
Core–multi-shell design: unlocking multimodal capabilities in lanthanide-based nanoparticles as upconverting, T2-weighted MRI and CT probes†
Nan
Liu
 a,
Christian
Homann
a,
Samuel
Morfin
b,
Meghana S.
Kesanakurti
a,
Nicholas D.
Calvert
ac,
Adam J.
Shuhendler
acd,
Tom
Al
b and
Eva
Hemmer
*a
a,
Christian
Homann
a,
Samuel
Morfin
b,
Meghana S.
Kesanakurti
a,
Nicholas D.
Calvert
ac,
Adam J.
Shuhendler
acd,
Tom
Al
b and
Eva
Hemmer
*a
aDepartment of Chemistry and Biomolecular Sciences, University of Ottawa, Ottawa, ON, Canada. E-mail: ehemmer@uottawa.ca
bDepartment of Earth and Environmental Sciences, University of Ottawa, Ottawa, ON, Canada
cUniversity of Ottawa Heart Institute, Ottawa, ON, Canada
dDepartment of Biology, University of Ottawa, Ottawa, ON, Canada
First published on 13th November 2023
Abstract
Multimodal bioimaging probes merging optical imaging, magnetic resonance imaging (MRI), and X-ray computed tomography (CT) capabilities have attracted considerable attention due to their potential biomedical applications. Lanthanide-based nanoparticles are promising candidates for multimodal imaging because of their optical, magnetic and X-ray attenuation properties. We prepared a set of hexagonal-phase (β)-NaGdF4:Yb,Er/NaGdF4/NaDyF4 core/shell/shell nanoparticles (Dy-CSS NPs) and demonstrated their optical/T2-weighted MRI/CT multimodal capabilities. A known drawback of multimodal probes that merge the upconverting Er3+/Yb3+ ion pair with magnetic Dy3+ ions for T2-weighted MRI is the loss of upconversion (UC) emission due to Dy3+ poisoning. Particular attention was paid to controlled nanoparticle architectures with tuned inner shell thicknesses separating Dy3+ and Er3+/Yb3+ to shed light on the distance-dependent loss of UC due to Yb3+ → Dy3+ energy transfer. Based on the Er3+ UC spectra and the excited state lifetime of Yb3+, a 4 nm thick NaGdF4 inner shell did not only restore but enhanced the UC emission. We further investigated the effect of the outer NaDyF4 shell thickness on the particles’ magnetic and CT performance. MRI T2 relaxivity measurements in vitro at a magnetic field of 7 T performed on citrate-capped Dy-CSS NPs revealed that NPs with the thickest outer shell thickness (4 nm) exhibited the highest r2 value, with a superior T2 contrast effect compared to commercial iron oxide and other Dy-based T2 contrast agents. In addition, the citrate-capped Dy-CSS NPs were demonstrated suitable for CT in in vitro imaging phantoms at X-ray energies of 110 keV, rendering them interesting alternatives to clinically used iodine-based agents that operate at lower energies.
Introduction
Biomedical imaging plays a critical role in health and life sciences, from basic research to diagnostics and treatment.1,2 Among the various imaging techniques currently available to healthcare providers and researchers, optical imaging allows for the investigation of time-dependent biological phenomena with high spatial imaging resolution and sensitivity. Yet, one of its major drawbacks is the low penetration depth of light.3 In contrast, magnetic resonance imaging (MRI) and X-ray computed tomography (CT) imaging come with the advantage to provide physiological and anatomical information with unlimited penetration depth. However, their relatively low spatial resolution and sensitivity remain a challenge.4 Merging the advantages of each imaging technique on its own, multimodal approaches have been proposed. In this vein, multifunctional nanoparticles (NPs) exhibiting properties suitable for two or more imaging modalities bear great potential as bioprobes enabling multimodal imaging.5–10 These multimodal imaging probes may provide more precise diagnosis and monitoring of disease progression through integrating several imaging modalities with different properties into one nanoparticle platform.11Lanthanide (Ln)-based materials are promising candidates for multimodal imaging probes because of their optical, magnetic, and X-ray attenuation properties.6,7,12,13 Ln-based materials exhibit unique optical properties due to their capability to emit UV and visible light (upconversion) as well as near-infrared (NIR) light (downshifting) when excited with NIR light (e.g. 980 or 808 nm).14–16 Compared to conventional optical probes excited with UV-visible light, the use of NIR light provides several advantages, such as deeper penetration depth, weak autofluorescence, minimal photobleaching, and low phototoxicity.16,17 In addition, gadolinium- (Gd3+) and dysprosium- (Dy3+) based materials have attracted growing attention as MRI contrast agents (CAs) because of their characteristic magnetic properties. Owing to the Gd3+ 4f7 electron configuration, Gd3+-based chelates are commonly used as MRI T1 CAs.18,19 Gd3+-based NPs have also been widely investigated as alternatives to chelates due to their longer blood circulation time and large density of Gd3+ ions on their surface, which dramatically increases the concentration of the contrast-providing species in the region of interest.20–23 On the other hand, clinically used MRI T2 CAs are based on iron oxides, which, however, suffer from magnetization saturation at high magnetic fields (greater than 3 T).24 Dy3+ is a suitable candidate for high-field T2 CAs due to its relatively large magnetic moment (10.6μB) and short relaxation time, without saturation of the magnetization even at high magnetic fields up to 11.7 T.25–28 High-field MRI is gaining increasing attention due to its higher spatial resolution and signal-to-noise ratio, holding potential for more effective diagnostic imaging.28,29 Therewith comes the raising demand for suitable high-field T2 CAs.
Ln-based NPs as optical/T1 imaging probes combining luminescent Ln3+ (e.g. Er3+, Tm3+, Nd3+, Ho3+) and magnetic Gd3+ ions have been widely studied.7,30–32 However, there are only a few reports about multimodal probes that combine optical and T2 imaging capabilities.5,6,9,33 A main reason for the slow development of optical/T2 probes based on Ln3+ ions may be the severe quenching effect of Dy3+ on the UC luminescence, thus, hampering the probe's optical performance.34 In order to circumvent the UC poisoning effect of Dy3+ ions, some studies suggested to physically separate Dy3+ from the luminescent Ln3+ ions, e.g. Er3+ or Tm3+.5,9,33–35 For instance, Su et al. synthesized NaGdF4:Yb,Er/NaDyF4 core/shell (CS) NPs and found that the UC emission intensity of Er3+ decreased upon shelling with NaDyF4 because of the quenching effect of the Dy3+ ions.34 To overcome this issue, Biju et al. proposed a strategy based on a lower Dy3+ concentration in the shell by preparing NaGdF4:Yb,Er/NaYbF4:Dy NPs with 38 mol% Dy3+ doped into the shell. While UC emission could be partially restored, the emission intensity still dropped by 27% compared to that of CS NPs with an undoped NaGdF4 shell.33 Moreover, reducing the amount of Dy3+ ions might not be advantageous when aiming for additional T2 imaging capabilities. The introduction of an intermediate CaF2 shell as a separating barrier between Dy3+ and the emitting Ln3+ ions was introduced by Li et al. A 2.3 nm thick inert shell between the upconverting Tm3+ ions in the core and the Dy3+ ions in the outer shell (NaYbF4:Tm/CaF2/NaDyF4 core/shell/shell (CSS) architecture) allowed for UC emission intensities of Tm3+ as high as those from NaYbF4:Tm/CaF2 CS NPs.5 This and related work demonstrated the high promise of the CSS architecture design as a strategy towards optical/T2-weighted MRI probes based on upconverting Ln3+ and Dy3+ ions.6
In addition, the potential of Ln-based NPs as CAs for CT imaging applications has been demonstrated,36 given their relatively higher X-ray attenuation coefficients compared to clinically used iodine-based CAs (I: 1.9, Gd: 3.1, Dy: 3.4, Yb: 3.9 cm2 g−1 at 100 keV![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 37). The higher X-ray attenuation coefficient of these Ln-based NPs can confer higher efficacy than that of iodine-based CAs, thus, expecting to provide an equivalent contrast at a lower dose compared to the iodine-based CAs. This reduced dose requirement is highly beneficial in a contrast agent as it may reduce potential adverse effects in patients in clinical applications.10,38
37). The higher X-ray attenuation coefficient of these Ln-based NPs can confer higher efficacy than that of iodine-based CAs, thus, expecting to provide an equivalent contrast at a lower dose compared to the iodine-based CAs. This reduced dose requirement is highly beneficial in a contrast agent as it may reduce potential adverse effects in patients in clinical applications.10,38
The overarching goal of this work was to design Ln3+-based nanoparticle probes that merge the manifold properties of the Ln3+ ions into one multimodal imaging agent that has UC, T2-modulating, and CT capabilities. Inspired by previous work, here, we designed a set of CSS NPs, i.e., hexagonal (β)-phase NaGdF4:Yb,Er/NaGdF4/NaDyF4 (Dy-CSS, Fig. 1A). Particular attention was paid to the establishment of new protocols for the controlled shell growth via a rapid and straightforward microwave-assisted route. The adjustment of the inner and outer shell thicknesses allowed to (i) shed light on distance-dependent Dy3+ quenching effects and energy transfer mechanisms as well as (ii) assess the effect of shell thickness on T2 MRI and CT performance. As a result, Dy-CSS NPs were obtained with not only restored but enhanced UC emission as well as superior T2 and CT capabilities compared to previously reported nanoparticle architectures and common CAs, holding potential as a multimodal imaging probe.
