
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow-power microwaves: a cell-compatible physical treatment to enhance the mechanical properties of self-assembling peptides†
Maria Gessica
Ciulla
 a,
Amanda
Marchini
ab,
Jacopo
Gazzola
c,
Manuel
Sambrotta
d and
Fabrizio
Gelain
*ab
a,
Amanda
Marchini
ab,
Jacopo
Gazzola
c,
Manuel
Sambrotta
d and
Fabrizio
Gelain
*ab
aInstitute for Stem-Cell Biology, Regenerative Medicine and Innovative Therapies, IRCCS Casa Sollievo della Sofferenza, 71013 San Giovanni Rotondo, Italy. E-mail: f.gelain@css-mendel.it
bCenter for Nanomedicine and Tissue Engineering (CNTE), ASST Grande Ospedale Metropolitano Niguarda, 20162 Milan, Italy
cDepartment of Biotechnology and Bioscience, University of Milan – Bicocca, 20125 Milan, Italy
dDepartment of Chemistry, Materials and Chemical Engineering “Giulio Natta”, Politecnico di Milano, 20133 Milan, Italy
First published on 25th September 2023
Abstract
Biomaterials designed for tissue engineering applications should, among other requirements, mimic the native extracellular matrix (ECM) of the tissues to be regenerated, both in terms of biomimetic and mechanical properties. Ideally, the scaffold stiffness and stress resistance should be tuned for each specific implantation therapy. Self-assembling peptides (SAPs) are promising synthetic bionanomaterials prone to easy multi-functionalization, bestowing biomimetic properties. However, they usually yield soft and fragile hydrogels unsuited for the regeneration of medium-to-hard tissues. For this purpose, chemical cross-linking of SAPs is an option, but it often requires a moderately toxic and expensive chemical compound and/or the presence of specific residues/reactive sites, posing issues for its feasibility and translational potential. In this work, we introduced, characterized by rheology, atomic force microscopy (AFM), Thioflavin-T assay (ThT), and Fourier transform infrared (FT-IR) tests, and optimized (by tuning the power, temperature and treatment time) a novel fast, green and affordable methodology using mild microwave (MW) irradiation to increase the mechanical properties of diverse classes of SAPs. Low-power MWs increase stiffness, resilience, and β-structuration, while high-power MW treatments partially denature the tested SAPs. Our pure-physical methodology does not alter the SAP biomimetic properties (verified via in vitro tests of viability and differentiation of human neural stem cells), is compatible with already seeded cells, and is also synergic with genipin-based cross-linking of SAPs; therefore, it may become the next standard for SAP preparation in tissue engineering applications at hand of all research labs and in clinics.
Introduction
In tissue engineering, several biomaterials are being developed with the ultimate goal of regenerating tissue injuries or whole organs. In general, promising candidates should feature low toxicity, micro-/nano-scaled morphology, selective biomimetic properties, and tuned biomechanics; final objectives would be to trigger endogenous cell-driven regeneration, foster transplanted cell engraftment (if present), precisely release one or more drugs (if present) and favor a seamless integration of the scaffold with the surrounding tissues.1,2 Also, negligible foreign body reaction, the absence of pathogen transfer, affordable costs of scaled-up production and minimum lot-to-lot variability are all features that have to be carefully weighted in order to develop a successful translational approach.1 Self-assembling biomaterials, yielding supramolecular nanostructures, can be bottom-up designed and produced at an affordable cost.2 Molecular self-assembling is ubiquitous in nature3 and it spontaneously occurs when chaotic molecules self-organize in ordered structures thanks to non-covalent interactions.4 Examples of self-assembled structures in biological systems are phospholipid-packed cell membranes,5 DNA double helixes,6 and actin filaments responsible for cellular motility.7,8 SAPs are synthetic bionanomaterials giving nanostructures upon exposure to trigger stimuli thanks to physical entanglements, hydrophobic forces, aromatic stacking and electrostatic/van der Waals interactions.9 SAPs can be readily synthesized and produced in large quantities with high purity, reproducibility, and no risk of pathogen transfer; moreover, they can be designed at the molecular level depending on the target application.10 Indeed, they have been widely investigated in drug delivery approaches,11–13 for targeting tumor sites with anticancer drugs14 and as treatments against bacterial infections.14,15 In tissue engineering, biomimetic (multi-functionalized) SAPs were demonstrated to favor nervous,16–18 liver,19 cartilage,20,21 and vascular regeneration.22 Nonetheless, with just weak interactions (non-covalent bonds) involved in the self-assembling phenomenon, modest stiffness and poor resilience have been the major drawbacks of SAPs, thus limiting their applications in many regenerative medicine projects. In previous works, we combined SAPs with cross-linkers to enhance their mechanical properties through covalent cross-linking by using homobifunctional, heterobifunctional, or photoreactive cross-linkers, such as genipin (GP),23,24 4-(N-maleimidomethyl) cyclohexane-1-carboxylic acid 3-sulfo-N-hydroxysuccinimide ester (Sulfo-SMCC),25,26 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide/N-hydroxysulfosuccinimide (EDC/sulfo-NHS),27 and Rubpy.28MW irradiation is already known to increase and accelerate reactions.29 Indeed, it is widely used in organic chemistry, and it is considered as a non-conventional method to perform organic synthesis with higher yields under milder reaction conditions.29 Furthermore, microwaves are used for several applications, such as in green chemistry,30 for solvent-free reactions,31 in fullerene chemistry,32 in polymer (and peptide) chemistry,33 in catalysis and bio-catalysis,34,35 in carbohydrates,36,37 and in medicinal chemistry.29 The resulting acceleration in terms of reaction time is likely due to both thermal and non-thermal effects of MWs that cannot be reproduced by conventional heating.33
Stupp et al. have already reported a heating approach suitable for a designed peptide amphiphile (PA) library, triggering two-dimensional plaque formation with filamentous patterns spontaneously aligning in nanofibers.38 However, their approach was limited to β-forming PA in systems (PA + solution) with carefully balanced cohesive and repulsive forces and required a strong heating procedure of the sample (e.g. 80 °C for 30 min).
On the other hand, no studies on the effect of MW energy on the mechanical properties of SAPs have been reported so far. We here developed and optimized a physical MW-based method suitable for a set of diverse SAPs to promote the over-formation of nanofibers (AFM), yielding stiffer and more resilient hydrogels (rheology), further increasing their β-structuration (ATR-FTIR and Thioflavin-T assay). This novel approach improved the stiffness of standard SAPs but was also synergic to genipin cross-linking of SAPs and allowed them to withstand multiple deformation cycles similarly to in vivo tissues. In in vitro tests with human neural stem cells (hNSCs), we also demonstrated that MW pre-treatment does not affect SAP biomimetic properties, neither pose issues of residual cytotoxicity (cell proliferation, viability, and differentiation test), laying the foundation for a general cheap and green pre-treatment suited for most SAPs. Lastly, we showed that low-power (or non-thermal) MW treatments can be applied to cells already embedded in SAP scaffolds without triggering significant cytotoxicity.
The important role of hydrogels as biomaterials
In the field of biomaterials research, hydrogels (water-swollen polymeric materials with a 3D structure)39 were the first materials designed for the human body.40,41 The tremendous potential of polymers is that they can mimic different roles of the extracellular matrix. Thus, hydrogels may be engineered to be utilized in regenerative medicine,42,43 tissue engineering,44 targeted-drug delivery,45 organ transplantation,46 and for brain imaging. However, the limitations of hydrogels are their poor mechanical properties and their elastic response to external stimuli.40 Many efforts have been made to improve the ability of these biomaterials to resist stresses and changes in the external environment. The use of cross-linkers can enhance the mechanical properties of various hydrogels, but it is often expensive, with long-lasting reactions and requires the presence of active sites (i.e. specific residues) in the polymeric sequences for selected cross-linkers to be effective. Also, even small amounts of unreacted cross-linkers may cause toxicity issues if released in vivo.Rheology, one of the optimal techniques to investigate the mechanical properties of hydrogels, is crucial for assessing the potential feasibility of a hydrogel-like biomaterial in biomedical applications.47 The storage modulus, G′, and the loss modulus, G′′, pointing to the solid-like and liquid-like behaviors of hydrogels, respectively, are here detailed to characterize different pre- and post-treated SAPs.
In this work we present an economic, green and fast enhancement of the mechanical properties of SAPs by using microwave irradiation with no chemical helper involved.
Results and discussion
Effect of MW power on SAPs
MWs are not only used in several daily applications,29 but also for food processing,48 sterilization,49 waste treatment,50 and so on. They are also capable of catalyzing chemical reactions51 and turned out to be helpful in protein unfolding and selective digestion.52 The most well-known effect of the MW electromagnetic field is ascribable to the dielectric properties of the target in which their power is dissipated, where heat is generated as a result of dipole rotation and oscillation along the applied field. It was postulated that MWs can excite torsional vibrations in protein backbones, thus altering their tertiary structure via unfolding, defragmentation, nucleation, and fibril growth.53 For this purpose, MWs were used to facilitate the formation of β-amyloid aggregates from β-lactoglobulin,53–56 or to obtain whey protein isolate nanofibrils useful for stabilizers of Pickering emulsions.53,54 Nonetheless, the overall MW thermal effect is considered to be insufficient for explaining their interactions and outcomes on biological systems,55 hence non-thermal effects of MWs have been introduced, but they are still under strong debate by the scientific community.53 Indeed, a considerable amount of mechanistic research work is still required for an appropriate interpretation of thermal and non-thermal effects of MWs. The thermal effect is due to vibrational energy that is converted into heat energy:56 this mechanism is different from conventional heating as it works mainly on the alignment of dipoles.57 On the other hand, the non-thermal effect is not related to a macroscopic temperature change, and it depends on the molecules and solvent involved.52 Indeed, MWs are more effective on polar biomolecules like water, DNA, and proteins as they force dipole oscillation and influence their alignment.52In the present work, we report a MW-based methodology to increase the β-structuration and mechanical properties of self-assembling peptides, without affecting their biomimetic properties and their suitability for subsequent genipin cross-linking reactions. In this case we suppose that the MW non-thermal effect has an impact on the aggregation of SAPs. Indeed, as described below, the most significant results were obtained with the use of low-power MW irradiation, with limited temperature increase.
Effects of MW treatments were assessed in terms of the mechanical, biomimetic and structural properties of SAPs. We found that small energy and prolonged doses of microwaves significantly improved the mechanical properties of a variety of peptide hydrogels (Table 1), representative of different classes of SAPs. LDLK12 is a non-functionalized SAP widely used in regenerative medicine applications;58,59 the functional motif of FAQ-LDLK12 was panned from a phage display against murine-NSC;58 BM3 is a BMHP1-derived SAP;60 HYDROSAP, comprising branched and linear SAPs, was developed to obtain an optimal 3D in vitro model of densely cultured hNSCs;17 CK1 belongs to co-assembling SAPs used for hetero-bifunctional cross-linking;26,27 Pal1 is an amphiphilic SAP conjugated with a palmitoyl moiety as hydrophobic tail.
| Peptide | Sequence | Net charge |
|---|---|---|
| LDLK12 | Ac-(LDLK)3-CONH2 | 0 |
| FAQ-LDLK12 | FAQRVPPGGG(LDLK)3-CONH2 | +2 |
| BM3 | Biotin-GGGAFASAKA-CONH2 | +1 |
| HYDROSAP | Ac-SSLSVNDGGG(LDLK)3-CONH2 | −1 |
| Ac-(LDLK)3-CONH2 | 0 | |
| Ac-KLPGWGGGG(LDLK)3-CONH2 | +1 | |
| [Ac-(LDLK)3G]2-KG(LDLK)3-CONH2 | 0 | |
| CK1 | Ac-CGG(LKLK)3GGC-CONH2 | +5,9 |
| Pal1 | Pal-GGGAFASAKA-CONH2 | +1 |
In order to understand this phenomenon, several MW conditions (Table 2 and Fig. 1) were investigated by varying the irradiation power, time and maximum temperature.
