
Phosphine-catalysed denitrative rearomatising (3 + 2) annulation of α,β-ynones and 3-nitroindoles†
Lona
Dutta
,
Anwita
Chattopadhyay
 ,
Nisha
Yadav
and
S. S. V.
Ramasastry
*
,
Nisha
Yadav
and
S. S. V.
Ramasastry
*
Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Mohali, Sector 81, S. A. S. Nagar, Punjab 140306, India. E-mail: ramsastry@iisermohali.ac.in; ramsastrys@gmail.com
First published on 20th December 2022
Abstract
We describe a metal-free strategy to access various α-arylidene cyclopenta[b]indoles via phosphine-catalysed (3 + 2) annulation of α,β-ynones and 3-nitroindoles. For the first time, the rearomatisation of the indole nucleus was observed in such an annulative transformation. The method was extended to the synthesis of an antimalarial natural product, bruceolline E.
The cyclopenta[b]indole moiety is a part structure of several bioactive natural products and pharmaceutically relevant compounds (Fig. 1).1 For example, Fischer indole L shows cytotoxicity against HCl-H460 cell lines,2 laropiprant exhibits cholesterol-lowering effect,3 bruceollines are traditionally used for treating malaria and other parasitic diseases,4 and yuehchukene shows anti-fertility and estrogenic activities.5 Due to the attractive biological properties of cyclopenta[b]indoles, the development of new methods for their preparation, especially under metal-free conditions,6 has become a major area of investigation for synthetic chemists.1d
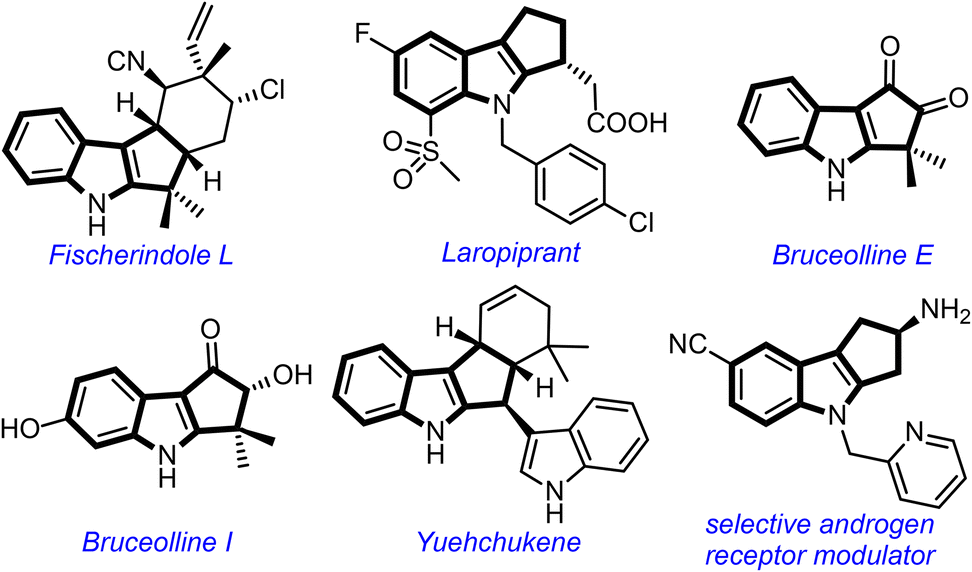 | ||
| Fig. 1 Selected natural products and medicinally important compounds possessing the cyclopenta[b]indole motif. | ||
Engaging the C2–C3 π-bond of indoles in cycloaddition reactions is a powerful approach to generate poly(hetero)-cyclic structures. For instance, this strategy has been exploited in constructing cyclopenta[b]indolines via the (3 + 2) cycloaddition of different types of three-carbon units and 3-nitroindoles under metal catalysis, Scheme 1a.7–9 Here, the nitro group imparts electrophilic character to the otherwise electron-rich indole moiety and dictates the regioselectivity of the cyclisation event.10
