
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccelerated solid-phase synthesis of glycopeptides containing multiple N-glycosylated sites†
Poriah
Strauss
a,
Francesca
Nuti
 b,
Michael
Quagliata
b,
Anna Maria
Papini
*b and
Mattan
Hurevich
*a
b,
Michael
Quagliata
b,
Anna Maria
Papini
*b and
Mattan
Hurevich
*a
aThe Institute of Chemistry, The Hebrew University of Jerusalem, Edmond J. Safra Campus, Givat Ram, Jerusalem, 91904, Israel. E-mail: mattan.hurevich@mail.huji.ac.il
bInterdepartmental Research Unit of Peptide and Protein Chemistry and Biology, Department of Chemistry “Ugo Schiff”, University of Florence, 50019 Sesto Fiorentino, Italy. E-mail: annamaria.papini@unifi.it
First published on 10th November 2022
Abstract
Peptide fragments of glycoproteins containing multiple N-glycosylated sites are essential biochemical tools not only to investigate protein–protein interactions but also to develop glycopeptide-based diagnostics and immunotherapy. However, solid-phase synthesis of glycopeptides containing multiple N-glycosylated sites is hampered by difficult couplings, which results in a substantial drop in yield. To increase the final yield, large amounts of reagents but also time-consuming steps are required. Therefore, we propose herein to utilize heating and stirring in combination with low-loading solid supports to set up an accelerated route to obtain, by an efficient High-Temperature Fast Stirring Peptide Synthesis (HTFS-PS), glycopeptides containing multiple N-glycosylated sites using equimolar excess of the precious glycosylated building blocks.
Introduction
Post-translational modifications (PTMs) are essential for diversifying protein functions.1 In fact, pattern-dependent multi-site modifications can have unpredictable effects because they are essential to study and understand their role at the molecular level. N- and O-glycosylations are the most common protein co- and post-translational modifications, respectively playing a crucial role in protein–protein interactions, cell communication, structure stabilization, etc.2 Interestingly, multi-site glycosylations on proteins have a huge importance in disease-related interactions and in particular in immunology.3–5 However, studying glycosylated proteins bearing multiple copies of sugars is extremely difficult since their isolation from biological material but also protein expression results in heterogeneous mixtures. Therefore, obtaining homogeneous proteins with a specific glycosylation pattern is a challenge.6 Libraries of peptide fragments bearing specific glycosylations serve as tools to determine the effect of the glycosylation pattern on interaction preferences and activity of the parent protein. The synthesis of glycopeptides with several copies of sugars not only on serine, threonine, and hydroxyproline (linked by an O-glycosidic bond) but also on asparagine (by an N-glycosidic i.e., amide bond) is essential to understand their function and utilize them for biomedical applications.Solid-Phase Peptide Synthesis (SPPS) is nowadays the most common strategy to produce fast peptides and glycopeptides.7 The two main approaches for synthesizing N-glycopeptides are based on either post-assembly glycosylation or on the building block approach in which specifically glycosylated amino-acid building blocks (Gbb), adequately protected for the selected orthogonal protection strategy, are used during the assembly. In post-assembly glycosylation, glycosylation takes place after the peptide is synthesized. Several post-assembly glycosylation methods have been developed over the years,8 typically via Lansbury aspartylation in which an aminosugar reacts with the activated aspartic acid side-chain to form the N-glycan.9–11
The main shortcoming of this method was the formation of several prominent side reactions such as cyclic aspartimide. New synthetic strategies utilize pseudoprolines to overcome aspartimide formation, providing N-glycopeptides via post-assembly glycosylation in higher yield.12,13 These synthetic glycoforms proved valuable for the preparation of glycoproteins aimed to study SARS-CoV-2 spike protein-related interactions. The second and more popular strategy for N-glycopeptide synthesis is the Gbb approach in which a glycosylated amino acid is prepared in advance with orthogonally protected reactive functions. The pre-glycosylated amino acid is then incorporated into the growing on-resin peptide chain using standard SPPS protocols, allowing streamlining of the process, as the only post-assembly step is the removal of the protecting groups.
