
The aromatic Claisen rearrangement of a 1,2-azaborine†
Hannah M.
Robichaud
a,
Jacob S. A.
Ishibashi‡
 a,
Tomoya
Ozaki
a,
Walid
Lamine
b,
Karinne
Miqueu
*b and
Shih-Yuan
Liu
*ab
a,
Tomoya
Ozaki
a,
Walid
Lamine
b,
Karinne
Miqueu
*b and
Shih-Yuan
Liu
*ab
aDepartment of Chemistry, Boston College, Chestnut Hill, Massachusetts 02467-3860, USA. E-mail: shihyuan.liu@ bc.edu
bUniversité de Pau et des Pays de l'Adour, E2S UPPA/CNRS, Institut des Sciences Analytiques et de Physico-Chimie pour l'Environnement et les Matériaux IPREM UMR 5254, Hélioparc, 2 avenue P. Angot, 64053 Pau cedex 09, France. E-mail: karinne.miqueu@univ-pau.fr
First published on 14th April 2023
Abstract
The first aromatic Claisen rearrangement of a 1,2-azaborine is described along with a quantitative kinetic comparison of the reaction of the azaborine with its direct all-carbon analogue. The azaborine A rearranged in a clean, regioselective fashion and reacted faster than the all-carbon substrate B at all temperatures from 140–180 °C. Activation free energies were extracted from observed first-order rate constants (A: ΔG‡298K = 32.7 kcal mol−1; B: ΔG‡298K = 34.8 kcal mol−1) corresponding to a twenty fold faster rate for A at observed reaction temperatures. DFT calculations show that the rearrangement proceeds via a concerted six-membered transition state and that the electronic structure of the BN and CC rings is mostly responsible for the observed regioselectivity and relative reactivity.
By exchanging a CC unit for an isoelectronic and isostructural BN unit in an organic scaffold, it is possible to diversify electronic structure and chemical space without significantly altering the geometric footprint of a given scaffold. BN/CC isosterism1 has shown potential in biomedical applications; for example, isosteric azaborines and benzenes can bind to biological targets in a similar structural way2 but may convey different function as a result of hydrogen bonding3 or other properties introduced by BN/CC isosterism.4 Uncovering the basic properties and reactivity of monocyclic azaborines should therefore continue inform future applications. Our group as well as others have investigated the energetic,5 magnetic,6 structural,7 and reactivity-related8 consequences of BN/CC isosterism for the monocyclic 1,2-azaborine motif. In 2015 we found that the lower aromatic character of azaborines relative to benzenes facilitated a Diels–Alder reaction.9 Thermal [4 + 2] cycloadditions of benzenes are rare,10 presumably because their high resonance stabilization energy (RSE) creates unfavorable kinetic and thermodynamic conditions. It follows, then, that other pericyclic reactions that break aromaticity in their mechanisms should, in principle, be faster for an azaborine than for a direct all-carbon analogue.
The aromatic Claisen rearrangement is one such pericyclic reaction that breaks aromaticity along its reaction coordinate.11 Since Claisen's first report of the reaction in 1912,12 many studies on its mechanism and scope have been reported, and the aromatic Claisen rearrangement has been executed in a number of complex molecule syntheses.13,14 A kinetic study by White15 in addition to that of Goering and Jacobson16 showed that the reaction likely proceeds through a concerted mechanism. The rearrangement of heteroaryl allyl ethers has been reported,17 but to our knowledge, there is only a single kinetic mechanistic study including heteroatoms in the aromatic moiety.18 In this work, we describe the first aromatic Claisen rearrangement of an 1,2-azaborine-derived allyl ether, and we provide a quantitative kinetics comparison of its rearrangement with the corresponding all-carbon analogue along with a computational mechanistic analysis.
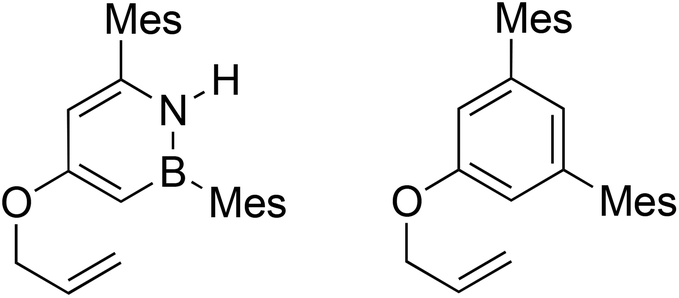
We designed the substrates for our study with the following points in mind: (1) The boron atom on the BN substrate A should be protected from undesired reactivity using a mesityl group. (2) For the purpose of clean kinetic measurements, the all-carbon substrate B should be symmetric to avoid generation of isomers. Our synthetic strategy to 1,2-azaborine A involves the functionalization of the BN-heterocyclic 1,2-azaborine core (Scheme 1).8f To this end, the known borylated 1,2-azaborine 119 undergoes Suzuki–Miyaura coupling to give mesityl-substituted 2. This compound is then borylated once more at the 4-position to yield 3.20 While the C–H borylation reaction to form 1 is quite facile and proceeds at room temperature, the second borylation to form 3 was more difficult and required heating for an extended period of time. Oxidation of the newly installed C–Bpin group with N-methylmorpholine-N-oxide20 yields phenolic compound 4, which is then allylated after treatment with potassium hydride and allyl bromide to furnish the target compound A. We synthesized the direct, all-carbon analogue, O-allyl-(3,5-dimesityl)phenol B using a route involving the cross coupling of 3,5-dibromophenol, followed by the allylation of the phenol (see ESI†).
