
Synthesis of isochromanone containing natural products from myxobacteria
Mina
Salimimarand
and
Mark A.
Rizzacasa
 *
*
School of Chemistry, The Bio21 Molecular Science and Biotechnology Institute, The University of Melbourne, Parkville, Victoria 3010, Australia. E-mail: masr@unimelb.edu.au
First published on 10th January 2023
Abstract
This review details the biological activity, biosynthesis and synthesis of isochromanone metabolites isolated from myxobacteria. Strategies towards the synthesis of the isochomanone and oxazole fragments of these natural products are highlighted.
Introduction
Among the many sources, microorganisms are a prolific source of compounds that have medicinal applications and around 130 current drugs have microbial origins.1 According to the latest study of all medications globally between 1981 and 2006, natural products or their related semisynthetic counterparts account for 34% of all small-molecule pharmaceuticals.2,3 Myxobacteria are Gram-negative, and rod-shaped eubacteria4 that are a rich source of biologically active natural products.5 These microorganisms dwell in a broad scope of environments such as soil, marine, compost and rotting plants and exhibit morphological and physiological properties that are practically identical with eukaryotic species.6 Myxcobacterial cells can exist in temperatures ranging from 4 to 45 °C, although vegetative cells may withstand temperatures as low as −60 °C and as high as 140 °C.4 Two distinct traits distinguish myxobacteria from other bacteria. Firstly, they can move by gliding on solid surfaces and, as such, are often known as “gliding bacteria”. Myxobacteria cells can therefore travel across surfaces by gliding or creeping, resulting in cell colonies that spread as thin swarms.7 The movement across solid surfaces is enabled by two virtually independent gliding motility processes. Social movement allows cells to migrate provided that the cells are within one cell length of one another and requires cells employing pili to drive the cell further which is vital in multicellular activities including fruiting body generation.6 The precise mechanism of adventurous movement is unclear but this involves cells moving independently within groups and without direct interaction with other cells.6The second distinguishing feature is that when a swarm is under starvation conditions, myxobacterial cells assemble into multicellular formations called fruiting bodies,11 which are conglomerations of thousands of cells that range in size from 20 to 200 μm. These bodies can possess up to ten dormant cells and are often vividly coloured, commonly yellow, orange, red brown, or black6 (Fig. 1). The fruiting bodies are resilient to severe temperatures, UV radiation, and dehydration, enabling the swarm to endure environmental adversities for longer durations, assuring survivability during periods in which growth is not favourable.6 The greater cell density that results from the formation of fruiting bodies is hypothesised to enhance cells longevity by minimizing the impact of dispersion on the swarm's hydrolytic enzymes and nutrients. The shape of the fruiting bodies varies by species, starting from a plain lump bearing myxospores in the genus Myxococcus continuing to miniature treelets with slime-made stems that split into sporangioles in the genus Chondromyces.7,12
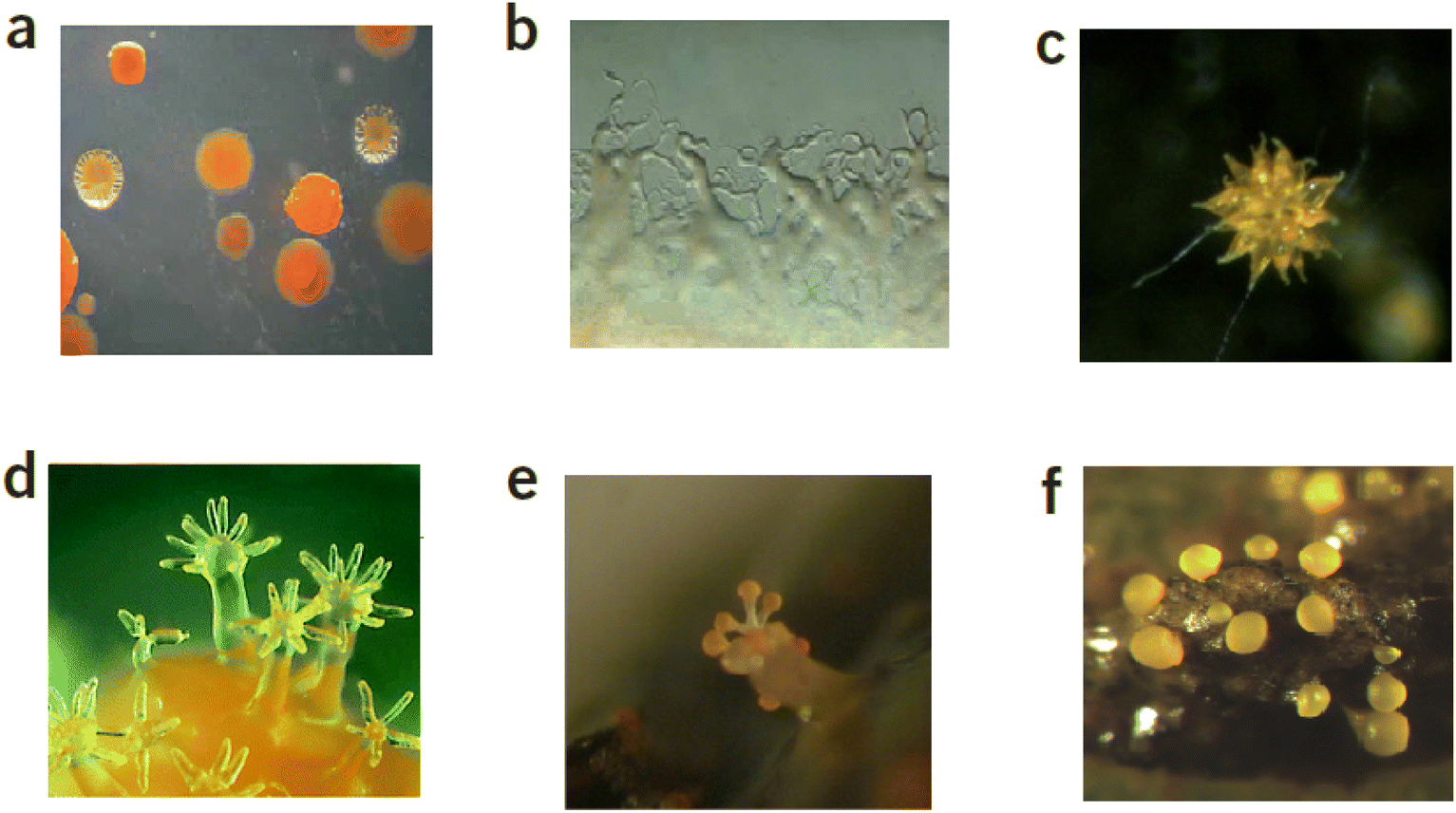 | ||
| Fig. 1 (a) Sorangium cellulosum colonies8 (b) Swarming growth patterns of Sorangium cellulosum So ce568 (c) Chondromyces apiculatus,8 (d) A variant of fruiting bodies in myxobacteria9 (e) Melittangium boletus, early stage of development, on a piece of plant material attached to rabbit dung8 (f) Fruiting body with spores of Myxococcus xanthus.10 | ||
Myxobacteria secondary metabolites
Myxobacteria are an abundant source of diverse bioactive secondary metabolites. Out of nearly 7500 reported myxobacterial strains, around 67 compound family classes have been identified in myxobacteria to date with more than 500 compounds recorded.8,13 A number of unique natural products are produced by myxobacteria with structural variety including polyketides, non-ribosomal peptides (NRPs), polypeptides, and their corresponding hybrids, as well as alkaloids, terpenoids, and phenyl-propanoids.13,14Myxobacteria account for around 5% of all reported microbial substances and the range of biological activities include antifungal, antibacterial, anticancer, antiviral, protein-synthesis inhibition, cytotoxicity and pheromone-like properties.13 For example, both epothilone B (1) (from Sorangium cellulosum) and tubulysin A (3)15–17 (from Archangium gephyra) are cytotoxic and operate on the cytoskeleton of eukaryotic cells. Compound 2 inhibits tubulin polymerization, while compound 1 regulates microtubules with sub-nanomolar IC50 values,18 and both cause the arrest of the cell cycle and trigger apoptosis.16,19 The natural compound 1 was too toxic for further investigation but the aza-analogue ixabepilone 2 (Ixempra®) became the first myxobacteria-derived drug licensed in 2007 for clinical use in the treatment of breast cancer in US (Fig. 2).20,21
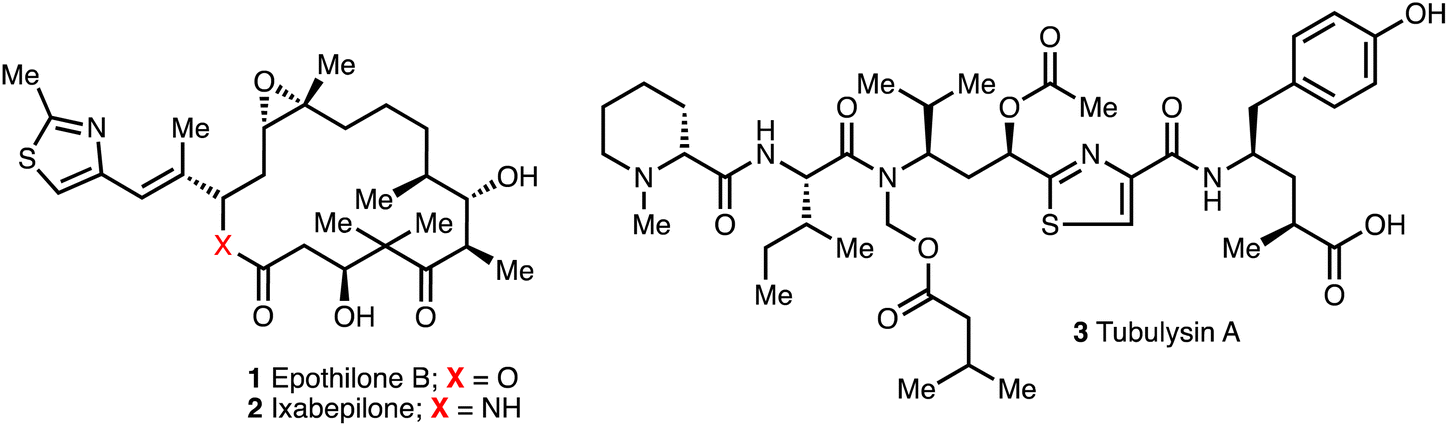 | ||
| Fig. 2 Epothilone B (1), the semi-synthetic derivative ixabepilone (2) and tubulysin A (3). | ||
Isochromanones from myxobacteria
The isochromanone containing ajudazols A (4) and B (6)22 were isolated from Sorangium cellulosum and possess potent antifungal and antibacterial activity. The biosynthetic precursors, deshydroxyajudazol A (5) and B (7) were identified as minor metabolites from extracts of the myxobacterium Sorangium cellulosum, strain So ce701 by Mueller and co-workers23 in 2012 but not fully characterised. The related isochromanones icumazoles A (8), B1 (9) and B2 (10) and noricumazoles A (11), B (12) and C (13),23,24 were isolated from the myxobacterium, Sorangium cellulosum, strain So ce701 by Mueller and co-workers23 and possess antifungal and antiviral activity (Fig. 3).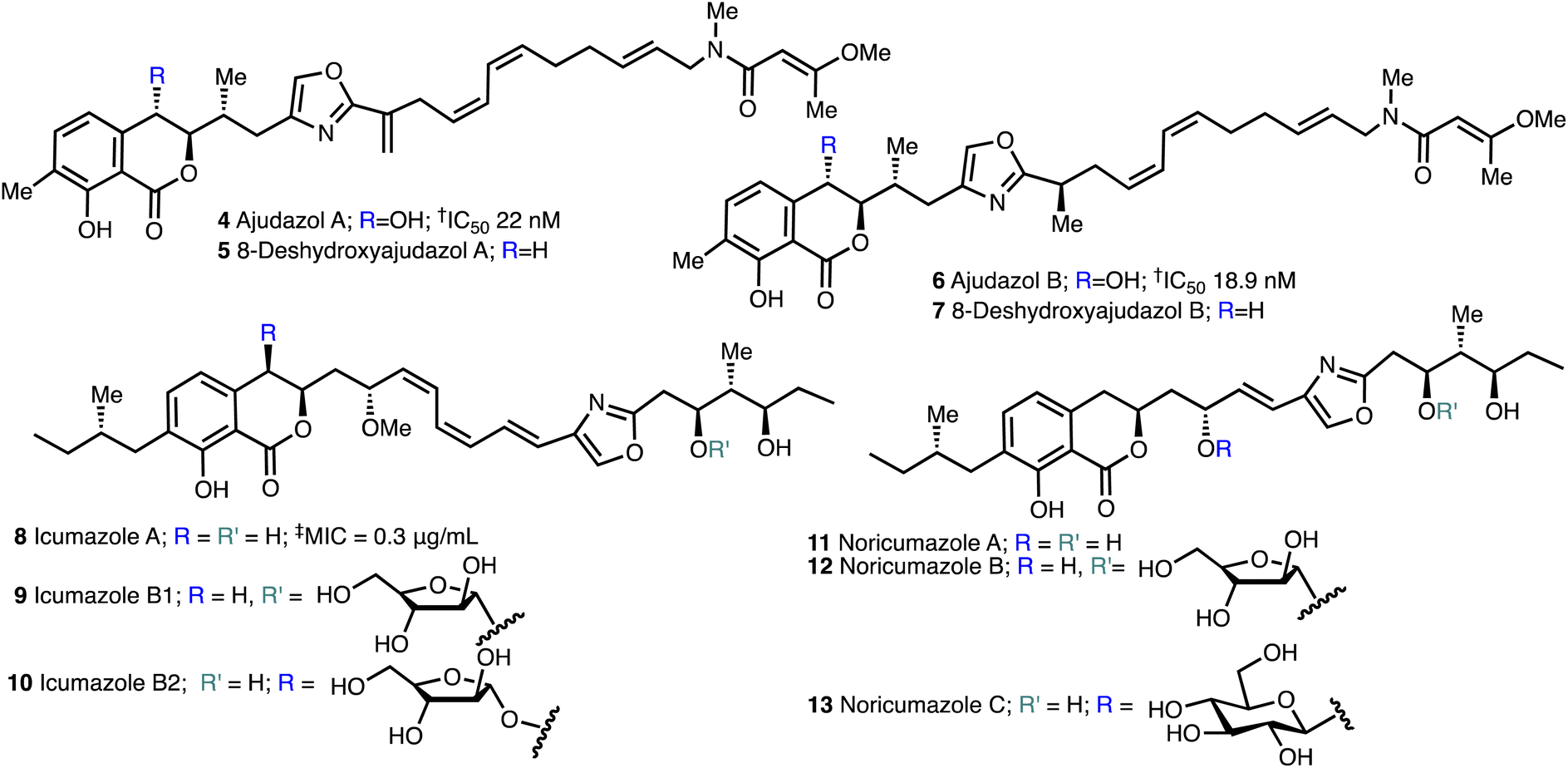 | ||
| Fig. 3 Structure of ajudazols, 8-deshydroxyajuazols, icumazoles and noricumazoles. †Inhibition of the oxidation of NADH in SMP. ‡Inhibition of the cell growth in R. glutinis. | ||
Structural assignment
Menche and co-workers25 employed a novel bioinformatics gene cluster analysis of a ketoreductase (KR) domain for assignment of a hydroxyl-bearing stereocentre together with enoylreductase (ER) assignment of methyl containing stereocentres to fully determine the stereochemistry of ajudazols A (4) and B (6) (Fig. 4). This analysis was then confirmed by the total synthesis of ajudazol B (6) using an asymmetric ortho-lithiation approach.25 A ketoreductase (KR)-mediated reduction of the ketone intermediate yields the hydroxyl-bearing stereocenter at C9 and the presence of an amino acid aspartate residue links to an S-configuration for the 2° alcohol asymmetric centre at C9 in the propionate residue. ER-catalysed reduction determines the stereochemistry of the methyl-bearing C10 and C15 centres and the absence of a tyrosine residue in the catalytic ER domain sets the R stereochemistries shown.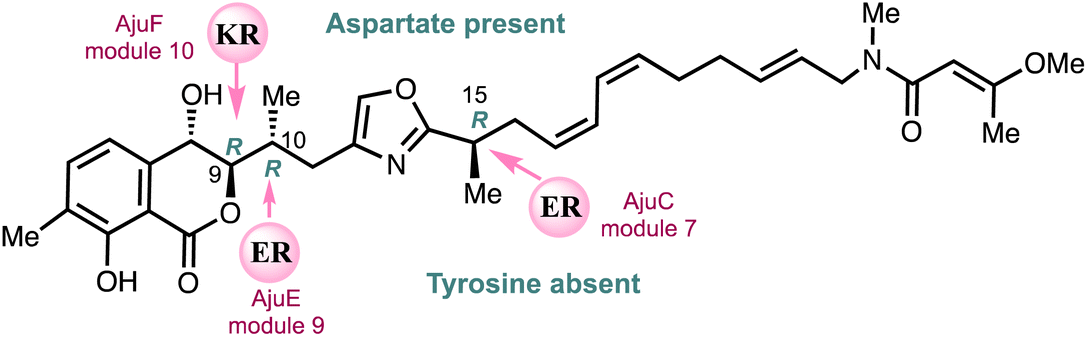 | ||
| Fig. 4 Assignment of the absolute configuration of ajudazol B (6) using proteomics.25 | ||
Key structural elements of the icumazoles include an isochromanone moiety, a Z,Z,E-configured triene chain, and a 2,4-disubstituted oxazole. The high-resolution mass spectrum (ESI) and the 13C and 1H NMR spectra suggested a molecular formula of C33H45NO7 and the structure of (8) was then assigned by COSY, HMQC, HMBC, and ROESY 2D NMR experiments (Fig. 5).