In previous studies, we reported the synthesis of a series of glycopeptides by introducing various glycosylated amino acids by a rapid microwave-assisted synthesis in high yield using N-glycosyl amino-acid building blocks orthogonally protected for Fmoc/tBu SPPS.14–16 This strategy enabled selective incorporation of the glycan on specific sites. However, the Gbb approach for SPPS has several drawbacks because of synthetic hurdles related to the protected N-glycosylated Asn (N-Sug). The steric hindrance of the building block results in a dramatic decrease in coupling efficiency. This problem further increases when several sites are glycosylated. It is assumed that the introduction of multiple bulky N-Sug moieties can result in aggregation, affecting further couplings. In common practice, a large excess of Gbb shall be used to enable the synthesis of multi-glycosylated peptides, the coupling time is usually very long and a higher temperature is often used to drive the reactions.17 These are major shortcomings since N-Sug are extremely difficult to synthesize in large quantities, which makes the preparation of libraries an expensive and time-consuming process.16,18,19
The standard SPPS reactors used today apply an inefficient mixing strategy (mainly N2 bubbling), which results in slow diffusion of reagents and deficient heat transfer. These mixing limitations prevent the acceleration of SPPS processes. Even when heating is applied, large excesses of reagents are used as a common practice to compensate for the mixing deficiency. Recently, our group developed the High-Temperature Fast Stirring Peptide Synthesis (HTFS-PS) strategy, which was proposed as a novel strategy to accelerate SPPS (Fig. 1).20 HTFS-PS utilizes high temperature (90 °C) and fast overhead stirring (1200 rounds per minute, rpm) to synthesize peptides in a short time, using low excesses. HTFS-PS proved to provide peptides in a record time with high efficiency. The strategy proved extremely useful for the synthesis of PTM-peptides, such as a set of O-glycopeptides that were assembled in minutes using equimolar amounts of Gbb.21 The strategy was also utilized for the synthesis of a set of multi-phosphorylated peptides in which the difficulty of the synthesis increased with the number and the proximity of the phosphorylation sites.22 For each PTM (e.g., phosphorylation), the same reactor was used but the chemistry was tailored to match the specific characteristics of the peptides.
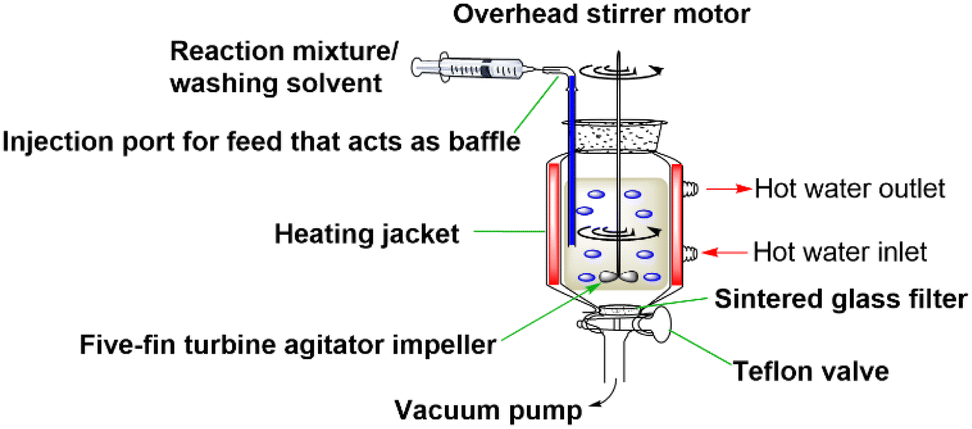 | ||
| Fig. 1 Schematic description of HTFS-PS reactor setup. | ||
In 2003 St Geme et al. first reported that the Non-Typeable Haemophilus influenzae (NTHi), a Gram-negative bacterium, was able to express on the cell surface the HMW Adhesin protein bearing N-glucosylation on several asparagines within the consensus sequence N–X–S/T.23 In particular, the cytoplasmic HMW1C glycosyltransferase, using UDP-glucose as the glycosyl donor, transfers glucose residues directly onto the possible asparagine sites. In 2016, our group demonstrated a strong connection between Multiple Sclerosis and the pathogen NTHi, which is a common cause of upper respiratory infection in humans. Our data showed that antibodies from Multiple Sclerosis patient sera preferentially recognize the hyperglucosylated adhesin protein HMW1 of NTHi.24–26 The hyperglucosylated protein can be considered a candidate for triggering pathogenic antibodies in Multiple Sclerosis. The development of adhesin peptides containing multiple N-glucosylated sites is important for understanding the role of NTHi infection in Multiple Sclerosis.
