
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing strongly exchange coupled magnetic behaviors in soft/hard Ni/CoFe2O4 core/shell nanoparticles†
J. K.
Han
 *,
A. A.
Baker
,
J. R. I.
Lee
and
S. K.
McCall
*,
A. A.
Baker
,
J. R. I.
Lee
and
S. K.
McCall
Critical Materials Institute, Lawrence Livermore National Laboratory, Livermore, California 94550, USA. E-mail: han10@llnl.gov
First published on 24th July 2023
Abstract
Exchange coupling in a model core–shell system is demonstrated as a step on the path to 3d exchange spring magnets. Employing a model system of Ni@CoFe2O4, high quality core–shell nanoparticles were fabricated using a simple two-step method. The microstructural quality was validated using TEM, confirming a well-defined interface between the core and the shell. A strongly temperature dependent two-phase magnetic hysteresis loop was measured, wherein an analysis of step heights indicates coupling of roughly 50% between the core and the shell. Element-specific XMCD hysteresis confirms the presence of exchange coupling, suppressing the superparamagnetism of the Ni core at room temperature, and reaching a coercivity of >6 kOe at 80 K. These results provide a pathway to the development of heterostructured metal-oxide exchange coupled nanoparticles with improved maximum energy product.
Introduction
The high cost and complicated supply chains of rare-earth elements (REE) have motivated extensive research efforts to develop permanent magnets with reduced or zero REE content.1,2 Exchange coupling, which provides a promising pathway to this goal, involves the coupling of the spins of a magnetically soft material with high saturation magnetization (Ms) to a neighboring magnetically hard phase with high coercivity (Hc) to create a composite system with an enhanced maximum energy product (BHmax). However, the nanometer length scale of the exchange interaction makes bulk realization of the concept extremely challenging, as does the requirement for high-quality interfaces between the two materials throughout the composite structure. Bi-component magnetic materials with the potential for strong exchange coupling have been extensively investigated in multilayer films,3–6 demonstrating the principle of exchange coupling in the case of heterostructures tens of nanometers thick. More recently, attempts to synthesize viable bulk exchange spring magnets (ESMs) have been extended to the development of nanoscale bi-magnetic core/shell heterostructures, seeking an efficient approach for tuning magnetic properties.7,8 Material composition, shape, and morphology are all parameters that have been explored.9–11 In particular, various bi-magnetic core/shell nanoparticle compositions have been extensively studied in ferromagnetic (FM)/antiferromagnetic (AFM) systems to investigate exchange bias.12–14 An additional benefit of core–shell nanomaterials for ESM applications is that they are likely to form single domain states, which negates the need for defect engineering to enhance hysteresis required in bulk systems.2Despite extensive research, the demonstration of strong exchange interactions between hard and soft phases in core/shell nanoparticles remains challenging, due in large part to the difficulties in fabricating high-quality nanoscale core/shell architectures, specifically crystalline materials, with a uniform shell coating and controlled interfacial structure. Research efforts to date have focused on, for example, FeCo/CoFe2O4, FeCoB/CoFe2O4, and FeCo/FeCoOx core/shell nanoparticles prepared by treatment (i.e. oxidation or reduction) of the core particles to yield a surface shell.9,15–17 This approach is hampered by poor structural quality and inhomogeneous interfaces, which weakens the exchange coupling between the hard and soft phases. A seeded-growth method is a promising synthetic route to address these challenges, as it offers independent synthetic control of the core and the shell, enabling excellent crystallinity and interface quality. Using the method, some hard/soft core/shell nanoparticles such as FePt/Fe3O4, MFe2O4/MFe2O4 (M = Ni, Mn, Fe), and FePt/Co have successfully demonstrated an enhanced energy product as compared with the single hard phases.8,10,18–20 Building on these works, “inverted” soft/hard core/shell systems have been proposed as an attractive alternative, having advantages such as improved resilience to local demagnetizing fields.16,20 Coating a soft metal core with a hard oxide shell also mitigates the increased surface reactivity associated with nanoscale systems. However, there has been only limited success in studying exchange interactions in mixed metal-oxide systems,11,15,18 due to the complex synthesis required.
Heterostructured “inverted” Ni core/CoFe2O4 shell nanoparticles with high structural and interfacial quality were synthesized and exchange coupling was observed at room temperature. This specific composition, with unique elements in the core and the shell, was chosen to enable separate measurement of the magnetic behavior of the magnetization reversal using X-ray magnetic circular dichroism. The synthesis approach described herein offers a pathway to similar core–shell structures with improved performance, for example, high-magnetization FeCo cores, and dramatically extends the range of materials that can be incorporated into nanoscale core–shell structures. The core/shell particles are prepared by seeding growth of CoFe2O4 on monodisperse Ni nanoparticles, which significantly improves the structural quality at the interface in comparison with alternative methods. This is the first report of CoFe2O4 and Ni in a core/shell structure, and the elemental contrast allows unambiguous demonstration of exchange coupling using element specific soft X-ray magnetic circular dichroism (XMCD). A comparison with bare Ni nanoparticles reveals a suppression of superparamagnetism by the addition of CoFe2O4 and provides insights into the Co-seeding that drives formation of the shell. The techniques used here are broadly applicable to a range of soft/hard materials and are anticipated to be of wide relevance when designing technologically relevant core–shell nanoparticles.