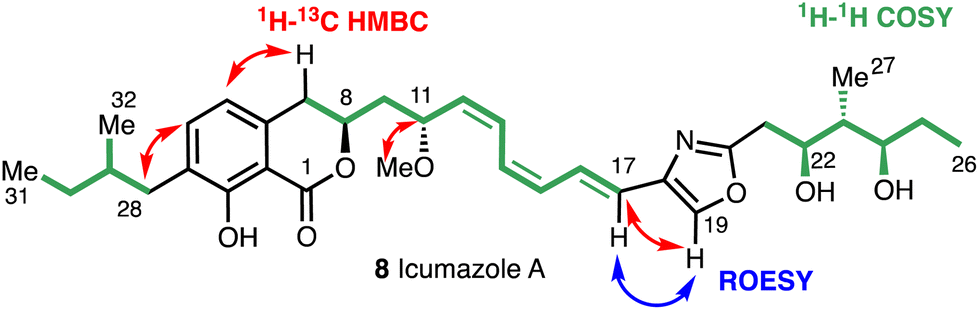 | ||
| Fig. 5 Key 2D NMR correlations for icumazole A (8). | ||
The 1H NMR and HSQC spectra of icumazole A (8) enabled the assignment of all protons. Correlations in the HSQC spectrum showed that H19 was a component of a heteroaromatic ring system (Fig. 4) and the HMBC correlations for H19 with C18 and C20 confirmed the presence of an oxazole ring. Correlations for H11 and the methoxy group C33 confirmed the existence of a methoxy group. The relative configuration of the 1,3-diol moiety (C22 and C24) was proposed using the method reported by Rychnovsky.26 The stereochemistry of triene chain was suggested as 12Z, 14Z, 16E by the vicinal coupling constants between H12 and H13, H14 and H15 (J = 11–12 Hz, cis), and H16 and H17 (J = 15.2 Hz, trans).23 The absolute stereochemistry of icumazole A (8) was proposed to be that shown in Fig. 3 by comparison with that of noricumazole A (11), the structure of which was confirmed by a total synthesis.23 This synthesis also allowed for the elucidation of the structures of icumazole B1 (9) and icumazole B2 (10).
Biological activity of icumazoles
Icumazole A (8) inhibited cell growth in the yeast-like bacterium R. glutinis, (MIC = 0.3 μg mL−1).4 Although no fungicidal behaviour was observed, inhibition DNA, RNA, and protein synthesis was observed immediately after the introduction of icumazole A (8) to R. glutinis. Icumazole A (8) inhibits NADH oxidation by 60–70% with a maximal inhibitory concentration of 5 μg mL−1 and it has been proposed that complete inhibition of NADH oxidation does not occur due to alternate NADH oxidation routes in fungi.23Proposed biosynthesis of the ajudazols
The myxobacterium Chondromyces crocatus produces six distinct classes of secondary metabolites, among them the oxazole-containing anti-fungal compounds ajudazols A (4) and B (6) (Fig. 3)22,27 as well as the minor metabolites, 8-deshydroxyajudazols A (4) and B (7). These natural products all contain an isochromanone moiety, a 2,4-disubstituted oxazole, and a Z,Z,E-configured triene chain terminating in a 3-methoxybutenamide and ajudazol B (6) 8-deshydroxyajudazol B (7) both have an additional asymmetric centre. The ajudazols A (4) and B (4) are potent inhibitors of the electron transport chain in beef heart submitochondrial particles (SMP) at the site of complex I (NADH uniquinone-oxidoreductase).27 Ajudazol A (4) inhibited the oxidation of NADH in SMP with an IC50 of 13.0 ng mL−1 (22 nM), whilst ajudazol B 6 had an IC50 of 10.9 ng mL−1 (18.39 nM).22,28The enzymes polyketide synthase (PKS) and non-ribosomal polypeptide synthase (NRPS) mediate the synthesis of the key polyketide components. Coenzyme-A activated short chain carboxylic acids and amino acids are embedded into products in these mixed systems in three fundamental stages.29
To achieve the molecular scaffolding, polyketide functions are transformed by enzymatic reductions, dehydrations, and cyclisation, which can then be subjected to post-PKS adjustments to yield metabolites. For the ajudazols, it was found that even though PKS-NRPS is responsible for the majority of the carbon structure, several functional groups, particularly the C8-hydroxyl group and the C15-exo-methylene, had no plausible polyketide biosynthetic source.
The gene cluster involved in ajudazol bioproduction was discovered during the study in the core biosynthetic pathways of Chondromyces crocatus. Remarkably, the PKS-NRPS sequence for ajudazol biosynthesis has a significant level of correlation between the order of biochemical transformations and the sequence of genes inside the cluster, which is an uncommon feature. Deshydroxyajudazol production is regulated by enzymes known as AjuX that are represented by 10 of the 12 genes in the ajudazol biosynthetic gene cluster. Eight type I PKS enzymes, one NRPS, and one hybrid NRPS-PKS enzyme are among the AjuX enzymes (Scheme 1).
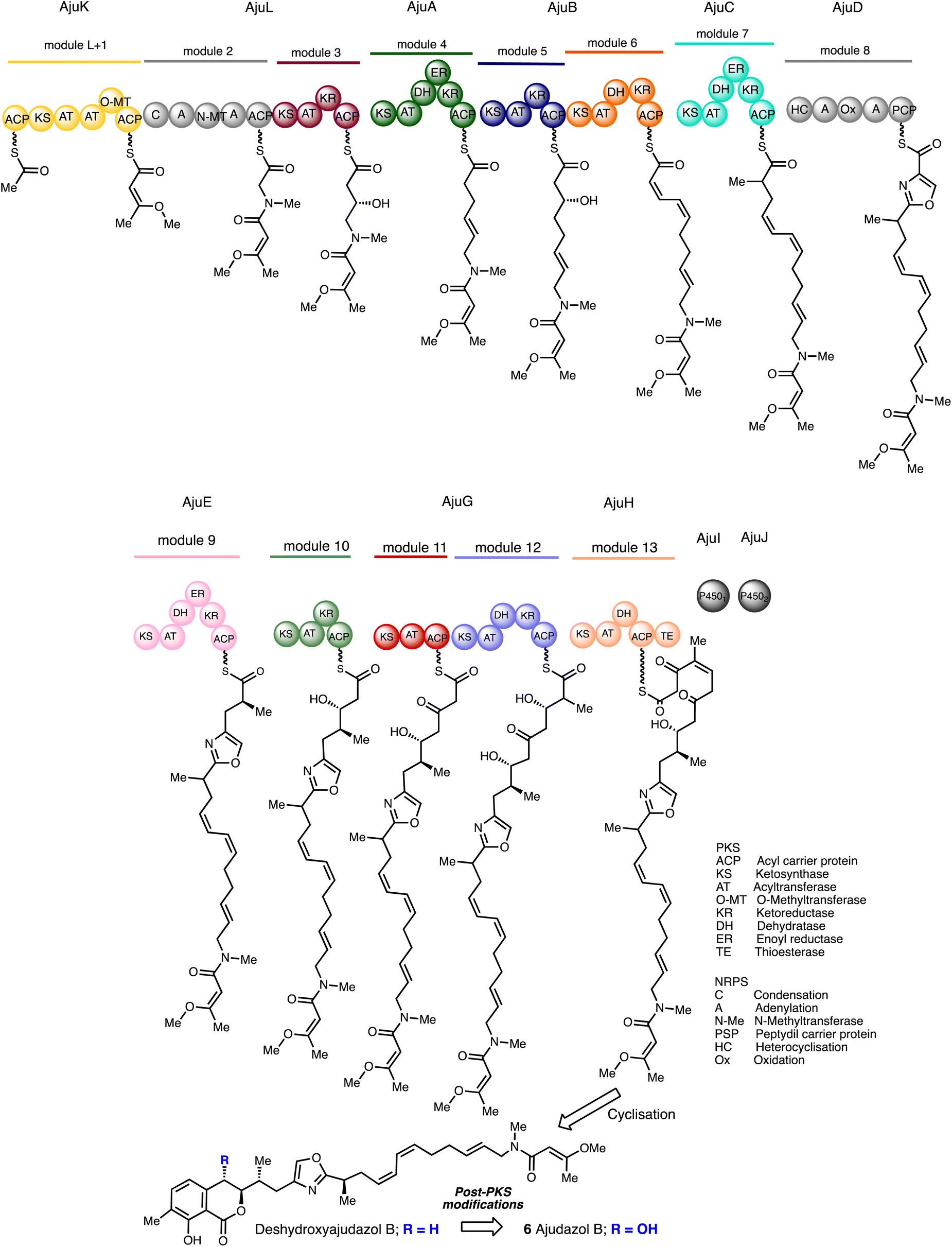 | ||
| Scheme 1 Postulated biosynthesis of the ajudazols.30 | ||
AjuK possesses an O-methyl transferase that utilises S-adenosyl methionine (SAM) to methylate diketone enols. The subsequent stages in the formation of the carbon backbone are conducted by acyltransferase or adenylation domains situated in the modules of subunits AjuA–AjuH. Numerous dehydratase domains are missing or deemed dormant, although other active dehydratase domains in downstream modules are expected to make up the difference. All the functionalisation steps are established ahead of cleavage from the acyl carrier protein (ACP) excluding the hydroxyl group at C8 and the generation of the isochromanone.
Mueller and co-workers30 proposed that an unusual thioesterase mediates isochromanone ring generation in ajudazol biosynthesis. It is believed that the biosynthetic machinery does not utilise a terminal cyclase but is a derivative of a thioesterase (TE) which is responsible for the construction of the isochromanone. The first proposed biosynthetic pathway (i) involves the attack of the C9 hydroxyl group on the acyl unit under TE catalysis to yield a ten-membered lactone, followed by C2/C7 aldol condensation and aromatisation to furnish the isochromanone. The second proposed route (ii) begins with aldol condensation and aromatisation with the TE domain still attached and further elaboration under TE-catalysis causes lactonisation and chain detachment. The final possible pathway (iii) entails spontaneous generation of the isochromanone entity by detachment of the ACP domain in absence of TE (Fig. 6).
