
Copper-catalyzed tandem cyclization/arylation of α,β-alkynic hydrazones with diaryliodonium salts: synthesis of N-arylpyrazoles†
Sushanta Kumar
Parida
,
Saurav
Joshi
and
Sandip
Murarka
 *
*
Department of Chemistry, Indian Institute of Technology Jodhpur, Karwar-342037, Rajasthan, India. E-mail: sandipmurarka@iitj.ac.in
First published on 4th July 2023
Abstract
A copper-catalyzed efficient method for the synthesis of a diverse variety of substituted N-aryl pyrazoles from readily available α,β-alkynic N-tosyl hydrazones and diaryliodonium triflates is realized. This one-pot multi-step methodology features a broad scope with good yields, scalability, and appreciable functional group tolerance. Detailed control experiments reveal that the reaction proceeds through tandem cyclization/deprotection/arylation events where the copper catalyst plays a distinct role.
Introduction
Pyrazoles belong to a privileged class of five-membered heterocycles and are ubiquitous in numerous drugs and natural products.1 In particular, N-aryl pyrazoles and their derivatives are important molecular motifs and key components of several drug molecules (apixaban, celecoxib, rimonabant, lonazolac, and tepoxalin) and insecticides (fipronil) (Fig. 1).2 Because of their profound applications and broad spectrum of pharmacological properties, the construction of N-aryl pyrazoles has attracted considerable attention in the last few decades. Conventional methods include condensation of 1,3-dielectrophiles, such as 1,3-diones, enones and ynones with aryl hydrazines,1a,3 and 1,3-dipolar cycloadditions of diazo compounds with olefins or alkynes.4 Despite these tremendous developments allowing convenient access to substituted N-arylated pyrazoles, bottlenecks, such as limited availability of aryl hydrazines, the hazardous nature of diazo compounds, and harsh reaction conditions, call for further developments. In this regard, α,β-alkynic N-tosyl hydrazones have evolved as useful building blocks for the construction of carbon–carbon and carbon–heteroatom bonds.5 Such moieties readily undergo electrophilic cyclizations in the presence of a variety of electrophiles and allow the introduction of trifluoromethyl, fluoro, iodo, sulfur and selenium substituents at the 4-position of the pyrazole ring (Scheme 1a).6 However, the construction of N-aryl pyrazoles requires the utilization of the corresponding N-aryl hydrazone precursor and limited availability of aryl hydrazines restricts the diversification of the desired pyrazoles. A copper iodide-mediated