Here, we describe the use of HTFS-PS for the synthesis of glucopeptides derived from the NTHi adhesin protein. In particular, we focused on peptides of the C-terminal fragment of the HMW1A adhesin (residues 1205–1536, termed HMW1ct) that contains up to 12 glycosylation sites on Asn in NX(S/T) sequons.27,28 To investigate the role of the HMW1 protein fragments in antibody recognition the synthesis of a set of various N-glycopeptide fragments of adhesin bearing multiple N-Glc moieties is necessary.
The effect of the introduction of multiple copies of N-Glc by HTFS-PS was evaluated and different loadings of the selected solid support were investigated. Optimization of the HTFS-PS protocol enabled an accelerated synthesis of glycopeptides containing multiple N-glucosylated sites using almost equimolar quantities of the N-Sug and in particular of the N-Glc building block Fmoc-L-Asn[Glc(OAc)4]-OH.
Results and discussion
Adhesin-derived N-glycopeptides were selected to demonstrate the efficiency of a High-Temperature Fast-Stirring Solid-Phase Synthesis (HTFS-PS).Synthesis of a model glycopeptide bearing one single N-Glc
HTFS-PS was applied first for the synthesis of the C-terminal HMW1ct(1349–1357), the minimal adhesin fragment with only one glycosylation site, i.e., [Asn1352(Glc)]HMW1ct(1349–1357) with the following Glc-protected precursor, VTLN[Glc(OAc)4]TTGTL-NH2 (GP-1).GP-1 was synthesized on a Rink Amide MBHA resin with 0.48 mmol g−1 loading. Fmoc-L-Asn[Glc(OAc)4]-OH was used as a building block in the process and was introduced using a standard HTFS-PS cycle (Fig. 2). The reaction was performed at 90 °C, using N,N-dimethylformamide (DMF) as a solvent and an overhead stirring of 1200 rpm. Equimolar quantities of amino acids, including Fmoc-L-Asn[Glc(OAc)4]-OH (1.2 equiv.), and of the coupling system 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU, 1.1 equiv.)/N,N-diisopropylethylamine (DIPEA, 2.4 equiv.) were used. One min coupling and 30 s deprotection cycles were applied. GP-1 was synthesized by HTFS-PS in an overall time of 22.5 min including washings, which provided the monoglucosylated peptide with a crude HPLC purity of 62% (Fig. S1 and S2†). The above results confirmed that an equimolar amount of Fmoc-L-Asn[Glc(OAc)4]-OH allowed synthesizing the N-glucopeptide with one glucosylation site in a short time and high efficiency, using a limited quantity of this precious building block.
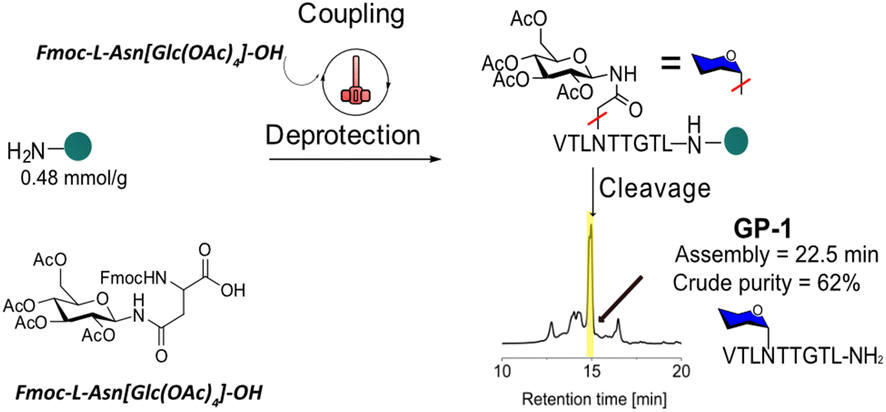 | ||
| Fig. 2 HTFS-PS of GP-1 on Rink Amide MBHA resin (0.48 mmol g−1 loading) solid support. Couplings were performed using 1.2 equiv. of all Fmoc-AAs, including Fmoc-L-Asn[Glc(OAc)4]-OH for 1 min at 90 °C using HATU/DIPEA as coupling system. Fmoc-deprotections were performed using 20% piperidine in DMF for 30 s. HPLC of the crude peptide was recorded at 220 nm. | ||
Synthesis of a model glycopeptide bearing multiple copies of N-Glc moieties
To evaluate the use of HTFS-PS for the synthesis of glycopeptides containing multiple copies of N-Glc moieties, we selected the peptide [Asn1348(Glc), Asn1352(Glc)]HMW1ct(1348–1357) with two N-glucosylation sites, and synthesized the precursor N[Glc(OAc)4]VTLN[Glc(OAc)4]TTGTL-NH2 (GP-2) bearing acetate (Ac)-protected N-Glc moieties.GP-2 was synthesized on Rink Amide MBHA resin with a loading of 0.48 mmol g−1via elongation of GP-1 (Fig. 3).
