
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
MOF-based heterogeneous catalysis in continuous flow via incorporation onto polymer-based spherical activated carbon supports†
Anthony
Griffiths
a,
Sarah L.
Boyall
 a,
Pia
Müller
a,
John P.
Harrington
b,
Anna M.
Sobolewska
c,
William R.
Reynolds
c,
Richard A.
Bourne
ad,
Kejun
Wu
ef,
Sean M.
Collins
d,
Mark
Muldowney
c and
Thomas W.
Chamberlain
*a
a,
Pia
Müller
a,
John P.
Harrington
b,
Anna M.
Sobolewska
c,
William R.
Reynolds
c,
Richard A.
Bourne
ad,
Kejun
Wu
ef,
Sean M.
Collins
d,
Mark
Muldowney
c and
Thomas W.
Chamberlain
*a
aInstitute of Process Research and Development, School of Chemistry, University of Leeds, Leeds, UK. E-mail: t.w.chamberlain@leeds.ac.uk
bLeeds Electron Microscopy and Spectroscopy Centre, LEMAS, Bragg Centre for Materials Research, University of Leeds, Leeds, LS2 9JT, UK
cSterling Pharma Solutions Limited, Dudley, Cramlington, Northumberland NE23 7QG, UK
dSchool of Chemical and Process Engineering and School of Chemistry, University of Leeds, Leeds, LS2 9JT, UK
eZhejiang Provincial Key Laboratory of Advanced Chemical Engineering Manufacture Technology, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, P. R. China
fInstitute of Zhejiang University-Quzhou, Quzhou 324000, P. R. China
First published on 23rd October 2023
Abstract
We present an approach to harnessing the tuneable catalytic properties of complex nanomaterials for continuous flow heterogeneous catalysis by combining them with the scalable and industrially implementable properties of carbon pelleted supports. This approach, in turn, will enable these catalytic materials, which largely currently exist in forms unsuitable for this application (e.g. powders), to be fully integrated into large scale, chemical processes. A composite heterogeneous catalyst consisting of a metal–organic framework-based Lewis acid, MIL-100(Sc), immobilised onto polymer-based spherical activated carbon (PBSAC) support has been developed. The material was characterised by focused ion beam-scanning electron microscopy-energy dispersive X-ray analysis, powder X-ray diffraction, N2 adsorption, thermogravimetric analysis, atomic absorption spectroscopy, light scattering and crush testing with the catalytic activity studied in continuous flow. The mechanically robust spherical geometry makes the composite material ideal for application in packed-bed reactors. The catalyst was observed to operate without any loss in activity at steady state for 9 hours when utilised as a Lewis acid catalyst for the intramolecular cyclisation of (±)-citronellal as a model reaction. This work paves the way for further development into the exploitation of MOF-based continuous flow heterogeneous catalysis.
Introduction
Wider adoption of continuous manufacturing within the fine chemical industry has the potential to deliver significant quality, efficiency and safety benefits, with studies suggesting that 30–50% of current batch processes could be carried out more efficiently if done continuously.1–3 The implementation of such technologies has also received support from regulatory bodies such as the Food and Drug Administration (FDA), which recognises continuous manufacturing as an ‘emerging technology that can enable pharmaceutical modernisation’, citing patient and industry benefits including improved quality control and reduced manufacturing costs.4Heterogeneously catalysed processes are routinely exploited in chemical industry, with 80% of all catalyst-containing chemical processes involving a heterogeneous species.5 However, due to the complexity of designing continuous, multi-phase systems, e.g. obtaining the required mixing of solids and liquids/gases in narrow tubing while avoiding blockages and pressure build-ups caused by catalyst breakdown, the exploitation of heterogeneous catalysis in continuous modes of operation has seen limited but increasing implementation to date.6,7 The translation of heterogeneously catalysed organic transformations to continuous flow therefore offers significant benefits for fine chemical manufacturing.
Progress in the commercial implementation of high-throughput and robust heterogeneously catalysed continuous processes for fine chemical manufacture requires the development of suitable catalyst materials designed across length scales. Both the form (e.g. pellet, powder) and bulk properties (e.g. surface area, immobilisation type) determine the suitability for continuous flow processes.
In their pure form, heterogeneous catalysts typically exist as fine powders and thus their direct application in flow reactors (e.g. packed-bed reactors) may in turn lead to blockages inside the reactor hindering process efficiency, or worse, producing catastrophic pressure build-up. The use of larger-sized shaped supports, such as monoliths, extrudates and porous beads prepared frequently from materials such as alumina, silica, polymers and carbon are used to manage these issues with the active species often anchored to the support.8,9
As well as packing regularly enough to prevent a large pressure drop across the reactor, catalyst supports that are resistant to both crushing and abrasion when inside of the reactor are desirable to avoid breakdown and plug formation. These mechanical considerations are balanced with the requirement of maximising the surface area of the active component of the catalysts and immobilising this species to the support in a manner that ensures sufficient stability for long-term industrial operation (e.g. to prevent leaching or physical detachment over time). Additional consideration must be given to immobilisation approaches as physisorption or the formation of a single covalent bond between the active catalyst and support may not be robust enough for continuous flow operations.10
One promising support material which provides a good balance between macro- and nano-scale properties is polymer-based spherical activated carbon (PBSAC).11 These mechanically strong, commercially available spherical activated carbon pellets have a regular, tuneable size and possess a high, graphitic carbon surface area for catalyst deposition with their successful operation in flow reactors established. Previous research into the use of PBSAC spheres as catalyst supports has primarily involved active species such as metal nanoparticles,12–18 metal oxides19–21 and ionic liquids.22–25 However, composite materials incorporating more complex catalytic nanomaterials, for example metal–organic frameworks (MOFs), is significantly less developed.26,27 MOFs have become attractive candidates as industrial catalysts28 and examples of their application as heterogeneous catalysts in flow reactors, typically packed-bed reactors, have been reported for a range of chemical transformations; such developments have been discussed in detail by Garcia and colleagues.29 Despite initial demonstration of their potential to serve as continuous flow heterogeneous catalysts, however, MOFs typically exist as powders which currently limits their more widespread usage in these reactor platforms.
The MIL-100(M) (M = metal centre) series of MOFs are an isostructural series of large-pore MOFs known to exhibit Lewis acid catalytic activity via metal-containing M3O trimers situated throughout the framework. MOFs of this type have a general stoichiometry of [M3O(BTC)2X], where M is a metal, BTC is benzene-1,3,5-tricarboxylic acid (trimesic acid) and X is a singly charged anion required to balance the positive charge of the framework.30–32 A scandium-based MOF, MIL-100(Sc), has previously been demonstrated to be an active Lewis acid catalyst for intermolecular carbonyl ene and Michael additions.33 MIL-100(Sc) has also been demonstrated to be more catalytically active for such addition reactions than a range of other MOFs on a number of occasions in both batch and continuous flow. Previous studies by Wright et al.33,34 and Čejka et al.35 detail the greater catalytic activity of MIL-100(Sc) when compared to both isostructural MOFs (MIL-100(M), M = Fe, Cr, Al, In) and other MOFs (i.e., MIL-101(Sc), MIL-101(Cr), HKUST-1(Cu), CPO-27(Ni), STA-12(Ni)) in batch. Upon studying MIL-100(Sc) in continuous flow for the carbonation of propylene oxide with CO2, Sanford et al.36 found that this species possessed the highest steady state catalytic activity when compared with a several other catalytically active species (MIL-88D(Sc), MIL-66(Sc), MIL-101(M) (M = Cr, Fe, Sc)) and was also found to exhibit minimal loss in reactivity over a 24 hours continuous operation study. It is important to note that this study focused on testing of powdered MIL-100(Sc) in small scale, continuous flow, with no discussion of routes to scale up. Whilst the formation of composite materials consisting of MIL-100(Fe) and activated carbon has previously been reported,37 the physical form of these materials have always been limited to powders. The formation of MIL-100 MOFs immobilised on macroscopic support materials such as PBSAC spheres is therefore a priority for incorporation into industrially focused, preparative scale, continuous flow reactors.
In this article we report the synthesis, characterisation and initial catalytic activity studies of a novel Lewis acidic heterogeneous catalyst for operation in continuous flow consisting of catalytically active MOF MIL-100(Sc) supported onto PBSAC spheres, termed MIL-100(Sc)@PBSAC. The Lewis acidic catalytic activity of the prepared MIL-100(Sc)@PBSAC composites was studied in continuous flow using the intramolecular cyclisation of (±)-citronellal as a model reaction.
This reaction, when carried out using enantiomerically pure (+)-citronellal, constitutes an important step in the Takasago process for the synthetic production of (−)-menthol, one of the most consumed flavour compounds worldwide with an estimated 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 metric tonnes of menthol consumed annually.38–40 We show that MIL-100(Sc)@PBSAC composites possesses properties suitable for implementation into industrial flow reactors (e.g. packed bed reactors) for fine chemical synthesis with promising results obtained in preliminary catalytic activity studies.
000 metric tonnes of menthol consumed annually.38–40 We show that MIL-100(Sc)@PBSAC composites possesses properties suitable for implementation into industrial flow reactors (e.g. packed bed reactors) for fine chemical synthesis with promising results obtained in preliminary catalytic activity studies.