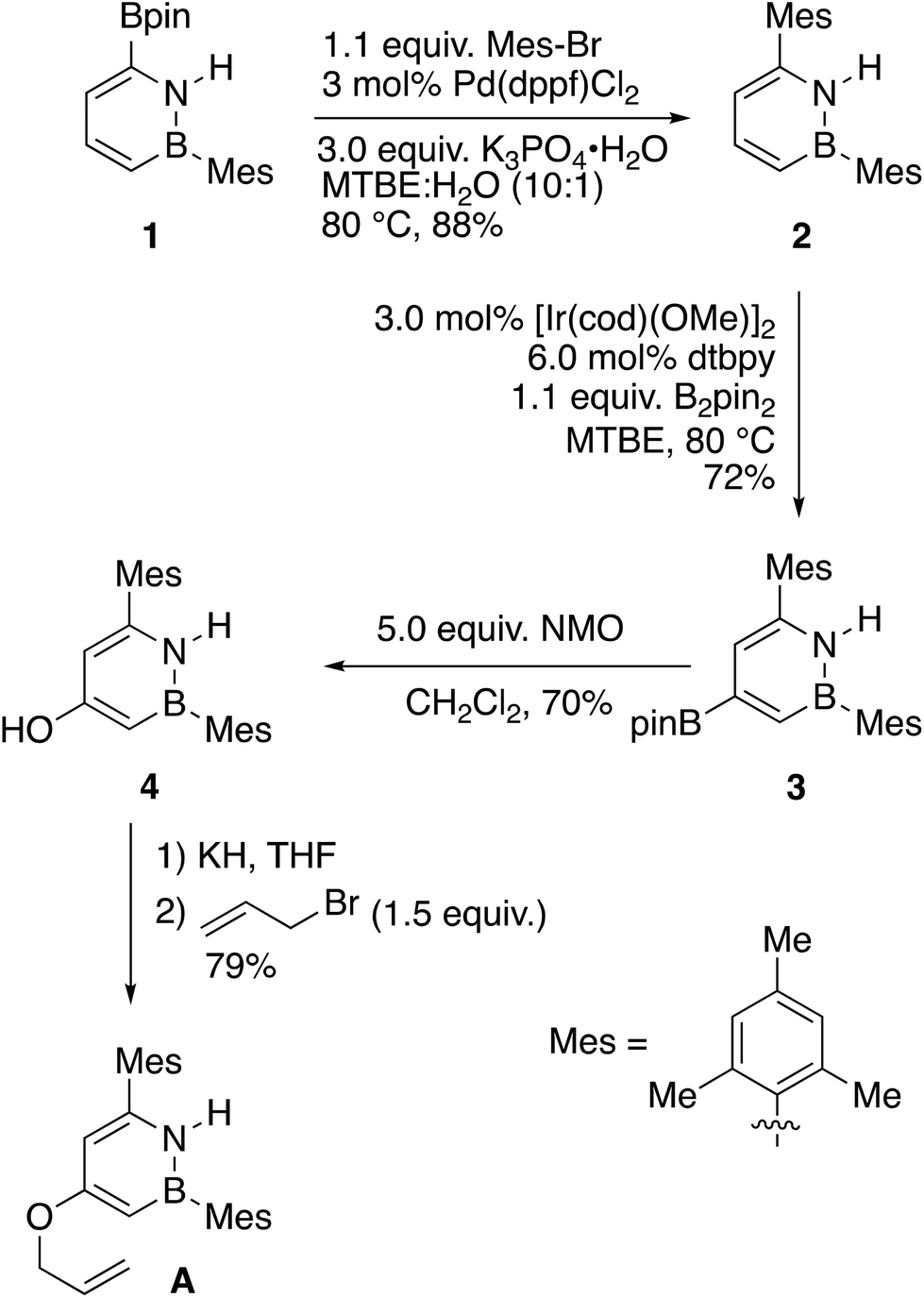 | ||
| Scheme 1 Synthesis of azaborine A. | ||
We observe full conversion of A regioselectively to PDT-A after heating A to 180 °C for 18 hours in ortho-dichlorobenzene-d4 (ODCB-d4) (Scheme 2, see ESI† for HSQC data). While meta-substituted phenyl allyl ethers generally do not undergo the aromatic Claisen rearrangement regioselectively,21 Moody has shown that heteroaromatic systems such as indoles and thiophenes can exhibit regioselectivity in this reaction.22 Bond length alternation caused by heteroatoms in an aromatic ring implies that some bonds in the heteroarene are more olefinic in nature than others. The Claisen rearrangement should proceed through these more olefinic bonds on the arene rather than more σ-like bonds.23 In the case for BN heterocycle A, the observed product may result from the reaction of the dominant resonance contributor more accurately described using the neutral valence bond structure A rather than the zwitterionic form Ares (Scheme 2). Our group has previously shown that the intra-ring C(3)–C(4) and C(5)–C(6) bonds are shorter than the C(4)–C(5) bond,7 consistent with the observed rearrangement regioselectivity for A.