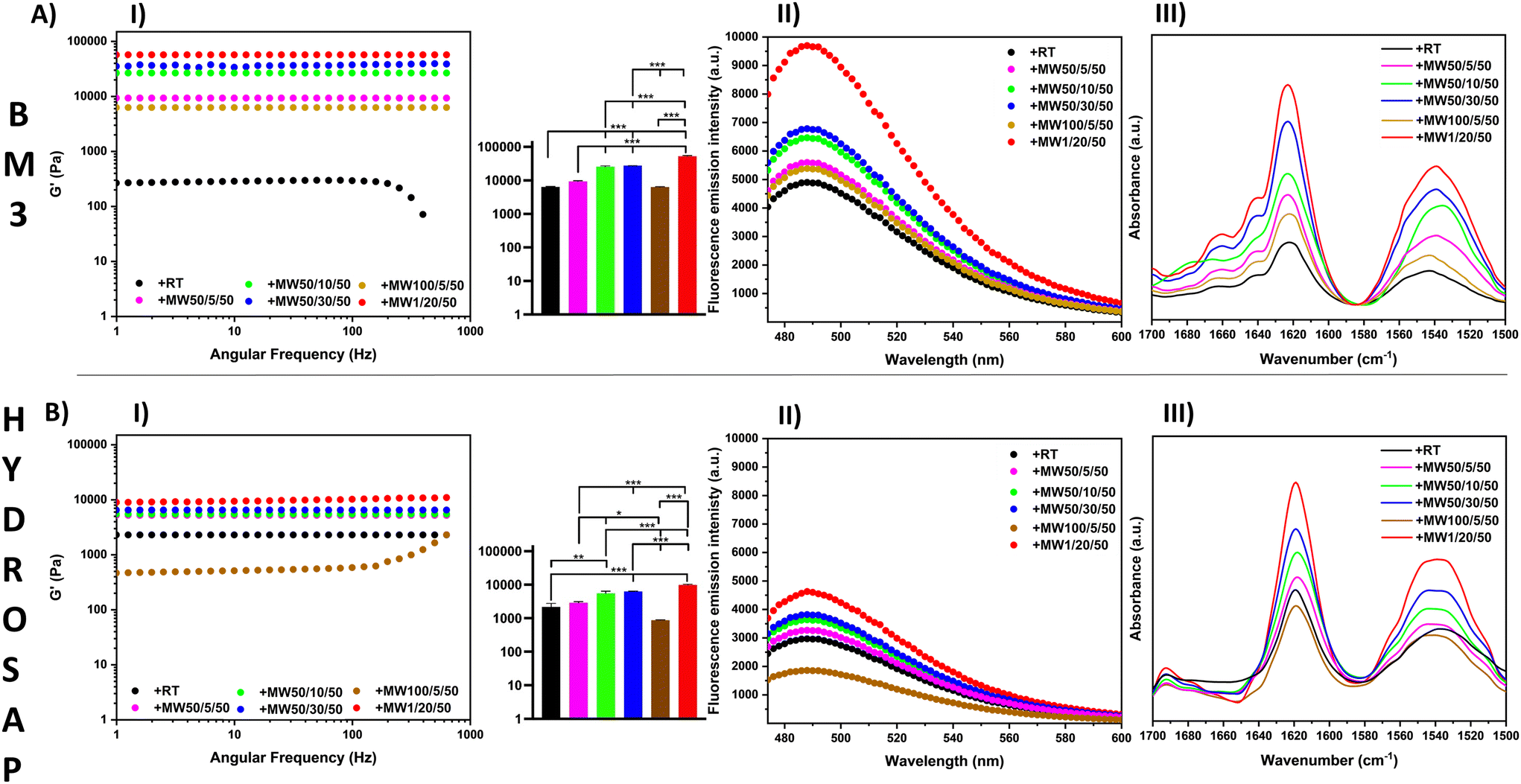 | ||
| Fig. 1 (A) BM3 and (B) HYDROSAP subjected to different MW treatments. (I) Rheology measurements of saps under different power energy conditions (frequency sweep test). G′ increased significantly along with power increments and for extended application times, but 100 W MW treatment caused a strong fall of G′ values. (II) ThT binding assay. ThT fluorescence peak intensity increased over time at 50 W, while peaked at 1 W after 20 min and fell after 100 W MW treatment. (III) In the ATR-FTIR absorption spectra, both BM3 and HYDROSAP showed typical β-sheet patterns in amides I and II, with the highest values for 1 W MW treatment after 20 min. ATR-FTIR spectra, ThT assays and rheological tests suggest a strong correlation between MW-induced β-structuration and higher scaffold stiffnesses. | ||
| Peptide | Conditions | Acronym | G′ (Pa) | G′′ (Pa) |
|---|---|---|---|---|
| BM3 | Without MW, RT | +RTcp | 6435 ± 587 | 692 ± 53 |
| BM3 | Without MW, 50 °C | hcp | 7233 ± 351 | 587 ± 98 |
| BM3 | MW, 1 W, 20′, 50 °C | +MW1/20/50 |
57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 250 ± 2975 250 ± 2975 |
3984 ± 274 |
| HYDROSAP | Without MW, RT | +RTcp | 2500 ± 705 | 140 ± 9 |
| HYDROSAP | Without MW, 50 °C | Hcp | 2603 ± 443 | 156 ± 28 |
| HYDROSAP | MW, 1 W, 20′, 50 °C | +MW1/20/50 |
10825 ± 630 |
585 ± 97 |
| LDLK12 | Without MW, RT | +RTcp | 1110 ± 130 | 105 ± 29 |
| LDLK12 | Without MW, 50 °C | hcp | 2710 ± 245 | 137 ± 36 |
| LDLK12 | MW, 1 W, 20′, 50 °C | +MW1/20/50 |
10150 ± 630 |
585 ± 97 |
| FAQ-LDLK12 | Without MW, RT | +RTcp | 442 ± 123 | 61 ± 13 |
| FAQ-LDLK12 | Without MW, 50 °C | hcp | 1132 ± 89 | 101 ± 38 |
| FAQ-LDLK12 | MW, 1 W, 20′, 50 °C | +MW1/20/50 | 3154 ± 225 | 159 ± 35 |
| CK1 | Without MW, RT | +RTcp | 9800 ± 354 | 618 ± 78 |
| CK1 | Without MW, 50 °C | hcp | 9845 ± 376 | 741 ± 83 |
| CK1 | MW, 1 W, 20′, 50 °C | +MW1/20/50 |
19440 ± 225 |
2399 ± 271 |
| Pal1 | Without MW, RT | +RTcp | 112 ± 21 | 34 ± 7 |
| Pal1 | Without MW, 50 °C | hcp | 285 ± 29 | 48 ± 12 |
| Pal1 | MW, 1 W, 20′, 50 °C | +MW1/20/50 | 1092 ± 87 | 96 ± 34 |
In rheology experiments the SAP sol-to-gel transition, revealing the occurrence of self-assembly, was triggered by the gradual addition of DPBS. We measured both the storage (G′) and loss (G′′) moduli, and the maximum strain % at failure, important rheological parameters to assess the mechanical properties of soft materials.61
A preliminary set of experiments was run to verify the effect of the standard overnight incubation (OI) at +4 °C after SAP dissolution in water, allowing for the initial spontaneous formation of loosely interacting nanofibers.62 As previously demonstrated,29 shortening the incubation time from the standard OI to a few mins was detrimental to the overall mechanical performance of SAPs (Table S3†). As shown in Table 2 and in Table S1,† we studied the effect of MW irradiation on self-assembling peptides with diverse backbones, β-structuration and functional motifs: BM3, HYDROSAP, LDLK12, FAQ-LDLK12, CK1 and Pal1. First, we MW treated them for 5 minutes at 50 W, and at a max. temperature of 50 °C. In all cases both G′ and G′′ (with G′ ≫ G′′) were higher in MW-treated samples. MW treatment was, first, compared with a simple heating treatment (heating, 50 °C, Table 2), demonstrating the poor efficiency of the latter on improving SAP mechanical performances.
Inspired by these data, we varied the power, time and temperature of MW treatments for BM3 and HYDROSAP in order to assess potential improvements or limits of our approach (Table 2, Fig. 1, AI and BI). By extending the duration of MW irradiation from 5 minutes to 10 or 30 minutes, we detected a conjoint increase of G′ and G′′. In contrast, power increments from 50 W to 100 W were detrimental to the overall scaffold stiffness. For example, 100 W MW treatment for 5 minutes caused a decrease in G′ possibly due to SAP degradation. Indeed, G′ differences were statistically significant for all conditions without MW treatment (+RT), except when 100 W MW treatment was applied.
Vice versa, 1 W MW irradiation for 20 min caused an impressive improvement of both storage and loss moduli.
For BM3, we detected a 9-fold increase of G′ when compared to untreated BM3 (57.250 Pa vs. 6.435 Pa), while the G′ value of HYDROSAP was ∼4 times higher than its untreated counterpart (10.825 Pa vs. 2.500 Pa). Rheological tests of MW-treated LDLK12, FAQ-LDLK12 and CK1 showed similar results (Fig. S1A–S3A†) and, as such, increase the range of applicability of MW treatments to a set of diverse SAPs.
Additionally, viscosities of all tested SAPs were measured prior gelation with DPBS and are reported in Fig. S1B–S4B, S5 and S6† to compare the degree of reticulation of these materials before and after MW treatments.63 All MW-treated samples demonstrated increased viscosity with respect to the untreated ones, suggesting the presence of somehow augmented nanofiber aggregation.
Since MW irradiation can exert both thermal and non-thermal effects, they may contribute to this phenomenon, i.e., both irradiation energy converted into thermal energy and direct interaction of the MW electric field with the SAP dissolved in a polar aqueous solution may have played a role.64
However, by demonstrating that main benefits come from low-power MW treatments we consider the non-thermal effect of MWs as most likely the main factor of such MW-facilitated self-assembly. Indeed, the contribution of a non-thermal effect was corroborated by the observation of a small variation of the sample temperature (<50 °C throughout all treatment) for 20 min application of 1 W MWs (temperature was continuously monitored using a fiber optic probe).
For this purpose, Belyaev reported how non-thermal effects are needed to justify the complex effects of MWs on biological systems.65
It was also suggested that MW treatment favors a higher stabilization of β-sheet conformation and a reduction of unordered structures in β-amyloid fibrils.66 Lee et al. demonstrated that MWs induced the formation of oligomeric amyloid aggregates, showing a significant increase in amyloid aggregation levels after MW irradiation compared to conventional heating.67 Recently, a discussion was presented by Ford and Kloxin regarding the use of MW heating in the synthesis of SAPs, in the disruption of peptide aggregation, in the formation of but β-aggregates, and in overcoming the steric hindrance.68
Here, ThT binding assay and ATR-FTIR experiments were performed to assess the effect of MWs on SAP secondary structures.
