
Asymmetric total synthesis of diosniponols A and B†
Deepak
Saini‡
,
Dashrath
Jangid‡
and
Rodney A.
Fernandes
 *
*
Department of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai 400076, Maharashtra, India. E-mail: rfernand@chem.iitb.ac.in; Fax: +9122 25767152; Tel: +91 22 25767174
First published on 21st July 2023
Abstract
A concise asymmetric total synthesis of diosniponols A and B has been achieved based on an enantioselective Jacobsen kinetic resolution of racemic epoxide and the important 2,3-dihydro-4H-pyran-4-one moiety being installed by the metal-free δ-hydroxyalkynone rearrangement catalyzed by p-TsOH. A diastereoselective catalytic hydrogenation set the required all-syn stereochemistry leading to diosniponol A, which then, under the Mitsunobu inversion conditions, provided diosniponol B. The structure and absolute stereochemistry of the natural products were further confirmed.
Introduction
Diaryl motif bearing compounds are ubiquitous in nature and because of their impressive bioactivities like anticancer,1 antidepressant,2 antioxidative,3 and antifungal activities,4 they have drawn significant attention; a few of the cyclic compounds 1a–e are depicted in Fig. 1. Lee and co-workers5 isolated two new cyclic diaryl heptanoids named diosniponols A (1a) and B (1b), having a tetrahydropyran core, from the rhizomes of Dioscorea nipponica, which is a perennial liana mainly distributed in Southeast Asia, Japan, China and Korea.6 Chen and co-workers7a isolated the cyclic diarylhaptanoids 1c–e from the roots and rhizomes of Dioscorea villosa L. (wild yam) and elucidated their structures and assigned their absolute configuration by the combination of HRESIMS, 1D and 2D NMR, 1H iterative full spin analysis (HiFSA), and the Mosher ester method. However, later they revised the structures of 1c–e to those of their enantiomers ent-1c–e.7b The diaryl heptanoids showed anti-inflammatory8 and anti-neuroinflammatory5 properties. The tetrahydropyran moiety is present in many natural products and synthetic compounds that have found application in the medicinal and pharmaceutical industries.9 Most commonly, the 2,6-cis- and 2,6-trans-disubstituted-THP moiety is found in many natural products,10 including those having the C-4 hydroxy functionality.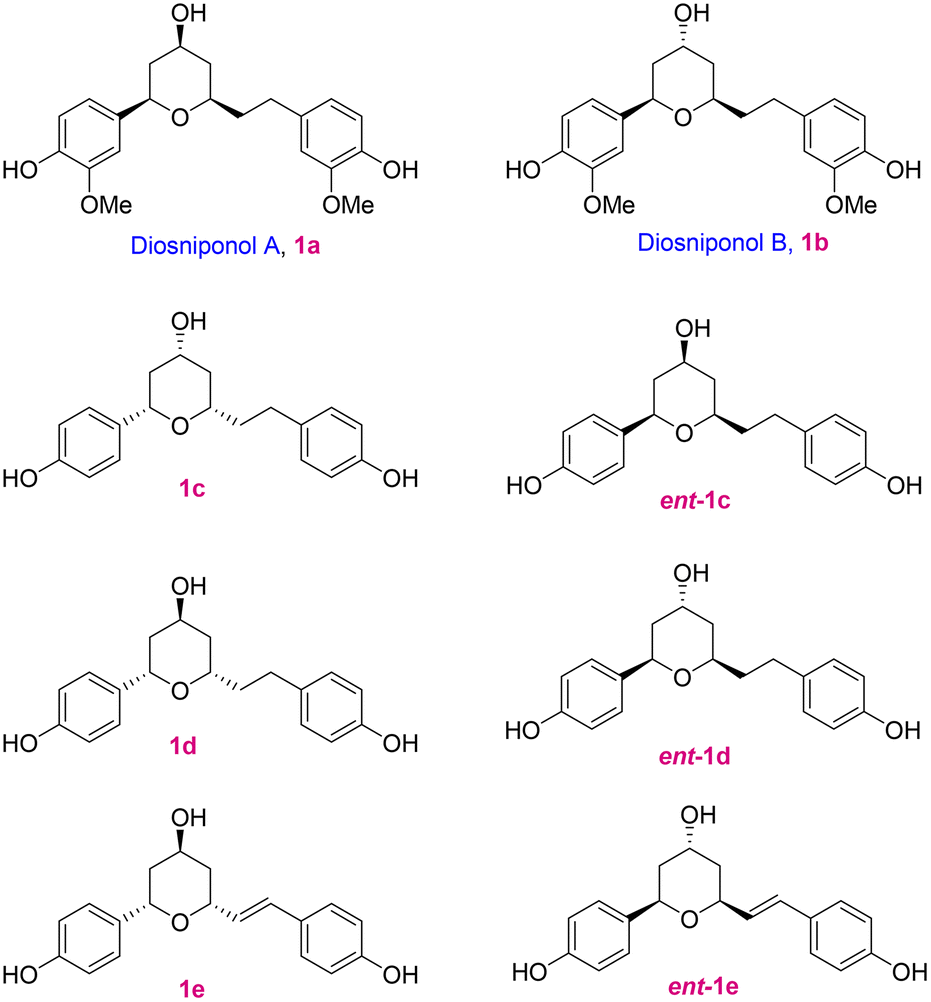 | ||
| Fig. 1 Examples of cyclic diaryl heptanoids including diosniponols. | ||
Initially, based on 2D NMR studies,11 the absolute stereochemistry of diosniponols A and B was assigned. In 2014, Yadav and co-workers12 synthesized diosniponols A and B using the Keck allylation and Prins cyclization reactions as the key steps. They assumed the configuration of the stereocentres given in the report on isolation to be true, i.e. 2S,4R,6S. However, the synthesis and comparison of the optical rotation values of the synthesized diosniponols with those of the isolates showed that they were opposite.5 Thus, the enantiomers of diosniponols A and B were synthesized and hence the absolute configuration reversal, i.e. 2R,4S,6R, was considered to be as shown in 1a and 1b for the natural products. In this work, we have considered the synthesis of the natural diosniponols A (2R,4S,6R) and B (2R,4R,6R) for the first time with the configurations as depicted in Fig. 1. With this we also confirm the absolute structure of these natural products.