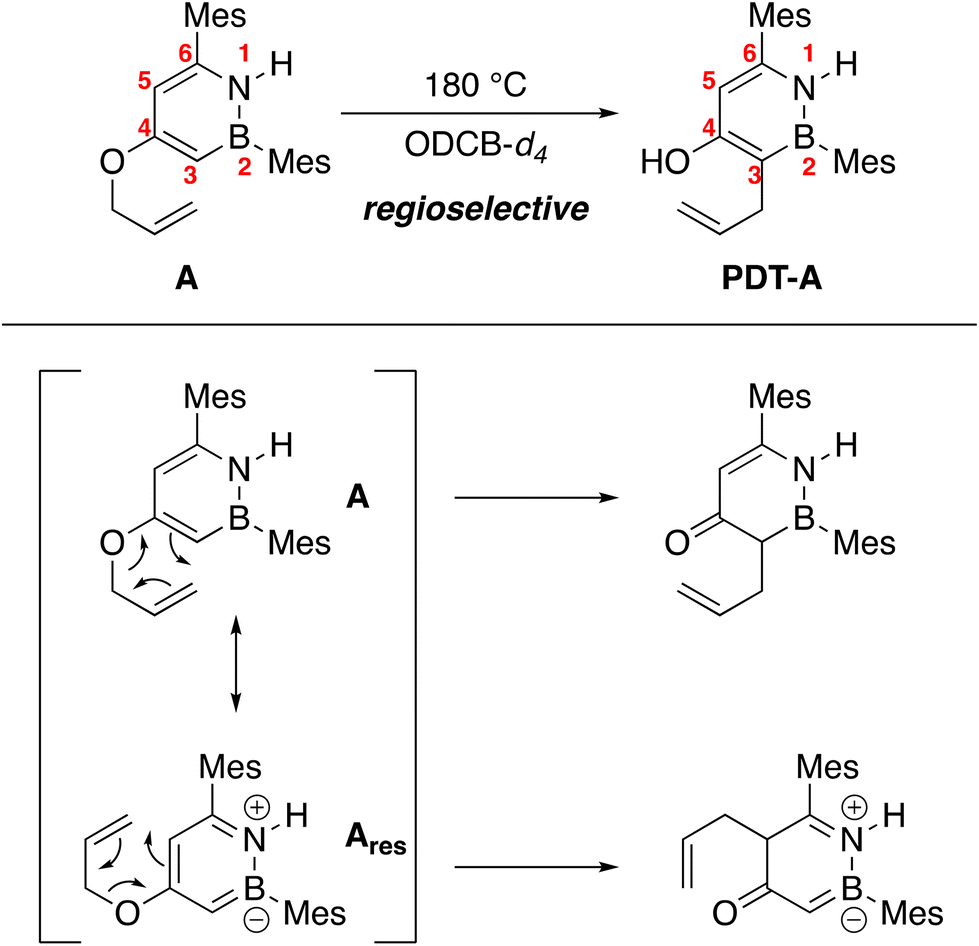 | ||
| Scheme 2 Claisen rearrangement of A is regioselective for the 3-position. | ||
We performed a kinetic comparison of the aromatic Claisen rearrangements of A and B using 1H NMR (see ESI† for details). Experiments were run in ODCB-d4 in the temperature range of 140–180 °C. The first-order rate constants for the aromatic Claisen rearrangements of A and B are reported in Table 1 as the average of five runs at each temperature. The aromatic Claisen rearrangement of A is about twenty-fold faster than the analogous reaction of B at all temperatures investigated. Thus, it appears that BN/CC isosterism accelerates the aromatic Claisen rearrangement, consistent with our original hypothesis.
|
|
||
|---|---|---|
| A | B | |
| T (°C) | k × 10−6 (s−1) | |
| 180 | 118 ± 12 | 6.5 ± 0.8 |
| 170 | 66 ± 5 | 3.6 ± 0.7 |
| 160 | 31 ± 5 | 1.6 ± 0.4 |
| 150 | 12 ± 3 | 0.50 ± 0.04 |
| 140 | 5.7 ± 0.3 | 0.30 ± 0.04 |
| Δ G ‡ 298K (kcal mol−1) | +32.7 ± 0.5 | +34.8 ± 0.6 |
Using an Eyring plot (Fig. 1), we determined that ΔG‡298K (ΔG‡298K = ΔH‡ − TΔS‡, for T = 298 K) for the rearrangement of A (+32.7 ± 0.5 kcal mol−1) is smaller by 2.1 kcal mol−1 than the all-carbon compound B (+34.8 ± 0.6 kcal mol−1). The experimentally determined activation enthalpy ΔH‡ for the carbonaceous compound B (+29 kcal mol−1) is apparently slightly larger than for BN heterocycle A (+28 kcal mol−1). Additionally, the observed activation entropy ΔS‡ for B (−18 e.u.) is apparently more negative than it is for A (−15 e.u.). These activation parameters24 indicate an ordered transition state for both reactions, consistent with a concerted cyclic rearrangement process. The error range of the activation parameters (ΔG‡, ΔH‡, ΔS‡) is reported as the standard deviation from the calculated average of five independent runs for each reaction temperature. As can be seen from Fig. 1, the experimentally determined ΔH‡ and ΔS‡ values for A and B are sufficiently similar so that any comparative interpretations are to be taken with extreme caution. On the other hand, the experimentally determined ΔG‡ values are significantly distinct25 for A and B (consistent with the clear observed slower reaction rates for B than A) to allow for these values to be used as a benchmark for a detailed computational mechanistic analysis.