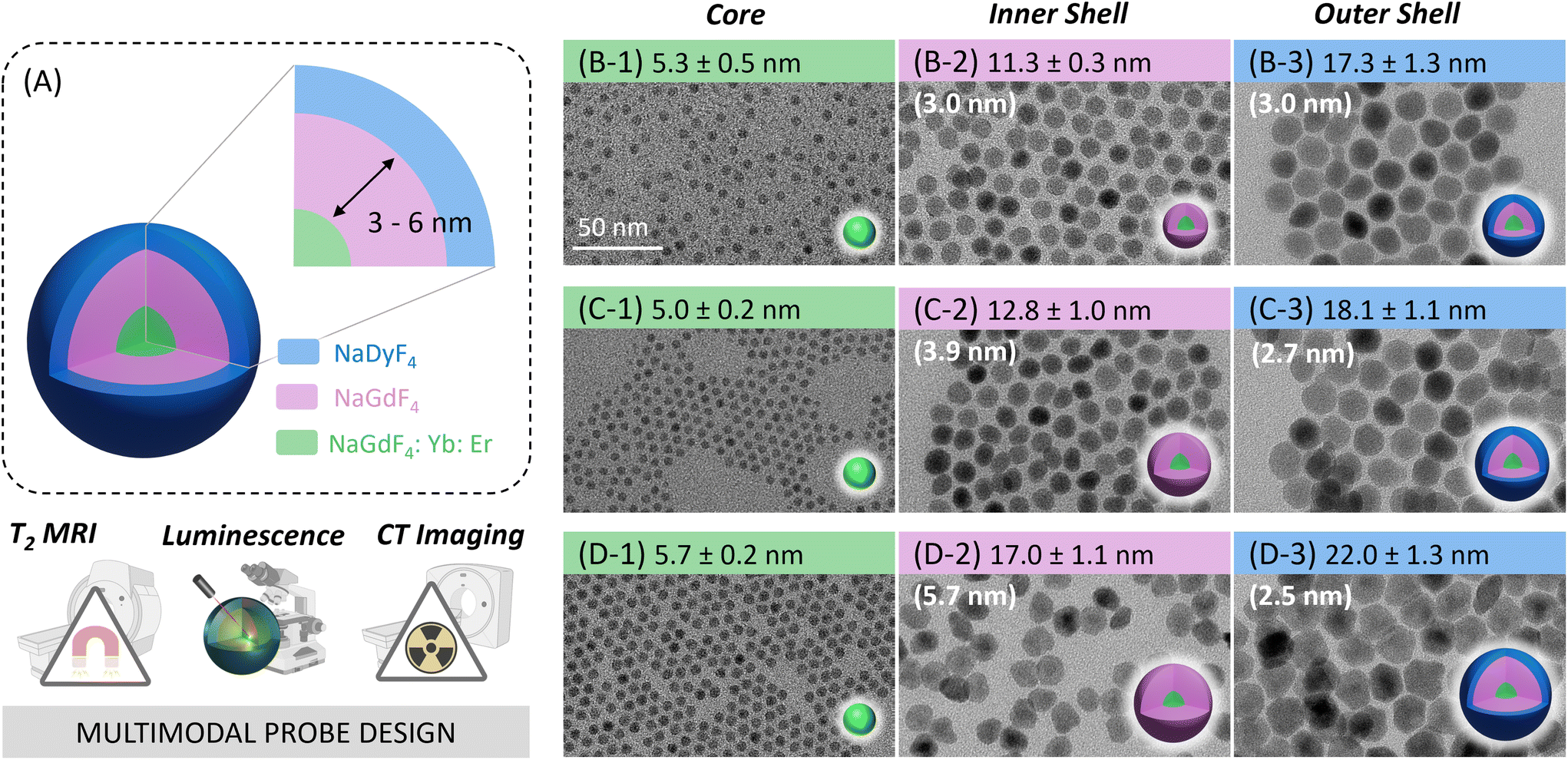 | ||
| Fig. 1 (A) Schematic illustration of the core/shell/shell Dy-NP architecture proposed as multimodal optical/T2/CT probe. (B–D) TEM images and sizes of OA-capped (1) core β-NaGdF4:Yb,Er, (2) core/shell β-NaGdF4:Yb,Er/NaGdF4, and (3) core/shell/shell β-NaGdF4:Yb,Er/NaGdF4/NaDyF4 NPs. The NaGdF4 inner shell thickness and NaDyF4 outer shell thickness are indicated in brackets. Core sizes and outer shell thicknesses were kept comparable, while the thickness of the inner shell increased from (B) to (D). The 50 nm scale bar applies to all TEM images. | ||
Results and discussion
Synthesis and structural characterization of Dy-CSS NPs
A set of oleate (OA)-capped Dy-CSS NPs was successfully synthesized using a modified version of the microwave-assisted protocol previously established by our group.21,39 Control over the shell thickness in multi-shell Ln-NPs is crucial to optimize their performance and to understand structure–property relationships. While the microwave-assisted route was previously reported as suitable for simple core/shell architectures, stringent control over shell thicknesses and the synthesis of core/multi-shell systems remained a challenge. Here, we demonstrate that the adjustment of the ratio between core NPs acting as seeds for shell growth and shell precursor (Gd-TFA) in the reaction mixture allowed to tune the thickness of the undoped NaGdF4 shell on the Er/Yb-co-doped core from ca. 3 to 4 and 6 nm, respectively (Fig. 1, Fig. S1 and Table S1†). Following the same strategy, the thickness of the outer NaDyF4 shell of the CSS Dy-NPs could be controlled (vide infra). A detailed description of the synthesis of the CSS NPs – hereafter labelled as 3nmDy-CSS3nm, 4nmDy-CSS3nm, and 6nmDy-CSS3nm, whereas the first and second superscript indicate the thickness of the inner and outer shell, respectively – is provided in the Experimental section. X-ray diffraction (XRD) analysis confirmed that all the Dy-CSS NPs were of pure hexagonal phase of NaGdF4 and NaDyF4 (Fig. S2†). The morphology and size of core, CS and CSS NPs were examined by transmission electron microscopy (TEM). As shown in Fig. 1B–D and Fig. S3,† all NPs were monodisperse with narrow size distributions. The obtained NaGdF4 core NPs, co-doped with 2 mol% Er3+ and 20 mol% Yb3+ to induce luminescent properties, exhibited a spherical morphology with an average diameter of ca. 5.4 nm. Growing an undoped NaGdF4 shell resulted in CS NPs of 11.3 ± 0.3 nm, 12.8 ± 1.0 nm, and 17.0 ± 1.1 nm, with controlled shell thicknesses of ca. 3, 4, and 6 nm. Further growth of a ca. 2.7 nm thick NaDyF4 outer shell to introduce Dy3+ ions for T2-weighted MRI led to overall NP sizes of 17.3 ± 1.3 nm, 18.1 ± 1.1 nm, and 22.0 ± 1.3 nm for 3nmDy-CSS3nm, 4nmDy-CSS3nm, and 6nmDy-CSS3nm, respectively. Importantly, the core size and outer shell thickness of the obtained Dy-CSS NPs were kept comparable among the three NP architectures, while three different inner shell thicknesses were introduced. Such control of the CSS architecture allows for the investigation of the efficiency of the inner barrier layer between the Er3+/Yb3+ ions in the core and the Dy3+ ions in the outer shell.Optical properties of Dy-CSS NPs
The upconversion (UC) luminescence properties of the obtained NPs, dispersed in toluene, were investigated under 980 nm excitation. As shown in Fig. 2(A-1), the emission spectra of 3nmDy-CSS3nm and their corresponding core/shell (3nmCS) and core NPs exhibited characteristic upconversion emission peaks in the green and red spectral regions ascribed to the 2H11/2 → 4I15/2 (520 nm), 4S3/2 → 4I15/2 (540 nm), and 4F9/2 → 4I15/2 (650 nm) Er3+ transitions. The low emission intensity obtained from core NPs was no surprise given the very small NP size (ca. 5 nm) resulting in severe surface quenching. When adding a 3 nm undoped NaGdF4 shell, as expected, the emission intensity increased, reducing surface quenching effects.40 However, the addition of an additional outer NaDyF4 shell (3nmDy-CSS3nm NPs) resulted in lower UC emission intensities than obtained for 3nmCS NPs without the NaDyF4. The observed loss (ca. 50% for both the green and red emissions) indicated that an inner shell of 3 nm thickness was not sufficient to protect the emitter pair Er3+/Yb3+ from the Dy3+ quencher. Previous studies reported that an inner CaF2 shell as thin as 2.3 nm was suitable to restore the UC emission,5 while in our study, emission loss was observed with a 3 nm inner NaGdF4 shell. This might be explained by potential cation intermixing in the Dy-CSS NPs taking place during shell growth, resulting in the distribution of Ln3+ dopant ions (i.e., Er3+ and Dy3+) into the nominally undoped shell.40,41 As expected, growing a thicker inner shell of 4 nm on core NPs significantly increased the UC emission intensity, 4nmCS NPs showing an overall 2000 times higher UC intensity than core NPs (Fig. 2(A-2)). Most interestingly, no loss of UC was observed upon the addition of an outer NaDyF4 shell. The obtained 4nmDy-CSS3nm NPs exhibited not only restored UC emission but an UC enhancement by more than 100% for the green and by ca. 270% for the red emission compared to their respective 4nmCS NPs. This result indicated that an inner NaGdF4 shell of at least 4 nm in thickness was required to overcome the Dy3+-induced loss of UC.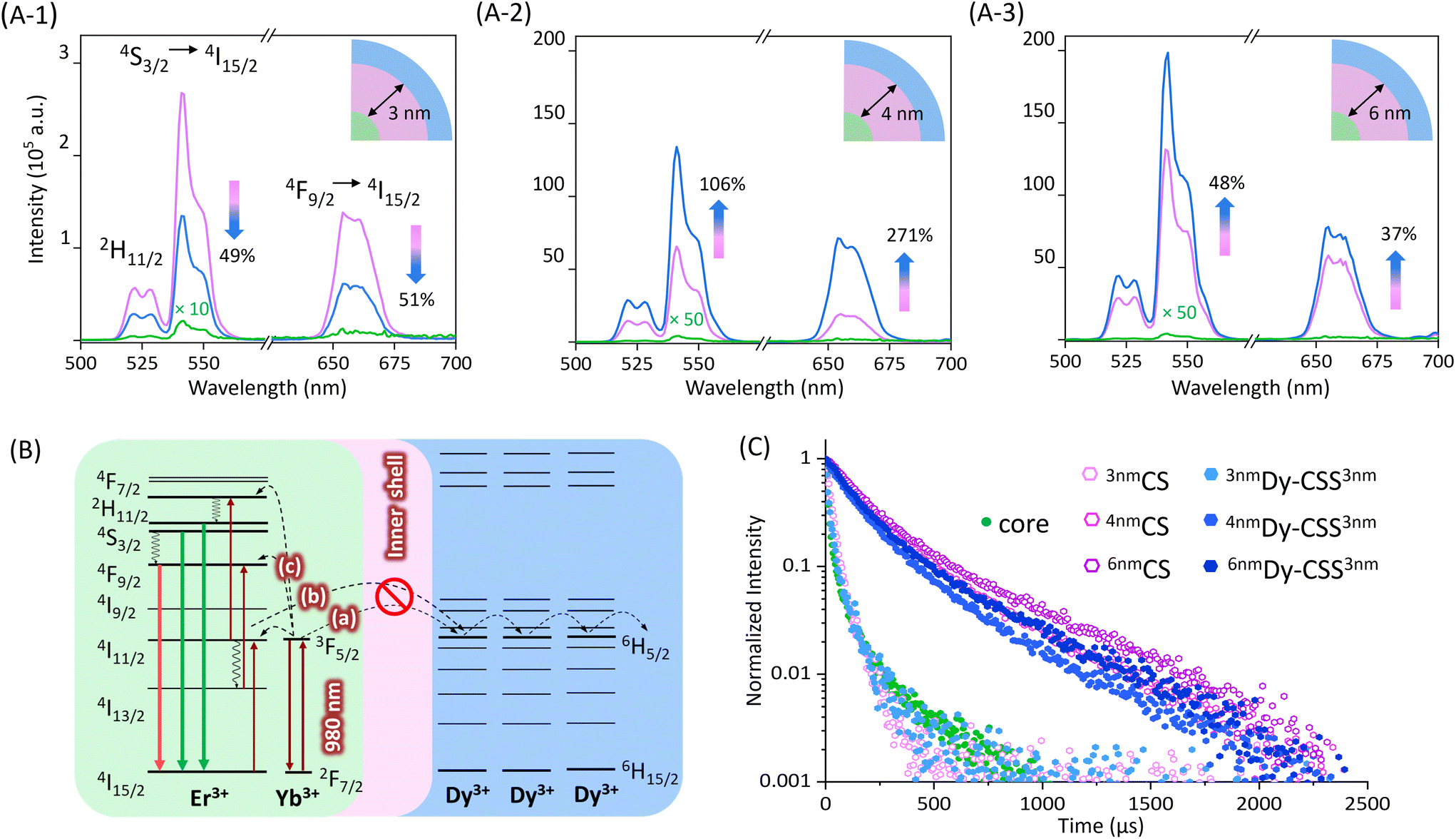 | ||
