2018, 14, 5256–5269 RSC.
- R. Rashmi, H. Hasheminejad, S. Herziger, A. Mirzaalipour, A. K. Singh, R. R. Netz, C. Böttcher, H. Makki, S. K. Sharma and R. Haag, Macromol. Rapid Commun., 2022, 43, 2100914–2100925 CrossRef CAS PubMed.
- M. P. Krafft and J. G. Riess, Curr. Opin. Colloid Interface Sci., 2015, 20, 192–212 CrossRef CAS.
- A. K. Singh, B. Schade, M. Rosati, R. Rashmi, V. Dichiarante, G. Cavallo, P. Metrangolo and R. Haag, Macromol. Biosci., 2022, 22, 2200108–2200115 CrossRef CAS PubMed.
- M. Rosati, A. Acocella, A. Pizzi, G. Turtù, G. Neri, N. Demitri, N. Nonappa, G. Raffaini, B. Donnio, F. Zerbetto, F. B. Bombelli, G. Cavallo and P. Metrangolo, Macromolecules, 2022, 55, 2486–2496 CrossRef CAS.
- M. Almgren, Soft Matter, 2010, 6, 1383–1390 RSC.
- K. Edwards, J. Gustafsson, M. Almgren and G. Karlsson, J. Colloid Interface Sci., 1993, 161, 299–309 CrossRef CAS.
- V. F. Motlaq, M. Ortega-Holmberg, K. Edwards, L. Gedda, J. Lyngsø, J. S. Pedersen and L. M. Bergström, Soft Matter, 2021, 17, 7769–7780 RSC.
- R. Kakehashi, G. Karlsson and M. Almgren, J. Colloid Interface Sci., 2009, 331, 484–493 CrossRef CAS PubMed.
- J. Liu, S. Xiao, J. Li, B. Yuan, K. Yang and Y. Ma, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 2234–2241 CrossRef CAS PubMed.
- H. Chen, X. Yu, Y. Fan, X. Xing, S. Trépout and M.-H. Li, CCS Chem., 2022, 4, 2651–2661 CrossRef CAS.
- K. Wang, G. Karlsson and M. Almgren, J. Phys. Chem. B, 1999, 103, 9237–9246 CrossRef CAS.
- I. D. Azmi, S. M. Moghimi and A. Yaghmur, Ther. Delivery, 2015, 6, 1347–1364 CrossRef CAS PubMed.
- N. Kundu, D. Banik and N. Sarkar, Langmuir, 2018, 34, 11637–11654 CrossRef CAS PubMed.
- H. Chen, Y. Fan, X. Yu, V. Semetey, S. Trépout and M.-H. Li, ACS Nano, 2021, 15, 884–893 CrossRef CAS PubMed.
- H. Oh, A. M. Ketner, R. Heymann, E. Kesselman, D. Danino, D. E. Falvey and S. R. Raghavan, Soft Matter, 2013, 9, 5025–5033 RSC.
- J.-K. Kim, E. Lee, Y.-B. Lim and M. Lee, Angew. Chem., Int. Ed., 2008, 47, 4662–4666 CrossRef CAS PubMed.
- X. Li, Y. Yang, J. Eastoe and J. Dong, ChemPhysChem, 2010, 11, 3074–3077 CrossRef CAS PubMed.
- Y. La, J. Song, M. G. Jeong, A. Cho, S.-M. Jin, E. Lee and K. T. Kim, Nat. Commun., 2018, 9, 5327 CrossRef CAS PubMed.
- C. Contini, W. Hu and Y. Elani, Chem. Commun., 2022, 58, 4409–4419 RSC.
- S. Li, H. Xie, F. Xie, Q. Yi and H. Tan, Microchim. Acta, 2022, 189, 358 CrossRef CAS PubMed.
- B. S. Cha, E. S. Lee, S. Kim, J. M. Kim, S. H. Hwang, S. S. Oh and K. S. Park, Microchem. J., 2020, 158, 105130–105146 CrossRef CAS.
- N. E. Dixon, J. A. Hinds, A. K. Fihelly, C. Gazzola, D. J. Winzor, R. L. Blakeley and B. Zerner, Can. J. Biochem., 1980, 58, 1323–1334 CrossRef CAS PubMed.
- H. L. T. Mobley, Helicobacter pylori: Physiology and Genetics, ASM Press, Washington (DC), 2001 Search PubMed.
- R. I. Krohn, Curr. Protoc. Cell Biol., 2002, 15, A3H1–A3H28 Search PubMed.
- M. Berthelot, Report. Chem. Appl., 1859, 1, 284 Search PubMed.
- B. Hammouda, J. Res. Natl. Inst. Stand. Technol., 2013, 118, 151–167 CrossRef CAS PubMed.
- Units Symbols, Useful Quantities and Relations, ed. J. N. Israelachvili, Academic Press, San Diego, 3rd edn, 2011 Search PubMed.
- Z. Xu and R. D. Oleschuk, J. Chromatogr. A, 2014, 1329, 61–70 CrossRef CAS PubMed.
- Q. Sun, Chem. Phys. Lett., 2017, 672, 21–25 CrossRef CAS.
- B. Kang, H. Tang, Z. Zhao and S. Song, ACS Omega, 2020, 5, 6229–6239 CrossRef CAS PubMed.
- M. Hishida, Y. Kaneko, M. Okuno, Y. Yamamura, T.-a. Ishibashi and K. Saito, J. Chem. Phys., 2015, 142, 171101 CrossRef PubMed.
- Y. Kawabata, R. Bradbury, S. Kugizaki, K. Weigandt, Y. B. Melnichenko, K. Sadakane, N. L. Yamada, H. Endo, M. Nagao and H. Seto, J. Chem. Phys., 2017, 147, 034905 CrossRef CAS PubMed.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- C. Has and S. Pan, J. Liposome Res., 2021, 31, 90–111 CrossRef CAS PubMed.
- R. Nagarajan, Langmuir, 2002, 18, 31–38 CrossRef CAS.
- I. I. Slowing, B. G. Trewyn and V. S.-Y. Lin, J. Am. Chem. Soc., 2007, 129, 8845–8849 CrossRef CAS PubMed.
- H. Zhang, Thin-Film Hydration Followed by Extrusion Method for Liposome Preparation, Springer, New York, 2017 Search PubMed.
- M. Garni, T. Einfalt, R. Goers, C. G. Palivan and W. Meier, ACS Synth. Biol., 2018, 7, 2116–2125 CrossRef CAS PubMed.
- A. C. Hortelão, S. García-Jimeno, M. Cano-Sarabia, T. Patiño, D. Maspoch and S. Sanchez, Adv. Funct. Mater., 2020, 30, 2002767–2002775 CrossRef.
- C. Billerit, G. D. M. Jeffries, O. Orwar and A. Jesorka, Soft Matter, 2012, 8, 10823–10826 RSC.
- M. Houbrechts, L. Caire da Silva, A. Ethirajan and K. Landfester, Soft Matter, 2021, 17, 4942–4948 RSC.
- B. G. De Geest, N. N. Sanders, G. B. Sukhorukov, J. Demeester and S. C. De Smedt, Chem. Soc. Rev., 2007, 36, 636–649 RSC.
- M. C. Smith, R. M. Crist, J. D. Clogston and S. E. McNeil, Anal. Bioanal. Chem., 2017, 409, 5779–5787 CrossRef CAS PubMed.
- J. B. Sumner and D. B. Hand, J. Am. Chem. Soc., 1929, 149, 1255–1260 CrossRef.
- Z. Zhang, W. Shen, J. Ling, Y. Yan and J. Hu, Nat. Commun., 2018, 9, 1377 CrossRef PubMed.
- B. Chaize, J.-P. Colletier, M. Winterhalter and D. Fournier, Artif. Cells, Blood Substitutes, Biotechnol., 2004, 32, 67–75 CrossRef CAS PubMed.
- A. Laouini, C. Jaafar-Maalej, I. Limayem-Blouza, S. Sfar, C. Charcosset and H. Fessi, J. Colloid Sci. Biotechnol., 2012, 1, 147–168 CrossRef CAS.
- M. Al-Amin, F. Bellato, F. Mastrotto, M. Garofalo, A. Malfanti, S. Salmaso and P. Caliceti, Int. J. Mol. Sci., 2020, 21, 1611 CrossRef CAS PubMed.
- W. Perkins, S. Minchey, P. Ahl and A. Janoff, Chem. Phys. Lipids, 1993, 64, 197–217 CrossRef CAS PubMed.
- X. Xu, M. A. Khan and D. J. Burgess, Int. J. Pharm., 2012, 423, 410–418 CrossRef CAS PubMed.
- P. G. Squire, P. Moser and C. T. O'Konski, Biochemistry, 1968, 7, 4261–4272 CrossRef CAS PubMed.
- M. Taha, F. Rahim, H. Ullah, A. Wadood, R. K. Farooq, S. Adnan, A. Shah, M. Nawaz and Z. A. Zakaria, Sci. Rep., 2020, 10, 10673 CrossRef CAS PubMed.
- S. D. Cesareo and S. R. Langton, FEMS Microbiol. Lett., 1992, 99, 15–21 CrossRef CAS.
- R. Dilrukshi, K. Nakashima and S. Kawasaki, Soils Found., 2018, 58, 894–910 CrossRef.
- D. Cogan, J. Cleary, C. Fay, A. Rickard, K. Jankowski, T. Phelan, M. Bowkett and D. Diamond, Anal. Methods, 2014, 6, 7606–7614 RSC.
- U. H. Yildiz, H.-P. M. De Hoog, Z. Fu, N. Tomczak, A. N. Parikh, M. Nallani and B. Liedberg, Small, 2014, 10, 442–447 CrossRef CAS PubMed.
- S. M. Christensen and D. Stamou, Soft Matter, 2007, 3, 828–836 RSC.
- J. Y. Kim, G. Y. Sung and M. Park, Biomedicines, 2020, 8, 596 CrossRef CAS PubMed.
- C. G. Palivan, R. Goers, A. Najer, X. Zhang, A. Car and W. Meier, Chem. Soc. Rev., 2016, 45, 377–411 RSC.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 8572–8610 RSC.
- M. V. Dinu, I. A. Dinu, S. S. Saxer, W. Meier, U. Pieles and N. Bruns, Biomacromolecules, 2021, 22, 134–145 CrossRef CAS PubMed.
- J. Zhang, Z. Wang, C. He, X. Liu, W. Zhao, S. Sun and C. Zhao, ACS Omega, 2019, 4, 2853–2862 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr00493g |
| This journal is © The Royal Society of Chemistry 2023 |