
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thiourea-catalysed conjugate additions of amines to vinyl phosphonates and phosphinates†
Peter E.
McDermott
 ab,
Martin P. Ó.
Fearraigh
b,
Alexandra M.
Horan
bc and
Eoghan M.
McGarrigle
*abc
ab,
Martin P. Ó.
Fearraigh
b,
Alexandra M.
Horan
bc and
Eoghan M.
McGarrigle
*abc
aA2P CDT in sustainable chemistry and BiOrbic Bioeconomy SFI Research Centre, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: eoghan.mcgarrigle@ucd.ie
bCentre for Synthesis & Chemical Biology, UCD School of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland
cSSPC, the SFI Research Centre for Pharmaceuticals, Centre for Synthesis & Chemical Biology, UCD School of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland
First published on 6th January 2023
Abstract
Thiourea catalysts activated α,β-unsaturated phosphonates and phosphinates toward conjugate addition by amines to give β-aminophosphonates and β-aminophosphinates. The organocatalytic methodology was used to synthesise 15 β-aminophosphonates and -phosphinates in yields up to 99%. A gram-scale example furnished the corresponding β-aminophosphonate in an isolated yield of 99% with 97% catalyst recovery. Based on mechanistic experiments, hydrogen bonding between the phosphoryl oxygen and thiourea are proposed to play a crucial role in substrate activation.
Introduction
Due to their high atom economy and versatility, conjugate additions have long been recognised as efficient carbon–carbon and carbon–heteroatom bond forming reactions.1 Thiourea catalysts have been used to catalyse a wide range of conjugate addition reactions via hydrogen bonding catalysis.2–7 Although vinylphosphoryl compounds have been used as substrates in other organocatalysed reactions,8–16 to the best of our knowledge, there are no examples of thioureas being used to catalyse conjugate additions to unactivated vinylphosphoryl compounds. There is evidence that phosphoryl compounds are significantly stronger H-bond acceptors than similar carbonyl compounds (Fig. 1A).17 We hypothesised that this H-bond acceptor ability could make α,β-unsaturated phosph(in/on)ates suitable substrates for H-bonding catalysis. However, α,β-unsaturated phosphoryl compounds show lower reactivity compared to other more commonly used conjugated electrophiles. Their reduced ability to stabilise α-anionic charges can be inferred from the relatively high pKa of the α-protons in phosphoryl compounds compared to similar nitro and carbonyl compounds (including esters) (Fig. 1B). Indeed, Alexakis and co-workers have noted that diethyl vinylphosphonate was an unsuitable substrate in enamine-catalysed conjugate additions.18 We hypothesised that the strong H-bonding interaction between phosphoryl compounds and thioureas could help to stabilise negative charge build up on the α-carbon during C–Nu bond formation and in the intermediate species formed (Fig. 1C). This would increase reactivity toward conjugate addition and enable the synthesis of valuable compounds in an efficient manner.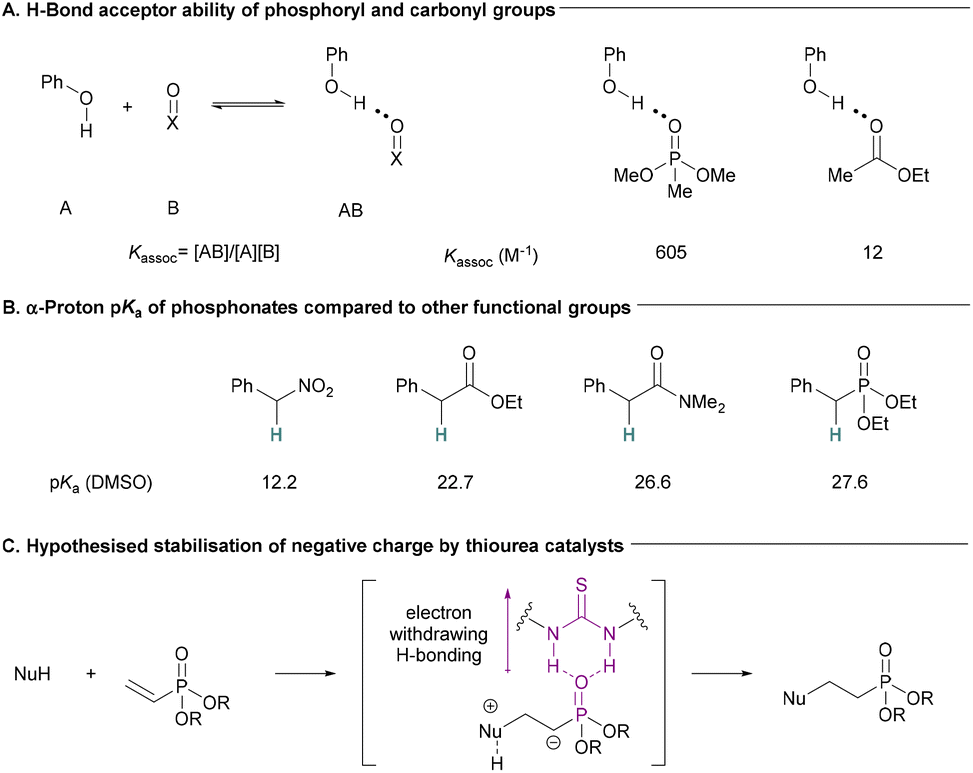 | ||
| Fig. 1 (A) Phosphonates make stronger H-bonds with phenol compared to carboxylic esters.17 (B) pKa of protons α to functional groups found in conjugate addition electrophiles.19–21 (C) Hypothesis for the activation of α,β-unsaturated phosphonates toward conjugate additions. | ||
β-Aminophosphonic acids and their esters are medicinally relevant molecules. They are bioisoteres of β-amino acids and can act as mimics of the tetrahedral intermediate formed during enzyme-catalysed hydrolysis of esters.22 These properties lead to a wide variety of biological activities.23 They have found use as drugs, such as receptor antagonist Perzinfotel which has been investigated as an anti-stroke agent,24 antibacterial agents,25 and as enzyme inhibitors26 (Fig. 2). Existing syntheses of these compounds include: Arbuzov reactions between phosphites and β-aminoalkyl halides27,28 and nucleophilic substitution between β-halophosphonates and amines.29,30 Both of these routes suffer from poor yields, a requirement for high temperatures and generation of stoichiometric waste products. Conjugate addition of H-phosphonates to α,β-unsaturated nitro compounds,31 followed by reduction could also be used. However, this route requires multiple steps and only gives primary amines. Routes involving nucleophilic addition to N-heterocycles have also been reported.32,33 However, these methods require the synthesis of complex starting materials.