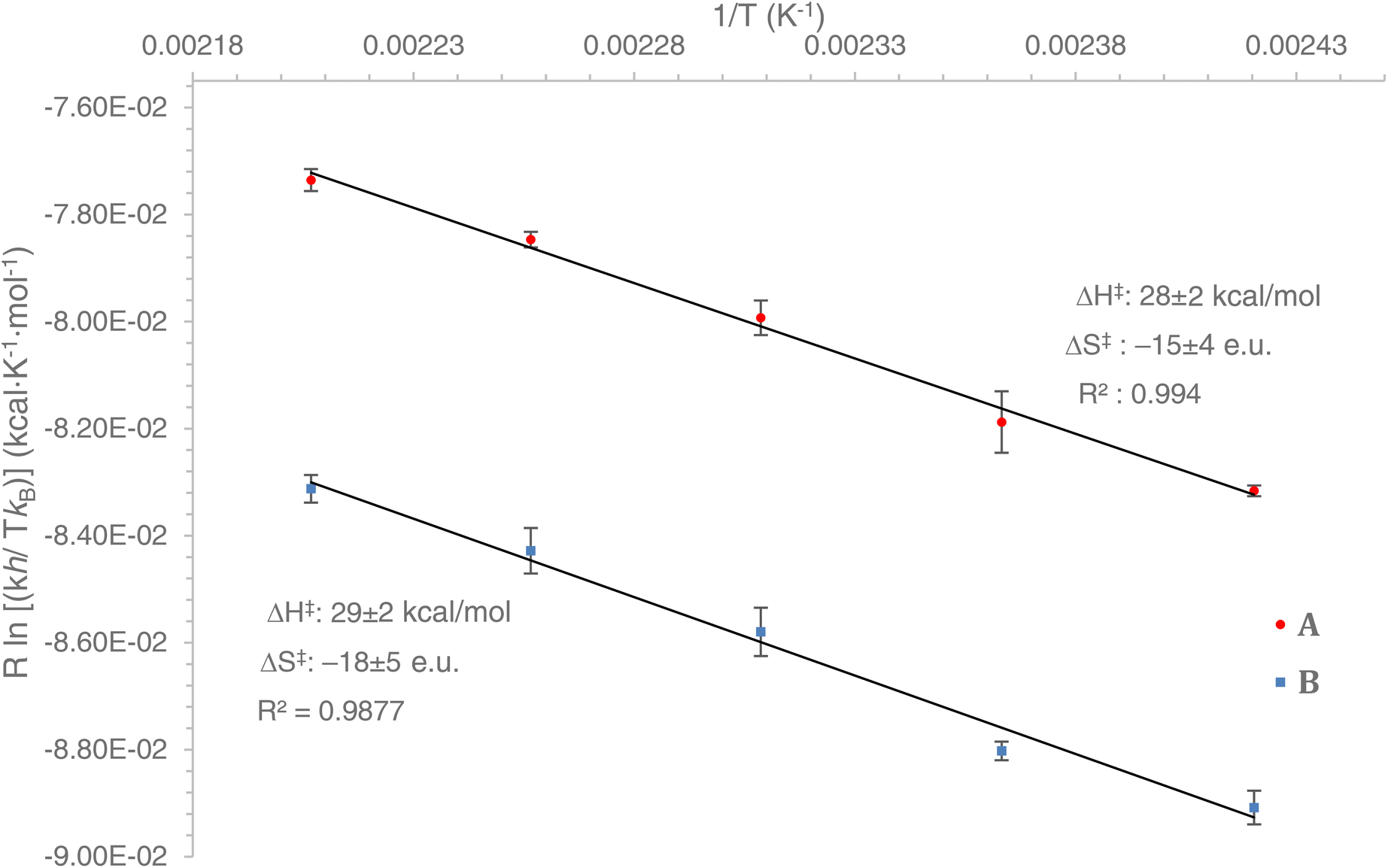 | ||
| Fig. 1 Eyring plots for A and B. Error bars are the standard deviation from the calculated average of five separate measurements. | ||
We carried out DFT calculations26 at SMD(o-DCB)-PBE0-D3(BJ)/6-31G** level of theory27 in order to obtain better insights into the observed regiochemistry (C(3) vs. C(5)) of the aromatic Claisen rearrangment of the BN heterocycle A and to analyze the impact of BN/CC isosterism on the rate of the rearrangement (Avs.B). As can be seen from Fig. 2, two rotamers (A and A′) were found on the potential energy surface (PES), with the double bond of the allyl group directed toward C3 or C5 atoms of the BN-heterocyclic ring, respectively. The energy difference between A and A′ is 0.8 kcal mol−1. In agreement with the experimental observations, DFT calculations reveal that the rearrangement to the C(3)-position from A is predicted to be 11.4 kcal mol−1 more favored than the rearrangement from A to the C(5)-position (ΔG‡DFTAC5 = 42.6 kcal mol−1). The computed activation barrier for the rearrangement from A to PDT-A (ΔG‡DFTA = 31.2 kcal mol−1) matches well with the value determined from the Eyring analysis (ΔG‡expA = +32.7 ± 0.5 kcal mol−1). The reaction occurs in a quasi-synchronous concerted pathway via a 6-membered transition state. Analysis of the geometrical parameters reveals that the breaking C–O bond is elongated about 35.4% for TSA-C3 and 43.4% for TSA-C5, compared to the C–O distance of the initial reactant A. On the other hand, the forming C–C bond is elongated by 39.9% for TSA-C3 and 32.1% for TSA-C5, compared to the corresponding dearomatized keto-intermediates IntA-C3 and IntA-C5, respectively. These bond distance variations indicate that TSA-C3 is an earlier transition state than TSA-C5. The regioselectivity is governed both by orbital considerations and the thermodynamic stabilities of the dearomatized intermediates IntA-C5vs.IntA-C3. The two orbitals involved in this rearrangement are the HOMO (Fig. 2), which is mainly localized on the π-system of the BN-ring, and the LUMO+5, which is the 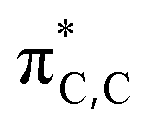 orbital of the O-allyl moiety. The HOMO of A shows