electrophilic cyclization of α,β-alkynic N-aryl hydrazones has been developed as well (Scheme 1b).6b Notably, N-aryl pyrazoles could also be prepared through N-arylation of 1H-pyrazoles.7,8 In this regard, Novák's transition-metal-free N-arylation of symmetrically-substituted pyrazoles utilizing diaryliodonium triflates under mild basic conditions is noteworthy.7d Recently, a copper-mediated cross-coupling between cyclopropanols and aryldiazonium salts has been disclosed (Scheme 1c).9 This method allows efficient access to regioisomerically pure pyrazoles; however, the requirement of potentially explosive aryldiazo compounds and stoichiometric amounts of a copper salt is less desirable. Hence, considering the widespread utility of N-arylated pyrazoles, the development of alternative, convenient and efficient one-pot multi-step processes enabling the synthesis of a diverse variety of N-arylated pyrazoles is highly desirable. | ||
| Fig. 1 Selected examples of biologically active N-arylated pyrazoles. | ||
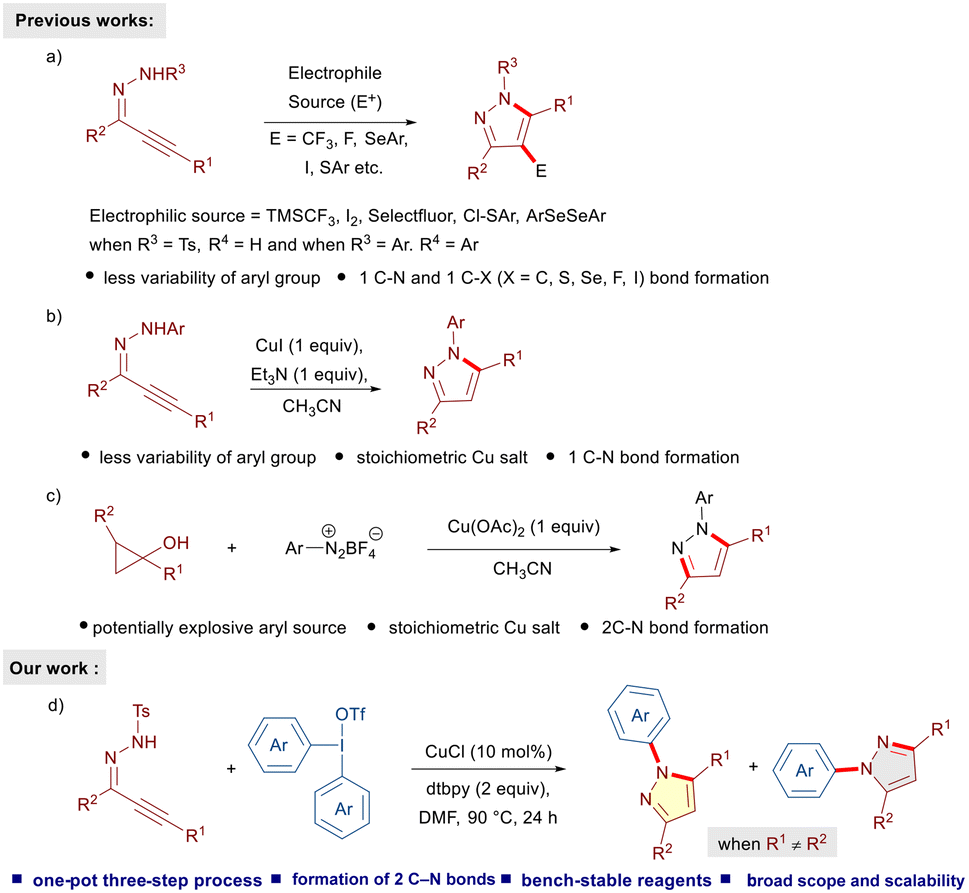 | ||
| Scheme 1 Strategies for the synthesis of N-aryl pyrazoles and our work. | ||
Lately, diaryliodonium reagents (DAIRs)10 have emerged as easily available, non-toxic and bench-stable arylating precursors for a variety of carbon- and heteroatom-centered nucleophiles under various conditions.11 We envisioned the possibility of reacting α,β-alkynic N-tosyl hydrazones with DAIRs under copper-catalyzed conditions to afford the corresponding N-arylated pyrazoles. We reasoned that N-tosyl pyrazoles generated through intramolecular cyclization of α,β-alkynic hydrazones should readily undergo N-arylation with DIARs in the presence of a suitable copper catalyst to furnish the desired products. Such a one-pot multi-step strategy would be novel, highly efficient, and fascinating from the perspective of step economy. As part of our program on hypervalent iodine(III) reagents,12 we disclose a copper-catalyzed reaction of α,β-alkynic N-tosyl hydrazones with DAIRs to furnish an array of highly substituted N-arylated pyrazoles in an efficient fashion.
Results and discussion
After preliminary studies, we established that the planned reaction was best conducted through heating a solution of α,β-alkynic hydrazone 1a (0.1 mmol, 1 equiv.), diphenyliodonium triflate 2a (0.12 mmol, 1.2 equiv.), CuCl (10 mol%) and 4,4′-di-tert-butyl-2,2′-dipyridyl (dtbpy, 0.2 mmol, 2 equiv.) in DMF at 90 °C under a nitrogen atmosphere (entry 1, Table 1, see the ESI for the complete optimization table†). To