ThT is a dye used to detect β-rich structures,68 since it increases fluorescence upon interacting with β-sheet-rich peptides and proteins, its fluorescence intensity is, therefore, indicative of samples with β-sheet content.69 ThT assay showed β-structuration in all samples (amyloid-binding emission peak at 490 nm), but the fluorescence intensity increased after MW treatments by varying the time and power energy, reaching the highest peaks for 20 min treatment at 1 W in both BM3 and HYDROSAP (Fig. 1, AII and BII). In accordance with rheology tests, 50 W MW treatment increased their effect over time (at 5, 10 and 30 min), while increasing the MW power from 50 W to 100 W was, instead, detrimental to SAP structuration.
A major presence of β-rich components was also found in ThT fluorescence curves of LDLK12, FAQ-LDLK12 and CK1 (Fig. S1C–S3C†) after MW treatments; all curves showed the highest values for 1 W MW treatment after 20 min.
All ATR-FTIR spectra showed typical peaks in amide I (1616 cm−1, 1623 cm−1) and in amide II (1532 cm−1) regions (Fig. 1, AIII and BIII), with greater intensities in MW-treated SAPs (+MW1/20/50 > +MW50/30/50 > +MW50/10/50 > +MW50/5/50 > +MW100/5/50 > +RT), along with ThT findings, thus confirming a high sensitivity of secondary structure vibrational modes to MW treatments (Fig. S12†).
Being such absorptions mainly indicative of β-sheets interaction in peptides,70,71 we performed FT-IR absorption spectral deconvolution (after preliminary second derivative analysis) to resolve the partially overlapped secondary structure bands contributing to amide I and amide II peaks (Fig. S7†). In all cases, area quantification (shown in Table S4†) suggested an increment of β-sheet components (∼15–20%) after MW irradiation with respect to the untreated samples.
Overall, the results were in accordance with the ThT assay data and showed the highest presence of β-sheets after 20 minutes of 1 W MW irradiation, while whenever the MW power was increased, it caused the intensities of amides I and II to fall.
From AFM analysis we detected morphological changes in MW-treated samples compared to untreated SAPs, in terms of fiber height and width (Fig. 2). Indeed, after MW treatment,72 a higher level of clustering of multiple nanofibers was favored, similarly to what is observed after the chemical cross-linking reactions of SAPs.27–29 In BM3, Fig. 2A, +RT nanofiber populations showed average width values of 22.3 ± 0.92 nm and, in the case of more entangled fibers, 39.1 ± 1.60, while the average height value was 2.7 ± 0.37 nm. Instead, BM3 +MW1/20/50 exhibited visible fasciculations of nanofibers with increased values of height and width; the average height value was 2.85 ± 0.37 nm, and width values showed four different distributions (peaks at 28.0 ± 0.92 nm, 43.1 ± 1.23 nm, 55.9 ± 1.74 nm and 86.7 ± 1.18 nm). On the other hand, HYDROSAP (Fig. 2B) +RT featured short nanofibers, as already reported,18 displaying average width and height values of 13.84 ± 1.08 nm and 1.9 ± 0.29 nm, respectively. HYDROSAP +MW1/20/50 showed an average fiber width of 21.15 ± 0.20 nm while the height average was 2.23 ± 0.23 nm. Statistical significance was achieved by comparing fiber width distribution in both BM3 and HYDROSAP (+RT vs. +MW1/20/50), with observed widths approximately ∼50–60% larger than those of MW-treated samples.
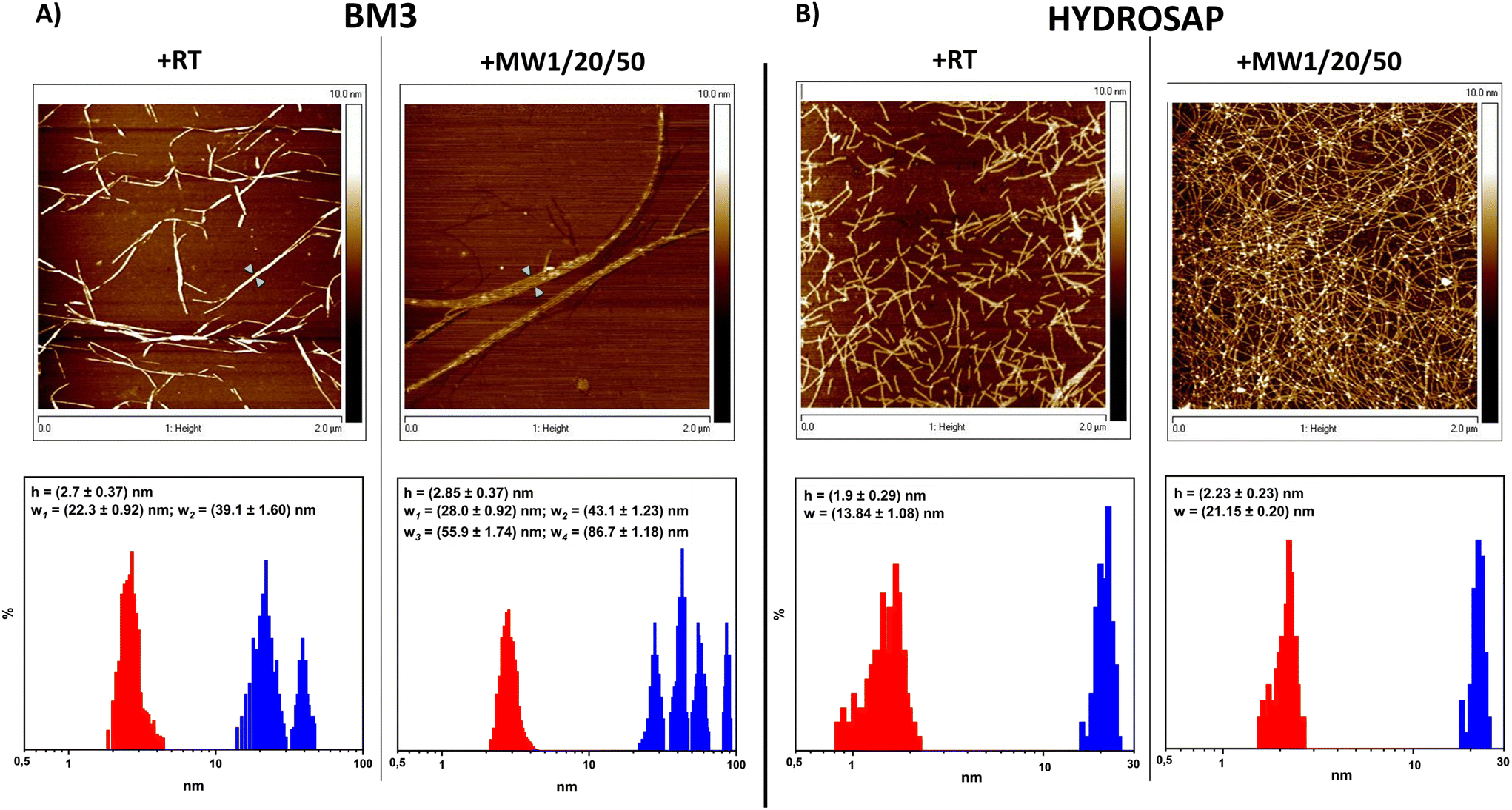 | ||
| Fig. 2 AFM analysis of BM3 (A) and HYDROSAP (B) with and without MW treatment; height (red) and width (blue) distributions are shown. In general, MW treatment increased the interactions among nanofibers, yielding higher and larger fiber bundles. In BM3, green arrowheads point to where the largest width profiles were measured. | ||
AFM images underlined the over-structuring of self-assembling fibers, resulting in large populations of entangled nanofibrous structures after MW treatment (Fig. 2). Morphological analysis revealed that MW irradiation does lead to the formation of a dense network of fasciculated fibers.
All these data suggest a significant contribution to stronger fiber fasciculation for MW-treated SAPs. In the case of 50 W and 100 W MW treatments we detected fiber fasciculation and defragmentation, respectively, confirming the detrimental effect of high-power MW treatment on the overall SAP structural stability (Fig. S9 and S12†) and the presumably major role of the non-thermal effect of MW treatments on their mechanical and β-structuration increments.
Lastly, 1 mL of SAP solutions were treated with the same protocol (1W/20/50) and yielded to comparable rheological values (Fig. S10†), demonstrating the scalability of the low power MW irradiation protocol to improve SAP performances for biomedical applications.
MW treatments and GP cross-linking on SAPs
The important need for novel biomimetic materials customized for each specific regenerative therapy requires SAPs featuring both functional moieties and mechanical properties tuned for each target tissue to be regenerated.73,74 In this regard, stress tolerance (i.e. resilience) and breaking point of these scaffolds have been among the major limitations of these biomaterials.We previously reported a covalent cross-linking approach using GP, a natural cross-linker with low cytotoxicity against hNSC.28 Since GP confers promising high G′ values on various SAP hydrogels,27 we investigated the effect of GP on SAPs pre-treated with the previously optimized MW irradiation protocol (i.e. 1 W, 20 min, 50 °C) (Table 3, Table S2,† and Fig. 3).