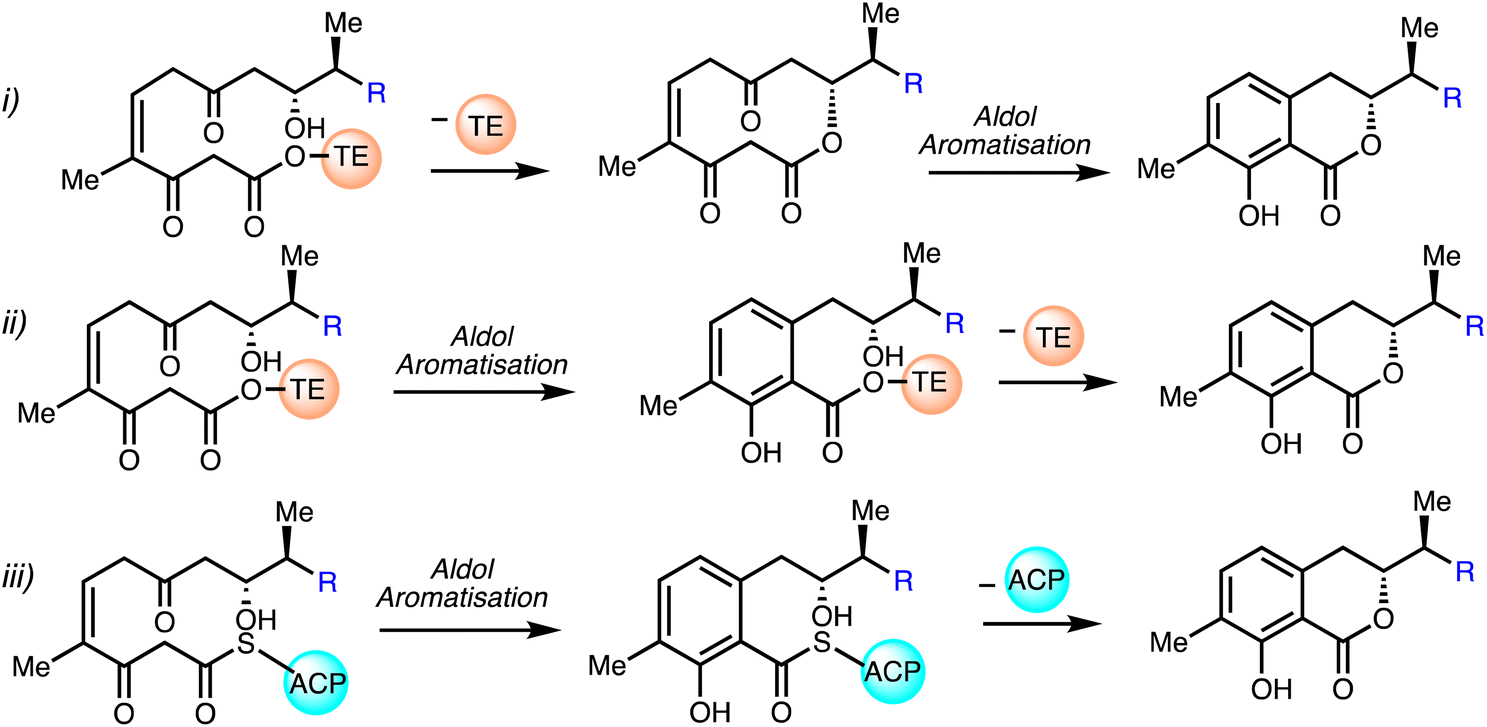 | ||
| Fig. 6 Proposed biosynthesis of the isochromanone.30 | ||
Synthetic approaches towards isochromanones from myxobacteria
Taylor and co-workers31 reported a synthesis of the eastern fragment 26 of ajudazol A (4) which utilised a double acetylene carbocupration starting with iodide 14. Coupling of the resultant cuprate with 2,3-dibromopropene 16 gave the polyene 17 and deprotection gave alcohol 18 (Scheme 2). Oxidation afforded aldehyde 19 and Wittig reaction with ylide 20 provided ester 21. DIBAL-H reduction followed by THP protection afforded 22 and halogen exchange gave the more reactive iodide 23 which was converted into amine 24. Coupling with acid 25 then gave the amine 26 and a Stille coupling with oxazole stannane 27 furnished the model oxazole 28 in 60% yield.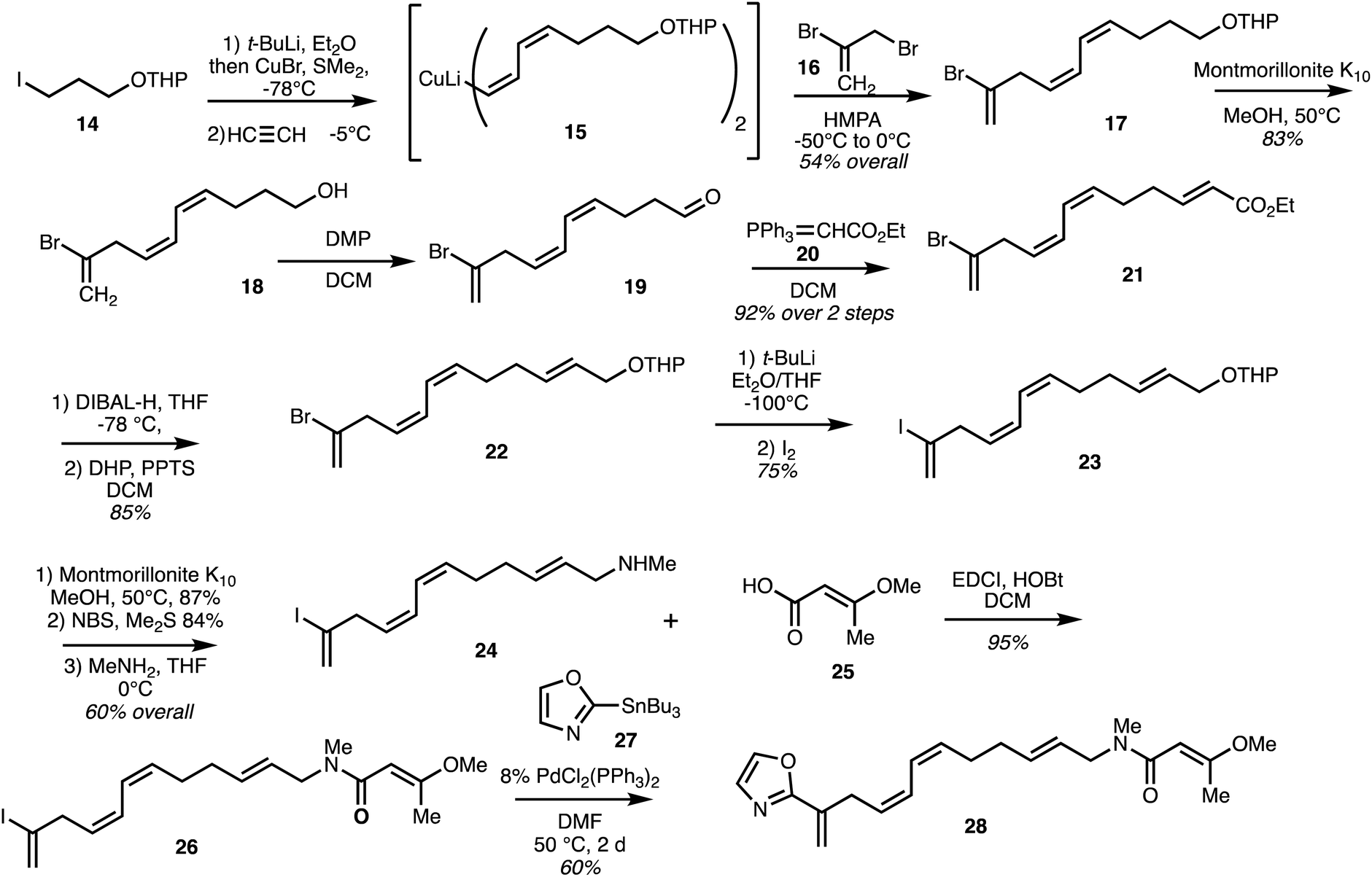 | ||
| Scheme 2 Synthesis of the model eastern fragment of ajudazol A (4) (Taylor).31 | ||
In 2007, Rizzacasa and co-workers reported the synthesis of the C9–C29 oxazole triene fragment of ajudazols A (1) and B (6).32 The synthetic strategy commenced with alkylation of dimethyl malonate 29 with propargyl bromide. Reduction then furnished diol 31 and subsequent monosilylation and two-staged oxidation provided acid 32 which underwent peptide coupling with the amine 33 followed by selective removal of the TBS group. Dess–Martin oxidation of the alcohol 34 afforded aldehyde which was cyclised under Wipf protocol to oxazole 35. Racemic amine 33 underwent peptide coupling with acid 36 to afford the amide. Silyl group removal, Dess–Martin oxidation, and oxazole formation afforded the model ajudazol B oxazole 37 in high yield (Scheme 3).
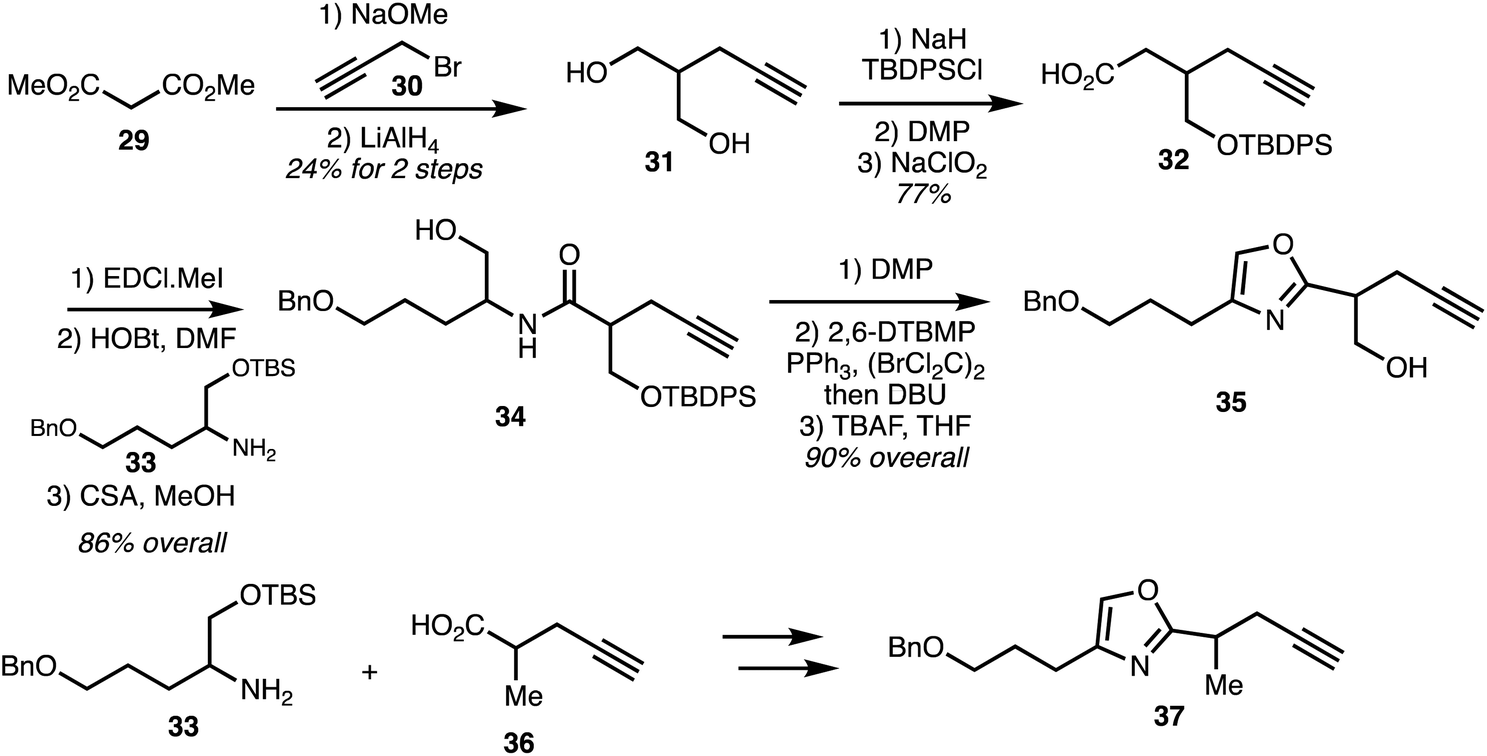 | ||
| Scheme 3 Synthesis of the oxazole fragment of ajudazols (Rizzacasa).32 | ||
The vinyl iodide-containing eastern fragment 41 of the ajudazols was constructed beginning with oxidation of the known alcohol 38 (Scheme 4).32 Wittig homologation furnished diene 39 and ester reduction together with bromide synthesis and methylamine substitution provided the secondary amine 40. The key eastern fragment 41 was then afforded through peptide coupling of 40 with acid 25.