Results and discussion
In retrosynthetic analysis, diosniponol B can be formally obtained in one step from diosniponol A by the Mitsunobu inversion13 of the C-4 hydroxyl group (Scheme 1). A diastereoselective hydrogenation of 2 was expected to deliver the all-syn configuration of diosniponol A. A metal-free δ-hydroxyalkynone rearrangement14 of 3 would give the desired dihydropyran 2. The former can be obtained by epoxide 4 opening with an alkyne. The Jacobsen kinetic resolution15 was planned to obtain the chiral epoxide 4, the racemate of which was considered to be obtained by epoxidation of requisite olefin derived from vanilin 5 by chain extension. The strategy is flexible and can be extended to other molecules, as shown in Fig. 1. | ||
| Scheme 1 Retrosynthesis of diosniponols A and B. | ||
The forward synthesis commenced from the commercially available vanillin 5 that on benzyl protection to 6 and aldehyde reduction led to benzyl alcohol 7 in good yields (Scheme 2). Next, the bromination of the alcoholic group using PBr3 at 0 °C gave the corresponding benzyl bromide which was then subjected to bromide displacement using freshly prepared allyl magnesium bromide to give alkene 8 in 68% yield over 2 steps. The latter on epoxidation with m-CPBA gave the racemic epoxide (±)-4 in 84% yield. Furthermore, the Jacobsen hydrolytic-kinetic resolution15 of the racemic epoxide using the (R,R)-Co-salen complex furnished the enantiopure epoxide 4 in 44% yield along with diol 9 (42%).16 The latter was efficiently recycled back to epoxide 4 under the conditions reported17 for similar compounds. These involved primary hydroxy protection as the pivaloate ester, the secondary alcohol to mesylate conversion and its displacement with the regenerated primary alcohol. The subsequent epoxide 4 opening in the presence of the Lewis acid BF3·OEt2 using trimethylsilyl–acetylene followed by desilylation using K2CO3 afforded alkynol 1016 in 85% yield (2 steps). Alkynol 10 upon a nucleophilic addition reaction with aldehyde 6 using n-BuLi as a base provided diol 11 in 78% yield as an inconsequential diastereomeric mixture. This was next subjected to selective benzylic oxidation using MnO2 to provide the desired δ-hydroxyalkynone 3 in 75% yield. The latter when subjected to metal-free rearrangement under p-TsOH (10 mol%) catalysis14 underwent smooth rearrangement in the MeOH solvent to give 2 in 84% yield. The mechanism of this step is shown in Fig. 2a. MeOH addition to alkynone 3 provides the allene intermediate A that tautomerizes to ketone B. This undergoes intramolecular hemiacetal formation to intermediate C. The latter on dehydration followed by hydrolysis delivers 2,3-dihydro-4H-pyran-4-ones 2. The ensuing Pd/C-catalyzed hydrogenation of 2,3-dihydro-4H-pyran-4-ones 2 furnished the thermodynamically more stable syn-2,4,6-trisubstituted-THP ring containing natural product diosniponol A 1a in 78% yield. Here, the enone functionality of 2,3-dihydro-4H-pyran-one 2 was selectively hydrogenated from the sterically less hindered side to deliver the thermodynamically more stable diosniponol A 1a having all bulky groups at the equatorial position (Fig. 2b). The advantageous benzyl protection was also concomitantly removed under the hydrogenation conditions. The spectral and analytical data of 1a were found to be similar to the reported data.12 The optical rotation of the synthesized 1a [α]28D = +43.8 (c 1.0, MeOH) matched well with that reported for the enantiomer [α]28D = −34.8 (c 0.4, MeOH)12 although the quantitative value varies with respect to the natural isolate [α]25D = +4.8 (c 0.06, MeOH).5 Finally, diosniponol A 1a was converted into diosniponol B 1b by using Mitsunobu inversion conditions13 in two steps and in 80% yield. The optical rotation of the synthesized 1b [α]28D = +32.4 (c 0.4, MeOH) matched well with that reported for the enantiomer [α]28D = −28.0 (c 0.4, MeOH)12 although the quantitative value varies with respect to the natural isolate [α]25D = +0.8 (c 0.005, MeOH).5 The spectral and analytical data match with those of the natural isolate,5 which also confirms the absolute stereochemistry of the natural products as 1a and 1b, respectively, for diosniponols A and B.
 | ||
| Scheme 2 Asymmetric total synthesis of diosniponols A and B. | ||
 | ||
| Fig. 2 (a) Mechanism of δ-hydroxyalkynone 3 rearrangement to 2,3-dihydro-4H-pyran-4-one 2. (b) Diastereoselective hydrogenation of 2,3-dihydro-4H-pyran-4-one 2. | ||
A comparative outlook of the optical rotations of diosniponols is given in Table 1. The sign of the optical rotation obtained by us was the same as that reported by Lee and co-workers5 for the isolated diosniponols. The values of the optical rotations were quantitatively matching and opposite to those reported by Yadav and co-workers12 for the synthesized enantiomers of diosniponols. Hence, we have achieved the first asymmetric synthesis of naturally occurring diosniponols. The lower values for the isolates may be attributed to the not enantiomerically pure compounds obtained during isolation. This is sometimes observed in the literature for some natural products.