our delight, the reaction achieved completion in 24 h and the desired N-phenyl pyrazole 3aa was obtained in 82% yield under these conditions (entry 1). Other copper salts such as CuBr, CuI, Cu(OAc)2 and Cu(OTf)2 were also effective, but diminished yields were obtained (entries 2–5). Subsequent screening revealed the crucial role of base, solvent and temperature in this tandem process. Depending on the reaction conditions, either N-aryl pyrazole (3aa) or N-tosyl pyrazole 4a in the pure form or a mixture of both was obtained. Organic bases such as Et3N and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) failed to better the yield, and potassium carbonate provided 3aa and 4a in 46% and 38% yields, respectively (entries 6–8). Further evaluation of the reaction conditions divulged that solvents (DMSO and C2H4Cl2) other than DMF provided a mixture of 3aa and 4a in similar yields (entries 9 and 10). Importantly, in the absence of dtbpy, 3aa was obtained in 63% yield with a trace amount of 4a (entry 11). However, in the absence of CuCl, the formation of 3aa was completely suppressed and 4a was isolated exclusively in 74% yield (entry 12). These experiments provided us an indication that the copper catalyst may not be indispensable to the initial cyclization event, but plays a crucial role in the subsequent de-tosylative N-arylation process. The yield dropped to 68% upon reducing the Cu-catalyst loading to 5 mol%, and increasing the catalyst loading to 20 mol% did not impart any beneficial effect (entries 13 and 14). Performing the reaction at room temperature instead of 90 °C ceased the formation of 3aa and the undesired N-tosyl pyrazole 4a was obtained in 64% yield (entry 15). On the other hand, increasing the reaction temperature to 110 °C did not improve the yield of the desired 3aa (entry 16). Increasing the amount of DAIR 2a to 1.5 equiv. furnished yields that were similar to those obtained in the case of 1.2 equiv. DAIR, and decreasing the loading of dtbpy to 1.5 equiv. had detrimental effects on the outcome of the reaction (entries 17 and 18). Pleasingly, in contrast to most of the copper-catalyzed processes, the reaction could also be carried out under an air atmosphere to provide the desired 3aa in 74% yield (entry 19).|
|
||
|---|---|---|
| Entry | Variation from the optimized conditions | Yield of 3aa and 4a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (%) b (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.12 mmol, 1.2 equiv.), Cu-catalyst (10 mol%), base (0.2 mmol, 2 equiv.), and solvent (1 mL) under a nitrogen atmosphere at 90 °C for 24 h. b Isolated yield. | ||