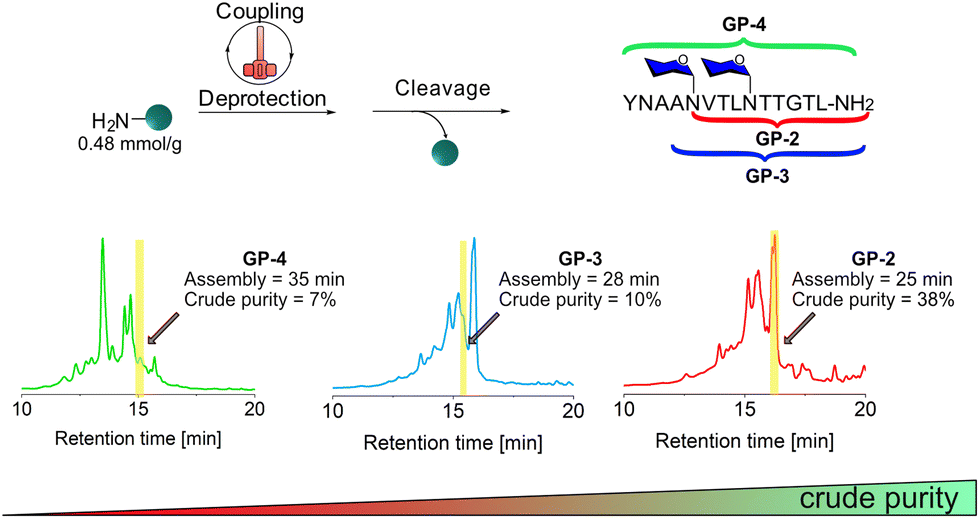 | ||
| Fig. 3 HTFS-PS of GP-2, GP-3, and GP-4 on Rink Amide MBHA resin (0.48 mmol g−1 loading). Couplings were performed using 1.2 equiv. of all Fmoc-AAs, including Fmoc-L-Asn[Glc(OAc)4]-OH and HATU/DIPEA as coupling system for 1 min at 90 °C. Fmoc-deprotections were performed using 20% piperidine in DMF for 30 s at 90 °C. HPLC analysis of the crude peptides were recorded at 220 nm. | ||
The synthesis of GP-2 was completed in 25 min and resulted in a crude purity of 38%, therefore lower than the one recorded for the monoglucosylated peptide GP-1 as an indication of the difficulty of the synthesis (Fig. S3 and S4†).
During the synthesis of AN[Glc(OAc)4]VTLN[Glc(OAc)4]TTGTL-NH2 (GP-3), after coupling Fmoc-Ala-OH to the second glucosylated Asn1348 in GP-2, a significant drop of crude purity to less than 10% was observed (Fig. 3, Fig. S5 and S6†). Further elongation of the peptide by addition of three additional amino acids to obtain YNAAN[Glc(OAc)4]VTLN[Glc(OAc)4]TTGTL-NH2 (GP-4), a Glc-protected precursor of [Tyr1344,Asn1348(Glc),Asn1352(Glc)]HMW1ct(1344–1357), resulted in a crude purity of 7% (Fig. S7 and S8†). Since the drop in purity was significant it was difficult to determine one specific deletion except the one of glycine, which is probably a sequence-dependent problem (see ESI†). These results clearly show that HTFS-PS encountered significant difficulties after the insertion of the second N-Glc.