| Fig. 2 (A) UC spectra of OA-capped 3nmDy-CSS3nm (1), 4nmDy-CSS3nm (2), and 6nmDy-CSS3nm (3) NPs (blue) as well as their corresponding core (green) and CS (pink) NPs. All spectra were obtained under 980 nm laser excitation. Power density = 6.0 W cm−2, [NP] = 5 mg mL−1 in toluene. (B) Partial energy level diagram for Er3+/Yb3+ and Dy3+, schematically showing how the addition of an inner shell physically separates Er3+/Yb3+ from Dy3+ ions, thus, preventing Dy3+-induced loss of the UC. Solid lines are radiative transitions, dotted lines indicate energy transfer, and wavy lines are non-radiative transitions. (C) Luminescence decay curves of the Yb3+ emission under 980 nm excitation obtained from core, CS, and CSS NPs. | ||
Possible quenching processes caused by Dy3+ were previously proposed by Zhang et al. based on steady-state UC spectroscopy.9 As schematically shown in Fig. 2B, in close vicinity to Dy3+ ions, the 2F5/2 excited state of Yb3+ and the 2I11/2 excited state of Er3+ can be depopulated via resonant energy transfer to the excited 6H5/2 level of Dy3+ (steps (a) and (b)). This depopulation of the excited Yb3+ and intermediate Er3+ levels compromise efficient UC to the emitting Er3+ levels (step (c)), ultimately resulting in loss of UC emission. The added inner shell acts as a barrier hampering the Yb3+ (and Er3+) → Dy3+ energy transfer, restoring Er3+ UC. In order to confirm the UC loss mechanism, two reference samples were synthesized, namely, cubic (α)-phase NaDyF4:Yb,Er core NPs and β-phase NaGdF4:Yb,Er/NaDyF4 NPs (Dy-CS) (Fig. S4†). As shown in Fig. S5,† not surprisingly, the emission of Er3+ was completely lost when the emitting Ln3+ ions were co-doped into the NaDyF4 host. UC loss still took place upon separation of Er3+/Yb3+ and Dy3+ into the β-NaGdF4:Yb,Er core and a NaDyF4 shell (Dy-CS).
Confirmation of the proposed quenching mechanism was obtained by determining the excited state lifetime of Yb3+ in the series of core, CS and CSS NPs under investigation. Therefore, the time-dependent emission of Yb3+ under 980 nm excitation was monitored at the shoulder of the ion's emission at 940 nm.42,43
The decay curves and respective lifetime values are given in Fig. 2C and Table 1. The small core NPs exhibited a relatively short lifetime of 25.5 μs due to surface quenching, which was slightly prolonged to 33.3 μs when an undoped shell of 3 nm was added (3nmCS). Yet, growth of an additional NaDyF4 shell shortened the lifetime to 26.1 μs (3nmDy-CSS3nm). This decrease in lifetime is in line with the reduced Er3+ emission (Fig. 2(A-1)). As expected, the addition of a thicker undoped NaGdF4 shell led to a significantly longer lifetime (4nmCS: 207.6 μs). The observed increase in UC emission intensity upon growth of an additional outer NaDyF4 shell (4nmDy-CSS3nm) suggests efficient shielding of the Yb3+/Er3+ ion pair from Dy3+ (Fig. 2(A-2)) as well as additional protection against surface loss processes. Hence, a prolonged Yb3+ lifetime might be expected. Conversely, the decay time decreased to 175.8 μs for 4nmDy-CSS3nm NPs, indicating continuous depopulation of the 2F5/2 Yb3+ level by Dy3+ ions. To assess whether an even thicker undoped shell could fully overcome energy transfer to Dy3+, the optical properties of 6nmDy-CSS3nm NPs with a ca. 6 nm thick inner shell were analyzed. As expected, emission spectra did not indicate any UC poisoning effect of Dy3+, and the sample exhibited the strongest emission intensity among all the investigated NPs due to its overall thick shell (NaGdF4 + NaDyF4: 8.2 nm) (Fig. 2(A-3)). Still, time-dependent measurements revealed that energy transfer to Dy3+ was not fully suppressed, as indicated by the shortening of the Yb3+ lifetime upon shelling with NaDyF4 (251.3 μs for 6nmCS versus 207.6 μs for 6nmDy-CSS3nm). The influence of the NaDyF4 shell on the UC emission was further demonstrated by power-dependent photoluminescence spectroscopy. Power plots and a brief discussion are provided in the ESI (Fig. S6†). Overall, based on the Er3+ emission spectra and Yb3+ lifetimes of 4nmDy-CSS3nm and 6nmDy-CSS3nm, a 4 nm inner shell thickness was deemed sufficiently efficient to suppress the most severe Dy3+-induced UC loss, resulting in bright UC emitters, despite some remaining energy transfer from Yb3+ to Dy3+. Herein, the increased shell thickness is suggested to play a dual role. First, the Yb3+ → Dy3+ energy transfer is hindered by the barrier layer, resulting in more efficient UC to the Er3+ emitting levels. Second, the overall shell (NaGdF4 + NaDyF4) around the emitting core reaches a thickness that was previously reported as highly efficient in reducing non-radiative depopulation of the emitting Er3+ levels, which is supported by increasing lifetimes of the green (4S3/2) and red (4F9/2) emitting excited states (Fig. S7 and S8†).44 Ultimately, both aspects together result in not only restored but enhanced UC emission, demonstrated here – to the best of our knowledge – for the first time.