a larger coefficient at C(3) position (∼21%) than the C(5) (∼12%) position, consistent with the observed regioselectivity of the rearrangement. Additionally, the formation of Int-C5 from A is endergonic by 18.5 kcal mol−1 in stark contrast to the formation of Int-C3 which is exergonic by 2.8 kcal mol−1. The difference in thermodynamic stabilities of Int-C3 and Int-C5 may be reflected in their corresponding transition states of formation according to Hammond's postulate,28 thus resulting in the observed C(3)-selective rearrangement.
orbital of the O-allyl moiety. The HOMO of A shows a larger coefficient at C(3) position (∼21%) than the C(5) (∼12%) position, consistent with the observed regioselectivity of the rearrangement. Additionally, the formation of Int-C5 from A is endergonic by 18.5 kcal mol−1 in stark contrast to the formation of Int-C3 which is exergonic by 2.8 kcal mol−1. The difference in thermodynamic stabilities of Int-C3 and Int-C5 may be reflected in their corresponding transition states of formation according to Hammond's postulate,28 thus resulting in the observed C(3)-selective rearrangement.
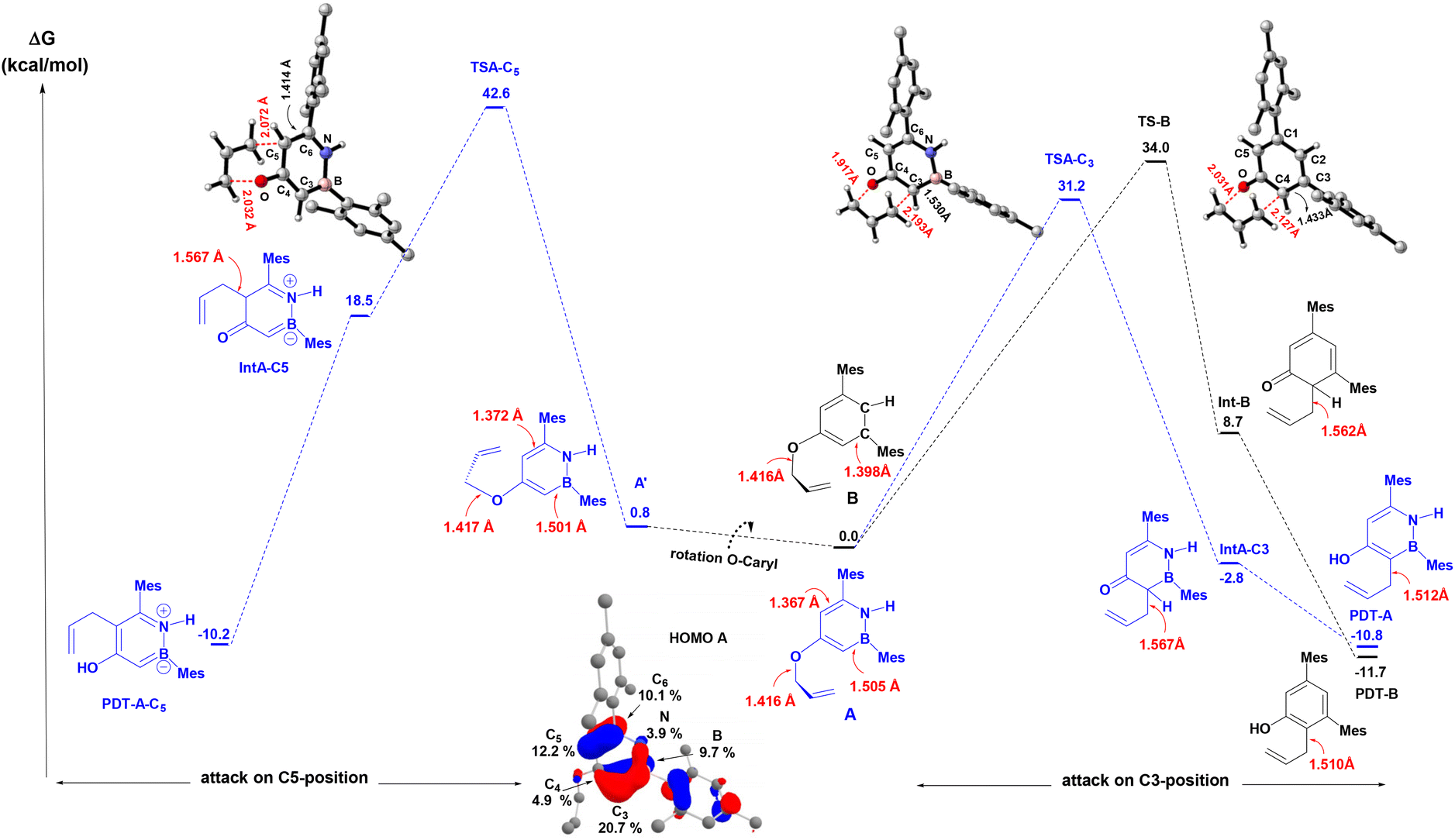 | ||
| Fig. 2 Calculated relative energy profiles for the aromatic Claisen rearrangement for A (C(3) and C(5)-positions, blue) and B (black) computed at SMD(o-DCB)-PBE0-D3(BJ)/6-31G** level of theory. Main distances in Å. Plot of the HOMO of A and main atomic orbital compositions in percents (%). | ||
We also computed the energy profile associated with the aromatic Claisen rearrangement for the carbonaceous compound B (Fig. 2, black trace). Consistent with the experimental Eyring data, the activation barrier for the rearrangement of B (ΔG‡DFTB = 34.0 kcal mol−1) was computed to be 2.8 kcal mol−1 higher in energy than that for the BN analogue A (ΔG‡DFTA = 31.2 kcal mol−1). Similar to the unfavorable rearrangement to PDT-A-C5, the formation of the dearomatized intermediate Int-B from B is endergonic by 8.7 kcal mol−1 (vs. an exergonic reaction for A), which is consistent with a faster aromatic Claisen rearrangement for the BN derivative A. The computed geometrical features of the transition states indicate a relatively early transition state for the rearrangement of A (Δ(C–Obreak) = 35.4% and Δ(C–Cform) = 43.4% compared to A and IntA-C3, respectively) and a relatively late transition state for B (Δ(C–Obreak) = 43.4% and Δ(C–Cform) = 36.2% compared to B and Int-B, respectively).