We assumed that the increase of steric hindrance significantly decreased the efficiency of couplings, especially of the amino acid following the N-glucosylated building block. The aggregation of heavily protected peptides on high-loading solid support is known to be more prominent with the size and complexity of the synthesized peptide. It was previously demonstrated that low-loading solid supports, in which the anchoring sites are distant, can favor the synthesis of prone-to-aggregate peptides.29,30
Effect of the loading of the resin on the efficiency of HTFS-PS of glycopeptides
The syntheses of GP-2, GP-3, and GP-4 were performed again but this time on TentaGel R RAM resin with a lower loading of 0.18 mmol g−1 using the standard HTFS-PS temperature and stirring rate (Fig. 4 and Fig. S9–S15†). The synthesis of GP-2 was performed in 25 min and resulted in a crude yield of 48%, higher than the one recorded with the 0.48 mmol g−1 loading. After further elongation to GP-3 and later to GP-4, HPLC analyses showed that the crude purity remains around 40% and did not significantly decrease after coupling of the second Fmoc-L-Asn[Glc(OAc)4]-OH building block, as previously observed with the high loading support.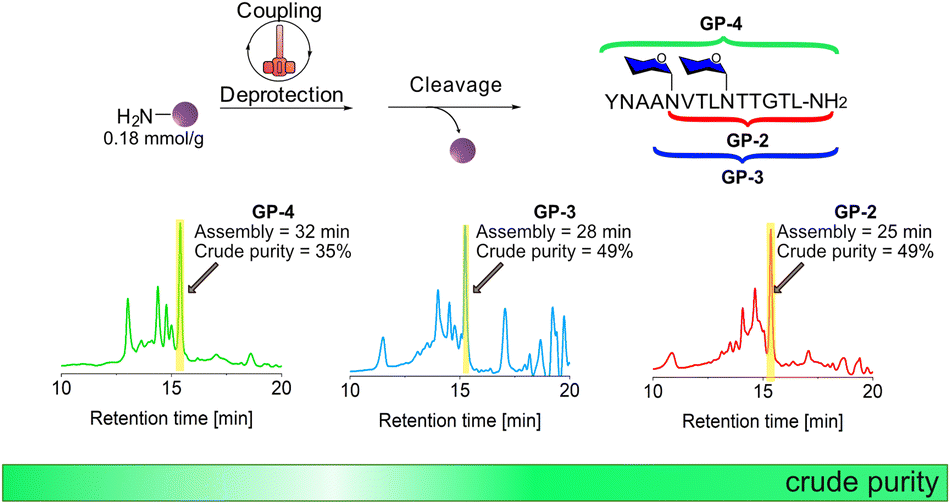 | ||
| Fig. 4 HTFS-PS of GP-2, GP-3, and GP-4 on TentaGel R RAM resin (0.18 mmol g−1 loading). Coupling was performed using 1.2 equiv. of all Fmoc-AAs, including Fmoc-L-Asn[Glc(OAc)4]-OH and HATU/DIPEA coupling system for 1 min at 90 °C. Fmoc-deprotections were performed using 20% piperidine in DMF for 30 s at 90 °C. HPLC analysis of the crude peptides were recorded at 220 nm. | ||
The HTFS-PS of glycopeptides containing multiple N-glycosylated sites suggests that its combination with low loading solid-support can be beneficial for other N-glycopeptides. Since Ala and Val have high aggregation potential compared to other amino acids, it was not surprising to find that, in addition to Fmoc-L-Asn[Glc(OAc)4]-OH, these amino acids introduce another major synthetic hindrance during the synthesis of glycopeptides containing multiple N-glycosylated sites.29 The ability of HTFS-PS performed on low-loading solid support, to overcome this combination of synthetic hurdles, suggests that our accelerated approach can be exploited successfully for other glycopeptide sequences.