| 1 | None | 82, 0 |
| 2 | CuBr instead of CuCl | 61, 0 |
| 3 | CuI instead of CuCl | 68, 0 |
| 4 | Cu(OAc)2 instead of CuCl | 79, 0 |
| 5 | Cu(OTf)2 instead of CuCl | 71, 0 |
| 6 | Et3N instead of dtbpy | 74, trace |
| 7 | DBU instead of dtbpy | 52, trace |
| 8 | K2CO3 instead of dtbpy | 46, 38 |
| 9 | DMSO instead of DMF | 64, 21 |
| 10 | C2H4Cl2 instead of DMF | 62, 28 |
| 11 | dtbpy was not used | 63, trace |
| 12 | CuCl was not used | 0, 74 |
| 13 | 5 mol% CuCl was used | 68, 0 |
| 14 | 20 mol% CuCl was used | 79, 0 |
| 15 | RT instead of 90 °C | Trace, 64 |
| 16 | 110 °C instead of 90 °C | 81, 0 |
| 17 | 1.5 equiv. instead of 1.2 equiv. of 2 | 83, 0 |
| 18 | 1.5 equiv. instead of 2 equiv. of dtbpy | 71, 0 |
| 19 | Under air instead of nitrogen | 74, 0 |
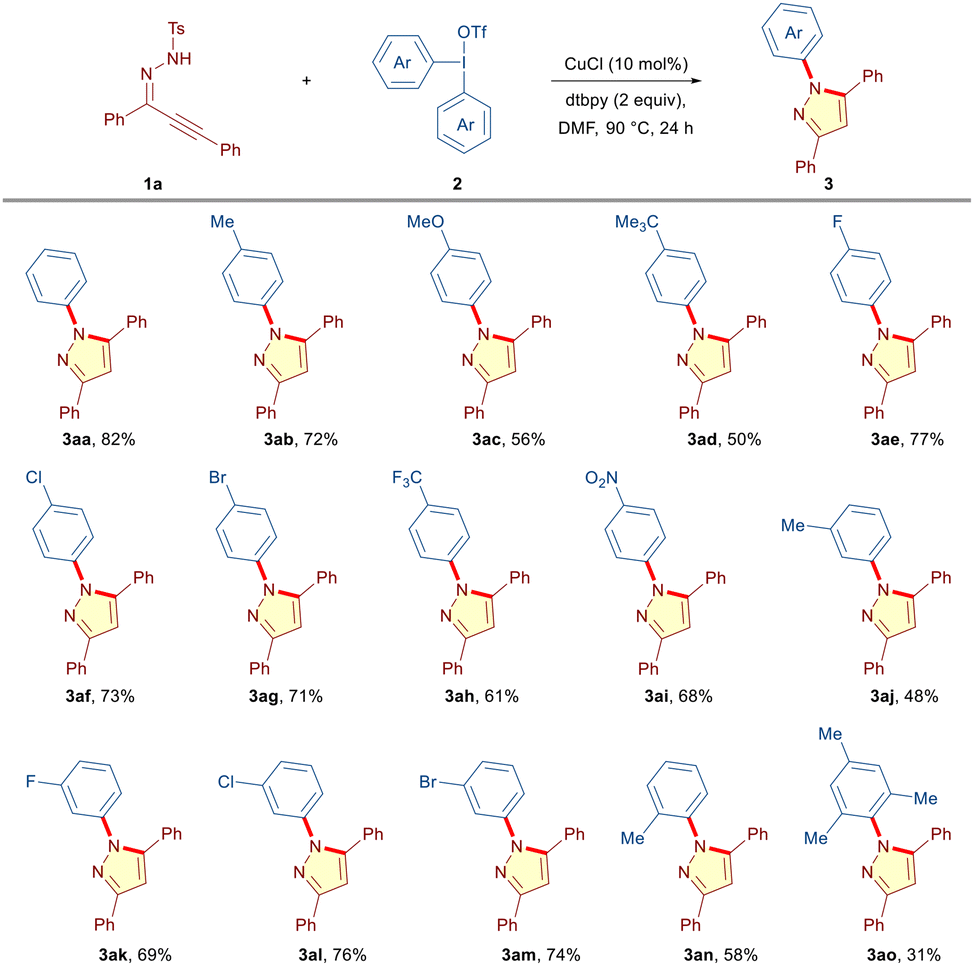
Next, we set out to explore the scope of the cascade manifold by reacting α,β-alkynic hydrazone 1a with a variety of symmetrical diaryliodonium triflates 2 under the optimized conditions (Table 2). The DAIRs having electron rich substituents (CH3, OMe, and t-Bu) at the para-position of the phenyl ring furnished the desired N-arylated pyrazoles (3ab–3ad) in moderate to good yields (50–72%). Pleasingly, the tandem process was found to be equally potent in the case of DAIRs having electron-withdrawing substituents (4-F, 4-Cl, and 4-Br) on the phenyl ring, delivering the corresponding products (3ae–3ag) in good yields (71–77%). Moreover, DAIRs containing reactive functional groups, such as –CF3 and –NO2, were well accommodated under the reaction conditions (3ah (61%), 3ai (68%)). Besides para substitution patterns, DAIRs embedded with electronically diverse meta substitution patterns (CH3, F, Cl, and Br) underwent smooth transformation into the expected products (3aj–3am) in moderate to good yields (48–76%). Notably, the presence of ortho (2-Me) and sterically constrained 2,4,6-trimethyl substituents on the phenyl ring of DAIR did not impose much steric threat and the respective products 3an and 3ao were obtained in 58% and 31% yields, respectively.
Surprisingly, the reaction between α,β-alkynic hydrazone 1b and diphenyl iodonium triflate 2a led to the formation of an inseparable mixture of regioisomeric pyrazoles 3ba and 3ba′ in a 0.3:1 ratio and 76% combined yield (Table 3). The regioisomeric ratio of products was determined from 1H NMR and the exact structure of both the isomers was established by comparing the 1H NMR data with the