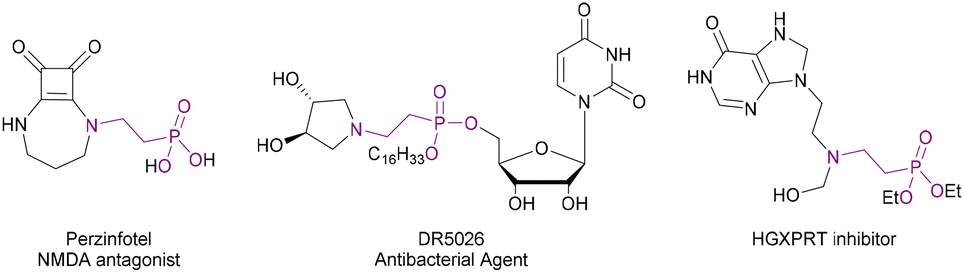 | ||
| Fig. 2 Examples of biologically active β-aminophosph(in/on)ates.24–26 | ||
In principle, conjugate additions of amines to α,β-unsaturated phosph(in/on)ates could provide efficient access to β-aminophosph(in/on)ates. However the efficiency of this approach is often reduced due to the poor electrophilicity of α,β-unsaturated phosph(in/on)ates. Excess of one reaction partner, stoichiometric additives, high temperatures or a combination of these drawbacks are usually required to compensate for the low electrophilicity.34–39 Use of water as solvent has been reported to give shorter reaction times in the conjugate addition of amines to α,β-unsaturated phosphonates.40–42 However sometimes higher temperatures (up to 100 °C) and/or extended reaction times are still required, and other literature reports using water as solvent sometimes require very long times and/or significant heating.43–45 This renders sensitive substrates incompatible with the methodology.
Reports of amines being used as nucleophiles in thiourea-catalysed conjugate additions are mostly limited to intramolecular versions or sp2-hybridised N-nucleophiles.46–50 Thiourea-catalysed intermolecular conjugate additions using other amines as nucleophiles are rare in the literature.51,52 Seidel and co-workers recently reported thiourea-catalysed conjugate additions of amines to α,β-unsaturated esters.53 They identified both reaction partners as challenging substrates and found that simple, more established thiourea catalysts were not suitable for this transformation. To achieve high yield and enantioselectivity, a complex bifunctional selenourea-thiourea catalyst was required. They attributed the need for a more complex catalyst to the low reactivity of the substrates.
To-date an efficient conjugate addition of amines to α,β-unsaturated phosphonates compatible with a variety of organic solvents has not been reported. Based on the strong hydrogen bonding acceptor ability of phosphoryl bonds and the hypothesis that water catalyses this reaction via H-bonding,40,54 we hypothesised that thiourea catalysts could be used to efficiently synthesise β-aminophosphonates via conjugate additions. To maximise the efficiency of the process, we aimed to avoid the use of high temperatures, and to use an equimolar amount of each staring material (Fig. 3).
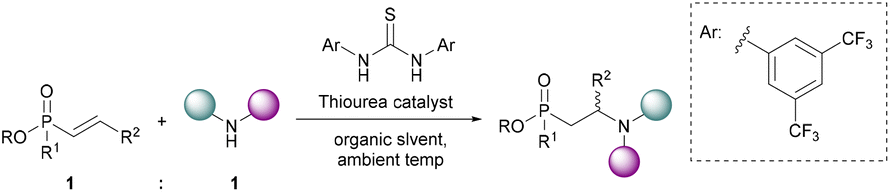 | ||
| Fig. 3 Target: an equimolar thiourea-catalysed conjugate addition of amines to α,β-unsaturated phosph(in/on)ates at ambient temperatures in organic solvents. | ||
Results and discussion
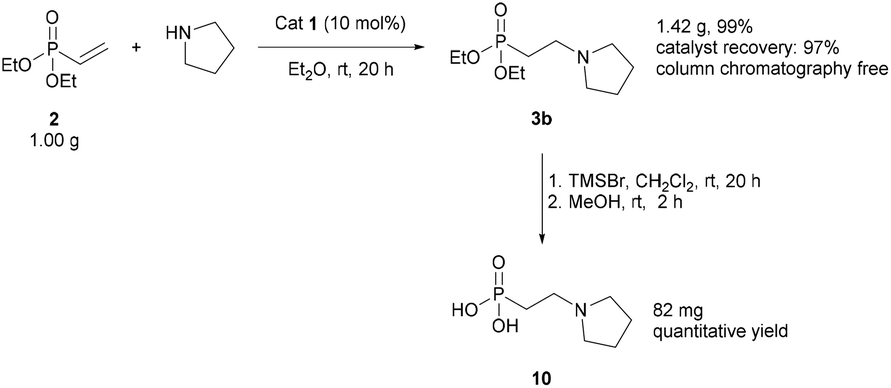
We began by establishing that Schreiner's catalyst 1 could accelerate the reaction of benzylamine with diethyl vinylphosphonate 2. In the presence of catalyst 1, 2 was converted to the corresponding β-aminophosphonate 3a in 73% yield, compared to 43% in the absence of catalyst (Scheme 1).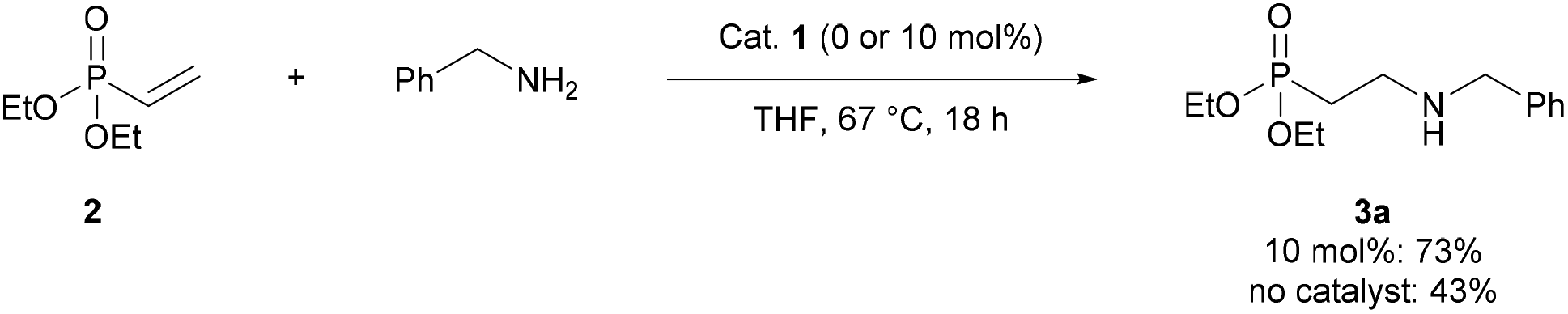 | ||
| Scheme 1 Proof-of-concept for thiourea-catalysed conjugate addition of amines to α,β-unsaturated phosphonates. Reaction conditions: diethyl vinylphosphonate (0.2 mmol), catalyst (0 or 0.02 mmol), benzylamine (0.2 mmol) and THF (0.25 mL). Yields measured by 31P NMR analysis based on conversion of starting material. | ||