Materials and methods
Reagents and materials
For MIL-100(Sc)@PBSAC synthesis, benzene-1,3,5-tricarboxylic acid (98%), scandium(III) nitrate hydrate (REacton, 99.9% (REO)), dimethylformamide (DMF) (≥99.5%) and ethanol (absolute) were obtained from Fisher Scientific Ltd and used without further purification. Water (deionised, 15.0 MΩ cm−1) was obtained from an Elga PURELAB Option purification system. Active carbon spheres (500 μm) were obtained from IBU-tec. For catalytic activity testing, n-decane (99%) was purchased from Thermo Scientific, toluene (≥99.8%) from Fisher Scientific Ltd and 2-propnaol (99.9% for HPLC), (±)-citronellal (≥95% (GC)) and glass spheres (500 μm diameter) from Sigma-Aldrich.Synthesis of MIL-100(Sc)@PBSAC composites
The synthesis of MIL-100(Sc)@PBSAC composites was based on previously reported procedures, i.e. the MOF supported on granular activated carbon.41 Benzene-1,3,5-tricarboxylic acid (0.043 mmol, 90.5 mg), PBSAC spheres (1.10 g) and DMF (20 mL) were stirred at 45 °C for 2 hours. Next, scandium nitrate hydrate (0.86 mmol, 245 mg) was added and the reaction mixture was stirred for another 2 hours at 45 °C. The reaction mixture was then refluxed with stirring overnight. All stirring was carried out at 500 rpm. Powdered MIL-100(Sc) was also observed to have formed inside of the reaction vessel independently of the PBSAC spheres. Following synthesis, the spheres were washed sequentially with excess water and ethanol and then stirred in methanol for 72 hours. The spheres were then filtered and dried at 65 °C under vacuum giving activated MIL-100(Sc)@PBSAC (1.18 g, ≈54% yield of MIL-100(Sc) immobilisation determined using TGA loading and maximum possible loading values). The appearance of the catalyst was identical to the unfunctionalised PBSAC spheres.(±)-Citronellal cyclisation in continuous flow
The flow system used to study this reaction consisted of a vertically placed stainless steel packed-bed reactor (1/2′′ OD, 0.065′′ wall thickness, length: 12 cm, empty volume with adapters: 11 mL), PTFE tubing (1/16′′ OD, 1/32′′ ID), a cartridge back pressure regulator (BPR) (75 psi) and a Jasco HPLC pump (model PU-986) (see ESI section 1.1† for setup). For a typical reaction, the bed was plugged at both ends with two 1 cm wide cotton wool balls and filled with a homogeneous mixture of MIL-100(Sc)@PBSAC spheres (1 g) and glass spheres (9 g) (500 μm). The packed-bed reactor volume filled with the catalyst and with adapters attached was 8.5 mL. This was calculated by measuring the increase in mass of the reactor upon filling the packed reactor with 2-propanol and subsequently calculating the volume by dividing by the solvent density (0.786 g cm−3 at 20 °C).Before beginning the reaction, toluene was flowed through the solvent-less system at 1 mL min−1 until a continuous liquid phase was observed at the outlet of the reactor and a stable pressure was achieved. The reactor, which was clamped vertically, was heated using an aluminium heating block which was connected to a Eurotherm temperature controller set to 110 °C. A solution of (±)-citronellal (0.43 M in toluene, n-decane internal standard) was pumped from bottom to top for ideal distribution throughout the packed-bed. The resultant outlet stream was analysed offline by gas chromatography. For the preparation of GC samples, 10 μL of the outlet stream was diluted to a total volume of 1 mL with toluene.
Characterisation
Powder X-ray diffraction (PXRD) was carried out on a Malvern PANalytical Empyrean diffractometer using Cu Kα radiation (1.54 Å) between 2–30° 2θ with a step size of 0.013°. Measurements were carried out at room temperature (20 °C). MIL-100(Sc)@PBSAC and PBSAC spheres were crushed into a powder with a pestle and mortar before analysis. Additional XRD were acquired on a Phaser D2 (Bruker) (Cu Kα radiation (1.54 Å)) – these data are labelled in the caption (Fig. SI8†).Thermogravimetric analysis (TGA) was carried out on a Shimadzu TGA-50. A typical analysis involved heating approximately 10 mg of the sample from 20 °C to 900 °C at 10 °C min−1 in flowing air (50 mL min−1).
Atomic absorption spectroscopy (AAS) was carried out using an Agilent 200 series AA system. Samples were prepared for analysis by heating in air a known amount at 600 °C for 90 minutes with the resultant white solid dissolved in nitric acid (10 mL, 67%). Following dissolution the sample was then diluted to a known volume (30 ppm) for analysis.
N2 adsorption analysis was carried out at −196 °C using a Micrometrics Tristar 3000. Samples (≈50 mg) were activated prior to analysis by heating at 150 °C overnight under a flow of N2. Brunauer–Emmett–Teller (BET) surface areas were calculated using the BETSI method.42 Pore volumes were obtained P/Po = 0.99. Micropore volumes were extracted from t-plots.
Gas chromatography (GC) analysis was carried out on a Hewlett Packard HP 6890 series GC system using a DB-624 column (length: 30 m, diameter: 0.25 m, film thickness: 1.40 μm) and FID detector at 300 °C. See ESI section 1.2† for specific method information.
PBSAC support particle size distributions were obtained using a Malvern Mastersizer 3000 laser diffraction system. PBSAC spheres, which were stirred in water using the Hydro MV automated wet dispersion unit with a stirring rate of 3500 rpm, were added until the laser obscuration reached between 10–20%.
Crush testing was done using an Instron 5566 according to a modified version of the ASTM D-4179 standard test for single pellet crush strength. Samples were heated to 250 °C under dynamic vacuum for 3 hours to remove water and then stored under nitrogen and in a desiccator until analysis. For measurements, increasing force at a uniform rate of 0.05 N s−1 was applied until crushing of the particle occurred. For each sample, 50 particles were crushed.
Focused ion beam-scanning electron microscopy (FIB-SEM) and energy dispersive X-ray (EDX) analysis of materials was carried out on an FEI Helios G4 CX DualBeam. Prior to imaging, 20 nm of carbon coating was evaporated onto the sample surface using a Quorum Q150TE coater to reduce charging. Images were collected using a standard ETD (Everhart–Thornley detector) in secondary electron mode or a CBS (circular backscattered electron detector) detector. The typical voltage used was 5 or 10 kV. EDX analysis was carried out using an Oxford instruments AZtec energy EDX system with a 150 mm X-Max SDD detector. The Ga source for the FIB was operated at 30 kV.
Results and discussion
MIL-100(Sc)@PBSAC composites were prepared under reflux conditions owing to the facile and scalable nature of this synthetic technique. At all stages of the synthesis stirring was carried out to provide contact between MIL-100(Sc) precursors and PBSAC spheres. Trimesic acid adsorption onto PBSAC spheres in the first stirring step of the synthesis was confirmed by carrying out TGA on a control sample in which PBSAC spheres and trimesic acid, in the quantities used in the synthesis (90.5 mg trimesic acid, 1.10 g PBSAC spheres), were stirred for 2 hours at 45 °C. In the TGA profile (see ESI section 1.3†) of this PBSAC-trimesic acid control sample (Fig. SI6†) there is a small weight loss at approximately 300 °C corresponding to the thermal decomposition of adsorbed trimesic acid (Fig. SI7† shows the TGA profile of trimesic acid) which confirms that this species adsorbs in the initial mixing step.The stoichiometry of MIL-100(Sc) in this work was determined to be [Sc3O(BTC)1.45OH2.65] by carrying out TGA analysis on a powdered MIL-100(Sc) sample and thus for catalyst synthesis reagent amounts which gave a maximum MIL-100(Sc) loading of 11.87% w/w (a scandium loading of 3.23% w/w) onto PBSAC spheres was selected (Table 1). This loading value was selected as it provides a balance between minimising cost in the initial catalyst development stage whilst also ensuring sufficient catalytic activity.
| Molar ratio (PBSAC:M:L:DMF)a |
Resultant maximum possible scandium loading (% w/w) | Resultant maximum possible MIL-100(Sc) loading (% w/w) |
|---|---|---|
| a Where PBSAC, M, L and DMF is the amount of PBSAC spheres, scandium nitrate hydrate, benzene-1,3,5-tricarboxylic acid and DMF respectively in moles. For molar ratio calculations it was assumed that PBSAC spheres are composed of 100% carbon, a molecular weight of 12.01 g mol−1. | ||
| 212:2:1:600 |
3.23 | 11.87 |
Under the synthetic conditions employed for the preparation of MIL-100(Sc)@PBSAC composites, the formation of loose, powdered MIL-100(Sc) independent of the PBSAC as a white solid was observed in the reaction vessel. This powder was confirmed as crystalline MIL-100(Sc) by powder X-ray diffraction (ESI section 1.4†) suggesting that not all MIL-100(Sc) that formed was immobilised onto PBSAC spheres. Similarly to previous observations with other MOF@AC composite materials,41 the appearance of MIL-100(Sc)@PBSAC was seemingly unchanged from the as received PBSAC spheres (Fig. 1a). Additionally, a small amount of material was lost from the PBSAC in the form of black powder owing to impact with the stirrer bar inside of the reaction vessel over time. After synthesis, the material was activated by stirring in methanol for 72 hours at 40 °C to replace residual solvent material present inside of the pores of the framework and then dried at 60 °C under vacuum.