To isolate the electronic influences and remove steric bias on the reactivity of A and B,29 we calculated the energy profiles for the unsubstituted compounds A–H and B–H (ESI, Fig. S1,† mesityl groups are replaced with H), and we found similar reactivity trends as with the mesityl substituted systems, i.e., the ΔG‡ for A–H (31.6 kcal mol−1) is smaller than the ΔG‡ for B–H (34.9 kcal mol−1). We analyzed the key molecular orbitals at play in the reactivity (i.e., πBN/CC-ring and 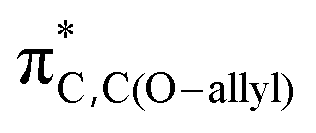 , see ESI Fig. S2†) for A, A–H, B, B–H, and we found a good correlation (ESI, Fig. S3†) between the energy of the πBN/CC-ring orbital and the computed activation barriers. The higher the energy of the πBN/CC-ring orbital, the smaller is the computed activation free energy ΔG‡ of the rearrangement, indicating that the reactivity difference between A and B is to a large extent governed by the electronic effects.
, see ESI Fig. S2†) for A, A–H, B, B–H, and we found a good correlation (ESI, Fig. S3†) between the energy of the πBN/CC-ring orbital and the computed activation barriers. The higher the energy of the πBN/CC-ring orbital, the smaller is the computed activation free energy ΔG‡ of the rearrangement, indicating that the reactivity difference between A and B is to a large extent governed by the electronic effects.
Finally, we explored the possibility of a radical pathway for the Claisen rearrangement by employing a radical scavenger, butylated hydroxytoluene (BHT), to trap potential radial intermediates. A stoichiometric amount BHT was added to the substrates A and B, and after heating the mixtures to 170 °C, we observe no BHT adduct formation.30 Furthermore, the addition of the scavenger did not affect the rate of the reaction for either analogue A or B (see ESI† for details). These observations are consistent with the proposed concerted six-membered cyclic rearrangement mechanism.