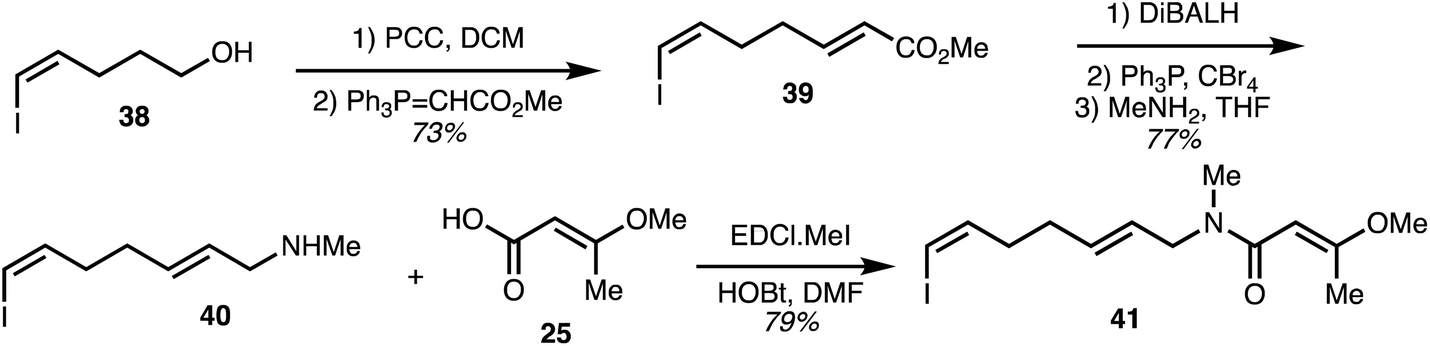 | ||
| Scheme 4 Synthesis of the eastern fragment of ajudazols (Rizzacasa).32 | ||
A Sonogashira cross-coupling step between the oxazole-isochromanone fragment alkynes 35 and vinyl iodide 41 forms the C18–C19 bond in 42 and partial reduction installed C17–C18 Z-alkene to give the model system 43 (Scheme 5).32
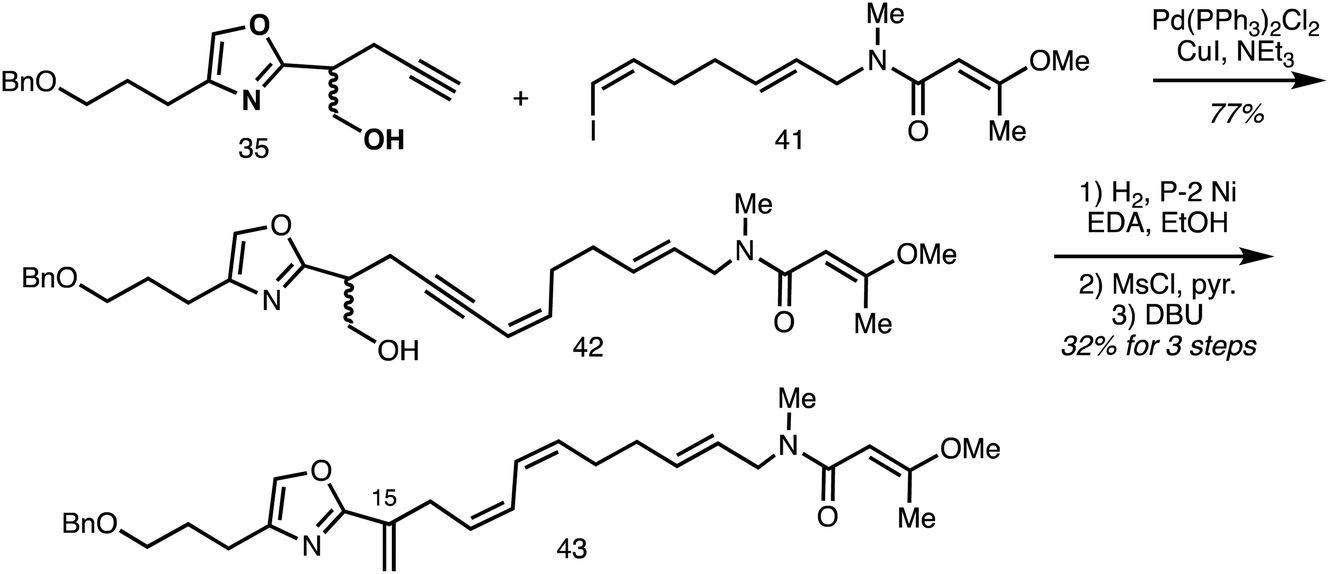 | ||
| Scheme 5 Synthesis of the C9–C29 fragment 43 of ajudazol A (Rizzacasa).32 | ||
In 2008, Marquez and co-workers,33 reported a synthetic approach to the western isochromaone fragment of the ajudazols by an oxidative rearrangement of an isobenzofuran. This involved reduction of commercially available phthalide 44 to alcohol 45 and this was transformed into the acetal 46. Treatment of acetal 46 with methyllithium and diisopropylamine gave the isobenzofuran anion 47, which upon treatment with aldehyde 48 generated the hydroxyisobenzofuran 49. Oxidative rearrangement of intermediate 49 afforded the unstable lactols 50 and further oxidation gave the isochromanone 51 (Scheme 6).33
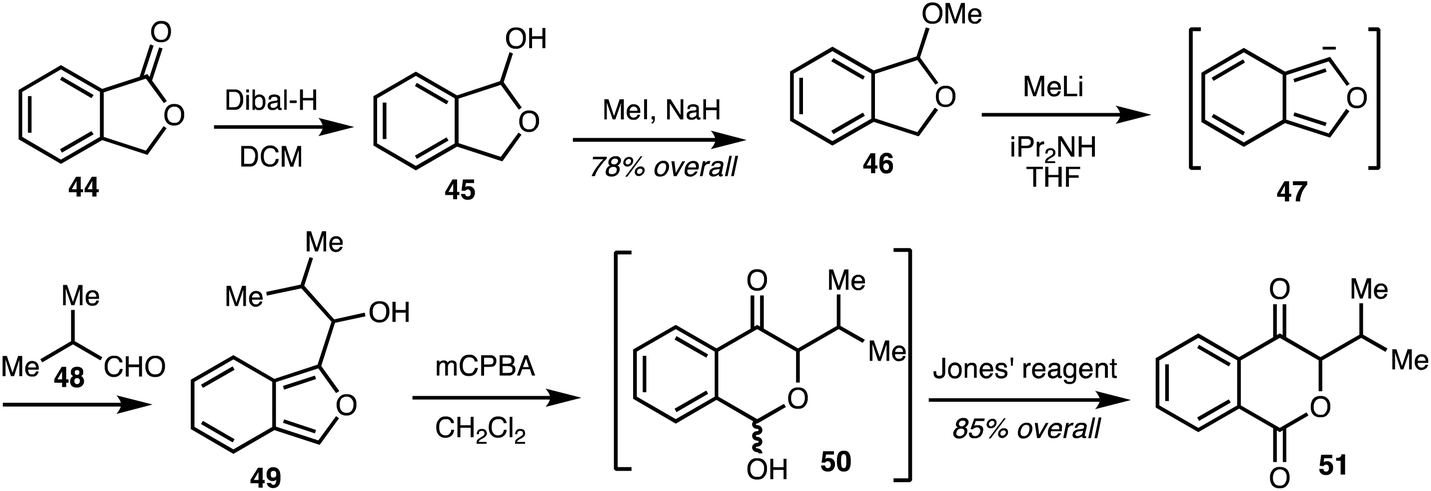 | ||
| Scheme 6 Synthesis isochromanones by oxidative rearrangement of isobenzofurans (Marquez).33 | ||
Coupling of the aldehyde 52 with the anion derived from isobenzofuran 46 followed by oxidative rearrangement of the carbinol intermediate and Jones oxidation of the subsequent lactols 53 gave the lactones 54 and 55 as a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mixture of diastereomers. Luche reduction furnished isochromones 56 and 57 and selective crystallisation afforded the major diastereomer 56 (Scheme 7).
:2 mixture of diastereomers. Luche reduction furnished isochromones 56 and 57 and selective crystallisation afforded the major diastereomer 56 (Scheme 7).
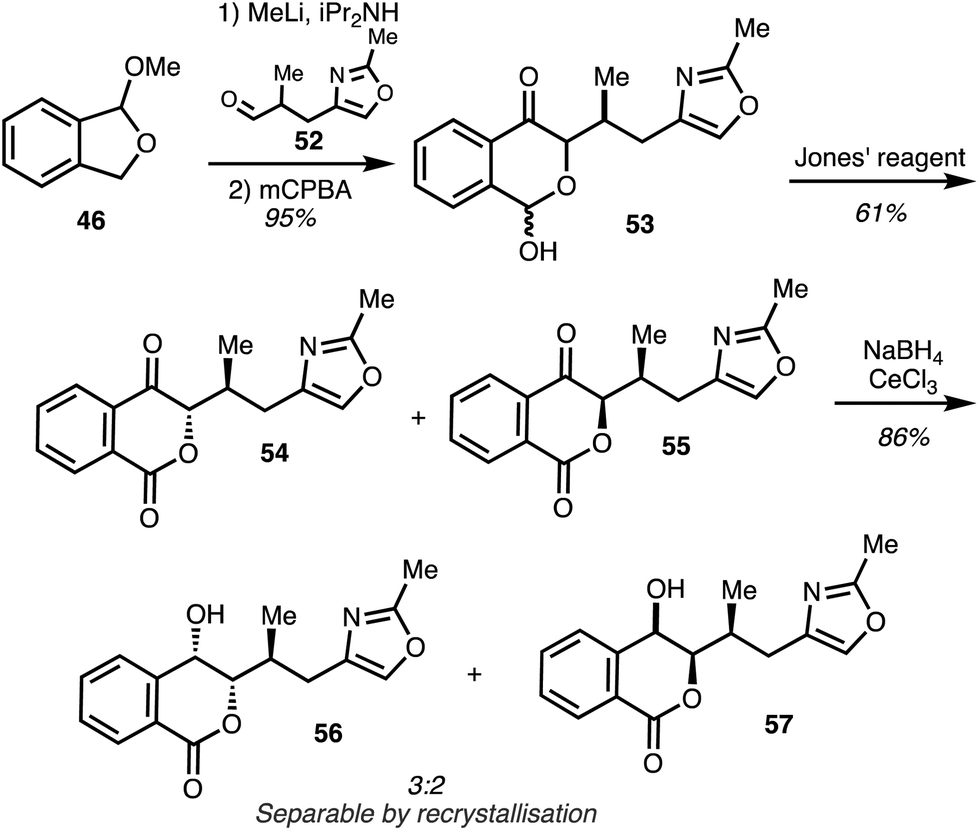 | ||
| Scheme 7 Synthesis of the western fragment of the ajudazols (Marquez).33 | ||
In two separate accounts, Rizzacasa and co-workers detailed the total synthesis of 8-dehydroxyajudazols A (5) and B (7).34,35 The isochromanone fragment was synthesised via an intramolecular Diel–Alder (IMDA) approach.36 The Diels–Alder precursor 63 was obtained by a Wittig olefination between optically pure aldehyde 58 and the ylide derived from the phosphonium salt 59. The diene 60 was obtained as a 3:1 mixture of Z,E diene as the major product. Acid-induced TBS deprotection followed by ester exchange with excess methyl propiolate 61 mediated by Otera's catalyst gave ester 62 and alkyne bromination afforded the IMDA precursor 63 (Scheme 8).
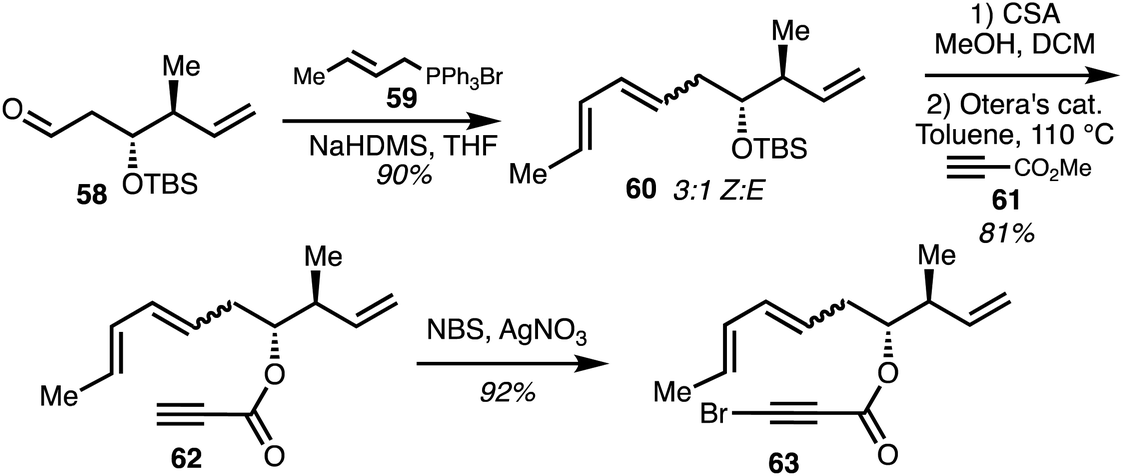 | ||
| Scheme 8 Synthesis of isochromaone precursor 63 (Rizzacasa).34 | ||
Heating a solution of the bromo dienyne 63 in toluene at 180 °C in a sealed tube induced the IMDA reaction and subsequent oxidative aromatisation afforded the isochromanone 64. Coupling of the bromide with pinacol borane under conditions reported by Buchwald and oxidation followed protection gave isochromanone 65. Compound 65 was then extended to afford 66 and dihydroxylated to give diol 67. Amide precursor 68 was formed from coupling between diol 67 and the known optically pure acid R-36. Azide formation and reduction along with concomitant O,N-acyl migration gave amide 69. Oxidation and cyclodehydration under Wipf conditions37,38 gave the oxazole 70. Sonogashira coupling of alkyne 70 with vinyl iodide 41 yielded 71 which was subjected to partial reduction with H2 gas and P2-Ni in the presence of ethylenediamine34 to give a diastereoisomer of 8-deshydroxyajudazol B (7) (Scheme 9).