| Sample | Inner shell thickness (nm) | Lifetime (μs) |
|---|---|---|
| Core | NA | 25.5 |
| 3nmCS | ca. 3 | 33.3 |
| 3nmDy-CSS3nm | 26.1 | |
| 4nmCS | ca. 4 | 207.6 |
| 4nmDy-CSS3nm | 175.8 | |
| 6nmCS | ca. 6 | 251.3 |
| 6nmDy-CSS3nm | 207.6 |
To explore the effect of the NaDyF4 shell on the probes’ performance, particularly MRI T2 and CT capabilities (vide infra), additional Dy-CSS NPs were synthesized. Given the good optical performance of 4nmDy-CSS3nm, exhibiting a ca. 4 nm thick inner shell, two samples with an outer NaDyF4 shell of varying thickness on 4nmCS NPs were synthesized. This resulted in a total set of three samples of comparable core size and inner shell thickness, yet, varying outer shells of ca. 1 nm (4nmDy-CSS1nm), 3 nm (4nmDy-CSS3nm), and 4 nm (4nmDy-CSS4nm), respectively (Table S1†). The morphology, size, crystalline phase, and UC spectra of these samples are shown in Fig. S9–S11.†
Rendering the NPs water-dispersible: citrate-capping
To explore MRI and CT capabilities, the OA-capped NPs were transferred to the aqueous milieu by ligand exchange with tri-sodium citrate molecules.21,45 The successful citrate capping was confirmed by Fourier transform infrared (FTIR) spectroscopy, and the morphology and the crystalline phase remained (Fig. S12†). The cytotoxicity of Cit-4nmDy-CSS3nm NPs against human lung adenocarcinoma cells (A549) was assessed using the flow cytometry assay. As shown in Fig. S13,† the cell viability remained 95% for all NP concentrations, indicating low cytotoxicity of citrate-capped NPs at concentrations below 500 μg mL−1. Despite some expected loss of photoluminescence intensity upon dispersion of the NPs in water compared to their OA-capped counterparts in toluene, Cit-4nmDy-CSS3nm NPs still showed bright UC emission and NIR emission at 1570 nm, which stems from the 4I13/2 → 4I15/2 Er3+ transition (Fig. S14†). Moreover, the NPs exhibited good luminescence stability after incubation in water and phosphate-buffered saline (PBS) for 24 h at 37 °C (Fig. S15†). Er3+-doped UC emitters are well-known for their suitability to act as ratiometric nanothermometers, using the luminescence intensity ratio (LIR) between the two green emission bands as the thermal parameter.46 While nanothermometry was not the major focus of this study, we stepped forward to briefly explore this avenue, assessing the potential of the prepared NPs for thermal sensing and imaging. Temperature-dependent measurements from 15 to 50 °C unveiled decent thermal sensing behavior in the physiologically relevant temperature range, with a relative thermal sensitivity Sr of 1.1% °C−1 at 40 °C. Data and a short discussion are provided in the ESI (Fig. S16†).Citrate-capped Dy-CSS NPs for MR imaging
The Dy3+ ion has the potential to be a T2-weighted MRI contrast agent due to its high magnetic moment. To study the effect of the NaDyF4 shell thickness of the obtained Cit-Dy-CSS NPs on T2 contrast performance, T2 and T1 relaxivities (r2 and r1) were measured at 7 T. As shown in Fig. 3A, the T2-weighted MRI signals obtained from Cit-Dy-CSS NPs changed from brighter to darker when the concentration of Dy3+ increased, indicating the potential of these NPs to act as T2 contrast agents. The relaxivity values were extracted from the slopes of the linear fits of the relaxation rates R2 and R1 (=1/T2 and 1/T1) against the molar concentration of Dy3+ ions of the NP dispersions as obtained from inductively coupled plasma optical emission spectroscopy (ICP-OES) (Fig. 3B – r2; Fig. S17† – r1; all values are summarized in Table 2). Similar r2 values were obtained for Cit-4nmDy-CSS1nm (260.0 mM−1 s−1) and Cit-4nmDy-CSS3nm NPs (256.9 mM−1 s−1), while a significant increase of r2 to 442.1 mM−1 s−1 was observed when the NaDyF4 shell thickness increased to 4 nm (Cit-4nmDy-CSS4nm). This increase in r2 was ascribed to the increasing amount of Dy3+ ions in the sample upon growth of a thicker outer shell, resulting in a higher Dy3+-to-Gd3+ ratio (Fig. S18†). Larger particle sizes have previously been reported to result in more pronounced T2 contrast enhancement, as demonstrated for different-sized NaDyF4 NPs.27,28 Even though the direct comparison with data from the literature is difficult given the different NP architectures under investigation (CSS in this study versus previously investigated core-only NPs), the larger size of the Cit-4nmDy-CSS4nm NPs might also contribute to the observed T2 contrast enhancement. On the contrary, the r1 values decreased with increasing NaDyF4 shell thickness (Fig. S17† and Table 2). It is important to recall that NdGdF4 was used as host matrix for the core and inner shell of the CSS-NPs. Gd3+ ions are well known for their T1 MRI capabilities when in close vicinity to water protons. Thus, the Gd3+ ions in the core and inner shell might influence the overall MRI performance of the CSS-NPs. Indeed, the highest r1 value was obtained for Cit-4nmDy-CSS1nm NPs with a very thin outer shell. Such thin shelling might be less efficient and prone to cation intermixing, resulting in Gd3+ ions close to the NP surface where they can interact with water protons, contributing to higher r1 value. Thicker NaDyF4 shells provided greater shielding of the Gd3+ ions in the inner shell, resulting in lower r1 values. Overall, the resultant r2/r1 ratios increased from 40.0 to 55.8 and 134.0 with increasing NaDyF4 shell thickness. As larger r2/r1 ratios provide for higher signal-to-noise and more robust contrast enhancement in T2-weighted images,27 Cit-4nmDy-CSS4nm NPs with the thickest outer shell are the most promising among the studied T2 MRI CA candidates.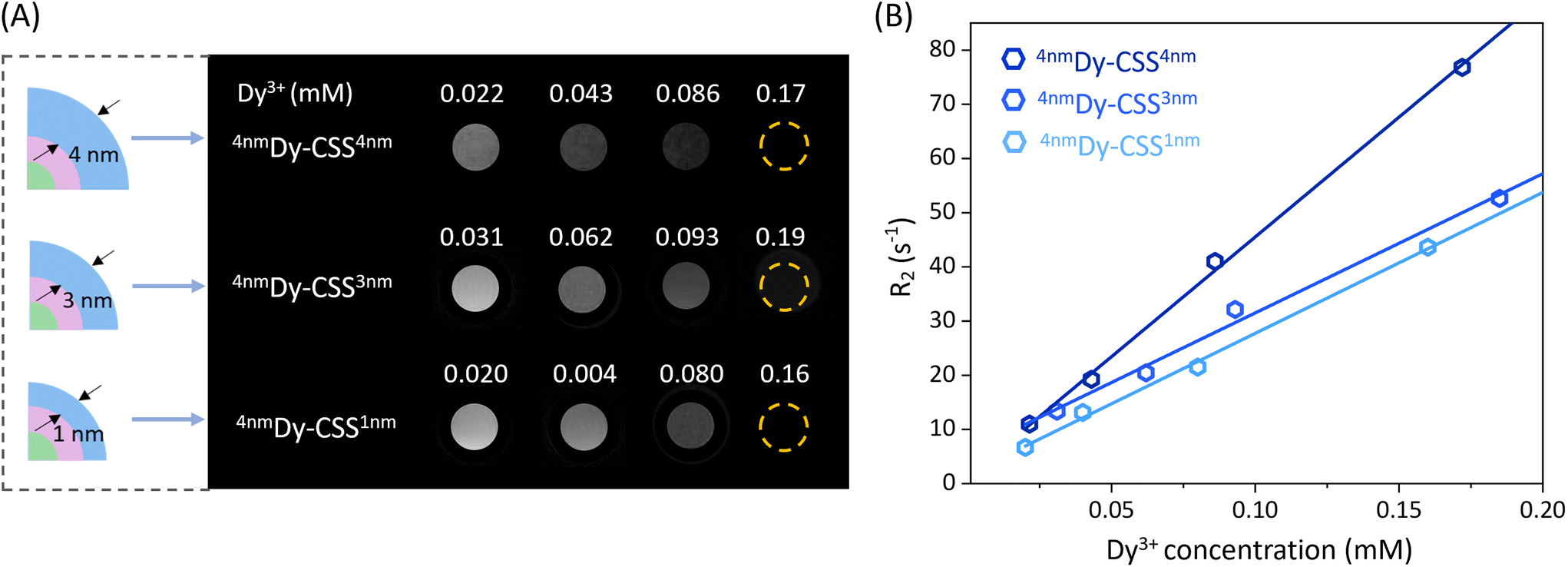 | ||
| Fig. 3 (A) T2-Weighted MR images of Cit-Dy-CSS NPs with varying NaDyF4 outer shell thickness (Cit-4nmDy-CSS1nm, Cit-4nmDy-CSS3nm and Cit-4nmDy-CSS4nm) as a function of the Dy3+ concentration. (B) Relaxation rate R2 (=1/T2) of water protons plotted against the molar concentration of Dy3+ at 7 T. Solid lines are linear fits. | ||
| Sample | Size (nm) | r 2 (mM−1 s−1) | r 1 (mM−1 s−1) | r 2/r1 |
|---|---|---|---|---|
| 4nmDy-CSS1nm | 16.9 | 260.0 ± 9.8 | 6.5 ± 0.1 | 40.0 |
| 4nmDy-CSS3nm | 18.1 | 256.9 ± 17.7 | 4.6 ± 0.1 | 55.8 |
| 4nmDy-CSS4nm | 21.9 | 442.1 ± 22.1 | 3.3 ± 0.2 | 134.0 |
| 6nmDy-CSS3nm | 22.0 | 116.0 ± 10.0 | 2.5 ± 1.1 | 46.4 |
The effect of the inner NaGdF4 shell thickness on the T2 contrast behavior was also investigated. As shown in Fig. S19,† Cit-4nmDy-CSS3nm NPs exhibited a higher r2 value (256.9 mM−1 s−1) than Cit-6nmDy-CSS3nm NPs (116.0 mM−1 s−1), which can be ascribed to the relatively higher Dy3+ content in the sample (Fig. S18†). In line, a larger r2/r1 ratio was obtained for Cit-4nmDy-CSS3nm (55.8) than for Cit-6nmDy-CSS3nm (46.4) NPs, demonstrating the importance to not only tune the outer NaDyF4 but also inner NaGdF4 shell.