Conclusion
In summary, we have completed the first asymmetric total synthesis of natural diosniponols A and B in 11 and 12 sequential steps with 7% and 5.5% overall yields, respectively. Our approach is concise, straightforward, reliable and convergent. The key steps involved are the Jacobsen kinetic resolution of racemic epoxide, p-TsOH-catalyzed rearrangement of δ-hydroxyalkynone to 2,3-dihydro-4H-pyran-3-one, all-syn catalytic hydrogenation and Mitsunobu inversion. The strategy can be further employed toward the synthesis of other diaryl cyclic heptanoids, as shown in Fig. 1.Experimental
General information
The solvents were dried by using standard procedures. Thin-layer chromatography was performed on EM 250 Kieselgel 60 F254 silica gel plates. The spots were visualized by staining with KMnO4 or by using a UV lamp. 1H NMR and 13C NMR spectra were recorded with a spectrometer operating at 400 and 100 MHz for proton and carbon nuclei, respectively. The chemical shifts are based on the TMS peak at δ = 0.00 ppm for proton NMR and the CDCl3 peak at δ = 77.00 ppm (t) for carbon NMR. IR spectra were obtained on an FT-IR spectrometer by evaporating compounds dissolved in CHCl3 on CsCl pellets. HRMS (ESI-TOF) spectra were recorded using positive electrospray ionization by the TOF method. Optical rotations were measured using a JASCO P-2000 digital polarimeter having sodium D-line (589 nm).4-(Benzyloxy)-3-methoxybenzaldehyde (6)
To a solution of vanillin 5 (600 mg, 3.943 mmol, 1.0 equiv.) in acetone (15 mL), benzyl bromide (742 mg, 4.338 mmol, 1.1 equiv.) was added, followed by anhydrous K2CO3 (817 mg, 5.915 mmol, 1.5 equiv.). The reaction mixture was refluxed for 3 h. After the completion of the reaction, the mixture was concentrated under reduced pressure and the residue was dissolved in EtOAc and water. The organic layer was separated and the aqueous layer was extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with water and brine, dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as an eluent to give aldehyde 6 (0.908 g, 95%) as a white solid. M.p. = 57–59 °C (lit.18 M.p. 60–61 °C). IR (CHCl3) υmax = 3064, 3032, 2939, 2834, 2730, 1681, 1590, 1508, 1464, 1424, 1388, 1235, 1160, 1134, 1020, 918, 866, 810, 737, 698 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.83 (s, 1H), 7.45–7.41 (m, 3H), 7.40–7.35 (m, 3H), 7.35–7.31 (m, 1H), 6.98 (d, J = 8.2 Hz, 1H), 5.25 (s, 2H), 3.95 (s, 3H) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ 190.7, 153.4, 149.9, 135.9, 130.1, 128.6, 128.0, 127.1, 126.4, 112.2, 109.2, 70.7, 55.8 ppm. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C15H14O3Na 265.0835, found 265.0840.
:1) as an eluent to give aldehyde 6 (0.908 g, 95%) as a white solid. M.p. = 57–59 °C (lit.18 M.p. 60–61 °C). IR (CHCl3) υmax = 3064, 3032, 2939, 2834, 2730, 1681, 1590, 1508, 1464, 1424, 1388, 1235, 1160, 1134, 1020, 918, 866, 810, 737, 698 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.83 (s, 1H), 7.45–7.41 (m, 3H), 7.40–7.35 (m, 3H), 7.35–7.31 (m, 1H), 6.98 (d, J = 8.2 Hz, 1H), 5.25 (s, 2H), 3.95 (s, 3H) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ 190.7, 153.4, 149.9, 135.9, 130.1, 128.6, 128.0, 127.1, 126.4, 112.2, 109.2, 70.7, 55.8 ppm. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C15H14O3Na 265.0835, found 265.0840.
4-Benzyloxy-3-methoxyphenyl-methanol (7)
To a stirred solution of aldehyde 6 (608 mg, 2.509 mmol, 1.0 equiv.) in MeOH (20 mL) was added NaBH4 (104.4 mg, 2.76 mmol, 1.1 equiv.) at 0 °C and the reaction mixture was stirred at 0 °C for 0.5 h. After the completion of the reaction, the mixture was quenched by the addition of saturated aq. NH4Cl and concentrated under reduced pressure. The residue was dissolved in EtOAc and water. The organic layer was separated and the aqueous layer was extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with water and brine, dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (7:3) as an eluent to give 7 (0.539 g, 88%) as a white solid. M.p. = 69–71 °C (lit.19 M.p. 70–71 °C). IR (CHCl3) υmax = 3283, 2905, 1609, 1583, 1511, 1249, 1108, 1085, 1015, 971, 811, 753, 737, 697 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.46–7.41 (m, 2H), 7.38–7.33 (m, 2H), 7.32–7.28 (m, 1H), 6.90 (d, J = 1.8 Hz, 1H), 6.82 (d, J = 8.1 Hz, 1H), 6.76 (d, J = 8.1, 1.9 Hz, 1H), 5.11 (s, 2H), 4.51 (s, 2H), 3.83 (s, 3H), 2.86 (br s, 1H) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ 149.4, 147.2, 136.9, 134.1, 128.3, 127.6, 127.0, 119.0, 113.7, 110.7, 70.8, 64.6, 55.6 ppm. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C15H16O3Na 267.0992, found 267.0989.