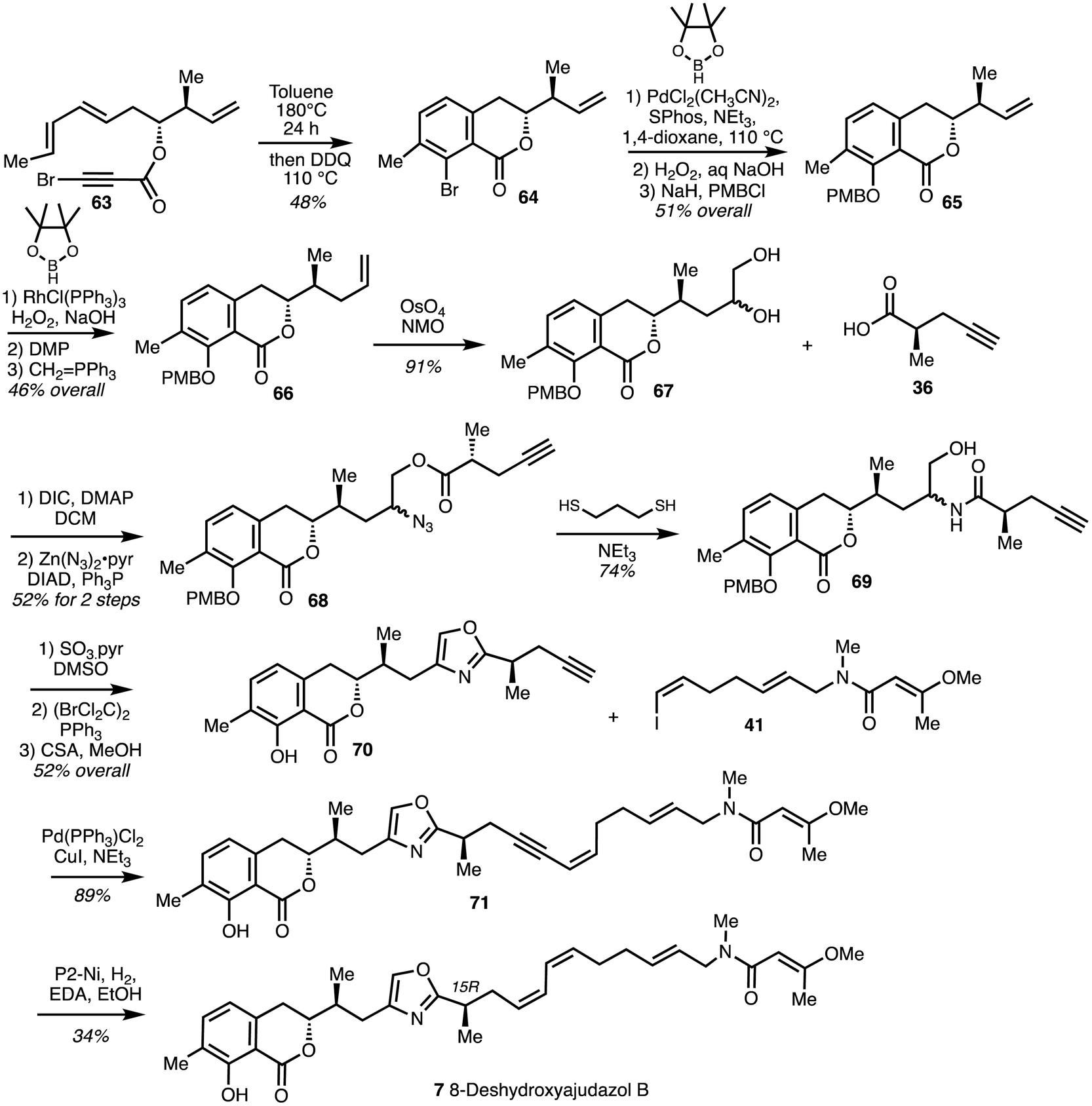 | ||
| Scheme 9 Total synthesis of 8-deshydroxyajudazol B (7) (Rizzacasa).34 | ||
In the synthesis of the enantiomer of 8-deshydroxyajudazol A (5) reported by Rizzacasa and co-workers,35 the double bond at C15 was installed before the Sonogashira coupling. Mesylation of alcohol 72 (derived from diol 67) provided 73 which was subjected to PMB deprotection and exposure to DBU gave the 1,1-disubstituted alkene 74 in high yield. Sonogashira coupling of alkyne 74 with iodide 41 and partial reduction with Cu/Ag activated Zn afforded the enantiomer of 8-deshydroxyajudazol A (5) (Scheme 10).
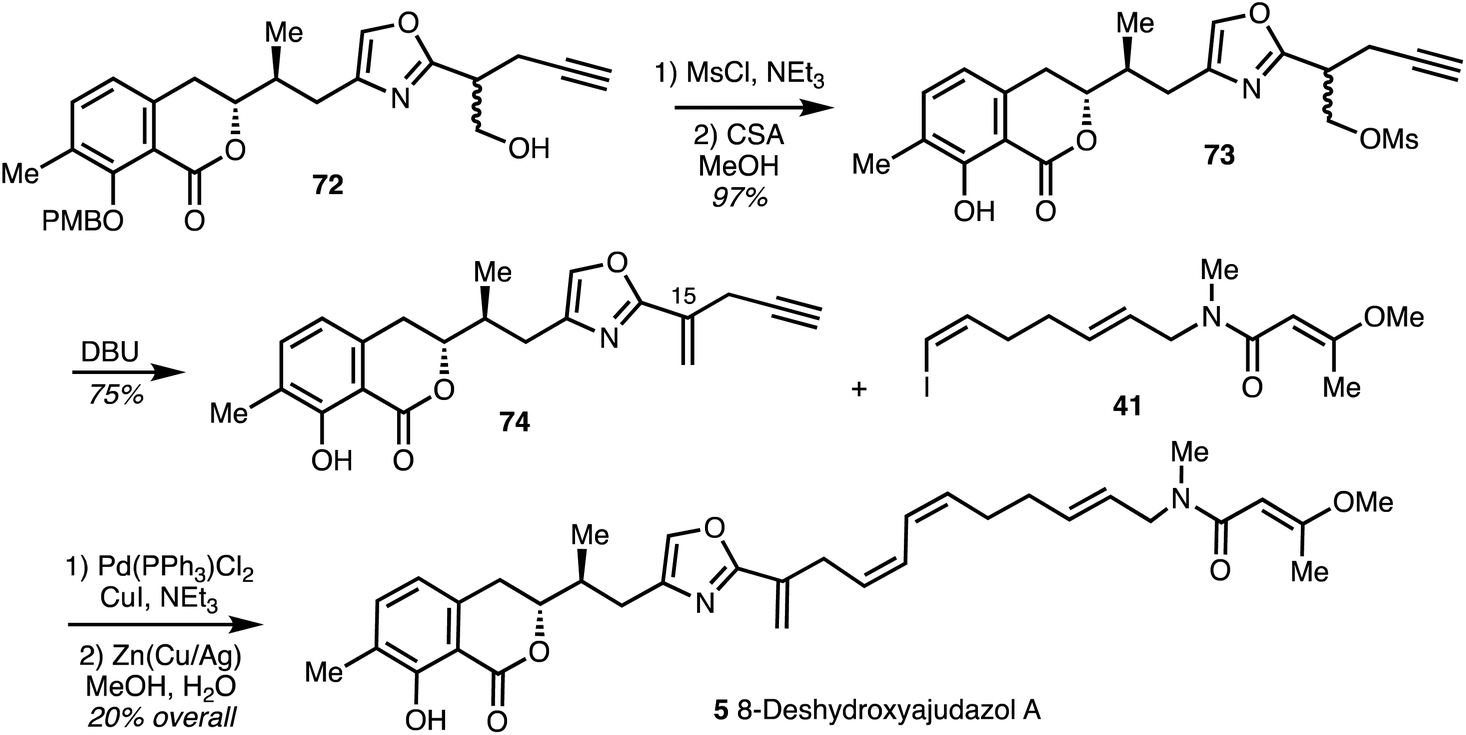 | ||
| Scheme 10 Total synthesis of 8-deshydroxyajudazol A (5) (Rizzacasa).35 | ||
Menche and co-workers25 utilised a synthetic strategy centered on a Suzuki coupling of the eastern and western fragments. The eastern fragment 80 was obtained starting with ester 75 which underwent enol methylation hydrolysis and coupling with allyl amine gave 79. Cross-metathesis using 77 followed Ohira–Bestmann homologation using 78 provided alkyne 79 in 4 steps 55% overall yield. Hydroboration with pinacol borane using a rhodium catalyst39 furnished the eastern fragment 80 (Scheme 11).
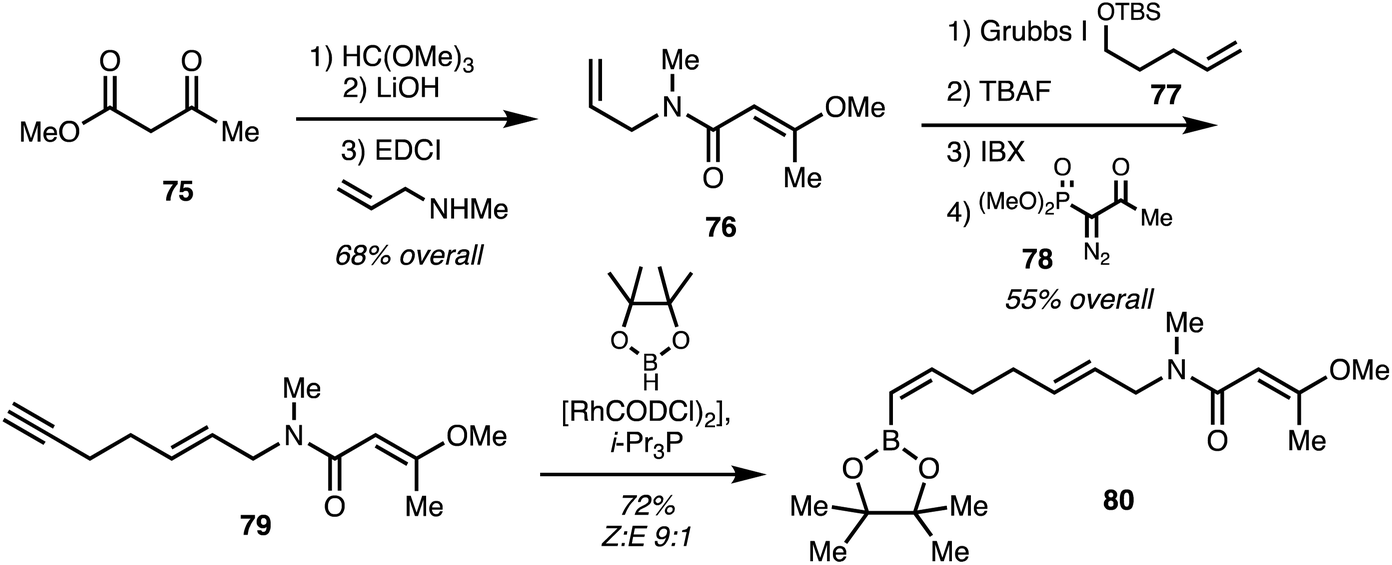 | ||
| Scheme 11 Synthesis of ajudazol B (6) eastern fragment (Menche).25 | ||
The synthesis of the western fragment began with a Brown crotylation of aldehyde 81 followed by TES protection to provide alkene 82. Homologation of 82 by hydroboration, oxidation and Wittig reaction afforded ester 83 which was converted into aldehyde 84. ortho-Lithiation of amide 85 followed by condsensation of the resultant anion with Andersen reagent1186 gave the sulfoxide 87. Lithiation of 87 and coupling with 84 furnished 88 in a high yield and good stereoselectivity. Deprotection of the phenol and lactonisation gave the isochromanone 89 and dihydroxylation, protection, azide substitution and reduction gave the amine 90 in 71% yield. Amide coupling of 90 with the acid 36 and cyclodehydration under modified Wipf conditions37 formed the oxazole 91. Alkyne iodination followed by cis-reduction using NBSH40 gave the Z-vinyl iodide 92. A Suzuki coupling of the iodide 92 with 80 followed by TBS-deprotection yielded (+)-ajudazol B (6) (Scheme 12).