reported compounds in the literature.13 The formation of regioisomeric mixtures could presumably be due to the non-symmetric substitution pattern (R1 ≠ R2) on the α,β-alkynic hydrazones 1, which led to the formation of tautomeric 1H-pyrazoles through tosyl deprotection and eventually delivered regioisomeric pyrazoles through N-arylation under the reaction conditions. Nevertheless, a similar reactivity pattern was observed and a regioisomeric mixture of pyrazoles was obtained when we investigated the scope of α,β-alkynic hydrazones 1 with R1 ≠ R2 (Table 3).
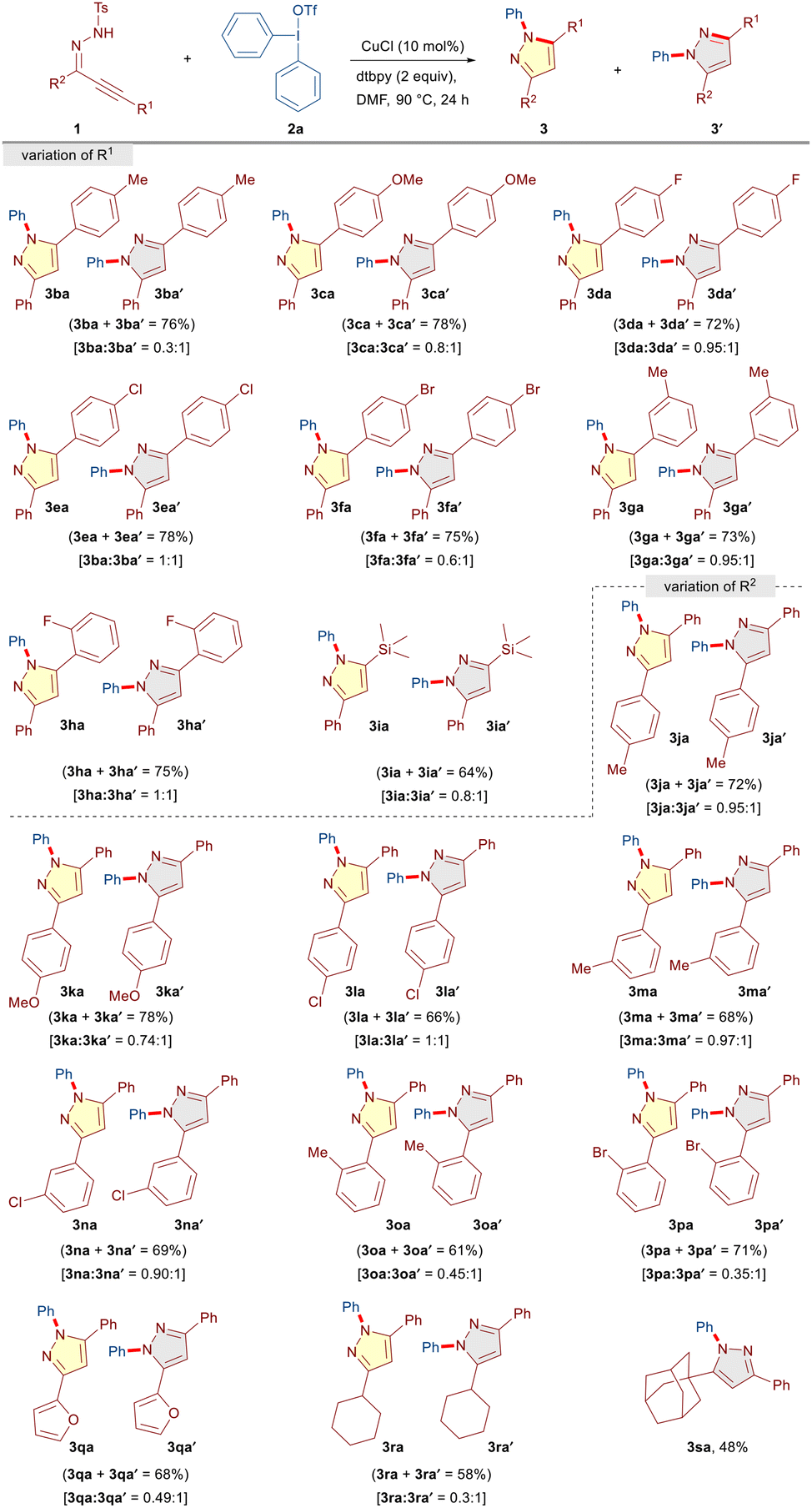
We began with the scope of R1 and realized that various aryl alkynyl moieties having electron-rich (CH3 and OMe) as well as electron-withdrawing (F, Cl, and Br) substituents at the para-position of the phenyl ring afforded the corresponding products (3ba/3ba′–3fa/3fa′) with varying levels of regioisomeric ratio in good yields (72–78%). Moreover, besides para-substituents, meta- and ortho-substituents (3-CH3 and 2-F) were well tolerated to furnish the corresponding products 3ga/3ga′ (0.95:1) and 3ha/3ha′ (1:1) in 73% and 75% yields, respectively. Notably, alkyl alkynyl functionalities, such as trimethylsilyl alkyne, also participated in the reaction to provide 3ia/3ia′ (0.8:1) in 64% yield. Subsequently, the influence of R2 substituents on the reaction outcome was investigated. Pleasingly, electronically diverse para (CH3, OMe, and Cl), meta (CH3 and Cl), and ortho (CH3 and Br) substitution patterns on the phenyl ring were well tolerated to furnish the corresponding products (3ja/3ja′–3pa/3pa′) as inseparable regioisomeric mixtures in moderate to good yields (61–78%). It is worth mentioning that heteroaryl (furan) and aliphatic (cyclohexyl and adamantyl) moieties as R2 substituents underwent facile transformation into the desired products (3qa/3qa′, 3ra/3ra′ and 3sa) in moderate to good yields (48–68%).
The scalability and practical applicability of the method were demonstrated by reacting 1 g of alkynic hydrazone 1a with diphenyliodonium triflate 2a under the optimized conditions to furnish the desired N-phenylated pyrazole 3aa in 66% yield (Scheme 2a). Notably, the final products were amenable for further synthetic modifications. Accordingly, 3aa was successfully subjected to bromination in the presence of N-bromosuccinimide to give 5 in 93% yield (Scheme 2b). Moreover, 5 was further transformed through Sonogashira coupling into the corresponding fully-substituted alkynylated pyrazole 6 in 55% yield (Scheme 2b).