In summary, we described the first example of an aromatic Claisen rearrangement of an 1,2-azaborine. We show that the rearrangement of a C-4 substituted 1,2-azaborine-derived O-allyl ether A occurs regioselectively to form the C(3)-allyl-C(4)-hydroxy-1,2-azaborine product PDT-A. A kinetic analysis of the Claisen rearrangement of A in direct comparison with its carbonaceous analogue B reveals that the rearrangement for the BN heterocyclic motif is more facile across the examined temperature ranges between 140 °C and 180 °C, correlating with the observed aromaticity trends. Reaction activation free energy for A (ΔG‡298K = 32.7 kcal mol−1) and B (ΔG‡298K = 34.8 kcal mol−1) derived from first-order rate constants via Eyring analysis are consistent with those predicted by DFT calculations. Computational mechanistic analyses show that the rearrangement proceeds via a concerted six-membered transition state and that the electronic structure of the BN and CC rings is mostly responsible for the observed regioselectivity and relative reactivity. In addition to fundamentally examining a classic reaction of arenes in the context of 1,2-azaborines, this work also provided a new synthetic tool box for the preparation of C(3),C(4)-disubstituted 1,2-azaborine derivatives.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health (NIGMS) under Award Number R01GM136920, the Excellence Initiative of Université de Pau et des Pays de l'Adour I-Site E2S UPPA, and by Boston College start-up funds. We also acknowledge the NIH-S10 (award: 1S10OD026910-01A1) and the NSF-MRI (award: CHE-2117246) for the support of Boston College's NMR facilities. The “Direction du Numérique” of the Université de Pau et des Pays de l'Adour (UPPA) and Mésocentre de Calcul Intensif Aquitain (MCIA) are acknowledged for the support of computational facilities. This work was also granted access to the HPC resources of IDRIS under the allocation 2022-[AD010800045R1] made by GENCI. We thank Drs Fredrik Haeffner and Andrew Baggett for helpful discussions and Lifeng Chen for the preparation of synthetic precursors. T. O. thanks the Takenaka Scholarship Foundation for support.References
- (a) M. J. Bosdet and W. E. Piers, Can. J. Chem., 2009, 87, 8–29 CrossRef; (b) Z. Liu and T. Marder, Angew. Chem., Int. Ed., 2008, 47, 242–244 CrossRef CAS PubMed; (c) P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS PubMed; (d) Z. X. Giustra and S. Y. Liu, J. Am. Chem. Soc., 2018, 140, 1184–1194 CrossRef CAS PubMed.
- L. Liu, A. J. V. Marwitz, B. W. Matthews and S.-Y. Liu, Angew. Chem., Int. Ed., 2009, 48, 6817–6819 CrossRef CAS PubMed.
- (a) H. Lee, M. Fischer, B. K. Shoichet and S.-Y. Liu, J. Am. Chem. Soc., 2016, 138, 12021–12024 CrossRef CAS PubMed; (b) Y. Liu and S.-Y. Liu, Org. Biomol. Chem., 2019, 17, 7002–7006 RSC.
- (a) D. H. Knack, J. L. Marshall, G. P. Harlow, A. Dudzik, M. Szaleniec, S.-Y. Liu and J. Heider, Angew. Chem., Int. Ed., 2013, 52, 2599–2601 CrossRef CAS PubMed; (b) P. Zhao, D. O. Nettleton, R. G. Karki, F. J. Zecri and S.-Y. Liu, ChemMedChem, 2017, 12, 358–361 CrossRef CAS PubMed.
- (a) P. G. Campbell, E. R. Abbey, D. Neiner, D. J. Grant, D. A. Dixon and S.-Y. Liu, J. Am. Chem. Soc., 2010, 132, 18048–18050 CrossRef CAS PubMed; (b) A. Chrostowska, S. Xu, A. N. Lamm, A. Mazière, C. D. Weber, A. Dargelos, P. Baylère, A. Graciaa and S.-Y. Liu, J. Am. Chem. Soc., 2012, 134, 10279–10285 CrossRef CAS PubMed.
- A. J. V. Marwitz, M. H. Matus, L. N. Zakharov, D. A. Dixon and S.-Y. Liu, Angew. Chem., Int. Ed., 2009, 48, 973–977 CrossRef CAS PubMed.
- E. R. Abbey, L. N. Zakharov and S.-Y. Liu, J. Am. Chem. Soc., 2008, 130, 7250–7252 CrossRef CAS PubMed.
- (a) A. J. Ashe, X. Fang, X. Fang and J. W. Kampf, Organometallics, 2001, 20, 5413–5418 CrossRef CAS; (b) J. Pan, J. W. Kampf and A. J. Ashe, Org. Lett., 2007, 9, 679–681 CrossRef CAS PubMed; (c) A. N. Lamm, E. B. Garner, D. A. Dixon and S.-Y. Liu, Angew. Chem., Int. Ed., 2011, 50, 8157–8160 CrossRef CAS PubMed; (d) A. N. Lamm and S.-Y. Liu, Mol. BioSyst., 2009, 5, 1303–1305 RSC; (e) A. N. Brown, L. N. Zakharov, T. Mikulas, D. A. Dixon and S.-Y. Liu, Org. Lett., 2014, 16, 3340–3343 CrossRef CAS PubMed; (f) C. R. McConnell and S.-Y. Liu, Chem. Soc. Rev., 2019, 48, 3436–3453 RSC.