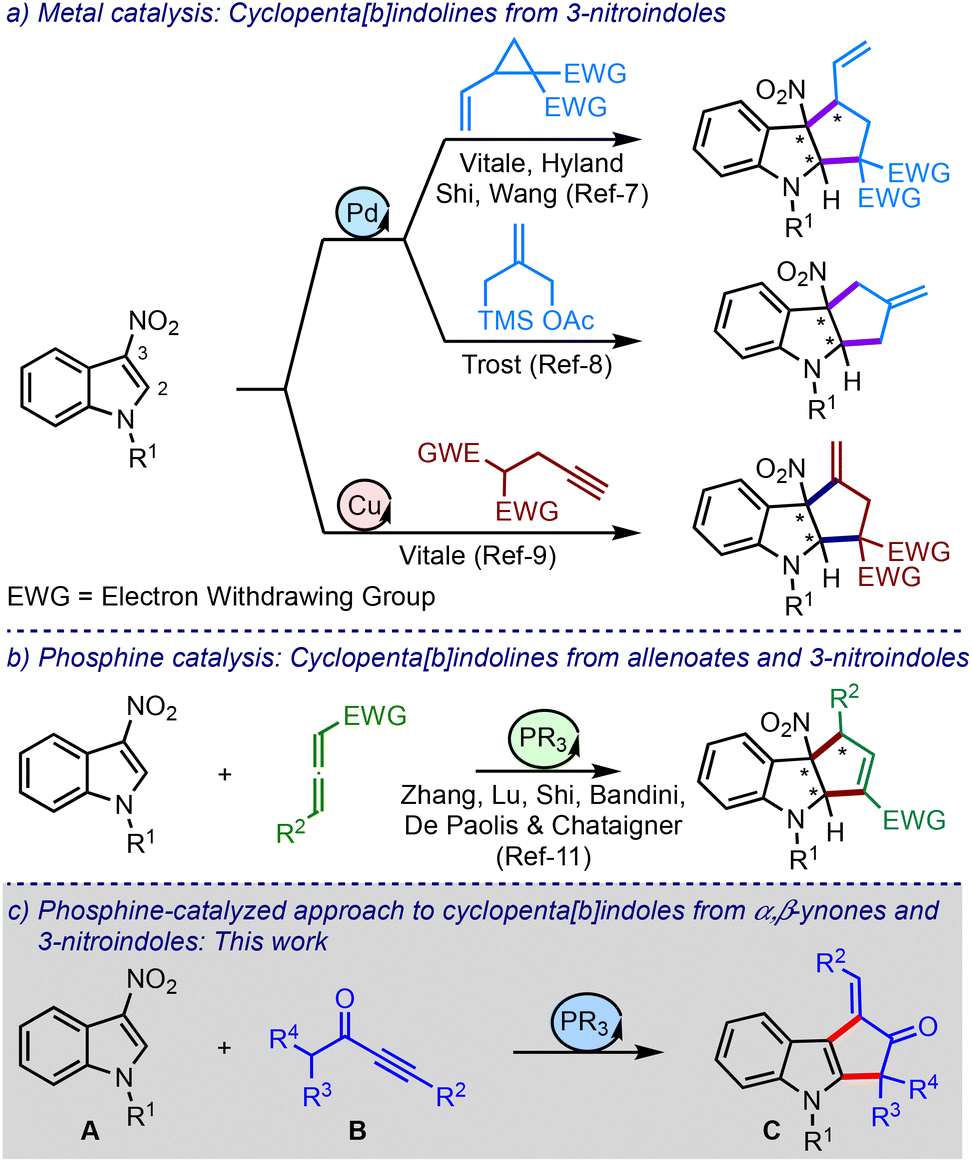 | ||
| Scheme 1 (a) Cyclopenta[b]indolines from 3-nitroindoles via metal-catalysis; (b) phosphine-catalysed (3 + 2) annulation of allenoates and 3-nitroindoles; (c) this work: phosphine-catalysed denitrative (3 + 2) annulation of α,β-ynones and 3-nitroindoles. | ||
On the other hand, the Zhang, Lu, Shi, Bandini, and Chataigner groups, all in 2019, have independently reported phosphine-catalysed (3 + 2) cycloaddition of allenoates and 3-nitroindoles to synthesise cyclopenta[b]indolines, Scheme 1b.11 To our knowledge, these are the only metal-free studies involving phosphines and 3-nitroindoles to construct cyclopentannulated indoles.12 Against this background and with our experience in phosphine chemistry,13 we intended to devise a novel protocol to access cyclopenta[b]indoles.
Phosphines readily participate in Michael-type additions to activated alkenes and alkynes, and trigger subsequent functionalisation reactions.14 For example, α,β-ynones having α′-hydrogens (such as B) have been demonstrated to undergo (3 + 2) cycloaddition reactions with a range of electron-deficient olefins.15 However, nitroolefins have not yet been employed in such transformations. Thus, we intended to explore 3-nitroindoles in the (3 + 2) annulation reactions involving α,β-ynones and phosphines, Scheme 1c.
It was hypothesised that allenoates D generated by the phospha-Michael addition of ynones B isomerise to enolates E, which undergo C2-addition to give 3-nitroindoles A, producing zwitterionic nitronates F, Scheme 2. A subsequent umpolung addition to the vinyl phosphonium moiety could generate ylides G. A 1,2-proton shift (leading to H) and regeneration of phosphine would liberate cyclopenta[b]indolines I. On the other hand, a 1,4-proton shift in G could trigger the elimination of nitrite from J and generate K. A subsequent phosphine elimination could provide cyclopenta[b]indoles C. Alternatively, the rearomatisation is also possible under thermal conditions.16 Interestingly, such in situ rearomatisations have not been observed in any reported cases (see Scheme 1).