Therefore we selected the sequence of the adhesin protein [Tyr1389]HMW1ct(1389-1403) and synthesized under the same conditions both the monoglucosylated peptide YTVVNATNAN[Glc(OAc)4]GSGSV-NH2 (GP-5) and the di-glucosylated YTVVN[Glc(OAc)4]ATNAN[Glc(OAc)4]GSGSV-NH2 (GP-6). GP-5 and GP-6 were synthesized via HTFS-PS on TentaGel resin in 38 min with a crude purity of 49% and 48%, respectively (Fig. 5 and Fig. S16–S21†).
 | ||
| Fig. 5 HTFS-PS of GP-5, GP-6, and GP-7 on TentaGel R RAM resin (0.18 mmol g−1 loading) solid support (A). Couplings were performed using 1.2 equiv.* of all Fmoc-AAs, including Fmoc-L-Asn[Glc(OAc)4]-OH and HATU/DIPEA as coupling system for 1 min at 90 °C. Fmoc-deprotections were performed using 20% piperidine in DMF for 30 s at 90 °C. HPLC of crude (B) and of purified peptides (C) were recorded at 220 nm. *The last six couplings in GP-7, excluding Fmoc-L-Asn[Glc(OAc)4]-OH, were performed using double couplings. | ||
Then, the efficency of HTFS-PS was demonstrated for the synthesis of the N-glycopeptide YALGN[Glc(OAc)4]HTVVN[Glc(OAc)4]ATNAN[Glc(OAc)4]GSGSV-NH2 (GP-7), a Glc-protected precursor of [Tyr1384,Asn1388(Glc),Asn1393(Glc),Asn1398(Glc)]HMW1ct(1384–1403), which has three Asn[Glc(OAc4)] moieties and is significantly longer than the previously synthesized peptides. The 20-amino acid long N-glycopeptide GP-7 was synthesized using HTFS-PS on TentaGEl resin in 63 min with a crude purity of 45% (Fig. 5 and Fig. S22–S24†). In the synthesis of the three above-mentioned peptides, deletion of threonine was observed, possibly because of the proximity to the two valine sequences, which tends to aggregate (see Fig. S17 and S23†).29 All peptides were purified using a standard preparative HPLC to provide the isolated glycopeptides in high purity.
The high crude purity testifies that the HTFS-PS approach has a major advantage in accessing these peptides in an accelerated fashion.
The synthesis of different adhesin fragments using the HTFS-PS approach showed that our strategy can be generalized and is not sequence-dependent. The relatively high crude purity proves that the combination of the low-loading solid support with HTFS-PS can give access to long N-glycopeptide sequences containing multiple N-glycosylated sites.
This combination allows to access these glycopeptides in a very short time and without using large molar excesses of a precious Gbb, such as Fmoc-L-Asn[Glc(OAc)4]-OH.
Conclusions
The importance of glycosylation patterns at multiple sites in biological systems suggests that any synthetic strategy that aims to provide sets of those molecules, have to be fast and with minimal waste of precious Gbb, such as Fmoc-L-Asn[Glc(OAc)4]-OH, which are expensive or not commercially available. The results that we describe here show unequivocally how the combination of HTFS-PS and low-loading solid supports provides a viable way to obtain glycopeptides containing multiple N-glycosylated sites in a short time, with high crude purity and therefore easier to be purified, and without waste of heavily protected Gbb. The new approach enabled overcoming the aggregation problems associated with sequences containing glycosylated and other prone-to-aggregate amino acids, such as Ala or Val. This suggests that the HTFS-PS approach that we propose can be exploited for the synthesis of collections of heavily glycosylated peptides, decreasing dramatically the time and effort to obtain peptides with multiple N-glycosylations at specific sites. This work adds N-glycopeptides to the previously described multi-phosphorylated peptides and O-glycopeptides, proving that HTFS-PS can become a prominent strategy to accelerate the synthesis of post-translationally modified peptides.Experimental
Reagents
All Fmoc-amino acids were obtained from GL Biochem (Shanghai, China), or Matrix Innovation (Quebec City, Canada), with the following side-chain protecting groups: Asn(Trt), His(Trt), Ser(tBu), Thr(tBu), Tyr(tBu). Rink Amide MBHA resin (0.48 mmol g−1) was purchased from Matrix Innovation (Quebec City, Canada). TentaGel R RAM resin (0.18 mmol g−1) was purchased from Rapp Polymere GmbH (Tübingen, Germany). 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) was purchased from Luxembourg Biotechnologies Ltd (Rehovot, Israel). Triisopropylsilane (TIPS) 98% was purchased from Alfa Aesar (Massachusetts, USA). N,N-Dimethylformamide (DMF), dichloromethane (DCM), acetonitrile (ACN), N,N-diisopropylethyl amine (DIPEA), trifluoroacetic acid (TFA), piperidine, methanol (MeOH), and diethyl ether (Et2O) were purchased from BioLab (Jerusalem, Israel) in either HPLC or peptide synthesis purity grade.Synthetic protocols for the synthesis of specifically glycosylated peptides by high-temperature fast stirring-solid phase synthesis (HTFS-PS)
The synthetic method used for the synthesis of glycopeptides GP-n follows the protocols set up for other post-translationally modified peptides by HTFS-PS.21,22 All syntheses were carried out in the same reactor, using 100 mg of solid support. All the steps in the process were performed at 90 °C with a stirring of 1200 rounds per minute (rpm).In the synthesis of GP-7 on the TentaGel resin, double couplings were performed for the last six amino acids except for Fmoc-L-Asn[Glc(OAc)4]-OH.