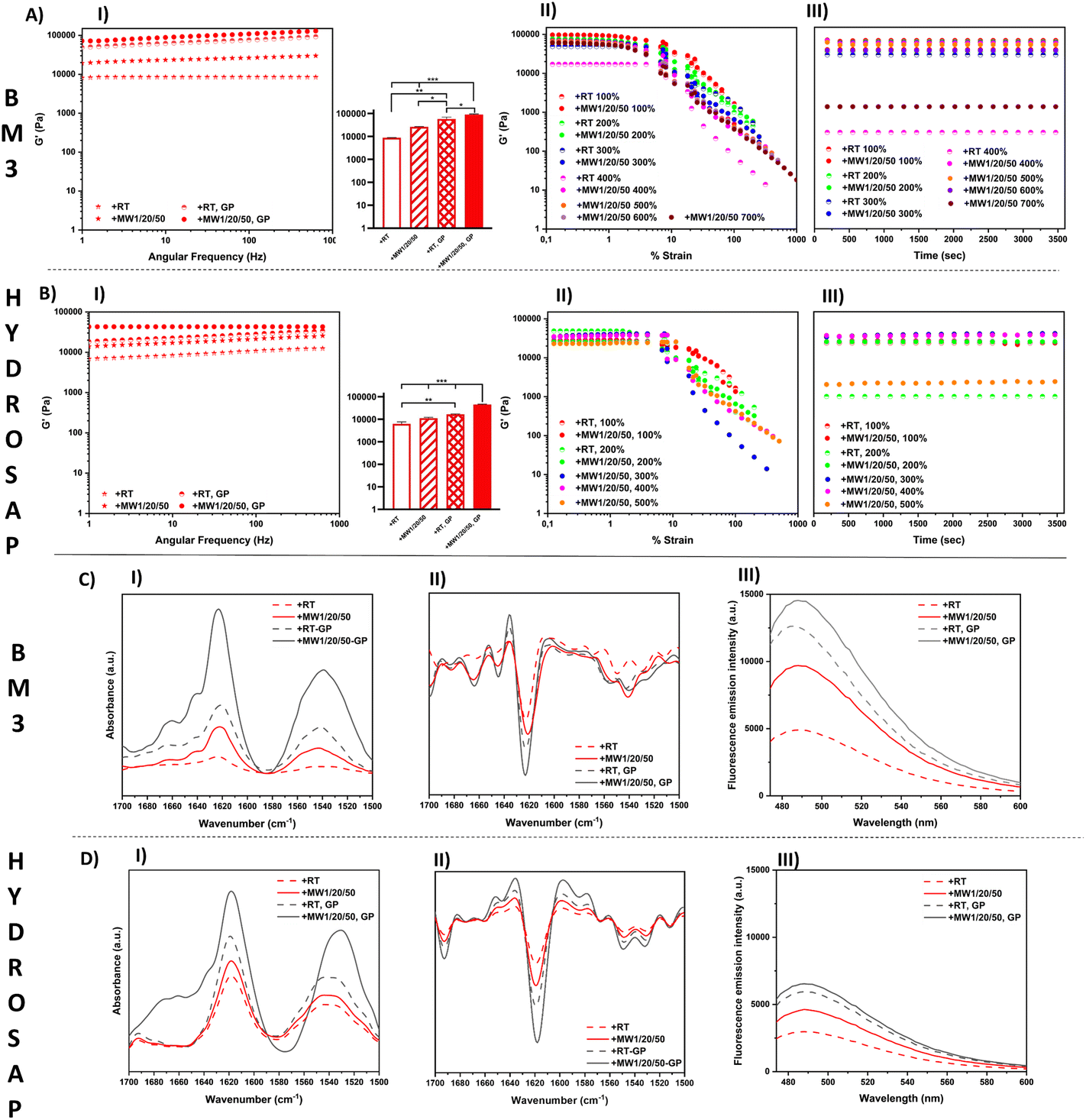 | ||
| Fig. 3 GP cross-linking of MW-treated BM3 and HYDROSAP. Rheological investigation of the flow/deformation properties of (A) BM3 and (B) HYDROSAP after MW treatment and GP cross-linking (parallel-plate geometry). (AI and BI) Frequency sweep tests: significant G′ increments were achieved for all treated vs. untreated saps (+RT). (AII and BII) Strain sweep tests of MW-treated BM3 and HYDROSAP after GP cross-linking. Tests were initially performed up to a max. strain of 100%, followed by time sweeps and subsequent strain sweeps at higher max. % strains until the biomaterial was broken without recovery. For +MW1/20/50 BM3 and +MW1/20/50 HYDROSAP cross-linked with GP failure was obtained at 600%–700% and 500%–600% strain, respectively. (AIII and BIII) Time-sweep tests for BM3 and HYDROSAP to assess failure/recovery after each strain sweep. Secondary structure assessment on MW-treated (C) BM3 and (D) HYDROSAP after GP cross-linking. (CI and DI) ATR-FTIR absorption spectra in the regions of amides I and II, and their second derivatives (CII and DII) used to identify unresolved peaks. (CIII and DIII) ThT assays for the evaluation of β-structuration. | ||
| Peptide | GP | Conditions | Acronym | G′ (Pa) | G′′ (Pa) | Max. strain (%) |
|---|---|---|---|---|---|---|
| BM3 | ✓ | Without MW, RT | +RTpp | 81![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 130 ± 3315 130 ± 3315 |
6359 ± 410 | 400–500 |
| BM3 | ✓ | MW, 1 W, 20′, 50 °C | +MW1/20/50pp |
96680 ± 3825 |
11110 ± 1389 |
600–700 |
| HYDROSAP | ✓ | Without MW, RT | +RTpp | 23984 ± 725 |
1796 ± 118 | 200–300 |
| HYDROSAP | ✓ | MW, 1 W, 20′, 50 °C | +MW1/20/50pp |
48000 ± 2150 |
5640 ± 358 | 500–600 |
| LDLK12 | ✓ | Without MW, RT | +RTpp | 19590 ± 334 |
2117 ± 113 | 100–200 |
| LDLK12 | ✓ | MW, 1 W, 20′, 50 °C | +MW1/20/50pp |
38160 ± 1560 |
4237 ± 237 | 200–300 |
| FAQ-LDLK12 | ✓ | Without MW, RT | +RTpp | 11640 ± 422 |
1428 ± 71 | 200–300 |
| FAQ-LDLK12 | ✓ | MW, 1 W, 20′, 50 °C | +MW1/20/50pp |
48860 ± 2345 |
4740 ± 280 | 400–500 |
| CK1 | ✓ | Without MW, RT | +RTpp | 96000 ± 2861 |
10530 ± 739 |
600–700 |
| CK1 | ✓ | MW, 1 W, 20′, 50 °C | +MW1/20/50pp |
234000 ± 4512 |
26131 ± 909 |
>1000 |
| Pal1 | ✓ | Without MW, RT | +RTpp | 767 ± 52 | 111 ± 23 | 100–200 |
| Pal1 | ✓ | MW, 1 W, 20′, 50 °C | +MW1/20/50pp | 11830 ± 314 |
516 ± 72 | 200–300 |
In other words, +MW1/20/50 SAP scaffolds were incubated with a cross-linking solution of GP, yielding to MW scaffolds cross-linked with GP (Fig. 3, AI and BI).
Since the GP diffusion protocol suitable for the cone-plate geometry setup requires considerably longer time sweeps for highly viscous MW-treated SAPs, for the sake of simplicity we preformed the MW-treated GP-crosslinked scaffolds prior to rheological measurements and used a parallel-plate geometry for their characterization (see ESI Methods for details†).75,76
First, we confirmed similar G′ and G′′ increments in MW-treated SAPs and +MW1/20/50 as the best MW irradiation protocol to achieve the highest mechanical properties (Table 3).
Interestingly GP cross-linking of MW-treated SAPs provided an additional boost to G′ and G′′ increments, significantly higher than standard GP-crosslinked SAPs (Fig. 3, AI and BI; Table 3).
+MW1/20/50 BM3 after cross-linking showed an average G′ of 96680 Pa while that without MW-treatment BM3-GP reached 81130 Pa (BM3, +RT-GP). The G′ of +MW1/20/50 HYDROSAP cross-linked with GP was 48000 Pa while its untreated counterpart scored a G′ value of 23984 Pa after GP cross-linking. Values of other tested SAPs are reported in Table 3.
We also performed consecutive strain sweep tests with incremental maximum strains in order to assess both the strain-dependent response and the maximum strain % before failure of each tested SAP after MW treatment and GP cross-linking (Fig. 3, AII and BII; Table 3).
Strain sweeps were stopped at maximum strains of 100%, 200%, 300% and so on, until material failure occurred. Each strain sweep was followed by 1 hour time sweep (Fig. 3, AIII and BIII) to track the SAP recovery at 1% strain. As shown in Fig. 3, AII and BII, all the samples provided a linear viscoelastic response up to approximately 10% of strain, but untreated SAPs usually showed a 50%–200% strain failure range (Table 3). After MW treatment BM3 cross-linked with GP showed a max. % strain range of 600%–700%, while for HYDROSAP it reached 500%–600%. Fig. 3, AIII and BIII, show the time sweep tests performed right after each strain sweep experiment, respectively for BM3 and HYDROSAP. Below the detected max. % strain threshold, G′ and G′′ went back to values similar to those found in strain sweep tests, and they were steady since the beginning of the time sweeps, thus suggesting a fast elastic response of the samples.
GP cross-linking increased G′, G′′ and max. % strain, but MW treatment was also effective alone and, most importantly, further boosted the GP effect and they together yielded values of crucial importance for tissue engineering applications.76–78 Notably, MW1W/20/50 CK1 cross-linked with GP showed higher max. % strain values that went beyond our instrumental capabilities (>1000%) (Table 3 and Table S4†).
The effect of GP cross-linking on MW-treated SAPs was also assessed with ATR-FTIR analysis and ThT binding assays (Fig. 3C and D). ATR-FTIR spectra showed typical peaks in the amide I and amide II regions. The presence of β-sheet vibrational modes was confirmed in amide I (1616–1623 cm−1 range) and amide II (∼1539 cm−1) bands. The ATR-FTIR spectra of GP cross-linked samples were very similar to those of non-cross-linked ones in terms of peak positions. On the other hand, the ATR-FTIR spectra of MW-treated GP cross-linked BM3 and HYDROSAP are dominated by the more intense bands of amides I and II and, in the case of HYDROSAP, a small shift of the amide II peak (Fig. 3, CI and DI). After the second derivative analysis (Fig. 3, CII and DII) and respective spectral deconvolutions we found the highest percentages of β-sheets in MW-treated GP cross-linked samples (Table S4†). As previously reported,27,28 GP crosslinking increased SAP β-structuration, and this was the case of MW-treated SAP as well.
Similarly, in ThT assays the highest fluorescence intensities were found in MW-treated GP cross-linked scaffolds (Fig. 3, CIII and DIII), further confirming that GP crosslinking and MW treatments synergically contribute to β-sheet formation in SAPs. GP cross-linking was already proved to be effective on SAP sequences featuring Lys in their self-assembling backbones, suggesting the need for a stable close proximity of primary amines.23,24 By enhancing β-structuration and nanofiber fasciculation per se, MW treatments very likely increase the overall concentration of such potential steady binding sites and, as such, favor a more efficient scaffold cross-linking, with superior elastic deformability. Indeed, the FTIR and ThT data also suggests that chemical GP cross-linking alone is more effective in “fastening” β-sheets than physical plain MW treatments. Overall, the rheological, FT-IR and ThT results indicate that MW irradiation can be a valid pre-treatment to improve cross-linking reactions in SAPs, as also demonstrated by the lowest percentages of free amines detected with TNBSA assay (Table S5 and ESI Method†) after GP cross-linking of MW pre-treated SAPs.