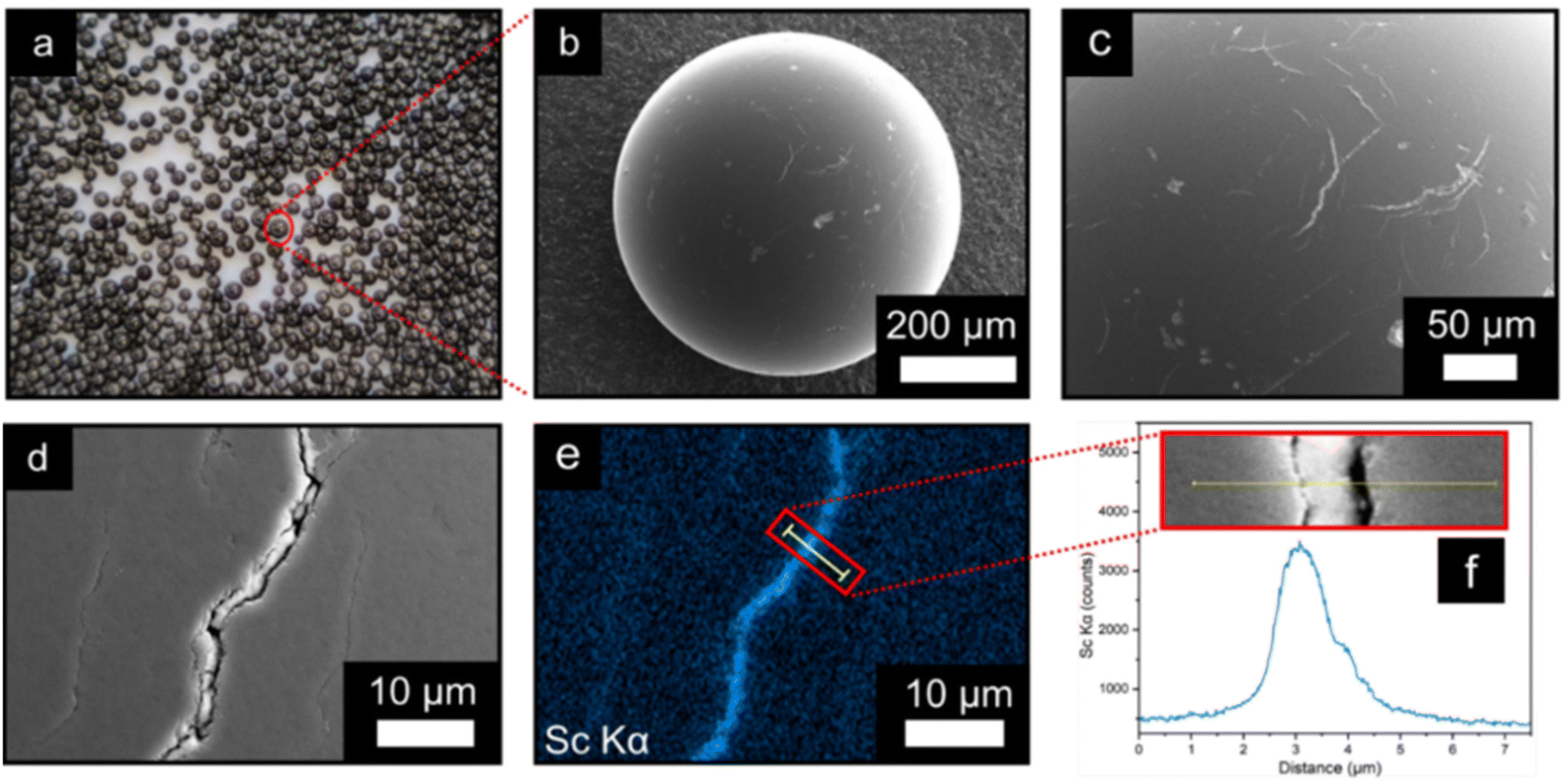 | ||
| Fig. 1 (a) Optical image of the MIL-100(Sc)@PBSAC composites. (b) SEM image of MIL-100(Sc)@PBSAC composite sphere (detector: ETD, mode: SE). (c and d) Magnified SEM images of the surface of a MIL-100(Sc)@PBSAC sphere illustrating the cracks and scratches on the surface with MIL-100(Sc) present inside of these cracks (1c detector: ETD, mode: SE). (e) Sc Kα EDX map of the surface of a MIL-100(Sc)@PBSAC sphere. (f) EDX line scan location across a crack on the surface of a MIL-100(Sc)@PBSAC sphere indicated by the yellow line in the inset image and subsequent scandium Kα counts across a crack on the surface of MIL-100(Sc)@PBSAC. | ||
The surface structure and location of MIL-100(Sc) on PBSAC spheres was studied using scanning electron microscopy and energy dispersive X-ray analysis (SEM-EDX) (Fig. 1b–f). Although not visible to the naked eye, imperfections such as scratches and cracks (≈3 μm in width) are situated throughout the outer surface of unfunctionalised PBSAC spheres (see ESI section 1.5†). MIL-100(Sc) appears to crystallise inside these cracks preferentially (Fig. 1b–d) and thus exploitation of this feature may be one approach to controlling the loading of MOFs on PBSAC spheres.
EDX mapping of the surface of MIL-100(Sc)@PBSAC suggests that the majority of MIL-100(Sc) is present within the cracks situated as indicated by the counts of the scandium Kα line scan across the surface and a crack (Fig. 1e and f).
To gain insight into both the depth of the cracks on the surface of the spheres and how far down MIL-100(Sc) resides into these cracks, FIB-SEM-EDX was used. A section of the surface of a MIL-100(Sc)@PBSAC bead was etched away to a depth of 10 μm and analysed by EDX analysis (Fig. 2a–d). This analysis revealed the cracks penetrate further than 10 μm into the surface of the PBSAC spheres, but that a majority of the MIL-100(Sc) that is immobilised in these cracks resides near the surface (approximately 2.5 μm from the surface).
 | ||
| Fig. 2 (a) SEM image showing a 10 μm FIB cross section to show the surface crack of a MIL-100(Sc)@PBSAC sphere (detector: ETD, mode: SE) (b–d) O Kα, C Kα and Sc Kα EDX map respectively of the etched sample. | ||
The presence of crystalline MIL-100(Sc) immobilised onto the PBSAC spheres was confirmed using powder X-ray diffraction (Fig. 3a and b). Despite significant diffuse background scattering in the powder pattern of MIL-100(Sc)@PBSAC as a result of the amorphous phases present in the activated carbon support,41,43 there are visible peaks at 2θ = 3–4.5, 9.5–11 and 17–20 present corresponding to the presence of crystalline MIL-100(Sc) as indicated by comparison with the underlying simulated pattern of MIL-100(Sc) (calculated by adjusting the unit cell parameter (a) of MIL-100(Cr), as reported previously44) and reflux prepared powdered MIL-100(Sc).30,33 This pattern confirms that crystalline MIL-100(Sc) was incorporated onto the PBSAC spheres.