1-Benzyloxy-4-(but-3-en-1yl)-2-methoxybenzene (8)
To a stirred solution of alcohol 7 (1.0 g, 4.093 mmol, 1.0 equiv.) in Et2O (40 mL) was added PBr3 (0.2 mL, 2.05 mmol, 0.5 equiv.) at 0 °C and allowed to stir for 0.5 h at 0 °C. After the completion of the reaction, the mixture was quenched with ice water. The organic layer was separated and the aqueous layer was extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with water and brine, dried (Na2SO4) and concentrated under reduced pressure. The crude residue was directly used for the next step without further purification.To the stirred solution of the above-prepared crude bromide in THF (40 mL) was added a freshly prepared solution of allylmagnesium bromide (4.5 mL, 1.0 M solution in Et2O, 4.502 mmol, 1.1 equiv.) at 0 °C and the resulting suspension was stirred for 16 h. The reaction mixture was quenched by the addition of saturated aq. NH4Cl and the organic layer was separated and the aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with water and brine, dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (19:1) as an eluent to give 8 (0.747 g, 68%) as a colorless oil. IR (CHCl3) υmax 3030, 2952, 2928, 2884, 2858, 1601, 1584, 1495, 1471, 1463, 1386, 1362, 1255, 1222, 1095, 1072, 1004, 937, 837, 780, 698, 669 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 7.3 Hz, 2H), 7.39–7.33 (m, 2H), 7.32–7.27 (m, 1H), 6.80 (d, J = 8.1 Hz, 1H), 6.75 (d, J = 1.9 Hz, 1H), 6.67 (dd, J = 8.1, 1.6 Hz, 1H), 5.94–5.80 (m, 1H), 5.13 (s, 2H), 5.08–4.98 (m, 2H), 3.89 (s, 3H), 2.66 (t, J = 7.8 Hz, 2H), 2.41–2.30 (m, 2H) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ 149.4, 146.3, 138.0, 137.3, 135.1, 128.4, 127.6, 127.2, 120.1, 114.8, 114.1, 112.3, 71.1, 55.8, 35.6, 34.9 ppm; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H21O2 269.1536, found 269.1536.
2-(4-Benzyloxy-3-methoxyphenethyl)oxirane (±)-4
To a stirred solution of compound 8 (4.5 g, 16.769 mmol, 1.0 equiv.) in CH2Cl2 (50 mL) was added m-CPBA (65% w/w, 4.9 g, 18.445 mmol, 1.1 equiv.) at 0 °C. The reaction mixture was stirred at room temperature for 12 h. After the completion of the reaction, the mixture was quenched with saturated aq. NaHCO3 and the organic layer was separated and the aqueous layer was extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were washed with water and brine, dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by silica gel column chromatography with petroleum ether/EtOAc (5:1) as an eluent to give pure compound (±)-4 (4.0 g, 84%) as a white solid. M.p. = 58–60 °C. IR (CHCl3) υmax 2949, 2928, 2884, 2854, 1635, 1601, 1491, 1471, 1454, 1256, 1191, 1132, 1090, 991, 868, 836, 810, 777, 731, 698 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 7.3 Hz, 2H), 7.38–7.33 (m, 2H), 7.32–7.27 (m, 1H), 6.81 (d, J = 8.2 Hz, 1H), 6.75 (d, J = 1.9 Hz, 1H), 6.68 (dd, J = 8.1, 1.9 Hz, 1H), 5.13 (s, 2H), 3.88 (s, 3H), 2.97–2.92 (m, 1H), 2.80–2.65 (m, 3H), 2.47 (dd, J = 4.9, 2.7 Hz, 1H), 1.89–1.76 (m, 2H) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ 149.5, 146.3, 137.2, 134.4, 128.4, 127.6, 127.2, 120.1, 114.1, 112.1, 71.0, 55.8, 51.7, 47.1, 34.3, 31.7 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C18H20O3Na 307.1305, found 307.1302.
(R)-2-(4-Benzyloxy-3-methoxyphenethyl)oxirane (4)
Glacial acetic acid (200 μL, 3.41 mmol) was added dropwise to a solution of [(R,R)-N,N′-bis(3,5-di-tert-butylsalicylidene)-1,2-cyclohexanediamino] cobalt-(II) (24 mg, 0.04 mmol, 0.5 mol%) in toluene (6 mL) and the mixture was stirred at room temperature for 2 h, when the colour change from red to brown was observed. The solvent and remaining acid were removed in vacuo and 2-(4-benzyloxy-3-methoxyphenethyl)oxirane (±)-4 (2.28 g, 8.018 mmol) dissolved in THF (5 mL) was added. The reaction mixture was cooled to 0 °C and H2O (80 μL, 4.41 mmol) was added and the mixture was warmed to room temperature and stirred for 24 h. The mixture was concentrated under reduced pressure and the residue was purified by silica gel column chromatography using petroleum ether/EtOAc (9:1) as an eluent to give the title compound 4 (1.0 g, 44%) as a white solid; further elution with petroleum ether/EtOAc (7:3) as an eluent gave diol 9 (1.02 g, 42%) as a white solid.
Data for 4: [α]25D = +14.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 7.2 Hz, 2H), 7.39–7.33 (m, 2H), 7.32–7.27 (m, 1H), 6.80 (d, J = 8.1 Hz, 1H), 6.75 (d, J = 1.7 Hz, 1H), 6.68 (dd, J = 8.1, 1.7 Hz, 1H), 5.13 (s, 2H), 3.88 (s, 3H), 2.99–2.91 (m, 1H), 2.80–2.65 (m, 3H), 2.48 (dd, J = 4.9, 2.7 Hz, 1H), 1.92–1.75 (m, 2H) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ 149.6, 146.4, 137.3, 134.5, 128.5, 127.7, 127.2, 120.2, 114.2, 112.2, 71.1, 55.9, 51.7, 47.2, 34.4, 31.8 ppm.
Data for 9: M.p. = 65–67 °C. [α]25D = −6.0 (c 1.0, CHCl3); IR (CHCl3): υmax 3380, 2929, 2865, 1589, 1514, 1454, 1418, 1378, 1333, 1260, 1227, 1158, 1137, 1099, 1031, 1003, 857, 804, 749 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 7.0 Hz, 2H), 7.38–7.32 (m, 2H), 7.31–7.27 (m, 1H), 6.79 (d, J = 8.1 Hz, 1H), 6.74 (d, J = 1.9 Hz, 1H), 6.65 (dd, J = 8.1, 1.9 Hz, 1H), 5.10 (s, 2H), 3.85 (s, 3H), 3.72–3.66 (m, 1H), 3.65–3.58 (m, 1H), 3.43 (dd, J = 11.1, 7.7 Hz, 1H), 3.10 (br s, 2H), 2.77–2.69 (m, 1H), 2.65–2.55 (m, 1H), 1.78–1.63 (m, 2H) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ 149.5, 146.3, 137.2, 134.9, 128.4, 127.7, 127.2, 120.1, 114.2, 112.3, 71.5, 71.1, 66.6, 55.9, 34.7, 31.3 ppm; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H23O4 303.1591, found 303.1583.