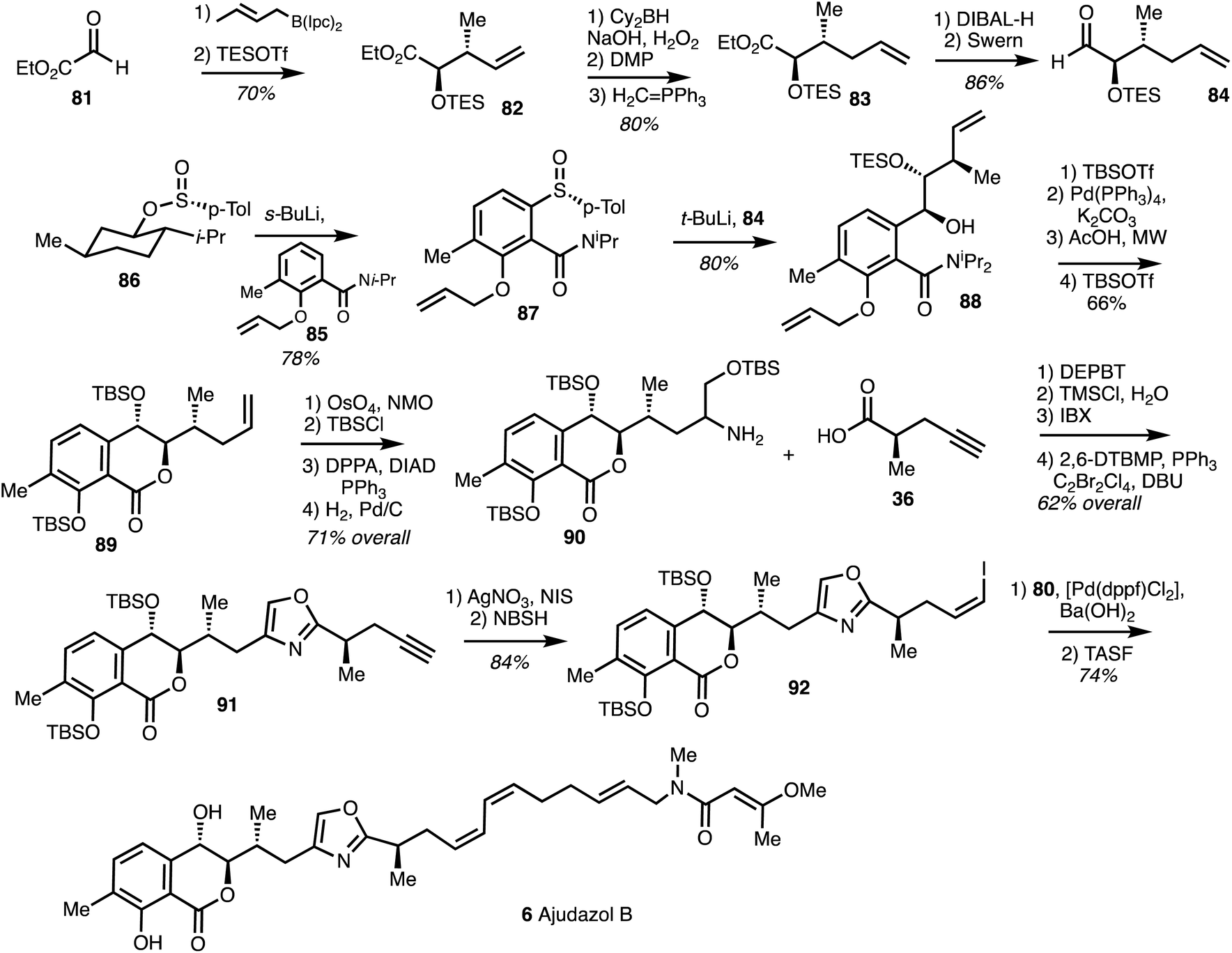 | ||
| Scheme 12 Total synthesis of ajudazol B (6) (Menche).25 | ||
Marquez and co-workers41 reported the synthesis of (−)-8-epi-ajudazol B (110) following their previously reported synthesis of isochromanones using isobenzofurans. The crucial components of this synthetic strategy include regioselective alkylation and oxidative rearrangement of an isobenzofuran derivative to form the isochromanone (Scheme 13). Iodide 93 was converted into terminal alkene 94 which was later subjected to dihydroxylation and mono protection to furnish alcohol 95 which was further elaborated into amine 96. Coupling 96 with acid 97 and deprotection gave amide 98. Oxidation and cyclodehydration followed by deprotection and oxidation gave aldehyde 99.
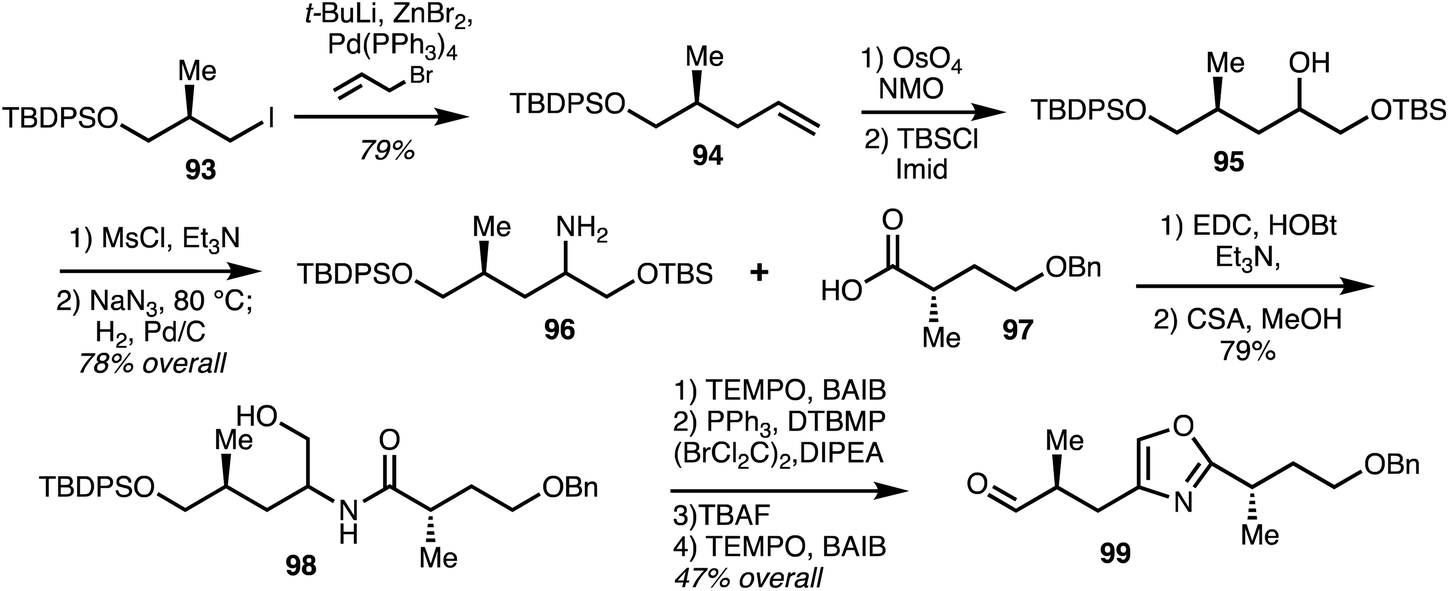 | ||
| Scheme 13 Synthesis of oxazole fragment of (−)-8-epi-ajudazol B (Marquez).41 | ||
Lithiation of 100 gave anion 101 and addition to aldehyde 102 gave alcohol 103. Oxidation and rearrangement then gave lactol 104 and further oxidation and reduction gave isochromanones 105 and 106 (Scheme 14).
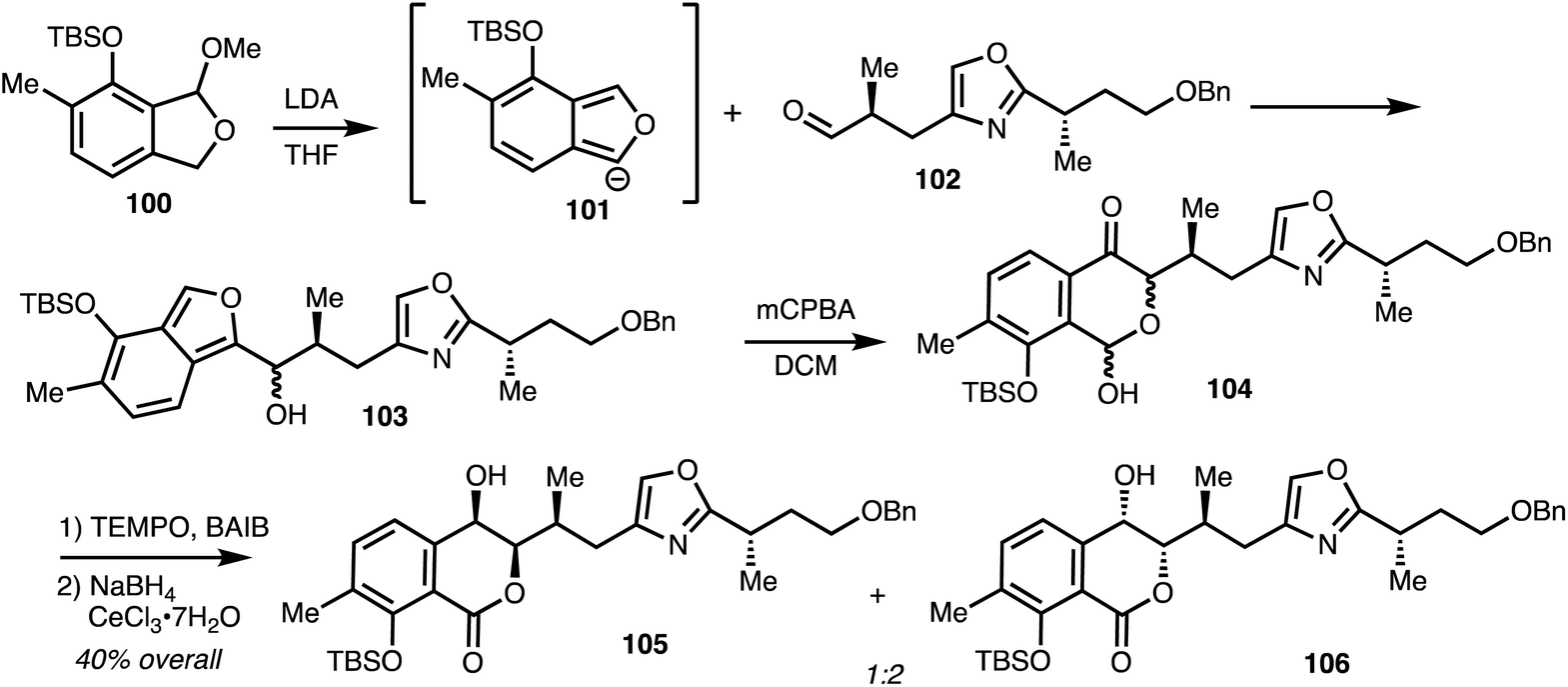 | ||
| Scheme 14 Synthesis of isochromanone fragment of (−)-8-epi-ajudazol B 110 (Marquez).41 | ||
The final stages mirrored those reported by Rizzacasa and began with conversion of 106 into aldehyde 107 followed by Bestman–Ohira reaction to give alkyne 109. Sonogashira coupling of alkyne 109 with the known iodide 4135 and partial hydrogenation of the resultant enyne yielded (−)-8-epi-ajudazol B (110) and the over reduced analogue 111 in good yield. Several analogues of ajudazol B (6) were produced and it was found that the isochromanone core plays a pivotal role in antifungal activity41 (Scheme 15).
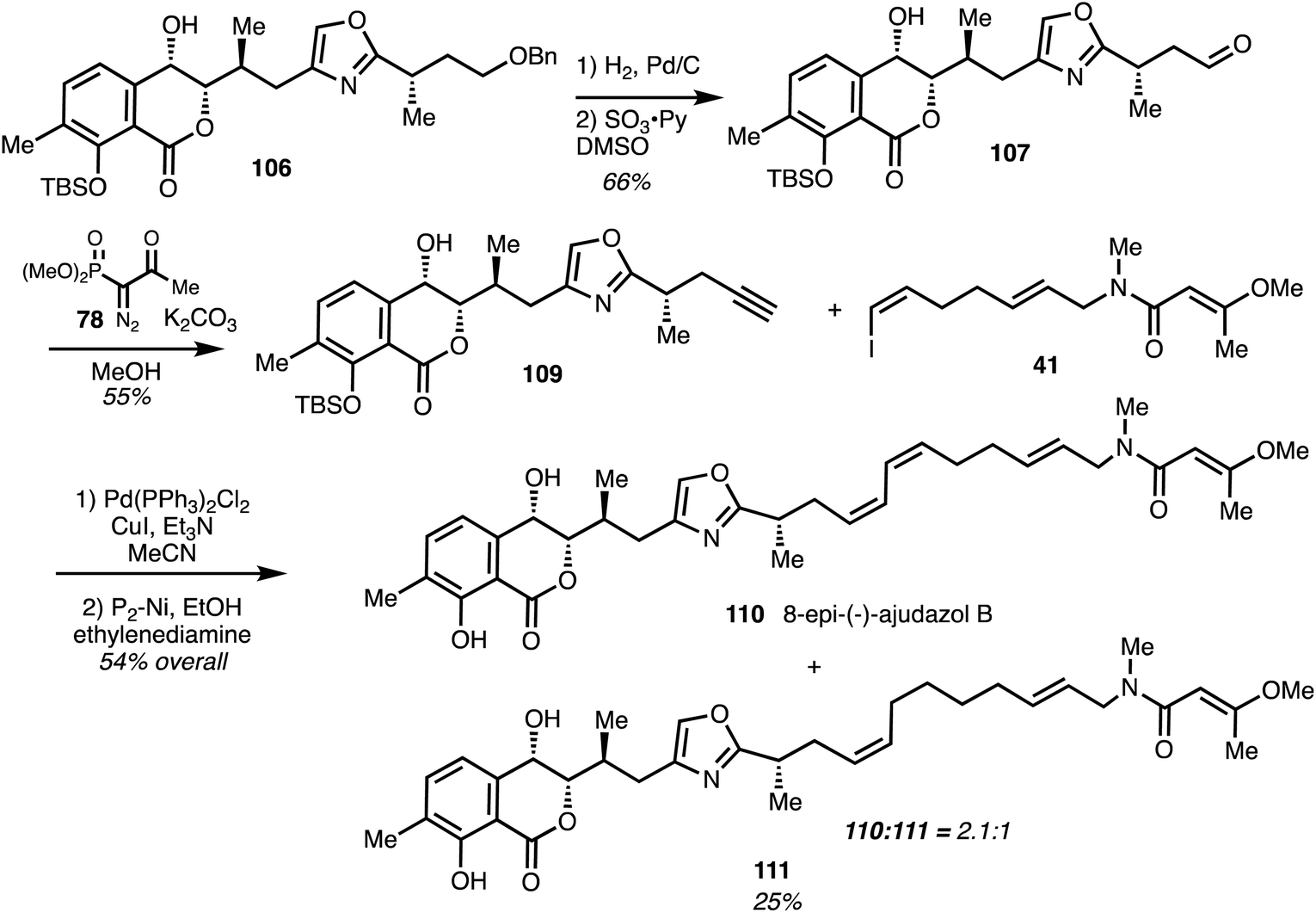 | ||
| Scheme 15 Synthesis of (−)-8-epiajudazol B 110 (Marquez).41 | ||
In 2020, Menche and co-workers42 reported the total synthesis of ajudazol A (4) in 17 steps. The route included a halogen dance sequence along with sp2–sp2 and sp2–sp3 Negishi cross coupling reactions. Lithiation of oxazole 112 followed by transmetalisation resulted in organozinc species 113 and the following Negishi cross coupling with the vinyliodide 114 gave oxazole 115 (Scheme 16). C5 iodination and a halogen dance reaction involving 4-iodooxazole 116, with the oxazole 117 as a catalyst to mediate lithium–iodide exchange,43 yielded iodide 118.