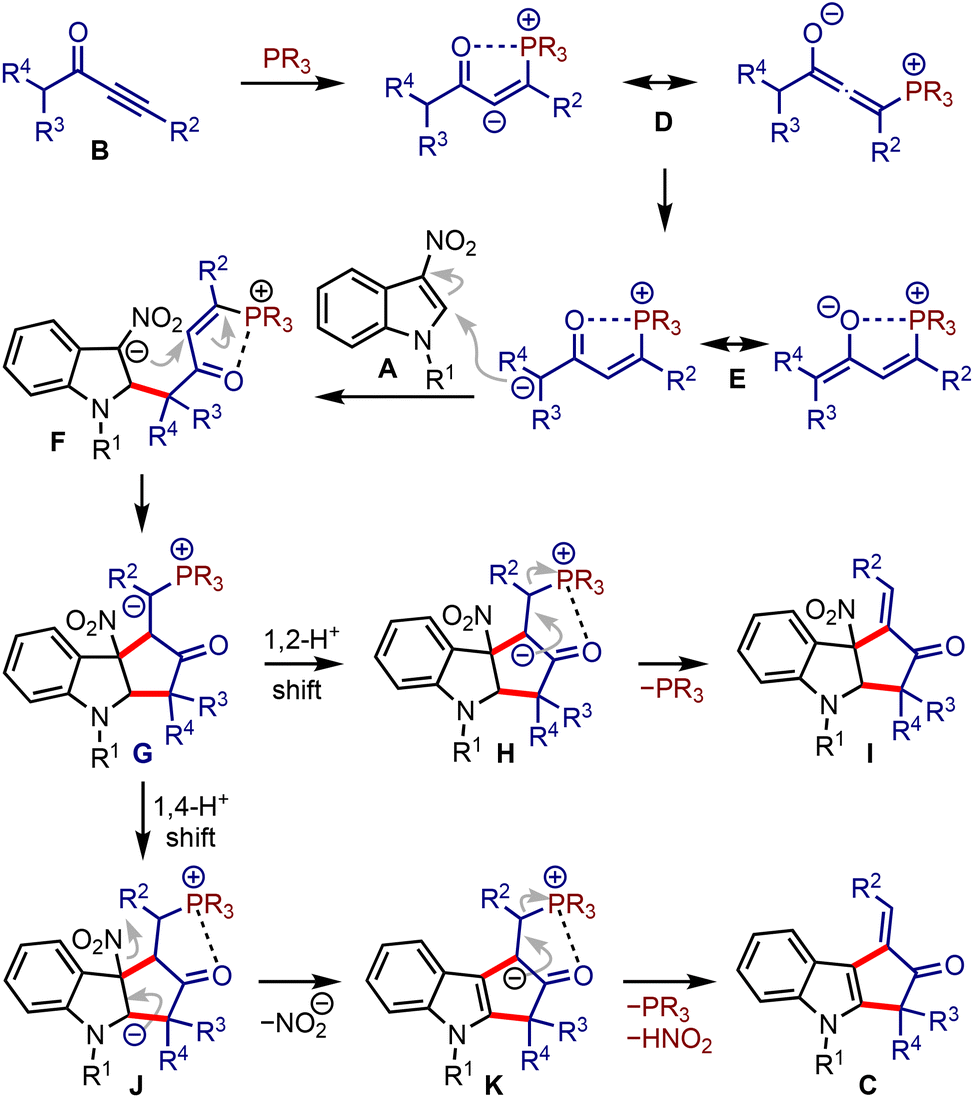 | ||
| Scheme 2 Plausible mechanistic scenario of the reaction between α,β-ynones and 3-nitroindoles leading to cyclopenta[b]indoles. | ||
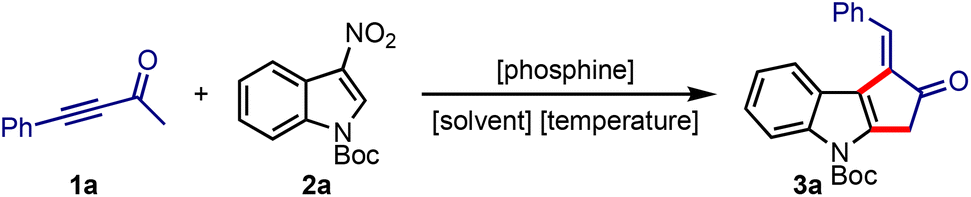
To validate the hypothesis, the ynone 1a and N-Boc-3-nitroindole 2a were treated with different phosphines under various conditions. Some of the significant results are presented in Table 1. We commenced the screening with highly nucleophilic phosphines such as PBu3 and PCy3, but the rearomatised product 3a was obtained in poor yields (entries 1–4). The reaction in the presence of PPh3 also gave similar results, although it indicated the influence of solvents (Table 1, entries 5–7). Subsequently, we observed a dramatic improvement when we switched to a combination of solvents (toluene–acetonitrile), in which the reaction delivered an excellent result even under catalytic conditions (entry 10). In the dual solvent system, we evaluated the other parameters in detail (entries 11–18). We studied the optimal requirement of the amount of 1a (entries 11–13), the influence of the temperature (entries 14–16), and the catalyst loading (entries 17 and 18). From these experiments, we realised that a catalytic amount of PPh3 in toluene–ACN combination at 100 °C efficiently generates 3a.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Phosphine (equiv.) | Solvent | Temp (°C) | Time (h) | Yieldb (%) |
| a Reaction conditions: see the ESI for details.† b Isolated yields after column chromatography. c 2 equiv. of 1a was used. d No reaction was observed under catalytic conditions. e 1.2 equiv. of 1a was used. f 3 equiv. of 1a was used. | |||||
| 1c,d | PBu3 (1.5) | Toluene | RT | 24 | 18 |
| 2e | PBu3 (1) | ACN | RT | 24 | Trace |
| 3c | PCy3 (1.5) | Toluene | RT | 24 | 26 |