(R)-2-(4-Benzyloxy-3-methoxyphenethyl)oxirane (4) from diol 9
To a solution of diol 9 (900 mg, 2.976 mmol, 1.0 equiv.) in pyridine (15 mL) at 0 °C was added trimethylacetyl chloride (0.5 mL, 3.87 mmol, 1.3 equiv.) dropwise and allowed to stir at room temperature for 8 h. The reaction mixture was diluted with EtOAc (50 mL) and washed with 1 N aq. HCI (3 × 10 mL). The organic layer was separated and the aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with water and brine, dried (Na2SO4) and concentrated under reduced pressure to give the pivaloyl protected alcohol as a colourless oil.The crude pivaloate was dissolved in pyridine (15 mL) followed by the addition of a catalytic amount of DMAP at 0 °C under a N2 atmosphere and the dropwise addition of methanesulfonyl chloride (0.35 mL, 4.464 mmol, 1.5 equiv.) and allowed to warm to room temperature and stirred for 24 h. The reaction mixture was diluted with Et2O (50 mL) and then washed with 1 N aq. HCI (3 × 10 mL). The organic layer was separated and the aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with water and brine, dried (Na2SO4) and concentrated under reduced pressure to give a light-yellow oil. The crude oil was dissolved in MeOH (25 mL) and K2CO3 (1.233 g, 8.928 mmol, 3.0 equiv.) was added and allowed to stir for 8 h at room temperature. The mixture was diluted by the addition of Et2O/hexane (1:1, 50 mL) followed by filtration through a short plug of silica gel. The solvent was removed under reduced pressure and the residue was purified by silica gel column chromatography using petroleum ether/EtOAc (9:1) as an eluent to give the title compound 4 (635 mg, 75%) as a white solid. The spectral and analytical data are the same as before.
(R)-1-(4-Benzyloxy-3-methoxyphenyl)hex-5-yn-3-ol (10)
To a solution of trimethylsilyl acetylene (0.3 mL, 2.11 mmol, 1.5 equiv.) in THF (20 mL) was added n-BuLi (1.3 mL, 1.6 M solution in n-hexane, 2.11 mmol, 1.5 equiv.) and stirred for 0.5 h at −78 °C. To the above reaction mixture, a solution of epoxide 4 (400 mg, 1.407 mmol, 1.0 equiv.) in THF (20 mL) was added followed by the addition of BF3·OEt2 (0.6 mL of 45% solution in Et2O, 2.11 mmol, 1.5 equiv.). The reaction mixture was further stirred for 2 h at 0 °C and then quenched with saturated aq. NaHCO3 and the organic layer was separated and the aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with brine, dried (Na2SO4) and concentrated. The residue was diluted with MeOH (20 mL) and K2CO3 (292 mg, 2.11 mmol, 1.5 equiv.) was added. After stirring at room temperature for 6 h, the mixture was concentrated under reduced pressure. The residue was again dissolved in water and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with brine, dried (Na2SO4) and concentrated. The residue was purified by silica gel column chromatography with petroleum ether/EtOAc (9:1) as an eluent to give pure compound 10 (371.2 mg, 85%) as a white solid. M.p. = 66–68 °C. [α]25D = +15 (c 1.0, CHCl3); IR (CHCl3) υmax 3372, 2952, 2930, 2883, 2857, 2234, 1603, 1493, 1471, 1462, 1454, 1427, 1392, 1255, 1134, 1081, 1004, 869, 836, 778, 698 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 6.9 Hz, 2H), 7.39–7.33 (m, 2H), 7.32–7.27 (m, 1H), 6.80 (d, J = 8.1 Hz, 1H), 6.76 (d, J = 1.9 Hz, 1H), 6.67 (dd, J = 8.1, 2.0 Hz, 1H), 5.12 (s, 2H), 3.88 (s, 3H), 3.82–3.74 (m, 1H), 2.79–2.70 (m, 1H), 2.68–2.59 (m, 1H), 2.48–2.41 (m, 1H), 2.39–2.30 (m, 1H), 2.07 (t, J = 2.6 Hz, 1H), 1.99 (s, 1H), 1.87–1.81 (m, 2H) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ 149.7, 146.5, 137.4, 134.9, 128.5, 127.8, 127.3, 120.3, 114.3, 112.4, 80.7, 71.2, 71.0, 69.2, 56.0, 37.9, 31.5, 27.5 ppm; HRMS (ESI-TOF) m/z: [M + K]+ calcd for C20H22O3K 349.1201, found 349.1197.
(5R)-1,7-Bis(4-benzyloxy-3-methoxyphenyl)hept-2-yne-1,5-diol (11)
To a solution of alkynol 10 (239 mg, 0.77 mmol, 1.0 equiv.) in THF (20 mL) cooled to −78 °C was added n-BuLi (1.0 mL, 1.6 M solution in n-hexane, 1.617 mmol, 2.1 equiv.) dropwise. The resulting mixture was stirred for 0.5 h and then a solution of 4-benzyloxy-3-methoxybenzaldehyde 6 (205 mg, 0.847 mmol, 1.1 equiv.) in THF (10 mL) was added in one portion. The reaction mixture was allowed to warm to 0 °C for 2 h and then quenched with saturated aq. NH4Cl. The organic layer was separated and the aqueous layer was extracted with EtOAc (3 × 15 mL). The combined organic layers were washed with brine, dried (Na2SO4), and concentrated under reduced pressure. The residue was purified by silica gel column chromatography with petroleum ether/EtOAc (4:1) as an eluent to give pure compound 11 (332 mg, 78%) as a yellow oil. IR (CHCl3) υmax 3443, 3005, 2935, 2222, 1627, 1592, 1507, 1466, 1454, 1419, 1381, 1344, 1271, 1220, 1138, 1081, 1031, 855, 812, 750, 697 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 7.3 Hz, 4H), 7.35 (t, J = 7.2 Hz, 4H), 7.32–7.27 (m, 2H), 7.08 (s, 1H), 6.97 (d, J = 8.2 Hz, 1H), 6.82 (d, J = 8.2, 1H), 6.78 (d, J = 8.2 Hz, 1H), 6.74 (s, 1H), 6.66–6.61 (m, 1H), 5.37 (s, 1H), 5.14 (s, 2H), 5.11 (s, 2H), 3.84 (s, 6H), 3.80–3.72 (m, 1H), 2.75–2.68 (m, 1H), 2.64–2.56 (m, 1H), 2.53–2.35 (m, 2H), 1.88–1.78 (m, 2H) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ 149.6, 149.4, 148.0, 146.3, 137.2, 136.9, 134.8, 134.0, 128.5, 128.4, 127.8, 127.7, 127.2, 127.1, 120.1, 118.8, 114.1, 113.4, 112.2, 110.2, 83.4, 82.7, 71.0, 70.8, 69.2, 64.4, 55.9, 37.9, 31.5, 27.8 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C35H36O6Na 575.2404, found 575.2405.