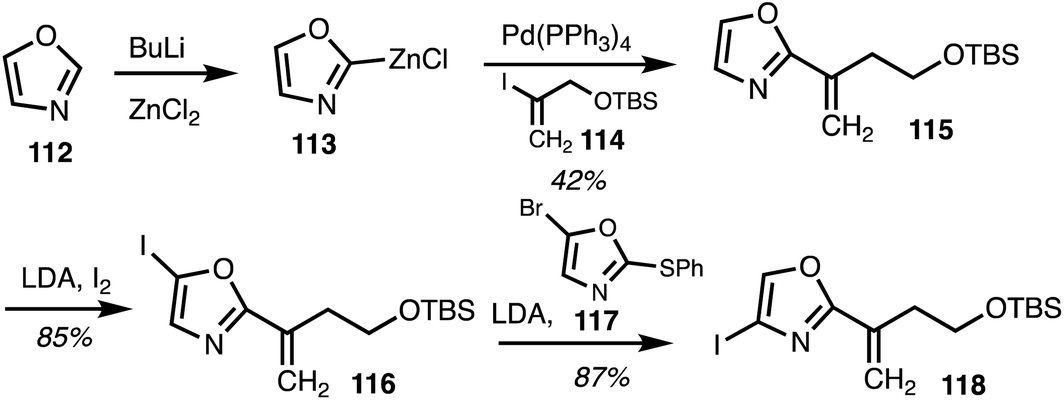 | ||
| Scheme 16 Selective oxazole functionalisation (Menche).42 | ||
The synthesis of the western fragment 122 began with TBS protection of isochromanone 119, followed by oxidative alkene cleavage, NaBH4 reduction to afford 120 which underwent Appel reaction and the Negishi cross coupling with 118 to yielded 121 in 66% yield.44 Compound 121 was then converted into vinyl iodide 122 which was then linked to the eastern fragment 80 by Suzuki coupling. Deprotection with HF·pyridine then furnished ajudazol A (4) (Scheme 17).
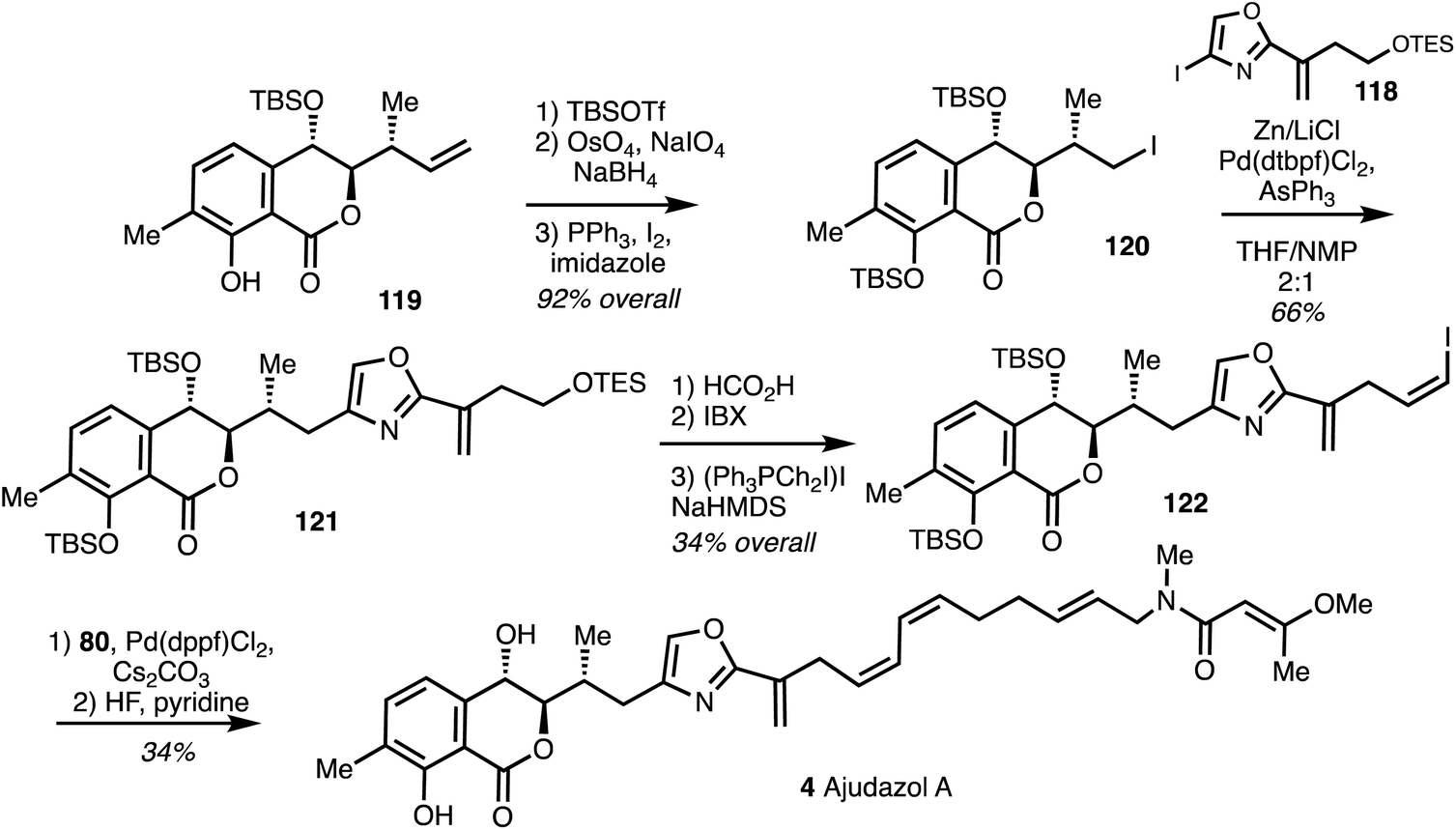 | ||
| Scheme 17 Total synthesis of ajudazol A (4) (Menche).42 | ||
Total synthesis of noricumazole A (11)
The first and only total synthesis of noricumzaole A (11) was reported by Müller.23 The synthesis of the oxazole fragment began with a Masamune anti aldol reaction between the enolate derived from 123 and propanal to give adduct 124 which underwent DIBAL reduction to furnish aldehyde 125. Subsequent asymmetric Nagao acetate aldol condensation of the derived aldehyde 125 employing the chiral auxiliary 126 afforded the adduct 127 that was further processed to give the acid 128 in 99% yield. Peptide coupling with amine 129 gave amide 130 and cyclodehydration/aromatisation then formed the oxazole 131. Reduction gave the aldehyde 132 and vinyl iodide formation gave the iodide 133 over 3 steps (Scheme 18).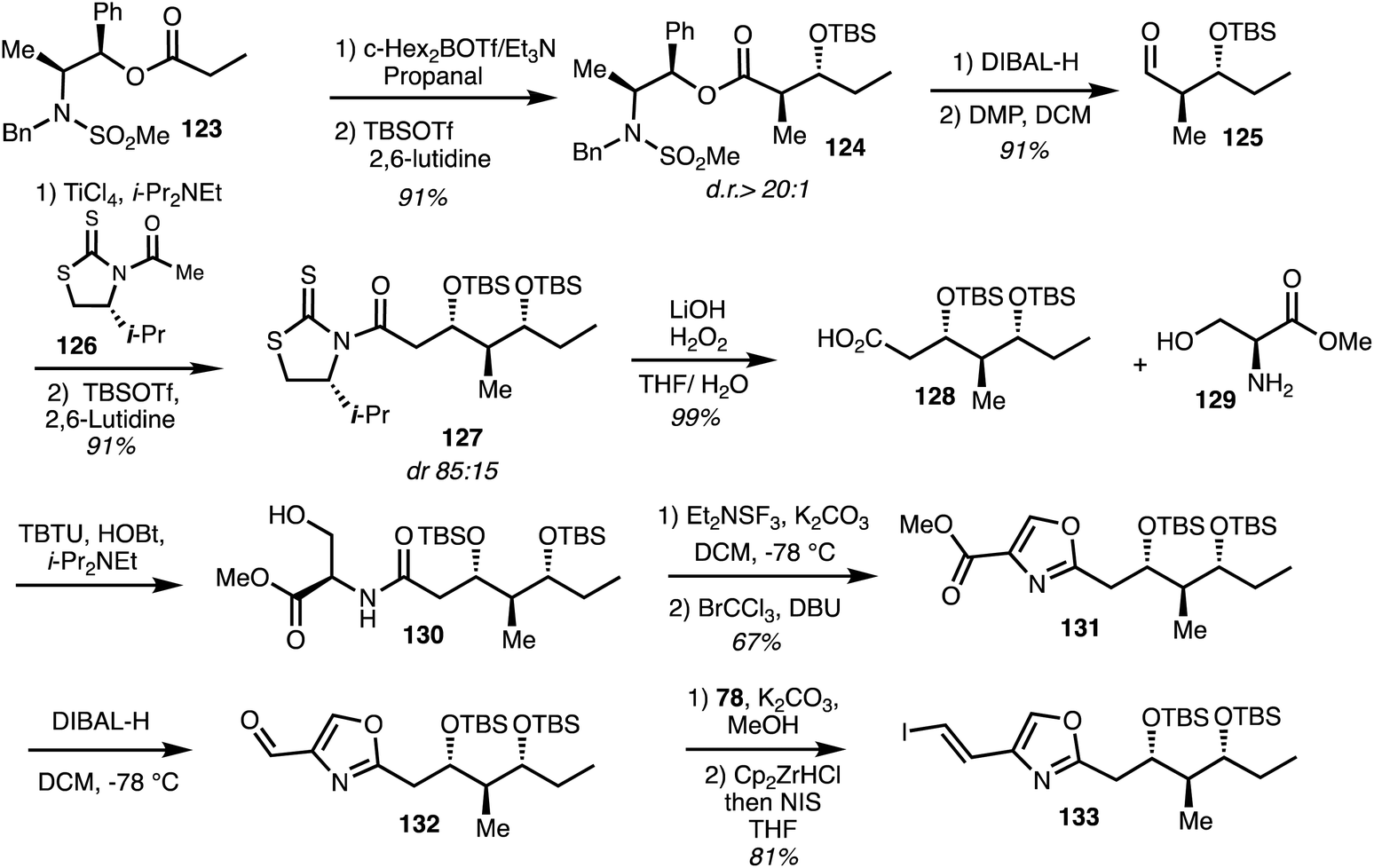 | ||
| Scheme 18 Total synthesis of eastern fragment of noricumazole A (11) (Müller).23 | ||
Iron mediated cross-coupling of the Grignard reagent derived from the bromide 134 with (S)-iodide 135 gave 136 and lithiation followed by addition to tert-butyl isocyanate gave amide 137 (Scheme 19).
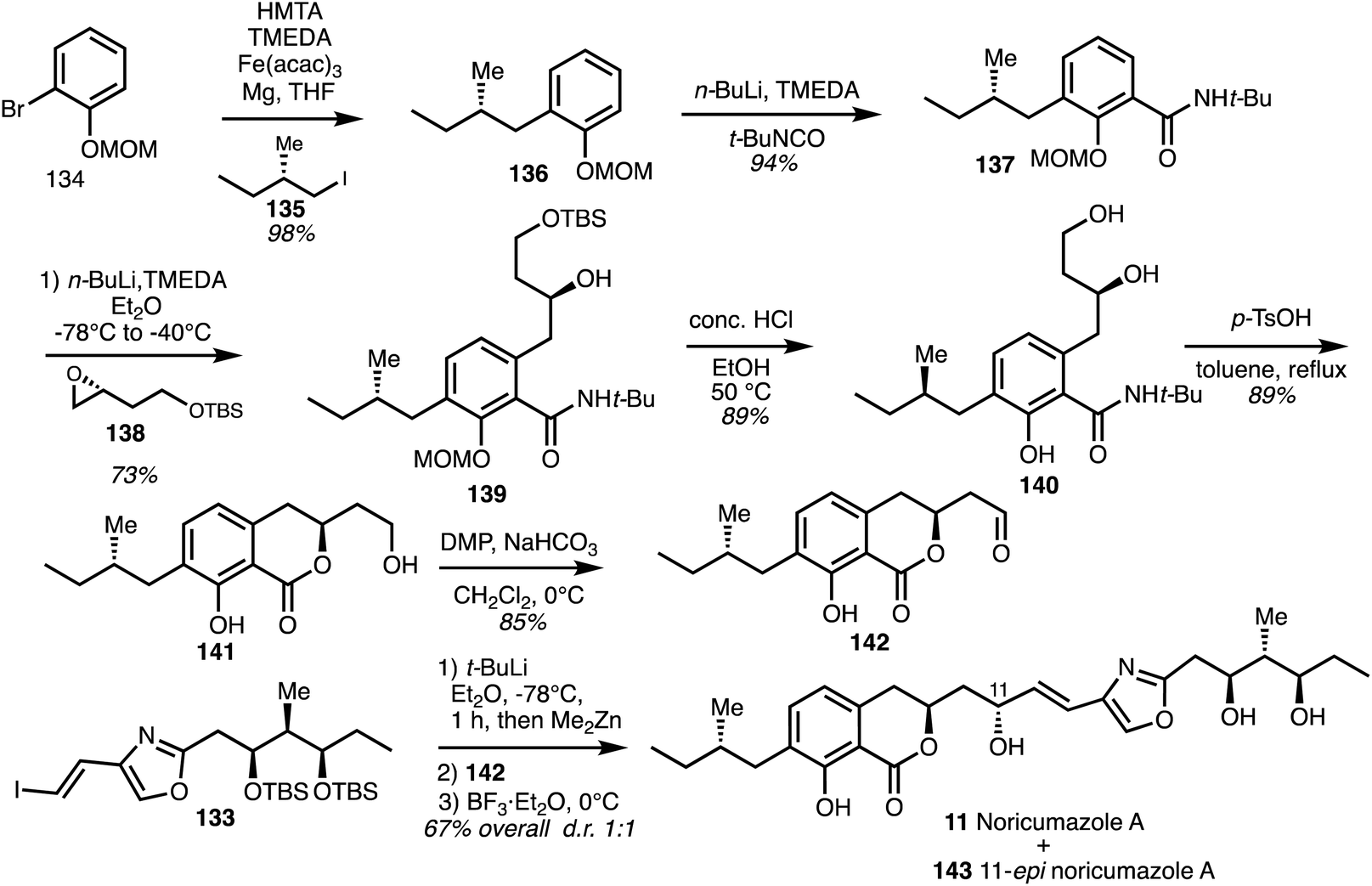 | ||
| Scheme 19 Total synthesis of noricumazole A (11) (Müller).23 | ||
ortho-Lithiation and addition to the epoxide 138 gave the intermediate 139 which was deprotected to yield alcohol 140 and cyclisation under acidic conditions provided the isochromanone 141. Oxidation gave aldehyde 142 and addition of the zincate derived from vinyl iodide 142 gave the alcohols in a 1:1 ratio of diaseteroisomers. Lewis's acid mediated cleavage of the TBS ethers then afforded noricumazole A (11) and the C11 epimer 143.