| 4c | PCy3 (1.5) | Dioxane | RT | 24 | 44 |
| 5c | PPh3 (1.5) | Toluene | RT | 24 | 22 |
| 6c | PPh3 (1.5) | DMF | RT | 24 | 15 |
| 7e | PPh3 (1) | ACN | RT | 24 | 37 |
| 8f | PBu3 (0.3) | Toluene–ACN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
100 | 15 | 40 |
| 9f | PCy3 (0.3) | Toluene–ACN (1:1) |
100 | 5 | — |
| 10f | PPh2Me (0.3) | Toluene–ACN (1:1) |
100 | 15 | 70 |
| 11f | PPh3 (0.3) | Toluene–ACN (1:1) |
100 | 17 | 77 |
| 12e | PPh3 (0.3) | Toluene–ACN (1:1) |
100 | 36 | 44 |
| 13c | PPh3 (0.3) | Toluene–ACN (1:1) |
100 | 17 | 59 |
| 14f | PPh3 (0.3) | Toluene–ACN (1:1) |
80 | 24 | 70 |
| 15f | PPh3 (0.3) | Toluene–ACN (1:1) |
50 | 36 | 47 |
| 16f | PPh3 (0.3) | Toluene–ACN (1:1) |
RT | 48 | 29 |
| 17f | PPh3 (0.2) | Toluene–ACN (1:1) |
100 | 24 | 63 |
| 18f | PPh3 (0.1) | Toluene–ACN (1:1) |
100 | 30 | 48 |
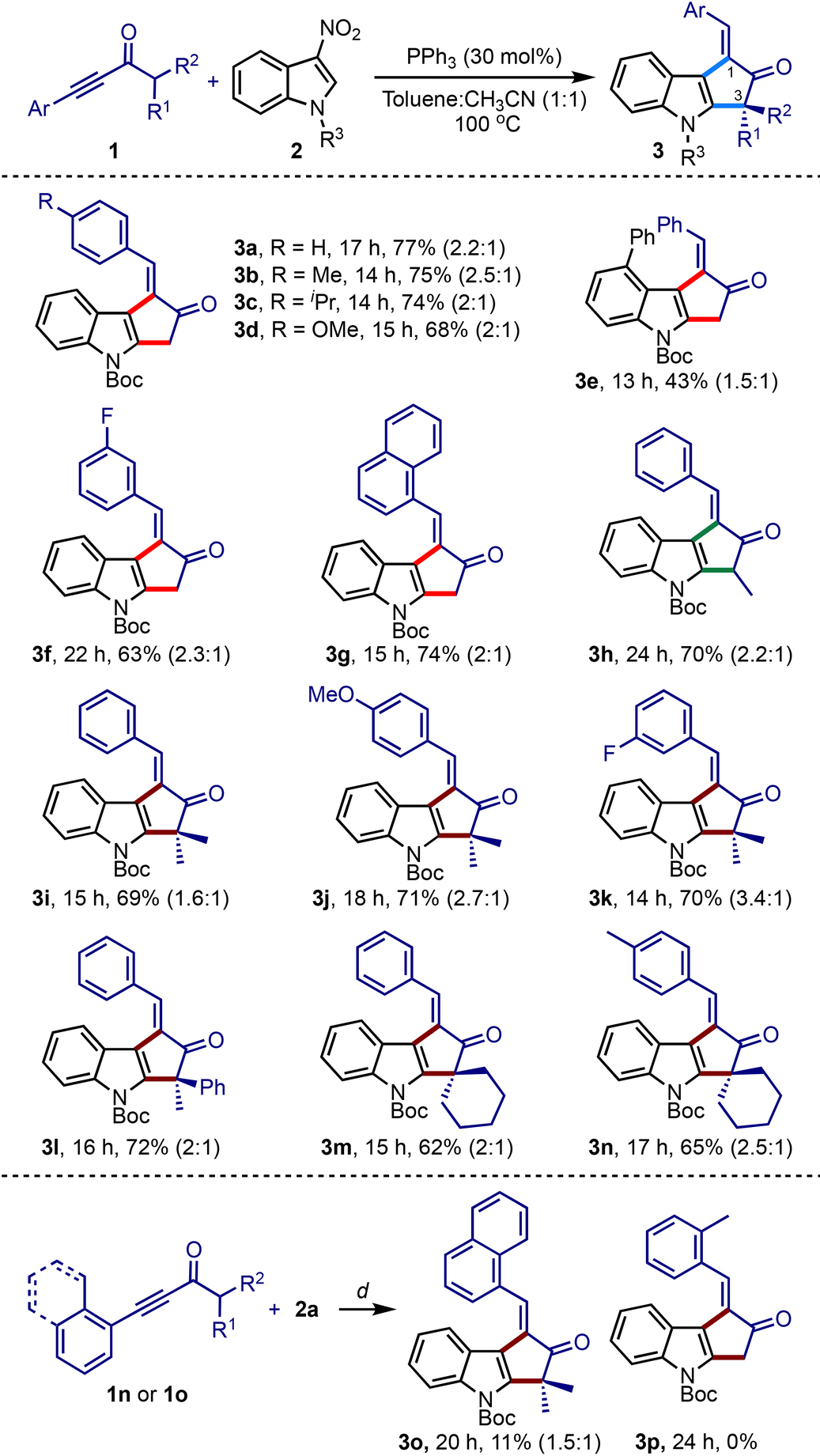
Under the optimised conditions, we studied the scope with respect to ynones and nitroindoles, Table 2. An array of synthetically challenging 1-arylidene-cyclopenta[b]indol-2-ones could be assembled in good yields (3b–3o).17 Substrates with electron-donating (3b–3d, 3j and 3n) and electron-withdrawing groups (3f, 3g, and 3k) across the ynone moiety showed only a marginal influence on the efficiency of the reaction. Of particular interest, α′-branched ynones successfully generated a tertiary carbon (3h) and an all-carbon quaternary center at the C3-position (3i–3l and 3o). This method was also extended to creating spiro centers (3m and 3n). However, the reaction of 1n or 1o, possessing ortho-substituted arenes on alkynes, did not give encouraging results. For instance, the 1-naphthyl adduct 3o was obtained in poor yield, while the ynone 2p with the o-tolyl group did not produce the desired product 3p.