- R. J. Burford, B. Li, M. Vasiliu, D. A. Dixon and S.-Y. Liu, Angew. Chem., Int. Ed., 2015, 54, 7823–7827 CrossRef CAS PubMed.
- For a leading reference, see: Y. Inagaki, M. Nakamoto and A. Sekiguchi, Nat. Commun., 2014, 5, 3018 CrossRef PubMed.
- For an overview of the Claisen rearrangement, see: A. M. Martín Castro, Chem. Rev., 2004, 104, 2939–3002 CrossRef PubMed.
- L. Claisen, Ber. Dtsch. Chem. Ges., 1912, 45, 3157–3166 CrossRef CAS.
- For an overview, see: (a) K. C. Majumdar and R. K. Nandi, Tetrahedron, 2013, 69, 6921–6957 CrossRef CAS; (b) The Claisen Rearrangement: Methods and Applications, ed. M. Hiersemann and U. Nubbemeyer, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2007.
- For select examples, see: (a) K. C. Nicolaou and J. Li, Angew. Chem., Int. Ed., 2001, 40, 4264–4268 CrossRef CAS; (b) E. J. Tisdale, I. Slobodov and E. A. Theodorakis, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12030–12035 CrossRef CAS PubMed; (c) T. R. R. Pettus, X.-T. Chen and S. J. Danishefsky, J. Am. Chem. Soc., 1998, 120, 12684–12685 CrossRef CAS; (d) X. Zhang, X. Li, H. Sun, X. Wang, L. Zhao, Y. Gao, X. Liu, S. Zhang, Y. Wang, Y. Yang, S. Zeng, Q. Guo and Q. You, J. Med. Chem., 2013, 56, 276–292 CrossRef CAS PubMed.
- W. N. White, D. Gwynn, R. Schlitt, C. Girard and W. Fife, J. Am. Chem. Soc., 1958, 80, 3271–3277 CrossRef CAS.
- H. L. Goering and R. R. Jacobson, J. Am. Chem. Soc., 1958, 80, 3277–3285 CrossRef CAS.
- For an overview, see: C. J. Moody, Adv. Heterocycl. Chem., 1987, 42, 203–244 CrossRef CAS.
- M. L. S. Cristiano and R. A. W. Johnstone, J. Chem. Soc., Perkin Trans. 2, 1997, 489–494 CAS.
- A. W. Baggett, M. Vasiliu, B. Li, D. A. Dixon and S.-Y. Liu, J. Am. Chem. Soc., 2015, 137, 5536–5541 CrossRef CAS PubMed.
- C. R. McConnell, F. Haeffner, A. W. Baggett and S.-Y. Liu, J. Am. Chem. Soc., 2019, 141, 9072–9078 CrossRef CAS PubMed.
- F. C. Gozzo, S. A. Fernandes, D. C. Rodrigues, M. N. Eberlin and A. J. Marsaioli, J. Org. Chem., 2003, 68, 5493–5499 CrossRef CAS PubMed.
- (a) C. J. Moody, J. Chem. Soc., Chem. Commun., 1983, 1129–1131 RSC; (b) C. J. Moody, J. Chem. Soc., Perkin Trans. 1, 1984, 1333–1337 RSC.
- Kruse and Cha hypothesized that bond alternation in 2-naphthyl allyl ethers may lead to the observed 1-selective aromatic Claisen rearrangement since the C(1)–C(2) bond in naphthalenes is generally shorter than the C(2)–C(3) bond, see: L. I. Kruse and J. K. Cha, J. Chem. Soc., Chem. Commun., 1982, 1333–1336 RSC.
- The activation parameters for the aromatic Claisen rearrangement of substituted tetrazoles (ΔH‡ = +24.8 to +18.9 kcal mol−1; ΔS‡ = −8.1 to −38.8 e.u.) are in similar range as observed for A and B, see ref. 18.
- For an overview of enthalpy-entropy compensation, see: L. Liu and Q.-X. Guo, Chem. Rev., 2001, 101, 673–695 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- (a) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6169 CrossRef CAS; (b) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154119 CrossRef PubMed; (c) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed; (d) A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- R. S. Macomber, J. Chem. Educ., 1978, 55, 449–449 CrossRef CAS.
- The (Mes)B–C(3) distance in BN-heterocycle A is longer than the corresponding C–C distance in B due to the larger covalent radius of boron. Thus a possible argument could be made for the faster rate for A based on sterics that is eliminated by comparing the unsubstituted compounds A–H and B–H.
- For leading references of BHT as a radical scavanger, see: (a) X.-Y. Cheng, Y.-F. Zhang, J.-H. Wang, Q.-S. Gu, Z.-L. Li and X.-Y. Liu, J. Am. Chem. Soc., 2022, 144, 18081–18089 CrossRef CAS PubMed; (b) S. Fujisawa, Y. Kadoma and I. Yokoe, Chem. Phys. Lipids, 2004, 130, 189–195 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterization data, computational information (PDF). Optimized cartesian coordinates (.XYZ). See DOI: https://doi.org/10.1039/d2ob02186b |
| ‡ Present address: Fluent BioSciences, Watertown, Massachusetts, United States. E-mail: E-mail: jacob.ishibashi@gmail.com |
| This journal is © The Royal Society of Chemistry 2023 |