Total synthesis of noricumazole B (12)
The reported total synthesis of noricumazole B (12)45 is based on the noricumazole A (11) synthesis and is depicted in Scheme 20. Compound 144 was taken through 5 steps to give acid 145 which was converted into vinyl iodide 146. Vinyl zinc addition to aldehyde 142 afforded alcohol 147 and the C11-epimer 148 in 1:1 diastereomeric ratio. The undesired isomer 148 could be converted into the correct alcohol 147 by Mitsunobu inversion and hydrolysis. Phenol acetylation and TBS group removal to provide acetate 149. A modified Koenigs–Knorr reaction employing commercially available bromo arabinose 150 under Helferich-type conditions installed the furanoside and subsequent deprotection gave noricumazole B (12) in an overall yield of 4% over 19 steps.
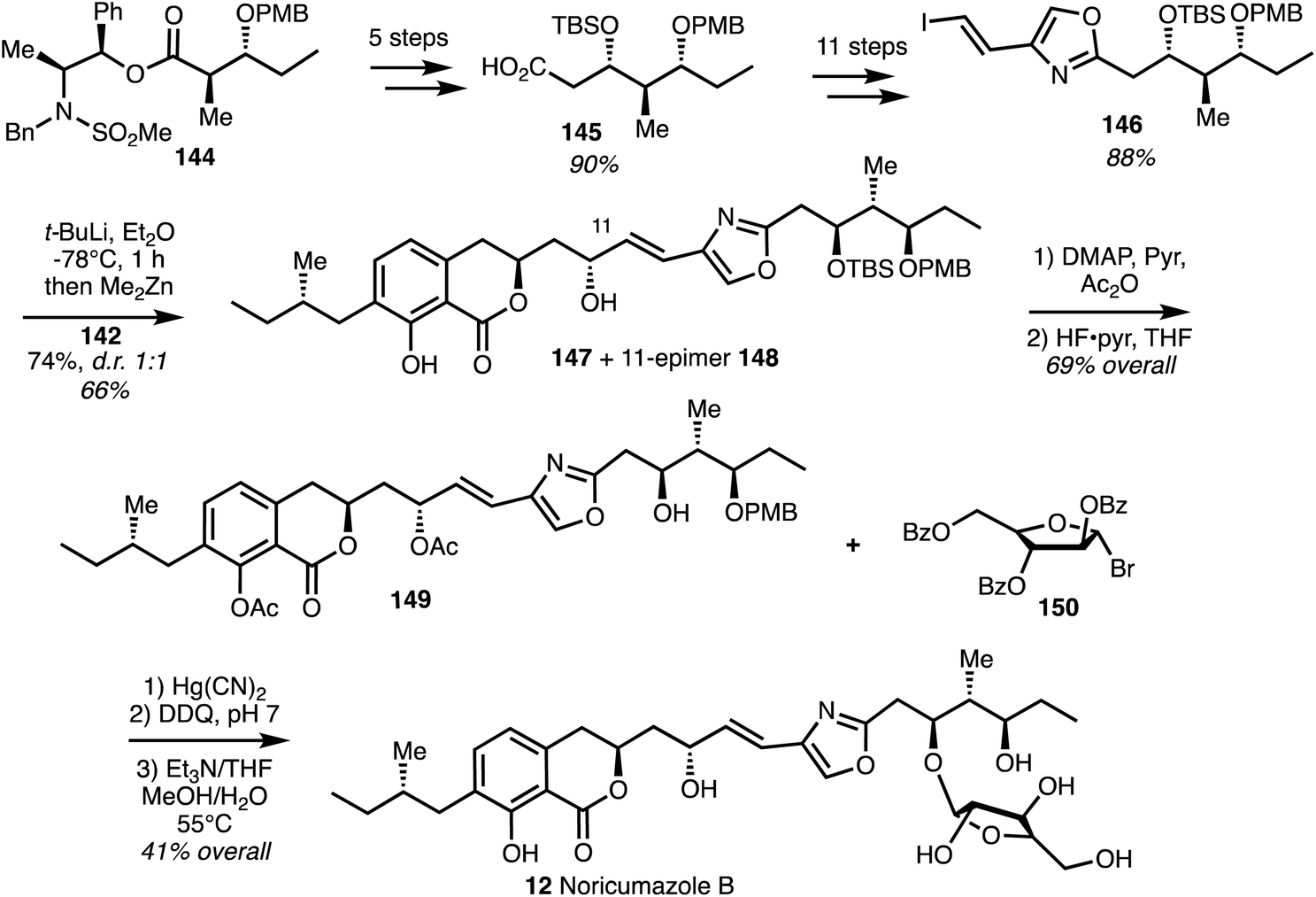 | ||
| Scheme 20 Total synthesis of noricumazole B (12) (Jansen and Kirschning).45 | ||
Conclusion
Myxobacteria have emerged as prolific source of diverse metabolites with a range of biologicalactivities. The isochromanone family of metabolites in particular presents a significant synthetic challenge and this has resulted in a number of efficient routes to this moiety as well as the oxazole fragments. The total synthesis of a number of these natural products has paved the way for further biological studies and analogue synthesis.Conflicts of interest
There are no conflicts to declare.References
- M. S. Butler, Nat. Prod. Rep., 2005, 22, 162–195 RSC.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2007, 70, 461–477 CrossRef CAS PubMed.
- M.-Q. Zhang and B. Wilkinson, Curr. Opin. Biotechnol., 2007, 18, 478–488 CrossRef CAS PubMed.
- H. Reichenbach, Environ. Microbiol., 1999, 1, 15–21 CrossRef CAS PubMed.
- H. B. Bode and R. Müller, J. Ind. Microbiol. Biotechnol., 2006, 33, 577–588 CrossRef CAS PubMed.
- M. Dworkin, S. Falkow, E. Rosenberg, K.-H. Schleifer and E. Stackebrandt, The Prokaryotes, Vol. 7:Proteobacteria: Delta and Epsilon Subclasses. Deeply Rooting Bacteria, 2006, pp. 31–35 Search PubMed.
- H. Reichenbach, J. Ind. Microbiol. Biotechnol., 2001, 27, 149–156 CrossRef CAS PubMed.
- R. O. Garcia, D. Krug and R. Müller, in Methods Enzymol., Academic Press, 2009, vol. 458, pp. 59–91 Search PubMed.
- J. Herrmann, A. A. Fayad and R. Müller, Nat. Prod. Rep., 2017, 34, 135–160 RSC.
- L. Gross, PLoS Biol., 2005, 3, e398 CrossRef.
- D. Claessen, D. E. Rozen, O. P. Kuipers, L. Søgaard-Andersen and G. P. van Wezel, Nat. Rev. Microbiol., 2014, 12, 115–124 CrossRef CAS PubMed.
- W. Dawid, FEMS Microbiol. Rev., 2000, 24, 403–427 CrossRef CAS PubMed.
- K. J. Weissman and R. Müller, Nat. Prod. Rep., 2010, 27, 1276–1295 RSC.
- M. Nett and G. M. König, Nat. Prod. Rep., 2007, 24, 1245–1261 RSC.
- F. Sasse, H. Steinmetz, J. Heil, G. Höfle and H. Reichenbach, J. Antibiot., 2000, 53, 879–885 CrossRef CAS PubMed.
- M. W. Khalil, F. Sasse, H. Lünsdorf, Y. A. Elnakady and H. Reichenbach, ChemBioChem, 2006, 7, 678–683 CrossRef CAS PubMed.
- H. Steinmetz, N. Glaser, E. Herdtweck, F. Sasse, H. Reichenbach and G. Höfle, Angew. Chem., Int. Ed. Engl., 2004, 43, 4888–4892 CrossRef CAS PubMed.
- H. Reichenbach and G. Höfle, Drugs R&D, 2008, 9, 1–10 CrossRef CAS PubMed.
- K.-H. Altmann, in The Epothilones: An Outstanding Family of Anti-Tumor Agents: From Soil to the Clinic, ed. K. H. Altmann, G. Höfle, R. Müller, J. Mulzer and K. Prantz, Springer Vienna, Vienna, 2009, pp. 157–220 Search PubMed.
- M. V. Cobham and D. Donovan, Cancer Manage. Res., 2009, 1, 69–77 CrossRef CAS.
- F. Y. F. Lee, R. Borzilleri, C. R. Fairchild, A. Kamath, R. Smykla, R. Kramer and G. Vite, Cancer Chemother. Pharmacol., 2008, 63, 157–166 CrossRef CAS PubMed.
- B. Kunze, R. Jansen, G. Höfle and H. Reichenbach, J. Antibiot., 2004, 57, 151–155 CrossRef CAS PubMed.
- J. Barbier, R. Jansen, H. Irschik, S. Benson, K. Gerth, B. Bohlendorf, G. Hofle, H. Reichenbach, J. Wegner, C. Zeilinger, A. Kirschning and R. Müller, Angew. Chem., Int. Ed., 2012, 51, 1256–1260 CrossRef CAS PubMed.
- J. Barbier, J. Wegner, S. Benson, J. Gentzsch, T. Pietschmann and A. Kirschning, Chem. – Eur. J., 2012, 18, 9083–9090 CrossRef CAS PubMed.
- S. Essig, S. Bretzke, R. Müller and D. Menche, J. Am. Chem. Soc., 2012, 134, 19362–19365 CrossRef CAS PubMed.
- S. D. Rychnovsky, B. N. Rogers and T. I. Richardson, Acc. Chem. Res., 1998, 31, 9–17 CrossRef CAS.
- R. Jansen, B. Kunze, H. Reichenbach and G. Höfle, Eur. J. Org. Chem., 2002, 917–921 CrossRef CAS.
- B. Kunze, R. Jansen, G. Höfle and H. Reichenbach, J. Antibiot., 2004, 57, 151 CrossRef CAS PubMed.
- K. Buntin, S. Rachid, M. Scharfe, H. Blocker, K. J. Weissman and R. Muller, Angew. Chem., Int. Ed., 2008, 47, 4595–4599 CrossRef CAS PubMed.
- K. Buntin, K. J. Weissman and R. Muller, ChemBioChem, 2010, 11, 1137–1146 CrossRef CAS PubMed.
- O. Krebs and R. J. K. Taylor, Org. Lett., 2005, 7, 1063–1066 CrossRef CAS PubMed.
- D. Ganame, T. Quach, C. Poole and M. A. Rizzacasa, Tetrahedron Lett., 2007, 48, 5841–5843 CrossRef CAS.
- S. J. Hobson, A. Parkin and R. Marquez, Org. Lett., 2008, 10, 2813–2816 CrossRef CAS PubMed.
- S. Birkett, D. Ganame, B. C. Hawkins, S. Meiries, T. Quach and M. A. Rizzacasa, Org. Lett., 2011, 13, 1964–1967 CrossRef CAS PubMed.
- S. Birkett, D. Ganame, B. C. Hawkins, S. Meiries, T. Quach and M. A. Rizzacasa, J. Org. Chem., 2013, 78, 116–123 CrossRef CAS PubMed.
- B. R. Graetz and S. D. Rychnovsky, Org. Lett., 2003, 5, 3357–3360 CrossRef CAS PubMed.
- P. Wipf and S. Lim, J. Am. Chem. Soc., 1995, 117, 558–559 CrossRef CAS.
- I. Saito, M. Takayama and S. Kawanishi, J. Am. Chem. Soc., 1995, 117, 559 Search PubMed.
- T. Ohmura, Y. Yamamoto and N. Miyaura, J. Am. Chem. Soc., 2000, 122, 4990–4991 CrossRef CAS.
- A. G. Myers, B. Zheng and M. Movassaghi, J. Org. Chem., 1997, 62, 7507 CrossRef CAS PubMed.
- L. Adair, B. A. Egan, C. M. Pearson, R. Lopez-Gonzalez, M. Kuchar, A. Mendoza-Mendoza, J. Prunet and R. Marquez, Eur. J. Org. Chem., 2020, 6661–6672 CrossRef CAS.
- P. Wollnitzke, S. Essig, J. P. Gölz, K. von Schwarzenberg and D. Menche, Org. Lett., 2020, 22, 6344–6348 CrossRef CAS PubMed.
- N. Proust, M. F. Chellat and J. P. Stambuli, Synthesis, 2011, 3083 CAS.
- S. Essig, B. Schmalzbauer, S. Bretzke, O. Scherer, A. Koeberle, O. Werz, R. Müller and D. Menche, J. Org. Chem., 2016, 81, 1333–1357 CrossRef CAS PubMed.
- J. Barbier, K. Gerth, R. Jansen and A. Kirschning, Org. Biomol. Chem., 2012, 10, 8298–8307 RSC.
| This journal is © The Royal Society of Chemistry 2023 |