Experimental
Synthesis of Ni nanoparticles
Ni nanoparticles were prepared using a modification of an existing literature protocol.21 Briefly, 1 g of nickel(II) acetylacetonate (Ni(acac)2, Sigma-Aldrich, 95%) was added to 10.4 g of oleylamine (Sigma-Aldrich, 70%) and the solution was degassed at room temperature, and then stirred for 30 min under an Ar atmosphere. Subsequently, in an air-sensitive glovebox environment, 3 mL of trioctylphosphine (Sigma-Aldrich, 97%) was injected into the mixture and then stirred for 30 min. Note that all reactions were performed under an Ar atmosphere. Thereafter, the mixture was heated under magnetic stirring, at a heating rate of 5 °C min−1 up to 220 °C and kept for 2 h. The mixture was later allowed to cool to room temperature and then washed with a solution of hexane and acetone 3 times under ambient conditions, prior to further dissolution in hexane.Synthesis of Ni/CoFe2O4 core/shell nanoparticles
Ni/CoFe2O4 core/shell nanoparticles were prepared through a facile thermal decomposition method. Immediately after the generation of Ni nanoparticles, ∼50 mg of the Ni nanoparticles in 5 mL of hexane was added to 20 mL of benzenyl ether in the presence of 1 mL of oleic acid (solution (1)). In parallel, 0.11 mmol cobalt(II) acetylacetonate (Co(acac)2, Sigma-Aldrich, 95%) and 0.22 mmol iron(III) acetylacetonate (Fe(acac)3, Sigma-Aldrich, 95%) were dissolved in 1 mL of oleylamine and the mixture was added to solution (1). Subsequently, the mixture was degassed at room temperature, and then heated at 120 °C for 30 min in order to remove the trace amounts of air, moisture and hexane. Note that all reactions were prepared under magnetic stirring and a static flow of Ar. Then, the solution was heated to 220 °C for 2 h at a heating rate of 5 °C min−1, and further to 290 °C for 1 h at a heating rate of 3 °C min−1. Thereafter, the mixture was cooled to room temperature, washed with hexane and acetone 3 times and stored in EtOH for future use.Characterization
TEM images were taken at an accelerating voltage of a 300 kV TITAN TEM, equipped with EDS capabilities. The capacity to obtain high-quality HRTEM images coupled with the ability to obtain reasonable selected area electron diffraction (SAED) patterns was rendered possible using a 300 kV TITAN TEM microscope equipped with a Gatan image filter (GIF) spectrometer, operating at an accelerating voltage of 300 kV. Specimens for all of these electron microscopy experiments were prepared by dispersing the as-prepared product in ethanol, sonicating it for 2 min to ensure an adequate dispersion of the nanostructures, and finally dropping one drop of the solution onto a Si wafer for SEM and a 300 mesh Cu grid, coated with a lacey carbon film, for TEM and HRTEM analyses. Magnetometry measurements were performed in a SQUID magnetometer (Quantum Design MPMS) over the temperature range 5–300 K. Samples were immobilized in polyethylene glycol (PEG) to prevent them from rotating during field sweeps. Powders of the nanoparticles were placed inside non-magnetic gelcaps along with PEG (average Mn 3350, melting point 58 °C = 331 K) in a 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The gelcap was vigorously shaken to mix the PEG and the sample, and then secured in a low-background holder (polypropylene tube). Once inside the chamber, the PEG was melted using the following protocol: heat to 350 K at 5 °C min−1, hold for 5 minutes, cool to 270 K at 5 °C min−1, hold for 10 min, and return to 300 K to begin the measurement profile. In this way, the PEG is fully liquid and able to fully surround and then immobilize the particles. The temperature dependence of magnetic hysteresis, M(H), with a maximum field of 40 kOe, and the temperature dependence of magnetization M(T) in a 50 Oe field were both investigated. X-ray magnetic circular dichroism (XMCD) measurements were performed on beamline I10 (BLADE) of the Diamond Light Source (UK), beamline 4.0.2 of the Advanced Light Source (US), and beamline 29 (BOREAS) of ALBA (Spain). XMCD provides element- and site-specific measurements of the electronic and magnetic character of the nanoparticles, allowing the determination of cation site occupancies, as well as direct detection of the exchange coupling between the Ni core and the CoFe2O4 shell. X-ray absorption spectroscopy (XAS) of the Fe, Co, and Ni L2,3 edges was measured in surface-sensitive total electron yield (TEY) mode and bulk-sensitive fluorescence yield (FY) mode. XMCD was obtained as the difference between two XAS spectra obtained with opposite helicity vectors; measurements were performed in normal incidence. Element resolved XMCD hysteresis loops were performed by recording the field dependence of the XMCD signal at energies corresponding to the maximum dichroism of the Co, Ni L3 edge (779.3 eV and 852.3 eV, respectively). The Ni L3 energy was chosen as the maximum dichroism of metallic Ni to avoid unwanted contributions from any oxide contamination. Note that due to experimental limitations the data presented at 300 K were gathered in the bulk-sensitive FY mode, while the data from 80 K were gathered in the surface-sensitive TEY mode, which gave increased prominence to the near-interface region of Ni. Measurements in the FY mode at 150 K are presented in the ESI,† and show strongly two-phase switching in Ni, arguing for the presence of two regions, strongly and weakly bound, within Ni. XMCD spectra of the CoFe2O4 shell were calculated within the atomic and crystal field theory to assess site occupancy using the program CTM4XAS. Spectra were calculated for 2+ and 3+ valences, in octahedral and tetrahedral symmetries, and the experimental spectrum fitting using a linear superposition of these calculations. For Ni2+ in an octahedral environment 10Dq was set to 1.2 eV. For Co2+ in an octahedral (tetrahedral) environment 10Dq was set to 0.9 eV (0.5 eV), and for Fe2+/3+ in an octahedral (tetrahedral) environment 10Dq was set to 1.2 eV (0.6 eV). The Slater integrals were reduced to 70% (d–d) and 80% (p–d), while an exchange field of 10 meV was applied to break the spin symmetry. For Fe, a Lorentzian broadening of 0.2 eV (0.3 eV) was applied to the L3 (L2) edges to account for lifetime effects. For Co, 0.2 eV and 0.35 eV were used. For Ni, 0.2 eV and 0.25 eV were used. In addition, a Gaussian broadening of 0.2 eV was applied to account for the finite resolving power of the instrumentation. These parameters are consistent with previous studies on similar materials.22–25