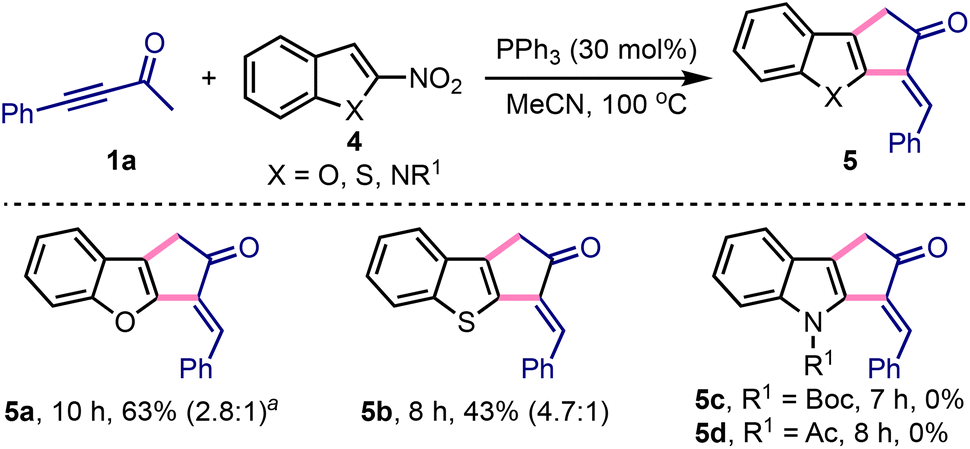
We extended this method to access 3-benzylidene-1,3-dihydro-2H-cyclopenta[b]benzofuran-2-one 5a by treating 1a with 2-nitrobenzofuran 4a under slightly modified conditions, Scheme 3.18,19 Similarly, 3-benzylidene-cyclopenta[b]-fused benzothiophene 5b was synthesised from 1a and 2-nitrobenzothiophene 4b. Interestingly, the synthesis of 5a or 5b or their analogues has not yet been achieved. On the other hand, despite several attempts, the synthesis of 3-benzylidene-cyclopenta[b]indol-2-ones 5c or 5d was elusive under these conditions.20
 | ||
Scheme 3 Synthesis of 3-arylidene-cyclopenta[b]-fused heteroarenes. aE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :Z ratios are given in the parentheses. :Z ratios are given in the parentheses. | ||
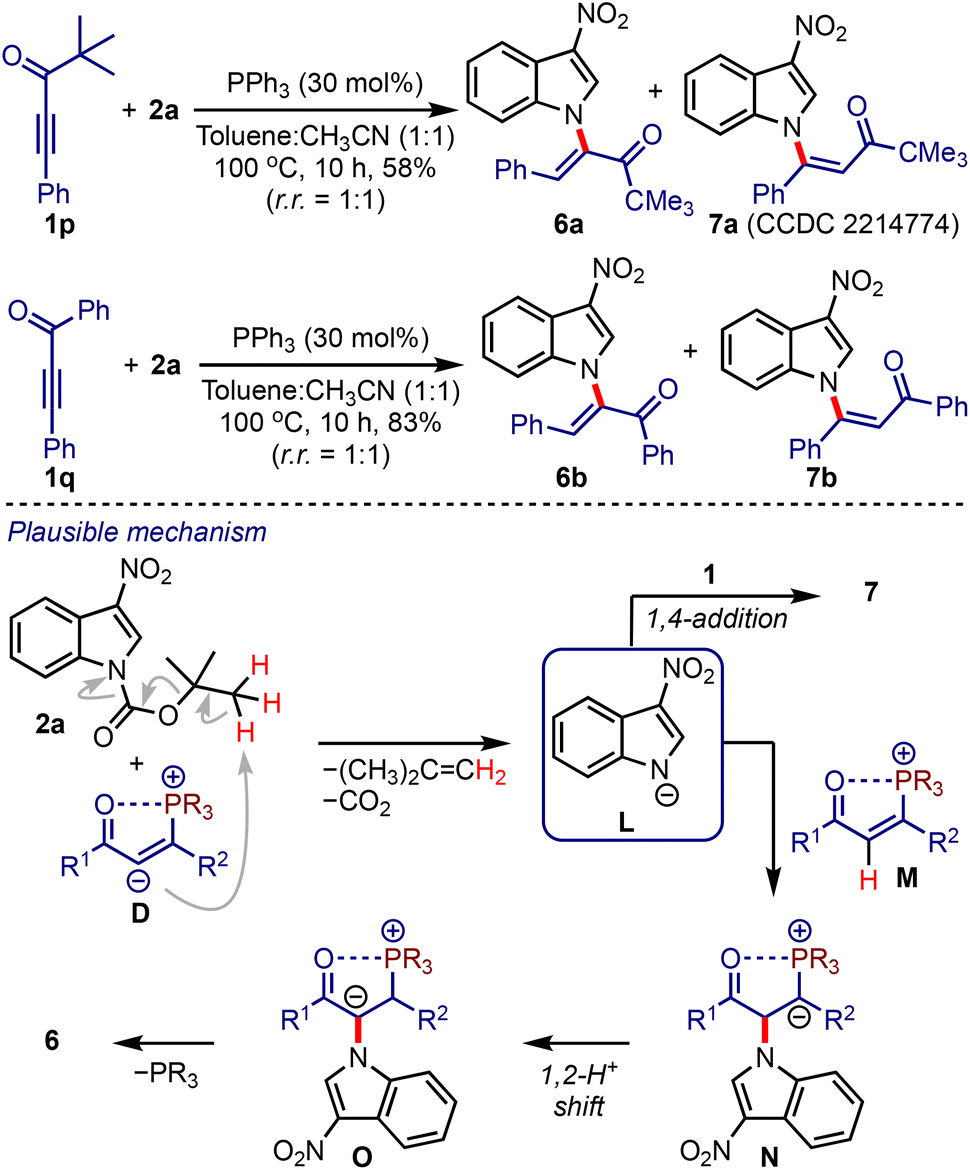
At this stage, we were curious about the outcome of a reaction between ynones lacking α′-hydrogens and 2-nitroindoles under the optimised conditions, Scheme 4. Accordingly, we considered the reaction between pivalylethynylbenzene 1p and 2a. The formation of a separable mixture of α- and β-N-indolyl enones (6a and 7a) was observed.21 Similarly, the enones 6b and 7b were obtained from 1q and 2a in good yields. A plausible mechanism leading to 6 and 7 is depicted in Scheme 4. In the absence of α′-hydrogens, the intermediate D facilitates the formation of L,22 which undergoes umpolung addition to M to generate N. A subsequent 1,2-proton shift (to O) followed by the elimination of phosphine provides 6. On the other hand, a 1,4-addition of L to 1 generates 7. It is noteworthy that the synthesis of enones of type 6 or 7 is not known yet and is otherwise difficult to achieve.