Following on from this initial hit, optimisation of the reaction conditions was carried out. In the absence of catalyst, at room temperature in toluene, a yield of less than 1% of 3a was observed after 20 h (Table 1, entry 1). With 10 mol% of catalyst 1, the yield improved to 33% (Table 1, entry 2). Next, a range of different thiourea catalysts were tested in the reaction. Catalyst 1 proved to be the most efficient in a screen (Table 1, entries 2–6). Increasing catalyst loading to 20 mol% brought the yield to 46% (Table 1, entries 7 and 8). A solvent screen showed Et2O to be the best solvent tested (Table 1, entry 11). Slightly increasing the temperature to 30 °C gave 3a in 78% yield after 20 h (Table 1, entry 14). Crucially these reactions were all carried out using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of amine:phosphonate and did not require high temperatures.
:1 molar ratio of amine:phosphonate and did not require high temperatures.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Catalyst loading (mol%) | Temp. (°C) | Solvent | Yieldb (%) |
| a Reaction conditions: diethyl vinylphosphonate (0.1 mmol), amine (0.1 mmol) and solvent (0.2 mL). b Measured by 31P NMR analysis based on conversion of starting material. | |||||
| 1 | — | — | rt | Toluene | <1 |
| 2 | 1 | 10 | rt | Toluene | 33 |
| 3 | 4 | 10 | rt | Toluene | 20 |
| 4 | 5 | 10 | rt | Toluene | 10 |
| 5 | 6 | 10 | rt | Toluene | 12 |
| 6 | 7 | 10 | rt | Toluene | 24 |
| 7 | 1 | 5 | rt | Toluene | 29 |
| 8 | 1 | 20 | rt | Toluene | 46 |
| 9 | 1 | 10 | rt | THF | 45 |
| 10 | 1 | 10 | rt | CH2Cl2 | 30 |
| 11 | 1 | 10 | rt | Et2O | 48 |
| 12 | 1 | 10 | rt | MeCN | 2 |
| 13 | 1 | 20 | rt | Et2O | 73 |
| 14 | 1 | 20 | 30 | Et2O | 78 |
| 15 | — | 20 | 30 | Et2O | 4 |
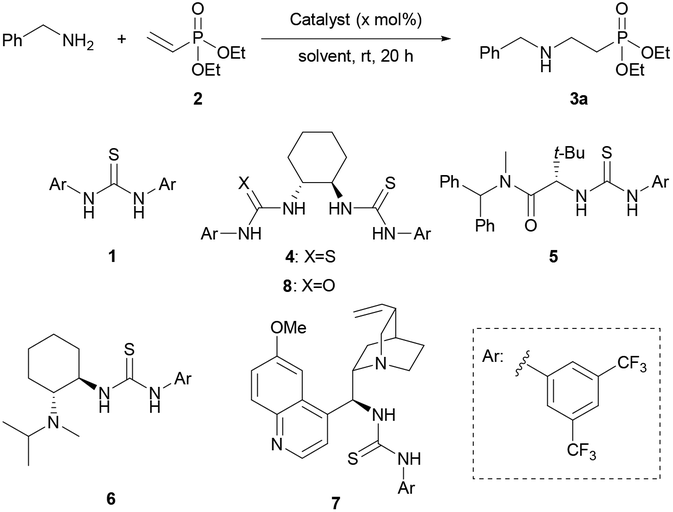
With suitable conditions in hand, a range of different amines were tested as nucleophiles (Table 2). Cyclic and acyclic secondary amines were well tolerated with pyrrolidine, piperidine, morpholine and diethylamine all giving excellent yields of the corresponding β-aminophosphonates (3b–e). Primary amines other than benzylamine were also well tolerated; 2-aminoethanol underwent conjugate addition with good yield and excellent site-selectivity for N-alkylation (based on 1H NMR spectroscopic analysis of the crude material). The more sterically encumbered cyclohexylamine, α-methylbenzylamine and tert-butylamine gave moderate yields of 3g–3i, respectively. We noted that when primary amines were used, double addition products were not observed by 31P NMR analysis. Even when excess equivalents of 2 were used dialkylation was not observed although yields of the monoalkylated product were increased. To investigate this observation, isolated 3a was resubjected to standard reaction conditions, again no dialkylation was observed. Addition of diethylamine to 2 was also attempted in the presence of 1 equiv. of 3a. A reduced yield of 62% of 3e was obtained. This result is consistent with product inhibition of catalysis. The presence of a H-bond accepting phosphoryl group in 3a could lead to non-constructive off-cycle catalyst-substrate interactions which reduce the rate of conjugate addition. Next the use of α,β-unsaturated phosphinates was investigated. Ethyl phenylvinylphosphinate reacted with pyrrolidine and morpholine to give 9a and 9b, respectively, in excellent yields. n-Butyl phenylphosphinate was also well tolerated furnishing 9c in excellent yield. Anilines were not tolerated and no product was observed by 1H NMR analysis even when the reaction was carried out in refluxing toluene using aniline or p-methoxyaniline.