:1 ratio. The gelcap was vigorously shaken to mix the PEG and the sample, and then secured in a low-background holder (polypropylene tube). Once inside the chamber, the PEG was melted using the following protocol: heat to 350 K at 5 °C min−1, hold for 5 minutes, cool to 270 K at 5 °C min−1, hold for 10 min, and return to 300 K to begin the measurement profile. In this way, the PEG is fully liquid and able to fully surround and then immobilize the particles. The temperature dependence of magnetic hysteresis, M(H), with a maximum field of 40 kOe, and the temperature dependence of magnetization M(T) in a 50 Oe field were both investigated. X-ray magnetic circular dichroism (XMCD) measurements were performed on beamline I10 (BLADE) of the Diamond Light Source (UK), beamline 4.0.2 of the Advanced Light Source (US), and beamline 29 (BOREAS) of ALBA (Spain). XMCD provides element- and site-specific measurements of the electronic and magnetic character of the nanoparticles, allowing the determination of cation site occupancies, as well as direct detection of the exchange coupling between the Ni core and the CoFe2O4 shell. X-ray absorption spectroscopy (XAS) of the Fe, Co, and Ni L2,3 edges was measured in surface-sensitive total electron yield (TEY) mode and bulk-sensitive fluorescence yield (FY) mode. XMCD was obtained as the difference between two XAS spectra obtained with opposite helicity vectors; measurements were performed in normal incidence. Element resolved XMCD hysteresis loops were performed by recording the field dependence of the XMCD signal at energies corresponding to the maximum dichroism of the Co, Ni L3 edge (779.3 eV and 852.3 eV, respectively). The Ni L3 energy was chosen as the maximum dichroism of metallic Ni to avoid unwanted contributions from any oxide contamination. Note that due to experimental limitations the data presented at 300 K were gathered in the bulk-sensitive FY mode, while the data from 80 K were gathered in the surface-sensitive TEY mode, which gave increased prominence to the near-interface region of Ni. Measurements in the FY mode at 150 K are presented in the ESI,† and show strongly two-phase switching in Ni, arguing for the presence of two regions, strongly and weakly bound, within Ni. XMCD spectra of the CoFe2O4 shell were calculated within the atomic and crystal field theory to assess site occupancy using the program CTM4XAS. Spectra were calculated for 2+ and 3+ valences, in octahedral and tetrahedral symmetries, and the experimental spectrum fitting using a linear superposition of these calculations. For Ni2+ in an octahedral environment 10Dq was set to 1.2 eV. For Co2+ in an octahedral (tetrahedral) environment 10Dq was set to 0.9 eV (0.5 eV), and for Fe2+/3+ in an octahedral (tetrahedral) environment 10Dq was set to 1.2 eV (0.6 eV). The Slater integrals were reduced to 70% (d–d) and 80% (p–d), while an exchange field of 10 meV was applied to break the spin symmetry. For Fe, a Lorentzian broadening of 0.2 eV (0.3 eV) was applied to the L3 (L2) edges to account for lifetime effects. For Co, 0.2 eV and 0.35 eV were used. For Ni, 0.2 eV and 0.25 eV were used. In addition, a Gaussian broadening of 0.2 eV was applied to account for the finite resolving power of the instrumentation. These parameters are consistent with previous studies on similar materials.22–25
Results and discussion
Fig. 1a and b shows TEM images of the Ni cores prior to shell synthesis and the Ni/CoFe2O4 core/shell nanoparticles, respectively. TEM image analysis reveals that the average diameter of the Ni core is 11.8 nm with a standard deviation of 2.1 nm and the thickness of the CoFe2O4 shell is 1.6 nm with a standard deviation of 0.3 nm. The core–shell nanoparticles are highly crystalline and of uniform size and shape, and the Ni cores are uniformly coated with CoFe2O4 shells. Examination of SAED patterns (Fig. S1†) along with interplanar distance estimation compared with theoretical values (Table S1†) reveal features that can be assigned to the Ni and CoFe2O4 nanostructures. | ||
| Fig. 1 TEM images of morphology and composition of particles. Panel (a) shows bare Ni nanoparticles while (b) shows Ni/CoFe2O4 core/shell structures. EDS maps in (c) reveal the well-defined interfaces and the presence of limited homogeneous nucleation of CoFe2O4 (top right of image). Linescans across a representative particle (d) confirm the formation of the desired core/shell architecture, with some interdiffusion of Co into the core. Note that Fe and Co are detected throughout the cross-section as these are 3-D particles. | ||
Following coating, no independent Ni nanoparticles are observed, and only a limited formation of pure CoFe2O4 nanoparticles (<5% amongst more than 100 surveyed). High-angle annular dark field (HAADF)-STEM or TEM images of representative particles are included in Fig. S2.† Suitable balancing of seed particles and reagents is essential to suppressing homogeneous nucleation of CoFe2O4 and ensuring the uniform coating necessary for high quality core–shell nanoparticles (Fig. S3 and S4†). This fabrication method is amenable for the high-yield production of Ni/CoFe2O4 core/shell nanoparticles and can be adapted to other core/shell nanostructure systems. Fig. 1c shows a representative HAADF image of the core/shell nanoparticles as well as their corresponding elemental maps. Ni dominates the particles in the images and the Co, Fe and O atoms are primarily located on the surface of the Ni particles. EDS line scan analysis across the diameter of a single core/shell nanoparticle and quantitative analysis of the nanoparticles are shown in Fig. 1d, and Fig. S5,† respectively. Ni is confined within the core area in the region of 8–20 nm, while a higher intensity of Co and Fe is found in the shell region (5–7 nm and 20–22.5 nm). Note that Co and Fe are detected throughout the core also, as sampling through the center also captures the coating on the top and bottom of the core. However, the flatter profile of the Co linescan suggests that some Co is present within the core.