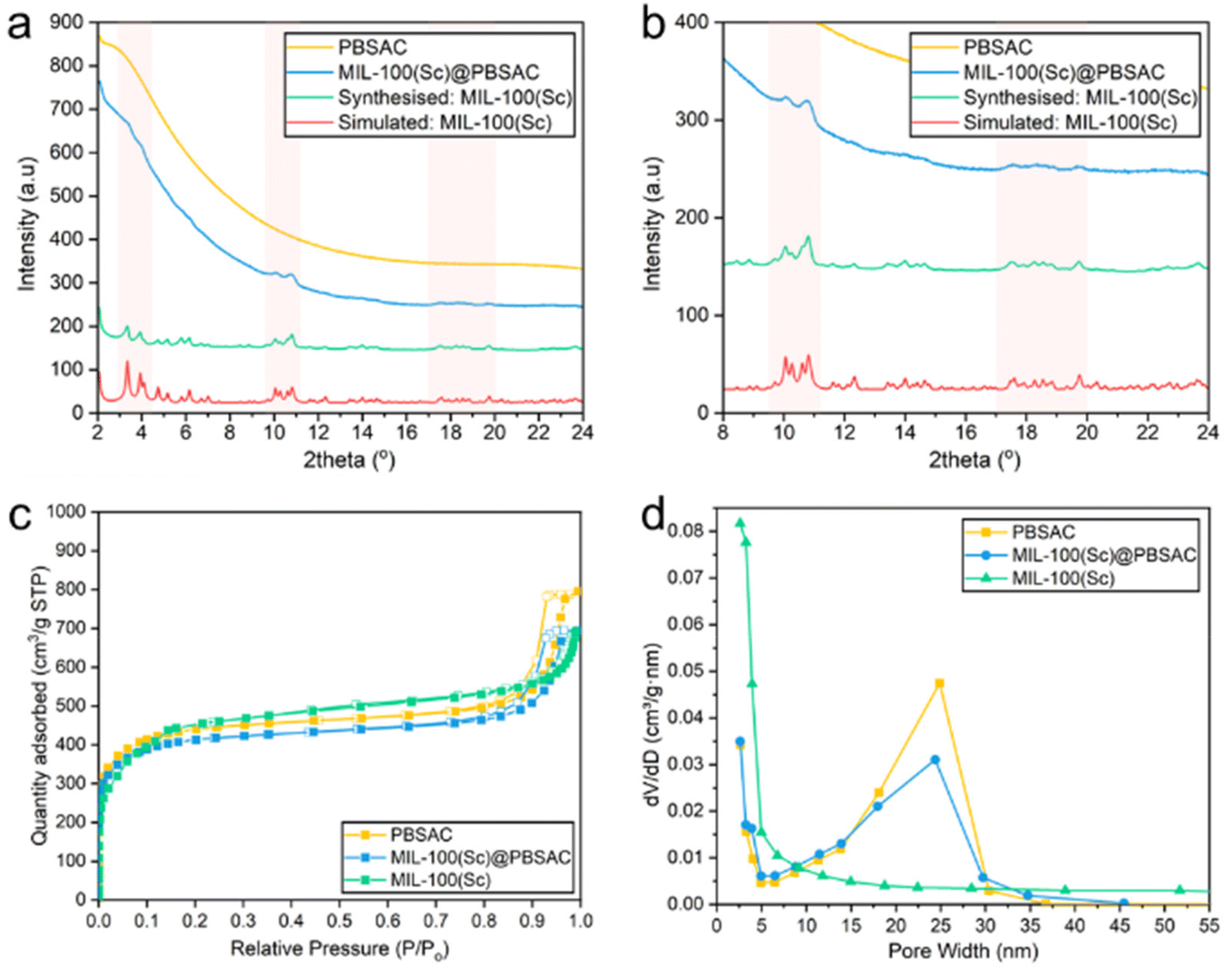 | ||
| Fig. 3 (a and b) Powder X-ray diffraction patterns of MIL-100(Sc) simulated by adjusting the unit cell parameter of MIL-100(Cr) to 74.518 Å (red), MIL-100(Sc) synthesized by reflux (green), MIL-100(Sc)@PBSAC composites (blue) and PBSAC spheres (control) (yellow). N.B. The data presented in (b) is identical to (a) but displays narrower y-axis and x-axis ranges to enable clearer MIL-100(Sc) peak visualisation. (c) N2 adsorption/desorption isotherms of PBSAC (yellow), MIL-100(Sc)@PBSAC (blue) and MIL-100(Sc) (green). The adsorption isotherm represented by filled points whilst the desorption isotherm is represented by hollow points. (d) Plot of pore width vs. BJH desorption differential pore volume for PBSAC (yellow), MIL-100(Sc)@PBSAC (blue) and MIL-100(Sc) (green). | ||
Studying porosity and metal loading
N2 adsorption/desorption analysis was used to characterise the surface and pores in the materials. Unfunctionalised PBSAC spheres displayed a type IV isotherm characteristic of mesoporosity, with a notably steep rise in adsorption at low P/Po values also suggesting the presence of micropores in PBSAC (Fig. 3c). This material also possesses a large BET surface area of 1648 m2 g−1 which is beneficial for a high dispersion of catalyst particles (Table 2). MIL-100(Sc) exhibited an N2 isotherm characteristically intermediate to type I and type IV isotherms including a secondary uptake at P/Po = 0.12, in line with previous reports for MIL-100(M, M = Fe, Cr) materials.30,32 The BET surface area of MIL-100(Sc) was calculated to be 1628 m2 g−1. When the constituent materials were combined to form MIL-100(Sc)@PBSAC, a slight decrease in BET surface area of approximately 100 m2 g−1 to 1544 m2 g−1 was recorded. We hypothesise that this is a result of some of the pores of the PBSAC spheres becoming blocked by the MOF material upon composite formation. Decreases in the surface area of similar magnitudes have been reported for other MOF@AC composite materials relative to the surface area of the constituent materials previously.45–47Upon immobilisation of MIL-100(Sc) onto PBSAC spheres, there is a decrease in the total pore volume of PBSAC from 1.23 cm3 g−1 to 1.07 cm3 g−1 suggesting that MIL-100(Sc) is partially occupying the pores of PBSAC spheres (Table 2). There is also no significant difference in the micropore volume of MIL-100(Sc)@PBSAC (0.39 cm3 g−1) compared to unfunctionalised PBSAC spheres (0.41 cm3 g−1). Fig. 3d shows the BJH desorption differential pore volumes of PBSAC, MIL-100(Sc)@PBSAC and MIL-100(Sc) in the range of 0–50 nm. This visualisation illustrates that there is a decrease in the pore volume of MIL-100(Sc)@PBSAC composites compared to unfunctionalised PBSAC spheres.
Insight into the thermal stability of the materials and the loading of MIL-100(Sc) on PBSAC spheres was obtained using thermogravimetric analysis (TGA) (Fig. 4). There are three characteristic mass changes in the TGA profiles of MIL-100 MOFs: at 0–100 °C is the removal of free solvent present in the pores of the framework, 100–350 °C shows loss of more tightly bound solvent to the metal centres throughout the framework and finally at ≈400 °C the breakdown of the MOF structure occurs.30 When analysed in air, the residual mass in the TGA profile of powdered MIL-100(Sc) corresponds to the mass of Sc2O3 that was formed following framework decomposition. By comparing the mass of residual scandium present after framework decomposition to the mass of MIL-100(Sc) present after solvent removal but before framework decomposition (at ≈400 °C), the stoichiometry of MIL-100(Sc) was inferred.
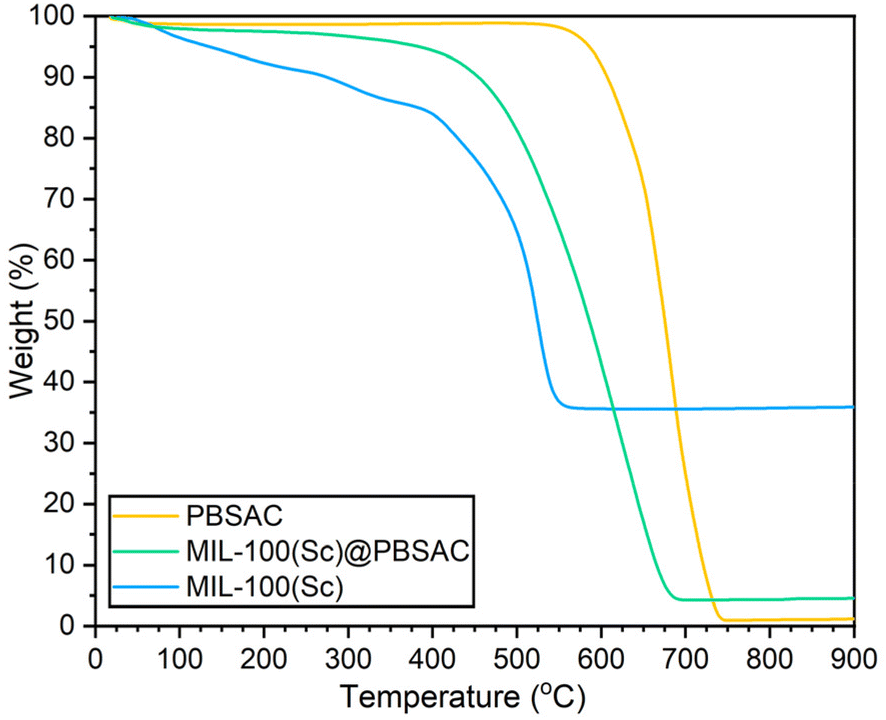 | ||
| Fig. 4 TGA profiles of PBSAC (yellow), MIL-100(Sc)@PBSAC (green) and MIL-100(Sc) (blue) in flowing air. | ||
When prepared by reflux, powdered MIL-100(Sc) possesses a scandium wt% of 27.2%, as inferred from the TGA data. This scandium wt% value may correspond to a ‘dry’ (i.e. following solvent removal) stoichiometry of [Sc3O(BTC)1.45OH2.65] where it is assumed that the anion used to balance the charge of the framework is OH− (see ESI section 1.7† for calculation). Upon the assumption that this is also the formula of MIL-100(Sc) that is incorporated onto PBSAC spheres, then full incorporation of the scandium used in the synthesis of MIL-100(Sc)@PBSAC composites gives a 11.87% MIL-100(Sc) loading (a scandium loading of 3.23% w/w).
By carrying out an analogous calculation with the mass values obtained in the TGA profile of MIL-100(Sc)@PBSAC composites but by adjusting the residual mass by subtracting the residual mass in the TGA profile of PBSAC spheres, the MIL-100(Sc) loading was calculated to be 6.42 ± 1.93% w/w (a scandium loading of 1.75 ± 0.52% w/w). The loading value obtained by carrying out these calculations was also similar in value to what was obtained with atomic absorption spectroscopy (AAS) where a MIL-100(Sc) loading value of 5.71 ± 0.93% w/w (a scandium loading of 1.55 ± 0.25% w/w) onto PBSAC spheres was measured. The loading values that were calculated were lower than the maximum value based on the mass of Sc used in the preparation (11.87% w/w MIL-100(Sc) loading), which is in keeping with the observed formation of loose powdered MIL-100(Sc) in the reaction vessel alongside MIL-100(Sc)@PBSAC spheres and thus was not included in loading calculations.