In vitro tests
Conventional 2D cell cultures offer a quick and easy opportunity to determine the biocompatibility of new biomaterials and/or advanced treatments in terms of cell proliferation, viability, and differentiation for disparate applications in biology, medicine, tissue engineering and so on.79 It is well known that substrate stiffness can influence the fate decisions of various stem cells.80,81 In our previous works, we demonstrated the negligible cytotoxicity of GP17 and Sulfo-SMCC26,27 cross-linkings of SAPs, as well as their limited effect on hNSC proliferation and differentiation. hNSCs are a well-standardized biological source for the in vitro screening of biomaterials in nervous tissue engineering and are also very sensitive to toxic compounds. Similarly, we investigated if MW treatments could affect hNSC behavior in term of cell proliferation, viability, and differentiation.hNSCs were seeded on the top surface of BM3 (Fig. 4A) and HYDROSAP (Fig. 4B) under different conditions (+RT; +MW1/20/50; GP, +RT; GP, +MW1/20/50) and cultured for 7 days in vitro. Cultrex-coated and untreated-bottom well surfaces were used as positive and negative controls, respectively. Cell proliferation assay was evaluated via MTS assay. Seeded and differentiated hNSCs on BM3 (Fig. 4, AI) and HYDROSAP (Fig. 4, BI) showed similar amounts of total cell population as demonstrated by statistical analysis. All samples resulted in comparable to gold-standard Cultrex (0.64 ± 0.04 a.u. (arbitrary units)), except for BM3 + RT (0.44 ± 0.02 a.u.), and significantly higher than untreated glass (0.19 ± 0.04 a.u.). As shown in Fig. 4, AII and BII, Fig. 5 and Fig. S13,† a similar trend was obtained in cell viability assays, where high percentages of viable cells were detected in all tested samples. Cultrex, with 96.97 ± 0.59% of live cells, did not show significant differences with respect to BM3 and HYDROSAP for all treatments, while hNSCs differentiated on untreated wells exhibited the lowest values (87.82 ± 1.83%) of live cells. In other words, MW treatments did not significantly alter the viability of hNSCs with respect to their untreated counterparts (Fig. 4, AII and BII). hNSC differentiation over the analyzed samples was evaluated by staining βIII-tubulin (neurons), GFAP (astrocytes) and GALC/O4 (oligodendrocytes) markers for 7 days in vitro (Fig. 4, AIII and AIV; Fig. 4, BIII and BIV). MW treatment did not affect the percentage of neurons, showing 19.22 ± 2.22% (+RT), 17.35 ± 1.34% (+MW1/20/50), 17.06 ± 1.39% (GP, +RT) and 15.37 ± 1.07% (GP, MW1/20/50) in BM3 (Fig. 5, AIII) and 15.61 ± 1.54% (+RT), 13.50 ± 1.33% (+MW1/20/50), 15.57 ± 0.97% (GP, +RT) and 18.76 ± 1.56% (GP, MW1/20/50) in HYDROSAP. On the other hand, astrocytes progeny resulted in more sensitive to stiffness increments conferred by GP (significances detected between GP cross-linked and untreated samples). Nonetheless, there was no statistical evidence of significant differences between MW-treated samples and their untreated counterparts. Percentages of astrocytes detected on BM3 MW-treated samples were 27.34 ± 1.52% for +MW1/20/50 and 37.21 ± 1.66% for GP + MW1/20/50 while 33.02 ± 3.74% and 45.02 ± 2.31% were detected for +RT and GP + RT, respectively. Similarly, the astrocyte population differentiated on HYDROSAP was not affected by MW irradiation either: values ranged from 39.71 ± 2.13% for +RT and 39.75 ± 3.19% for +MW1/20/50 to 29.51 ± 4.18% for GP + RT and 35.69 ± 2.74% for GP + MW1/20/50. Like βIII-Tubulin, a comparison between all conditions in BM3 and HYDROSAP samples revealed a statistically similar percentage of oligodendrocytes (all results are comparable to 11.94 ± 0.88% of Cultrex). Lastly, qualitative images of differentiated hNSC progeny (Fig. 4, AIV and BIV) suggest how increased stiffness of the MW-treated samples may modulate the cell morphology by affecting cell anchoring and adhesion.82 Cells became more spread and more adhesive on stiffer MW-treated substrates and this peculiar feature was more noticeable with HYDROSAP (Fig. 4, BIV). In general, microwave treatment (and GP cross-linking) ameliorate cell sprouting and cell branching.
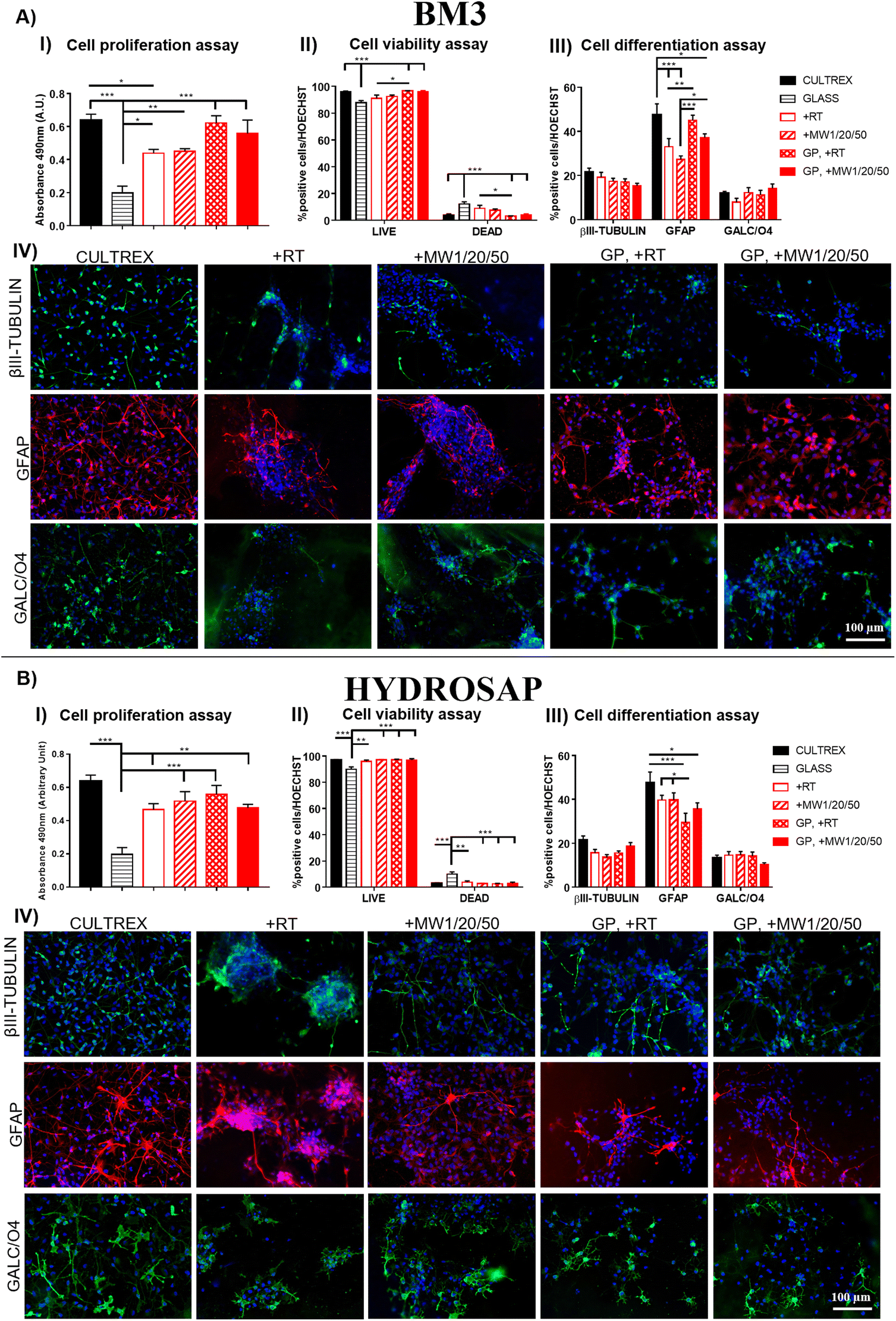 | ||
| Fig. 4 Proliferation, viability and differentiation assays of hNSCs seeded on BM3 (A) and HYDROSAP (B) for 7 days in vitro. (I) MTS assay for the evaluation of proliferation rate. Absorbance values were detected at 490 nm (a.u., arbitrary units). (II) Cell viability assay was performed with a Live/Dead kit. (III) Quantification of positive cells for βIII-tubulin (neurons), GFAP (astrocytes) and GALC/O4 (oligodendrocytes). (IV) Representative fluorescence images show neural differentiation of neurons stained with βIII-tubulin marker in green, astrocytes with GFAP marker in red, and oligodendrocytes with GALC/O4 marker in green. Cell nuclei were stained with Hoechst (in blue). Cultrex and glass (untreated-bottom well surface) were used as the positive and negative controls, respectively. In all tested samples MW treatments did not significantly alter the proliferation, viability and differentiation of seeded cells. Data are represented as the mean ± SEM. Statistical analysis shows significant differences between different groups (*p < 0.05; **p < 0.01; ***p < 0.001). All measures were performed in triplicate. Scale bar, 100 μm. | ||
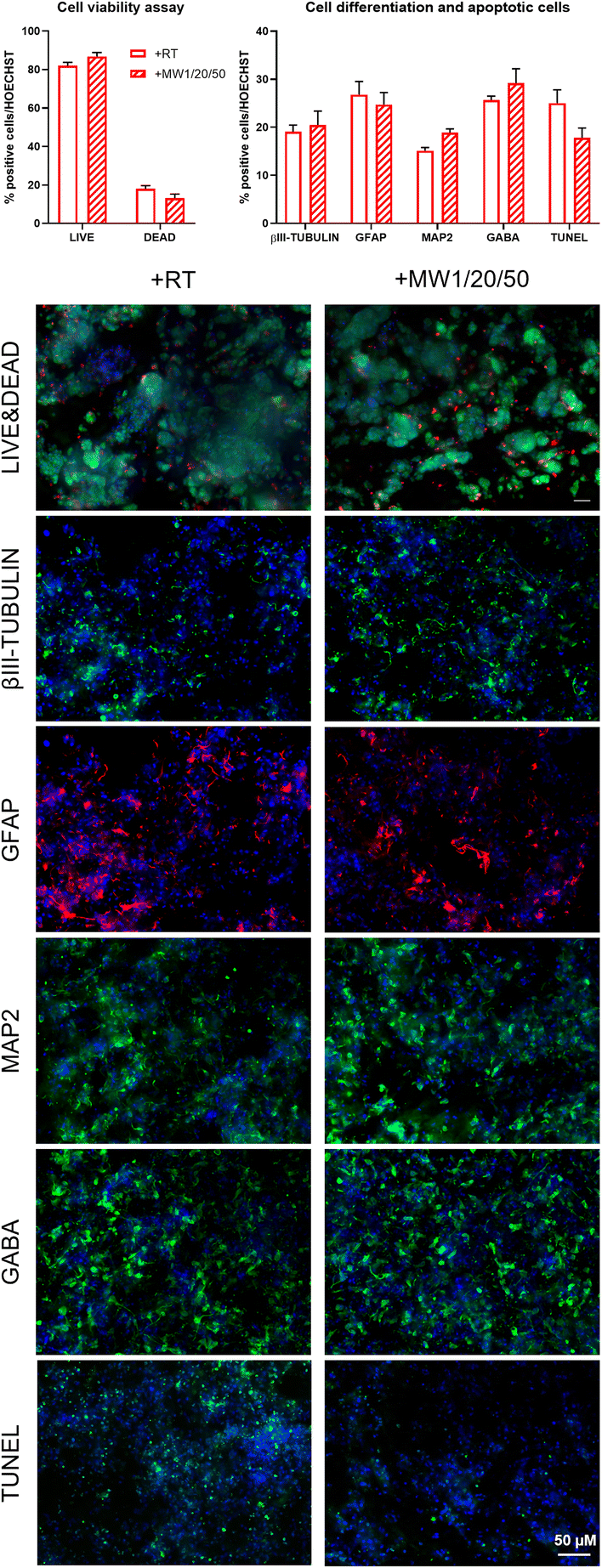 | ||
| Fig. 5 Viability and differentiation assays on the 3d constructs of hNSCs embedded in HYDROSAP hydrogels after low power MW treatment (+MW1/20/50) and in the control group (+RT). Quantification of live and dead cells after viability assay performed at t0 (1 hour after sap gelation) with the Live/Dead kit shows high cell viability in both +RT and +MW1/20/50, without significant differences between the control and treated groups. Quantification of positive cells for βIII-tubulin (immature neurons), GFAP (astrocytes), MAP2 (mature neurons), GABA (GABAergic neurons) and TUNEL (apoptotic cells) after 7 days of hNSC differentiation confirmed that low power MW treatment did not affect cell differentiation and maturation. Representative fluorescence images of hNSCs embedded in HYDROSAP in the +RT and +MW1/20/50 groups for Live/Dead assay with live cells stained in green and dead cells stained in red, immature neurons stained with βIII-tubulin marker in green, astrocytes with GFAP marker in red, mature neurons with MAP2 marker in green, GABAergic neurons with GABA marker in green and apoptotic cells with TUNEL assay kit in green. Cells nuclei are stained with Hoechst in blue. Scale bar 50 μm. Data are represented as the mean ± SEM. Graphs show no significant differences using two-way ANOVA with the Bonferroni correction between the +RT and +MW1/20/50 groups. | ||
Since MW treatment does not significantly affect hNSC behavior in terms of proliferation, viability and differentiation, our data demonstrate that low-power MW irradiation does not affect the biomimetic properties of the tested SAPs, and, as such, may be a valid technique for already developed SAPs in regenerative medicine applications and others to come in the future.