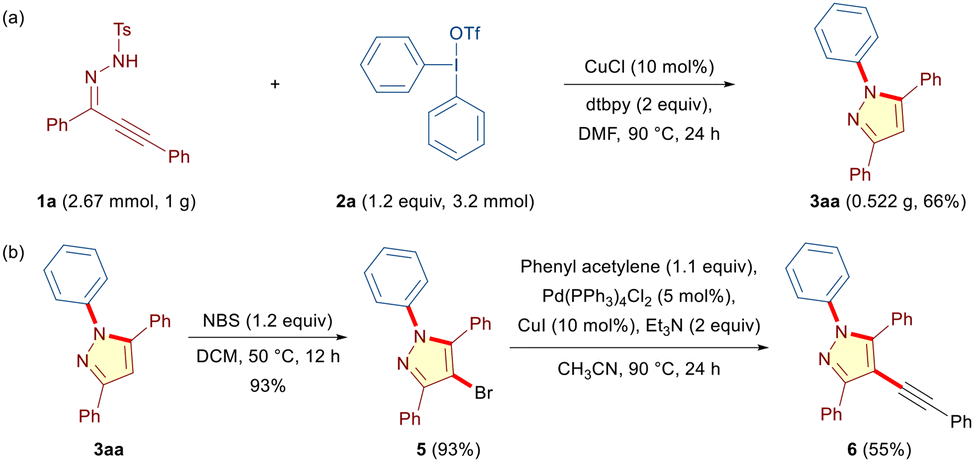 | ||
| Scheme 2 Scale-up experiment and post-synthetic modifications. | ||
Subsequently, we synthesized a set of unsymmetrical DAIRs (2p–2s) and reacted them with 1a under the optimized conditions to evaluate whether or not chemoselective transfer of one aryl moiety over the other is possible (Scheme 3). In the case of DAIRs 2p and2q, a predominant transfer of the electron-deficient aryl group providing the corresponding compounds 3aa (56%, Ph over 4-t-Bu-Ph) and 3ai (49%, 4-NO2-Ph over Ph) was observed (Scheme 3a and b). This is somewhat different from the earlier transition-metal-catalyzed reactions involving DAIRs, where more electron-rich aryl groups were generally transferred.11h,14 However, several exceptions to this observation have been documented as well.14b,15 Expectedly, an exclusive transfer of the sterically less demanding phenyl group was observed (3aa, 66%) when the mesityliodonium salt 2r was employed (Scheme 3c). Moreover, in the case of DAIR 2s, the phenyl group was transferred in exclusivity (3aa, 62%), and the thienyl moiety served as a dummy group (Scheme 3d). These findings indicate that under our reaction conditions, the transfer of the electron-deficient and sterically less encumbered aryl ring in DAIR is preferred.
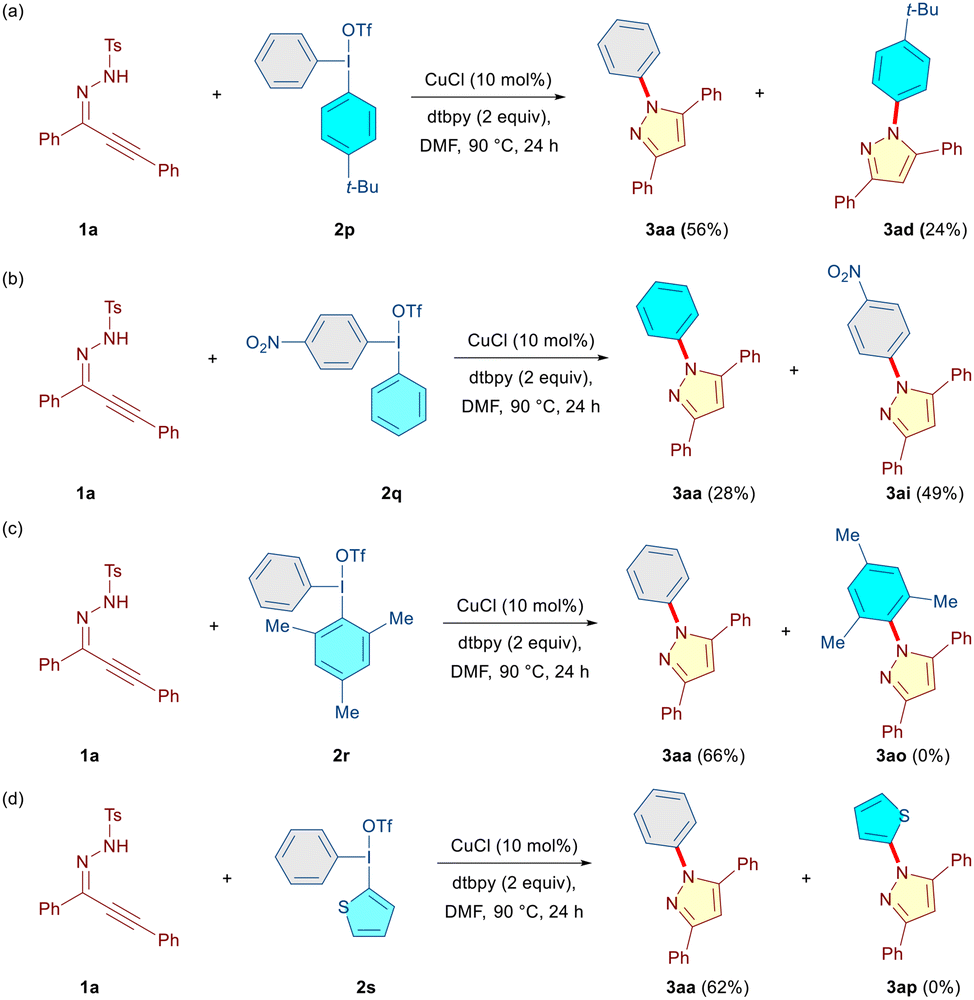 | ||
| Scheme 3 Selectivity studies for unsymmetrical diaryliodonium triflates. | ||
Subsequently, we performed several carefully designed control experiments to get insight into the reaction mechanism. Based on the optimization studies (Table 1, entries 12 and 13), we reasoned that 1a initially could undergo a base-promoted 5-endo-dig cyclization to furnish N-tosyl pyrazole 4a, which then reacts with DAIR 2a under copper-catalyzed conditions to provide N-phenylated pyrazole 3aa through detosylation. Although copper is not essential in the initial cyclization step, it certainly could coordinate to the alkyne moiety and provide the necessary impetus.6g Initially, we reacted 1a and 2a in the absence of a copper catalyst and dtbpy under otherwise optimized conditions to obtain 4a in 66% yield (Scheme 4a). To probe if the conjugate anion in DAIR 2a is steering the