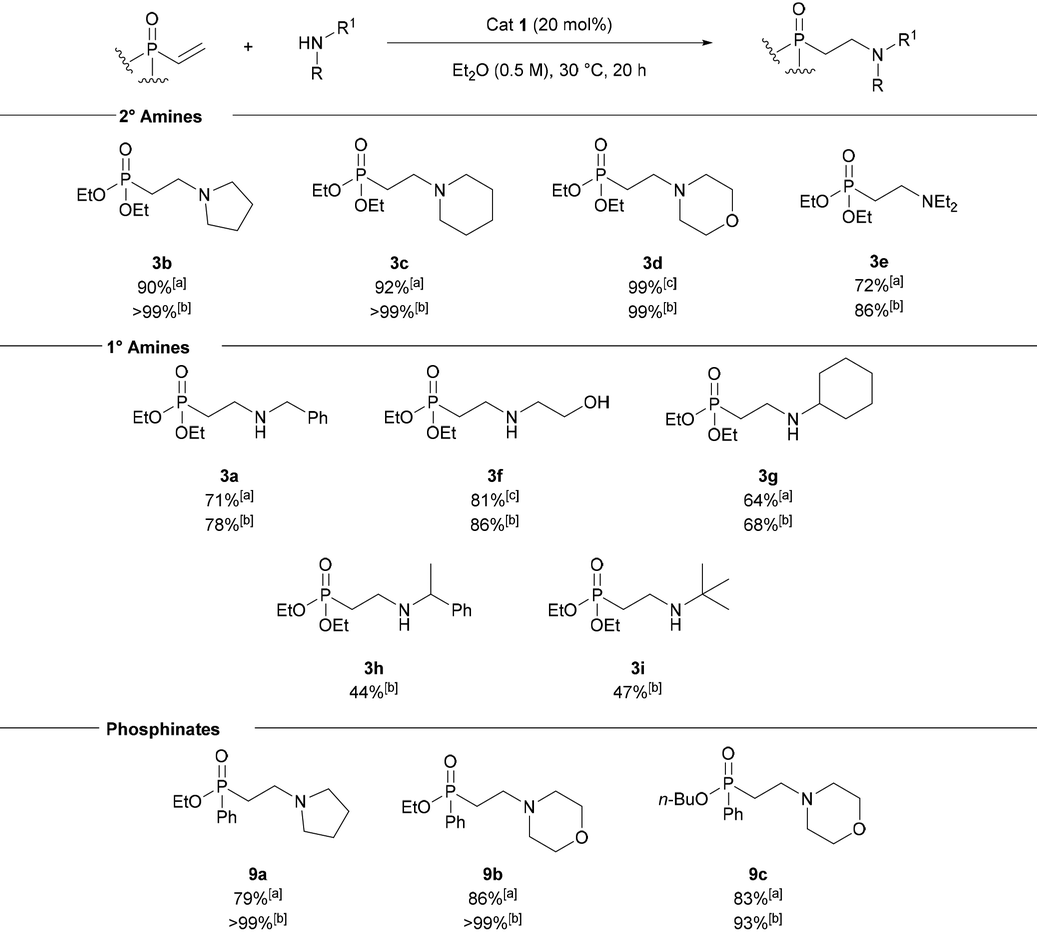
The robustness of the reaction was demonstrated on a 1 g scale (Scheme 2). The addition of pyrrolidine to diethyl vinylphosphonate 2 gave 3b in a 99% isolated yield using only liquid/liquid extractions. Avoiding column chromatography was particularly useful as the β-aminophosph(in/on)ate products were typically challenging substrates for column chromatography. The catalyst could also be recovered in 97% yield using only liquid/liquid extractions and was used in subsequent reactions without compromising the yield. A portion of 3b synthesised using this methodology was then converted to phosphonic acid 10 using TMSBr (Scheme 2).55 This route provides access to 10 in 99% yield over three steps, using commercially available starting materials and avoiding column chromatography.
 | ||
| Scheme 2 Gram-scale synthesis of 3b and synthesis of phosphonic acid 10. | ||
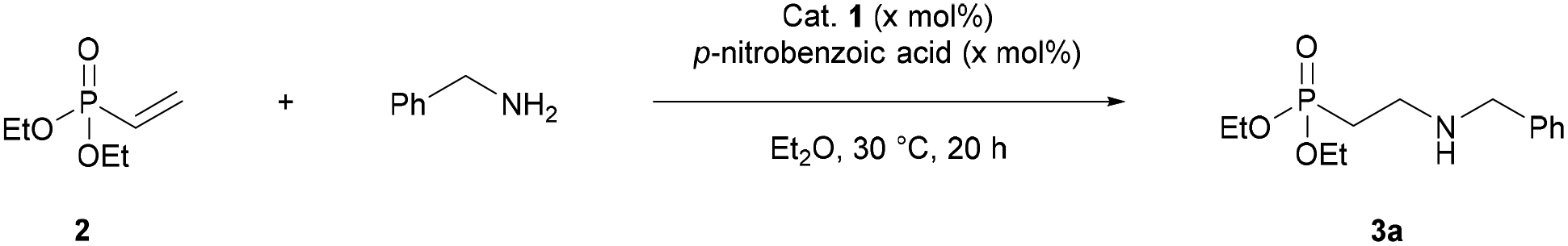
Thiourea catalysts are known to act as both hydrogen bonding and Brønsted acid catalysts.56–58 For example, in Seidel's selenourea–thiourea-catalysed conjugate addition of amines to α,β-unsaturated esters, the catalyst is proposed to act as a Brønsted acid, protonating the α-carbon after addition of the amine.53 A control experiment was designed to elucidate the mode of catalysis occurring under our reaction conditions (Table 3). p-Nitrobenzoic acid, which has a pKa similar to catalyst 1 (9.1 vs. 8.5 in DMSO),21,57 was tested as a catalyst in the addition of benzylamine to diethyl vinylphosphonate 2. Some catalysis was observed, however yields were significantly lower than those obtained using catalyst 1. When p-nitrobenzoic acid and catalyst 1 were used as co-catalysts, the yield was comparable to the yield obtained when just catalyst 1 was used (Table 3, entry 4). These results suggest that catalyst 1 has a mode of action beyond simply acting as a Brønsted acid catalyst. The dual H-bonding ability of catalyst 1 is likely the cause of the significantly increased yield compared to a similarly acidic single H-bond donor such as p-nitrobenzoic acid.