Based on these results, a mechanism for the growth of CoFe2O4 shells on Ni core particles is proposed. In EDS mapping (Fig. 1c and d), Co atoms were found not only at the surface but also at the center of the Ni core, while Fe and O were more segregated. This may be attributed to the generation of a NiCo alloy (∼77 at% Ni and ∼23 at% Co based on EDS line scan and analysis (Fig. S5†)) by the inter-diffusion of Ni and Co. We hypothesized that the reduction of Co2+ could be achieved first using Ni nanoparticles as a catalyst and then the nucleation of Co0 could take place on the surface of the Ni nanoparticles to form Ni–Co (core–shell) nanoparticles. These Co0 can act as anchoring sites to reduce the lattice mismatch between Ni and CoFe2O4 (e.g. aCoFe2O4 = 8.4 A, d(311) of CoFe2O4 = 2.52 A, aNi 3.5A, aCo = 2.5 A) and thus promote the heteroepitaxial nucleation of CoFe2O4, thereby resulting in the uniform growth of the CoFe2O4 shells on the Ni nanoparticles. During the CoFe2O4 shell formation (i.e. oxidation steps from Co0 to CoFe2O4), interdiffusion between Ni and Co takes place, resulting in Co atom distribution all over the core/shell (Fig. 1c and d) due to a lower inter-diffusion barrier for Co–Ni among transition metals, which has been observed in oxidation steps in catalytic studies of Co and Ni composites.26,27 Heteroepitaxial nucleation is further viable because the decomposition temperatures (Tdecompose) of the precursors of Co (i.e. Co(acac)2, Tdecompose ∼215 °C) are lower than those of Fe (i.e. Fe(acac)3, Tdecompose ∼235 °C) and the synthetic reaction was performed between these two thresholds (220 °C for 2 h). Similar synthetic approaches were attempted at temperatures without precursor decomposition to form a seeding layer and these yielded primarily homogeneous nucleation that left the majority of the Ni particles uncoated (Fig. S3 and S4†). The proposed mechanism is consistent with reports on the synthesis of Ni/Co28 and Co/CoFe2O4,29,30 and its nucleation and growth step is likely critical for the formation of high-quality core–shell nanoparticles, as direct growth is suppressed in the absence of favorable linkages (i.e. seeds and ligands) due to a large difference of lattice parameters. The detailed characteristics for the unsuccessful core/shell formation shown in Fig. S3 and S4† are helpful to clarify the formation of the CoFe2O4 spinel shell and so the details of the homo- and hetero-nucleation of Ni and CoFe2O4 in the core/shell and related interdiffusion theory and simulation works will likely be needed in future studies.
The local electronic characters of Ni, Co, and Fe were assessed using XMCD21 in total electron yield (TEY) mode to allow accurate interpretation of the spectral lineshapes. Fig. 2 compares experimental and calculated spectra,22 along with the constituent spectra due to different oxidation states and local symmetries of the component elements. The Ni (Fig. 2a) XMCD exhibits a mixed metallic-oxide lineshape,23 attributed to reactions at the Ni/CoFe2O4 interface. A comparison with the calculated spectrum yields a 2:1 ratio of Ni 2+ Oh to Ni metal. It is important to note that these measurements are inherently surface sensitive (with a typical sampling depth of ∼5 nm), which emphasizes the signal from the near-surface (interface) region. Allowing for an exponential decrease in the contribution to the signal with increasing depth, this is consistent with a thin Ni oxide region at the shell interface. This assignment is supported by the TEM-EDS (Fig. 1c), which indicates that the oxygen is concentrated in the shell region. On the other hand, despite the role of Co as a nucleation site for the CoFe2O4 shell, there is little evidence for metallic Co (Fig. 2b) at the interface region of the core/shell, indicating that it is consumed in the thermal decomposition reaction to generate CoFe2O4 shells. The metallic Co in the core region suggested by TEM is not detected here, likely due to the signal being dominated by the shell region in the extremely surface-sensitive TEY XMCD. The Fe XMCD reveals the presence of 2+ and 3+ valences, in octahedral and tetrahedral symmetries. These sites are occupied in the ratio 0.9:1.0:0.85 (2+ Oh:3+ Oh:3+ Th), consistent with previous XMCD studies of cobalt ferrite.24,31,32 The contribution of 2+ Fe in CoFe2O4 has previously been observed in thin films32 or biogenic samples,25 and can arise from variations in the synthesis route including stress or oxygen availability.31 Furthermore, if some Co has diffused into the core during synthesis, this could leave unoccupied 2+ Oh sites, which the Fe readily occupies.