Noteworthy, all the r2 values of Cit-Dy-CSS NPs are higher than those of commercial iron oxide-based T2 contrast agents (Combidex: 60.0 mM−1 s−1, Feridex: 93.0 mM−1 s−1) and other comparable sized Dy-based NPs (Table S2†).5,10,26,27,35,47–54 These observations are promising with respect to the potential use of Dy-CSS NPs T2 CAs. Particularly, high relaxivity CAs are generally considered beneficial as they can allow for the administration of lower doses, ultimately contributing to lower toxicity risks.55
Citrate-capped Dy-CSS NPs for CT imaging
Owing to the relatively high K-edge values and X-ray attenuation coefficients of the Ln ions, Cit-Dy-CSS NPs hold potential for CT imaging. To assess their CT contrast performance, we first used X-ray spectrometry (Fig. S20†) to compare the X-ray absorption efficacy of Cit-Dy-CSS NPs to that of iohexol (a widely used clinical CT CA). With the iodine K-edge at 33.3 keV, iohexol absorbs X-ray photons most effectively in the range 35–50 keV.56 In comparison, Cit-Dy-CSS NPs absorb more photons in a higher energy region (50–120 keV) due to the higher K-edge values of Gd (50.2 keV) and Dy (53.8 keV). In an alternative approach to demonstrate the X-ray absorption behavior of iohexol and Cit-Dy-CSS NPs (Cit-4nmDy-CSS3nm, Cit-6nmDy-CSS3nm), radiography imaging was performed across a range of X-ray source voltages from 70 to 120 kV. These voltages are consistent with conditions used in clinical applications. As shown in Fig. S21A,† CT values (Hounsfield units, HU) for iohexol were highest at a source voltage of 70 kV, followed by a continuous decrease with voltage increase to 120 kV. This is expected given the optimized performance of iohexol at lower energies.57 On the contrary, the CT values of Cit-Dy-CSS NPs displayed an initial increase in CT values when the voltage increased from 70 to 110 kV, followed by a decrease at 120 kV (Fig. S21B/C†). This decrease in CT values at higher voltage was caused by the mismatch of the K-edge values of Gd and Dy in the high-energy region. The highest CT value obtained for Cit-6nmDy-CSS3nm NPs at 110 kV was 1080 HU (Ln3+ ion concentration, i.e., sum of Gd3+, Dy3+, Yb3+ and Er3+ = 94.2 mM) compared to 850 HU of iohexol (I− ion concentration = 157.6 mM). Normalization of the CT values to the concentration of the corresponding contrast agent, revealed more than two-times higher values for the Cit-Dy-CSS NPs than for iohexol between 100 and 120 kV (Fig. S21D†). These findings demonstrate the potential of Cit-Dy-CSS NPs as CT CA candidates with higher contrast efficacy compared to iohexol at 110 kV and the advantage that X-rays optimized for the K-edge of the Ln3+ have lower potential for tissue damage than those optimized for the K-edge of iodine.36Encouraged by the results of radiography imaging, CT phantom imaging measurements of Cit-Dy-CSS NPs and iohexol were performed at 110 kV. Herein, samples with thinner and thicker NaDyF4 shells were investigated to assess any potential effect that the outer shell thickness might have on CT performance. As shown in Fig. 4A, with similar CA concentrations, all Cit-Dy-CSS NPs produced brighter CT signals than iohexol. The obtained CT values increased linearly with the concentration for Cit-Dy-CSS NPs and iohexol (Fig. 4B), and the slopes extracted from the linear fits are shown in Table 3. Among the three Cit-Dy-CSS samples, the slope values increased with the NaDyF4 shell thickness increasing from 1 nm to 4 nm (9.4 ± 1.3 HU mM−1 for Cit-4nmDy-CSS1nm, 11.4 ± 0.5 HU mM−1 for Cit-4nmDy-CSS3nm and 13.1 ± 0.05 HU mM−1 for Cit-4nmDy-CSS4nm). These findings can be ascribed to the relatively larger amount of Dy3+ with increasing NaDyF4 shell thickness, resulting in a higher Dy3+-to-Gd3+ ratio (Fig. S18†), ultimately allowing for higher X-ray attenuation. Noteworthy, all the slope values obtained from Cit-Dy-CSS NPs were higher than that of iohexol (5.8 ± 1.3 HU mM−1), indicating that Cit-Dy-CSS NPs exhibit higher contrast efficacy than iohexol.
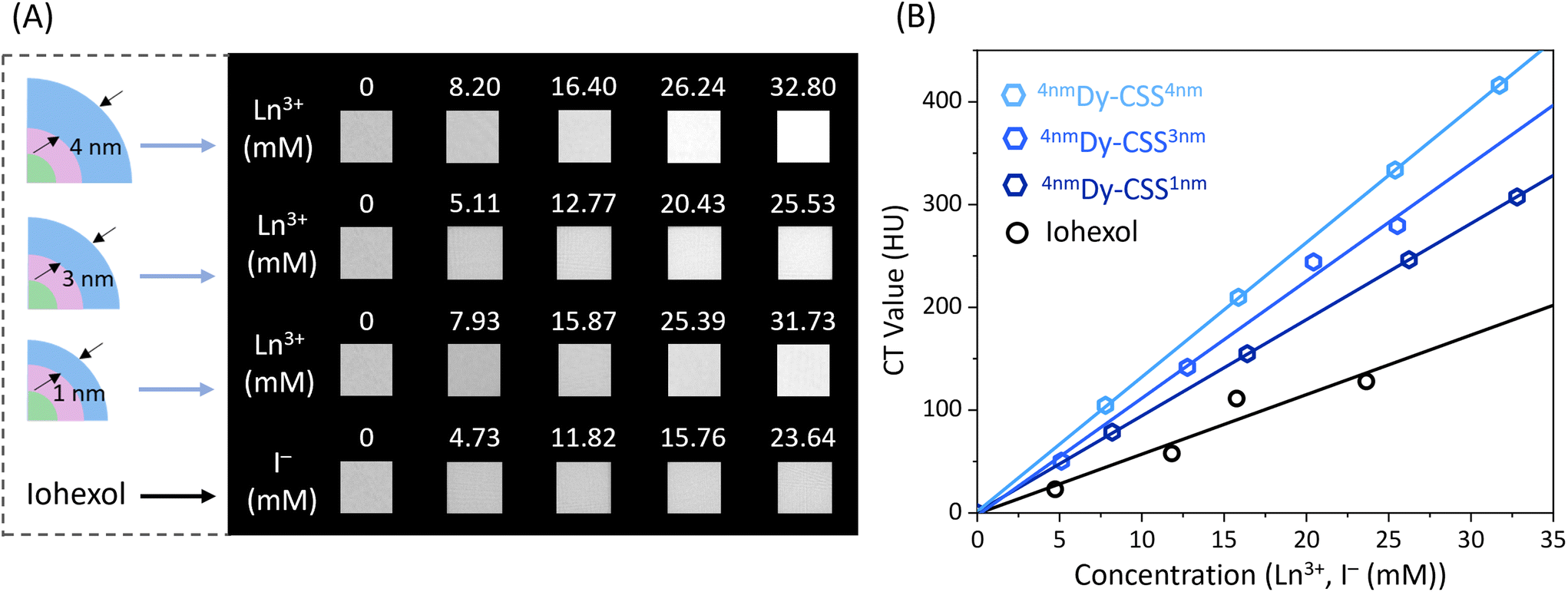 | ||
| Fig. 4 (A) CT images of Cit-Dy-CSS (Cit-4nmDy-CSS1nm, Cit-4nmDy-CSS3nm, and Cit-4nmDy-CSS4nm) NPs and clinically used CT CA iohexol at different concentrations. (B) CT values for different concentrations of Cit-Dy-CSS NPs dispersed in water and iohexol solutions. Solid lines are linear fits. | ||
| Sample | Size (nm) | Slope of the linear fit (HU mM−1) |
|---|---|---|
| 4nmDy-CSS1nm | 16.9 | 9.4 ± 0.03 |
| 4nmDy-CSS3nm | 18.1 | 11.4 ± 0.5 |
| 4nmDy-CSS4nm | 21.9 | 13.1 ± 0.05 |
| Iohexol | — | 5.8 ± 1.3 |
Conclusions
With the goal to design a NP candidate to act as multimodal probe that merges upconversion and T2 MRI capabilities, a set of Dy-CSS NPs doped with Er and Yb was synthesized using a microwave-assisted approach. Careful tuning of the microwave reaction conditions allowed for controlled inner shell thicknesses from ca. 3 to 6 nm. Such control is crucial to physically separate the luminescent Er3+/Yb3+ ion pair in the core from the magnetic Dy3+ ions in the outer shell, ultimately preventing UC loss. The mechanism of UC loss was investigated in detail by evaluating the Er3+ upconversion intensities and the excited state lifetimes of Yb3+ and Er3+ as a function of the inner shell thickness. Based on the Er3+ emission spectra and Yb3+ lifetimes, an isolating 4 nm inner shell between the emitting core and the NaDyF4 outer shell suppressed energy transfer from Yb3+ to Dy3+, followed by Dy3+-mediated energy migration and loss at the NP surface. As a result, UC excitation of the Er3+ ions was restored, and enhanced UC emission was observed upon addition of the outer NaDyF4 shell. Noteworthy, the microwave-assisted strategy led to CSS architectures, while keeping the overall NP size close to 20 nm. In addition, the effect of the outer NaDyF4 shell thickness on the particles’ magnetic and CT performance was investigated. MRI T2 relaxivity measurements performed in vitro at 7 T on Cit-Dy-CSS NPs revealed that the Cit-4nmDy-CSS4nm NPs with the thickest outer shell (4 nm) exhibited the highest r2 value and r2/r1 ratio, indicating a superior T2 contrast effect compared to commercial iron oxide-based CAs and other Dy-based T2 CAs. In addition, the CT values of Cit-Dy-CSS NPs increased with increasing outer NaDyF4 shell thickness. Interestingly, all investigated Cit-Dy-CSS NPs exhibited advanced CT contrast efficacy compared to commercially used iohexol at an X-ray energy of 110 keV. Our results demonstrate the promise of the designed Dy-CSS NPs to act as potential multimodal imaging probe. The concept of fabricating multilayer NPs by use of microwave-assisted routes that offer control over shell thicknesses also provides insights for the future architecture design of Ln-based materials for their e.g., biomedical applications and beyond.Experimental
Chemicals
Gadolinium oxide (Gd2O3, 99.999%), ytterbium oxide (Yb2O3, 99.998%) and erbium oxide (Er2O3, 99.99%) were purchased from Alfa Aesar. Dysprosium oxide (Dy2O3, 99.9%) was purchased from Sigma Aldrich. Trifluoroacetic acid (CF3COOH, H-TFA, 98%), sodium trifluoroacetate (CF3COONa, Na-TFA, 98%), oleic acid (CH3(CH2)7CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(CH2)7COOH, OA, 90%), oleylamine (CH3(CH2)7CHCH(CH2)7CH2NH2, OAm, 70%), 1-octadecene (CH3(CH2)15CHCH2, ODE, 90%) and sodium citrate dihydrate (99%), 1,3-diphenylisobenzofuran (C20H14O, 97%) were purchased from Sigma Aldrich. Iohexol (C19H26I3N3O9, 300 mg I per mL) was purchased from GE Healthcare. Ethanol (99%) was purchased from Commercial Alcohols. Acetone and hexane (analytical grade) were purchased from Fischer Chemicals. Toluene (99.8%) and hydrochloric acid (HCl, 36.5%) were purchased from Fisher Scientific. All chemicals were used as received.