Lastly, we assessed the feasibility of the optimized low-power MW treatment for hNSCs encapsulated inside HYDROSAP hydrogels. It is well established that direct MW treatments on different type of cells induce cell apoptosis,83–85 but low-power MW treatments may be tolerable for encapsulated cells. HYDROSAP gels seeded with hNSCs were treated with low-power MW irradiation (1 W) for 20 minutes at the monitored temperature (<37 °C). In this case, Live/Dead and TUNEL assays of MW-treated samples showed similar results to control samples and, as such, negligible cytotoxicity effects (Fig. 5). Similarly, hNSC differentiation was not significantly influenced by low-power MW irradiation, both in terms of the percentages and morphology of neuronal cells (βIII-Tubulin), their maturation (MAP2, GABA), and astrocytes (GFAP) (Fig. 5). Therefore, low-power MW treatments can be considered as a feasible option for 3D stem cell cultures of SAPs as well.
Conclusion
In this work, we introduced, characterized and optimized a MW-based protocol aimed at boosting the mechanical properties of peptide scaffolds, and we demonstrated its feasibility for a wide range of diverse SAP sequences.Inspired by the urgent demand for new high-performing biomimetic biomaterials, we developed a facile method lending SAP hydrogels significantly higher stiffness, resilience and increased β-structuration, likely thanks to their non-thermal MW effects. We have described an accessible, fast, cheap and green approach based on the irradiation of microwaves, which can be taken into account in several biomedical applications. Although the mechanism involved is not completely clear, MW treatments improved β-structuration, nanofiber fasciculation and supramolecular stability of the assembled SAPs; in turn, the mechanical properties of SAPs were improved without the use of enzymatic or chemical cross-linking. On the other hand, when used as pre-treatments, mild MWs could actually help in subsequent SAP cross-linking reactions as well by stabilizing the close proximity of “reactive sites” and increasing the concentration of newly formed chemical bonds. By demonstrating that low-power MW pre-treatments do not affect SAP biomimetic properties, nor add any cytotoxicity during and after treatments, we paved the way for a standard methodology to be widely used in regenerative medicine applications with SAP biomaterials and probably with proteinaceous scaffolds as well.
Experimental
Peptide synthesis and purification
Reagents for peptide synthesis were purchased from Merck (Merck Millipore, Darmstadt, Germany). CEM (Matthews, NC, USA) was the vendor for all Fmoc-protected amino acids and Rink amide resin. Solvents for chromatography (VWR, Radnor, PA, USA) were all technical grade. A Liberty Blue system (CEM Corp., Matthews, NC, Canada) with solid-phase Fmoc chemistry was used to synthesize all peptides.59 The first amino acid was loaded on Rink amide resin (0.19–0.56 mmol g−1, 100–200 mesh) and peptides were cleaved from the resin with a freshly prepared cocktail mixture of 92.5% TFA, 2.5% H2O, 2.5% DODt, and 2.5% TIS. All syntheses were carried out at the 0.25 mmol scale in excess of a 0.2 M amino acid solution (in DMF) and in the presence of 1 M DIC (in DMF) and 1 M Oxyma (in DMF). A 10% v/v of piperazine in 9:1 NMP/EtOH was used as the deprotection solution for the removal of Fmoc-protecting groups. N-terminal acetylation (for LDLK12, CK1, and pure-HYDROSAP components) was performed using 20% v/v Ac2O in DMF solution. BM3 was capped with biotin at the N-terminal group. The crude peptides were purified via semi-preparative Waters binary RP-HPLC (>96%) using a C18 Restek™ column. The mobile phase consisted of a gradient over 30 minutes of 75% H2O with 0.1% TFA and 25% CH3CN with 0.1% TFA (0 min) until 75% CH3CN with 0.1% TFA (20 min). The pure fractions were reunited and lyophilized (Labconco, Kansas City, MO, USA). To confirm purity and peptide identification, mass spectra were recorded on a LC-MS via single quadrupole mass detection (Waters LC-MS Alliance 3100) using a nebulizing nitrogen gas at 800 L min−1 and a temperature of 150 °C, 10 mL min−1 cone flow, 3.11 kvolts capillary and 52 V cone voltage.
Peptide selection
We selected a set of diverse SAPs featuring diverse self-assembly backbones to test the effect of MW irradiation on different classes of SAPs. For this purpose, the tested SAPs are reported in Table 1 with their respective net charge. LDLK12 is a non-functionalized SAP widely used in regenerative medicine applications;58 the functional motif of FAQ-LDLK12 was panned from a phage display against murine neural stem cells;58 BM3 is a BMHP1-derived SAP,60 linked with biotin at the N-terminal group; HYDROSAP was developed to obtain a 3D in vitro model of densely cultured hNSCs;17 CK1 belongs to co-assembling SAPs used for hetero-bifunctional cross-linking,26,27 and a representative example of cationic peptides. As previously reported,17 the purified linear sequences Ac-(Leu-Asp-Leu-Lys)3-CONH2, Ac-Lys-Leu-Pro-Gly-Trp-Gly-Gly-Gly-Gly-(Leu-Asp-Leu-Lys)3-CONH2, and Ac-Ser-Ser-Leu-Ser-Val-Asn-Asp-Gly-Gly-Gly(Leu-Asp-Leu-Lys)3-CONH2 and branched tris(Leu-Asp-Leu-Lys)3-CONH2 were combined and dissolved in distilled H2O to obtain the so-called pure HYDROSAP. Pal1 is a novel fatty acid-conjugated SAP, formed by linking with palmitic acid and a BMHP1-dreived SAP, a representative example of peptide amphiphiles.86Microwave irradiation protocol
All purified selected SAPs were subjected to MW irradiation using a Discover single-mode microwave reactor (CEM, Matthews, NC) that provides self-tuning with a continuous power delivery in the range of 0–300 Watts (W). The temperature is continuously monitored using a fiber optic probe. All experiments were conducted by selecting MW power and keeping the temperature below a selected threshold value over time. SAPs were initially dissolved in a stock solution of distilled H2O at appropriate concentrations (1% w/v LDLK12, 2% w/v FAQ-LDLK12, 2% w/v BM3, 1% w/v HYDROSAP and 5% w/v CK1) and incubated overnight at +4 °C prior to usage18,29 unless specifically stated (ESI†). The selected SAP stock solution was divided in vials (100 μL each) and treated for 1, 2, 5, 10, 20, or 30 minutes with microwave irradiation at RT, 37 °C and 50 °C max. temperature. Untreated and MW-irradiated samples were used for the subsequent experiments and characterization.Mechanical characterization
The mechanical behavior and viscoelastic properties were investigated using a rotational AR-2000ex rheometer (TA Instruments, Waters Corp., Milford, CT, USA). For cone-plate measurements, we used an acrylic cone-plate geometry with a diameter of 20 mm, a truncation gap of 34 μm and 1% angle to evaluate the storage (G′) and loss moduli (G′′) in a linear regime. Time sweep experiments were carried out for 3 hours at a constant angular frequency (ω = 1 Hz) in the presence of DPBS (1×). Subsequently, a frequency sweep test (0.1–1000 Hz) was performed as a function of angular frequency at a constant strain of 1%.Viscosity measurements were performed using a cone-plate geometry via a continuous shear ramp. Curves were fitted with the Carreau function to calculate values at approximate zero and infinite shear rates. Additionally, all samples were also tested at RT using a parallel-plate configuration with a diameter of 20 mm and a gap of 100 μm. A frequency sweep experiment (0.1–1000 Hz, constant strain of 1%) followed by a strain sweep test (from 0.01% to 1000%) were performed. For parallel-plate geometry tests, scaffolds were pre-formed in a plastic mold before rheological measurements; 1 ml of DPBS (1×) was added to 100 μL of the selected SAP (previously dissolved and incubated overnight at +4 °C) and incubated for 3 hours at RT to allow for complete hydrogel self-assembling. DPBS was then removed and 1 ml of 170 mM GP cross-linker solution (95:5 DPBS/EtOH) was added and incubated at 37 °C for additional 72 hours. Right before rheology measurements the cross-linked scaffold was thoroughly washed with DPBS to remove the unreacted GP.
All results were processed with OriginPro 2019 software (OriginLab Corporation, Northampton, MA, USA).
Attenuated total reflection/Fourier transform infrared analysis
To assess secondary structure formation, transition intensities and vibrational frequencies, a PerkinElmer Spectrum Two IR spectrometer (PerkinElmer Ltd, Beaconsfield, United Kingdom) equipped with a PerkinElmer single-reflection diamond ATR was used. All samples were measured in the wavelength range between 400–4000 cm−1 at room temperature, after dissolution in water at the respective working concentration, and with the addition of DPBS above the ATR-diamond to investigate the presence of amides I and II. ATR-FTIR spectra were analyzed with OriginPro 2019 software (OriginLab Corporation, Northampton, MA, USA) after baseline correction. Deconvolution of ATR-FTIR absorption spectra was performed by using the second derivative method followed by smoothing with a 7–9 point Savitsky–Golay function (polynomial order of 2). Peak fitting/deconvolution was obtained with the Voigt function from OriginPro.87Preparation of Thioflavin-T assay and fluorescence measurement
All peptides were treated with a solution of the benzothiol dye ThT previously dissolved in DPBS buffer and mixed in a ThT/peptide ratio of 0.5:1. For the detection of β-sheets, the ThT fluorescence was measured at room temperature, right after the reaction, using an Infinite M200 Pro plate reader (Tecan, Mennedorf, Switzerland) with excitation at 440 nm and emission at 482 nm.
Atomic force microscopy
AFM images were captured in tapping mode using a Multimode Nanoscope V (Digital Instruments, Veeco, Plainview, NY, USA), with single-beam silicon cantilever probes (Veeco RFESP MPP-21100-10: resonance frequency 76–90 kHz, nominal tip radius of curvature 8 nm). All peptides were dissolved in distilled water at a final concentration of 0.01% w/v. After 30 min sonication, 1 μL of peptide solution was mixed with 20 μL of distilled water. 1 μL of this final solution was deposited on freshly cleaved mica and rinsed with 100 μL of distilled water.88Because of the tip convolution effect, fiber measurements were performed. Being the observed nanofiber heights far lower than the tip radius, the observed widths were corrected with the following formula:89
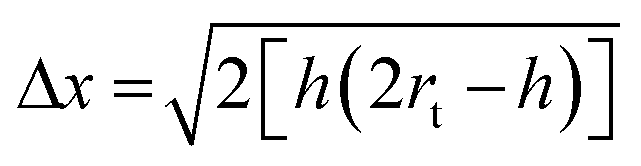
Image flattening was performed prior to image analysis. Height profiles were traced using FiberApp software,90 while width profiles were measured using Nanoscope software.