 | ||
| Scheme 4 Reaction between ynones lacking α′-hydrogens and 2a (r.r. = regioisomeric ratio). | ||
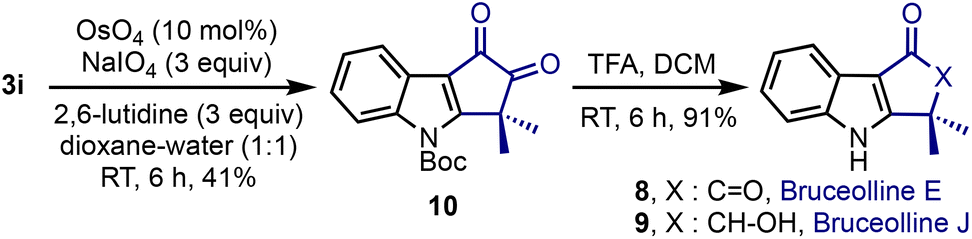
Bruceolline E 8 was isolated by Ohmoto and co-workers in 1994 from the root wood of Brucea mollis var. ronkinensis.4a We explored the feasibility of extending the current method to synthesise 8, Scheme 5. Accordingly, the oxidative cleavage of the benzylidene moiety in 3i was accomplished under OsO4/NaIO4 conditions. The N-Boc-group in 10 was deprotected using trifluoroacetic acid and bruceolline E 8 was isolated in an excellent yield. It constitutes a short 3-step synthesis of 8 with a 26% overall yield.23 It is noteworthy that the synthesis of 8 also constitutes the formal synthesis of bruceolline J 9.23b
 | ||
| Scheme 5 Total synthesis of bruceolline E. | ||
We tested the practicality of the method by conducting 1 mmol scale reactions of 2a with 1a and 1h, Scheme 6. Both the reactions delivered the respective products 3a and 3i in consistent yields, indicating that the reaction is scalable.
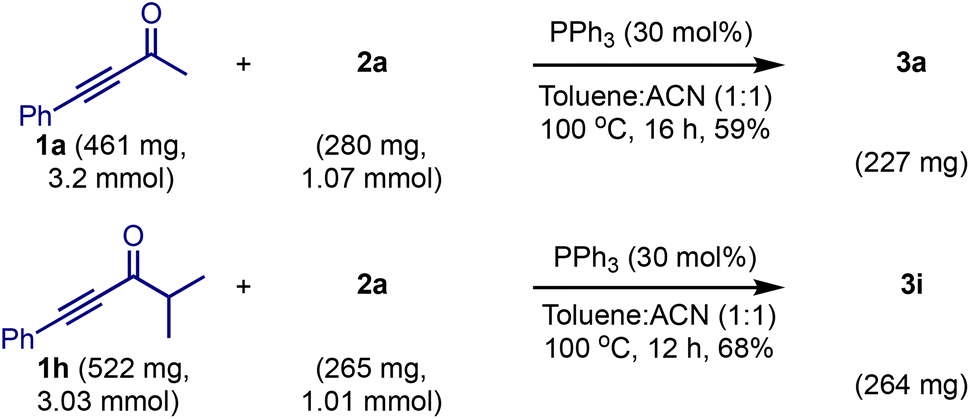 | ||
| Scheme 6 Scale-up reactions. | ||
In summary, we have developed the first phosphine-catalysed rearomatising (3 + 2) annulation of α,β-ynones and 3-nitroindoles to access unprecedented α-arylidene cyclopenta[b]indoles. The reaction was promising in creating a tertiary center and sterically encumbered all-carbon quaternary- and spiro- centers. The strategy was extended to synthesise 3-benzylidene-cyclopenta[b]benzofuran-2-ones, 3-benzylidene-cyclopenta[b]-fused benzothiophenes, α- and β-N-indolyl enones, the synthesis of which is not known yet. The utility of the method was demonstrated by synthesising the antimalarial natural product, bruceolline E. Other salient features of this work are: (i) easily accessible starting compounds and reagents, (ii) straightforward mechanism, and (iii) efficient and practical reaction conditions. We are extending this strategy to synthesise other complex natural products listed in Fig. 1. The results will be communicated in due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank IISER Mohali for the NMR, mass spectrometry, and X-ray facilities. S. S. V. R. thanks SERB for the Swarnajayanti fellowship (DST/SJF/CSA-01/2017-18) and IISER Mohali for funding. L. D. thanks the IISER Mohali, A. C. thanks the KVPY and N. Y. thanks the CSIR for research fellowships.References
- (a) M. Ishikura, T. Abe, T. Choshi and S. Hibino, Nat. Prod. Rep., 2013, 30, 694–752 RSC; (b) I. S. Marcos, R. F. Moro, I. Costales, P. Basabe and D. Diez, Nat. Prod. Rep., 2013, 30, 1509–1526 RSC; (c) M. Petrovic and E. G. Occhiato, Chem. – Asian J., 2016, 11, 642–659 CrossRef CAS; (d) T. Vivekanand, B. Satpathi, S. K. Bankar and S. S. V. Ramasastry, RSC Adv., 2018, 8, 18576–18588 RSC.