CH(CH2)7COOH, OA, 90%), oleylamine (CH3(CH2)7CHCH(CH2)7CH2NH2, OAm, 70%), 1-octadecene (CH3(CH2)15CHCH2, ODE, 90%) and sodium citrate dihydrate (99%), 1,3-diphenylisobenzofuran (C20H14O, 97%) were purchased from Sigma Aldrich. Iohexol (C19H26I3N3O9, 300 mg I per mL) was purchased from GE Healthcare. Ethanol (99%) was purchased from Commercial Alcohols. Acetone and hexane (analytical grade) were purchased from Fischer Chemicals. Toluene (99.8%) and hydrochloric acid (HCl, 36.5%) were purchased from Fisher Scientific. All chemicals were used as received.
Microwave-assisted synthesis of core β-NaGdF4:Yb,Er NPs
The synthesis of the NPs was performed using a modified version of the microwave-assisted thermal decomposition of lanthanide trifluoroacetate, [Ln(TFA)3], precursors (Ln = Gd3+, Yb3+, Er3+ and Dy3+) previously reported by our group.39 Briefly, for the synthesis of β-NaGdF4 NPs doped with 20 mol% Yb3+ and 2 mol% Er3+, 0.625 mmol of [Ln(TFA)3] were prepared mixing Gd2O3 (0.244 mmol), Yb2O3 (0.0625 mmol), and Er2O3 (0.00625 mmol) in a 50 mL three-neck round-bottom flask. A 10 mL amount of a 1:1 TFA-to-H2O mixture was added, the slurry was refluxed at 95 °C until it became clear and dried at 60 °C overnight. Subsequently, 1.875 mmol of Na-TFA (3:1 Na+-to-Ln3+ ion ratio) was added to the synthesized [Ln(TFA)3] along with 2.5 mL of oleic acid, 2.5 mL of oleylamine and 5 mL of 1-octadecene. This mixture was degassed at 120 °C under vacuum for 30 min. Subsequently, the degassed precursor solution was transferred to a 35 mL microwave vessel, purged with N2 and tightly sealed. The vessel was inserted into a CEM Discovery SP microwave reactor. The precursor solution was heated to 270 °C for 1 s, followed by rapid cooling to 250 °C, the temperature at which the reaction was held for 10 min, and subsequent cooling to 50 °C. Following the synthesis, the reaction mixture was washed with a 1:3 hexane-to-ethanol mixture and centrifuged at RCF = 6595g for 20 min. The product was then washed with a 1:3 toluene-to-acetone mixture and centrifuged using the same conditions. After purification, the NPs were dispersed and stored in 5 mL of toluene for further use. NaF as a potential by-product can be removed by redispersing the NPs in 5 mL of ethanol prior to the addition of 5 mL of water, followed by precipitation via centrifugation. Obtained NPs were washed one more time with 10 mL of ethanol before being stored in 5 mL of toluene for further characterization. In case of loss of dispersion stability of the NPs following this washing procedure, dispersibility in toluene can be restored by adding 0.2 mL of OA to the NP dispersion and subsequent stirring at room temperature overnight.
Microwave-assisted synthesis of β-NaGdF4:Yb,Er/NaGdF4 core/shell NPs
An undoped NaGdF4 shell was grown onto the doped core NPs by subjecting the NPs to a second microwave-assisted treatment, adding [Gd(TFA)3] as shell precursor. 0.625 mmol of the shell precursor was prepared as described above, using Gd3+ as sole lanthanide. Subsequently, 1.875 mmol of Na-TFA (3:1 Na+-to-Gd3+ ratio), 4 mL of ODE and 5 mL of OA were added to the shell precursor. The reaction mixture was degassed under vacuum at 120 °C for 30 min. The shell thickness was controlled by adjusting the ratio between the core NPs acting as seeds for shell growth and the shell precursor. More specifically, 100 mg, 40 mg, or 30 mg of the core NPs, prepared in the previous step, were used as seeds to grow shells of ca. 3, 4, and 6 nm, respectively. Therefore, the respective amount of core NPs was firstly dispersed in 1 mL of ODE (note: in contrast to the core synthesis, no OAm was used to grow the NaGdF4). Subsequently, this 1 mL of ODE containing core NPs and 9 mL of the degassed shell precursor solution were transferred to a 35 mL microwave vessel, purged with N2, and tightly sealed. The vessel was inserted into a CEM Discover SP microwave reactor, and the mixture was heated to 250 °C and held for 10 min under stirring. The cooling and washing procedure as well as storage of the obtained core/shell NPs were as described above for the core NPs.
Microwave-assisted synthesis of β-NaGdF4:Yb,Er/NaGdF4/NaDyF4 core/shell/shell NPs
The procedure to grow the outer shell of NaDyF4 was similar to that used for the synthesis of the core/shell NPs, except that [Dy(TFA)3] was used as precursor. The shell precursor, 0.625 mmol of [Dy(TFA)3], was prepared as described above, using Dy3+ as sole lanthanide. Then, 1.25 mmol of Na-TFA (2:1 Na+-to-Dy3+ ratio), 4 mL of ODE and 5 mL of OA were added to the precursor. The reaction mixture was degassed under vacuum at 120 °C for 30 min. The shell thickness was controlled by adjusting the ratio between the core/shell NPs acting as seeds for shell growth and the shell precursor. More specifically, 160 mg, 120 mg, or 90 mg of the core/shell NPs, prepared in the previous step, were used as seeds to grow shells of ca. 1, 3, and 4 nm, respectively. Subsequently, the 1 mL of ODE containing core/shell NPs and 9 mL of the degasses shell precursor solution were transferred to a 35 mL microwave vessel, purged with N2, and tightly sealed. The vessel was inserted into a CEM Discover SP microwave reactor, the mixture was heated to 250 °C and held for 10 min under stirring. The cooling and washing procedure as well as storage of the obtained core/shell/shell NPs were as described above for the core NPs.
Surface modification with citrate groups
Trisodium citrate was used to replace oleate (OA) groups at the surface of the as-prepared NaGdF4:Yb,Er/NaGdF4/NaDyF4 NPs following a modified version of previously reported protocols.21 Briefly, 70 mg of NPs was dispersed in 5 mL of hexane. Then, 5 mL of 0.2 M trisodium citrate buffer (pH = 2.7) was added to the NP colloidal dispersion. The two-phase mixture was stirred for 4 h at 45 °C. Subsequently, the aqueous/organic mixture was poured into a separatory funnel, and the aqueous phase now containing the NPs was isolated. The NPs were precipitated with acetone (aqueous to organic phase ratio: 1:3) and centrifuged at 6595g for 20 min. The organic solvent was removed, and the recovered NPs were re-dispersed in 5 mL of trisodium citrate buffer (pH = 7). The dispersion was stirred for 1 h, followed by three washing steps using water and acetone for precipitation and subsequent centrifugation under aforementioned conditions. The obtained citrate-capped NPs were stored in 5 mL of water.