2D cell culture assays
HNSCs were obtained according to good manufacturing practice (GMP) protocols, in agreement with the European Medical Agency (EMA) guidelines and Agenzia Italiana del Farmaco (AIFA), protocol number aM 101/2010 (updated in 2018 after AIFA inspection to number aM 54/2018). Leukemia growth factor (LIF) was purchased from Merck (Merck Millipore, Darmstadt, Germany) and brain derived neurotrophic factor (BDNF) was obtained from Peprotech. Colorimetric MTS assay (Promega) to evaluate cell proliferation and LIVE/DEAD™ cell imaging kit (Invitrogen) were acquired from Thermo Fisher (Fischer Scientific GmbH, Schwerte, Germany). hNSCs were expanded and cultured as previously described.19 hNSCs were seeded on the top surface of SAP hydrogels at a density of 3 × 104 cells per cm2 and then cultured for 7 days in vitro. A Cultrex-BME substrate (from now on called just Cultrex) and an untreated-bottom well surface (namely glass) were used as the gold standard and negative control, respectively. hNSCs were differentiated in basal medium supplemented with basic fibroblast growth factor (bFGF, 10 ng ml−1). After two days, bFGF medium was replaced with basal medium supplemented with leukemia growth factor (LIF, 20 ng ml−1) and brain-derived neurotrophic factor (BDNF, 20 ng ml−1). Fresh medium was shifted after 3 days. Cell cultures were maintained at 37 °C, 20% O2 and 5% CO2. After 7 days, cell proliferation was evaluated by using colorimetric CellTiter96® AQueous One Solution Cell Proliferation Assay (MTS assay, Promega, following manufacturer's instructions). Briefly, 20% of MTS solution was added to the culture media and incubated for 1 h at 37 °C. The absorbance was detected at 490 nm using Infinite M200 Pro plate reader (Tecan, Mennedorf, Switzerland). A LIVE/DEAD™ viability/cytotoxicity kit (Invitrogen) was used to discriminate between live (stained with green calcein-AM) and dead (identified by nucleic acid red dye ethidium homodimer-1) cells. For immunofluorescence tests, samples were fixed with 4% paraformaldehyde, washed with DPBS, permeabilized with 0.3% Triton X-100 and blocked with 10% normal goat serum. To assess hNSC differentiation, the following antibodies were used: rabbit anti-GFAP (1:500, DakoCytomation), mouse anti-βIIITubulin (1:500, Biolegend), mouse anti-GalC (1:200, Merck), mouse anti-O4 (1:200, Merck), goat anti-mouse Alexa 488 (1:1000, Molecular Probes), goat anti-rabbit Alexa 488, goat anti-mouse Cy3 and goat anti-rabbit Cy3 (1:1000, Jackson Immunoresearch). Cell nuclei were stained with Hoechst 33342 (1:500, Molecular Probes). Fluorescence images were acquired using a Zeiss microscope ApoTome system and processed with ImageJ software. Three independent experiments were performed for each sample.
Cell viability and differentiation assay after MW treatment of hNSC-HYDROSAP scaffolds
HNSCs were encapsulated in HYDROSAP hydrogels as previously reported.17,91 Briefly, pure HYDROSAP (previously dissolved at 1% w/v in distilled water and incubated overnight at +4 °C) was mixed with 280 mM sucrose solution, 2.5 mM NaOH and 4.5 × 104 cells per μL hNSCs. The mixing solution was treated for 20 minutes at 1 W (+MW1/20/50), reaching the maximum temperature of 37 °C. As a control, the same hNSCs + hydrogel mixing solution was incubated at controlled temperature (≤37 °C) for 20 minutes (+RT). After incubation, a droplet (40 μl) was placed in 24-well plates, and serum-free medium supplemented with bFGF (20 ng ml−1, Peprotech) was added to trigger SAP gelation. After 1 hour, a LIVE/DEAD™ viability/cytotoxicity kit (Invitrogen) was used to evaluate the cytotoxicity effect of MW irradiation. Fluorescence images were acquired using a Zeiss microscope ApoTome system and processed with ImageJ software (see Fig. 5 and Fig. S13†). For cell differentiation assay, after 2 days in serum-free medium supplemented with bFGF, the medium was shifted to the basal medium supplemented with leukemia growth factor (LIF, 20 ng ml−1, Merck Millipore) and brain-derived neurotrophic factor (BDNF, 20 ng ml−1, Peprotech). Fresh medium was added after 3 days. Cell cultures were maintained at 37 °C, 20% O2 and 5% CO2 for 1 week. After 7 days, 3D hNSC-HYDROSAP scaffolds (+MW1/20/50 and +RT) were fixed with 4% paraformaldehyde overnight, incubated in sucrose 30% (in DPBS) and cryopreserved in OCT. Samples were cryo-sectioned at 100 μm thick slices using a cryostat (Histo-Line Laboratories). For immunofluorescence analyses, slices were washed in DPBS, permeabilized with 0.3% Triton X-100 for 10 minutes at 4 °C and blocked with 10% normal goat serum (NGS, GIBCO) for 1 h at room temperature. The following primary and secondary antibodies were used: mouse antiβIII-tubulin (1:500, BioLegend), rabbit anti-glial fibrillary acidic protein (GFAP) (1:500, DAKO), rabbit anti-nestin (1:500, Millipore), mouse anti-microtubule-associated protein 2 (MAP2) (1:300, Invitrogen), and rabbit anti-g-aminobutyric acid (GABA) (1:500, Sigma-Aldrich). To reveal primary antibodies, the following secondary antibodies were used: goat anti-rabbit Cy3 (1:1000, Jackson), goat anti-mouse Cy3 (1:1000, Jackson), goat anti-rabbit Alexa 488 (1:1000, Invitrogen), and goat anti-mouse Alexa 488 (1:1000, Invitrogen). Apoptotic cells were analysed via TUNEL assay (in situ cell death detection kit fluorescein, Roche), following the manufacturer's instructions. Briefly, sections were permeabilized with 0.3% Triton X-100 for 10 minutes at 4 °C and incubated with a TUNEL reaction mixture (1:10 in enzyme solution) for 1 h at 37 °C. Cell nuclei were stained with DAPI (Molecular Probes). A minimum of three fields for three independent experiments were chosen randomly. Acquisitions were performed at 20× and 40× magnification using a Zeiss Microscope with Apotome system. Quantitative analyses were performed by counting manually positive cells for each marker using NIH ImageJ software.
Statistics and reproducibility
Statistical analysis was performed using Prism 9 software (GraphPad, Inc.). One-way ANOVA followed by Tukey's multiple comparison test was used to determine statistical significance. *p < 0.01 for all tests, with the exception of in vitro tests *p < 0.05. All experiments were conducted in triplicate.Author contributions
M. G. C. and F. G. conceived the idea, planned and directed the studies, and wrote and reviewed the manuscript. M. G. C. designed the experiments, performed chemical synthesis, analyzed the data, and conducted the Thioflavin-T binding assay, rheological, AFM, and FT-IR experiments. A. M. conducted cellular assays, analyzed in vitro data with the support of M. S., and wrote the manuscript. J. G. investigated and analyzed the data. F. G. funded and led the work. All author checked the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the support by INAIL (BRIC2019-ID25), the Italian Ministry of Health (Ricerca Corrente 2022–2024) and the “5X1000” voluntary contributions. Financial support also came from Revert Onlus.References
- E. Mazzoni, M. R. Iaquinta, C. Lanzillotti, C. Mazziotta, M. Maritati, M. Montesi, S. Sprio, A. Tampieri, M. Tognon and F. Martini, Front. Bioeng. Biotechnol., 2021, 19, 1–17 Search PubMed.
- T. T. Tran, Z. A. Hamid and K. Y. Cheong, J. Phys.: Conf. Ser., 2018, 1082, 012080 CrossRef.
- C. A. Palma, P. Samorì and M. Cecchini, J. Am. Chem. Soc., 2010, 132, 17880–17885 CrossRef CAS PubMed.
- D. Mandal, A. Nasrolahi Shirazi and K. Parang, Org. Biomol. Chem., 2014, 12(22), 3544–3561 RSC.
- J. M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef CAS PubMed.
- D. Marsh, Biophys. J., 2012, 102, 1079–1087 CrossRef CAS PubMed.
- A. Sharma, K. Vaghasiya, R. K. Verma and A. B. Yadav, Chapter 3 - DNA nanostructures: chemistry, self-assembly, and applications, Emerging Applications of Nanoparticles and Architecture Nanostructures, Elsevier Inc., 2018 Search PubMed.
- A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef CAS.
- E. Granger, G. McNee, V. Allan and P. Woodman, Semin. Cell Dev. Biol., 2014, 31, 20–29 CrossRef CAS PubMed.
- P. M. Kharkar, K. L. Kiick and A. M. Kloxin, Chem. Soc. Rev., 2013, 42, 7335 RSC.
- T. T. Wang, Y. Y. Xia, J. Q. Gao, D. H. Xu and M. Han, Pharmaceutics, 2021, 13, 753 CrossRef CAS PubMed.
- J. B. Matson and S. I. Stupp, Chem. Commun., 2011, 47, 7962–7964 RSC.
- J. B. Matson, C. J. Newcomb, R. Bitton and S. I. Stupp, Soft Matter, 2012, 8, 3586–3595 RSC.
- V. Torchilin, Eur. J. Pharm. Biopharm., 2009, 71, 431–444 CrossRef CAS PubMed.
- Z. Oren and Y. Shai, Biochemistry, 2000, 39, 6103–6114 CrossRef CAS PubMed.
- A. Makovitzki, J. Baram and Y. Shai, Biochemistry, 2008, 47, 10630–11063 CrossRef CAS PubMed.
- A. Marchini, A. Raspa, R. Pugliese, M. A. El Malek, V. Pastori, M. Lecchi, A. L. Vescovi and F. Gelain, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 7483–7492 CrossRef CAS PubMed.
- J. Guo, K. K. Leung, H. Su, Q. Yuan, L. Wang, T. H. Chu, W. Zhang, J. K. Pu, G. K. Ng, W. M. Wong, X. Dai and W. Wu, Nanomedicine, 2009, 5, 345–351 CrossRef CAS PubMed.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352–1355 CrossRef CAS PubMed.
- C. E. Semino, J. R. Merok, G. G. Crane, G. Panagiotakos and S. Zhang, Differentiation, 2003, 71, 262–270 CrossRef CAS PubMed.
- J. D. Kisiday, M. Jin, M. A. Di Micco, B. Kurz and A. J. Grodzinsk, J. Biomech., 2004, 37, 595–604 CrossRef PubMed.
- A. Mujeeb, A. F. Miller, A. Saiani and J. E. Gough, Acta Biomater., 2013, 9, 4609–4617 CrossRef CAS PubMed.