Author contributions
Poriah Strauss performed the experiments, synthesized, analyzed the compounds, and wrote the paper. Francesca Nuti conceived the idea, performed the experiments, synthesized, analyzed the compounds, and wrote the paper. Michael Quagliata performed the experiments, synthesized, analyzed the compounds, and wrote the paper. Anna Maria Papini conceived the idea, supervised the work, analyzed the data, and wrote the paper. Mattan Hurevich conceived the idea, supervised the work, analyzed the data, and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by the Rita Levi Montalcini Prize for scientific cooperation between Italy (University of Florence) and Israel (Weizmann Institute of Science, Hebrew University, and Bar Ilan University) to A. M. P. (Israel Council for Higher Education grant 2018, attributed in 2019). P. S. thanks the Hebrew University Center for Nanoscience and Nanotechnology for the excellent M.Sc. student scholarship.References
- I.-R. Tak, F. Ali, J. S. Dar, A. R. Magray, B. A. Ganai and M. Z. Chishti, in Protein Modificomics, ed. T. A. Dar and L. R. Singh, Academic Press, 2019, pp. 1–35 Search PubMed.
- A. Varki, Glycobiology, 1993, 3, 97–130 CrossRef CAS.
- S. Esmail and M. F. Manolson, Eur. J. Cell Biol., 2021, 100, 151186 CrossRef CAS.
- F. M. Tuccillo, A. de Laurentiis, C. Palmieri, G. Fiume, P. Bonelli, A. Borrelli, P. Tassone, I. Scala, F. M. Buonaguro, I. Quinto and G. Scala, BioMed Res. Int., 2014, 742831 Search PubMed.
- A. Vasconcelos-dos-Santos, I. A. Oliveira, M. C. Lucena, N. R. Mantuano, S. A. Whelan, W. B. Dias and A. R. Todeschini, Front. Oncol., 2015, 5, 138 Search PubMed.
- M. Slovakova and Z. Bilkova, Catalysts, 2021, 11, 1007 CrossRef CAS.
- J. M. Palomo, RSC Adv., 2014, 4, 32658–32672 RSC.
- A. B. Taresh and C. A. Hutton, Angew. Chem., Int. Ed., 2022, 61(43), e202210367 CrossRef CAS PubMed.
- S. T. Anisfeld and P. T. Lansbury, Angew. Chem., Int. Ed., 1990, 55, 4561–4561 Search PubMed.
- T. Conroy, K. A. Jolliffe and R. J. Payne, Org. Biomol. Chem., 2009, 7, 2255–2258 RSC.
- V. Ullmann, M. Rädisch, I. Boos, J. Freund, C. Pöhner, S. Schwarzinger and C. Unverzagt, Angew. Chem., Int. Ed., 2012, 51, 11566–11570 CrossRef CAS.
- P. Wang, B. Aussedat, Y. Vohra and S. J. Danishefsky, Angew. Chem., Int. Ed., 2012, 51, 11571–11575 CrossRef CAS.
- F. R. Ye, J. Zhao, P. Xu, X. L. Liu, J. Yu, S. G. Wei, J. Z. Liu, X. S. Luo, C. Li, T. L. Ying, J. Wang, B. Yu and P. Wang, Angew. Chem., Int. Ed., 2021, 60, 12904–12910 CrossRef CAS.