(R)-1,7-Bis(4-benzyloxy-3-methoxyphenyl)-5-hydroxyhept-2-yn-1-one (3)
To a solution of diol 11 (259 mg, 0.469 mmol, 1.0 equiv.) in CH2Cl2 (20 mL) was added MnO2 (408 mg, 4.689 mmol, 10.0 equiv.) and the reaction mixture was stirred at room temperature for 12 h. After the completion of the reaction, the MnO2 residue was separated by filtration through Celite and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (4:1) as an eluent to give 3 (194 mg, 75%) as a yellow oil. [α]25D = +5.6 (c 1.0, CHCl3); IR (CHCl3) υmax 3433, 2949, 2925, 2861, 2227, 1645, 1598, 1466, 1449, 1313, 1261, 1115, 1103, 1095, 1080, 836, 805, 776, 758, 702 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.74 (dd, J = 8.4, 2.0 Hz, 1H), 7.61 (d, J = 1.9 Hz, 1H), 7.45–7.40 (m, 4H), 7.39–7.28 (m, 6H), 6.89 (d, J = 8.4 Hz, 1H), 6.80 (d, J = 8.1 Hz, 1H), 6.76 (d, J = 1.9 Hz, 1H), 6.67 (dd, J = 8.1, 1.9 Hz, 1H), 5.22 (s, 2H), 5.11 (s, 2H), 3.94–3.89 (m, 1H), 3.91 (s, 3H), 3.85 (s, 3H), 2.84–2.60 (m, 5H), 1.96–1.86 (m, 2H) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ 176.7, 153.5, 149.7, 149.4, 146.5, 137.3, 136.0, 134.6, 130.3, 128.7, 128.5, 128.1, 127.7, 127.2, 127.15, 125.5, 120.2, 114.3, 112.3, 112.0, 110.6, 92.0, 81.3, 71.2, 70.8, 68.9, 56.0, 55.9, 38.2, 31.4, 28.2 ppm; HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C35H34O6Na 573.2248, found 573.2247.
(R)-2-(4-Benzyloxy-3-methoxyphenethyl)-6-(4-benzyloxy-3-methoxyphenyl)-2,3-dihydro-4H-pyran-4-one (2)
To a solution of δ-hydroxyalkynone 3 (190 mg, 0.345 mmol, 1.0 equiv.) in MeOH (10 mL) was added p-TsOH·H2O (7 mg, 0.0345 mmol, 10 mol%) at room temperature. The reaction mixture was stirred at room temperature for 12 h and then MeOH was evaporated under reduced pressure. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (3:2) as an eluent to give 2,3-dihydro-4H-pyran-4-one 2 (159.6 mg, 84%) as a yellow oil. [α]25D = +135.2 (c 1.0, CHCl3); IR (CHCl3) υmax 2922, 2854, 1630, 1601, 1573, 1451, 1337, 1241, 1054, 763, 701 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 7.1 Hz, 4H), 7.26–7.40 (m, 8H), 6.92 (d, J = 8.4 Hz, 1H), 6.82 (d, J = 8.1 Hz, 1H), 6.76 (d, J = 1.8 Hz, 1H), 6.69 (dd, J = 8.1, 1.9 Hz, 1H), 5.95 (s, 1H), 5.22 (s, 2H), 5.13 (s, 2H), 4.56–4.47 (m, 1H), 3.93 (s, 3H), 3.84 (s, 3H), 2.94–2.77 (m, 2H), 2.60–2.45 (m, 2H), 2.32–2.23 (m, 1H), 2.06–1.98 (m, 1H) ppm; 13C{1H} NMR (100 MHz, CDCl3) δ 193.1, 169.9, 151.3, 149.7, 149.5, 146.7, 137.2, 136.3, 133.9, 128.6, 128.5, 128.1, 127.8, 127.2, 127.15, 125.5, 120.2, 120.1, 114.4, 113.1, 112.3, 109.6, 101.1, 78.4, 71.2, 70.8, 56.0, 55.96, 41.3, 36.3, 31.0 ppm; HRMS (ESI-TOF) m/z: [M + K]+ calcd for C35H34O6K 589.1987, found 589.1992.