Next a series of NMR experiments were carried out to probe the nature of the catalyst-substrate interactions. NMR analysis has previously been used to provide evidence for H-bonding interactions between thiourea catalysts and substrates.59–61 We noted that the solubility of catalyst 1 in CDCl3 was increased dramatically in the presence of diethyl vinylphosphonate which is consistent with formation of an H-bonded complex between the two species. H-Bonding interactions between thioureas and phosphoryl compounds have been studied using 31P NMR and used to create a scale of catalyst H-bonding ability.62–64 As expected, a downfield shift was observed in the 31P NMR spectrum of diethyl vinylphosphonate with increasing amounts of catalyst 1 (Fig. S4†). 13C NMR spectra were also recorded as the proposed mechanism requires activation of the alkene of the vinylphosphoryl compounds (Fig. 4). Consistent with a reduction in electron density, the signal for the β-carbon was shifted downfield with increasing catalyst 1 loading. Meanwhile the α-carbon signal was shifted upfield, consistent with a more polarised alkene. Similar changes in chemical shift were observed when a 13C NMR spectrum was taken in the presence of benzylamine (Fig. S6†), suggesting that the proposed H-bonding interaction is not disrupted by the presence of amine. These experiments are consistent with literature studies on N-crotonylpyrrolidinone complexes with Schreiner's thiourea61 and our own results obtained using methyl acrylate (Fig. S5†). With the latter, the same pattern in shift changes was observed however the Δδ was smaller – which is in line with evidence suggesting phosphoryl bonds are significantly stronger H-bond acceptors than carbonyl bonds.17
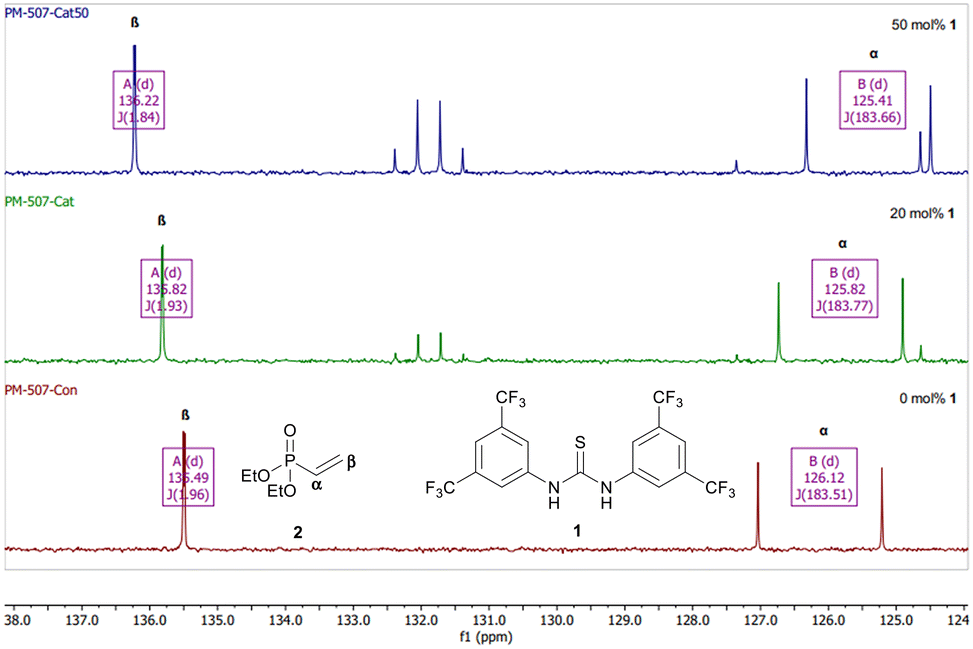 | ||
| Fig. 4 Changes in 13C NMR spectra of 2 (CDCl3, 101 MHz) with increasing equivalents of catalyst 1: 0 mol% (bottom), 20 mol% (middle), 50 mol% (top). | ||
Based on these mechanistic results, we propose the following catalytic cycle (Scheme 3). Firstly, the thiourea catalyst will H-bond with the phosphoryl oxygen of diethyl vinylphosphonate forming catalyst substrate complex I. This interaction increases the polarity of the alkene, activating the phosphonate toward conjugate addition. Next the amine adds to the β-carbon forming a zwitterionic intermediate II which again is stabilized by electron-withdrawing H-bonding interactions with the catalyst. Proton exchange then occurs to give the β-aminophosphonate which dissociates from the catalyst. The catalyst may also be involved in these proton shuttling steps in line with Seidel's observations using α,β-unsaturated esters.53
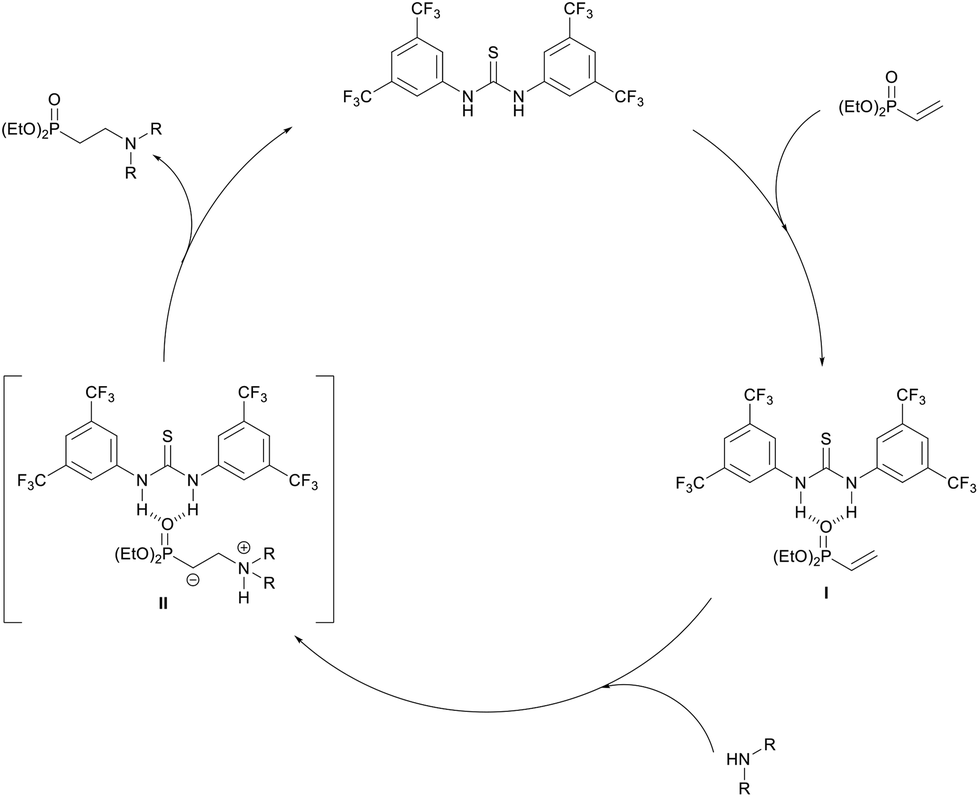 | ||
| Scheme 3 Proposed catalytic cycle for thiourea-catalysed addition of amines to diethyl vinylphosphonate. | ||