In the TGA profile of the MIL-100(Sc)@PBSAC composite (Fig. 4), the material exhibits a steady decrease in mass of approximately 5% (corresponding to the loss of material bound to the Sc3+ sites) before framework decomposition occurred at ≈380 °C. The composite MIL-100(Sc)@PBSAC was found to be less thermally stable than unfunctionalised PBSAC spheres where thermal decomposition occurred at ≈550 °C; with the thermal decomposition temperature being more similar in value to that of powdered MIL-100(Sc) where framework decomposition begins to occur at ≈380 °C. Upon incorporation into PBSAC spheres, the thermal stability of MIL-100(Sc) was shown not to improve, despite previous reports of enhanced MOF stability upon incorporation onto AC substrates.37 Inspection of the derivative weight loss curves of MIL-100(Sc) and MIL-100(Sc)@PBSAC (Fig. SI13†) shows that material decomposition occurs at a very similar temperature (maximum rate of weight loss at ≈520 °C).
Mechanical and physical properties for flow chemistry
The high mechanical strength of PBSAC spheres in relation to other shaped forms of activated carbon is one attractive property that has led to their exploration as catalyst supports in continuous flow due to the high pressures and long reaction times employed in this mode of operation. Accordingly, the crush strength of PBSAC and MIL-100(Sc)@PBSAC was measured to assess the mechanical strength of the composite material (Table 3). Both the crush strength of MIL-100(Sc)@PBSAC and unfunctionalised PBSAC spheres were similar in value (Table 3); the lack of change in mechanical strength that was observed following MIL-100(Sc)@PBSAC synthesis in conditions involving refluxing in DMF for 16 hours also demonstrates the chemical robustness of the PBSAC spheres.As well as the crush strength, the particle size of PBSAC spheres was determined in order to calculate reactor tube diameter (Dt) to particle diameter (Dp) ratio (Dt/Dp ratio) as this may have implications on the uniformity of liquid flow through the column. Generally speaking, a Dt/Dp ratio of ≥15 is desirable to minimise flow maldistribution across the reactor.48 With a D10, D50 and D90 (corresponding to the diameter below which 10%, 50% or 90% of particles are smaller than respectively) of 359 μm, 465 μm and 602 μm respectively (see ESI section 1.9† for volume distribution graph), 1/2′′ tubing with an internal diameter of 3/8′′ was selected for the packed bed reactor. This ensures uniform flow across the catalyst bed upon operation in continuous flow as the Dt/Dp ratio varied from approximately 16–27 (between the D10 to D90 value range) (Table 3).
Catalytic activity in continuous flow
Following their fabrication, the catalytic activity of MIL-100(Sc)@PBSAC was studied in continuous flow using the intramolecular cyclisation of (±)-citronellal as a model reaction. In this reaction, (±)-citronellal undergoes an intramolecular carbonyl–ene reaction resulting in the formation of one of four isomers of isopulegol (Scheme 1); industrially the formation of (−)-isopulegol is the most desirable as this species is further hydrogenated to produce the (−)-menthol isomer responsible for the characteristic mint flavour.40 For the cyclisation step, Takasago report the use of a homogeneous tris(2,6-diarylphenoxy)aluminum catalyst offering a benchmark diastereoselectivity towards (−)-isopulegol formation of 99.3%.49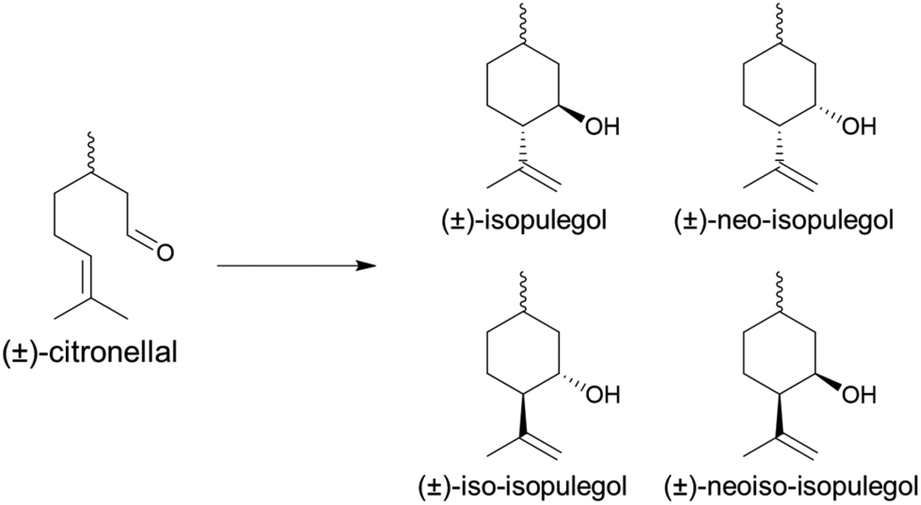 | ||
| Scheme 1 The intramolecular cyclisation of (±)-citronellal, the model reaction that was studied in this work with novel MIL-100(Sc)@PBSAC composites. Four isopulegol isomers can be produced with (±)-isopulegol the most desirable from an industrial perspective due to the relevance in synthetic menthol production. | ||
Due to the industrial significance of this reaction, it is frequently used to quantify and compare the Lewis acidic catalytic activity of novel catalysts, both homogeneous and heterogeneous,50 with the use of the more economical racemic equivalent, (±)-citronellal, often preferred. When studying this reaction using a novel Lewis acid catalyst, which is most frequently done in a batch mode of operation, important metrics include the rate of the reaction as well as the selectivity towards the formation of (±)-isopulegols and diastereoselectivity towards (±)-isopulegol formation.
To date, there are examples regarding the implementation of this reaction into continuous flow systems,51–58 but none (to the best of the authors knowledge) that employ a MOF-based catalyst in this mode of operation. Many examples still exist regarding the catalysis of this reaction by MOFs in batch and discontinuous flow, Table 5 summarises some of the most popular examples to date. For this reason, and due to the industrial importance of this reaction we therefore sought to explore the catalytic activity of novel MIL-100(Sc)@PBSAC heterogeneous catalyst composites in continuous flow.
A packed-bed reactor was filled with a combination of MIL-100(Sc)@PBSAC composite spheres and an inert packing material (500 μm glass beads) and heated to 110 °C. A solution of (±)-citronellal (0.43 M) was flowed through at 0.25 mL min−1 (Fig. 5a). The catalytic activity, selectivity and efficiency was studied over the course of 9 hours time-on-stream (Fig. 5b–d). The average results of the 9 hours time-on-stream are summarised in Table 4 (see ESI section 2† for calculations of reaction metrics).