(+)-Diosniponol A (1a)
To a solution of compound 2 (46 mg, 0.0835 mmol, 1.0 equiv.) in EtOAc (10 mL) was added 10% Pd/C (4.5 mg, 0.0042 mmol, 5 mol%) and the mixture was hydrogenated for 10 h with a bladder balloon (4 atm pressure) at room temperature. Then, the reaction mixture was filtered through a Celite pad and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (1:1) as an eluent to give (+)-diosniponol A 1a (24.4 mg, 78%) as a colorless oil. [α]28D = +43.8 (c 1.0, CH3OH), lit.12 [α]28D = −34.8 (c 0.4, CH3OH) for the enantiomer; IR (CHCl3) υmax 3401, 2929, 2853, 1601, 1516, 1463, 1378, 1275, 1154, 1122, 1080, 1033, 858, 817, 699 cm−1; 1H NMR (400 MHz, CD3COCD3) δ 7.50 (s, 1H), 7.31 (s, 1H), 7.01 (d, J = 1.7 Hz, 1H), 6.84 (dd, J = 8.1, 1.6 Hz, 1H), 6.80 (d, J = 1.7 Hz, 1H), 6.79 (d, J = 4.5 Hz, 1H), 6.72 (d, J = 8.0 Hz, 1H), 6.64 (dd, J = 8.0, 1.8 Hz, 1H), 4.28 (dd, J = 11.4, 1.6 Hz, 1H), 3.94 (br s, 1H), 3.84 (s, 3H), 3.78 (s, 3H), 3.48–3.40 (m, 1H), 2.74–2.60 (m, 2H), 2.15–2.08 (m, 1H), 2.00–1.93 (m, 1H), 1.88–1.73 (m, 2H), 1.41–1.36 (m, 1H), 1.26–1.20 (m, 1H); 13C{1H} NMR (100 MHz, CD3COCD3) δ 148.1, 148.0, 146.5, 145.4, 135.6, 134.4, 121.5, 119.4, 115.6, 115.3, 112.8, 110.5, 78.1, 75.5, 68.4, 56.2, 56.1, 44.3, 42.1, 39.0, 32.0 ppm; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H27O6 375.1802, found 375.1797.
(+)-Diosniponol B (1b)
To a solution of diosniponol A 1a (50 mg, 0.134 mmol, 1.0 equiv.) in benzene (10 mL) were added PPh3 (176 mg, 0.667 mmol, 5.0 equiv.), p-nitrobenzoic acid (111.5 mg, 0.667 mmol, 5.0 equiv.) and DEAD (0.1 mL, 0.667 mmol, 5.0 equiv.) under a N2 atmosphere. The resultant mixture was stirred at room temperature for 4 h. After the completion of the reaction, the reaction mixture was concentrated under reduced pressure. The residue was dissolved in MeOH (5 mL) and K2CO3 (55.6 mg, 0.402 mmol, 3.0 equiv.) was added and allowed to stir for 6 h at room temperature. Then, the reaction mixture was filtered on a small pad of Celite and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography with petroleum ether/EtOAc (1:1) as an eluent to give compound 1b (40 mg, 80%) as a colourless oil. [α]28D = +32.4 (c 0.4, CH3OH), lit.12 [α]28D = –28.0 (c 0.4, CH3OH) for the enantiomer; IR (CHCl3) υmax 3407, 2925, 2854, 1603, 1515, 1464, 1430, 1395, 1274, 1153, 1124, 1083, 1032, 859, 817, 771 cm−1; 1H NMR (400 MHz, CD3COCD3) δ 7.47 (s, 1H), 7.30 (s, 1H), 6.99 (d, J = 1.8 Hz, 1H), 6.84 (ddd, J = 8.1, 2.3, 0.5 Hz, 1H), 6.80 (d, J = 1.9 Hz, 1H), 6.78 (d, J = 8.1 Hz, 1H), 6.71 (d, J = 8.0 Hz, 1H), 6.64 (dd, J = 8.0, 1.9 Hz, 1H), 4.75 (dd, J = 11.6, 2.0 Hz, 1H), 4.26–4.22 (m, 1H), 3.95–3.90 (m, 1H), 3.84 (s, 3H), 3.78 (s, 3H), 2.71–2.62 (m, 2H), 1.89–1.84 (m, 1H), 1.81–1.75 (m, 1H), 1.74–1.67 (m, 3H), 1.55–1.50 (m, 1H) ppm; 13C{1H} NMR (100 MHz, CD3COCD3) δ 147.8, 147.75, 146.1, 145.1, 136.2, 134.4, 121.3, 119.0, 115.3, 115.1, 112.6, 110.2, 73.9, 71.4, 64.5, 55.9, 55.8, 41.5, 39.1, 39.0, 31.7 ppm; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H27O6 375.1802, found 375.1800.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank DST-SERB New Delhi (grant CRG/2021/000427) for financial support. D. S. and D. J. thank the Council of Scientific and Industrial Research (CSIR), New Delhi, India for research fellowships.References
- M. T. Huang, R. C. Smart, C. Q. Wong and A. H. Conney, Cancer Res., 1988, 48, 5941 CAS.
- D. Ameen and T. J. Snape, MedChemComm, 2013, 4, 893 RSC.
- A. Jitoe, T. Masuda and T. J. Mabry, Tetrahedron Lett., 1994, 35, 981 CrossRef CAS.
- K. Endo, E. Kanno and Y. Oshima, Phytochemistry, 1990, 29, 797 CrossRef CAS.
- K. W. Woo, E. Moon, O. W. Kwon, S. O. Lee, S. Y. Kim, S. Z. Choi, M. W. Son and K. R. Lee, Bioorg. Med. Chem. Lett., 2013, 23, 3806 CrossRef CAS PubMed.
- M. H. Yang, K. D. Yoon, Y. W. Chin and J. W. Kim, Korean J. Pharmacogn., 2009, 40, 257 Search PubMed.
- (a) S.-H. Dong, D. Nikolić, C. Simmler, F. Qiu, R. B. van Breemen, D. D. Soejarto, G. F. Pauli and S.-N. Chen, J. Nat. Prod., 2012, 75, 2168 CrossRef CAS PubMed; (b) S.-H. Dong, D. Nikolić, C. Simmler, F. Qiu, R. B. van Breemen, D. D. Soejarto, G. F. Pauli and S.-N. Chen, J. Nat. Prod., 2013, 76, 2005 CrossRef CAS.