Thiourea-catalysed additions to prochiral phosphonates were also attempted. A range of α- or β-substituted phosph(in/on)ates were synthesised and tested as electrophiles (Fig. S1†). Of the electrophiles tested only 11 underwent conjugate addition. Following optimisation studies (Table S2†) the best result found was using catalyst 8 at −20 °C gave β-aminophosphonate 12 in 22% ee (Scheme 4, top). Kinetic resolution of chiral vinylphosphinates via conjugate addition was also attempted. Following screening (Table S3†), 9e was synthesised in 37% yield and 14% de (Scheme 4, bottom). Given the disappointing selectivities for both asymmetric variations of the methodology, asymmetric studies were ceased.
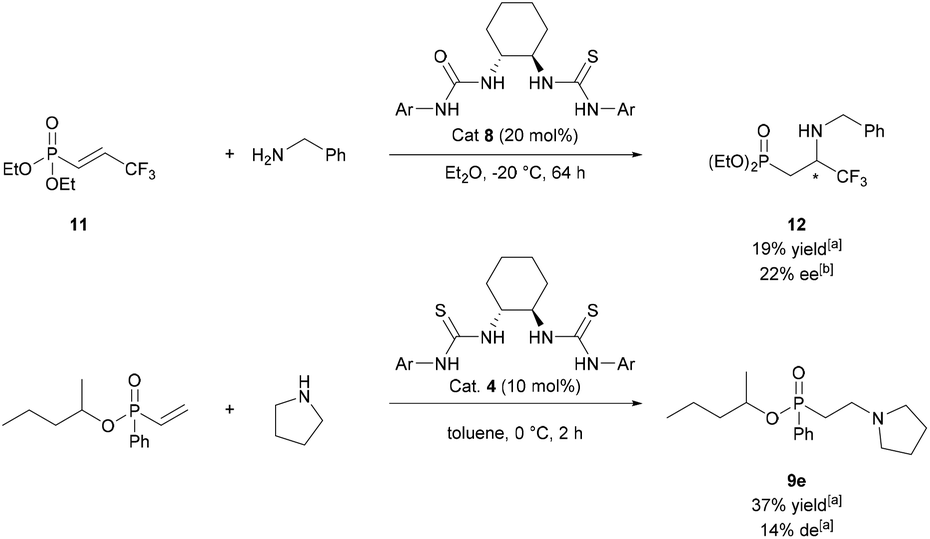 | ||
| Scheme 4 Top: attempted asymmetric addition of benzylamine to 11. Bottom: attempted kinetic resolution of racemic vinyl phosphinates. ameasured by 31P NMR analysis. bMeasured by chiral phase HPLC. | ||
Conclusions
In conclusion, thiourea catalysts have been used for the first time to catalyse conjugate additions to α,β-unsaturated phosph(in/on)ates. This novel catalysis was used to synthesise 15 β-aminophosph(on/in)ates using a simple, commercially available, thiourea catalyst in yields up to 99%. The methodology does not require an excess of either reaction partner, high temperatures or strong bases and is compatible with several commonly used organic solvents. On larger scales column chromatography is not required and the catalyst can easily be recovered and reused. A β-aminophosphonate synthesised on gram scale was converted to the corresponding β-aminophosphonic acid using TMSBr with a 99% yield over three steps. Additional mechanistic experiments suggest that H-bonding interactions play a key role in the catalysis.Author contributions
P. E. M.; conceptualization, investigation, methodology, writing. M. O. F. and A. M. H.; resources. E. M. M.; conceptualization, supervision, writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. E. M. thanks the A2P CDT which is supported by Science Foundation Ireland (SFI) and the Engineering and Physical Sciences Research Council (EPSRC) under Grant No. 18/EPSRC-CDT/3582 and BiOrbic, the Bioeconomy SFI Research Centre, which is funded by Ireland's European Structural and Investment Programmes, Science Foundation Ireland (16/RC/3889) and the European Regional Development Fund. A. M. H. and M. O. F. thank the Irish Research Council for Postgraduate Scholarships (GOIPG/2017/1306 and GOIPG/2014/528). We thank SFI (12/RC/2275_p2) and (18/RI/5702) for MS infrastructure.References
- P. Perlmutter, in Tetrahedron Organic Chemistry Series, ed. P. Perlmutter, Elsevier, 1992, vol. 9, pp. 1–373 Search PubMed.
- D. Almaşi, D. A. Alonso and C. Nájera, Tetrahedron: Asymmetry, 2007, 18, 299–365 CrossRef.
- Y. L. Sun, Y. Wei and M. Shi, ChemCatChem, 2017, 9, 718–727 CrossRef CAS.
- T. Parvin, R. Yadav and L. H. Choudhury, Org. Biomol. Chem., 2020, 18, 5513–5532 RSC.
- A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713–5743 CrossRef CAS PubMed.
- M. Tsakos and C. G. Kokotos, Tetrahedron, 2013, 69, 10199–10222 CrossRef CAS.
- G. Koutoulogenis, N. Kaplaneris and C. G. Kokotos, Beilstein J. Org. Chem., 2016, 12, 462–495 CrossRef CAS PubMed.
- H. Park, C. W. Cho and M. J. Krische, J. Org. Chem., 2006, 71, 7892–7894 CrossRef CAS PubMed.
- C. Garzon, M. Attolini and M. Maffei, Tetrahedron Lett., 2010, 51, 3772–3774 CrossRef CAS.
- S. Sulzer-Mossé, M. Tissot and A. Alexakis, Org. Lett., 2007, 9, 3749–3752 CrossRef PubMed.
- L. Albrecht, A. Albrecht, H. Krawczyk and K. A. Jørgensen, Chem. – Eur. J., 2010, 16, 28–48 CrossRef CAS PubMed.
- M. Capuzzi, D. Perdicchia and K. A. Jørgensen, Chem. – Eur. J., 2008, 14, 128–135 CrossRef CAS PubMed.
- M. T. Barros and A. M. F. Phillips, Eur. J. Org. Chem., 2008, 2525–2529 CrossRef CAS.
- H. Ishikawa, T. Suzuki and Y. Hayashi, Angew. Chem., Int. Ed., 2009, 48, 1304–1307 CrossRef CAS PubMed.
- T. Minami and J. Motoyoshiya, Synthesis, 1992, 333–349 CrossRef CAS.
- T. Janecki, J. Kêdzia and T. W
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) sek, Synthesis, 2009, 1227–1254 CrossRef CAS.
sek, Synthesis, 2009, 1227–1254 CrossRef CAS. - A. M. Modro and T. A. Modro, Can. J. Chem., 1999, 77, 890–894 CrossRef CAS.
- S. Sulzer-Mossé, A. Alexakis, J. Mareda, G. Bollot, G. Bernardinelli and Y. Filinchuk, Chem. – Eur. J., 2009, 15, 3204–3220 CrossRef PubMed.
- F. G. Bordwell and H. E. Fried, J. Org. Chem., 1981, 46, 4327–4331 CrossRef CAS.
- F. G. Bordwell, J. E. Bares, J. E. Bartmess, G. J. McCollum, M. Van Der Puy, N. R. Vanier and W. S. Matthews, J. Org. Chem., 1977, 42, 321–325 CrossRef CAS.
- H. Reich, Hans Reich's Collection. Bordwell pKa Table, https://organicchemistrydata.org/hansreich/resources/pka/pka_data/pka-compilation-reich-bordwell.pdf, (accessed 8 August 2022).
- F. Palacios, C. Alonso and J. M. de los Santos, Chem. Rev., 2005, 105, 899–931 CrossRef CAS PubMed.
- P. Kafarski and B. Lejczak, Phosphorus, Sulfur Silicon Relat. Elem., 1991, 63, 193–215 CrossRef CAS.
- W. A. Kinney, M. Abou-Gharbia, D. T. Garrison, J. Schmid, D. M. Kowal, D. R. Bramlett, T. L. Miller, R. P. Tasse, M. M. Zaleska and J. A. Moyer, J. Med. Chem., 1998, 41, 236–246 CrossRef CAS PubMed.
- G. Seydlová, R. Pohl, E. Zborníková, M. Ehn, O. Šimák, N. Panova, M. Kolář, K. Bogdanová, R. Večeřová, R. Fišer, H. Šanderová, D. Vítovská, P. Sudzinová, J. Pospíšil, O. Benada, T. Křížek, D. Sedlák, P. Bartůněk, L. Krásný and D. Rejman, J. Med. Chem., 2017, 60, 6098–6118 CrossRef PubMed.
- D. Hocková, D. T. Keough, Z. Janeba, T. H. Wang, J. De Jersey and L. W. Guddat, J. Med. Chem., 2012, 55, 6209–6223 CrossRef PubMed.
- G. M. Kosolapoff and L. Maier, Aust. J. Chem., 1979, 32, 463–464 CrossRef.
- H. Gali, K. R. Prabhu, S. R. Karra and K. V. Katti, J. Org. Chem., 2000, 65, 676–680 CrossRef CAS.
- M. J. Sparkes, H. B. Dixon, K. F. Geoghegan and E. Visedo-Gonzalez, Biochem. J., 1989, 260, 296–297 CrossRef CAS PubMed.
- Y. J. Chun, J. H. Park, G. M. Oh, S. I. Hong and Y. J. Kim, Synthesis, 1994, 909–910 CrossRef CAS.
- V. Rai and I. N. N. Namboothiri, Tetrahedron: Asymmetry, 2008, 19, 2335–2338 CrossRef CAS.
- F. Palacios, A. M. Ochoa De Retana and J. M. Alonso, J. Org. Chem., 2006, 71, 6141–6148 CrossRef CAS PubMed.