- M. A. Bonache, F. Nuti, A. Le Chevalier-Isaad, F. Real-Fernández, M. Chelli, P. Rovero and A. M. Papini, Tetrahedron Lett., 2009, 50, 4151–4153 CrossRef CAS.
- F. Nuti, E. Peroni, F. Real-Fernández, M. A. Bonache, A. Le Chevalier-Isaad, M. Chelli, N. Lubin-Germain, J. Uziel, P. Rovero, F. Lolli and A. M. Papini, Biopolymers, 2010, 94, 791–799 CrossRef CAS.
- I. Paolini, F. Nuti, M. de la Cruz Pozo-Carrero, F. Barbetti, B. Kolesinska, Z. J. Kaminski, M. Chelli and A. M. Papini, Tetrahedron Lett., 2007, 48, 2901–2904 CrossRef CAS.
- F. Rizzolo, G. Sabatino, M. Chelli, P. Rovero and A. M. Papini, Int. J. Pept. Res. Ther., 2007, 13, 203–208 CrossRef CAS.
- S. Pandey, M. C. Alcaro, M. Scrima, E. Peroni, I. Paolini, S. Di Marino, F. Barbetti, A. Carotenuto, E. Novellino, A. M. Papini, A. M. D'Ursi and P. Rovero, J. Med. Chem., 2012, 55, 10437–10447 CrossRef CAS PubMed.
- F. Real-Fernández, M. Di Pisa, G. Rossi, N. Auberger, O. Lequin, M. Larregola, A. Benchohra, C. Mansuy, G. Chassaing, F. Lolli, J. Hayek, S. Lavielle, P. Rovero, J. M. Mallet and A. M. Papini, Biopolymers, 2015, 104, 560–576 CrossRef PubMed.
- J. N. Naoum, I. Alshanski, G. Mayer, P. Strauss and M. Hurevich, Org. Process Res. Dev., 2022, 26, 129–136 CrossRef CAS.
- D. Ben Abba Amiel and M. Hurevich, Eur. J. Org. Chem., 2022, 2022(38), e202200623 CrossRef.
- D. Grunhaus, E. R. Molina, R. Cohen, T. Stein, A. Friedler and M. Hurevich, Org. Process Res. Dev., 2022, 26, 2492–2497 CrossRef CAS PubMed.
- S. Grass, A. Z. Buscher, W. E. Swords, M. A. Apicella, S. J. Barenkamp, N. Ozchlewski and J. W. St Geme, Mol. Microbiol., 2003, 48, 286–286 CrossRef PubMed.
- M. Ieronymaki, F. Nuti, D. Brancaccio, G. Rossi, F. Real-Fernández, Y. Cao, O. Monasson, M. Larregola, E. Peroni, J. Uziel, G. Sabatino, E. Novellino, A. Carotenuto, A. M. Papini and P. Rovero, ChemMedChem, 2017, 12, 751–759 CrossRef CAS PubMed.
- A. Mazzoleni, F. Real-Fernández, F. Nuti, R. Lanzillo, V. Brescia Morra, P. Dambruoso, M. Bertoldo, P. Rovero, J.-M. Mallet and A. M. Papini, ChemBioChem, 2022, 23, e202100515 CrossRef CAS PubMed.
- A. M. Papini, J. Pept. Sci., 2009, 15, 621–628 CrossRef CAS.
- M. T. C. Walvoort, C. Testa, R. Eilam, R. Aharoni, F. Nuti, G. Rossi, F. Real-Fernández, R. Lanzillo, V. Brescia Morra, F. Lolli, P. Rovero, B. Imperiali and A. M. Papini, Sci. Rep., 2016, 6, 39430 CrossRef CAS PubMed.
- L. Yakovlieva, C. Ramírez-Palacios, S. J. Marrink and M. T. C. Walvoort, ACS Chem. Biol., 2021, 16, 165–175 CrossRef CAS.
- V. K. Z. Flegelova and J. Vagner, Int. J. Pept. Protein Res., 1993, 42, 450–454 Search PubMed.
- A. J. Mijalis, D. A. Thomas, M. D. Simon, A. Adamo, R. Beaumont, K. F. Jensen and B. L. Pentelute, Nat. Chem. Biol., 2017, 13, 464–466 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob01886a |
| This journal is © The Royal Society of Chemistry 2023 |