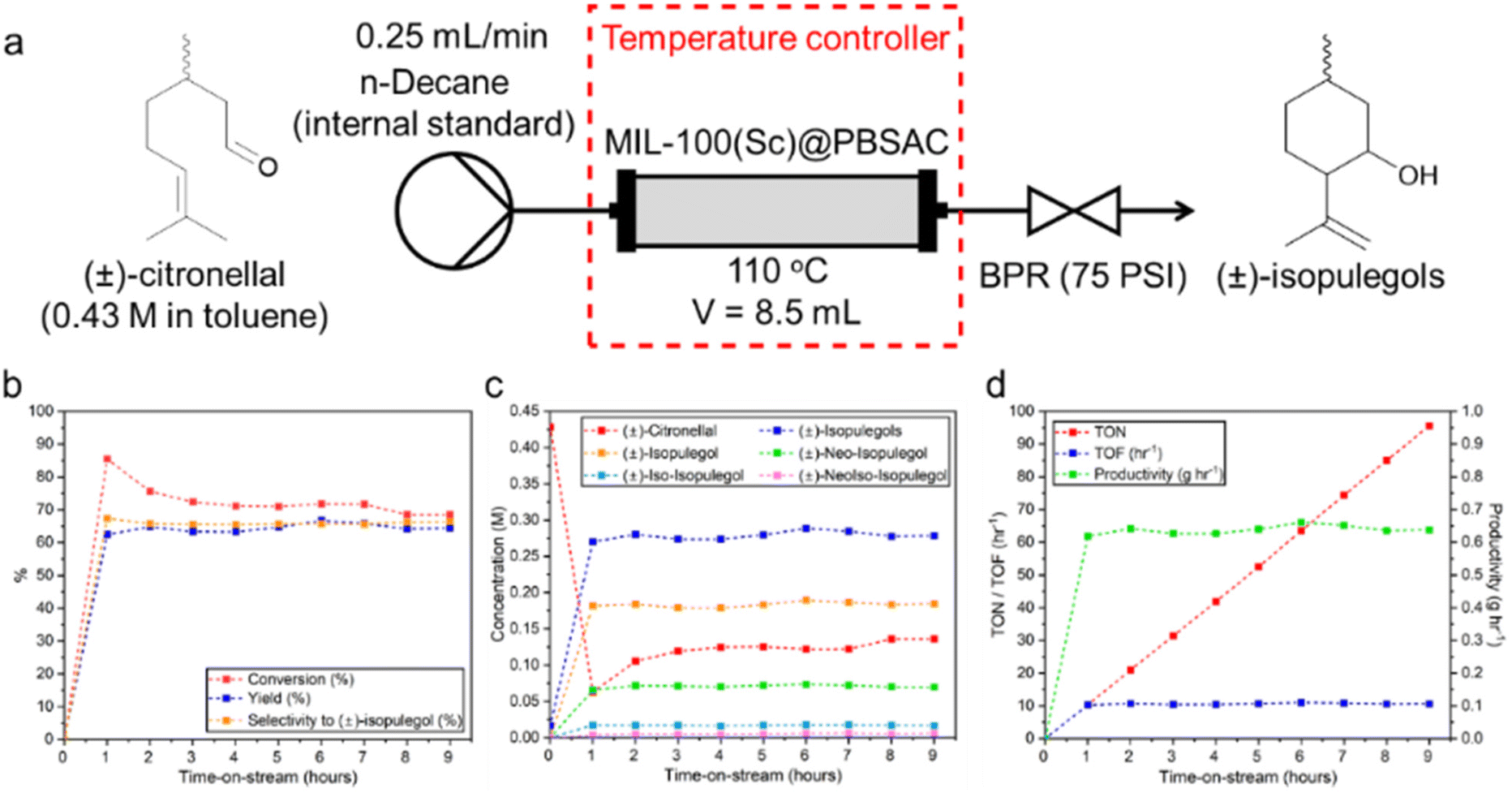 | ||
| Fig. 5 (a) The flow system used in this work in which (±)-citronellal (0.43 M in toluene, n-decane internal standard) was pumped, at 0.25 mL min−1, through a packed-bed reactor containing 1 g of MIL-100(Sc)@PBSAC and maintained at 110 °C. (b) Conversion, yield and selectivity to (±)-isopulegol vs. time plot for 9 hours time-on-stream. (c) Concentration vs. time plots for (±)-citronellal and four (±)-isopulegol isomers that are produced in the reaction for the 9 hours time-on-stream with the most abundant isomers that were formed being (±)-isopulegol, (±)-neo-isopulegol, (±)-iso-isopulegol and (±)-neoiso-isopulegol respectively. (d) TON, TOF and productivity of the reaction for the formation of (±)-isopulegols vs. time over the 9 hours on stream. | ||
| Conversion (%) | Yield of (±)-isopulegols (%) | Selectivity to (±)-isopulegols (%) | Selectivity to (±)-isopulegol (%) | Productivitya (g h−1) | TON after 9 hoursb | TOFc (h−1) |
|---|---|---|---|---|---|---|
| a Productivity represents the average amount of (±)-isopulegols formed, in g, per hour over the 9 hours on stream. b TON after 9 hours = amount of (±)-isopulegols formed after 9 hours (in moles)/amount of catalyst (in moles). c TOF (h−1) = TON per hour/time (in hours) for the 9 hours time-on-stream. Calculation of TON and TOF assumed that all scandium atoms in the MIL-100(Sc) framework participated in catalysis. | ||||||
| 72.9 ± 5.2 | 64.4 ± 1.3 | 88.8 ± 6.5 | 66.0 ± 0.6 | 0.64 ± 0.01 | 95.6 | 10.6 ± 0.2 |
Based on the liquid flow rate (0.25 mL min−1) and volume of the packed reactor (8.5 mL), the residence time was calculated to be 34 minutes. Sampling was carried out using offline gas chromatography, with an initial spike in the conversion of (±)-citronellal (86%) after one hour observed when compared to the steady conversion value that was measured over the next 8 hours time-on-stream between 69–76% (Fig. 5b). Conversion was determined by comparison of the initial (±)-citronellal concentration with the concentration that was present upon exiting the reactor. Thus, this initial spike may be indicative of phenomena such as (±)-citronellal adsorption onto the catalyst or the initial mass transfer of (±)-citronellal molecules into the pore network of the MIL-100(Sc)@PBSAC composites which, at the start of the reaction, were only filled with toluene and unoccupied by reactant molecules.
The catalytic activity (i.e. the amount of (±)-citronellal converted per gram of catalyst per hour) was measured over the course of the 9 hours time-on-stream and compared with the current literature (Table 5, column 5). MIL-100(Sc)@PBSAC composites were found to be the second most active MOF-based species to catalyse this reaction (72.9 ± 5.2 mmol gcat−1 h−1) to date after superacidic MOF-808-2.5SO4 (83.1 mmol gcat−1 h−1),64 with an Sn-β zeolite catalyst found overall to be the most active (341.2 mmol gcat−1 h−1). In some cases, the catalytic activity of MIL-100(Sc)@PBSAC composites was found to be two orders of magnitude greater than other species (i.e. Cu3(BTC)2, MIL-100(Fe), MIL-101(Cr), Pd@MIL-100(Cr), Zr(Ti)-NDC).
| Catalyst | Mode | Temperature (°C) | Solvent | Activitya (mmol gcat−1 h−1) | Selectivity to (±)-isopulegols (%) | Selectivity to (±)-isopulegol (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a Data for other catalysts taken from work carried out by Wade et al.59 Calculated as mmol of citronellal converted per gram of catalyst (MIL-100(Sc)) per hour. b Coincidentally conversion and activity values were identical for MIL-100(Sc)@PBSAC composites. c UiO-66 synthesised with different equivalents of TFA. d For hydrated and dehydrated UiO-66 respectively. e Obtained following treatment with SO4: MOF-808-2.5SO4. f Decreased with increasing acid activation. g PNNNP-Pd pincer complexes immobilised onto MFU-4l-OH. h Carried out in microwave oven. i Zeolite. j Mesoporous silicate. | |||||||
| MIL-100(Sc)@PBSAC | Flow | 110 | Toluene | 72.9 ± 5.2b | 88.8 ± 6.5 | 66.0 ± 0.6 | (This work) |
| UiO-66-NO2 | Batch | 100 | Toluene | 4.9 | — | 81 | 61 |
| UiO-66 | Batch | 110 | Toluene | — | — | 75–77c | 62 |
| (De)hydrated UiO-66 | Batch | 80 | Cyclohexane | 0.4 and 1.3d | — | 75 and 86d | 63 |
| MOF-808 | Batch | 60 | Toluene | 83.1e | — | 55–85f | 64 |
| Cu3(BTC)2 | Batch | 110 | Toluene | 0.6 | >99 | 65–69 | 65 |
| MIL-100(Fe) | Batch | 50 | Acetone | 0.2 | — | — | 66 |
| MIL-101(Cr) | Batch | 80 | Cyclohexane | 0.6 | >99 | 74 | 60 |
| Pd@MIL-101(Cr) | Batch | 80 | Cyclohexane | 0.9 | >99 | 73 | 60 |
| PdBF4g |
Batch | 100 | Toluene | 4.1–12.2g | — | 65 | 59 |
| Zr(Ti)-NDC | Batch | 150h | Toluene | 0.6 | 99 | 76 | 67 |
| Sn-βi | Flow | 80 | Acetonitrile | 341.2 | 99 | 83 | 55 |
| H-βi | Batch | 80 | Cyclohexane | — | >99 | 75 | 68 |
| Ir/H-βi | Batch | 80 | Cyclohexane | — | >99 | 75 | 68 |
| Zr-TUD-1j | Batch | 80 | Toluene | — | >99 | 65 | 69 |
For the duration of the experiment there was a stable yield (62–67%) towards the formation of (±)-isopulegols (Fig. 5b and c) meaning that, on average, 89% of the (±)-citronellal that was converted (conversion measured as difference in (±)-citronellal concentration at start and outlet of reactor) was forming (±)-isopulegols. The diastereoselectivity of the reaction, that is, the selectivity towards the formation of (±)-isopulegol compared to other (±)-isopulegols, also remained very consistent at between 65–67% over the 9 hours-on-stream (Fig. 5b and c). This value is in a similar range with what has previously been obtained with other powdered, unsupported MOF catalysts in batch (Table 5, column 8). The fact that the diastereoselectivity remained constant over the course of the 9 hours means that consistent amounts of (±)-isopulegol were reliably synthesised inviting potential for the introduction of downstream reaction modules to the system (e.g. tandem hydrogenation to produce (±)-menthol) which is enabled intrinsically by the continuous mode of operation used.
A total amount of 5.7 g of (±)-isopulegols were produced with an average productivity of 0.64 g h−1 (Table 4 and Fig. 5d); such numbers translate to a cumulative turnover number (TON) of 92.9 and turnover frequency (TOF) of 10.3 h−1.
Over the 9 hours on stream, the reaction ran smoothly with no reactor blockages or downtime. Additionally, no physical destruction of the catalyst (e.g. attrition, crushing) was observed following visual inspection after use. These positive results briefly showcase the high potential and robustness of this support material for use in continuous systems in the future. The consistent rate of reaction over 9 hours on stream as indicated by the consistent TON values indicated minimal loss of catalyst from the reactor. However, after longer reaction times (26 hours) some loss of scandium from the support (7.5–13.6%) was observed (ESI section 2.1.1†) with the conversion also decreasing to 41% while SEM examination of the catalyst after 26 hours of reaction showed no observable changes (ESI section 2.1.2†).
Conclusion
In conclusion, we have synthesised a novel catalyst consisting of the Lewis acidic catalytically active MOF, MIL-100(Sc), immobilised onto activated carbon spheres ideally shaped for use in packed-bed, continuous flow reactors. It is observed that MIL-100(Sc) resides both in the pores and within cracks situated throughout the surface of the carbon spheres. Exploitation of the new material in continuous flow revealed promising performance, with a stable conversion obtained over 9 hours on stream, with no crushing of the catalyst observed. This demonstrates the potential for MOF-based heterogeneous catalysis in continuous flow for chemical synthesis in line with the trend from batch to continuous manufacturing in the fine chemical industry. Additionally, this work may provide the basis for a general approach to exploit the high versatility of MOF-based catalysts in continuous systems and improve their practicality due to the easier handling of these materials.Author contributions
AG performed the experiments, interpreted the data and drafted the original manuscript. Expert help with continuous flow experiments and data was supplied by SB and PM, and by JH for electron microscopy. TC conceived the original idea, secured funding to perform the research and supervised the experiments and data analysis with KW (flow experiments), SC (MOF characterisation) and RB (continuous flow experiments) at the University of Leeds and the Industrial team at Sterling pharma Solutions, AS, WR and MM (API related heterogeneous catalysis). All authors contributed to drafting the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported through a studentship supported by the EPSRC Centre for Doctoral Training in Molecules to Product (EP/SO22473/1), in collaboration with Sterling Pharma Solutions. The authors greatly acknowledge their support of this work. The authors also gratefully acknowledge the help of Faye Esat and Chris Pask for the acquisition of powder X-ray diffraction data, the former within the X-ray diffraction facility as part of the Bragg Centre of Materials Research, at the University of Leeds. FIB-SEM-EDX was supported via the Bragg Centre for Materials Research, Faculty of Engineering and Physical Sciences, The University of Leeds. The authors also acknowledge Alex Kulak for SEM, thermogravimetric analysis provided by Adrian Cunliffe and Karine Alves, N2 adsorption and laser diffraction (Mastersizer 3000) provided by Benjamin Douglas, gas chromatography support from Mary Bayana, Instron 5566 for mechanical testing supplied by Mojtaba Ghadiri and Wei Pin Goh and atomic absorption spectroscopy provided by Karine Alves and Simon Lloyd at the University of Leeds.References
- C. Badman, C. L. Cooney, A. Florence, K. Konstantinov, M. Krumme, S. Mascia, M. Nasr and B. L. Trout, J. Pharm. Sci., 2019, 108, 3521–3523 CrossRef CAS PubMed.
- S. K. Teoh, C. Rathi and P. Sharratt, Org. Process Res. Dev., 2016, 20, 414–431 CrossRef CAS.
- D. M. Roberge, L. Ducry, N. Bieler, P. Cretton and B. Zimmermann, Chem. Eng. Technol., 2005, 28, 318–323 CrossRef CAS.
- FDA, Quality Considerations for Continuous Manufacturing Guidance for Industry, US, 2019 Search PubMed.
- S. Wacławek, V. V. T. Padil and M. Černík, Ecol. Chem. Eng. S, 2018, 25, 9–34 Search PubMed.
- K. Masuda, T. Ichitsuka, N. Koumura, K. Sato and S. Kobayashi, Tetrahedron, 2018, 74, 1705–1730 CrossRef CAS.
- R. Ciriminna, M. Pagliaro and R. Luque, Green Energy Environ., 2021, 6, 161–166 CrossRef CAS.
- P. Unnikrishnan and D. Srinivas, in Industrial Catalytic Processes for Fine and Specialty Chemicals, ed. S. S. Joshi and V. V. Ranade, Elsevier, Amsterdam, 2016, pp. 41–111, DOI:10.1016/B978-0-12-801457-8.00003-3.
- A. Basrur and D. Sabde, in Industrial Catalytic Processes for Fine and Specialty Chemicals, ed. S. S. Joshi and V. V. Ranade, Elsevier, Amsterdam, 2016, pp. 113–186, DOI:10.1016/B978-0-12-801457-8.00004-5.
- S. Hübner, J. G. de Vries and V. Farina, Adv. Synth. Catal., 2016, 358, 3–25 CrossRef.
- B. Böhringer, O. Guerra Gonzalez, I. Eckle, M. Müller, J.-M. Giebelhausen, C. Schrage and S. Fichtner, Chem. Ing. Tech., 2011, 83, 53–60 CrossRef.
- E. Fernandez-Puertas, A. J. Robinson, H. Robinson, S. Sathiyalingam, H. Stubbs and L. J. Edwards, Org. Process Res. Dev., 2020, 24, 2147–2156 CrossRef CAS.
- A. Feiz, A. Bazgir, A. M. Balu and R. Luque, Sci. Rep., 2016, 6, 32719 CrossRef PubMed.
- G. Orsy, F. Fülöp and I. M. Mándity, Green Chem., 2019, 21, 956–961 RSC.
- A. J. Garcia-Olmo, A. Yepez, A. M. Balu, A. A. Romero, Y. Li and R. Luque, Catal. Sci. Technol., 2016, 6, 4705–4711 RSC.
- L. I. Godina, A. V. Tokarev, I. L. Simakova, P. Mäki-Arvela, E. Kortesmäki, J. Gläsel, L. Kronberg, B. Etzold and D. Y. Murzin, Catal. Today, 2018, 301, 78–89 CrossRef CAS.
- M. Munoz, V. Kolb, A. Lamolda, Z. M. de Pedro, A. Modrow, B. J. M. Etzold, J. J. Rodriguez and J. A. Casas, Appl. Catal., B, 2017, 218, 498–505 CrossRef CAS.
- H. Klefer, M. Munoz, A. Modrow, B. Böhringer, P. Wasserscheid and B. J. M. Etzold, Chem. Eng. Technol., 2016, 39, 276–284 CrossRef CAS.
- M.-H. Baek, J.-W. Yoon, J.-S. Hong and J.-K. Suh, Appl. Catal., A, 2013, 450, 222–229 CrossRef CAS.
- S. Fichtner, J. Hofmann, A. Möller, C. Schrage, J. M. Giebelhausen, B. Böhringer and R. Gläser, J. Hazard. Mater., 2013, 262, 789–795 CrossRef CAS PubMed.
- P. C. With, N. Wilde, A. Modrow, S. Fichtner, B. Böhringer and R. Gläser, Chem. Eng. Technol., 2015, 38, 1671–1676 CrossRef CAS.
- M. J. Schneider, M. Haumann, M. Stricker, J. Sundermeyer and P. Wasserscheid, J. Catal., 2014, 309, 71–78 CrossRef CAS.
- R. Castro-Amoedo, Z. Csendes, J. Brünig, M. Sauer, A. Foelske-Schmitz, N. Yigit, G. Rupprechter, T. Gupta, A. M. Martins, K. Bica, H. Hoffmann and K. Kirchner, Catal. Sci. Technol., 2018, 8, 4812–4820 RSC.
- F. T. U. Kohler, S. Popp, H. Klefer, I. Eckle, C. Schrage, B. Böhringer, D. Roth, M. Haumann and P. Wasserscheid, Green Chem., 2014, 16, 3560–3568 RSC.
- A. Weiß, M. Munoz, A. Haas, F. Rietzler, H.-P. Steinrück, M. Haumann, P. Wasserscheid and B. J. M. Etzold, ACS Catal., 2016, 6, 2280–2286 CrossRef.
- R. Gil-San-Millan, P. Delgado, E. Lopez-Maya, J. D. Martin-Romera, E. Barea and J. A. R. Navarro, ACS Appl. Mater. Interfaces, 2021, 13, 50491–50496 CrossRef CAS PubMed.
- W. Xiang, S. Gebhardt, R. Gläser and C.-J. Liu, Microporous Mesoporous Mater., 2020, 300, 110152 CrossRef CAS.
- D. Yang and B. C. Gates, ACS Catal., 2019, 9, 1779–1798 CrossRef CAS.
- A. Dhakshinamoorthy, S. Navalon, A. M. Asiri and H. Garcia, Chem. Commun., 2020, 56, 26–45 RSC.
- P. Horcajada, S. Surblé, C. Serre, D.-Y. Hong, Y.-K. Seo, J.-S. Chang, J.-M. Grenèche, I. Margiolaki and G. Férey, Chem. Commun., 2007, 2820–2822 RSC.
- G. Zhong, D. Liu and J. Zhang, Cryst. Growth Des., 2018, 18, 7730–7744 CrossRef CAS.
- G. Férey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surblé, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296–6301 CrossRef PubMed.
- L. Mitchell, B. Gonzalez-Santiago, J. P. S. Mowat, M. E. Gunn, P. Williamson, N. Acerbi, M. L. Clarke and P. A. Wright, Catal. Sci. Technol., 2013, 3, 606–617 RSC.
- L. Mitchell, P. Williamson, B. Ehrlichová, A. E. Anderson, V. R. Seymour, S. E. Ashbrook, N. Acerbi, L. M. Daniels, R. I. Walton, M. L. Clarke and P. A. Wright, Chem. – Eur. J., 2014, 20, 17185–17197 CrossRef CAS PubMed.
- G. Gómez-Pozuelo, C. P. Cabello, M. Opanasenko, M. Horáček and J. Čejka, ChemPlusChem, 2017, 82, 152–159 CrossRef PubMed.
- B. R. James, J. A. Boissonnault, A. G. Wong-Foy, A. J. Matzger and M. S. Sanford, RSC Adv., 2018, 8, 2132–2137 RSC.
- J. C. Muñoz-Senmache, S. Kim, R. R. Arrieta-Pérez, C. M. Park, Y. Yoon and A. J. Hernández-Maldonado, ACS Appl. Nano Mater., 2020, 3, 2928–2940 CrossRef.
- M. Emura and H. Matsuda, Chem. Biodiversity, 2014, 11, 1688–1699 CrossRef CAS PubMed.
- G. P. P. Kamatou, I. Vermaak, A. M. Viljoen and B. M. Lawrence, Phytochemistry, 2013, 96, 15–25 CrossRef CAS PubMed.
- D. Dylong, P. J. C. Hausoul, R. Palkovits and M. Eisenacher, Flavour Fragrance J., 2022, 37, 195–209 CrossRef CAS.
- L. N. McHugh, A. Terracina, P. S. Wheatley, G. Buscarino, M. W. Smith and R. E. Morris, Angew. Chem., Int. Ed., 2019, 58, 11747–11751 CrossRef CAS PubMed.
- J. W. M. Osterrieth, J. Rampersad, D. Madden, N. Rampal, L. Skoric, B. Connolly, M. D. Allendorf, V. Stavila, J. L. Snider, R. Ameloot, J. Marreiros, C. Ania, D. Azevedo, E. Vilarrasa-Garcia, B. F. Santos, X.-H. Bu, Z. Chang, H. Bunzen, N. R. Champness, S. L. Griffin, B. Chen, R.-B. Lin, B. Coasne, S. Cohen, J. C. Moreton, Y. J. Colón, L. Chen, R. Clowes, F.-X. Coudert, Y. Cui, B. Hou, D. M. D'Alessandro, P. W. Doheny, M. Dincă, C. Sun, C. Doonan, M. T. Huxley, J. D. Evans, P. Falcaro, R. Ricco, O. Farha, K. B. Idrees, T. Islamoglu, P. Feng, H. Yang, R. S. Forgan, D. Bara, S. Furukawa, E. Sanchez, J. Gascon, S. Telalović, S. K. Ghosh, S. Mukherjee, M. R. Hill, M. M. Sadiq, P. Horcajada, P. Salcedo-Abraira, K. Kaneko, R. Kukobat, J. Kenvin, S. Keskin, S. Kitagawa, K.-I. Otake, R. P. Lively, S. J. A. DeWitt, P. Llewellyn, B. V. Lotsch, S. T. Emmerling, A. M. Pütz, C. Martí-Gastaldo, N. M. Padial, J. García-Martínez, N. Linares, D. Maspoch, J. A. Suárez del Pino, P. Moghadam, R. Oktavian, R. E. Morris, P. S. Wheatley, J. Navarro, C. Petit, D. Danaci, M. J. Rosseinsky, A. P. Katsoulidis, M. Schröder, X. Han, S. Yang, C. Serre, G. Mouchaham, D. S. Sholl, R. Thyagarajan, D. Siderius, R. Q. Snurr, R. B. Goncalves, S. Telfer, S. J. Lee, V. P. Ting, J. L. Rowlandson, T. Uemura, T. Iiyuka, M. A. van der Veen, D. Rega, V. Van Speybroeck, S. M. J. Rogge, A. Lamaire, K. S. Walton, L. W. Bingel, S. Wuttke, J. Andreo, O. Yaghi, B. Zhang, C. T. Yavuz, T. S. Nguyen, F. Zamora, C. Montoro, H. Zhou, A. Kirchon and D. Fairen-Jimenez, Adv. Mater., 2022, 34, 2201502 CrossRef CAS PubMed.
- Z. Zhu, A. Li, S. Zhong, F. Liu and Q. Zhang, J. Appl. Polym. Sci., 2008, 109, 1692–1698 CrossRef CAS.
- D. F. Sava Gallis, K. W. Chapman, M. A. Rodriguez, J. A. Greathouse, M. V. Parkes and T. M. Nenoff, Chem. Mater., 2016, 28, 3327–3336 CrossRef CAS.
- K. L. B. Solis, Y.-H. Kwon, M.-H. Kim, H.-R. An, C. Jeon and Y. Hong, Chemosphere, 2020, 238, 124656 CrossRef CAS PubMed.
- S.-Y. Lee and S.-J. Park, Int. J. Hydrogen Energy, 2011, 36, 8381–8387 CrossRef CAS.
- O. Abuzalat, D. Wong and M. A. Elsayed, J. Inorg. Organomet. Polym. Mater., 2022, 32, 1924–1934 CrossRef CAS.
- R. K. Reddy and J. B. Joshi, Particuology, 2010, 8, 37–43 CrossRef CAS.
- H. Itoh, H. Maeda, S. Yamada, Y. Hori, T. Mino and M. Sakamoto, RSC Adv., 2014, 4, 61619–61623 RSC.
- E. J. Lenardão, G. V. Botteselle, F. de Azambuja, G. Perin and R. G. Jacob, Tetrahedron, 2007, 63, 6671–6712 CrossRef.
- A. Zuliani, C. M. Cova, R. Manno, V. Sebastian, A. A. Romero and R. Luque, Green Chem., 2020, 22, 379–387 RSC.
- H. Chen, H. Zou, Y. Hao and H. Yang, ChemSusChem, 2017, 10, 1989–1995 CrossRef CAS PubMed.
- Z. Vajglová, N. Kumar, P. Mäki-Arvela, K. Eränen, M. Peurla, L. Hupa, M. Nurmi, M. Toivakka and D. Y. Murzin, Ind. Eng. Chem. Res., 2019, 58, 18084–18096 CrossRef.
- Z. Vajglová, N. Kumar, P. Mäki-Arvela, K. Eränen, M. Peurla, L. Hupa and D. Y. Murzin, Org. Process Res. Dev., 2019, 23, 2456–2463 CrossRef.
- A. Corma and M. Renz, Chem. Commun., 2004, 550–551 RSC.
- V. I. Anikeev, I. V. Il'ina, K. P. Volcho and N. F. Salakhutdinov, Russ. J. Phys. Chem. A, 2012, 86, 1917–1919 CrossRef CAS.
- M. Azkaar, P. Mäki-Arvela, Z. Vajglová, V. Fedorov, N. Kumar, L. Hupa, J. Hemming, M. Peurla, A. Aho and D. Y. Murzin, React. Chem. Eng., 2019, 4, 2156–2169 RSC.
- S. Chatterjee, V. Degirmenci and E. V. Rebrov, Chem. Eng. J., 2015, 281, 884–891 CrossRef CAS.
- J. S. Hilliard and C. R. Wade, Dalton Trans., 2023, 52, 1608–1615 RSC.
- F. G. Cirujano, F. X. Llabrés i Xamena and A. Corma, Dalton Trans., 2012, 41, 4249–4254 RSC.
- F. Vermoortele, M. Vandichel, B. Van de Voorde, R. Ameloot, M. Waroquier, V. Van Speybroeck and D. E. De Vos, Angew. Chem., Int. Ed., 2012, 51, 4887–4890 CrossRef CAS PubMed.
- F. Vermoortele, B. Bueken, G. Le Bars, B. Van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. Van Speybroeck, C. Kirschhock and D. E. De Vos, J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- F. G. Cirujano and F. X. Llabrés i Xamena, J. Phys. Chem. Lett., 2020, 11, 4879–4890 CrossRef CAS PubMed.
- J. Jiang, F. Gándara, Y.-B. Zhang, K. Na, O. M. Yaghi and W. G. Klemperer, J. Am. Chem. Soc., 2014, 136, 12844–12847 CrossRef CAS PubMed.
- L. Alaerts, E. Séguin, H. Poelman, F. Thibault-Starzyk, P. A. Jacobs and D. E. De Vos, Chem. – Eur. J., 2006, 12, 7353–7363 CrossRef CAS PubMed.
- F. Vermoortele, R. Ameloot, L. Alaerts, R. Matthessen, B. Carlier, E. V. R. Fernandez, J. Gascon, F. Kapteijn and D. E. De Vos, J. Mater. Chem., 2012, 22, 10313–10321 RSC.
- A. M. Rasero-Almansa, M. Iglesias and F. Sánchez, RSC Adv., 2016, 6, 106790–106797 RSC.
- F. Neaţu, S. Coman, V. I. Pârvulescu, G. Poncelet, D. De Vos and P. Jacobs, Top. Catal., 2009, 52, 1292–1300 CrossRef.
- A. Ramanathan, M. Carmen Castro Villalobos, C. Kwakernaak, S. Telalovic and U. Hanefeld, Chem. – Eur. J., 2008, 14, 961–972 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr03634k |
| This journal is © The Royal Society of Chemistry 2023 |