- M. E. Duggan and D. S. Karanewsky, Tetrahedron Lett., 1983, 24, 2935–2938 CrossRef CAS.
- L. Maier and P. J. Diel, Phosphorus, Sulfur Silicon Relat. Elem., 1989, 45, 165–176 CrossRef CAS.
- W. Cen and Y. Shen, J. Fluor. Chem., 1995, 72, 107–110 CrossRef CAS.
- C. Bolzati, N. Salvarese, B. Spolaore, A. Vittadini, D. Forrer, S. Brunello, S. Ghiani and A. Maiocchi, Mol. Pharm., 2022, 19, 876–894 CrossRef CAS PubMed.
- D. H. Suk, D. Rejman, C. C. Dykstra, R. Pohl, K. W. Pankiewicz and S. E. Patterson, Bioorg. Med. Chem. Lett., 2007, 17, 2811–2816 CrossRef CAS PubMed.
- M. Schmider, E. Müh, J. E. Klee and R. Mülhaupt, Macromolecules, 2005, 38, 9548–9555 CrossRef CAS.
- N. Khusainova, O. Mostovaya, E. Berdnikov, S. Rybakov and R. Cherkasov, Phosphorus, Sulfur Silicon Relat. Elem., 2009, 184, 865–871 CrossRef CAS.
- E. V. Matveeva, P. V. Petrovskii and I. L. Odinets, Tetrahedron Lett., 2008, 49, 6129–6133 CrossRef CAS.
- E. V. Matveeva, P. V. Petrovskii, Z. S. Klemenkova, N. A. Bondarenko and I. L. Odinets, C. R. Chim., 2010, 13, 964–970 CrossRef CAS.
- N. Bou Orm, Y. Dkhissi, S. Daniele and L. Djakovitch, Tetrahedron, 2013, 69, 115–121 CrossRef CAS.
- V. Bernat, T. H. Admas, R. Brox, F. W. Heinemann and N. Tschammer, ACS Chem. Biol., 2014, 9, 2664–2677 CrossRef CAS PubMed.
- K. Malhotra, R. Fuku, T. S. Chan, N. Kraljevic, A. Sedighi, P. A. E. Piunno and U. J. Krull, Chem. Mater., 2020, 32, 4002–4012 CrossRef CAS.
- W. Gu, S. Martinez, A. K. Singh, H. Nguyen, J. Rozenski, D. Schols, P. Herdewijn, K. Das and S. De Jonghe, Eur. J. Med. Chem., 2021, 225, 113785 CrossRef CAS PubMed.
- R. Upadhyay, R. Rana and S. K. Maurya, ChemCatChem, 2021, 13, 1867–1897 CrossRef CAS.
- Y. C. Wu, Y. Jhong, H. J. Lin, S. P. Swain, H. H. G. Tsai and D. R. Hou, Adv. Synth. Catal., 2019, 361, 4966–4982 CrossRef CAS.
- U. K. Bhagat and R. K. Peddinti, J. Org. Chem., 2018, 83, 793–804 CrossRef CAS PubMed.
- R. Miyaji, K. Asano and S. Matsubara, Org. Lett., 2013, 15, 3658–3661 CrossRef CAS PubMed.
- X. F. Wang, J. An, X. X. Zhang, F. Tan, J. R. Chen and W. J. Xiao, Org. Lett., 2011, 13, 808–811 CrossRef CAS PubMed.
- C. De Fusco, T. Fuoco, G. Croce and A. Lattanzi, Org. Lett., 2012, 14, 4078–4081 CrossRef CAS PubMed.
- J. Wang, H. Li, L. Zu and W. Wang, Org. Lett., 2006, 8, 1391–1394 CrossRef CAS PubMed.
- Y. Lin, W. J. Hirschi, A. Kunadia, A. Paul, I. Ghiviriga, K. A. Abboud, R. W. Karugu, M. J. Vetticatt, J. S. Hirschi and D. Seidel, J. Am. Chem. Soc., 2020, 142, 5627–5635 CrossRef CAS PubMed.
- B. C. Ranu and S. Banerjee, Tetrahedron Lett., 2007, 48, 141–143 CrossRef CAS.
- C. E. McKenna, M. T. Higa, N. H. Cheung and M. C. McKenna, Tetrahedron Lett., 1977, 18, 155–158 CrossRef.
- G. A. Bradshaw, A. C. Colgan, N. P. Allen, I. Pongener, M. B. Boland, Y. Ortin and E. M. McGarrigle, Chem. Sci., 2019, 10, 508–514 RSC.
- G. Jakab, C. Tancon, Z. Zhang, K. M. Lippert and P. R. Schreiner, Org. Lett., 2012, 14, 1724–1727 CrossRef CAS PubMed.
- X. Li, H. Deng, B. Zhang, J. Li, L. Zhang, S. Luo and J. P. Cheng, Chem. – Eur. J., 2010, 16, 450–455 CrossRef CAS PubMed.
- T. Ghosh, A. Mukherji and P. K. Kancharla, J. Org. Chem., 2021, 86, 1253–1261 CrossRef CAS PubMed.
- N. Spiliopoulou, N. F. Nikitas and C. G. Kokotos, Green Chem., 2020, 22, 3539–3545 RSC.
- K. M. Lippert, K. Hof, D. Gerbig, D. Ley, H. Hausmann, S. Guenther and P. R. Schreiner, Eur. J. Org. Chem., 2012, 5919–5927 CrossRef CAS.
- K. M. Diemoz and A. K. Franz, J. Org. Chem., 2019, 84, 1126–1138 CrossRef CAS PubMed.
- A. R. Nödling, G. Jakab, P. R. Schreiner and G. Hilt, Eur. J. Org. Chem., 2014, 6394–6398 CrossRef.
- R. R. Walvoord, P. N. H. Huynh and M. C. Kozlowski, J. Am. Chem. Soc., 2014, 136, 16055–16065 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob02116a |
| This journal is © The Royal Society of Chemistry 2023 |