- J. M. Richter, Y. Ishihara, T. Masuda, B. W. Whitefield, T. Llamas, A. Pohjakallio and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 17938–17954 CrossRef CAS.
- E. Lai, I. De Lepeleire, T. M. Crumley, F. Liu, L. A. Wenning, N. Michiels, E. Vets, G. O'Neill, J. A. Wagner and K. Gottesdiener, Clin. Pharmacol. Ther., 2007, 81, 849–857 CrossRef CAS PubMed.
- (a) Y. Ouyang, K. Koike and T. Ohmoto, Phytochemistry, 1994, 37, 575–578 CrossRef CAS; (b) D. Scarpi, C. Faggi and E. G. Occhiato, J. Nat. Prod., 2017, 80, 2384–2388 CrossRef CAS PubMed.
- Y. C. Kong, K. H. Ng, K. H. Wat, A. Wong, I. F. Saxena, K. F. Cheng, P. P. H. But and H. T. Chang, Planta Med., 1985, 51, 304–307 CrossRef CAS PubMed.
- For selected reviews, see: (a) Y.-C. Zhang, F. Jiang and F. Shi, Acc. Chem. Res., 2020, 53, 425–446 CrossRef CAS PubMed; (b) M.-S. Tu, K.-W. Chen, P. Wu, Y.-C. Zhang, X.-Q. Liu and F. Shi, Org. Chem. Front., 2021, 8, 2643–2672 RSC.
- (a) M. Laugeois, J. Ling, C. Feŕard, V. Michelet, V. Ratovelomanana-Vidal and M. R. Vitale, Org. Lett., 2017, 19, 2266–2269 CrossRef CAS; (b) Y. S. Gee, D. J. Rivinoja, S. M. Wales, M. G. Gardiner, J. H. Ryan and C. J. T. Hyland, J. Org. Chem., 2017, 82, 13517–13529 CrossRef CAS; (c) M. Sun, Z.-Q. Zhu, L. Gu, X. Wan, G.-J. Mei and F. Shi, J. Org. Chem., 2018, 83, 2341–2348 CrossRef CAS PubMed; (d) J.-Q. Zhang, F. Tong, B.-B. Sun, W.-T. Fan, J.-B. Chen, D. Hu and X.-W. Wang, J. Org. Chem., 2018, 83, 2882–2891 CrossRef CAS.
- B. M. Trost, V. Ehmke, B. M. O'Keefe and D. A. Bringley, J. Am. Chem. Soc., 2014, 136, 8213–8216 CrossRef CAS PubMed.
- J. Ling, D. Mara, B. Roure, M. Laugeois and M. R. Vitale, J. Org. Chem., 2020, 85, 3838–3848 CrossRef CAS.
- For some selected articles on 3-nitroindoles as electrophiles, see: (a) A. Cerveri and M. Bandini, Chin. J. Chem., 2020, 38, 287–294 CrossRef CAS; (b) B. Rkein, A. Bigot, L. Birbaum, M. Manneveau, M. De Paolis, J. Legros and I. Chataigner, Chem. Commun., 2021, 57, 27–44 RSC; (c) B. Rkein, M. Manneveau, L. Noel-Duchesneau, K. Pasturaud, M. Durandetti, J. Legros, S. Lakhdar and I. Chataigner, Chem. Commun., 2021, 57, 10071–10074 RSC; (d) Y.-L. Li, K.-K. Wang and X.-L. He, Adv. Synth. Catal., 2022, 364, 3630–3650 CrossRef CAS.
- Arranged in the order of the date of submission: (a) H. Wang, J. Zhang, Y. Tu and J. Zhang, Angew. Chem., Int. Ed., 2019, 58, 5422–5426 CrossRef CAS; (b) K. Li, T. P. Goncalves, K.-W. Huang and Y. Lu, Angew. Chem., Int. Ed., 2019, 58, 5427–5431 CrossRef CAS; (c) L.-W. Jin, F. Jiang, K.-W. Chen, B.-X. Du, G.-J. Mei and F. Shi, Org. Biomol. Chem., 2019, 17, 3894–3901 RSC; (d) A. Cerveri, O. N. Faza, C. S. Loṕez, S. Grilli, M. Monari and M. Bandini, J. Org. Chem., 2019, 84, 6347–6355 CrossRef CAS; (e) L. Birbaum, L. Gillard, H. Gerard, H. Oulyadi, G. Vincent, X. Moreau, M. De Paolis and I. Chataigner, Chem. – Eur. J., 2019, 25, 13688–13693 CrossRef CAS.