cyclization in this case, we reacted 1a with NaOTf in the absence of 2a, CuCl and dtbpy. Indeed, 4a was obtained in 63% yield (Scheme 4b). Next, 4a was subjected to detosylative arylation both in the presence and absence of the copper catalyst under otherwise identical conditions. Interestingly, although no transformation was observed in the absence of the Cu-catalyst, 4a underwent smooth transformation into the desired 3aa in the presence of 2a and CuCl (Scheme 4c and d). These experiments unequivocally establish the crucial role of CuCl in the transformation of 4a into 3aa. We anticipated that 4a may undergo detosylation under the reaction conditions to 1H-pyrazole 7a, which upon arylation delivers the desired 3aa. Considering the possibility of tautomeric structures and formation of regioisomers thereof in 3,5-unsymmetrical 1H-pyrazoles, this hypothesis indeed explains the formation of regioisomeric N-arylated pyrazoles that was observed for alkynic hydrazones 1b–1q (Table 3). Accordingly, the reaction between 4a and 2a was intercepted in 4 h, and expectedly, 1H-pyrazole 7a was isolated in 52% yield along with the desired N-arylated pyrazole 3aa in 33% yield, thereby confirming our hypothesis of detosylation preceding the final N-arylation en route to the desired N-arylated pyrazole 3aa (Scheme 4e). Notably, detosylated pyrazole 7a was not detected in the absence of CuCl (Scheme 4d).6g In an attempt to understand the role of CuCl in the final N-arylation step, we reacted 7a with 2a under the optimized conditions, albeit in the absence of the copper catalyst (Scheme 4f). The substantially lower yield of 3aa (15%) explicitly revealed the essential role of CuCl in both the detosylation and N-arylation step. Finally, to explore the possibility of a radical mechanism,161a and 2a were reacted under the optimized conditions in the presence of 2,2,6,6-tetramethyl-1-piperidine-1-oxyl (TEMPO) and 2,6-di-tert-butyl-4-methylphenol (BHT) as the radical scavengers (Scheme 4g). The tandem annulation was unaffected by the presence of both the radical scavengers and product 3aa was formed in 71% and 78% yields, respectively. These experiments exclude the possibility of an underlying radical mechanism.
