
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of 1,2-diaminotruxinic δ-cyclobutanes by BF3-controlled [2 + 2]-photocycloaddition of 5(4H)-oxazolones and stereoselective expansion of δ-cyclobutanes to give highly substituted pyrrolidine-2,5-dicarboxylates†
Sonia
Sierra
 a,
Rosa
López
b,
Enrique
Gómez-Bengoa
b,
Larry R.
Falvello
c and
Esteban P.
Urriolabeitia
*a
a,
Rosa
López
b,
Enrique
Gómez-Bengoa
b,
Larry R.
Falvello
c and
Esteban P.
Urriolabeitia
*a
aInstituto de Síntesis Química y Catálisis Homogénea, ISQCH (CSIC - Universidad de Zaragoza), 50009 Zaragoza, Spain. E-mail: esteban@unizar.es
bDepartamento de Química Orgánica I, Universidad del País Vasco, UPV-EHU, CP-20080 Donostia-San Sebastián, Spain
cInstituto de Nanociencia y Materiales de Aragón, INMA (CSIC - Universidad de Zaragoza), 50009 Zaragoza, Spain
First published on 24th March 2023
Abstract
The irradiation of (Z)-4-arylidene-5(4H)-oxazolones 1a–1u with blue light (465 nm) in the presence of the photosensitizer [Ru(bpy)3](BF4)2 (2.5 mol%) and the Lewis acid BF3·OEt2 (2 equiv.) in deoxygenated methanol at room temperature affords the corresponding 1,2-diaminotruxinic cyclobutane bis-amino esters 2a–2u stereoselectively as the δ-isomer. Characterization of cyclobutanes 2 shows that the photocycloaddition takes place by the coupling of two Z-oxazolones in a head-to-head 1,2-anti way. This change in the orientation of the coupling is promoted by O- or/and N-bonding of the BF3 additive. The δ-cyclobutanes 2 undergo a ring expansion when heated in methanol in the presence of NaOMe (1/1 molar ratio) to give densely substituted pyrrolidine-2,5-dicarboxylates 3 in a regio- and stereoselective way. The mechanism of the cyclobutane-to-pyrrolidine ring expansion has been elucidated using DFT methods.
Introduction
Amino acids are the basic components of life. The nature of their basic skeleton, the presence of alkyl or aryl lateral groups, the types of intra- and intermolecular contacts they can establish, and the type and number of substituents they have determine their role in the proteins where they are incorporated and, finally, the role of the proteins themselves.1 Among them, amino acids with the cyclobutane skeleton are an important family of compounds due to their remarkable applications. For instance, 18F-fluciclovine (1-amino-3-18F-fluorocyclobutane-1-carboxylic acid), shown in Fig. 1a, is extensively use as a PET tracer in prostate and breast cancer imaging.2 Despite this interest, the number of synthetic methods to access these substrates is scarce.3 This situation is even more limited if cyclobutane-bis(amino acids) are considered, and the only known method to access 1,3-diaminotruxillic (Fig. 1b) or 1,2-diaminotruxinic derivatives (Fig. 1c) is the [2 + 2]-photocycloaddition of oxazolones.4–6 This is surprising, taking into account the high pharmacological relevance of 1,3-diaminotruxillic and 1,2-diaminotruxinic derivatives.7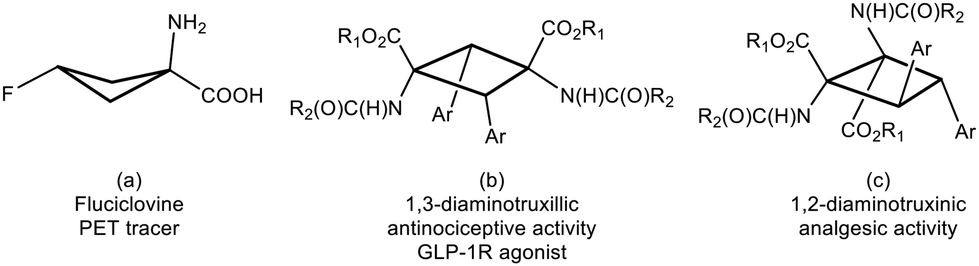 | ||
| Fig. 1 Structures and activities of some relevant cyclobutane-amino acids and bis(amino acids). | ||
Our group4,5a,b,6a and other groups5c,6b have made some contributions in this field. We have achieved the fully stereoselective synthesis of the esters of 1,3-diaminotruxillic acid as single isomers (epsilon isomer, Fig. 1b) by irradiation with blue light (465 nm) of orthopalladated complexes of (Z)-4-arylidene-5(4H)-oxazolones as templates.4 We have also reported the highly efficient synthesis of 1,3-diaminotruxillic derivatives (four different isomers) in quantitative yields by the direct photoirradiation of simple 4-arylidene-5(4H)-oxazolones with blue light.5a,b In this respect, the group of Wang described a much less efficient reaction (10% yield) using high-power UV irradiation.5c In contrast to this relative success, the synthesis of 1,2-diaminotruxinic derivatives is almost unexplored, and there are only two published contributions up to now.6 We have recently achieved the complete regio- and stereoselective synthesis of the esters of 1,2-diaminotruxinic acid as a single isomer (mu-isomer, Fig. 2, past work) by the irradiation of 4-arylidene-5(4H)-oxazolones with blue light in the presence of the photosensitizer [Ru(bpy)3](BF4)2 (bpy = 2,2′-bipyridine).6a The exact role of the photosensitizer has been determined by laser flash photolysis, and it is the generation of the triplet excited state of the oxazolones by energy transfer from the ruthenium complex. The change of the multiplicity of the excited state from singlet to triplet changes the orientation of the reaction (head-to-tail to head-to-head) and provides full selectivity.6a Along the same lines, Amarante and coworkers reported in 2018 the synthesis of a different isomer (zeta-isomer) of the esters of 1,2-diaminotruxinic acid using eosin-Y as the photocatalyst (Fig. 2, past work).6b However, these are only precursors of the 1,2-diaminotruxinic derivatives, because one of the oxazolone rings does not undergo the necessary ring opening reaction. Despite these advancements, the synthesis of cyclobutanes as a single isomer is still a challenge, and controlled access to other isomers of 1,2-diaminotruxinic cyclobutanes is not available yet.
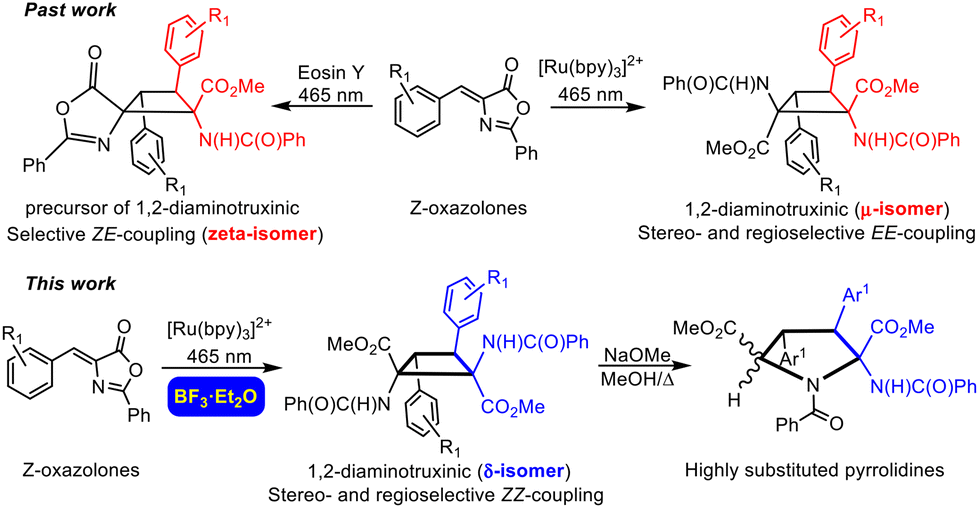 | ||
| Fig. 2 Context of this research with respect to previous contributions. | ||
In this contribution we have described the regio- and stereoselective synthesis of 1,2-diaminotruxinic derivatives as the delta (δ) isomer by the [2 + 2]-cycloaddition of (Z)-4-arylidene-5(4H)-oxazolones photocatalysed by the triplet sensitizer [Ru(bpy)3](BF4)2 in the presence of BF3 as a Lewis acid. It is well-known that the presence of a Lewis acid can change the orientation and selectivity of a photochemical process.8 We show here how the presence of BF3 maintains the global orientation of the process, which takes place in a head-to-head way so 1,2-diaminotruxinic species are obtained, but changes the relative orientation of the substituents affording selectively a new isomer (Fig. 2, this work). Moreover, we also show here how further treatment of the stable 1,2-diaminotruxinic delta-cyclobutanes with a base (NaOMe) and gentle warming promotes an unexpected expansion of the cyclobutane ring giving densely substituted pyrrolidine-2,5-dicarboxylic derivatives, which can be considered as proline-like compounds (Fig. 2, this work). This new method for the synthesis of pyrrolidines is unprecedented. The importance of pyrrolidines is undeniable, not only as synthetic targets, but also due to their applications.9 As far as we know, the expansion of aminocyclobutanes to give pyrrolidines has not been reported until now.10
Results and discussion
Synthesis and characterization of the methyl esters of delta-1,2-diaminotruxinic acids 2 in the presence of BF3
The starting (Z)-4-(het)arylidene-2-phenyl-5(4H)-oxazolones 1a–1u (Fig. 3) have been prepared using the known Erlenmeyer–Plöchl method.11 Substituents of the 4-arylidene ring have been selected to cover the widest scope of electronic and steric requirements. Therefore, electron-releasing (OMe and Me) or electron-withdrawing (CN, F, Cl, Br, and CF3) groups in the ortho-, meta- or para-positions of the 4-arylidene ring, as well as one example of heterocycles (thienyl-1u), are present in the starting materials.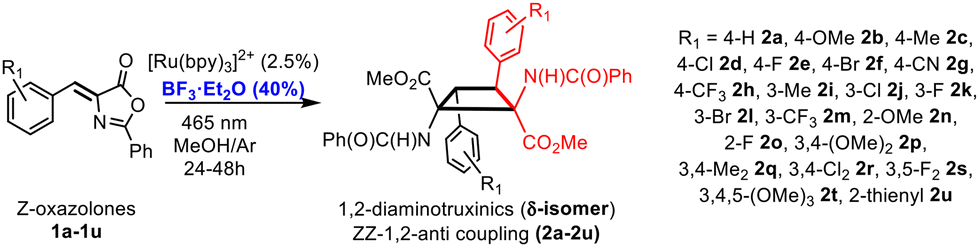 | ||
| Fig. 3 Synthesis of delta isomers of cyclobutanes 2a–2u (1,2-diaminotruxinic esters) by the [2 + 2]-photocycloaddition of (Z)-oxazolones 1a–1u in the presence of BF3. | ||
The oxazolone 1a was irradiated under the experimental conditions previously reported by us (465 nm, [Ru(bpy)3](BF4)2 5%, deoxygenated CH2Cl2, Ar, 48 h),6a but now in the presence of different amounts of the Lewis acid BF3·OEt2. In all attempted cases the precipitation of variable amounts of a white solid was observed, which could not be characterized due to its complete insolubility in the usual organic solvents. Therefore, the reaction needs to be optimized. A short screening of the conditions showed that the diester of the 1,2-diaminotruxinic diacid cyclobutane 2a was formed when using other solvents, and that the best results and the cleanest 2a were obtained under Ar in dry and deoxygenated methanol (Fig. 3). The optimization of the amount of BF3·OEt2 showed that a substoichiometric amount (40 mol%) was enough to achieve the maximum yield of 2a. The amount of the photosensitizer [Ru(bpy)3](BF4)2 was re-optimized to 2.5 mol% and the optimal reaction time was set to 24 h by monitoring the reaction by 1H NMR. The irradiation wavelength (465 nm) was kept the same because it was the excitation wavelength of the ruthenium photocatalyst. No other photocatalysts were used (for instance, Ir(III) species) as we previously determined that this Ru(II) photosensitizer gave the best results with oxazolones.6a The use of other Lewis acids was also attempted (AlCl3, TiCl4, and BBr3) but these reactions were much less selective and a higher variety of byproducts were detected, so all reactions were performed with BF3·OEt2.
Complete conversion of 1a was observed under the optimized reaction conditions, affording cyclobutane 2a in a stereo- and regioselective way. Despite the selectivity in the formation of 2a other byproducts were detected in the crude in amounts of up to 20% (1H NMR), with the corresponding dehydrophenylalanine being the main one. Washing with water efficiently removed BF3, but not dehydrophenylalanine (see the ESI†); therefore, chromatographic purification was mandatory. For this reason, the yield of pure isolated 2a was only 34% due to the partial loss of the product during column chromatography. As is evident from Fig. 3, the reaction of the (Z)-oxazolone 1a to give the diester of 1,2-diaminotruxinic acid 2a has to involve two different steps. We propose that the [2 + 2]-photocycloaddition takes place first to give the cyclobutane, which is followed then by the ring-opening reaction of the oxazolone by methanolysis (vide infra).12 Once the optimal conditions were determined, the scope of the reaction was examined. The reaction works with both electron-donating and electron-withdrawing substituents (Fig. 4), and although full conversions were determined by 1H NMR of the crude after 24 h of irradiation, the isolated yields of analytically pure products were only moderate to low (20–50%) due to mandatory chromatographic purification.
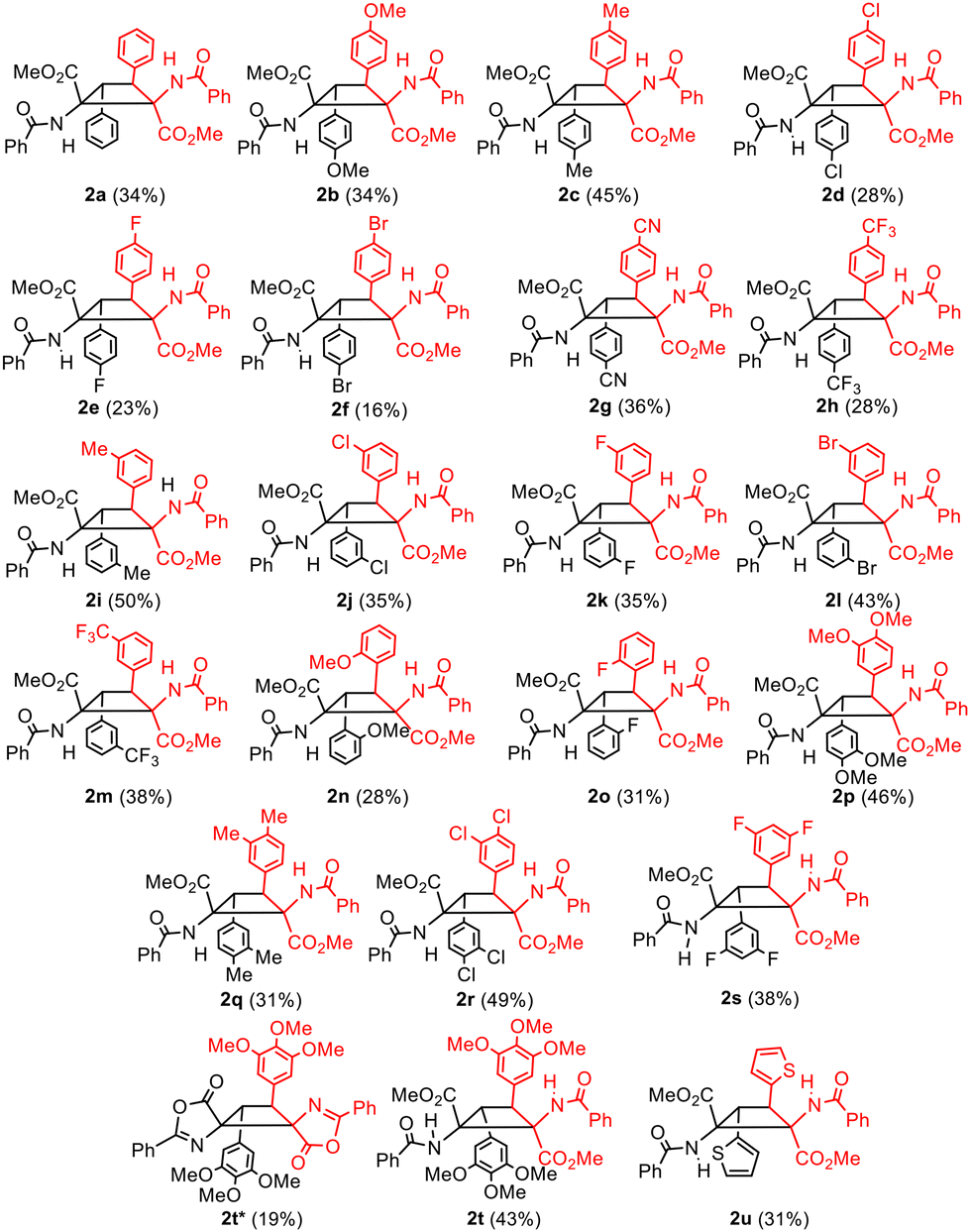 | ||
| Fig. 4 Scope of the formation of cyclobutanes 2a–2u. | ||
As is clear from Fig. 4, the diesters of 1,2-diaminotruxinic derivatives 2 were obtained directly as unique products in all cases except for 2t. In this case, a mixture of dispyrocyclobutane 2t* and cyclobutane 2t was obtained, and they were separated by column chromatography. This gives proof that the reaction takes place sequentially, first the [2 + 2]-photocycloaddition and then the ring-opening reaction.
While it seems that the extent of the [2 + 2]-photocycloaddition does not depend on the electronic characteristics of the substituents at the 4-arylidene ring, the steric hindrance – mainly determined by the ortho-, meta- or para-position of the substituent in the aromatic ring – appears to be much more critical. In fact, para-substituents at the 3- or 4-aryl rings of the cyclobutanes are well tolerated (2b–2h in Fig. 3 and 4), but those at the meta-position showed a more restricted scope (2i–2m). When the substituents were at the ortho position, only the cyclobutanes containing OMe (2n) or F (2o) could be obtained. As expected, two or more substituents were allowed in such 3- and 4-aryl rings, provided that they are at the meta/para positions, regardless of being electron-releasing (OMe 2p, 2t; Me 2q) or electron-withdrawing (Cl 2r, F 2s) in nature. The reaction also takes place when heterocycles are at 3- and 4-cyclobutane positions (2u).
The characterization of cyclobutanes 2a–2u has been carried out by spectroscopic and crystallographic methods. The mass spectra (ESI+) of 2a–2u show the presence of peaks with m/z values and isotopic distributions in agreement with the formation of the cyclobutane. In addition, the 1H and 13C NMR spectra of 2a–2u show the presence of peaks reflecting the formation of a cyclobutane of high symmetry as a single isomer. This allows us to discard any isomer coming from the coupling of a Z-oxazolone with an E-oxazolone (4 isomers).6 The comparison of the chemical shifts of cyclobutanes 2a–2u with those reported in previous studies also allows us to discard the formation of ε-, α- and μ-isomers, already characterized.5a,6a Therefore, a new isomer was formed in this reaction, and there are several structures (peri, β, δ, and ω)12 which can match with the NMR data. The determination of the crystal structures of the cyclobutanes 2n, 2o and 2s allowed us to fully characterize the new species, and their structures are shown in Fig. 5, 6 and 7, respectively.
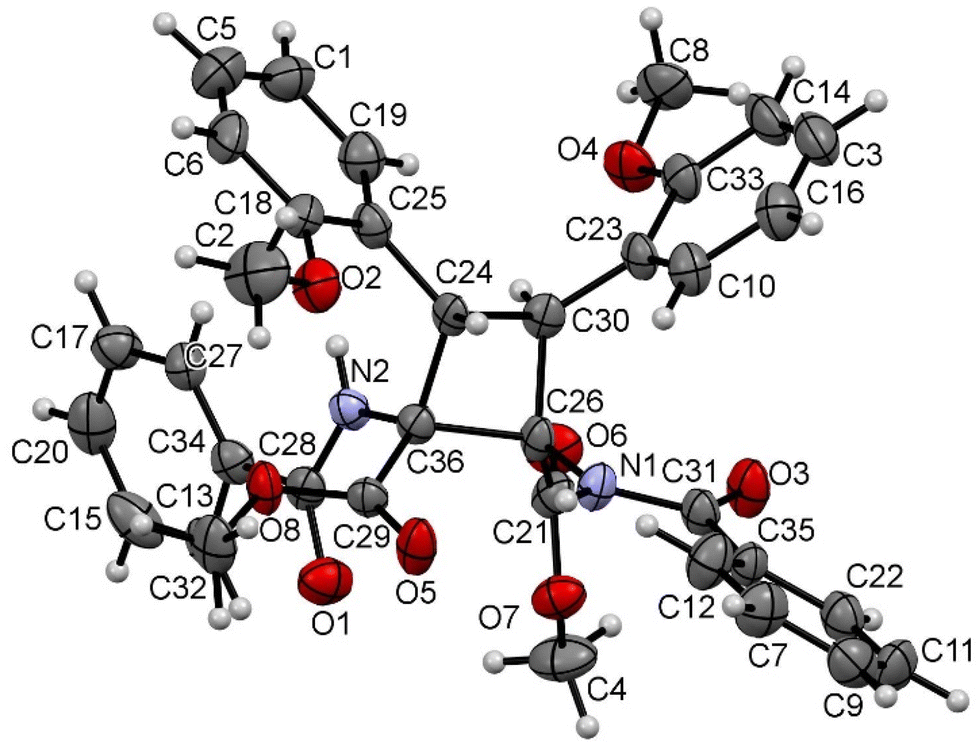 | ||
| Fig. 5 X-ray molecular structure of cyclobutane 2n. Ellipsoids are drawn at the 30% probability level. | ||
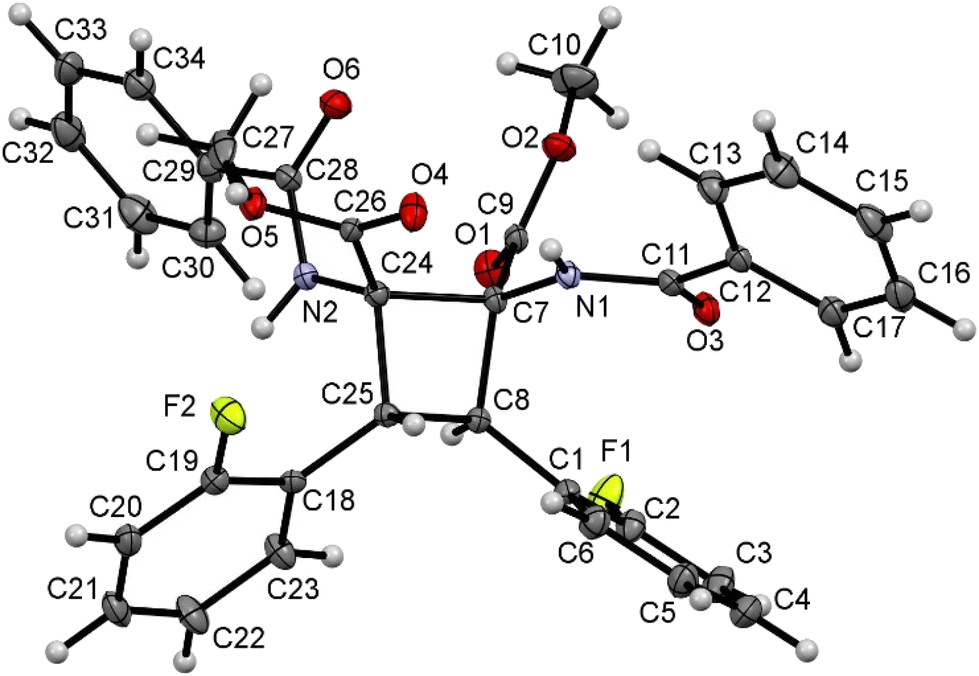 | ||
| Fig. 6 X-ray molecular structure of cyclobutane 2o. Ellipsoids are drawn at the 30% probability level. | ||
 | ||
| Fig. 7 X-ray molecular structure of cyclobutane 2s. Ellipsoids are drawn at the 30% probability level. | ||
The three structures are isostructural and show the cyclobutane core formed by the head-to-head (1,2)-anti photocycloaddition of two (Z)-oxazolones. That is, the new isomer obtained in this reaction is the delta (δ) isomer.13
The three cyclobutanes show two trans aryl rings (2-C6H4OMe in 2n, 2-C6H4F in 2o, and 3,5-C6H3F2 in 2s) in adjacent C atoms of the cyclobutane. Each of the other two C atoms shows one CO2Me and one N(H)C(O)Ph group, arising from the ring opening by methanolysis of the oxazolone. The relative arrangement of the two benzamido groups is trans, as well as that of the esters. In addition, each aryl ring is cis with respect to the benzamido group located in the adjacent carbon, showing that the oxazolone is not isomerized and that the geometries of the Z-oxazolones 1 are preserved in the cyclobutanes 2. Therefore, the configuration of the molecule is 1,2-trans-2,3-cis-3,4-trans. Other internal parameters are identical, within the experimental error, to those reported by us recently,6a and also to those found in the literature for related systems.14
The comparison of the different reaction conditions used for the synthesis of the different isomers (μ-isomer: Ru, CH2Cl2, blue light;6a δ-isomer: Ru, BF3, MeOH, blue light) suggests that the Lewis acid BF3 is mainly responsible for the divergent behaviour observed. We have discarded the effect of the solvent in the obtention of different isomers because we have observed previously that the change of the solvent does not change the orientation of the photochemical reaction,5b,6a even if it participates in the whole process5b as in this case. We suggest that the role of methanol in this reaction is just the promotion of the ring-opening reaction under very smooth conditions, probably assisted by BF3, as it has been observed by us recently for related thiazolones.5b In order to determine the precise role of the Lewis acid we attempted the study of the interaction between the oxazolones and BF3 in methanol as the solvent, both in the ground state and in the excited state. The 1H NMR spectra of solutions of 1a in CD3OD do not show changes in the shape of the signals due to the oxazolone, nor in the chemical shifts, after the addition of different amounts of BF3·OEt2, suggesting a very weak interaction between BF3 and 1a, not detectable by NMR. A similar conclusion can be derived from the analysis of the excitation–emission spectra of 1a in methanol in the absence or presence of BF3.5b Although the oxazolone–BF3 interaction in methanol seems to be too weak to characterize, the different isomers obtained in the presence (δ) and absence (μ) of BF3 suggest that this interaction exists at some point of the reaction. We can even propose that the interaction is plausible along the whole reaction path (Fig. 8), considering that the μ-isomer is not detected in presence of BF3.
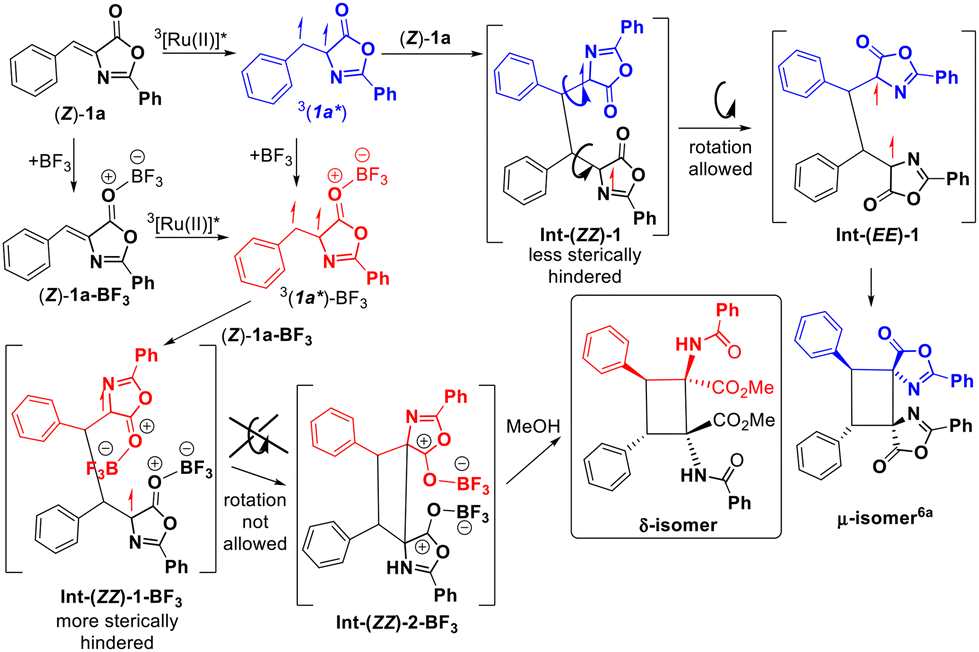 | ||
| Fig. 8 Role of BF3 in the synthesis of delta-isomers of 1,2-diaminotruxinic-cyclobutanes 2. | ||
The role of BF3 in this reaction is shown in Fig. 8 and seems to be merely steric, based on our recent study of the Ru-sensitized photocycloaddition of (Z)-oxazolones, where the 1,4-diradical Int-(ZZ)-1 undergoes free rotation to give Int-(EE)-1 before the ring closing step and formation of the μ-isomer.6a We propose here that the new 1,4-diradical intermediate Int-(ZZ)-1-BF3 would be much more sterically hindered than Int-(ZZ)-1, not allowing further rotation and giving Int-(ZZ)-2-BF3. Release of BF3 at this point should give dispyrocyclobutanes. In this respect, isolation of 2t* is indirect evidence of the formation of the species Int-(ZZ)-2-BF3. In addition, bonding of BF3 to Int-(ZZ)-2-BF3 increases the electrophilic character of the carbonyl carbon, making it more prone to undergo nucleophilic attack from the solvent and promoting the ring opening reaction of the oxazolone, giving finally the δ-isomer of 2.
Expansion of δ-1,2-diaminotruxinic cyclobutanes 2. Synthesis of highly substituted esters of pyrrolidine-2,5-dicarboxylic acids 3
During the synthesis of cyclobutanes 2a–2u at short reaction times we experienced in a few cases difficulties in promoting the complete ring-opening reaction of the oxazolone at room temperature, and therefore mixtures of cyclobutanes with the heterocycles in the closed and open forms were obtained, as explained for the case of 2t and 2t* (see Fig. 4). In such cases we forced the complete ring-opening reaction by treatment of the mixture with a catalytic amount of NaOMe in refluxing MeOH at the end of the reaction. Surprisingly, in addition to the expected 1,2-diaminotruxinic diesters 2 we obtained a second product related to the deprotonation of such cyclobutanes 2. Due to this unexpected additional reactivity, we decided to investigate in-depth the reactions of 1,2-diaminotruxinic diesters 2 with NaOMe in refluxing methanol. The obtained results are shown in Fig. 9.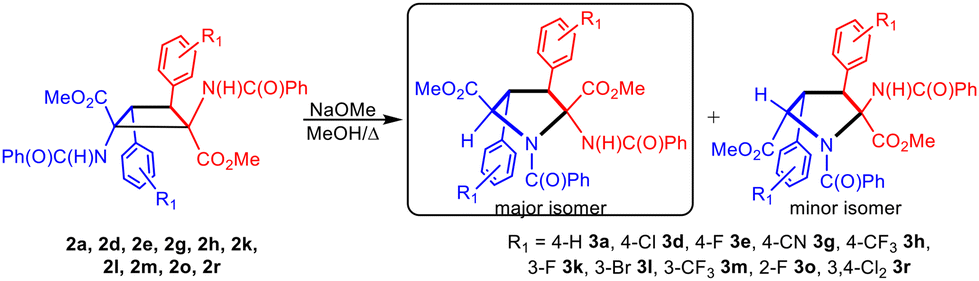 | ||
| Fig. 9 Synthesis of pyrrolidines 3a–3r by the ring expansion of cyclobutanes 2a–2r. | ||
The treatment of cyclobutanes 2 with a stoichiometric amount of NaOMe (1/1 molar ratio) in methanol at 110 °C resulted in the formation of the corresponding dimethyl 2-benzamido-3,4-diaryl-N-benzoyl-pyrrolidine-2,5-dicarboxylates 3, as shown in Fig. 9. We attempted this reaction for the full set of cyclobutanes 2a–2u, and we observed full conversion in all cases. The respective crude of the reactions showed (1H NMR) a mixture of compounds, in which pyrrolidines 3 were the main components (more than 90%). In turn, pyrrolidines 3 were present as a mixture of two diastereomers in different molar ratios, one of them being clearly more abundant than the other. We attempted the separation and purification of this mixture by column chromatography. We were successful in the separation of pyrrolidines 3 into pure products in the cases where electron-withdrawing substituents (F, Cl, Br, CN, and CF3) were present on the aryl rings (Fig. 9 and 10). In most of these cases, the separation afforded the major isomer as a single isomer (3d, 3e, 3h, 3l, 3m, and 3r), while the minor isomer was always contaminated with the major one. In the other cases (3a, 3g, 3k, and 3o) pyrrolidines could be purified, but not separated (molar ratio in the mixture ranging from 3.3/1 to 2/1).
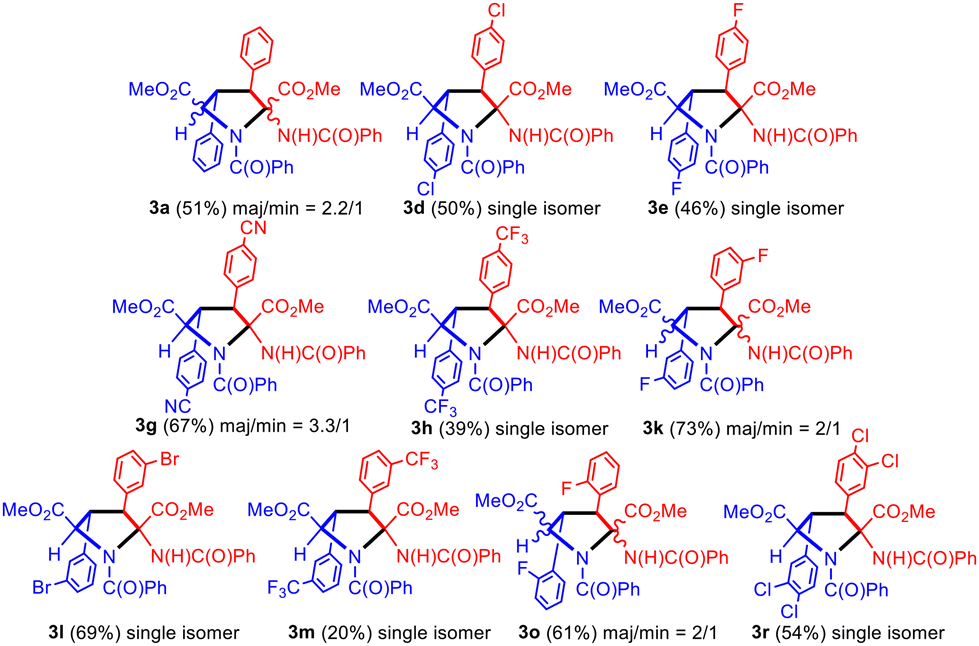 | ||
| Fig. 10 Scope of the synthesis of pyrrolidine-2,5-dicarboxylates 3. | ||
In general, the obtained yields were moderate to good (39% to 73%) except for 3m (20%). Attempts to optimize the reaction showed that a lower amount of NaOMe or a lower reaction temperature resulted in lower yields of 3. The nature of the base is critical to promote the formation of the pyrrolidines 3. Using the couple NaOMe/MeOH (pKa = 15.5),15 an efficient deprotonation of the NH unit from the benzamido group can be achieved, which is the starting point of the reaction (see below). However, the use of carbonate as the base (Cs2CO3, pKa = 10) in refluxing methanol results in the formation of a mixture of dehydrophenylalanine (DHPA) and pyrrolidine 3, with DHPA being the main product of the reaction. This fact can be interpreted as a competition between the deprotonation of cyclobutane 2, leading to pyrrolidine 3, and the thermal retro-[2 + 2] of 2, which affords the DHPA moiety. In line with this result, the use of acetate as the base (NaOAc, pKa = 4.76) in refluxing methanol produces only DHPA as the reaction product. Clearly, the base has to be strong enough to promote a fast and efficient deprotonation of cyclobutane 2, otherwise, a thermal retro-[2 + 2] takes place and the DHPA byproduct is obtained.
As is clear from Fig. 9, the reaction implies formally the expansion of a substituted cyclobutane ring, with four stereogenic centers, to give a five-membered pyrrolidine ring, also with four stereogenic centers. As far as we know, the expansion of a benzamidocyclobutane to build a pyrrolidine scaffold has not been reported previously. This methodology provides an additional and alternative synthetic pathway for the obtention of highly substituted pyrrolidine-2,5-dicarboxylates.
Due to the presence of four stereogenic centers, an important aspect of this reaction is the stereoselectivity with which it takes place considering that, a priori, up to 8 different stereoisomers of 3 can be obtained from 2. As explained, pyrrolidine-2,5-dicarboxylates 3 were obtained as two diastereomers in all of the cases, from which the major diastereomer could be isolated as a unique isomer for 3d, 3e, 3h, 3l, 3m and 3r, while for 3a, 3g, 3k and 3o the mixture could not be separated.
The spectroscopic characterization of pyrrolidines 3 by NMR shows that the diastereomer obtained as a unique isomer (or as the major isomer for 3a, 3g, 3k and 3o) has 2S, 3S, 4S, 5R/2R, 3R, 4R, 5S configurations (Fig. 9 and 10). This arrangement can be inferred from the observation of a strong NOE effect between the H at C3 and the H at C5 (5.81 ppm and 5.30 ppm in 3o, see the ESI†), as well as the NOE between the H at C3 and the NH of the benzamido group at C2 (8.06 ppm in 3o), meaning that these three fragments are on the same side of the molecular plane. This was further confirmed through the determination of the crystal structure of pyrrolidine 3r by X-ray diffraction methods (Fig. 11). Unfortunately, the determination of the structure of the minor isomers of 3a, 3g, 3k and 3o could not be performed experimentally, because no NOEs were observed after the inversion of signals associated with such species (even though the NOE was clear for the major isomer of 3o), neither single crystals of these species could be obtained. The structural proposal for these compounds is based on DFT calculations (see below).
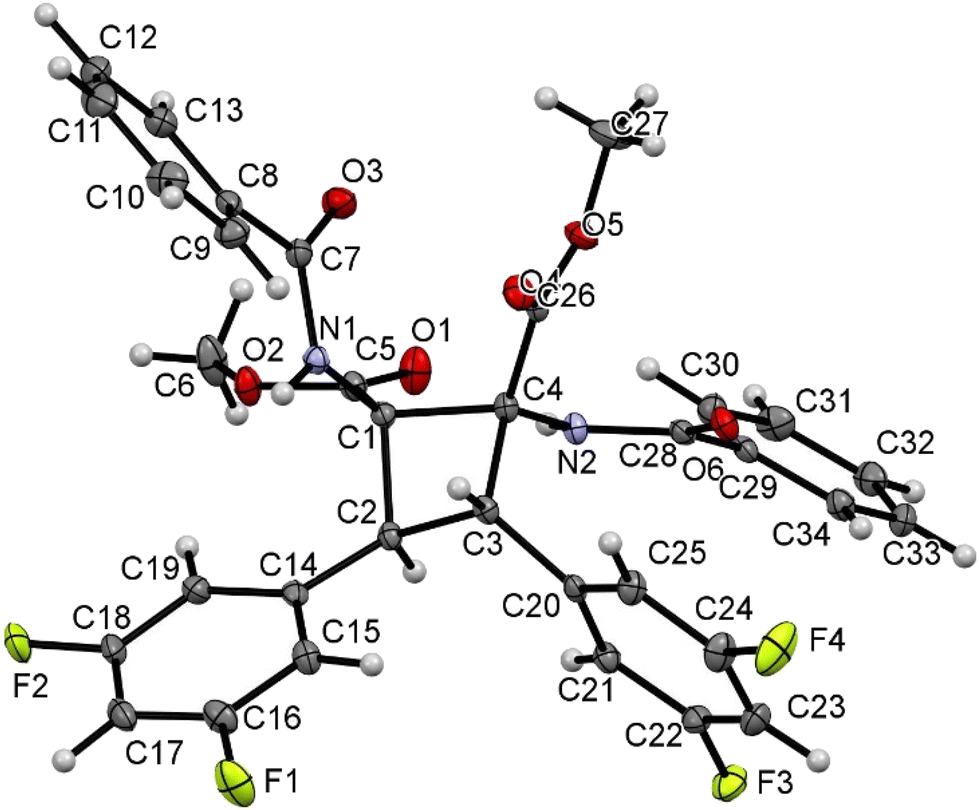
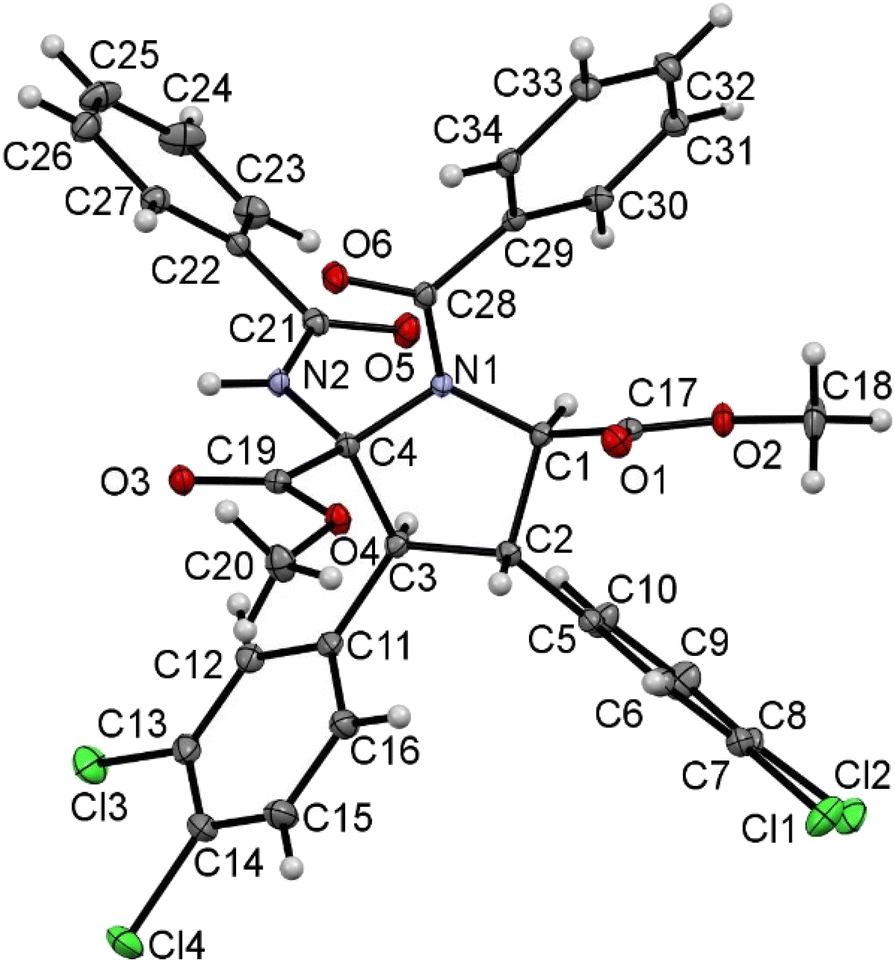 | ||
| Fig. 11 X-ray molecular structure of pyrrolidine 3r. Ellipsoids are drawn at the 50% probability level. | ||
A molecular drawing of pyrrolidine 3r is shown in Fig. 11. The structure confirms the formation of a five-membered pyrrolidine ring, being N-substituted by a C(O)Ph moiety and having a CO2Me group at C1, a 3,4-C6H3Cl2 fragment at both C2 and C3, and one ester and one benzamide functional group at C4. The five-membered ring displays the expected envelope shape, aiming to minimize intramolecular steric contacts. The relative arrangements of the substituents at each C atom (C1 to C4) show interesting changes when compared with the structure of the cyclobutane precursors 2. The two 3,4-C6H3Cl2 aryl rings at C2 and C3 are in mutual trans positions, as are in the starting cyclobutanes, and therefore it seems that this part of the molecule has not experienced changes during the ring expansion. However, the benzamido group at C4 and the 3,4-C6H3Cl2 aryl ring at C3 are also in trans, while they are in cis in cyclobutanes 2 (see Fig. 5–7). This strongly suggests a free rotation around the C3–C4 bond during the cyclobutane expansion. The trans arrangement of the benzamido group and the aryl ring in the neighbour carbon was already observed in the μ-isomers of the 1,2-diaminotruxinic cyclobutanes.6a As a result of these geometrical arrangements, the absolute configurations of the stereogenic centers at C1, C2, C3 and C4 are RC1, SC2, SC3 and SC4.
However, the molecule crystallized in the monoclinic space group I2/a, which is centrosymmetric, and therefore both enantiomers (RC1, SC2, SC3 and SC4 and SC1, RC2, RC3 and RC4) have to be present in the unit cell. The internal C–C, C–N and C–O bond distances are identical, within the experimental error, to those found in other N-acyl-pyrrolidine-2,5-carboxylates.16 There are only slight differences between the bond distances N1–C1 (1.455(3) Å) and N1–C4 (1.473(3) Å) on the one hand, and C1–C2 (1.545(3) Å) and C3–C4 (1.568(4) Å) on the other hand. Other internal parameters are in the usual ranges of distances and angles found in the literature for these types of compounds.14
A plausible mechanism for this reaction is shown in Fig. 12, based on DFT calculations.17 The reaction is initiated by the methoxide anion, which deprotonates one of the two equivalent N–H bonds of the benzamide fragment in 2 and generates the corresponding N-anionic form (2 anion in Fig. 12), which was taken as the reference system for the energy in the potential energy surface (G = 0 kcal mol−1). The N-anion charge would trigger the cyclobutane ring opening through a C–C bond cleavage in TS1, a step that involves an energy barrier of 22.4 kcal mol−1, forming the open intermediate intA. This high-energy species contains the formal anionic charge at the carbon atom, and can be slightly stabilized by 1,2-shift tautomerization, to locate the negative charge at the nearby N-atom (intB, G = 13.6 kcal mol−1). The open intermediates present free rotation allowed around the C–C bonds of the remaining carbon skeleton. From intB, four diastereoisomeric attack options arise, where the N-anion reacts with the iminic C atom, closing to the pyrrolidine 3-anion adduct. Among the four diastereomeric final adducts, 3-anion (R,S) is the most stable species with an energy 6.3 kcal mol−1 lower than the initial situation in 2a-anion. Thus, the process is exergonic. The remaining 3a isomers are at least 3 kcal mol−1 less stable than the major R,S-isomer, in agreement with our experimental results. The activation energy for the pyrrolidine ring formation in TS2 is 16.9 kcal mol−1, affordable under the reaction conditions, and interestingly, the four computed TS2 transition states also show a clear preference in favour of the RS-isomer. In fact, an energy difference of at least 5 kcal mol−1 was noted between TS2-R,S and the rest of the TS2 transition states. Thus, the formation of the R,S-isomer could be explained with either kinetic or thermodynamic conditions, according to the calculations.
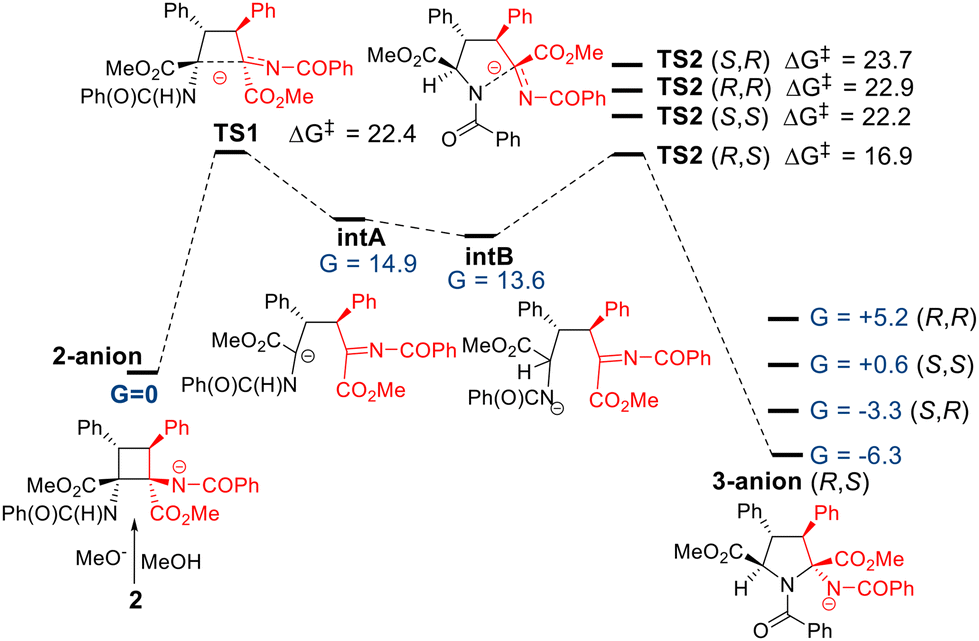 | ||
| Fig. 12 Mechanism of the ring expansion of cyclobutanes 2 to give pyrrolidines 3. | ||
However, relative energies of the transition states and the final adducts differ in the explanation of the formation of the minor experimental SR isomer. The energy distribution of the four isomers indicates that the second most stable adduct is the 3a-anion (S,R), corroborating that it is formed under thermodynamic conditions, since it is kinetically the least favoured adduct TS2 (S,R) (ΔG‡ = +23.7 kcal mol−1). As a further confirmation, the other two anions, the 3-anion (R,R) and the 3-anion (S,S) are much higher in energy and, as expected, they could not be obtained experimentally.
Experimental
General methods
[2 + 2]-photocycloaddition reactions were carried out under an inert (Ar) atmosphere, using dry and deoxygenated methanol from a Pure Solv MD5 solvent purification system. Purification of the compounds was carried out by flash column liquid chromatography using silica gel (70–230 μm) as the support. 1H, 13C and 19F NMR spectra of the isolated products 2 and 3 were recorded in CDCl3 or CD2Cl2 solutions at 25 °C (other temperatures were specified) on Bruker AV300 and Bruker AV500 spectrometers (δ in ppm, J in Hz) at 1H operating frequencies of 300.13 and 500.13 MHz, respectively. 1H and 13C NMR spectra were referenced using the solvent signal as the internal standard, while 19F NMR spectra were referenced to CFCl3. The assignment of 1H NMR peaks has been performed through standard 2D 1H-COSY and selective 1D 1H-SELNOE experiments. Typical mixing times in the case of selective 1D-SELNOE experiments were in the range of 1.2–1.8 s, as a function of the irradiated signal. These values of optimized mixing times were set equal to the longitudinal relaxation time T1, determined using the inversion–recovery sequence. 13C NMR peaks were identified using standard 1H–13C edited-HSQC and 1H–13C HMBC 2D-experiments. In both cases spectral widths of 10 ppm (1H) and 200 ppm (13C) were used, with average values of the coupling constants 1JCH = 145 Hz and long-range nJCH = 10 Hz. ESI (ESI+) mass spectra were recorded using an Esquire 3000 ion-trap mass spectrometer (Bruker Daltonic GmbH) equipped with a standard ESI/APCI source. HRMS and ESI (ESI+) mass spectra were recorded using a MicroToF Q, API-Q-ToF ESI with a mass range from 20 to 3000 m/z and mass resolution 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (FWHM). The oxazolones 1 used as starting materials were synthesized according to published methods.11 The compound [Ru(bpy)3](BF4)2 was also prepared following published procedures,18 and it was stored under a protecting atmosphere (Ar) at 4 °C.
000 (FWHM). The oxazolones 1 used as starting materials were synthesized according to published methods.11 The compound [Ru(bpy)3](BF4)2 was also prepared following published procedures,18 and it was stored under a protecting atmosphere (Ar) at 4 °C.
Irradiation setup
The irradiation setup used in this case was a metallic cylindrical recipient (16 cm internal diameter and 10.5 cm high) whose internal surface was covered with blue LEDs (222 diodes, 465 nm). This home-made system provided an electrical power of 18 W and used a 12 V power supply.Crystallography
Crystals of cyclobutanes 2n, 2o, 2s and pyrrolidine 3r of quality for X-ray measurements were grown by slow diffusion of n-pentane into CH2Cl2 (2n, 2o, and 2s) or CHCl3 (3r) solutions of the respective crude products at −18 °C for several weeks. One selected single crystal of each compound was mounted at the end of a quartz fiber in a random orientation, covered with perfluorinated oil and placed under a cold stream of N2 gas. The data collection was performed at 123 K (2s and 3r) or 153 K (2n and 2o) on Oxford Diffraction Xcalibur Sapphire 3 (2n and 3r), Bruker P4 (2o) and Bruker APEX Duo (2s) diffractometers, using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). A hemisphere of data were collected based on ω-scan and ϕ-scan runs. The diffraction frames were integrated using the programs CrysAlis RED19 or SAINT20 and the integrated intensities were corrected for absorption with SADABS.21 The structures were solved and developed using Fourier methods.22 All non-hydrogen atoms were refined with anisotropic displacement parameters. The H atoms were placed at idealized positions and treated as riding atoms. Each H atom was assigned an isotropic displacement parameter equal to 1.2 times the equivalent isotropic displacement parameter of its parent atom. For structure solving and refinement the SHELX-9723 software package was used. The structures were refined to Fo2 and all reflections were used in least-squares calculations.24 CCDC 2177354 (2o), 2177355 (2s), and 2176207 (3r)† contain the supplementary crystallographic data for this paper. The structure of cyclobutane 2n could be solved, but the low quality of the data prevented a complete refinement, so the structure has to be considered only as a connectivity scheme. For the structure of 3r a disordered pentane molecule in the (1/2, 1/2, 1/2) plane was found, which was refined using geometrical restraints and constraints for the anisotropic thermal displacement parameters.Computational details
All structures were optimized using density functional theory (DFT) calculations using Gaussian 16,25 with M06-2X26 as the functional and 6-311+G(d,p) as the basis set, introducing solvation factors with the IEF-PCM27 method, and methanol as the solvent, as in the optimized experimental conditions. The stationary points were characterized using frequency calculations in order to verify that they have the right number of imaginary frequencies.Synthesis of the bis(amino acid)s δ-1,2-diaminotruxinics 2a–2u
All cyclobutane bis(amino acid)s containing the 1,2-diaminotruxinic core 2a–2u have been prepared following the same experimental method, which is exemplified here for 2a. See the ESI† for the details of all compounds.Synthesis of dimethyl-1,2-bis(benzamido)-3,4-diphenylcyclobutane-1,2-dicarboxylate 2a
The oxazolone 1a (149.45 mg, 0.60 mmol) and [Ru(bpy)3](BF4)2 (11.2 mg, 0.015 mmol) were suspended in deoxygenated and dry methanol (4 mL) under an Ar atmosphere at room temperature. This suspension was treated with BF3·Et2O (34.1 mg, 0.24 mmol), and the resulting mixture was irradiated with blue light (465 nm) for 24 h at room temperature. After the reaction time the solvent was evaporated to dryness, and the solid residue was suspended in chloroform (10 mL). This suspension was washed with water (3 × 5 mL) and the organic phase was dried with anhydrous MgSO4. The resulting solution, which contained cyclobutane 2a, was purified by column chromatography using silica as the support and a mixture of ethyl acetate/n-hexane in a gradient from 1/9 to 3/7 ratio. Cyclobutane 2a was isolated as a white solid by solvent evaporation. Obtained: 56.8 mg (34% yield). 1H NMR (CDCl3, 300.13 MHz) δ 8.30 (s, 1H, NH), 7.56 (m, 2H, Ho, CO–Ph), 7.46 (m, 3H, Ho, Ph + Hp, CO–Ph), 7.39–7.27 (m, 5H, Hm, Ph + Hm, CO–Ph + Hp, Ph), 4.93 (s, 1H, CH), 3.73 (s, 3H, OCH3). 13C{1H} NMR (CDCl3, 75.5 MHz) δ 171.6, 166.7, 134.7, 133.3, 131.9, 129.1, 128.7, 128.6, 128.2, 127.0, 64.1, 53.1, 47.6. HRMS (ESI+) m/z calcd for C34H30N2NaO6 [M + Na]+: 585.2002, found: 585.1987.Expansion of the cyclobutane of δ-1,2-diaminotruxínics: synthesis of pyrrolidines 3
All pyrrolidine-2,5-dicarboxylate derivatives 3 have been prepared following the same experimental method, which is discussed in detail here for 3a. See the ESI† for the details of all compounds.Synthesis of dimethyl-2-benzamido-1-benzoyl-3,4-diphenylpyrrolidine-2,5-dicarboxylate 3a
A solution of δ-cyclobutane 2a (41.8 mg, 0.074 mmol) in methanol (5 mL) was treated with NaOMe (4.06 mg, 0.075 mmol), and the resulting mixture was heated at 110 °C for 1 h in a J-Young sample flask. Once cooled, the resulting solution was evaporated to dryness and the solid residue was extracted with CH2Cl2 (3 × 5 mL). Any insoluble solid was removed at this point by filtration through Celite. The resulting clear solution was evaporated to dryness to give impure pyrrolidine 3a, which was purified by crystallization from CH2Cl2/pentane to give 3a as a white solid. Obtained: 21.2 mg (51% yield). Pyrrolidine 3a was characterized using NMR methods as a mixture of two diastereomers in a 2.2:1 molar ratio. Only the major isomer could be fully characterized by NMR. 1H NMR (CDCl3, 300.13 MHz) δ 8.14 (s, 1H, NH), 7.91 (m, 2H, Ho, NHCO–C6H5), 7.59–7.12 (m, 18H, C6H5), 5.28 (d, 3JHH = 10 Hz, 1H, H–C5), 5.27 (d, 3JHH = 13.1 Hz, 1H, H–C3), 4.27 (dd, 3JHH = 13.1 Hz, 3JHH = 10 Hz, 1H, H–C4), 3.70 (s, 3H, C2–COOCH3), 3.10 (s, 3H, C5–COOCH3). 13C{1H}NMR (CDCl3, 75.5 MHz) δ 170.9, 170.8, 169.0, 167.4, 136.2, 135.2, 134.6, 133.0, 132.2, 130.9, 129.0, 129.0, 128.9, 128.7, 128.6, 128.6, 128.4, 128.2, 128.2, 127.4, 81.4, 69.7, 53.4, 53.4, 51.9, 51.8. HRMS (ESI+) m/z calc for C34H30N2NaO6 [M + Na]+ 585.2002, found 585.2003.
Conclusions
In conclusion, the addition of the BF3 Lewis acid changes the outcome of the Ru-photosensitized [2 + 2]-cycloaddition of (Z)-4-aryliden-5(4H)-oxazolones. In the presence of BF3 the δ-isomer of the 1,2-diaminotruxinic cyclobutanes 2 is obtained in moderate to low yields by the anti-1,2-head-to-head cycloaddition of two oxazolones, instead of the μ-isomer obtained in a free-Lewis acid reaction. The role of the Lewis acid seems to be steric in nature, hindering the free rotation in the transition state. Treatment of the δ-1,2-diaminotruxinic cyclobutanes 2 with a strong base promotes an unprecedented expansion of the cyclobutane ring affording highly substituted pyrrolidine-2,5-dicarboxylates 3 in moderate yields with a high degree of stereoselectivity.Author contributions
S. S. synthesized the compounds, performed the chemical characterization (investigation and validation) and wrote part of the original manuscript (visualization, writing – original draft, review and editing). R. L. and E. G. B. performed the computational calculations (formal analysis and methodology) and wrote part of the original manuscript (visualization, writing, original draft, review and editing). L. R. F. performed part of the crystallographic work (formal analysis and methodology) and corrected the original manuscript (visualization, writing – original draft, review and editing). E. P. U. conceived the original idea (conceptualization), directed the research (project administration and supervision), acquired the funding (funding acquisition and resources) and wrote the original manuscript (visualization, writing – original draft, review and editing). All authors analyzed and discussed the results and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
EPU and SS thank the Spanish Government (Grant PID2019-106394GB-I00/AEI/10.13039/501100011033, funded by MCIN/AEI/10.13039/501100011033) and Gobierno de Aragón-FSE (Spain, research group Aminoácidos y Péptidos E19_20R) for funding. S. S. thanks Gobierno de Aragón-FSE for a PhD fellowship. L. R. F. thanks Spanish Ministerio de Ciencia e Innovación (Grant PGC2018-093451-B-I00 funded by MCIN/AEI/10.13039/501100011033/ and FEDER “Una manera de hacer Europa”), the European Union Regional Development Fund (FEDER), and the Diputación General de Aragón, Project M4, E11_20R, for funding. R. L. and E. G. B. thank the Spanish Government (Grant PID2019-110008GB-I00/AEI/10.13039/501100011033, funded by MCIN/AEI/10.13039/501100011033) and SGIker (UPV/EHU) for providing human and computational resources.References
- (a) Amino Acids, Peptides and Proteins in Organic Chemistry, Volumes 1–5 (Volume 1: Origins and Synthesis of Amino Acids; Volume 2: Modified Amino Acids, Organocatalysis and Enzymes; Volume 3: Building Blocks, Catalysis and Coupling Chemistry; Volume 4: Protection Reactions, Medicinal Chemistry, Combinatorial Synthesis; Volume 5: Analysis and Function of Amino Acids and Peptides), ed. A. B. Hughes, Wiley–VCH Verlag GmbH & Co. KGaA, 2009–2011 Search PubMed; (b) Amino Acids, Peptides and Proteins, ed. M. Ryadnov and F. Hudecz, Royal Society of Chemistry, 2021, vol. 44 Search PubMed.