- Selected references on the synthesis of fused indolines containing a heteroatom under metal-free conditions from 3-nitroindoles: (a) J.-Q. Zhao, M.-Q. Zhou, Z.-J. Wu, Z.-H. Wang, D.-F. Yue, X.-Y. Xu, X.-M. Zhang and W.-C. Yuan, Org. Lett., 2015, 17, 2238–2241 CrossRef CAS PubMed; (b) K.-K. Wang, W. Du, J. Zhu and Y.-C. Chen, Chin. Chem. Lett., 2017, 28, 512–516 CrossRef CAS; (c) X.-M. Chen, C.-W. Lei, D.-F. Yue, J.-Q. Zhao, Z.-H. Wang, X.-M. Zhang, X.-Y. Xu and W.-C. Yuan, Org. Lett., 2019, 21, 5452–5456 CrossRef CAS PubMed; (d) H.-Z. Tian, S.-F. Wu, G.-Q. Lin and X.-W. Sun, Tetrahedron Lett., 2022, 103, 153969 CrossRef CAS.
- B. Satpathi, A. Mondal and S. S. V. Ramasastry, Chem. – Asian J., 2018, 13, 1642–1653 CrossRef CAS PubMed.
- For selected reviews, see: (a) X. Lu, C. Zhang and Z. Xu, Acc. Chem. Res., 2001, 34, 535–544 CrossRef CAS; (b) J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035–1050 CrossRef CAS; (c) H. Ni, W.-L. Chan and Y. Lu, Chem. Rev., 2018, 118, 9344–9411 CrossRef CAS PubMed; (d) H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049–10293 CrossRef CAS.
- (a) H. Kuroda, I. Tomita and T. Endo, Org. Lett., 2003, 5, 129–131 CrossRef CAS PubMed; (b) Y. K. Chung and G. C. Fu, Angew. Chem., Int. Ed., 2009, 48, 2225–2227 CrossRef CAS PubMed; (c) Z. Lian and M. Shi, Eur. J. Org. Chem., 2012, 581–586 CrossRef CAS; (d) L. Yang, P. Xie, E. Li, X. Li, Y. Huang and R. Chen, Org. Biomol. Chem., 2012, 10, 7628–7634 RSC; (e) D. B. Ramachary, C. Venkaiah and P. M. Krishna, Org. Lett., 2013, 15, 4714–4717 CrossRef CAS; (f) L. Zhu, Y. Xiong and C. Li, J. Org. Chem., 2015, 80, 628–633 CrossRef CAS PubMed; (g) J. Zhang and Z. Miao, Org. Biomol. Chem., 2018, 16, 9461–9471 RSC; (h) K. Zhang, L. Cai, S. Hong and O. Kwon, Org. Lett., 2019, 21, 5143–5146 CrossRef CAS; (i) Y. Zhang, Y. Sun, Y. Wei and M. Shi, Adv. Synth. Catal., 2019, 361, 2129–2135 CrossRef CAS; (j) T. P. Reddy, J. Gujral, P. Roy and D. B. Ramachary, Org. Lett., 2020, 22, 9653–9657 CrossRef CAS PubMed.
- S. M. Wales, D. J. Rivinoja, M. G. Gardiner, M. J. Bird, A. G. Meyer, J. H. Ryan and C. J. T. Hyland, Org. Lett., 2019, 21, 4703–4708 CrossRef CAS PubMed and references cited therein.
- There are no synthetic approaches reported to date to access 1-arylidene-cyclopenta[b]indol-2-ones.
- See the ESI for optimisation studies.†.
- 3-Arylidene-cyclopenta[b]-fused heteroarenes have found great relevance in medicinal chemistry. For example, see: (a) A. Ekebergh, A. Borje and J. Mårtensson, Org. Lett., 2012, 14, 6274–6277 CrossRef CAS PubMed; (b) A. Ekebergh, J. Mårtensson and C. L. Ekebergh, ACS Omega, 2020, 5, 33455–33460 CrossRef CAS PubMed.
- The reactivity of 2-nitroindoles, which is unlike 2-nitrobenzofurans or 2-nitrobenzothiophenes, is consistent with literature precedence. For example, see: J.-Q. Zhao, L. Yang, X.-J. Zhou, Y. You, Z.-H. Wang, M.-Q. Zhou, X.-M. Zhang, X.-Y. Xu and W.-C. Yuan, Org. Lett., 2019, 21, 660–664 CrossRef CAS.
- X-ray crystallographic data of CCDC 2214774 (for 7(a)) are provided in the ESI.†.
- Alternatively, the conversion of 2a to L may be thermally-driven. In such a case, the conversion of D to M may have been assisted by adventitious water.
- For other synthetic approaches toward bruceolline E, see: (a) J. A. Jordan, G. W. Gribble and J. C. Badenock, Tetrahedron Lett., 2011, 52, 6772–6774 CrossRef CAS; (b) J. M. Lopchuk, I. L. Green, J. C. Badenock and G. W. Gribble, Org. Lett., 2013, 15, 4485–4487 CrossRef CAS PubMed; (c) E. Li, C. Li, J. Wang, J. Wang, L. Dong, X. Guo, C. Song and J. Chang, Tetrahedron, 2014, 70, 874–879 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterisation data for new compounds. CCDC 2214774 (7a). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ob02180c |
| This journal is © The Royal Society of Chemistry 2023 |