- (a) C. J. Lee, S. S. Lee, S. C. Chen, F. M. Ho and W. W. Lin, Br. J. Pharmacol., 2005, 146, 378 CrossRef CAS PubMed; (b) M. A. Lee, H. K. Lee, S. H. Kim, Y. C. Kim and S. H. Sung, Planta Med., 2010, 76, 1007 CrossRef CAS PubMed; (c) K. Y. Lee, S. H. Kim, S. H. Sung and Y. C. Kim, Planta Med., 2008, 74, 867 CrossRef CAS PubMed.
- (a) D. J. Faulkner, Nat. Prod. Rep., 1998, 15, 113 RSC; (b) E. J. Kang and E. Lee, Chem. Rev., 2005, 105, 4348 CrossRef CAS PubMed; (c) P. Singh and A. Bhardwaj, J. Med. Chem., 2010, 53, 3707 CrossRef CAS PubMed; (d) T. Voigt, C. Gerding-Reimers, T. T. N. Tran, S. Bergmann, H. Lachance, B. Schölermann, A. Brockmeyer, P. Janning, S. Ziegler and H. Waldmann, Angew. Chem., Int. Ed., 2013, 52, 410 CrossRef CAS PubMed; (e) A. Lorente, J. Lamariano-Merketegi, F. Albericio and M. Álverez, Chem. Rev., 2013, 113, 4567 CrossRef CAS PubMed; (f) S. L. Capim, G. M. Gonçalves, G. C. M. dos Santos, B. G. Marinho and M. L. A. A. Vasconcellos, Bioorg. Med. Chem., 2013, 21, 6003 CrossRef CAS PubMed; (g) N. Ahmed, N. K. Konduru, S. Ahmad and M. Owais, Eur. J. Med. Chem., 2014, 75, 233 CrossRef CAS PubMed.
- (a) V. Baliah, R. Jeyaraman and L. Chandrasekaran, Chem. Rev., 1983, 83, 379 CrossRef CAS; (b) T. L. B. Boivin, Tetrahedron, 1987, 43, 3309 CrossRef CAS; (c) M. M. Faul and B. E. Huff, Chem. Rev., 2000, 100, 2407 CrossRef CAS PubMed; (d) M. C. Elliott, J. Chem. Soc., Perkin Trans. 1, 2002, 2301 RSC; (e) K.-S. Yeung and I. Paterson, Chem. Rev., 2005, 105, 4237 CrossRef CAS PubMed; (f) J. Muzart, Tetrahedron, 2005, 61, 5955 CrossRef CAS; (g) M. Shindo, Top. Heterocycl. Chem., 2006, 5, 179 CAS; (h) P. A. Clarke and S. Santos, Eur. J. Org. Chem., 2006, 2045 CrossRef CAS; (i) I. Larrosa, P. Romea and F. Urpí, Tetrahedron, 2008, 64, 2683 CrossRef CAS; (j) N. Zaware, M. G. LaPorte, R. Farid, L. Liu, P. Wipf and P. E. Floreancig, Molecules, 2011, 16, 3648 CrossRef CAS PubMed; (k) H. Fuwa, Heterocycles, 2012, 85, 1255 CrossRef CAS; (l) N. M. Nasir, K. Ermanis and P. A. Clarke, Org. Biomol. Chem., 2014, 12, 3323 RSC; (m) H. Fuwa, Mar. Drugs, 2016, 14, 65 CrossRef PubMed.
- (a) S.-H. Dong, D. Nikolić, C. Simmler, F. Qiu, R. B. Van Breemen, D. D. Soejarto, G. F. Pauli and S.-N. Chen, J. Nat. Prod., 2012, 75, 2168 CrossRef CAS PubMed; (b) H. Kikuzaki and N. Nakatani, Phytochemistry, 1996, 43, 273 CrossRef CAS; (c) Q. F. Tao, Y. Xu, R. Y. Y. Lam, B. Schneider, H. Dou, P. S. Leung, S. Y. Shi, C. X. Zhou, L. X. Yang, R. P. Zhang, Y. C. Xiao, X. Wu, J. Stöckigt, S. Zeng, C. H. K. Cheng and Y. Zhao, J. Nat. Prod., 2008, 71, 12 CrossRef CAS PubMed; (d) H.-C. Wu, M.-J. Cheng, C.-F. Peng, S.-C. Yang, H.-S. Chang, C.-H. Lin, C.-J. Wang and I.-S. Chen, Phytochemistry, 2012, 82, 118 CrossRef CAS PubMed.
- J. S. Yadav, V. K. Singh, B. Thirupathaiah and A. B. Reddy, Tetrahedron Lett., 2014, 55, 4427 CrossRef CAS.
- O. Mitsunobu, Synthesis, 1981, 1 CrossRef CAS.
- S. P. Gholap, D. Jangid and R. A. Fernandes, J. Org. Chem., 2019, 84, 3537 CrossRef CAS PubMed.
- (a) J. F. Larrow, S. E. Schaus and E. N. Jacobsen, J. Am. Chem. Soc., 1996, 118, 7420 CrossRef CAS; (b) S. E. Schaus, B. D. Brandes, J. F. Larrow, M. Tokunaga, K. B. Hansen, A. E. Gould, M. E. Furrow and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 1307 CrossRef CAS PubMed.
- The racemic epoxide (±)-4 and the next product (±)-10 could not be resolved by HPLC on several chiral columns tried for enantiomeric excess determination. Similar compounds have been resolved by us and have above 98% ee. See: P. Kumar, R. A. Fernandes, M. N. Ahmad and S. Chopra, Tetrahedron, 2021, 96, 132375 CrossRef CAS.
- G. J. McGarvey, J. A. Mathys and K. J. Wilson, J. Org. Chem., 1996, 61, 5704 CrossRef CAS.
- P. Ploypradith, P. Cheryklin, N. Niyomtham, D. R. Bertoni and S. Ruchirawat, Org. Lett., 2007, 9, 2637 CrossRef CAS PubMed.
- R. Singh, G. C. Singh and S. K. Ghosh, Eur. J. Org. Chem., 2007, 5376 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob00863k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |