
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Site-selective introduction of thiols in unprotected glycosides†
Niels R. M.
Reintjens
,
Martin D.
Witte
 * and
Adriaan J.
Minnaard
*
* and
Adriaan J.
Minnaard
*
Stratingh Institute for Chemistry, University of Groningen, Groningen 9747 AG, The Netherlands. E-mail: a.j.minnaard@rug.nl; m.d.witte@rug.nl
First published on 2nd June 2023
Abstract
Thioglycosides or S-linked-glycosides are important glycomimetics. These thioglycosides are often prepared by glycosylating deoxythio sugar acceptors, which are synthesized via elaborate protecting group manipulations. We discovered that a carbonyl group, formed by site-selective oxidation of unprotected saccharides, can be converted into a thiol moiety. The transformation involves SN1-substitution of a chloro-azo intermediate, formed by oxidation of the corresponding trityl hydrazone, with a thiol. The prepared deoxythio sugars provide, in combination with the recently developed protecting group-free glycosylation of glycosyl fluorides, a protecting group-free synthesis of thioglycosides.
Introduction
Sulfur-substituted glycosides play an important role in carbohydrate chemistry and glycobiology. S-Linked-glycosides are more stable towards hydrolysis than their O-linked-glycoside counterparts, and have more conformational freedom, and sulfur therefore acts as a bioisostere in glycomimetics.1,2 Enzymatic3–5 or chemical6–11 glycosylation of deoxythio sugar acceptors is commonly used to prepare S-glycosides. The acceptors used in the glycosylation reactions typically have a secondary thiol group either at the C2, C3 or C4 position, or a primary thiol at C6. The introduction of these thiol groups is mostly achieved via substitution of a triflate, tosylate, or iodide with thioacetate as the nucleophile. A different route uses a diazonium intermediate, prepared by oxidative deamination of amino sugars, and has been used specifically for N-acetylneuraminic acid derivatives.12–14 The introduction of these leaving groups is designed in such a way that all-but-one hydroxy groups are protected.15–24 This requires several protecting group manipulations, which are well-developed for monosaccharides but are challenging for larger carbohydrates.The field of site-selective and late-stage modification of protection-group free mono- and oligosaccharides, however, is rapidly expanding and this should lead to alternative approaches.25–29 Substitution reactions in unprotected carbohydrates are complicated, however, because of intramolecular competition of the hydroxy groups.30 Another drawback is that the substitution reactions follow an SN2-pathway and lead to inversion of configuration. Replacing a secondary hydroxy group in a glycoside with retention of configuration requires therefore either double inversion, or prior inversion of the stereochemistry of the hydroxy group.
Recently, we reported a method to combine palladium-catalyzed site-selective oxidation of carbohydrates31–34 with subsequent trityl hydrazone formation, followed by conversion into the corresponding deoxychloro sugars (Scheme 1).35 Subjecting trityl hydrazone 1 to tert-butyl hypochlorite (tBuOCl) leads to formation of a chloro-azo intermediate I. Upon thermolysis in the presence of a thiol as H-atom donor, chlorosugar 2 is formed and isolated as an epimeric mixture.
 | ||
| Scheme 1 Functionalization of carbohydrate-derived trityl hydrazones after treatment with tBuOCl via direct thermolysis with a sterically hindered H-atom donor to exclusively form deoxychloro sugar 2, as reported previously, or via the here reported substitution-thermolysis sequence to form deoxythio sugar 3. | ||
We noted that the use of ethanethiol occasionally led to the formation of the corresponding thioether 3a as a side product. This unintentional thioether formation inspired us to study this side reaction for the formation of deoxythio glycosides. The possibility to capitalize on the well-developed site-selective oxidation of glycopyranosides in a formal deoxy-thiolation reaction would allow site-selective introduction of a thiol group in an unprotected carbohydrate. This potentially alleviates the need to pre-install a stereochemically defined leaving group.
We report here a new method to introduce a thioether or thioacetyl group in unprotected glycosides (Scheme 1) and its application in the synthesis of thioglycosides.
Results and discussion
Initially, we hypothesized that during the dehydroxy-chlorination reaction of 1 with EtSH as H-atom donor, thioether 3a was formed from 3-chloro-3-deoxy-glucoside 2. In an attempt to reproduce this substitution reaction, we heated a solution of 2 and EtSH in dimethyl acetamide (DMA, Scheme 2). | ||
| Scheme 2 Substitution reactions of 3-chloro-deoxy-glucoside 2 and the structure of epoxide 6. | ||
No conversion of 2 was observed, however, even upon heating to 130 °C. In a second attempt, sodium ethanethiolate was used, but heating to 50 °C resulted in full conversion of 2 into a mixture of unidentifiable products. These results strongly indicated that thioether 3a is formed via a different reaction pathway and not via SN2 substitution of 2.
Although the introduction of a thiol moiety was not possible in this way, we were able to use the chloride as a leaving group in the synthesis of azide 4 (ESI†).36 In accordance with earlier studies,37 also the intramolecular substitution reaction of equatorial 3-chloro-deoxy-glycosides was feasible, even on disaccharides. Addition of tert-butoxide as a base afforded the corresponding 2,3-epoxides 5 and 6 cleanly.
Since the substitution reactions on 2 with thiols did not give the thioether products, we focused our attention on the thermolysis step (Scheme 3, ESI Table S2†). In the dehydroxy-chlorination reaction, we did not observe the thioether product when we used a sterically hindered hydrogen atom donor (tert-butyl thiol) and quickly raised the temperature to 60 °C after addition of the thiol (Table S2,† entry 1). However, the thioether was the major product, when thermolysis of the chloro-azo intermediate was performed with an excess of ethanethiol at 40 °C for two hours. Under these conditions, chloride 2 was isolated in only 19% and thioether 3a was obtained in 74% (Table S2,† entry 2). Recent work by Creary shows that chloro-azo compounds generate resonance-stabilized carbocations that undergo SN1 substitution reactions.38,39 This stabilization is also the reason why chloro-azo compounds undergo rapid solvolysis compared to regular alkyl chlorides. Based on Creary's work, we reasoned that the yield of the thioether product could be increased by slowly raising the temperature after addition of the thiol. We were pleased to observe that by decreasing the temperature of the thermolysis step to 10 °C and prolonging the reaction time to five hours, thioether 3a was indeed formed in a near quantitative yield (97%) (Scheme 1).
 | ||
| Scheme 3 Direct thermolysis of I with tBuSH during the dehydroxy-chlorination reaction and the proposed mechanism for the deoxy-thiolation reaction by performing the thermolysis at lower temperature. The ratio for 3a is given as an equatorial/axial ratio. | ||
We hypothesize that at lower temperature, the thermolysis of chloro-azo intermediate I is sufficiently slowed down to allow the formation of cation II, which readily reacts with the thiol, present in excess, to form the thioether-azo compound III (Scheme 3). At slightly elevated temperature, intermediate III undergoes subsequent thermolysis to produce thioether 3a. Like in the dehydroxy-chlorination reaction,35 the stereochemical outcome of this reaction is determined in the hydrogen atom-transfer step. The H-atom donor approaches the radical preferentially from the less hindered equatorial face of the substrate, which leads to an axial thioether substituent.
Selecting the appropriate hydrazone turned out to be crucial for this deoxy-thiolation reaction. Treatment of a readily prepared dinitrophenyl chloro-azo glucoside35 with EtSH did not give any conversion, whereas the use of the unsubstituted phenyl hydrazone led to decomposition (ESI Scheme S3†). This shows that trityl hydrazones provide the right balance between stability and reactivity in this reaction.
This new deoxy-thiolation reaction nicely complements the scarce methods available in literature for this transformation. A recent and comprehensive overview by Li et al. describes the deoxygenative functionalization of aldehydes, ketones and carboxylic acids.40 Reductive thioetherification (or “reductive sulfanylation”) of ketones has been elaborated by Roth et al. and for benzylic compounds recently by Perrio et al.41,42 Both methods apply a combination of a thiol and a Brønsted – (triflic acid, CF3CO2H) or Lewis acid (BF3) with either Et3SiH or BH3 as a reducing agent. As expected, these reagents are not compatible with unprotected carbohydrates and our attempts to use these methods for the deoxy-thiolation of methyl 3-ketoglucose met with failure. Copper- or photochemical mediated oxidation of hydrazones to the corresponding diazo intermediate and subsequent carbene insertion into the S–H bond of a thiol has been reported, but is restricted to benzylic ketones.43–45 Conversion of tosylhydrazones under thermal basic conditions in the presence of thiols, can also lead to thioether formation.46 The group of König reported that tosylhydrazones can be used in an umpolung difunctionalisation reaction with photoredox catalysis.47,48 Although focusing on aromatic aldehydes as starting materials, also cyclic ketones were successfully explored and we expect that this approach can be adapted for the synthesis of thioethers. Currently, the chlorination of trityl hydrazone followed in situ by nucleophilic substitution of the chloride and subsequent thermolysis, is the only viable method for deoxy-thiolation in carbohydrates and therefore we continued by exploring its scope.
With the optimized conditions for the deoxy-thiolation reaction in hand, we first investigated the introduction of different thiol moieties at the C3 position of glucose. Performing the reaction with thiophenol (PhSH) and benzyl thiol (BnSH) gave the corresponding thioethers 3b and 3c in high yields (Scheme 4), again favoring the products with an axial thio-substituent. The excess of PhSH and BnSH was readily removed by extraction which eased column purification. Pleasingly, substitution with thioacetic acid (AcSH) gave 3d as an epimeric mixture, with a slight preference for the equatorial thioacetate product. This tendency of thioacetic acid to provide the equatorial substituted product had already been observed in the dehydroxy-chlorination reaction.35 The stereochemical outcome of the reaction is determined during the H-atom transfer step. As is in the dehydroxy-chlorination reaction, sterically hindered H-atom donors gave primarily the axial products, while small H-atom donors, such as thioacetic acid gave a slight preference for the equatorial product. Although the level of stereoselectivity is currently suboptimal, the result is important as it means that the C3 hydroxy group in glucose can be substituted for a C3 thiol group with overall retention of configuration. The acetyl group is readily removed by hydrolysis, liberating the thiol for use as a nucleophile in further glycosylation reactions, vide infra. This reaction was scaled to 6.0 mmol, which gave 3d in 58% yield. The yield was somewhat lower than expected, which is attributed to competing acetyl migration.23,49 The substitution reaction was also attempted with ten equivalents of 2,3,4,6-tetra-O-acetyl-β-thioglucose. Unfortunately, exclusively chloride 2 was isolated, indicating that the substitution with a sterically hindered thiol under the used reaction conditions is unsuccessful. Increasing the equivalents of the nucleophile to 80 was not considered for practical reasons.
 | ||
Scheme 4 Synthesis of deoxythio sugars. The major products are drawn. In brackets the isolated yields and the equatorial/axial ratio. Reactions with monosaccharides were performed on 0.4 mmol scale, with disaccharides on 0.25 mmol scale. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Raised to 10 °C; braised to 15 °C; cRaised to 5 °C. Raised to 10 °C; braised to 15 °C; cRaised to 5 °C. | ||
Subsequently, the carbohydrate scope was expanded (Scheme 4). Xylose derivative 7 was equipped with several thio-substituents in moderate yields (8a–c). Installing a thio-substituent at the C4 position turned out to be challenging due to side product formation and the desired thioether product 10 was isolated in 22% yield. The synthesis of C3′-thio-substituted maltose 12 in 57% yield shows the versatility of this novel strategy which is not limited to monosaccharides. The axial and equatorial epimer were separated by column chromatography.
In cases in which carbocation formation is slow or does not occur, the chlorosugar instead of the thiosugar is obtained. The trityl hydrazone of N-acetyl glucosamine for example, solely provided the chlorosugar. We hypothesize that the electron withdrawing effect of the acetamido group prevents carbocation formation. To substantiate our hypothesis, acetylated glucose 1 was subjected to the deoxy-thiolation conditions with EtSH. Also here, the thioether was not formed and only the corresponding chlorosugar was obtained (ESI Scheme S5†).
β-Glycosides give mixed results in the reaction. Performing the deoxy-thiolation reaction on methyl-β-glucoside 13 gave the expected benzyl thiol 14 in 21% yield, together with 54% of the corresponding chloride 15 and a small amount of a ring-contracted side product.50,51 With cellobiose 16 as substrate, only the deglycosylated product 17 was isolated. Based on recent work by Hansen et al.,52 we hypothesize that in alpha-glycosides a driving force for carbocation formation is the release of steric strain upon departure of chloride. The destabilization by the 1,3-diaxial interaction between the axial azobenzene substituent and the anomeric position is less pronounced in beta-glycosides and therefore, we reason that these substrates are less reactive.
An important application of unprotected thiosugars resides in protection-group free glycosylation reactions.25 Whereas, the group of Pedersen used in situ generated thiosugars for the synthesis of thiotrehalose,53 Withers and co-workers reported in the early 2000s the chemoenzymatic synthesis of thioglycosides from deoxythio sugars and glycosyl fluoride donors.3–5 The recent advances in development of methods to chemically activate the anomeric position in unprotected donors led to a steep growth in the development of protection-group free glycosylation reactions.54–59 In particular, the finding by Schepartz and Miller that unprotected glycosyl fluoride donors can be activated with calcium hydroxide and selectively coupled to sucrose acceptors60 or phenolic acceptors61 paved the path for the synthesis of S-linked glycosides. Both Tang et al. and Zhang et al. independently showed that the same activation conditions can be used for the synthesis of S-linked disaccharides and oligosaccharides.7,62 We here illustrate the versatility of the combination of, now readily available, deoxythio sugars and glucosyl fluorides by preparing disaccharides from thiosugars 18 and 21. To separate the epimeric mixture obtained from the deoxy-thiolation reaction, 3d was per-acetylated. Column chromatography gave pure fractions of the axial and the equatorial isomer, together with a mixed fraction due to the minor difference in Rf value. Thiosugars 18 and 21 were obtained by full deacetylation of the pure fractions and these thiosugars were then coupled to donor 19, which gave S-glycosides 20 and 22 (Scheme 5). Interestingly, we found that the products could be isolated without derivatization, making protecting groups in these reactions obsolete.
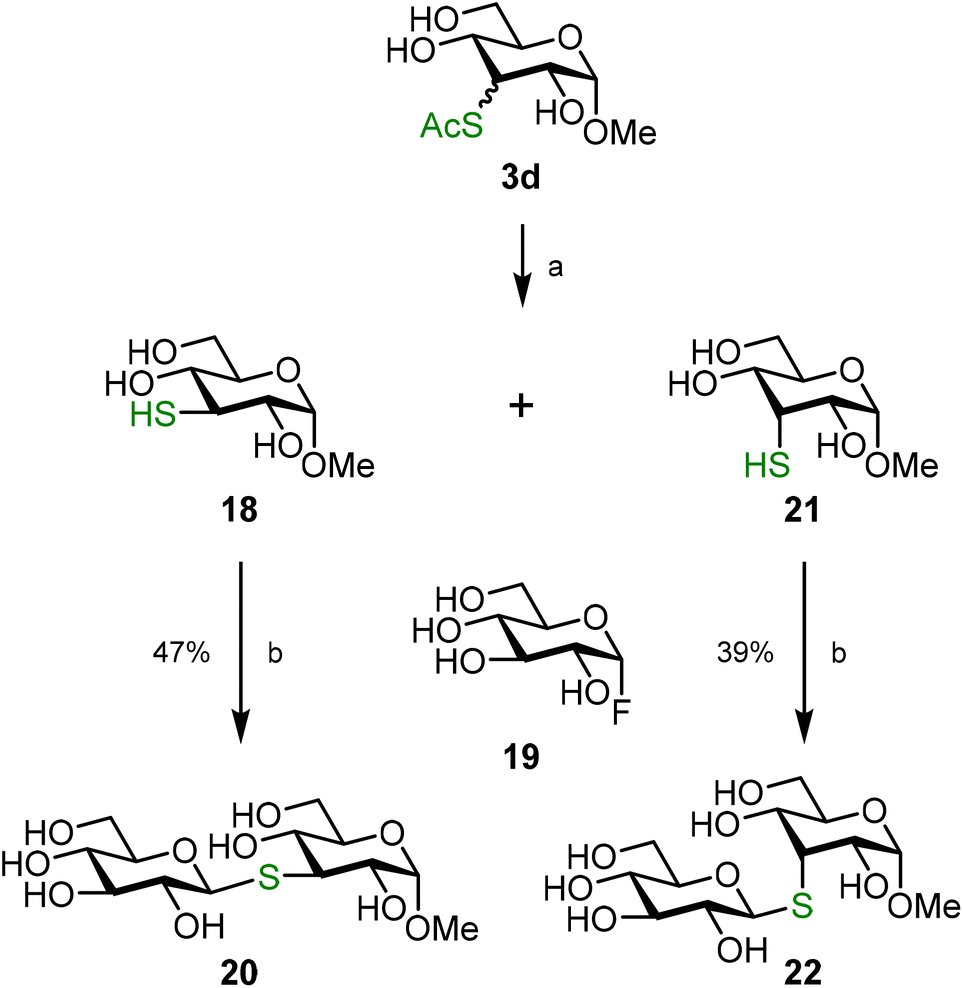 | ||
| Scheme 5 Synthesis of disaccharides 20 and 22. Reagents and conditions: (a) i. Ac2O, pyridine, 1.5 h, 98%; ii. NaOMe, MeOH, 1 h, 18: 83%, 21: 70%; (b) Ca(OH)2, H2O, 5 h. | ||
Conclusions
The discovery of a new deoxy-thiolation reaction allows the protecting group-free synthesis of deoxythio sugars. In combination with the site-selective oxidation of unprotected carbohydrates, which can be carried out in several ways, this satisfies an unmet need. Trityl-hydrazones, prepared from the corresponding keto-saccharides, provide chloro-azo intermediates upon treatment with tBuOCl. By selecting the appropriated thermolysis conditions, these chloro-azo intermediates can be converted either into chlorides, as shown previously, or into thioethers and thioacetates. The use of thioacetate as nucleophile gives mainly the product with overall retention of configuration. This versatile deoxy-thiolation reaction provides an efficient entry to the synthesis of thioglycosides with minimal use of protecting groups.The chlorosugars allow the introduction of an azide via SN2 substitution and in addition allow the introduction of an epoxide. This is versatile in particular for disaccharides as it avoids elaborate protecting group strategies. The current protocol is effective in particular for alpha-glucosides, and current studies aim to control the formation of the intermediate resonance-stabilized carbocation. In our opinion this is the key to expand the scope of this reaction further.
Author contributions
N. R. M. R. performed the experiments and analysed the data. M. D. W., A. J. M. conceived and directed the project. N. R. M. R., M. D. W., and A. J. M. prepared the manuscript. All authors discussed the experimental results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.References
- S. Zhu, G. Samala, E. T. Sletten, J. L. Stockdill and H. M. Nguyen, Chem. Sci., 2019, 10, 10475–10480 RSC.
- H. Driguez, ChemBioChem, 2001, 2, 311–318 CrossRef CAS PubMed.
- M. Jahn, J. Marles, R. A. J. Warren and S. G. Withers, Angew. Chem., Int. Ed., 2003, 42, 352–354 CrossRef CAS PubMed.
- M. Jahn, H. Chen, J. Müllegger, J. Marles, R. A. J. Warren and S. G. Withers, Chem. Commun., 2004, 274–275 RSC.
- Y.-W. Kim, A. L. Lovering, H. Chen, T. Kantner, L. P. McIntosh, N. C. J. Strynadka and S. G. Withers, J. Am. Chem. Soc., 2006, 128, 2202–2203 CrossRef CAS PubMed.
- V. Kelemen and A. Borbás, in Recent Trends in Carbohydrate Chemistry, Elsevier, 2020, pp. 161–215 Search PubMed.
- G.-L. Zhang, M. R. Gadi, X. Cui, D. Liu, J. Zhang, V. Saikam, C. Gibbons, P. G. Wang and L. Li, Green Chem., 2021, 23, 2907–2912 RSC.
- L. Lázár, A. Borbás and L. Somsák, Carbohydr. Res., 2018, 470, 8–12 CrossRef PubMed.
- I. Cumpstey, D. S. Alonzi and T. D. Butters, Carbohydr. Res., 2009, 344, 454–459 CrossRef CAS PubMed.
- O. Norberg, B. Wu, N. Thota, J.-T. Ge, G. Fauquet, A.-K. Saur, T. Aastrup, H. Dong, M. Yan and O. Ramström, Carbohydr. Res., 2017, 452, 35–42 CrossRef CAS PubMed.
- T. Xiong, R. Xie, C. Huang, X. Lan, N. Huang and H. Yao, J. Carbohydr. Chem., 2021, 40, 401–439 CrossRef CAS.
- V. A. Sarpe and D. Crich, Carbohydrate Chemistry: Chemical and Biological Approaches, 2022, vol. 44, pp. 1–30 Search PubMed.
- M. Hawsawi, A. Wickramasinghe and D. Crich, J. Org. Chem., 2019, 84, 14688–14700 CrossRef CAS PubMed.
- K. Qin, H. Zhang, Z. Zhao and X. Chen, J. Am. Chem. Soc., 2020, 142, 9382–9388 CrossRef CAS PubMed.
- S. Knapp, A. B. J. Naughton, C. Jaramillo and B. Pipik, J. Org. Chem., 1992, 57, 7328–7334 CrossRef CAS.
- C. Simiand, E. Samain, O. R. Martin and H. Driguez, Carbohydr. Res., 1995, 267, 1–15 CrossRef CAS PubMed.
- R. V. Stick and K. A. Stubbs, Tetrahedron: Asymmetry, 2005, 16, 321–335 CrossRef CAS.
- L. Gabrielli, I. Calloni, G. Donvito, B. Costa, N. Arrighetti, P. Perego, D. Colombo, F. Ronchetti, F. Nicotra and L. Cipolla, Eur. J. Org. Chem., 2014, 5962–5967 CrossRef CAS.
- Y. Zhou, X. Zhang, B. Ren, B. Wu, Z. Pei and H. Dong, Tetrahedron, 2014, 70, 5385–5390 CrossRef CAS.
- B. Wu, J. Ge, B. Ren, Z. Pei and H. Dong, Tetrahedron, 2015, 71, 4023–4030 CrossRef CAS.
- A. A. Lee, Y.-C. S. Chen, E. Ekalestari, S.-Y. Ho, N.-S. Hsu, T.-F. Kuo and T.-S. A. Wang, Angew. Chem., Int. Ed., 2016, 55, 12338–12342 CrossRef CAS PubMed.
- D. S.-E. Koffi Teki, A. Bil, V. Moreau, V. Chagnault, B. Fanté, A. Adjou and J. Kovensky, Org. Chem. Front., 2019, 6, 2718–2725 RSC.
- T. Luo, Y. Zhang, J. Xi, Y. Lu and H. Dong, Front. Chem., 2020, 8, 319 CrossRef CAS PubMed.
- W. Song, J. Cai, X. Zou, X. Wang, J. Hu and J. Yin, Chin. Chem. Lett., 2018, 29, 27–34 CrossRef CAS.
- M. D. Witte and A. J. Minnaard, ACS Catal., 2022, 12, 12195–12205 CrossRef CAS PubMed.
- N. Marinus, N. Tahiri, M. Duca, L. M. C. M. Mouthaan, S. Bianca, M. van den Noort, B. Poolman, M. D. Witte and A. J. Minnaard, Org. Lett., 2020, 22, 5622–5626 CrossRef CAS PubMed.
- C. E. Suh, H. M. Carder and A. E. Wendlandt, ACS Chem. Biol., 2021, 16(10), 1814–1828 CrossRef CAS PubMed.
- V. Dimakos and M. S. Taylor, Chem. Rev., 2018, 118, 11457–11517 CrossRef CAS PubMed.
- D. J. Gorelik, J. A. Turner and M. S. Taylor, Org. Lett., 2022, 24, 5249–5253 CrossRef CAS PubMed.
- J. G. Buchanan, J. Chem. Soc., 1958, 2511–2516 RSC.
- D. J. Gorelik, V. Dimakos, T. Adrianov and M. S. Taylor, Chem. Commun., 2021, 57, 12135–12138 RSC.
- M. Jäger, M. Hartmann, J. G. de Vries and A. J. Minnaard, Angew. Chem., Int. Ed., 2013, 52, 7809–7812 CrossRef PubMed.
- J. Zhang, N. N. H. M. Eisink, M. D. Witte and A. J. Minnaard, J. Org. Chem., 2019, 84, 516–525 CrossRef CAS PubMed.
- K. Chung and R. M. Waymouth, ACS Catal., 2016, 6, 4653–4659 CrossRef CAS.
- J. Zhang, N. R. M. Reintjens, J. Dhineshkumar, M. D. Witte and A. J. Minnaard, Org. Lett., 2022, 24, 5339–5344 CrossRef CAS PubMed.
- B. T. Lawton, W. A. Szarek and J. K. N. Jones, Carbohydr. Res., 1970, 15, 397–402 CrossRef CAS.
- J. K. N. Jones, M. B. Perry and J. C. Turner, Can. J. Chem., 1960, 38, 1122–1129 CrossRef CAS.
- X. Creary, J. Org. Chem., 2022, 87, 2241–2254 CrossRef CAS PubMed.
- R. E. MacLeay and C. S. Sheppard, PENNWALT CORP OP, US 45344474A OP – US 14904171A, 1977 Search PubMed.
- J. Li, C. Huang and C. Li, Angew. Chem., Int. Ed., 2022, 61, e202112770 CAS.
- B. A. Gellert, N. Kahlcke, M. Feurer and S. Roth, Chem. – Eur. J., 2011, 17, 12203–12209 CrossRef CAS PubMed.
- D. Deschamps, J.-F. Lohier, C. J. Richards, A.-C. Gaumont and S. Perrio, J. Org. Chem., 2021, 86, 507–514 CrossRef CAS PubMed.
- S. Chand, A. K. Pandey, R. Singh, S. Kumar and K. N. Singh, Chem. – Asian J., 2019, 14, 4712–4716 CrossRef CAS PubMed.
- A. K. Pandey, S. Chand, A. K. Sharma and K. N. Singh, J. Org. Chem., 2023, 88, 475–482 CrossRef CAS PubMed.
- X. Gao, Z. Song, J. Hu and H. Zhang, Polymers, 2022, 248, 124825 CrossRef CAS.
- T. Kaszás, M. Tóth and L. Somsák, New J. Chem., 2017, 41, 13871–13880 RSC.
- S. Wang, B.-Y. Cheng, M. Sršen and B. König, J. Am. Chem. Soc., 2020, 142, 7524–7531 CrossRef CAS PubMed.
- S. Wang and B. König, Angew. Chem., Int. Ed., 2021, 60, 21624–21634 CrossRef CAS PubMed.
- Y. Zhou, X. Zhang, B. Ren, B. Wu, Z. Pei and H. Dong, Tetrahedron, 2014, 70, 5385–5390 CrossRef CAS.
- T. Tsuchiya, K. Ajito, S. Umezawa and A. Ikeda, Carbohydr. Res., 1984, 126, 45–60 CrossRef CAS.
- A. El Nemr and T. Tsuchiya, Carbohydr. Res., 1997, 303, 267–281 CrossRef CAS.
- T. Hansen, P. Vermeeren, F. M. Bickelhaupt and T. A. Hamlin, Chem. Commun., 2022, 58, 12050–12053 RSC.
- J. Defaye, A. Gadelle and C. Pedersen, Carbohydr. Res., 1991, 217, 51–58 CrossRef CAS PubMed.
- N. Yoshida, M. Noguchi, T. Tanaka, T. Matsumoto, N. Aida, M. Ishihara, A. Kobayashi and S. Shoda, Chem. – Asian J., 2011, 6, 1876–1885 CrossRef CAS PubMed.
- A. Novoa, S. Barluenga, C. Serba and N. Winssinger, Chem. Commun., 2013, 49, 7608–7610 RSC.
- S. Köhling, M. P. Exner, S. Nojoumi, J. Schiller, N. Budisa and J. Rademann, Angew. Chem., Int. Ed., 2016, 55, 15510–15514 CrossRef PubMed.
- S. R. Alexander, D. Lim, Z. Amso, M. A. Brimble and A. J. Fairbanks, Org. Biomol. Chem., 2017, 15, 2152–2156 RSC.
- A. J. Fairbanks, Carbohydr. Res., 2021, 499, 108197 CrossRef CAS PubMed.
- X. Qiu, A. L. Garden and A. J. Fairbanks, Chem. Sci., 2022, 13, 4122–4130 RSC.
- G. Pelletier, A. Zwicker, C. L. Allen, A. Schepartz and S. J. Miller, J. Am. Chem. Soc., 2016, 138, 3175–3182 CrossRef CAS PubMed.
- T. J. Wadzinski, A. Steinauer, L. Hie, G. Pelletier, A. Schepartz and S. J. Miller, Nat. Chem., 2018, 10, 644–652 CrossRef CAS PubMed.
- P. Wen, P. Jia, Q. Fan, B. J. McCarty and W. Tang, ChemSusChem, 2022, 15, e202102483 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob00817g |
| This journal is © The Royal Society of Chemistry 2023 |