- (a) G. A. Ulaner and D. M. Schuster, Amino Acid Metabolism as a Target for Breast Cancer Imaging, PET Clin., 2018, 13, 437 CrossRef PubMed; (b) T. Seierstad, K. Hakon Hole, A. J. Tulipan, H. Stromme, W. Lilleby, M. E. Revheim and E. Hernes, 18F-Fluciclovine PET for Assessment of Prostate Cancer with Histopathology as Reference Standard - A Systematic Review, PET Clin., 2021, 16, 167 CrossRef PubMed.
- (a) M. Lasa and C. Cativiela, Synthesis of Enantiomerically Pure 1-Amino-2-Phenylcycloalkane carboxylic Acids (cnPhe), Synlett, 2016, 2517 Search PubMed; (b) M. M. Goodman, W. Yu and N. Jarkas, Synthesis and biological properties of radiohalogenated a,a-disubstituted amino acids for PET and SPECT imaging of amino acid transporters (AATs), J. Labelled Compd. Radiopharm., 2018, 61, 272 CrossRef CAS PubMed; (c) A. F. M. Noisier and M. A. Brimble, C-H Functionalization in the Synthesis of Amino Acids and Peptides, Chem. Rev., 2014, 114, 8775 CrossRef CAS PubMed.
- (a) D. Roiban, E. Serrano, T. Soler, I. Grosu, C. Cativiela and E. P. Urriolabeitia, Unexpected [2 + 2] C–C bond coupling due to photocycloaddition on orthopalladated (Z)-2-aryl-4-arylidene-5(4H)-oxazolones, Chem. Commun., 2009, 4681 RSC; (b) E. Serrano, A. Juan, A. García-Montero, T. Soler, F. Jiménez-Marquez, C. Cativiela, M. V. Gómez and E. P. Urriolabeitia, Stereoselective Synthesis of 1,3-Diaminotruxillic Acid Derivatives: An Advantageous Combination of C-H–ortho–Palladation and On–Flow [2 + 2]–Photocycloaddition in Microreactors, Chem. – Eur. J., 2016, 22, 144 CrossRef CAS PubMed; (c) C. Carrera, A. Denisi, C. Cativiela and E. P. Urriolabeitia, Functionalized 1,3-diaminotruxillic acids by Pd-mediated C-H activation and [2 + 2]-photocycloaddition of 5(4H)-oxazolones, Eur. J. Inorg. Chem., 2019, 3481 CrossRef CAS; (d) E. P. Urriolabeitia, P. Sánchez, A. Pop, C. Silvestru, E. Laga, A. I. Jiménez and C. Cativiela, Synthesis of esters of diaminotruxillics bis-amino acids by Pd-mediated photocycloaddition of analogs of the Kaede protein chromophore, Beilstein J. Org. Chem., 2020, 16, 1111 CrossRef CAS PubMed.
- (a) A. García-Montero, A. M. Rodríguez, A. Juan, A. H. Velders, A. Denisi, G. Jiménez-Osés, E. Gómez-Bengoa, C. Cativiela, M. V. Gómez and E. P. Urriolabeitia, Metal-Free [2 + 2]-Photocycloaddition of (Z)-4-Aryliden-5(4H)-Oxazolones as Straightforward Synthesis of 1,3-Diaminotruxillic Acid Precursors: Synthetic Scope and Mechanistic Studies, ACS Sustainable Chem. Eng., 2017, 5, 8370 CrossRef; (b) S. Sierra, D. Dalmau, S. Higuera, D. Cortés, O. Crespo, A. I. Jimenez, A. Pop, C. Silvestru and E. P. Urriolabeitia, Reactivity of (Z)-4-Aryliden-5(4H)-thiazolones: [2 + 2]-Photocycloaddition, Ring-Opening Reactions, and Influence of the Lewis Acid BF3, J. Org. Chem., 2021, 86, 12119 CrossRef CAS PubMed; (c) Q. Liu, N. Li, Y. Yuan, H. Lu, X. Wu, C. Zhou, M. He, H. Su, M. Zhang, J. Wang, B. Wang, Y. Wang, D. Ma, Y. Ye, H. C. Weiss, E. R. F. Gesing, J. Liao and M. W. Wang, Cyclobutane Derivatives As Novel Nonpeptidic Small Molecule Agonists of Glucagon-Like Peptide-1 Receptor, J. Med. Chem., 2012, 55, 250 CrossRef CAS PubMed.
- (a) S. Sierra, M. V. Gómez, A. I. Jiménez, A. Pop, C. Silvestru, M. L. Marín, F. Boscá, G. Sastre, E. Gómez-Bengoa and E. P. Urriolabeitia, Stereoselective, Ruthenium-Photocatalyzed Synthesis of 1,2-Diaminotruxinic Bis-amino Acids from 4-Arylidene-5(4H)-oxazolones, J. Org. Chem., 2022, 87, 3529 CrossRef CAS PubMed; (b) I. F. S. Marra, A. M. de Almeida, L. P. Silva, P. P. de Castro, C. C. Corrêa and G. W. Amarante, Stereoselective Intermolecular [2 + 2] Cycloadditions of Erlenmeyer–Plöchl Azlactones Using Visible Light Photoredox Catalysis, J. Org. Chem., 2018, 83, 15144 CrossRef CAS PubMed.
- (a) M. He, N. Guan, W. W. Gao, Q. Liu, X. Y. Wu, D. W. Ma, D. F. Zhong, G. B. Ge, C. Li, X. Y. Chen, L. Yang, J. Y. Liao and M. W. Wang, A continued saga of Boc5, the first non-peptidic glucagon-like peptide-1 receptor agonist with in vivo activities, Acta Pharmacol. Sin., 2012, 33, 148 CrossRef CAS PubMed; (b) W. T. Berger, B. P. Ralph, M. Kaczocha, J. Sun, T. E. Balius, R. C. Rizzo, S. Haj-Dahmane, I. Ojima and D. G. Deutsch, Targeting Fatty Acid Binding Protein (FABP) Anandamide Transporters – A Novel Strategy for Development of Anti-Inflammatory and Anti-Nociceptive Drugs, PLoS One, 2012, 7, e50968 CrossRef CAS PubMed; (c) S. Yan, M. W. Elmes, S. Tong, K. Hu, M. Awwa, G. Y. H. Teng, Y. Jing, M. Freitag, Q. Gan, T. Clement, L. Wei, J. M. Sweeney, O. M. Joseph, J. Che, G. S. Carbonetti, L. Wang, D. M. Bogdan, J. Falcone, N. Smietalo, Y. Zhou, B. Ralph, H. C. Hsu, H. Li, R. C. Rizzo, D. G. Deutsch, M. Kaczocha and I. Ojima, SAR studies on truxillic acid mono esters as a new class of antinociceptive agents targeting fatty acid binding proteins, Eur. J. Med. Chem., 2018, 154, 233 CrossRef CAS PubMed.
- (a) F. D. Lewis, D. K. Howard and J. D. Oxman, Lewis Acid Catalysis of Coumarin Photodimerization, J. Am. Chem. Soc., 1983, 105, 3344 CrossRef CAS; (b) F. D. Lewis, J. D. Oxman, L. L. Gibson, H. L. Hampsch and S. L. Quillen, Lewis Acid Catalysis of Photochemical Reactions. 4. Selective Isomerization of Cinnamic Esters, J. Am. Chem. Soc., 1986, 108, 3005 CrossRef CAS; (c) F. D. Lewis, S. L. Quillen, P. D. Hale and J. D. Oxman, Lewis Acid Catalysis of Photochemical Reactions. 7. Photodimerization and Cross-Cycloaddition of Cinnamic Esters, J. Am. Chem. Soc., 1988, 110, 1261 CrossRef CAS; (d) F. D. Lewis and S. V. Barancyk, Lewis Acid Catalysis of Photochemical Reactions. 8. Photodimerization and Cross-Cycloaddition of Coumarin, J. Am. Chem. Soc., 1989, 111, 8653 CrossRef CAS; (e) F. D. Lewis, J. E. Elbert, A. L. Upthagrove and P. D. Hale, Lewis Acid Catalysis of Photochemical Reactions. 9. Structure and Photoisomerization of (E)- and (Z)-Cinnamamides and Their Lewis Acid Complexes, J. Org. Chem., 1991, 56, 553 CrossRef CAS; (f) R. Brimioulle, H. Guo and T. Bach, Enantioselective Intramolecular [2 + 2] Photocycloaddition Reactions of 4-Substituted Coumarins Catalyzed by a Chiral Lewis Acid, Chem. – Eur. J., 2012, 18, 7552 CrossRef CAS PubMed; (g) R. Brimioulle, A. Bauer and T. Bach, Enantioselective Lewis Acid Catalysis in Intramolecular [2 + 2] Photocycloaddition Reactions: A Mechanistic Comparison between Representative Coumarin and Enone Substrates, J. Am. Chem. Soc., 2015, 137, 5170–5176 CrossRef CAS PubMed; (h) T. P. Yoon, Photochemical Stereocontrol Using Tandem Photoredox–Chiral Lewis Acid Catalysis, Acc. Chem. Res., 2016, 49, 2307 CrossRef CAS PubMed; (i) M. E. Daub, H. Jung, B. J. Lee, J. Won, M. H. Baik and T. P. Yoon, Enantioselective [2 + 2] Cycloadditions of Cinnamate Esters: Generalizing Lewis Acid Catalysis of Triplet Energy Transfer, J. Am. Chem. Soc., 2019, 141, 9543 CrossRef CAS PubMed; (j) M. Leverenz, C. Merten, A. Dreuw and T. Bach, Lewis Acid Catalyzed Enantioselective Photochemical Rearrangements on the Singlet Potential Energy Surface, J. Am. Chem. Soc., 2019, 141, 20053 CrossRef CAS PubMed.
- (a) G. Pandey, P. Banerjee and S. R. Gadre, Construction of Enantiopure Pyrrolidine Ring System via Asymmetric [3 + 2]-Cycloaddition of Azomethine Ylides, Chem. Rev., 2006, 106, 4484 CrossRef CAS PubMed; (b) C. J. Wang, G. Liang, Z. Y. Xue and F. Gao, Highly Enantioselective 1,3-Dipolar Cycloaddition of Azomethine Ylides Catalyzed by Copper(I)/TF-BiphamPhos Complexes, J. Am. Chem. Soc., 2008, 130, 17250 CrossRef CAS PubMed; (c) A. T. Parsons, A. G. Smith, A. J. Neel and J. S. Johnson, Dynamic Kinetic Asymmetric Synthesis of Substituted Pyrrolidines from Racemic Cyclopropanes and Aldimines: Reaction Development and Mechanistic Insights, J. Am. Chem. Soc., 2010, 132, 9688 CrossRef CAS PubMed; (d) S. Lee, H. Lei and T. Rovis, A Rh(III)-Catalyzed Formal [4 + 1] Approach to Pyrrolidines from Unactivated Terminal Alkenes and Nitrene Sources, J. Am. Chem. Soc., 2019, 141, 12536 CrossRef CAS PubMed; (e) S. N. Greszler, G. Zhao, M. Buchman, X. B. Searle, B. Liu and E. A. Voight, General Asymmetric Synthesis of Densely Functionalized Pyrrolidines via Endo-Selective [3 + 2] Cycloaddition of β-Quaternary-Substituted Nitroalkenes and Azomethine Ylides, J. Org. Chem., 2020, 85, 7620 CrossRef CAS PubMed; (f) M. J. Meyers, J. Liu, Z. Liu, H. Ma, L. Dai, D. Adah, S. Zhao, X. Li, X. Liu, Y. Lu, Y. Huang, Z. Tu, X. Chen and M. D. Tortorella, 4-Aryl Pyrrolidines as Novel Orally Efficacious Antimalarial Agents. Part 2: 2-Aryl-N-(4-arylpyrrolidin-3-yl)acetamides, ACS Med. Chem. Lett., 2019, 10, 966 CrossRef CAS PubMed; (g) K. P. Melnykov, A. N. Artemenko, B. O. Ivanenko, Y. M. Sokolenko, P. S. Nosik, E. N. Ostapchuk, O. O. Grygorenko, D. M. Volochnyuk and S. V. Ryabukhin, Scalable Synthesis of Biologically Relevant Spirocyclic Pyrrolidines, ACS Omega, 2019, 4, 7498 CrossRef CAS PubMed; (h) V. I. Savych, V. L. Mykhalchuk, P. V. Melnychuk, A. O. Isakov, T. Savchuk, V. M. Timoshenko, S. A. Siry, S. O. Pavlenko, D. V. Kovalenko, O. V. Hryshchuk, V. A. Reznik, B. A. Chalyk, V. S. Yarmolchuk, E. B. Rusanov and P. K. Mykhailiuk, Bicyclic Pyrrolidines for Medicinal Chemistry via [3 + 2]-Cycloaddition, J. Org. Chem., 2021, 86, 13289 CrossRef CAS PubMed.
- Selected references for examples of cyclobutane expansion: (a) E. Lee-Ruff and G. Mladenova, Enantiomerically Pure Cyclobutane Derivatives and Their Use in Organic Synthesis, Chem. Rev., 2003, 103, 1449 CrossRef CAS PubMed; (b) A. T. Parsons, A. G. Smith, A. J. Neel and J. S. Johnson, Dynamic Kinetic Asymmetric Synthesis of Substituted Pyrrolidines from Racemic Cyclopropanes and Aldimines: Reaction Development and Mechanistic Insights, J. Am. Chem. Soc., 2010, 132, 9688 CrossRef CAS PubMed; (c) D. J. Mack and J. T. Njardarson, Recent Advances in the Metal-Catalyzed Ring Expansions of Three- and Four-Membered Rings, ACS Catal., 2013, 3, 272 CrossRef CAS; (d) L. K. B. Garve, A. Kreft, P. G. Jones and D. B. Werz, Synthesis of 2-Unsubstituted Pyrrolidines and Piperidines from Donor–Acceptor Cyclopropanes and Cyclobutanes: 1,3,5-Triazinanes as Surrogates for Formylimines, J. Org. Chem., 2017, 82, 9235 CrossRef CAS PubMed; (e) Y. Miki, N. Tomita, K. Ban, H. Sajiki and Y. Sawama, Synthesis of 1-Pyrroline by Denitrogenative Ring Expansion of Cyclobutyl Azides under Thermal Conditions, Adv. Synth. Catal., 2021, 363, 3481 CrossRef CAS; (f) B. Biletskyi, P. Colonna, K. Masson, J.-L. Parrain, L. Commeiras and G. Chouraqui, Small rings in the bigger picture: ring expansion of three- and four-membered rings to access larger all-carbon cyclic systems, Chem. Soc. Rev., 2021, 50, 7513 RSC; (g) J. Sietmann, M. Ong, C. Mück-Lichtenfeld, C. G. Daniliuc and J. M. Wahl, Desymmetrization of Prochiral Cyclobutanones via Nitrogen Insertion: A Concise Route to Chiral γ-Lactams, Angew. Chem., Int. Ed., 2021, 60, 9719 CrossRef CAS PubMed; (h) M. Nandy, S. Das and S. Nanda, Cyclobutane based “overbred intermediates” and their exploration in organic synthesis, Org. Biomol. Chem., 2022, 20, 1582 RSC. Surprisingly, it has been reported very recently the contraction of a pyrrolidine to give a cyclobutane: (i) C. Hui, L. Brieger, C. Strohmann and A. P. Antonchick, Stereoselective Synthesis of Cyclobutanes by Contraction of Pyrrolidines, J. Am. Chem. Soc., 2021, 143, 18864 CrossRef CAS PubMed.
- (a) J. Plöchl, Ueber Phenylglycidasäure (Phenyloxacrylsäure), Chem. Ber., 1883, 16, 2815 CrossRef; (b) J. Plöchl, Ueber einige Derivate der Benzoylimidozimmtsäure, Chem. Ber., 1884, 17, 1623 CrossRef; (c) E. Erlenmeyer, Ueber die Condensation der Hippursäure mit Phtalsäureanhydrid und mit Benzaldehyd, Justus Liebigs Ann. Chem., 1893, 275, 1 CrossRef; (d) H. E. Carter, Azlactones, Chapter 5 of the book series Organic Reactions, 1946, vol. 3, p. 198 Search PubMed; (e) R. Filler, Advances in Heterocyclic Chemistry, ed. A. R. Katrizky, Academic Press, New York, 1954, ch. 4, p. 75 Search PubMed; (f) Y. S. Rao and R. Filler, Geometric Isomers of 2-Aryl(Aralkyl)-4-arylidene(alkylidene)-5(4H)-oxazolones, Synthesis, 1975, 749 CrossRef CAS; (g) C. Cativiela, M. D. Díaz de Villegas and E. Meléndez, On the synthesis of geometric isomers of 2-methyl (or phenyl)-4-[α-arylethylidene]-5(4H)-oxazolones, J. Heterocycl. Chem., 1985, 22, 1655 CrossRef CAS; (h) F. M. Bautista, J. M. Campelo, A. García, D. Luna, J. M. Marinas and A. A. Romero, Study on dry-media microwave azlactone synthesis on different supported KF catalysts: influence of textural and acid–base properties of supports, J. Chem. Soc., Perkin Trans. 2, 2002, 227 RSC.
- The photochemical reactivity of dehydrophenylalanine generated by the initial ring opening reaction before plausible photocycloaddition does not lead to the formation of cyclobutanes. In fact, no reaction at all was observed under the reaction conditions.
- (a) R. Stoermer and F. Bachér, über die Konfiguration der Truxin- und Truxillsäuren (VI), Ber. Dtsch. Chem. Ges. B, 1922, 55, 1860 CrossRef; (b) R. Stoermer and F. Bachér, Zur Stereoisomerie der Truxillsäuren und über die Auffindung der letzten Säure dieser Gruppe (VIII), Ber. Dtsch. Chem. Ges. B, 1924, 57, 15 CrossRef.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds, J. Chem. Soc., Perkin Trans. 2, 1987, S1 RSC.
- K. P. C. Vollhardt and N. E. Schore, Organic Chemistry, Structure and Function, W. H. Freeman&Co, NY, USA, 2005, 5th edn, p. 60 Search PubMed.
- (a) V. Kubyshkin and N. Budisa, Amide rotation trajectories probed by symmetry, Org. Biomol. Chem., 2017, 15, 6764 RSC; (b) S. Reboredo, E. Reyes, J. L. Vicario, D. Badía, L. Carrillo, A. de Cózar and F. Cossío, An Amine-Catalyzed Enantioselective [3 + 2] Cycloaddition of Azomethine Ylides and α,β-Unsaturated Aldehydes: Applications and Mechanistic Implications, Chem. – Eur. J., 2012, 18, 7179 CrossRef CAS PubMed; (c) M. Shi, J. K. Jiang, Y. M. Shen, Y. S. Feng and G. X. Lei, An Unexpected Carbon Dioxide Insertion in the Reaction of Trans-2,4-Disubstituted Azetidine, Trans-2,5-Disubstituted Pyrrolidine, or Trans-2,6-Disubstituted Piperidine with Diphenylthio-phosphinic Chloride and Diphenylselenophosphinic Chloride, J. Org. Chem., 2000, 65, 3443 CrossRef CAS PubMed.
- Computational studies were carried out using the Gaussian 16 suite of programs at the M06-2X/6-311+G(d,p) level, with the IEF-PCM implicit solvent method. For further details, see the ESI.†.
- (a) F. H. Burstall, Optical activity dependent on co-ordinated bivalent ruthenium, J. Chem. Soc., 1936, 173 RSC; (b) J. A. Broomhead, C. G. Young and P. Hood, Tris(2,2′-Bipyridine)Ruthenium(II) Dichloride Hexahydrate, Inorg. Synth., 1982, 21, 127 CAS.
- CrysAlis RED, version 1.171.27p8, Oxford Diffraction Ltd., Oxford, U.K., 2005 Search PubMed.
- SAINT; Version 5.0 ed, Bruker Analytical X-Ray Systems, Madison, WI, 1998 Search PubMed.
- G. M. Sheldrick, SADABS, Program for absorption and other corrections, Göttingen University, 1996 Search PubMed.
- G. M. Sheldrick, SHELXS-86, Phase annealing in SHELX-90: direct methods for larger structures, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef.
- G. M. Sheldrick, SHELXL-97, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- (a) E. Cancés, B. Mennucci and J. Tomasi, A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics, J. Chem. Phys., 1997, 107, 3032 CrossRef; (b) M. Cossi, V. Barone, B. Mennucci and J. Tomasi, Ab initio study of ionic solutions by a polarizable continuum dielectric model, Chem. Phys. Lett., 1998, 286, 253 CrossRef CAS; (c) J. Tomasi, B. Mennucci and E. Cancès, The IEF version of the PCM solvation method: an overview of a new method addressed to study molecular solutes at the QM ab initio level, J. Mol. Struct.: THEOCHEM, 1999, 464, 211 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2176207, 2177354 and 2177355. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob00284e |
| This journal is © The Royal Society of Chemistry 2023 |