Characterization techniques
To determine the crystalline phase of the NPs, powder X-ray diffraction (XRD) analysis was performed using a Rigaku Ultima IV Diffractometer or a Bruker D8 Endeavor (Cu Kα, λ = 1.5401 Å), operating at 44 kV and 40 mA (step size: 0.02°, scan speed: 1° min−1). Therefore, the NPs were deposited on a glass slide from their suspension. The morphology and size distribution of the obtained NPs were investigated by transmission electron microscopy (TEM, FEI Tecnai Spirit). The samples were dispersed on a Formvar/carbon film supported on a 300-mesh copper TEM grid. Size distributions (mean size ± standard deviation) of the samples were derived from TEM images using the software ImageJ. The respective size distributions were obtained analysing 200 particles per sample. The presence of the citrate ligands at the particles’ surface was investigated by Fourier-transform infrared (FTIR) spectroscopy, using a Cary 630 FTIR spectrometer in ATR mode (Diamond ATR crystal), with acquisition of 128 scans and resolution of 4 cm−1. Dynamic light scattering (DLS) and zeta (ζ-) potential measurements were carried out on NPs dispersed in water using a Malvern Zetasizer Nano-ZS.Investigation of the steady-state photoluminescence properties of the NPs in suspension (5 mg mL−1 in toluene or water) was carried out in a quartz cuvette (suitable spectral range from 200 to 2500 nm, 1 cm optical path) inserted into the Peltier temperature sample holder of a QuantaMaster 8075-21 spectrofluorometer from HORIBA, equipped with double grating emission monochromators, a red-extended photomultiplier detector R13456 PMT (185 to 980 nm) and a liquid nitrogen cooled InAs detector (1000 to 3450 nm). The NIR continuous excitation was performed using a 980 nm continuous-wave laser diode (power densities used to trigger upconversion and NIR emission were 6.0 W cm−2 and 33.3 W cm−2, respectively). Power-dependent studies were performed using the same setup described above, including different neutral density filters to stepwise reduce the incident laser power. For temperature-dependent studies, the temperature of the sample was changed from 15 °C to 50 °C in 5° steps, using the Peltier temperature sample holder with a liquid cooling system. At each step, the temperature was allowed to stabilize for 15 min prior to the measurements. Photoluminescence lifetimes were obtained by time-resolved spectroscopy, using the 980 nm laser diode in pulsed mode. Lifetime values were obtained by integration of the area under the normalized decay curves. For all the optical measurements, filters were placed in front of detectors. A short-pass 775 nm filter was used for visible emission acquisition, while a long-pass 1025 nm filter was used for NIR emission detection.
The Ln3+ ion concentration in the samples was determined with an Agilent ICP-OES spectrometer. Therefore, 10 mg of Dy-CSS NPs was added to a mixture of 0.5 mL of concentrated HNO3 and 1.5 mL of concentrated HCl, followed by heating at 80 °C for 5 h ensuring that all NPs were digested. Flow conditions of the Agilent ICP-OES spectrometer were as follows: nebulizer flow = 0.7 L min−1, plasma flow = 12 L min−1, auxiliary flow = 1 L min−1.
MRI relaxivity measurements were performed on a 7.0 T preclinical MRI instrument (Discovery MR901, Agilent/General Electric). T1 measurements were measured using a classical inversion recovery method with the following parameters: repetition time TR = 15000 ms, echo time TE = 6.4 ms, eight inversion recovery times (TI = 50, 100, 400, 800, 1200, 2000, 3000 and 4000 ms), field of view (FOV) = 8 × 8 mm2, slice thickness = 3 mm, matrix = 128 × 128. T2 measurements were performed using a Carr–Purcell–Meiboom–Gill (CPMG) sequence with the following parameters: TR = 5000 ms, FOV = 10 × 10 mm2, slice thickness = 3 mm, matrix = 128 × 256, TE = 13, 26.1, 39.1, 52.2, 65.2, 78.2, 91.3 and 104 ms. T2-Weighted images were obtained on the same MR instrument using the following parameters: TR = 5000 ms, FOV = 10 × 10 mm2, slice thickness = 3 mm, matrix = 128 × 256, TE = 52 ms. T1 and T2 relaxation times (R1 and R2) were calculated using single exponential fitting of the data extracted from mapping images for different concentrations of Cit-Dy-CSS NPs in water.
X-ray absorption spectroscopy measurements were conducted using a Hamamatsu L12161-07 tungsten source collimated to a 1 mm beam. A 0.6 mm Cu filter was used to harden the beam. The source current was 150 μA and the source voltage was 150 kV. An Amptek Cd–Te 25 mm2 spectrometer was used in combination with Amptek PX-5 Digital Pulse Processor. Each spectrum has an acquisition time of 120 s. Radiograph imaging measurements were performed with a PXS MicroCT system (Pinnacle X-ray Solutions Inc.) consisting of a Hamamatsu L12161-07 source coupled with a Varian PaxScan 1313DX digital image detector. Conditions of the source were 150 μA current with different voltages, i.e., 70, 80, 90, 100, 110 and 120 kV. The X-ray beam was hardened using a 3.3 mm Al filter. Each image consists of the average of 20 frames taken at 2 frame per second for a total acquisition time per image of 10 seconds. ImageJ 1.52c was used to determine an average attenuation value from an area within the frame. Images of water and an empty (air) vial were obtained as reference to calculate the Hounsfield unit (HU) values following eqn (1):58
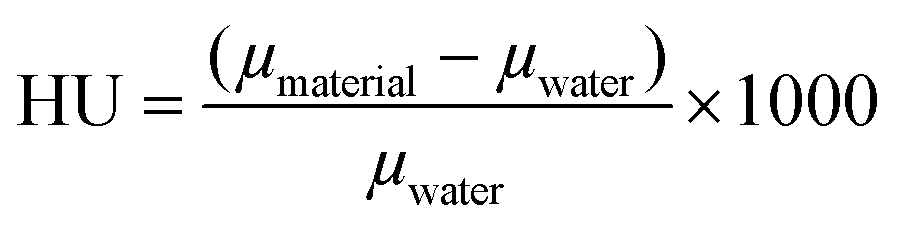 | (1) |
| I = I0 × e−μx | (2) |
Cytotoxicity of Cit-4nmDy-CSS3nm NPs
Human lung adenocarcinoma (A549) cells were seeded (75000 cells) in a 6-well plate to a final volume of media of 1 mL. After 48 h the media was removed from wells, and fresh media containing Cit-4nmDy-CSS3nm NPs (50 μg mL−1, 250 μg mL−1, 500 μg mL−1) was added. The Cit-4nmDy-CSS3nm NPs were incubated at 37 °C with the cells for 24 h. After the incubation, the cells were washed 3 times with PBS, followed by the addition of 250 μL of trypsin, diluted with 750 μL phenol red-free media and centrifuged (1000 RCF, 6 min). After the cells were decanted, 1 mL of PBS with 0.2% DMSO, 1 μM calcein-AM, and 2 μM ethidium homodimer-1 were added. The mixture was vortexed briefly three times. All cell experiments including controls were performed in triplicates. Flow cytometry (Beckman Coulter Gallios Flow Cytometer) was performed doing standard compensation and gating. The flow cytometry data analysis was performed with Kaluza software.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support provided by the University of Ottawa, the Canadian Foundation for Innovation (CFI), and the Natural Sciences and Engineering Research Council of Canada (NSERC). NL is thankful for financial support provided by China Scholarship Council (CSC). Schemes shown in Fig. 1 and the ToC figure were made in BioRender (https://www.biorender.com).References
- X. Y. Wong, A. Sena-Torralba, R. Alvarez-Diduk, K. Muthoosamy and A. Merkoci, ACS Nano, 2020, 14, 2585–2627 CrossRef CAS PubMed.
- C. Duan, L. Liang, L. Li, R. Zhang and Z. P. Xu, J. Mater. Chem. B, 2018, 6, 192–209 RSC.
- Z. Li, Y. Zhang, H. La, R. Zhu, G. El-Banna, Y. Wei and G. Han, Nanomaterials, 2015, 5, 2148–2168 CrossRef CAS PubMed.
- L. Prodi, E. Rampazzo, F. Rastrelli, A. Speghini and N. Zaccheroni, Chem. Soc. Rev., 2015, 44, 4922–4952 RSC.
- Y. Li, Y. Gu, W. Yuan, T. Cao, K. Li, S. Yang, Z. Zhou and F. Li, ACS Appl. Mater. Interfaces, 2016, 8, 19208–19216 CrossRef CAS PubMed.
- F. S. Ferreira, A. J. de Morais, C. M. Santos Calado, F. Iikawa, O. D. D. Couto Junior, G. Brunet, M. Murugesu, I. O. Mazali and F. A. Sigoli, Nanoscale, 2021, 13, 14723–14733 RSC.
- C. Cressoni, F. Vurro, E. Milan, M. Muccilli, F. Mazzer, M. Gerosa, F. Boschi, A. E. Spinelli, D. Badocco, P. Pastore, N. F. Delgado, M. H. Collado, P. Marzola and A. Speghini, ACS Appl. Mater. Interfaces, 2023, 15, 12171–12188 CrossRef CAS PubMed.
- Y. I. Park, K. T. Lee, Y. D. Suh and T. Hyeon, Chem. Soc. Rev., 2015, 44, 1302–1317 RSC.
- Y. Zhang, G. K. Das, V. Vijayaragavan, Q. C. Xu, P. Padmanabhan, K. K. Bhakoo, S. T. Selvan and T. T. Tan, Nanoscale, 2014, 6, 12609–12617 RSC.
- X. Jin, F. Fang, J. Liu, C. Jiang, X. Han, Z. Song, J. Chen, G. Sun, H. Lei and L. Lu, Nanoscale, 2015, 7, 15680–15688 RSC.