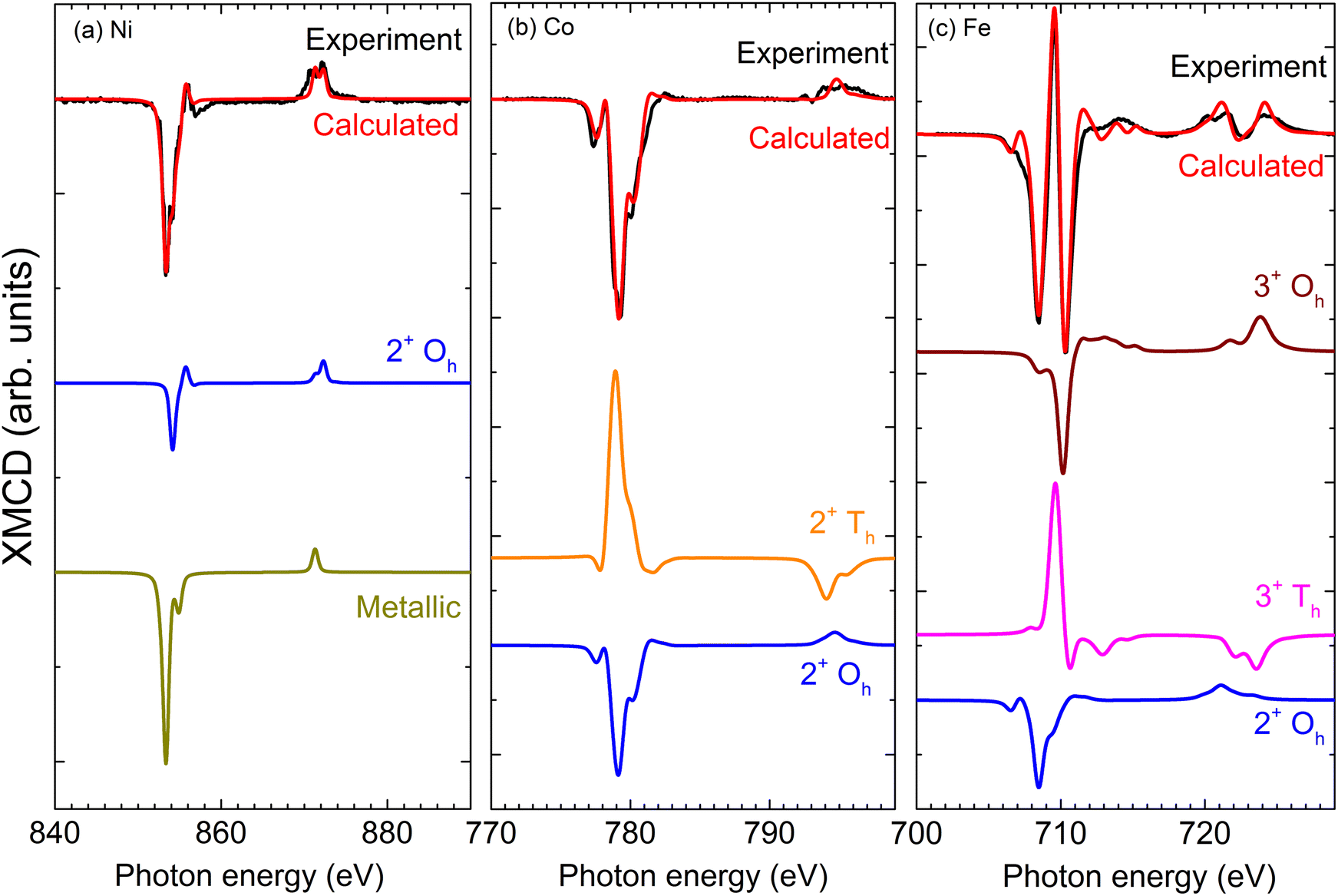 | ||
| Fig. 2 Experimental XMCD spectra for Ni@CoFe2O4 nanoparticles dispersed on the Si substrate gathered in the surface-sensitive total electron yield mode, along with calculated spectra from CTM4XAS. Mixed oxidation states can be calculated from a linear combination of the reference spectra, yielding a 2:1 ratio of Ni2+ Oh to Ni metal (a), a 6.25:1 ratio of 2+ Oh to 2+ Th for Co (b), and a 0.9:1.0:0.85 (2+ Oh:3+ Oh:3+ Th) ratio for Fe (c). Spectra have been vertically offset for clarity. | ||
Bulk magnetic hysteresis loops (Fig. 3) show the increase in coercivity as temperature decreases. At 300 K, the hysteresis loop is single-phase, with a coercivity of 850 Oe. As the temperature decreases, a pronounced two-step switching develops, indicative of a two-phase magnetic system. While the coercivity of the hard phase exceeds 10 kOe at low temperatures, that of the soft phase does not change significantly. These results initially indicate an uncoupled system, wherein the soft Ni core is independent of the hard CoFe2O4 shell. However, examination of the relative height of the two components of the loop indicates that the low-coercivity step does not account for the full magnetization of the Ni core. From a simple volume fraction argument, the Ni core contributes 49% of the total magnetization of the system (saturation moment/volume), but the step only accounts for 21% of the height of the loop (the Ni core is 48% of the total volume) while the CoFe2O4 contributes 51% of the magnetization (saturation moment/volume). This implies that some portions of the cores are coupled to the shell. These results suggest that some fractions of the Ni cores are well coupled to the shells, showing an increased coercivity due to the exchange coupling mechanism, but some remain uncoupled, likely due to disruptions at the interface arising from oxidation of Ni. The critical length scale for the exchange extending into the soft phase is given by: 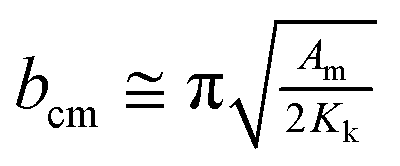 where Am is the spin stiffness in the soft phase (ANi ∼8 × 10–11 J m−1)1 and Kk is the magnetocrystalline exchange constant of the hard phase (KCo-ferrite = 250 kJ m−3)2 resulting in bcm ∼39 nm at 300 K, which will increase as temperature decreases. Provided the coremagnetically soft phase nanoparticles are smaller than this diameter, the entire core of the nanoparticle can be coupled to the enveloping hard phase material. Since exchange requires overlap of relevant orbitals, the presence of a few magnetically dead atomic layers negates the exchange coupling between the hard and soft phases, as could happen if antiferromagnetic Ni oxide forms at the interface. The domain wall width in CoFe2O4 is 20 nm and therefore the entirety of Ni should couple;2 it is possible that the length scale of coupling is reduced due to structural imperfections at the interface. Indeed, bare Ni cores are found to be superparamagnetic, with a blocking temperature of ∼60 K (Fig. S6†); a comparison of the M(H) loops of bare cores and the core–shell nanoparticles at 100 K is shown in Fig. 3 (b), where the bare cores display no coercivity.
where Am is the spin stiffness in the soft phase (ANi ∼8 × 10–11 J m−1)1 and Kk is the magnetocrystalline exchange constant of the hard phase (KCo-ferrite = 250 kJ m−3)2 resulting in bcm ∼39 nm at 300 K, which will increase as temperature decreases. Provided the coremagnetically soft phase nanoparticles are smaller than this diameter, the entire core of the nanoparticle can be coupled to the enveloping hard phase material. Since exchange requires overlap of relevant orbitals, the presence of a few magnetically dead atomic layers negates the exchange coupling between the hard and soft phases, as could happen if antiferromagnetic Ni oxide forms at the interface. The domain wall width in CoFe2O4 is 20 nm and therefore the entirety of Ni should couple;2 it is possible that the length scale of coupling is reduced due to structural imperfections at the interface. Indeed, bare Ni cores are found to be superparamagnetic, with a blocking temperature of ∼60 K (Fig. S6†); a comparison of the M(H) loops of bare cores and the core–shell nanoparticles at 100 K is shown in Fig. 3 (b), where the bare cores display no coercivity.
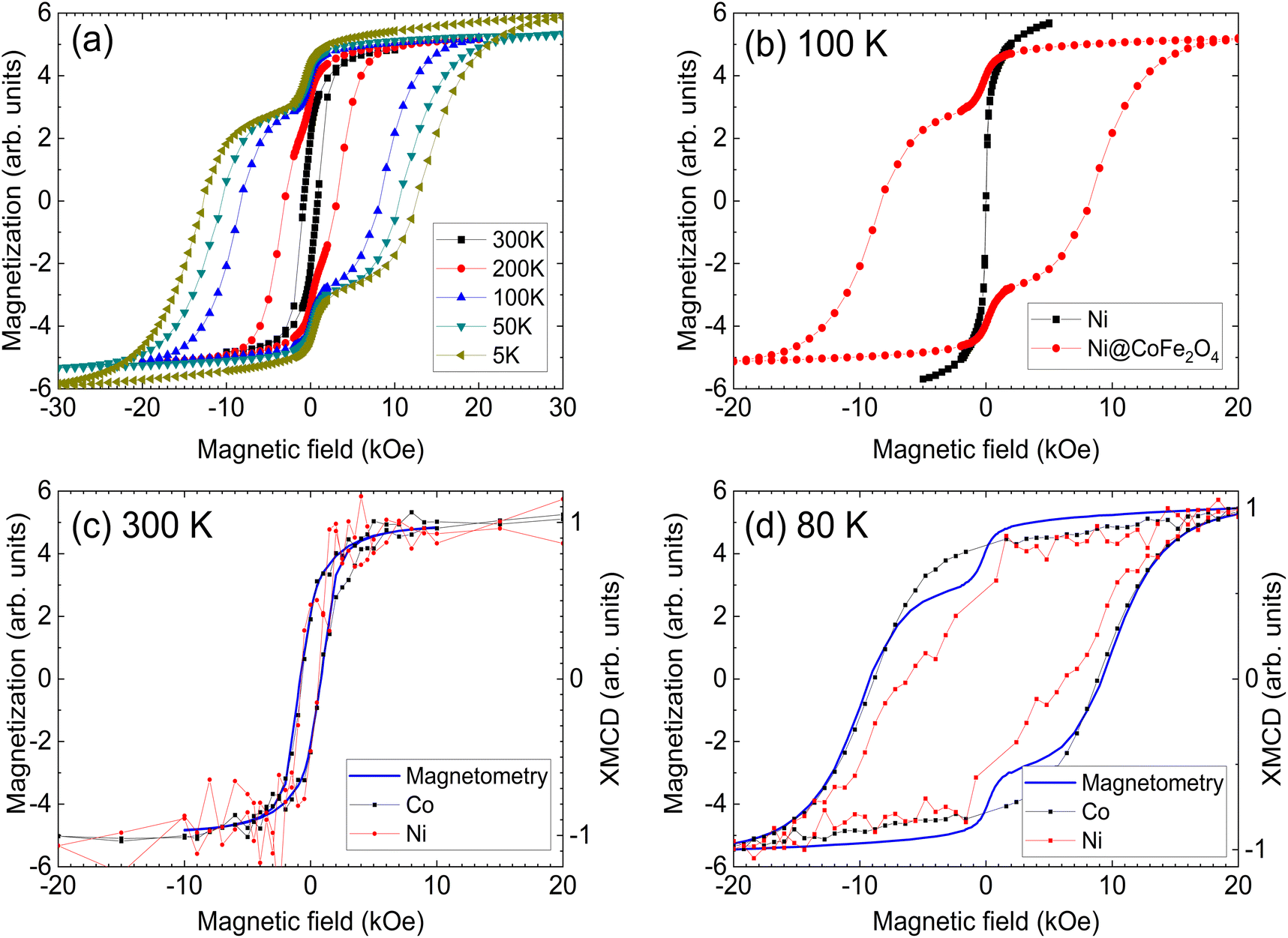 | ||
| Fig. 3 Hysteresis loops of the Ni@CoFe2O4 nanoparticles (a), and a comparison with bare Ni nanoparticles at 100 K (b). The presence of coercivity in the Ni signal shows that the particles are no longer superparamagnetic and are coupled to the cobalt ferrite. Element-specific XMCD-hysteresis at 300 K (c) and 80 K (d) reveals the presence of exchange coupling, as the Ni core rotates with the CoFe2O4 shell. Magnetization is presented in arbitrary units due to the difficulty of obtaining accurate mass measurements for the sub-milligram quantities of the nanoparticles. | ||
The contribution of each element to the hysteresis loop can be directly assigned using element-specific XMCD-hysteresis, separating the magnetic behavior of the core and the shell. Hysteresis loops for the Co and Ni L3 edges at 300 K and 80 K are overlaid with bulk magnetization (Fig. 3c and d). At 300 K, Ni and Co rotate together with a coercive field of 850 Oe, matching the bulk hysteresis loop. The non-zero coercivity of Ni implies the suppression of superparamagnetism due to coupling between the core and the shell. This interaction is more evident at 80 K, where Co shows a single reversal step with a coercivity coinciding with that of the bulk measurements. Strikingly, the coercivity of Ni exceeds 6 kOe, albeit with some evidence of an uncoupled soft phase. Since XMCD averages over all particles present in the beam, this soft phase is likely composed of Ni particles that are uncoupled from their CoFe2O4 shells. Nevertheless, the measured coercivity value far exceeds the theoretical coercivity for a monodomain Ni nanoparticle of 3.7 kOe, as predicted by the Stoner–Wohlfarth model.2 Such a high coercivity provides striking evidence of exchange coupling, especially when considering that experimentally measured coercivities rarely exceed 30% of the theoretical coercivity, as noted by Brown's paradox.33,34 Magnetization measurements at 150 K are presented alongside XMCD data for Co and Ni collected in the bulk-sensitive FY mode in Fig. S7.† Despite the lower data quality compared to the TEY of Fig. 3, a significant coercivity is visible in Ni, with two-stage switching evident, similar to what was observed at 80 K. There is some evidence of similar multi-stage reversal in Co also, which could indicate that Co present in the cores rotates coherently with Ni. Since Co was present in the core, one might expect the coercivity to increase slightly, but the anisotropy of NiCo is still significantly less than that of CoFe2O4, and less than the coercivity observed in the element-specific Ni hysteresis loops. It is possible that partial Co diffusion into the core aids the exchange coupling by providing a less abrupt gradient between the metallic core and the oxide shell, and that Co rotates together with the shell even though some of it has diffused into the core.
While FY is more bulk sensitive than TEY, signals are still dominated by the near-surface region at these energies. The two-phase, broad reversal suggests a range of coercivities across the Ni cores. This could arise due to the length scale of coupling not covering the entirety of the core, or alternatively from a range of strengths of exchange couplings over the different particles sampled by the X-ray beam. In either event, improvements could be achieved with a smaller core, enabling a fully coupled system. Furthermore, the strength of the exchange coupling of core/shell nanostructures could be varied by tuning the core and shell size, the volume ratio of the core and shell, and the shape of core/shell nanoparticles, a promising area for further study.
Conclusion
A model core–shell nanoparticle system for the study of exchange coupling has been developed, demonstrating room-temperature exchange interactions between a Ni core and a CoFe2O4 shell, wherein the magnetization reversal can be resolved using element-specific techniques. Thermal decomposition reactions allow the homogeneous nucleation of an oxide shell across the metal core, providing both crystalline materials and a controlled interface, as confirmed by TEM and XMCD. Magnetization measurements show that a significant portion of the Ni couples to CoFe2O4, although an uncoupled portion remains. Element-specific XMCD hysteresis loops provide unambiguous evidence for exchange coupling, with coercive Ni nanoparticles at room temperature and a coercivity in an excess of 6 kOe at 80 K. This model system provides a strategy for the rational design of exchange spring materials based upon mixed metal-oxide nanoparticles with tunable properties.Conflicts of interest
There are no conflicts of interest.Acknowledgements
This research was sponsored by the Critical Materials Institute, an Energy Innovation Hub funded by the U.S. Department of Energy (DOE), the Office of Energy Efficiency and Renewable Energy and Advanced Materials and Manufacturing Technologies Office by the Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344. Portions of this work were performed at the Advanced Light Source, Lawrence Berkeley National Laboratory, which is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. DOE under Contract DE-AC02–05CH11231. Some of these experiments were performed at BOREAS beamline at ALBA Synchrotron with the collaboration of ALBA staff. This work was carried out with the support of Diamond Light Source, I10 (proposal SI17516). We thank L. B. Duffy, E. Arenholz, and M. Valvidares for their assistance with XMCD measurements.References
- D. Bauer, D. Diamond, J. Li, D. Sandalow, P. Telleen and B. Wanner, U.S. Department of Energy Critical Materials Strategy, 1000846, 2010 Search PubMed.
- J. M. D. Coey, Magnetism and Magnetic Materials, Cambridge University Press, Cambridge, 2010 Search PubMed.
- E. E. Fullerton, J. S. Jiang, M. Grimsditch, C. H. Sowers and S. D. Bader, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 12193–12200 CrossRef CAS.
- E. E. Fullerton, J. S. Jiang and S. D. Bader, J. Magn. Magn. Mater., 1999, 200, 392–404 CrossRef CAS.
- V. Neu, K. Häfner, A. K. Patra and L. Schultz, J. Phys. D: Appl. Phys., 2006, 39, 5116 CrossRef CAS.
- M. Shindo, M. Ishizone, A. Sakuma, H. Kato and T. Miyazaki, J. Appl. Phys., 1997, 81, 4444–4446 CrossRef CAS.
- E. Lottini, A. Lopez-Ortega, G. Bertoni, S. Turner, M. Meledina, G. Van Tendeloo, C. de Julian Fernandez and C. Sangregorio, Chem. Mater., 2016, 28, 4214–4222 CrossRef CAS.
- Q. Song and Z. J. Zhang, J. Am. Chem. Soc., 2012, 134, 10182–10190 CrossRef CAS PubMed.
- A. López-Ortega, E. Lottini, C. de J. Fernández and C. Sangregorio, Chem. Mater., 2015, 27, 4048–4056 CrossRef.
- F. Liu, J. Zhu, W. Yang, Y. Dong, Y. Hou, C. Zhang, H. Yin and S. Sun, Angew. Chem., Int. Ed., 2014, 53, 2176–2180 CrossRef CAS PubMed.
- W. Yang, W. Lei, Y. Yu, W. Zhu, T. A. George, X. Z. Li, D. J. Sellmyer and S. Sun, J. Mater. Chem. C, 2015, 27, 7075–7080 RSC.
- G. Lavorato, E. Winkler, A. Ghirri, E. Lima Jr., D. Peddis, H. E. Troiani, D. Fiorani, E. Agostinelli, D. Rinaldi and R. D. Zysler, Phys. Rev. B, 2016, 94, 054432 CrossRef.
- M. Estrader, A. López-Ortega, S. Estrade, I. V. Golosovsky, G. Salazar-Alvarez, M. Vasilakaki, K. N. Trohidou, M. Varela, D. C. Stanley, M. Sinko, M. J. Pechan, D. J. Keavney, F. Peiro, S. Surinach, M. D. Baro and J. Nogues, Nat. Commun., 2013, 4, 2960 CrossRef CAS PubMed.
- E. Lima Jr., E. L. Winkler, D. Tobia, H. E. Troiani, R. D. Zysler, E. Agostinelli and D. Fiorani, Chem. Mater., 2012, 24, 512–516 CrossRef.
- R. Tang, T. Li, Z. Wu, W. Cai, H. Jiang, Z. Yang, W. Liu, Z. Zhang, B. Yang, X. Liu, Z. Zou and R. Yu, IEEE Trans. Magn., 2011, 47, 3456–3459 CAS.
- A. López-Ortega, M. Estrader, G. Salazar-Alvarez, A. G. Roca and J. Nogués, Phys. Rep., 2015, 553, 1–32 CrossRef.
- A. López-Ortega, E. Lottini, G. Bertoni, C. de Julián Fernández and C. Sangregorio, Chem. Mater., 2017, 29, 1279–1289 CrossRef.
- H. Zeng, J. Li, Z. L. Wang, J. P. Liu and S. Sun, Nano Lett., 2004, 4, 187–190 CrossRef CAS.
- D. J. Carnevale, M. Shatruk and G. F. Strouse, Chem. Mater., 2016, 28, 5480–5487 CrossRef CAS.
- A. Lopez-Ortega, M. Estrader, G. Salazar-Alvarez, S. Estrade, I. V. Golosovsky, R. K. Dumas, D. J. Keavney, M. Vasilakaki, K. N. Trohidou, J. Sort, F. Peiro, S. Surinach, M. D. Baroj and J. Nogues, Nanoscale, 2012, 4, 5138–5147 RSC.
- S. Carenco, C. Boissière, L. Nicole, C. Sanchez, P. Le Floch and N. Mézailles, Chem. Mater., 2010, 22, 1340–1349 CrossRef CAS.
- G. van der Laan and A. I. Figueroa, Coord. Chem. Rev., 2014, 277–278, 95–129 CrossRef CAS.
- E. Stavitski and F. M. F. de Groot, Micron, 2010, 41, 687–694 CrossRef CAS PubMed.
- C. Klewe, M. Meinert, A. Boehnke, K. Kuepper, E. Arenholz, A. Gupta, J. M. Schmalhorst, T. Kuschel and G. Reiss, J. Appl. Phys., 2014, 115, 123903 CrossRef.
- V. S. Coker, N. D. Telling, G. van der Laan, R. A. D. Pattrick, C. I. Pearce, E. Arenholz, F. Tuna, R. E. P. Winpenny and J. R. Lloyd, ACS Nano, 2009, 3, 1922–1928 CrossRef CAS PubMed.
- J. Li, R. Doig, J. Camardese, K. Plucknett and J. R. Dahn, Chem. Mater., 2015, 27, 7765–7773 CrossRef CAS.
- J. Helbig, T. Beuse, V. Siozios, T. Placke, M. Winter and R. Schmuch, J. Electrochem. Soc., 2020, 167, 060519 CrossRef CAS.
- T. Yamauchi, Y. Tsukahara, K. Yamada, T. Sakata and Y. Wada, Chem. Mater., 2011, 23, 75–84 CrossRef CAS.
- B. Aslibeiki, Ceram. Int., 2016, 42, 6413–6421 CrossRef CAS.
- L. J. Zhao and Q. Jiang, Mater. Lett., 2010, 64, 677–679 CrossRef CAS.
- J. A. Moyer, D. P. Kumah, C. A. F. Vaz, D. A. Arena and V. E. Henrich, Appl. Phys. Lett., 2012, 101, 021907 CrossRef.
- J. A. Moyer, C. A. F. Vaz, D. P. Kumah, D. A. Arena and V. E. Henrich, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 174404 CrossRef.
- W. F. Brown, Micromagnetics, J. Wiley, New York; London, 1963 Search PubMed.
- R. Skomski, J. Phys.: Condens. Matter, 2003, 15, R841–R896 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary texts and images with synthetic parameters and correlated structural and morphological analysis (i.e. TEM, EDS, and SAED). See DOI: https://doi.org/10.1039/d3nr03478j |
| This journal is © The Royal Society of Chemistry 2023 |