- R. Pugliese, L. Moretti, M. Maiuri, T. Romanazzi, G. Cerullo and F. Gelain, Mater. Des., 2020, 194, 108901 CrossRef CAS.
- R. Pugliese, M. Maleki, R. N. Zuckermann and F. Gelain, Biomater. Sci., 2019, 7, 76–91 RSC.
- R. Pugliese, A. Marchini, G. A. A. Saracino, R. N. Zuckermann and F. Gelain, Nano Res., 2018, 11, 586–602 CrossRef CAS.
- M. G. Ciulla, R. Pugliese and F. Gelain, Nanomaterials, 2022, 12, 320 CrossRef CAS PubMed.
- R. Pugliese and F. Gelain, Int. J. Mol. Sci., 2020, 21, 4261 CrossRef CAS PubMed.
- R. Pugliese, M. Montuori and F. Gelain, Nanoscale Adv., 2022, 4, 447–456 RSC.
- H. M. T. Albuquerque, D. C. G. A. Pinto and A. M. S. Silva, Molecules, 2021, 26, 6293 CrossRef CAS PubMed.
- A. de la Hoz, A. Díaz-Ortiz and A. Moreno, Chem. Soc. Rev., 2005, 34, 164–178 RSC.
- A. K. Bose, M. S. Manhas, S. N. Ganguly, A. H. Sharma and B. K. Banik, Synthesis, 2002, 1578 CrossRef CAS.
- R. S. Varma, Tetrahedron, 2002, 58, 1235 CrossRef CAS.
- F. Langa, P. de la Cruz, E. Espíldora and A. de la Hoz, Electrochem. Soc., 2000, 9, 168–178 Search PubMed.
- D. Bogdal and A. Prociak, Polymer Sci Tech, Gen. Blackwell Pub., 2007 Search PubMed.
- H. Will, P. Scholz and B. Ondruschka, Chem. Ing. Tech., 2002, 74, 1057 CrossRef CAS.
- C. C. Chen and J. F. Hsieh, Sci. Rep., 2016, 6, 39040 CrossRef CAS PubMed.
- A. Corsaro, U. Chiacchio, V. Pistara and G. Romeo, Curr. Org. Chem., 2004, 8, 511 CrossRef CAS.
- S. Zhang, M. A. Greenfield, A. Mata, L. C. Palmer, R. Bitton, J. R. Mantei, C. Aparicio, M. O. de La Cruz and S. I. Stupp, Nat. Mater., 2010, 9, 594–601 CrossRef CAS PubMed.
- J. Kopeček, Biomaterials, 2007, 28, 5185–5192 CrossRef PubMed.
- O. Wichterle and D. Lím, Nature, 1960, 185, 117–118 CrossRef.
- J. Kopeček and J. Yang, Polym. Int., 2007, 56, 1078–1098 CrossRef.
- Y. Xiang, X. Qi, E. Cai, C. Zhang, J. Wang, Y. Lan, H. Deng, J. Shen and R. Hu, Chem. Eng. J., 2023, 460, 141852 CrossRef CAS.
- B. V. Slaughter, S. S. Khurshid and O. Z. Fisher, Adv. Mater., 2009, 21, 32–33 CrossRef PubMed.
- M. Hamidi, A. Azadi and P. Rafiei, Adv. Drug Delivery Rev., 2008, 60, 1638–1649 CrossRef CAS PubMed.
- R. Narayanaswamy and V. P. Torchilin, Hydrogels and Their Applications in Targeted Drug Delivery, Molecules, 2019, 24(3), 603, DOI:10.3390/molecules24030603.
- C. Yu, Z. Yue, H. Zhang, M. Shi, M. Yao, Q. Yu, M. Liu, B. Guo, H. Zhang, L. Tian, H. Sun, F. Yao and J. Li, Adv. Funct. Mater., 2023, 33(15), 2211023 CrossRef CAS.
- C. Yan and D. J. Pochan, Chem. Soc. Rev., 2019, 39, 3528 RSC.
- D. Aparna and K. B. Bimal, Microwaves in Chemistry Applications Fundamentals, Methods and Future Trends, Elsevier, 1st edn, 2021 Search PubMed.
- S. Chandrasekaran, S. Ramanathan and T. Basak, Food Res. Int., 2013, 52, 243–261 CrossRef CAS.
- R. R. Mishra and A. K. Sharma, Crit. Rev. Solid State Mater. Sci., 2016, 41, 217–255 CrossRef CAS.
- G. Kłosowski, D. Mikulski and A. Menka, Catalysts, 2019, 9, 753 CrossRef.
- C. O. Kappe and A. Stadler, Microwaves in Organic and Medicinal Chemistry, Wiley-VCH, Weinheim, 2005 Search PubMed.
- C. A. Hettiarachchi, L. D. Melton, J. A. Gerrard and S. M. Loveday, Biomacromolecules, 2012, 13, 2868–2880 CrossRef CAS PubMed.
- Q. Jiao, Z. Liu, B. Li, B. Tian, N. Zhang, C. Liu, Z. Feng and B. Jiang, Foods, 2021, 10, 1892 CrossRef CAS PubMed.
- Y. Cao and R. Mezzenga, Adv. Colloid Interface Sci., 2019, 269, 334–356 CrossRef CAS PubMed.
- P. Shaw, N. Kumar and S. Mumtaz, Sci. Rep., 2021, 11, 14003 CrossRef CAS PubMed.
- T. Mishra, P. Kushwah, K. Dholiya and V. Kothari, Adv Microw. Wirel. Technol., 2013, 1, 4–11 CrossRef.
- F. Gelain, D. Cigognini, A. Caprini, D. Silva, B. Colleoni, M. Donegá, S. Antonini, B. E. Cohen and A. Vescovi, Nanoscale, 2012, 4, 2946 RSC.
- A. L. Sieminski, C. E. Semino, H. Gong; and R. D. Kamm, J. Biomed. Mater. Res., Part A, 2008, 87, 494–504 CrossRef CAS PubMed.
- D. Silva, A. Natalello, B. Sanii, R. Vasita, G. Saracino, R. N. Zuckermann, S. M. Doglia and F. Gelain, Nanoscale, 2013, 5, 704–718 RSC.
- S. Sathaye, A. Mbi, C. Sonmez, Y. Chen, D. L. Blair, J. P. Schneider and D. J. Pochan, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol., 2015, 7, 34–68 CrossRef CAS PubMed.
- N. V. Zelentsova, S. V. Zelentsov and Y. D. Semchikov, Chem. Inf., 2006, 37 DOI:10.1002/chin.200608267.
- F. Gelain, Z. Luo and S. Zhang, Chem. Rev., 2020, 120, 13434–13460 CrossRef CAS PubMed.
- A. Puisto, X. Illa, M. Mohtaschemi and M. J. Alava, Eur. Phys. J. E: Soft Matter Biol. Phys., 2012, 35, 1–7 CrossRef PubMed.
- I. Y. Belyaev, Eur. J. Oncol., 2010, 5 Search PubMed.
- G. Prasanna and P. Jing, Biochem. Biophys. Res. Commun., 2020, 534, 950–956 CrossRef PubMed.
- W. Lee, Y. Choi, S. W. Lee, I. Kim, D. Lee, Y. Hong, G. Lee and D. S. Yoon, Nanotechnology, 2018, 29, 345604 CrossRef PubMed.
- E. M. Ford and A. M. Kloxin, ACS Biomater. Sci. Eng., 2021, 7, 4175–4195 CrossRef CAS PubMed.
- C. Xue, T. Y. Lin, D. Chang and Z. R. Guo, Soc. Open Sci., 2017, 4, 160696 CrossRef PubMed.
- L. P. Jameson, N. W. Smith and S. V. Dzyuba, ACS Chem. Neurosci., 2012, 3, 807–819 CrossRef CAS PubMed.
- S. Seshadri, R. Khurana and A. L. Fink, Methods Enzymol., 1999, 309, 559–576 CAS.
- G. Lee, W. Lee, H. Lee, C. Y. Lee, K. Eom and T. Kwon, Sci. Rep., 2015, 5, 16220 CrossRef CAS PubMed.
- L. D. Black, P. G. Allen, S. M. Morris, P. J. Stone and B. Suki, Biophys. J., 2008, 94, 1916–1929 CrossRef CAS PubMed.
- C. Q. Yan and D. J. Pochan, Chem. Soc. Rev., 2010, 39, 3528–3540 RSC.
- H. Y. Song, R. Salehiyan, X. Li, S. H. Lee and K. Hyun, Korea Aust. Rheol. J., 2017, 29, 281–294 CrossRef.
- H. Abé, K. Hayashi and M. Sato, Data Book of Mechanical Properties of Living Cells, Tissues, and Organs, in Blood Vessels, Springer-Verlag, Tokyo, 1996, ch. 2.2, pp. 25–114 Search PubMed.
- N. Gjorevski and C. M. Nelson, Biophys. J., 2012, 103, 152–162 CrossRef CAS PubMed.
- G. A. Holzapfel and R. W. Ogden, Mechanics of Biological Tissue, Springer, 2006 Search PubMed.
- D. McManus, S. Vranic and F. Withers, Nat. Nanotechnol., 2017, 12, 343–350 CrossRef CAS PubMed.
- M. Sun, G. Chi, P. Li, S. Lv, J. Xu, Z. Xu, Y. Xia, Y. Tan, J. Xu, L. Li and Y. Li, Int. J. Med. Sci., 2018, 15, 257–268 CrossRef CAS PubMed.
- B. Yi, Q. Xu and W. Liu, Bioact. Mater., 2022, 15, 82–102 CrossRef CAS PubMed.
- S. Pandamooz, A. Jafari, M. S. Salehi, B. Jurek, A. Ahmadiani, A. Safari, S. Hassanajili, A. Borhani-Haghighi, M. Dianatpour, H. Niknejad, N. Azarpira and L. Dargahi, Biotechnol. Bioeng., 2020, 117, 305–317 CrossRef CAS PubMed.
- E. E. Fesenko, V. R. Makar, E. G. Novoselova and V. B. Sadovnikov, Bioelectrochem. Bioenerg., 1999, 49, 29–35 CrossRef CAS PubMed.
- H. D. Baille, Symposium Proceedings, ed. S. F. Cleary, 2010, vol. 38, pp. 728–736 Search PubMed.
- A. Cutz, Lens Eye Toxic. Res., 1989, 6, 729–736 Search PubMed.
- M. P. Hendricks, K. Sato, L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2017, 50, 2440–2448 CrossRef CAS PubMed.
- Y. Ji, X. Yang, Z. Ji, L. Zhu, N. Ma, D. Chen, X. Jia, J. JunTang and C. Yilin, ACS Omega, 2020, 5, 8572–8578 CrossRef CAS PubMed.
- H. Yokoi, T. Kinoshita and S. Zhan, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8414–8419 CrossRef CAS PubMed.
- H. O. Di Rocco, D. I. Iriarte and J. Pomarico, Appl. Spectrosc., 2001, 55, 822–826 CrossRef CAS.
- I. Usov and R. Mezzenga, Macromolecules, 2015, 48, 1269–1280 CrossRef CAS.
- A. Raspa, G. A. A. Saracino, R. Pugliese, D. Silva, D. Cigognini, A. Vescovi and F. Gelain, Adv. Funct. Mater., 2014, 24, 6317–6328 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr02738d |
| This journal is © The Royal Society of Chemistry 2023 |