- G. Chen, I. Roy, C. Yang and P. N. Prasad, Chem. Rev., 2016, 116, 2826–2885 CrossRef CAS PubMed.
- M. Raab, A. Skripka, J. Bulmahn, A. Pliss, A. Kuzmin, F. Vetrone and P. Prasad, ACS Appl. Bio Mater., 2022, 5, 4948–4954 CrossRef CAS PubMed.
- Z. Yi, Z. Luo, X. Qin, Q. Chen and X. Liu, Acc. Chem. Res., 2020, 53, 2692–2704 CrossRef CAS PubMed.
- J. Zhou, Z. Liu and F. Li, Chem. Soc. Rev., 2012, 41, 1323–1349 RSC.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS PubMed.
- E. Hemmer, N. Venkatachalam, H. Hyodo, A. Hattori, Y. Ebina, H. Kishimoto and K. Soga, Nanoscale, 2013, 5, 11339–11361 RSC.
- A. Gnach, T. Lipinski, A. Bednarkiewicz, J. Rybka and J. A. Capobianco, Chem. Soc. Rev., 2015, 44, 1561–1584 RSC.
- P. Verwilst, S. Park, B. Yoon and J. S. Kim, Chem. Soc. Rev., 2015, 44, 1791–1806 RSC.
- J. Wahsner, E. M. Gale, A. Rodriguez-Rodriguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS PubMed.
- N. J. J. Johnson, W. Oakden, G. J. Stanisz, R. Scott Prosser and F. C. J. M. van Veggel, Chem. Mater., 2011, 23, 3714–3722 CrossRef CAS.
- N. Liu, R. Marin, Y. Mazouzi, G. O. Cron, A. Shuhendler and E. Hemmer, Nanoscale, 2019, 11, 6794–6801 RSC.
- H. Chen, X. Li, F. Liu, H. Zhang and Z. Wang, Mol. Pharm., 2017, 14, 3134–3141 CrossRef CAS PubMed.
- H. Zhang, Y. Wu, J. Wang, Z. Tang, Y. Ren, D. Ni, H. Gao, R. Song, T. Jin, Q. Li, W. Bu and Z. Yao, Small, 2018, 14, 1702951 CrossRef PubMed.
- S. Liu, J. C. Brisset, J. Hu, E. M. Haacke and Y. Ge, J. Magn. Reson. Imaging, 2018, 47, 621–633 CrossRef PubMed.
- S. Viswanathan, Z. Kovacs, K. N. Green, S. J. Ratnakar and A. Dean Sherry, Chem. Rev., 2010, 110, 2960 CrossRef CAS PubMed.
- J. Zhao, H. Hu, W. Liu and X. Wang, Nanoscale Adv., 2021, 3, 463–470 RSC.
- G. K. Das, N. J. Johnson, J. Cramen, B. Blasiak, P. Latta, B. Tomanek and F. C. van Veggel, J. Phys. Chem. Lett., 2012, 3, 524–529 CrossRef CAS PubMed.
- X. Zhang, B. Blasiak, A. J. Marenco, S. Trudel, B. Tomanek and F. C. J. M. van Veggel, Chem. Mater., 2016, 28, 3060–3072 CrossRef CAS.
- A. Banerjee, B. Blasiak, A. Dash, B. Tomanek, F. C. J. M. van Veggel and S. Trudel, Chem. Phys. Rev., 2022, 3, 011304 CrossRef CAS.
- Y. Deng, H. Wang, W. Gu, S. Li, N. Xiao, C. Shao, Q. Xu and L. Ye, J. Mater. Chem. B, 2014, 2, 1521–1529 RSC.
- W. Fan, B. Shen, W. Bu, F. Chen, K. Zhao, S. Zhang, L. Zhou, W. Peng, Q. Xiao, H. Xing, J. Liu, D. Ni, Q. He and J. Shi, J. Am. Chem. Soc., 2013, 135, 6494–6503 CrossRef CAS PubMed.
- S. He, N. J. J. Johnson, V. A. Nguyen Huu, E. Cory, Y. Huang, R. L. Sah, J. V. Jokerst and A. Almutairi, Nano Lett., 2017, 17, 4873–4880 CrossRef CAS PubMed.
- S. Biju, M. Harris, L. V. Elst, M. Wolberg, C. Kirschhock and T. N. Parac-Vogt, RSC Adv., 2016, 6, 61443–61448 RSC.
- Y. Su, L.-N. Hao, K. Liu, J. Zhang, L. Dong, Y. Xu, Y. Lu and H.-S. Qian, RSC Adv., 2018, 8, 12944–12950 RSC.
- W. Yuan, D. Yang, Q. Su, X. Zhu, T. Cao, Y. Sun, Y. Dai, W. Feng and F. Li, Adv. Funct. Mater., 2016, 26, 8631–8642 CrossRef CAS.
- Y. Liu, K. Ai, J. Liu, Q. Yuan, Y. He and L. Lu, Angew. Chem., Int. Ed., 2012, 51, 1437–1442 CrossRef CAS PubMed.
- J. H. Hubbell and S. M. Seltzer, X-ray mass attenuation coefficients, NIST standard reference database 126, DOI: DOI:10.18434/T4D01F, accessed 07 June 2023.
- K. Ai, Y. Liu, J. Liu, Q. Yuan, Y. He and L. Lu, Adv. Mater., 2011, 23, 4886–4891 CrossRef CAS PubMed.
- I. Halimi, E. M. Rodrigues, S. L. Maurizio, H.-Q. T. Sun, M. Grewal, E. M. Boase, N. Liu, R. Marin and E. Hemmer, J. Mater. Chem. C, 2019, 7, 15364–15374 RSC.
- R. Shi, C. D. S. Brites and L. D. Carlos, Small Struct., 2022, 3, 2100194 CrossRef CAS.
- S. Dühnen and M. Haase, Chem. Mater., 2015, 27, 8375–8386 CrossRef.
- S. M. Kaczmarek, T. Tsuboi, M. Ito, G. Boulon and G. Leniec, J. Phys.: Condens. Matter, 2005, 17, 3771–3786 CrossRef CAS PubMed.
- Q. Jin, S. Gao, Y. Xu and A. Caggiano, Adv. Mater. Sci. Eng., 2021, 2021, 1–9 Search PubMed.
- C. Homann, L. Krukewitt, F. Frenzel, B. Grauel, C. Wurth, U. Resch-Genger and M. Haase, Angew. Chem., Int. Ed., 2018, 57, 8765–8769 CrossRef CAS PubMed.
- R. Naccache, P. Chevallier, J. Lagueux, Y. Gossuin, S. Laurent, L. Vander Elst, C. Chilian, J. A. Capobianco and M. A. Fortin, Adv. Healthcare Mater., 2013, 2, 1478–1488 CrossRef CAS PubMed.
- C. D. S. Brites, A. Millán and L. D. Carlos, Lanthanides in Luminescent Thermometry, in Handbook on the Physics and Chemistry of Rare Earths, ed. J. C. G. Bunzli and V. K. Pecharsky, Elsevier Science, B. V., Amsterdam, 2016, vol. 49, p. 339 Search PubMed.
- Y. X. Wang, Quant. Imaging Med. Surg., 2011, 1, 35–40 Search PubMed.
- M. Rohrer, H. Bauer, J. Mintorovitch, M. Requardt and H. J. Weinmann, Invest. Radiol., 2005, 40, 715–724 CrossRef PubMed.
- M. Harris, L. Vander Elst, S. Laurent and T. N. Parac-Vogt, Dalton Trans., 2016, 45, 4791–4801 RSC.
- P. Caravan, M. T. Greenfield and J. W. Bulte, Magn. Reson. Med., 2001, 46, 917–922 CrossRef CAS PubMed.
- Y. Zhang, J. D. Lin, V. Vijayaragavan, K. K. Bhakoo and T. T. Tan, Chem. Commun., 2012, 48, 10322–10324 RSC.
- S. Biju, J. Gallo, M. Banobre-Lopez, B. B. Manshian, S. J. Soenen, U. Himmelreich, L. Vander Elst and T. N. Parac-Vogt, Chem. – Eur. J., 2018, 24, 7388–7397 CrossRef CAS PubMed.
- M. Norek, E. Kampert, U. Zeitler and J. A. Peters, J. Am. Chem. Soc., 2008, 130, 5335–5340 CrossRef CAS PubMed.
- J. Yin, F. Xu, H. Qu, C. Li, S. Liu, L. Liu and Y. Shao, Phys. Chem. Chem. Phys., 2019, 21, 11883–11891 RSC.
- E. Gianolio, E. D. Gregorio and S. Aime, Eur. J. Inorg. Chem., 2019, 137–151 CrossRef CAS.
- P. L. McCormack, Clin. Drug Invest., 2013, 33, 155–166 CrossRef CAS PubMed.
- B. Johnson, J. L. Hinshaw, J. B. Robbins and P. J. Pickhardt, AJR, Am. J. Roentgenol., 2016, 206, 1202–1207 CrossRef PubMed.
- T. E. Reeves, P. Mah and W. D. McDavid, Dentomaxillofac. Radiol., 2012, 41, 500–508 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, including shell thickness control; additional structural analysis by XRD and TEM; additional optical characterization of OA-capped NPs; characterization of citrate-capped Dy-CSS NPs; nanothermometry of Cit-Dy-CSS NPs; MRI T1 relaxivity measurements; X-ray spectroscopy and radiography imaging measurements. See DOI: https://doi.org/10.1039/d3nr05380f |
| This journal is © The Royal Society of Chemistry 2023 |