
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNative extracellular matrix probes to target patient- and tissue-specific cell–microenvironment interactions by force spectroscopy†
H.
Holuigue
 a,
L.
Nacci
b,
P.
Di Chiaro
b,
M.
Chighizola
a,
I.
Locatelli
d,
C.
Schulte
*ac,
M.
Alfano
*d,
G. R.
Diaferia
*b and
A.
Podestà
*a
a,
L.
Nacci
b,
P.
Di Chiaro
b,
M.
Chighizola
a,
I.
Locatelli
d,
C.
Schulte
*ac,
M.
Alfano
*d,
G. R.
Diaferia
*b and
A.
Podestà
*a
aCIMAINA and Dipartimento di Fisica “Aldo Pontremoli”, Università degli Studi di Milano, Milano, Italy. E-mail: carsten.schulte@unimi.it; alessandro.podesta@mi.infn.it
bDepartment of Experimental Oncology, IEO, European Institute of Oncology IRCCS, Milano, Italy. E-mail: giuseppe.diaferia@ieo.it
cDepartment of Biomedical and Clinical Sciences “L. Sacco”, Università degli Studi di Milano, Milano, Italy
dDivision of Experimental Oncology/Unit of Urology, URI, IRCCS San Raffaele Hospital, Milan, Italy. E-mail: alfano.massimo@hsr.it
First published on 7th September 2023
Abstract
Atomic Force Microscopy (AFM) is successfully used for the quantitative investigation of the cellular mechanosensing of the microenvironment. To this purpose, several force spectroscopy approaches aim at measuring the adhesive forces between two living cells and also between a cell and an appropriate reproduction of the extracellular matrix (ECM), typically exploiting tips suitably functionalised with single components (e.g. collagen, fibronectin) of the ECM. However, these probes only poorly reproduce the complexity of the native cellular microenvironment and consequently of the biological interactions. We developed a novel approach to produce AFM probes that faithfully retain the structural and biochemical complexity of the ECM; this was achieved by attaching to an AFM cantilever a micrometric slice of native decellularised ECM, which was cut by laser microdissection. We demonstrate that these probes preserve the morphological, mechanical, and chemical heterogeneity of the ECM. Native ECM probes can be used in force spectroscopy experiments aimed at targeting cell–microenvironment interactions. Here, we demonstrate the feasibility of dissecting mechanotransductive cell–ECM interactions in the 10 pN range. As proof-of-principle, we tested a rat bladder ECM probe against the AY-27 rat bladder cancer cell line. On the one hand, we obtained reproducible results using different probes derived from the same ECM regions; on the other hand, we detected differences in the adhesion patterns of distinct bladder ECM regions (submucosa, detrusor, and adventitia), in line with the disparities in composition and biophysical properties of these ECM regions. Our results demonstrate that native ECM probes, produced from patient-specific regions of organs and tissues, can be used to investigate cell–microenvironment interactions and early mechanotransductive processes by force spectroscopy. This opens new possibilities in the field of personalised medicine.
1. Introduction
Cells and their microenvironment have a strong intercommunication and interplay, influencing each other. The extracellular matrix (ECM) surrounding the cells is mainly composed of a three-dimensional network of crosslinked proteins, such as collagens, fibronectin and laminins; ECM from various tissues can, however, have strong differences in their composition and biophysical properties (i.e., rigidity and structural features, such as nanotopography).1–4 Cells use the ECM as a scaffold for their anchorage and interact with it in a reciprocal manner. In fact, cells can convert external mechanical and topographical stimuli into biochemical signalling, which often determine changes in the mechanical properties of the cell through the reorganisation of the cytoskeleton, which eventually leads to modulations of gene/protein expression, a process called mechanotransduction. Cells are therefore able to perceive changes in the physical properties of their surrounding ECM and respond to them. In addition, the cell itself uses force to sense the biophysical characteristics of the ECM. The study of these reciprocal interactions pertains to the fields of cellular mechanobiology.5–13Different techniques are used to study these cell–ECM interactions, among which Atomic Force Microscopy (AFM) stands out due to its ability to both sense and apply forces at the nanoscale, with sub-nanonewton sensitivity.14–18 In the context of biophysical investigations, many AFM-based force spectroscopy (FS) configurations can be found:19 a single-cell probe approaching a substrate coated with proteins;20–25 an ECM-mimicking probe approaching an adherent cell;11,26–30 a single-cell probe approaching an adherent cell.16,31,32 Both cell–cell and cell–ECM interactions can be investigated, through the direct measurement of weak (10–100 pN) interaction forces, which are mostly cadherin or integrin related, respectively.33–37
The main advantage of FS techniques using tips functionalised with single proteins29,30,38 is the simplification of the complex cell–microenvironment interface, allowing us to study one specific molecular interaction at a time. An example for this approach for the study of cell–ECM interactions consists in the functionalisation of AFM tips with collagen, a major component of the ECM.30,39 While these methods enable an accurate characterisation of specific molecular interactions, the reconstituted interface is poorly mimicking the complexity of the native interface in physiological conditions.
Aiming at reproducing the native cell–ECM interface within a typical AFM-based FS experiment, we developed native ECM probes and demonstrated that they can be reliably used to scrutinise integrin-related adhesive interactions between cells and their microenvironment. Attaching the ECM to the cantilever and ramping it against adherent living cells, instead of using single-cell functionalised cantilevers against an ECM sample, is advantageous, among other reasons, because it allows to test (i) many different cells with the same probe, that can be used and re-used repeatedly, and (ii) cells seeded on different substrates, i.e. polarized or with different phenotypes such as type 1 and type 2 macrophages or epithelial cancer cells or cancer cells during and after epithelial–mesenchymal transition. Indeed, the ECM is more stable and easier to handle than living cells, and each single-cell probe requires a different cantilever and a new calibration; overall, using ECM probes more likely provides improved reliability and statistics of the FS experiments.
Here we describe the fabrication approach of novel native ECM probes and the results of their characterisation, showing that the chemico-physical properties of the native ECM are preserved, also after intense and prolonged use. We also report on the application of these new probes in adhesion force spectroscopy experiments, showing that the peculiar patterns of adhesive molecular interactions observed in FS experiments were reproduced. As a proof-of-principle, we focused on rat bladder-derived ECM and used the rat bladder tumour cell line AY-27 as a suitable interaction partner; the results demonstrate the capability to dissect specific cellular adhesive patterns against different tissue layers of the same organ. The described protocol is, however, universally applicable, and allows to reliably fabricate probes out of any ECM, and even from specific regions of the same ECM, to be tested against both immortalised and primary cells, paving the way to the investigation of patient-specific cell–ECM interactions.
2. Results and discussion
2.1. Production of native ECM probes for adhesion force spectroscopy
To produce native ECM probes, we developed a novel approach based on the cutting of ECM pieces from a glass-supported, decellularised ECM slice; these pieces will then be detached from the glass slide and attached to tipless functionalised cantilevers (Fig. 1A–D).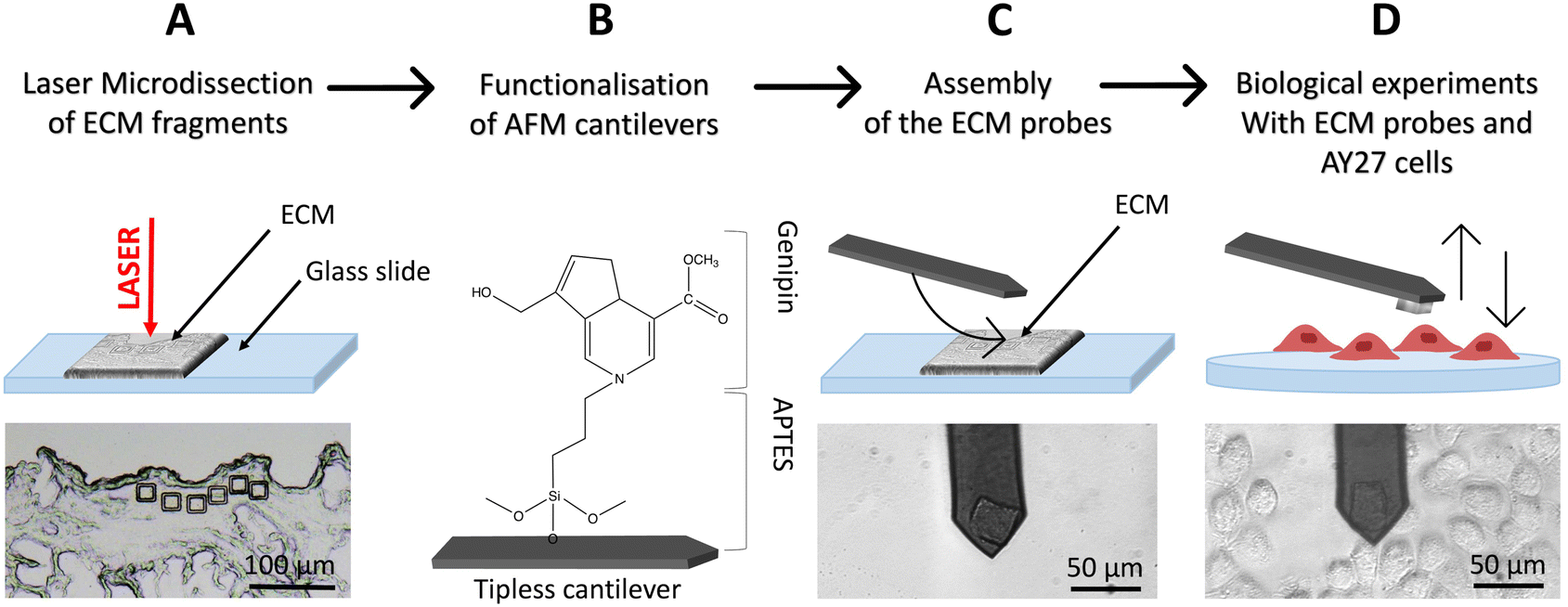 | ||
| Fig. 1 Schematics of the ECM probe production. (A) Optimized use of the LMD system to produce native ECM probes: mounting the decellularized tissue directly on a standard microscope glass slide, allows the UV laser to physically separate ECM fragments from the surrounding matrix, without detaching them from the glass support. (B) Tipless cantilevers are functionalised with APTES and genipin (covalently bonded), and then used for the attachment of the ECM probes (C), exploiting the XY micro-translation stage of the optical microscope that includes the AFM (stand-alone stages can be used equivalently). (D) The resulting ECM probes are used for biological experiments, such as force spectroscopy measurements on top of living cells. | ||
To cut pieces with predefined and reproducible dimensions and shapes from the ECM slice, we used an LMD apparatus (in Fig. 1A, square cuts with 20 μm × 20 μm area are shown).
LMD is typically used to cut a selected area from a dehydrated tissue section attached on the upper surface of a glass slide, where a thin plastic membrane is cemented to the glass at its edges leaving a rectangular area with air trapped between the membrane and the glass backbone (Fig. S1†). A focused UV laser then cuts the selected regions of the tissue, together with the plastic membrane allowing the micro-dissected pieces to fall by gravity in a reservoir located below. These samples are then typically digested for genomics or proteomics assays,40,41 where the presence of the rigid plastic membrane does not interfere with the subsequent molecular reactions.
For our application, given that the objective is to transfer and attach intact fragments of ECM to either AFM tipless cantilevers or spherical tips, the presence of the plastic membrane is detrimental. We therefore modified the standard LMD procedure to develop an optimised approach, based on the elimination of the plastic membrane from the glass slide and the introduction of suitable procedures to detach the cut pieces from the slide and to transfer them to the AFM cantilever or tip.
The protocol is represented schematically in Fig. 1. Here, the laser is still used for the cut, but the ECM slices are attached directly on the glass slide, without the plastic membrane in between (Fig. 1A and S2A†) and placed on the microscope, with the tissue facing up. After the cut, the micro-dissected pieces remain on the glass surface (Fig. 1A and S2B–D†), separated from the surrounding ECM by a narrow empty region vaporised by the laser, where they can be picked up by, and deposited onto, the functionalised cantilever used for the assembling of the ECM probe, exploiting the XY motorised stage of the AFM integrated in the optical microscope (see ESI Movies SM1–SM3†). This procedure takes place in liquid. After the attachment, the cantilever is withdrawn from the surface and kept at rest for 10 min. ECM probes were typically stored in PBS at 37 °C in a humid environment (for example in the cell incubator) and used in the following days.
The fact that the ECM pieces remain on the glass slide after the cut facilitates the investigation of same or twin specimens by other techniques, like immunofluorescence. Moreover, having the ECM attached to the cantilever and testing it against a cell population cultured on a suitable support is very convenient compared to having a single cell probe that interrogates an ECM sample; indeed, in the first case many cells can be tested with the same ECM probes, while in the standard configuration each new tested cell requires the preparation of a new probe.
In this work, 20 μm × 20 μm, 10 μm thick squares (Fig. 1A and 3A–E) were cut in different regions of decellularised rat bladder: submucosal, detrusor and adventitia.18,42,43
2.2. Characterisation of ECM and ECM probes
LMD provides many advantages including the possibility to precisely select the ROI, cut ECM pieces with predefined, reproducible geometry and dimensions, and collect the samples without damaging them. To ensure that the ECM pieces retain the properties of the pristine ECM, we performed several assessments of biophysical and compositional parameters.Firstly, we controlled the possible effect of the laser cut on morphology, mechanics and composition of the ECM, respectively, through combined AFM topographic and nanomechanical measurements and relative quantitative immunofluorescence staining (Fig. 2A–G). For the nanomechanical assessment of the ECM, we measured the region inside and outside the micro-dissected piece; the inner region is the one that is then attached to the AFM cantilever, while the outer region is taken as reference for the untreated ECM. For the immunofluorescence analysis, we measured the fluorescent signals detected after immunostaining from the inner regions of the micro-dissected pieces and the same regions from a consecutive section obtained from the same ECM specimen not subjected to laser ablation.
 | ||
| Fig. 2 (A) Optical image, (B) Young's modulus and (C) topographic maps of an ECM slice with the cut of the laser clearly visible and highlighted in C. (D) Distribution of the measured Young's modulus values for the internal and external regions of the cut. Immunostaining for Collagen IV (COL-IV, green), laminin β1 (LAMB1, red), and fibronectin (FN, cyan) before (E, pre-LMD) and after (F, post-LMD) the cut. The white boxes in the pre-LMD image represent the matched area subjected to microdissection. (G) Quantification of the fluorescent signal associated to the different proteins in each ROI (white squares). The data represent normalised fluorescent signal (a.u., arbitrary units) scaled between 0 and 1 (n = 30, 5 ROIs per each field, 6 fields acquired distributed along the layers of the bladder). | ||
Fig. 2A–C shows the optical image of a cut ECM piece together with AFM topographic and mechanical maps. The borders of the cut region are clearly visible in both optical image and topographic AFM maps. No evident differences between the inner and the outer regions are visible, regarding both morphological and mechanical properties (the median Young's modulus values are 280 and 289 kPa for the internal and external parts, respectively). Besides the medians, also the distributions of Young's modulus values E (Fig. 2D) are similar (the distribution of the external part is slightly broader, which can be explained by the larger covered area, encompassing a larger heterogeneity of the sample).
These results are in line with the results obtained by relative quantitative staining of major ECM proteins such as collagen, laminin, and fibronectin on different cut pieces of ECM, demonstrating that the composition of the matrix is not affected by the cut (Fig. 2E–G). The UV laser of the LMD apparatus could induce thermal damage and autofluorescence,44 and fibronectin inactivation45 around the edge of the ablated region; nevertheless, the central area used to produce the probes and interacting with the cells is not affected by these phenomena.
Stress tests were performed to assess the firm attachment of the matrix to the cantilever and the force spectroscopy functionality after repeated use. The ECM remained attached to the cantilever after scanning continuously in Contact Mode on a glass slide for 1 h (Fig. S3A†); similar adhesion forces were measured during continuous acquisition of 400 FCs (Fig. S3B†). Dehydration and rehydration processes before the LMD cut have been checked and no significant effect has been detected (Fig. S3C†).
To further assess the lifetime of ECM probes, in particular the structural resistance of the ECM fragments, the firmness of their attachment to the cantilevers and their capability to sense reliably molecular interactions, we performed adhesion force spectroscopy experiments during two consecutive days (day 1 and day 2) on cells from the same passage plated on two different Petri dishes. We observed that the probe detected similar mean number of jumps Nj (Fig. S4A†), with some discrepancies at higher contact times, and very similar force per jump 〈Fj〉 (Fig. S4B†). Remarkably, besides the satisfactory agreement of the mean Nj and 〈Fj〉 values, the distributions of the measured single-bonds forces for the two days for the same contact times agree very well (Fig. S4C and D†), and also clearly show that there is an increase of the force at higher contact times. This leads us to conclude that the same probe in different days is able to measure the same adhesive events, i.e. the probe maintains its ability to capture both qualitatively and quantitatively the biological picture of the adhesive cell–ECM interaction. The slight difference observed from day to day in the measured parameters could be explained by the well-known heterogeneity of cells, as well as by their high sensitivity to slight changes in the environmental conditions.
2.3. Use of tailored native ECM probes in adhesion force spectroscopy experiments
To demonstrate the potential of native ECM probes for the study of cell–microenvironment interactions, we carried out adhesion force spectroscopy experiments with these probes against AY-27 cells (Fig. 1D).For the proof-of-principle presented here, we selected the three main bladder regions for the creation of the ECM probes: the submucosal region, closer to the lumen of the bladder, rich in basement membrane components to support the attachment of the urothelium, a type of stratified epithelium made of transitional epithelial cells, the detrusor region, a central layer of the bladder made of muscle fibres that allow contraction,42,43 and the adventitia, the outer area of the bladder, as shown in Fig. 3A–E. The ECM probes, named after their corresponding region of origin (probe SM from submucosal, probe D from detrusor, probe A from adventitia) are interesting because, albeit deriving from adjacent areas of the same bladder tissue, they are characterised by major differences in the composition and mechanical properties.18,42
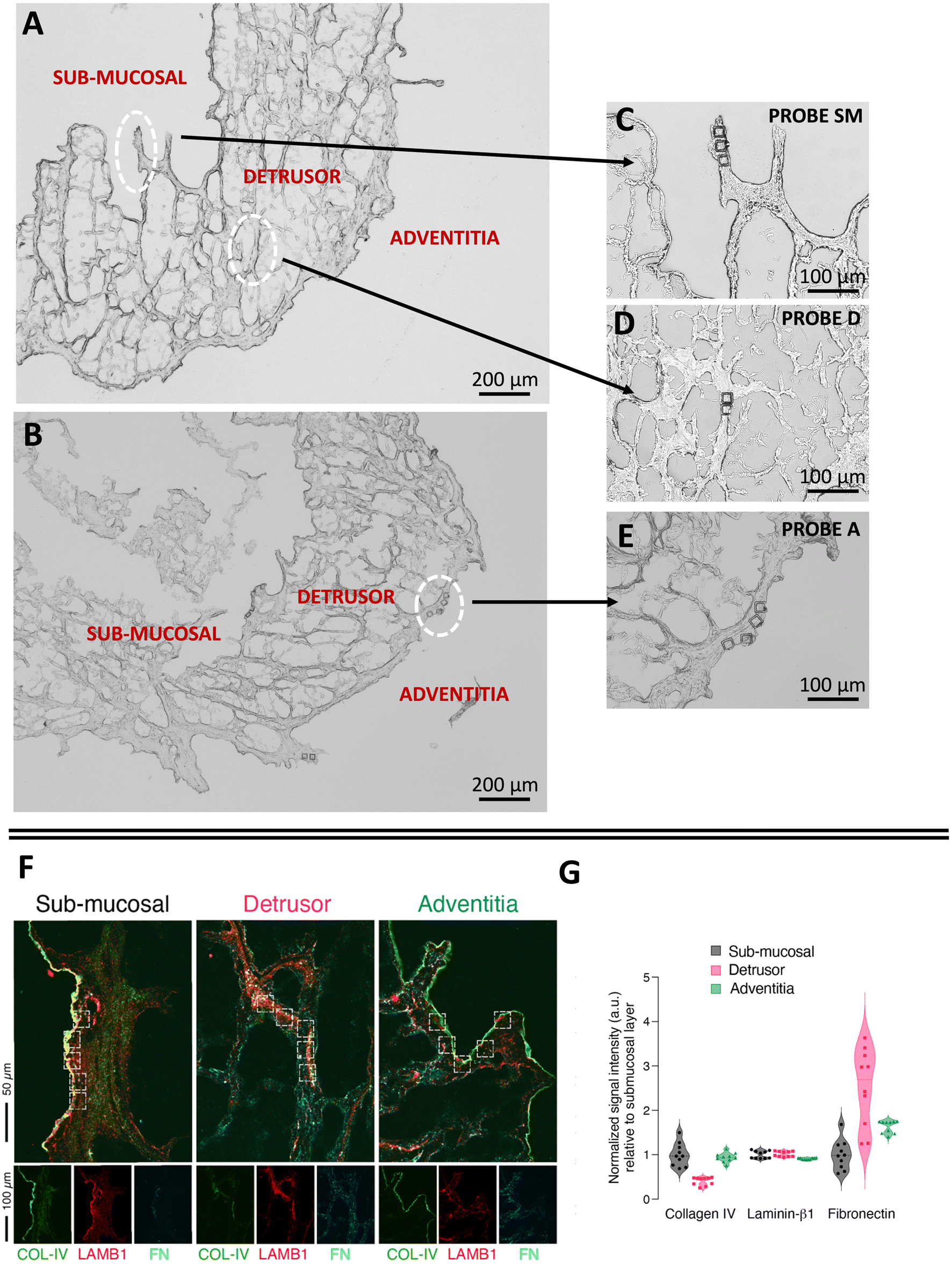 | ||
| Fig. 3 Optical images of the ECM (A) before, and (B–E) after the LMD cut. The white circles highlight the regions of the cuts, from where the ECM probes have been produced: probe SM from the submucosal, probe D from the detrusor, probe A from the adventitia. The denomination of the specific regions of the bladder in (A) follows ref. 42 and 43. (F) Immunostaining of different regions of the decellularised bladder (submucosal, detrusor and adventitia) with collagen IV (COL-IV, green), laminin β1 (LAMB1, red), and fibronectin (FN, cyan). (G) Quantification of the fluorescent signal associated to the different proteins in each ROI (white squares). The data represent normalised fluorescent signal (a.u., arbitrary units) relative to the mean value of the submucosal layer for each matrix (n = 10, 5 ROIs per each field, 2 fields acquired per each layer). | ||
We performed immunostaining and quantification of the abundance of principal ECM proteins, such as collagen, laminin, and fibronectin, which are responsible for major integrin-mediated cell/ECM interaction in the bladder tissue (Fig. 3F and G).
While laminin β1 seems to be equally distributed among all the layers, fibronectin and collagen IV are respectively enriched and depleted in the detrusor compartment (Fig. 3G). This is in line with the known contractive function of the detrusor layer, in which cells are distributed in an elastic connective tissue mainly composed of fibres of fibronectin, collagen type I and III and devoid of components of the basement membrane such as collagen IV. On the other hand, collagen IV is clearly visible, together with laminin β1, in the lamina propria of the submucosal layer (Fig. 3F), where we observed the lowest signal for fibronectin, as expected (Fig. 3G). The adventitia is composed of a loose connective tissue of thin collagen and fibronectin fibres and showed the lowest enrichment of laminin β1 and a higher amount of fibronectin compared to the submucosal layer.
We furthermore observed different mechanical properties of the ECM regions (Fig. S5†), with adventitia being the softest and detrusor the most rigid of the three, in agreement with the results recently reported for the three tissue layers of the rat bladder (i.e., urothelium, lamina propria and muscle layer).18 These assessments confirm that the different parts of the bladder ECM reproduced by probes SM, D and A have significant distinctions in their mechanical and compositional features.
We then performed adhesion force spectroscopy experiments comparing probe SM, probe D and probe A against AY-27 cells.
For all substrates, Nj rose initially (0–20 s) and then remained nearly constant (Fig. 4C). No clear difference is seen in the number of jumps among ECM probes D and SM, while A shows higher Nj values. We observed important differences in both 〈Fj〉 (Fig. 4D) and Fa (Fig. 4A). Concerning Fa, the probe A stands between the values of SM and D; the values were higher for the SM substrate at almost all time points, the maximum adhesion force for probe SM reached ∼1.5 nN, compared to ∼0.5 nN for probe D and ∼1.2 nN for probe A. After 20 s, the adhesion force on all substrates tends to remain constant. A similar outcome was detected for 〈Fj〉, with the only difference at contact time at 0 s. Moreover, we observe that the forces of the jumps in the adventitia region are weaker than in the two other regions, although they reach the detrusor value at longer contact time (120 s). The work of adhesion W (Fig. 4B) shows a dependence on the contact time very similar to the total adhesion force.
 | ||
| Fig. 4 The figure shows the results of adhesion force spectroscopy experiments performed on AY-27 cells of the same passage with three different ECM probes: probe SM (green), D (yellow) and A (blue), obtained using ECM fragments from submucosal, detrusor and adventitia regions, respectively. At least four cells for each contact times and each probe have been tested (ct = 0 s, 20 s, 60 s, 120 s). (A–D). The total adhesion force Fa, the work of adhesion W, the mean number of events (jumps) Nj, the mean force per jump 〈Fj〉 as a function of the contact time. (E, F) The distributions of the force per single jump at two contact times (ct = 60 s and 120 s, respectively). Error bars in A–D represent effective standard deviations of the mean, as explained in the Methods. The statistical significances of both adventitia and detrusor results compared to submucosal are marked by black asterisks. The line connecting the experimental data are a guide for the eye. | ||
The plateau observed after 20 s for all quantities could be due to increasing integrin clustering and thus a redistribution of forces among integrin nanoclusters in larger focal adhesions, which leads to a global stabilization of the existing adhesion contacts (where the microenvironmental conditions allow it) at nearly constant total force.46
The distribution of the forces of jump unbinding events (Fig. 4E and F) are very different for the probe SM, a more ample spectrum with occurrence of higher forces per jump was noted, whereas for probe D and A the forces are restricted to lower values. To be noticed, at longer contact times we observed a moderate shift towards lower forces for probe D, while in the distribution of probe SM the frequency of both smaller and higher forces increased slightly.
2.4. Correlation between force spectroscopy data and mechanical and chemical properties of the ECM
It is well known that cellular adhesion to different surfaces depends on several parameters:13,47 the number Nj of bonds detected correlate with the relative abundancies of specific ligands,28 while the force of single events detected is mainly due to the growth of the adhesion spots and reinforcement in the cytoskeletal domain.21,48 Such reinforcement is strictly related to the spatial distribution of the adhesion spots49–52 and the mechanical properties of the substrate. These two factors, or to be more precise, their specific combination, determine the maturation and growth of integrin-mediated adhesion sites.52,53To get a better understanding of these interrelationships in our experiments, we considered the correlation of the adhesive parameters with the biophysical and compositional properties of the different layers.
Fig. 5 shows the correlation of the mean force per jump 〈Fj〉 and the number of detected jumps Nj (for the contact time ct = 120 s) with both the Young's modulus of the matrix and the relative abundancies of ECM proteins, as quantified by immunostaining (Fig. 3F and G) (the complete correlation analysis at all contact times is shown in Fig. S6†).
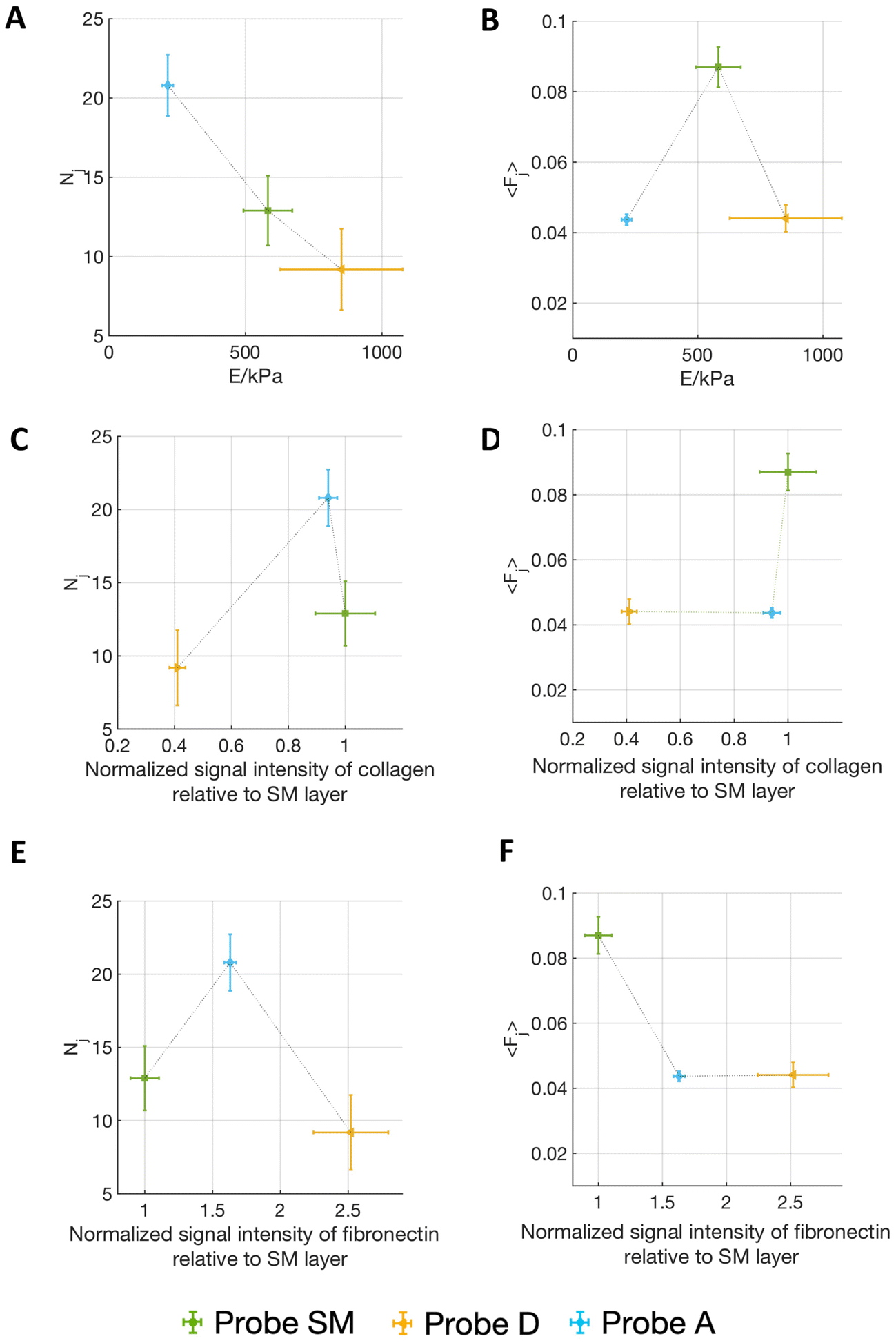 | ||
| Fig. 5 The figure shows (A, C and E) the number of detected jumps Nj and (B, D and F) the mean force per jump 〈Fj〉 resulting from adhesion force spectroscopy measurements performed on AY-27 cells with three different ECM probes: (green) SM, (yellow) D and (blue) A, obtained using ECM fragments from submucosal, detrusor and adventitia regions, respectively, set in relation with the mechanical and compositional features of the different ECMs. Nj and 〈Fj〉 are plotted versus (A and B) the Young's modulus of the matrix, and the normalized signal intensity relative to the SM layer in immunostaining maps of (C and D) fibronectin, and (E and F) collagen. Contact times was 120 s. Vertical error bars represent effective standard deviations of the mean for the 〈Fj〉, and Nj, and horizontal error bars represent the effective standard deviations of the mean of the Young's modulus of the matrix, as explained in the Materials and Methods section. The line connecting the experimental results are guides for the eye. | ||
Interestingly, Fig. 5A and B shows that on the softest matrix, adventitia, the highest number of jumps and the lowest force per jump are measured, while on the stiffest matrix, the detrusor, the lowest number of jumps is measured, although the force per jump is comparable to the one of the adventitia (Table 1). This behaviour is consistent with the common observation that a stiffer substrate promotes the assembly/maturation of numerous integrin nanoclusters into fewer, yet more stable, focal adhesions, whereas soft substrates do not foster this maturation and the adhesion sites remain small, unstable, and numerous.13,36,47 This might also be responsible for the biphasic force/ECM rigidity relationship between the different probes seen for 〈Fj〉, i.e. low values for adventitia and detrusor and high values for the submucosa. Indeed, on the softer adventitia, binding force is distributed and dissipated, the force transmission is thus less effective, leading to reduced reinforcement of molecular clutches and integrin nanoclusters, which disassemble as they do not reach the force thresholds that are necessary for reinforcement (this leads to high Nj and low 〈Fj〉). The more rigid detrusor, instead, enables a faster force transmission, which favours molecular clutch reinforcement and consequentially distribution of the force over more and stable integrin nanoclusters within maturing focal adhesion structures (this leads to both low Nj and 〈Fj〉). The situation for the submucosa is in between, therefore higher forces per single jump (i.e., adhesion spots) and an intermediate number of jumps are observed.13,36,47,53
| Contact time (s) | 0 | 20 | 60 | 120 |
|---|---|---|---|---|
| Probe SM | 63 ± 7 | 77 ± 4 | 85 ± 5 | 87 ± 6 |
| Probe D | 36 ± 9 | 51 ± 5 | 45 ± 3 | 45 ± 4 |
| Probe A | 23 ± 1 | 38 ± 2 | 40 ± 2 | 44 ± 2 |
However, it is known that different integrins (binding to different ligands) can contribute to mechanotransductive processes in a diverse manner, therefore the abundance of the different ECM components might add another layer that has to be considered, i.e., cell–fibronectin or cell–collagen IV interaction, depending on the ECM portion attached to the probe.
Since AY-27 rat bladder tumour cells are derived from a urothelium transitional carcinoma,54 they likely express those receptors that bind ECM components present in the submucosal layer, since this region of the bladder allows urothelial cells attachment, as mentioned above. The probe derived from the fibronectin-dominated, muscular type detrusor layer is probably less ideal for AY-27 attachment, which could also explain the lower overall Fa and W that we observed in contrast to the collagen-dominated SM matrix. The increase of the frequency of higher forces at longer contact times for probe SM is congruent with the transition from single to cooperative α2β1 integrin receptor binding to collagen I during the early steps of adhesion, shown by Taubenberger et al.55 In the fibronectin-prevalent probe D condition, instead, this increase does not occur (the trend is more in the opposite direction). However, the forces measured for the first pristine adhesion events to probe D at 0 s are in the range of what was measured for α5β1 integrin/fibronectin binding by Sun et al.38 (39 ± 8 pN versus 36 ± 9 pN in our measurements, see Table 1). The lower binding force and the lower adhesion pattern measured with probe D are consistent with the invading character of cancer cells in the inner muscular detrusor layers of the bladder,56,57 which is necessary for the tumour to become muscle invasive.
In addition, although the adventitia is characterised by high abundance of collagen IV and medium abundance of fibronectin, the probe A shows lower forces (both 〈Fj〉 and Fa), coupled, however, with a high number of binding events. This can be explained by the softer matrix, as abovementioned, which does not allow for the reinforcement of molecular clutches and thus the maturation of the cells’ focal adhesions.13,36,47,53
These results demonstrate that our approach allows to produce ECM probes from different regions of a tissue with tailored adhesive properties, capable to sense qualitatively and quantitatively different molecular interactions.
Altogether, the specific adhesive features measured with the three ECM probes are in line with both, the mechanical properties and the composition of the native ECM portions (Fig. 3F and G) and distinct types of cell–ECM interactions.
2.5. Evidence of integrin related unbinding events
To validate the association of measured jumps to integrin-related events, we performed a control experiment treating the cells with an allosteric inhibitory antibody against β1 integrin (4b4, Beckman Coulter), at fixed contact time (ct = 60 s), using two ECM probes made from a submucosal region (probe CTRL1 and CTRL2, Fig. S7†). The major component of the submucosa is collagen IV (Fig. 3F and G), which is bound by α1β1 and α2β1 integrins, targeting β1 was then strategical (Fig. 6A). We measured force of adhesion, mean force of jumps and tethers and mean number of events before and after the addition of 4b4 (Fig. 6B). Here, we considered both jumps and tethers together as representative events in these experiments, since the inhibitory antibody targets all β1 subunits of the integrins, either linked to the cell cytoskeleton (jumps related events) or not (tethers).55,58–60 The two ECM probes CTRL1 and CTRL2 (Fig. S7†) were used on two consecutive days and data were grouped together, as it was observed that the force of events were similar and displayed similar distributions (Fig. S8†).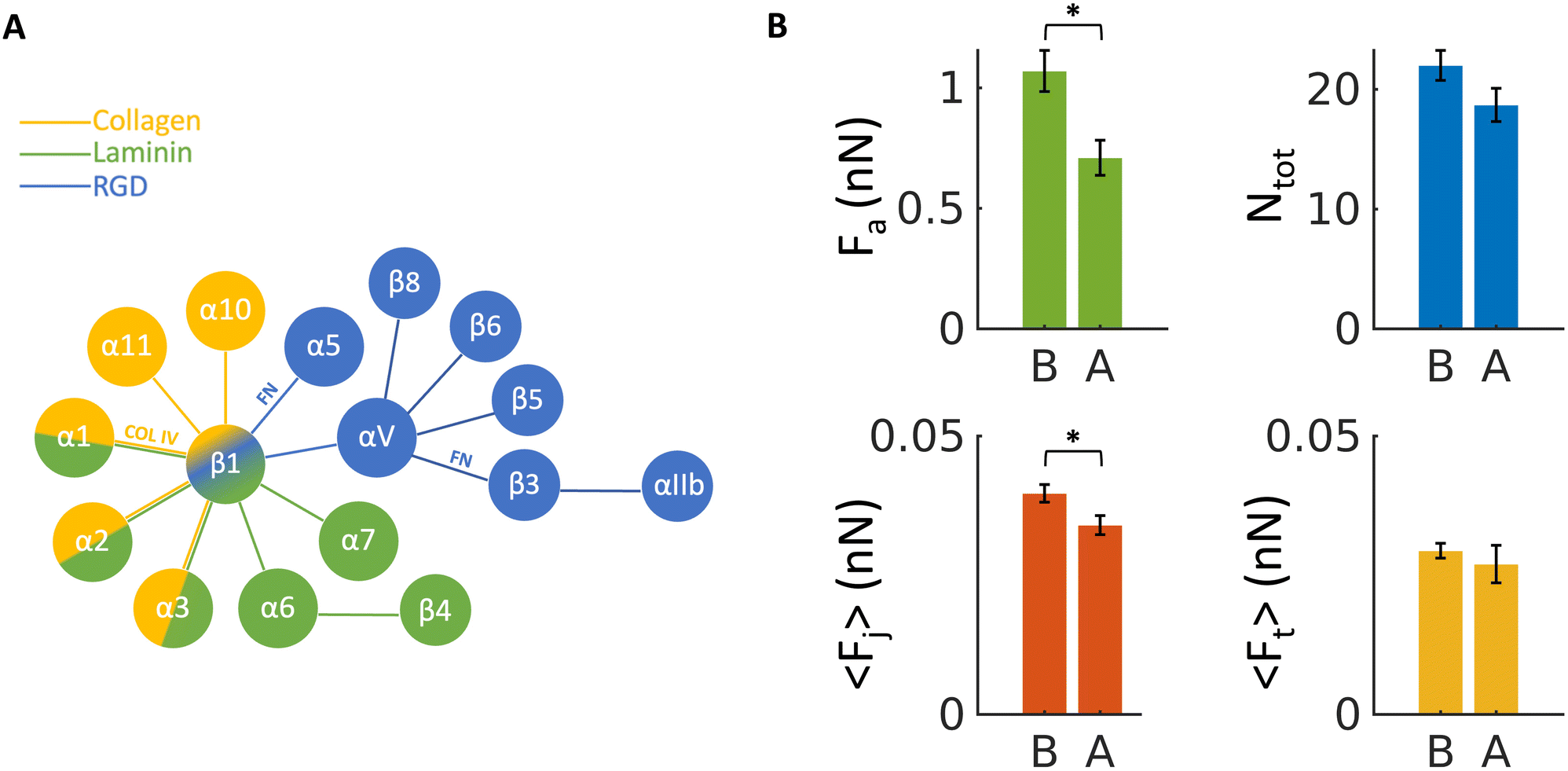 | ||
| Fig. 6 (A) Schematics of the possible combinations (represented by lines) of α and β integrin sub-units for binding to laminin, collagen and RGD domains, respectively; combinations that are particularly relevant for this study (for binding to collagen IV, COL IV, and fibronectin, FN) are highlighted. (B) The measured force of adhesion Fa, mean number of events Ntot (both jumps and tethers) and mean force per jump and tethers 〈Fj〉 and 〈Ft〉, respectively, measured using control probes CTRL1 and CTRL2 (B) before, and (A) after the addition of the 4b4 inhibitory antibody against β1 integrin, at concentration of 5 μg ml−1. Errors represent effective standard deviations of the mean, as explained in the Methods. | ||
Fig. 6B shows a significant decrease upon exposure of cells to the antibody of the total adhesion force Fa as well as the mean force of jumps 〈Fj〉. A decrease of the total number of events Ntot (jumps and tethers) and the mean force of tethers 〈Ft〉 was also observed, although not significant. These results confirm that the measured interaction forces depend at least partially on β1 subunits of integrin proteins.
3. Conclusions
In the current work, we presented a novel adhesion force spectroscopy approach for the study of the cell–microenvironment interaction obtained by attaching selected pieces of native ECM to an AFM tipless cantilever, exploiting laser microdissection. These probes reproduce the full complexity of the cell–ECM interface in the physiological condition, in terms of both biophysical cues and chemical composition.We demonstrated, as proof-of-principle, the functionalisation of AFM cantilevers with native rat bladder ECM pieces from different regions of the tissue, and the use of these novel probes in adhesion force spectroscopy experiments against rat bladder cancer cells AY-27. With this novel strategy, we could discriminate fine differences in the adhesive patterns of the cells with three different ECM layers.
The robustness and durability of the ECM probes were demonstrated by stress tests performed both in ramping and contact scanning modes. In addition, we showed the ability of this approach to detect specific integrin-related events reliably and repeatedly, as well as to assess significant differences in the mechanotransductive parameters such as the mean number and force of unbinding events, and their distribution, depending on the ECM region used to produce the probe. The observed differences correlate with the differences in the ECM protein and collagen composition, biophysical and mechanical properties, revealed respectively by the immunostaining and AFM mechanical analysis. The lower detected adhesion force, when the detrusor ECM was used, highlights the relevance of our strategy to investigate the impact of cell–ECM adhesion on tumour invasion.56,57 An added value of our approach is that its spatial resolution allows measurements at the single cell level, and thus it could be used to assess the heterogeneity of neoplastic clones.
Our results demonstrate the potential of these novel AFM probes to perform highly specific force spectroscopy investigations. Indeed, the adhesive properties of the probes can be tailored by selecting the type and the specific region of the ECM. The target cells can also be selected to probe specific mechanotransductive and mechanobiological interactions.
For example, experiments can be performed probing different types of matrices against a wide variety of cells (both healthy, tumoural, and from metastatic sites). Both ECM and cells could be extracted from different organs and tissues of the same patient, which might find application in identifying biomarkers for early diagnosis of diseases (particularly the context of cancer and metastasis, but also other diseases). This could allow in-depth understanding of disparities in the cell/microenvironment interaction between healthy, tumoural and metastatic cells confronted with different ECM substrates. The approach can also be used to test the effects of chemotherapeutic drugs targeting mechanotransduction-associated key players (such as integrins or Rho signalling-related proteins) and structures (e.g., components of the ECM, glycocalyx, and cytoskeleton) in pathophysiological conditions, with a very high precision at the level of forces and number of adhesion sites.
The main limitations of our approach are the relatively complex production procedure, which requires a laser microdissection apparatus and some delicate micromanipulation steps, and the not straightforward interpretation of the complex readout of the adhesion force experiment down to single molecular events, given that the signatures of several molecular specific unbinding events are coupled within the same force curve. Nevertheless, we think that the advantages coming from the durability, reliability, and application potential of ECM probes in detecting cell–microenvironment interactions easily exceed these drawbacks. A further improvement could be performing force spectroscopy experiments with ECM probes at variable loading rate, a common practice in the force spectroscopy community;61 however, the parallel nature of bonds formed between ECM and adhesive proteins, as well as the viscoelastic properties of the ECM, make this approach challenging and call for further work to fully assess the applicability of this approach.
In summary, ECM probes are a novel enabling feature of adhesion force spectroscopy and could become a key element in the nanotechnological toolkit of nascent personalised medicine.
4. Materials and methods
4.1. Cells
The rat bladder cancer cell line AY-27 (Sigma-Aldrich, cat. number SCC254) was authenticated for lack of cross-contamination by analyzing 9 short tandem repeats DNA (IDEXX Bioanalytics, Ludwigsburg, Germany).62 The cell line was cultured in RPMI 2 mM L-glutamine with 10% FBS, 1% penicillin/streptomycin and 1% amphotericin; cells were cultured in an incubator at 37 °C and 5% CO2 (Galaxy S, RS Biotech). All reagents and materials are from Sigma Aldrich, if not stated otherwise.For AFM measurements, the cells were plated one day before on glass bottom Petri dishes (∅ 40 mm Willco Wells) coated with poly-L-lysine (0.1% w/v for 30 min at room temperature), in the same RPMI medium without phenol red.
4.2. Extracellular matrices
The bladder from healthy rat was decellularised following the protocol from Genovese et al.,63 validated also for bladder.64 The ECM samples were embedded in optimal cutting temperature compound (OCT), frozen in dry ice and kept at −80 °C. All procedures and studies involving mice were approved by the Institutional Animal Care and Use Committee of San Raffaele Scientific Institute, and performed according to the prescribed guidelines (IACUC, approval number 942). Adult (9–10 weeks old) female Fischer rats were from Charles River Laboratories, Italy.For the laser microdissected slices, 10 μm thick ECMs were cut with a cryostat and mounted onto classical optical microscopy glass slides. The sample was then immerged in PBS to remove OCT and then in Ethanol 70% to dehydrate it prior to laser microdissection.
We tested the effect of dehydration of the ECM, and we confirmed that this process does not alter the mechanical properties of the matrix. For the comparison of mechanical properties before and after dehydration, an ECM slice was first characterised after gently washing away the OCT with PBS, then dehydrated in 70% EtOH (following the same procedure used for LMD) and measured again (Fig. S3C†).
Two different slices were used for the comparison of morphological and mechanical properties of the inner and outer regions of the LMD cut.
For AFM mechanical experiments, 50 μm thick ECMs were cut with a cryostat and attached to super-frost microscope glass slides. The prepared ECM slices were kept at −20 °C prior to the AFM experiments. Before starting the AFM measurements, the slices were washed with PBS to remove the OCT.
4.3. Reagents
For the functionalisation of the probes, 3-aminopropyl triethoxysilane (APTES) and Genipin were used, both purchased from Sigma. Genipin was diluted in 1.5% (v/v) in PBS. 4b4 antibody was purchased from Beckman Coulter.4.4. Laser microdissection (LMD)
400 μm2 (20μm × 20μm) regions were cut using a UV-based LMD7 laser microdissection system (Leica Microsystems) at 20× magnification placing the slide with the ECM section facing up. This setup allowed to separate the regions of interest (ROIs) from the rest of the tissue without detaching them from the glass. The engraved sections were kept dry at 4 °C until further processing.4.5. Atomic force microscopy
All the experiments have been performed using a Bioscope Catalyst AFM (Bruker), mounted on top of an inverted optical microscope (Olympus X71). The system was isolated from the ambient noise placing the AFM on top of an active anti-vibration base (DVIA-T45, Daeil Systems) and enclosing it in an acoustic box (Schaefer, Italy).Mechanical measurements , aimed to characterise the Young's modulus of elasticity of the ECM were performed by recording force versus distance curves (shortly force curves, FCs), then transformed into force vs. indentation curves, as described in ref. 14, 65 and 66. We used custom colloidal probes, produced by attaching borosilicate glass spheres to tipless cantilevers (MikroMasch HQ:CSC38/Tipless/No Al or NanoandMore TL-FM), as described in ref. 67. The tip radius was calibrated by means of reverse AFM imaging.67 The cantilever spring constant was calibrated using the thermal noise method,68,69 and fine corrections were applied to account for geometrical and dimensional issues.70,71 The deflection sensitivity (or inverse optical lever sensitivity, invOLS) of the optical beam deflection apparatus was measured as the inverse of the slope of the deflection vs. z-piezo displacement curves acquired on a stiff substrate.66 The deflection sensitivity was monitored and if necessary corrected during AFM experiments using the contactless SNAP method,72 assuming as reference spring constant the intrinsic spring constant previously calibrated.
The probes used had spring constants of 3–4 N m−1, radii of 5.5 and 9 μm. 10 sets of force–distance curves (force volumes, FVs), consisting of 20 × 20 curves each, spanning typically an area of 50 μm × 50 μm, were acquired on regions separated by more than 200 μm on each slice. All measurements were carried out in a droplet of PBS, confined on the glass slide by means of hydrophobic ink (Sigma).
The precise alignment of AFM and optical images was possible thanks to the Bruker MIRO software, which allowed to choose the ROIs on the ECMs for the analysis (as during the force spectroscopy experiments). The transparency of the slices with thickness below 50 μm allowed us to identify and select regions where pieces of ECM had been cut by LMD. The ECM probe can be placed precisely on top of a cell using the optical alignment, ensuring that contact is established with the ECM, and not with another part of the cantilever.
Adhesion force spectroscopy measurements were performed according to the protocol described in ref. 11 and 26, by acquiring several FCs per cell on at least 4 cells per each contact time, using the ECM probes. In force spectroscopy experiments, the adhesion pattern in the retracting portion of the FC is studied (Fig. 7); upon contact with the sample, the tip was retracted after a contact time (ct) of 0, 20, 60 and 120 s, during which the z-sensor position was kept constant by the feedback of the AFM. These time points were chosen as they cover the critical window in which nascent adhesions mature to focal complexes or even focal adhesions.26,73 A typical retraction portion of a FC is shown in Fig. 7, where the adhesive features (jumps and tethers) can be clearly observed, and all relevant adhesive parameters are indicated (details in the figure caption). We typically used the following values for the ramp parameters: ramp frequency, 1 Hz; ramp size, 10 μm; maximum force, 1 nN.
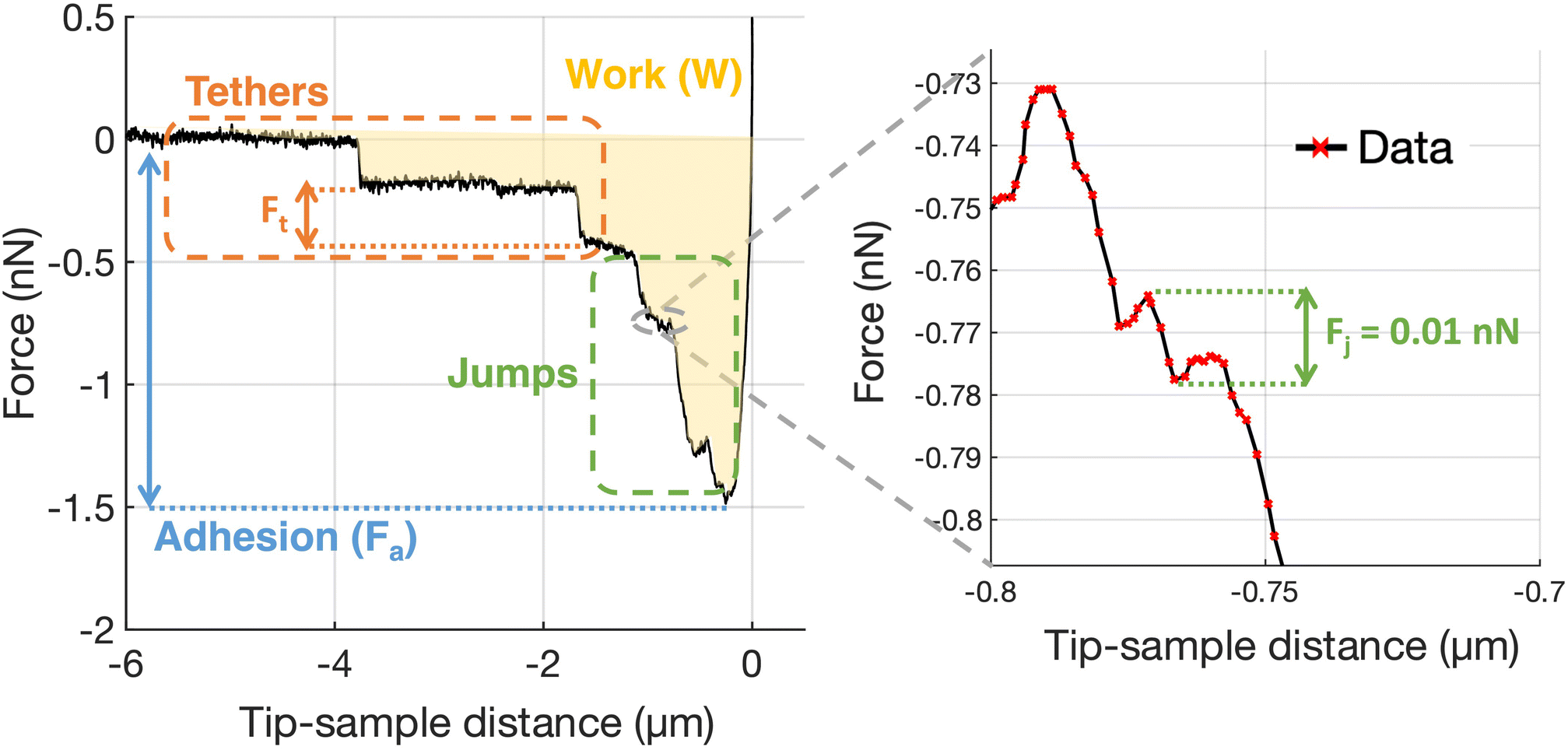 | ||
| Fig. 7 Representative force curve acquired during an adhesion force spectroscopy experiment using a native ECM probe with a zoom on a specific jump region. The relevant features and parameters are indicated: unbinding events (jumps and tethers); the total adhesion force Fa; the forces Fj and Ft necessary to break integrin(s)–ECM bond in jumps and tethers, respectively; the work of adhesion W. From the distribution of jumps and tethers forces, the mean forces 〈Fj〉 and 〈Ft〉, as well as the mean number of jump and tether events per force curves, Nj and Nt, respectively, and the total number of integrin-related events Ntot = Nj + Nt, are calculated. The adhesion force Fa is calculated as the absolute difference between the minimum force value and the baseline value. The work of adhesion W is calculated as the integral of adhesion force over distance; graphically, it is the area enclosed by the retraction part of the force curve. | ||
For the control (inhibition) experiment, the 4b4 antibody, an inhibitor of β1 integrin, was added to the medium, at a concentration of 5 μg mL−1, 20 minutes before the AFM measurement. Experiments were carried out at 37 °C using a thermostatic fluid cell and a temperature controller (Lakeshore 331, Ohio, USA).
The elastic properties of cells and ECMs were characterised through their Young's modulus (YM) of elasticity, extracted by fitting the Hertz model to the 20%–80% indentation range of the FCs (details in ref. 14 and 65),
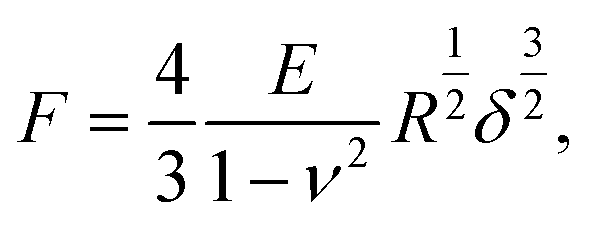 | (1) |
Finite thickness correction for cells was applied as described in ref. 65, 79 and 80. In the case of ECM, despite the relatively small thickness of the slices, this correction was not applied because the thickness is not known with good accuracy, being the underlying substrate not always exposed; nevertheless, despite a systematic overestimation of the YM of the ECM, we were able to carry out a comparative characterisation of ECM elasticity in different conditions.
For the analysis of adhesion force spectroscopy data, a custom MATLAB routine was used to detect specific adhesion events in the FCs (jumps and tethers23,26) and to calculate the values of the relevant parameters (see Fig. 7), as described in Chighizola et al.,26,81 like the mean number of jumps and tethers per force curve, Nj and Nt, respectively, the total number of events Ntot = Nj + Nt, and the mean total adhesion force Fa, all these quantities being averaged across all FCs for a given condition; the mean force per jump 〈Fj〉 and the mean force per tether 〈Ft〉 represent the mean values from the distribution of Fj and Ft values measured in a specific condition. The associated errors were calculated by summing in quadrature the standard deviation of the mean to an instrumental error of 3%, calculated by propagating the calibration uncertainties in the fitting procedure through a Monte Carlo simulation.65
4.6. Immunofluorescence
10 μm thick snap-frozen OCT-embedded cryosections were mounted on positively charged glass slide (Superfrost plus adhesion slide, #J1800AMNZ, Epredia Inc) and processed as described above for laser microdissection. Engraved and non-engraved sections were fixed in 4% Paraformaldehyde (#15710 EM grade, Electron Microscopy Sciences) at room temperature for 10 min. After washing in PBS, sections were incubated for 1 hour with blocking buffer (2% Donkey serum, 1.5% BSA, 0.25% Fish Gelatin, PBS pH = 7.2) and then incubated overnight at 4 °C with the following primary antibodies: anti-laminin β1 (4 μg ml−1, sc-17810, Santa Cruz Biotech), anti-fibronectin (10 μg ml−1, NBP1-91258SS, NovusBio), anti-collagen IV (20 μg ml−1, AB769, Millipore).After washing, anti-goat Alexa Fluor® 488, anti-mouse Alexa Fluor® 647 and anti-rabbit Alexa Fluor 555 conjugated fluorophore-labeled F(ab)2 donkey secondary antibodies were used (Jackson Immunoresearch). Sections were DAPI counterstained to verify proper tissue decellularization and 3 × 3 Z-stacked large images (9 μm depth, 11 stacks) were acquired using a Yokogawa Spinning Disk Field Scanning Confocal System (CSU-W1, Nikon Europe BV, Amsterdam, Netherlands) equipped with 405, 488, 561, 640, 785 nm lines of solid-state lasers, 40×/1.15NA water immersion objective lens and a Prime BSI sCMOS camera (Teledyne Photometrics, Tucson, AZ).
Mean fluorescent intensity signal was quantified in multiple ROIs (175 μm2 each; n = 10, 5 ROIs per each field, 2 field acquired per each layer) and normalised to the respective average pixel intensity of the entire stained area. Data represent the normalised signal intensity compared to the average value of the submucosal layer for each matrix. For pre- and post-cut evaluation, fluorescent signal intensity was quantified in the central portion of the engraved region (post-LMD) and the corresponding area on a consecutive section not subjected to laser cut (pre-LMD). The normalised signal intensity was averaged for each ECM and scaled between 0 and 1 (n = 30, 5 ROIs per each field, 6 fields acquired distributed along the layers of the bladder). Mann–Whitney test was used to compare differences in the signal intensity.
Author contributions
Conceptualisation: AP, CS; methodology – ECM preparation: IL; methodology – probe fabrication and characterisation: HH, GRD, AP; methodology – laser microdissection and staining: LN, PDC, GRD; methodology – cell culture and preparation: HH, CS; methodology – AFM and force spectroscopy: HH, MC, AP; data curation and analysis: HH, MC, GRD, PDC, AP; original draft writing and editing: HH, MC, AP; draft revision: all authors; supervision: MC, GRD, CS, AP; resources, funding, and project administration: AP, GRD, MA. Authors’ contributions were allocated adopting the terminology of CRediT Contributor Roles Taxonomy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie Action grant agreement no. 812772, project Phys2Biomed, and under FET Open grant agreement no. 801126, project EDIT. PDC was supported by an AIRC fellowship for Italy, grant no. 25542.References
- B. Alberts, Essential cell biology, Garland Science Pub., New York NY, 2nd edn, 1998, vol. 51 Search PubMed.
- C. Frantz, K. M. Stewart and V. M. Weaver, J. Cell Sci., 2010, 123, 4195–4200 CrossRef CAS PubMed.
- A. D. Theocharis, S. S. Skandalis, C. Gialeli and N. K. Karamanos, Adv. Drug Delivery Rev., 2016, 97, 4–27 CrossRef CAS PubMed.
- T. R. Cox, Nat. Rev. Cancer, 2021, 21, 217–238 CrossRef CAS PubMed.
- N. C. Gauthier and P. Roca-Cusachs, Curr. Opin. Cell Biol., 2018, 50, 20–26 CrossRef CAS PubMed.
- J. Alcaraz, J. Otero, I. Jorba and D. Navajas, Semin. Cell Dev. Biol., 2018, 73, 71–81 CrossRef CAS PubMed.
- Z. Sun, S. S. Guo and R. Fässler, J. Cell Biol., 2016, 215, 445–456 CrossRef CAS PubMed.
- D. E. Jaalouk and J. Lammerding, Nat. Rev. Mol. Cell Biol., 2009, 10, 63–73 CrossRef CAS PubMed.
- M. Chighizola, T. Dini, C. Lenardi, P. Milani, A. Podestà and C. Schulte, Biophys. Rev., 2019, 11, 701–720 CrossRef CAS PubMed.
- Z. Lansky, Y. Mutsafi, L. Houben, T. Ilani, G. Armony, S. G. Wolf and D. Fass, J. Struct. Biol.: X, 2019, 1, 100002 CAS.
- M. Chighizola, T. Dini, S. Marcotti, M. D'Urso, C. Piazzoni, F. Borghi, A. Previdi, L. Ceriani, C. Folliero, B. Stramer, C. Lenardi, P. Milani, A. Podestà and C. Schulte, J. Nanobiotechnol., 2022, 20, 418 CrossRef CAS PubMed.
- L. Martinez-Vidal, V. Murdica, C. Venegoni, F. Pederzoli, M. Bandini, A. Necchi, A. Salonia and M. Alfano, Commun. Biol., 2021, 4, 1–16 CrossRef PubMed.
- C. Schulte, Mechanotransduction, in Mechanics of Cells and Tissues in Diseases, ed. M. Lekka, D. Navajas, M. Radmacher and A. Podestà, DeGruyter, 2023, vol. 2 Search PubMed.
- H. Holuigue, E. Lorenc, M. Chighizola, C. Schulte, L. Varinelli, M. Deraco, M. Guaglio, M. Gariboldi and A. Podestà, Sensors, 2022, 22, 2197 CrossRef CAS PubMed.
- M. Krieg, G. Fläschner, D. Alsteens, B. M. Gaub, W. H. Roos, G. J. L. Wuite, H. E. Gaub, C. Gerber, Y. F. Dufrêne and D. J. Müller, Nat. Rev. Phys., 2019, 1, 41–57 CrossRef.
- R. Omidvar, M. Tafazzoli-shadpour, M. A. Shokrgozar and M. Rostami, J. Biomech., 2014, 47, 3373–3379 CrossRef PubMed.
- A. Alessandrini and P. Facci, Meas. Sci. Technol., 2005, 16, 65–92 CrossRef.
- L. Martinez-Vidal, M. Chighizola, M. Berardi, E. Alchera, I. Locatelli, F. Pederzoli, C. Venegoni, R. Lucianò, P. Milani, K. Bielawski, A. Salonia, A. Podestà and M. Alfano, Commun. Biol., 2023, 6, 217 CrossRef PubMed.
- D. J. Müller, A. C. Dumitru, C. lo Giudice, H. E. Gaub, P. Hinterdorfer, G. Hummer, J. J. de Yoreo, Y. F. Dufrêne and D. Alsteens, Chem. Rev., 2021, 121, 11701–11725 CrossRef PubMed.
- J. Friedrichs, J. Helenius and D. J. Muller, Nat. Protoc., 2010, 5, 1353–1361 CrossRef CAS PubMed.
- N. Strohmeyer, M. Bharadwaj, M. Costell, R. Fässler and D. J. Müller, Nat. Mater., 2017, 16, 1262–1270 CrossRef CAS PubMed.
- M. Yu, J. Wang, D. J. Muller and J. Helenius, Sci. Rep., 2015, 5, 8206 CrossRef CAS PubMed.
- J. Friedrichs, K. R. Legate, R. Schubert, M. Bharadwaj, C. Werner, D. J. Müller and M. Benoit, Methods, 2013, 60, 169–178 CrossRef CAS PubMed.
- S. A. Maynard, A. Gelmi, S. C. Skaalure, I. J. Pence, C. Lee-Reeves, J. E. Sero, T. E. Whittaker and M. M. Stevens, ACS Nano, 2020, 14, 17321–17332 CrossRef CAS PubMed.
- L. Jiang, Z. Sun, X. Chen, J. Li, Y. Xu, Y. Zu, J. Hu, D. Han and C. Yang, ACS Nano, 2016, 10, 207–217 CrossRef CAS PubMed.
- M. Chighizola, A. Previdi, T. Dini, C. Piazzoni, C. Lenardi, P. Milani, C. Schulte and A. Podestà, Nanoscale, 2020, 12, 14708–14723 RSC.
- Z. Sun, L. A. Martinez-Lemus, M. A. Hill and G. A. Meininger, Am. J. Physiol.: Cell Physiol., 2008, 295, C268–C278 CrossRef CAS PubMed.
- R. Harjumäki, X. Zhang, R. W. N. Nugroho, M. Farooq, Y.-R. Lou, M. Yliperttula, J. J. Valle-Delgado and M. Österberg, ACS Appl. Bio Mater., 2020, 3, 1406–1417 CrossRef PubMed.
- Z. Li, T. Liu, J. Yang, J. Lin and S. X. Xin, J. Mol. Recognit., 2020, 33, e2823 CrossRef CAS PubMed.
- L. Chièze, A. le Cigne, M. Meunier, A. Berquand, S. Dedieu, J. Devy and M. Molinari, J. Mol. Recognit., 2019, 32, e2767 CrossRef PubMed.
- J. Helenius, C. P. Heisenberg, H. E. Gaub and D. J. Muller, J. Cell Sci., 2008, 121, 1785–1791 CrossRef CAS PubMed.
- P. K. Viji Babu, U. Mirastschijski, G. Belge and M. Radmacher, Eur. Biophys. J., 2021, 1, 3 Search PubMed.
- Y. Takada, X. Ye and S. Simon, Genome Biol., 2007, 8, 215 CrossRef PubMed.
- T. Orré, O. Rossier and G. Giannone, Exp. Cell Res., 2019, 379, 235–244 CrossRef PubMed.
- J. D. Humphries, M. R. Chastney, J. A. Askari and M. J. Humphries, Curr. Opin. Cell Biol., 2019, 56, 14–21 CrossRef CAS PubMed.
- R. Changede and M. Sheetz, BioEssays, 2017, 39, 1–12 CrossRef CAS PubMed.
- I. D. Campbell and M. J. Humphries, Cold Spring Harbor Perspect. Biol., 2011, 3, a004994–a004994 Search PubMed.
- Z. Sun, L. A. Martinez-Lemus, A. Trache, J. P. Trzeciakowski, G. E. Davis, U. Pohl and G. A. Meininger, Am. J. Physiol.: Heart Circ. Physiol., 2005, 289, 2526–2535 CrossRef PubMed.
- R. W. N. Nugroho, R. Harjumäki, X. Zhang, Y.-R. Lou, M. Yliperttula, J. J. Valle-Delgado and M. Österberg, Colloids Surf., B, 2019, 173, 571–580 CrossRef CAS PubMed.
- S. Datta, L. Malhotra, R. Dickerson, S. Chaffee, C. K. Sen and S. Roy, Histol. Histopathol., 2015, 30(11), 1255–1269 CAS.
- C. Bevilacqua and B. Ducos, Mol. Aspects Med., 2018, 59, 5–27 CrossRef CAS PubMed.
- F. Ajalloueian, G. Lemon, J. Hilborn, I. S. Chronakis and M. Fossum, Nat. Rev. Urol., 2018, 15, 155–174 CrossRef PubMed.
- H. Orabi, S. Bouhout, A. Morissette, A. Rousseau, S. Chabaud and S. Bolduc, Sci. World J., 2013, 2013, 1–13 CrossRef PubMed.
- L. J. Mortensen, C. Alt, R. Turcotte, M. Masek, T.-M. Liu, D. C. Côté, C. Xu, G. Intini and C. P. Lin, Biomed. Opt. Express, 2015, 6, 32 CrossRef PubMed.
- T. Gudzenko and C. M. Franz, Front. Mol. Biosci., 2020, 7, 149 CrossRef CAS PubMed.
- R. Changede, H. Cai, S. J. Wind and M. P. Sheetz, Nat. Mater., 2019, 18, 1366–1375 CrossRef CAS PubMed.
- J. Z. Kechagia, J. Ivaska and P. Roca-Cusachs, Nat. Rev. Mol. Cell Biol., 2019, 20, 457–473 CrossRef CAS PubMed.
- L. B. Case and C. M. Waterman, Nat. Cell Biol., 2015, 17, 955–963 CrossRef CAS PubMed.
- M. Arnold, E. A. Cavalcanti-Adam, R. Glass, J. Blümmel, W. Eck, M. Kantlehner, H. Kessler and J. P. Spatz, ChemPhysChem, 2004, 5, 383–388 CrossRef CAS PubMed.
- E. A. Cavalcanti-Adam, T. Volberg, A. Micoulet, H. Kessler, B. Geiger and J. P. Spatz, Biophys. J., 2007, 92, 2964–2974 CrossRef CAS PubMed.
- Y. Liu, R. Medda, Z. Liu, K. Galior, K. Yehl, J. P. Spatz, E. A. Cavalcanti-Adam and K. Salaita, Nano Lett., 2014, 14, 5539–5546 CrossRef CAS PubMed.
- R. Oria, T. Wiegand, J. Escribano, A. Elosegui-Artola, J. J. Uriarte, C. Moreno-Pulido, I. Platzman, P. Delcanale, L. Albertazzi, D. Navajas, X. Trepat, J. M. García-Aznar, E. A. Cavalcanti-Adam and P. Roca-Cusachs, Nature, 2017, 552, 219–224 CrossRef CAS PubMed.
- A. Elosegui-Artola, R. Oria, Y. Chen, A. Kosmalska, C. Pérez-González, N. Castro, C. Zhu, X. Trepat and P. Roca-Cusachs, Nat. Cell Biol., 2016, 18, 540–548 CrossRef CAS PubMed.
- T. C. M. Zuiverloon, F. C. de Jong, J. C. Costello and D. Theodorescu, Bladder Cancer, 2018, 4, 169–183 Search PubMed.
- A. Taubenberger, D. A. Cisneros, J. Friedrichs, P. H. Puech, D. J. Muller and C. M. Franz, Mol. Biol. Cell, 2007, 18, 1634–1644 CrossRef CAS PubMed.
- M. Janiszewska, M. C. Primi and T. Izard, J. Biol. Chem., 2020, 295, 2495–2505 CrossRef CAS PubMed.
- U. Cavallaro and G. Christofori, Biochim. Biophys. Acta, Rev. Cancer, 2001, 1552, 39–45 CrossRef CAS PubMed.
- D. J. Müller, J. Helenius, D. Alsteens and Y. F. Dufrne, Nat. Chem. Biol., 2009, 5, 383–390 CrossRef PubMed.
- E. A. Evans and D. A. Calderwood, Science, 2007, 316, 1148–1153 CrossRef CAS PubMed.
- A. Mescola, S. Vella, M. Scotto, P. Gavazzo, C. Canale, A. Diaspro, A. Pagano and M. Vassalli, J. Mol. Recognit., 2012, 25, 270–277 CrossRef CAS PubMed.
- E. Evans, Faraday Discuss., 1998, 1–16 CAS.
- W. K. Alfred Yung, Neuro Oncol., 2012, 14, 675 CrossRef PubMed.
- L. Genovese, L. Zawada, A. Tosoni, A. Ferri, P. Zerbi, R. Allevi, M. Nebuloni and M. Alfano, Tissue Eng., Part A, 2014, 20, 2005–2018 CrossRef CAS PubMed.
- M. Alfano, M. Nebuloni, R. Allevi, P. Zerbi, E. Longhi, R. Lucianò, I. Locatelli, A. Pecoraro, M. Indrieri, C. Speziali, C. Doglioni, P. Milani, F. Montorsi and A. Salonia, Sci. Rep., 2016, 6, 36128 CrossRef CAS PubMed.
- L. Puricelli, M. Galluzzi, C. Schulte, A. Podestà and P. Milani, Rev. Sci. Instrum., 2015, 86, 33705 CrossRef PubMed.
- H. J. Butt, B. Cappella and M. Kappl, Surf. Sci. Rep., 2005, 59, 1–152 CrossRef CAS.
- M. Indrieri, A. Podestà, G. Bongiorno, D. Marchesi and P. Milani, Rev. Sci. Instrum., 2011, 82, 023708 CrossRef CAS PubMed.
- J. L. Hutter and J. Bechhoefer, Rev. Sci. Instrum., 1993, 64, 1868–1873 CrossRef CAS.
- H. J. Butt and M. Jaschke, Nanotechnology, 1995, 6, 1–7 CrossRef.
- M. Chighizola, L. Puricelli, L. Bellon and A. Podestà, J. Mol. Recognit., 2021, 34, e2879 CrossRef CAS PubMed.
- J. Laurent, A. Steinberger and L. Bellon, Nanotechnology, 2013, 24, 225504 CrossRef PubMed.
- H. Schillers, C. Rianna, J. Schäpe, T. Luque, H. Doschke, M. Wälte, J. J. Uriarte, N. Campillo, G. P. A. Michanetzis, J. Bobrowska, A. Dumitru, E. T. Herruzo, S. Bovio, P. Parot, M. Galluzzi, A. Podestà, L. Puricelli, S. Scheuring, Y. Missirlis, R. Garcia, M. Odorico, J.-M. Teulon, F. Lafont, M. Lekka, F. Rico, A. Rigato, J.-L. Pellequer, H. Oberleithner, D. Navajas and M. Radmacher, Sci. Rep., 2017, 7, 5117 CrossRef PubMed.
- M. Vicente-Manzanares and A. R. Horwitz, J. Cell Sci., 2011, 124, 3923–3927 CrossRef CAS PubMed.
- R. G. Acres, A. V. Ellis, J. Alvino, C. E. Lenahan, D. A. Khodakov, G. F. Metha and G. G. Andersson, J. Phys. Chem. C, 2012, 116, 6289–6297 CrossRef CAS.
- S. Fiorilli, P. Rivolo, E. Descrovi, C. Ricciardi, L. Pasquardini, L. Lunelli, L. Vanzetti, C. Pederzolli, B. Onida and E. Garrone, J. Colloid Interface Sci., 2008, 321, 235–241 CrossRef CAS PubMed.
- I. Jorba, G. Beltrán, B. Falcones, B. Suki, R. Farré, J. M. García-Aznar and D. Navajas, Acta Biomater., 2019, 92, 265–276 CrossRef PubMed.
- G. G. Genchi, G. Ciofani, I. Liakos, L. Ricotti, L. Ceseracciu, A. Athanassiou, B. Mazzolai, A. Menciassi and V. Mattoli, Colloids Surf., B, 2013, 105, 144–151 CrossRef CAS PubMed.
- K. Reinhardt, Handbook of Silicon Wafer Technology, 3rd edn, 2018 Search PubMed.
- P. D. Garcia and R. Garcia, Biophys. J., 2018, 114, 2923–2932 CrossRef CAS PubMed.
- E. K. Dimitriadis, F. Horkay, J. Maresca, B. Kachar and R. S. Chadwick, Biophys. J., 2002, 82, 2798–2810 CrossRef CAS PubMed.
- M. Chighizola, T. Dini, S. Marcotti, M. D'Urso, C. Piazzoni, F. Borghi, A. Previdi, L. Ceriani, C. Folliero, B. Stramer, C. Lenardi, P. Milani, A. Podestà and C. Schulte, J. Nanobiotechnol., 2022, 20, 418 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr01568h |
| This journal is © The Royal Society of Chemistry 2023 |