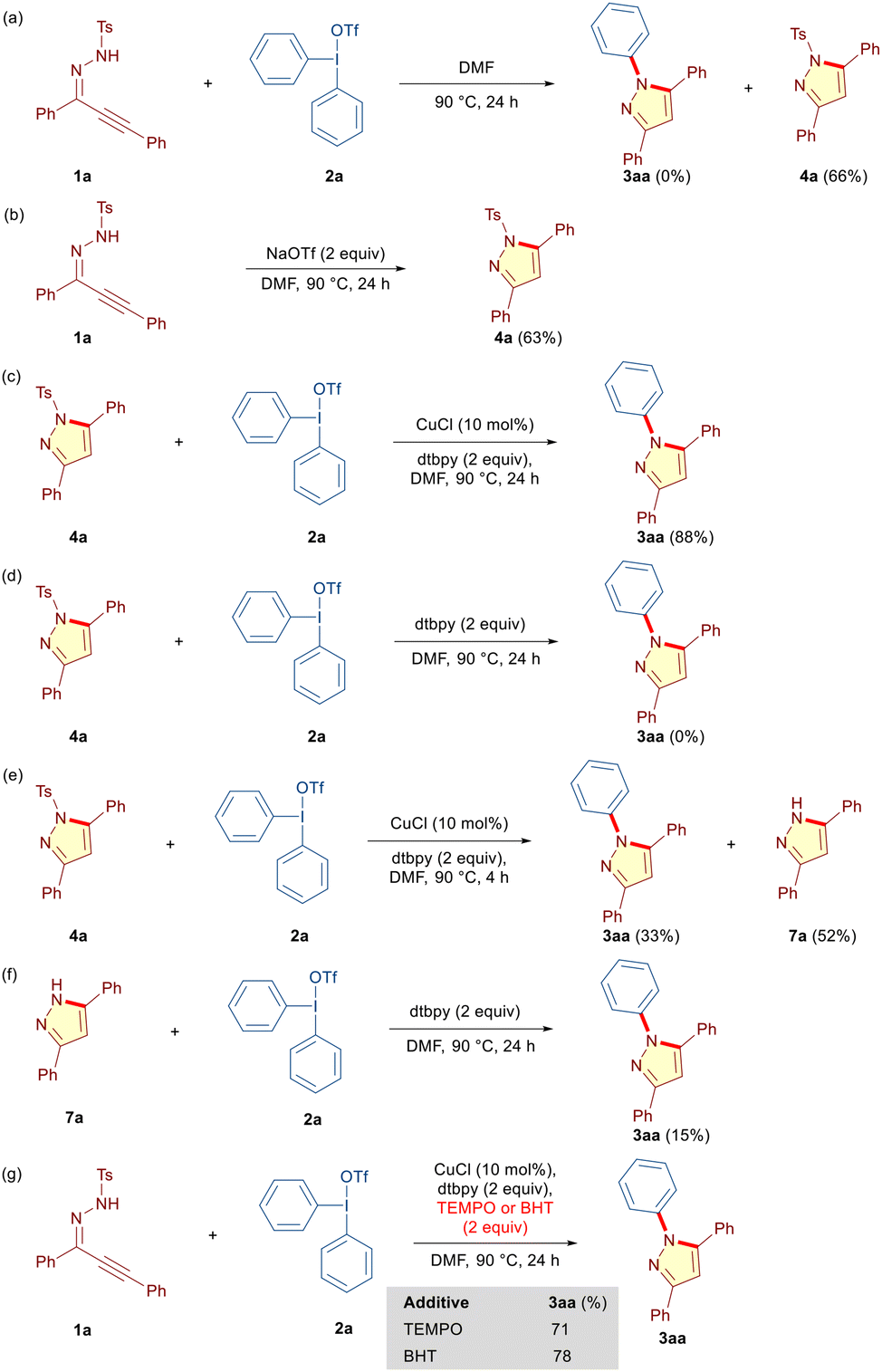 | ||
| Scheme 4 Control experiments. | ||
Based on our control experiments, we propose that the reaction begins by a base-mediated cyclization of alkynic hydrazones 1 to N-tosyl pyrazoles 4 with or without the assistance of a copper catalyst (Scheme 5). Subsequently, 4 leads to 1H-pyrazoles 7 under the copper-catalyzed reaction conditions. The N-arylation step is expected to proceed in a way similar to that reported for Cu(III)-catalyzed oxidative couplings involving DAIRs.11e,f,14a Initially, the Cu(I) catalyst gets oxidized by DAIRs 2 resulting in the formation of an Ar–Cu(III) species, which interacts with 7 to furnish the desired N-arylated pyrazole 3 with concomitant generation of the Cu(I) catalyst.
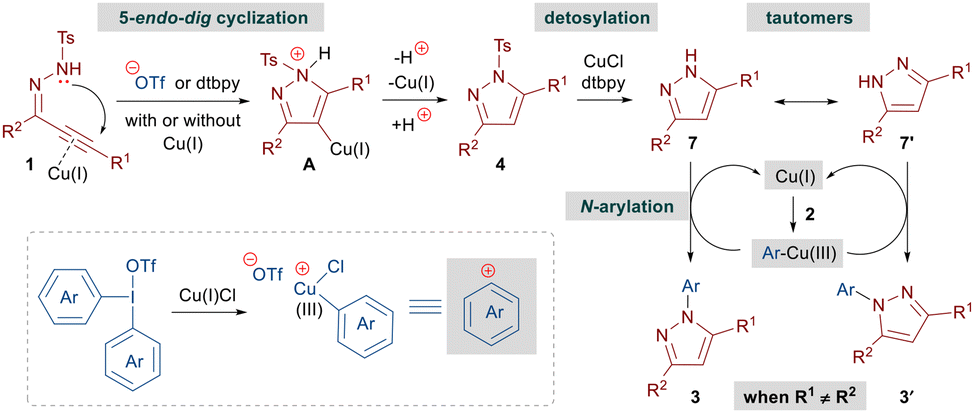 | ||
| Scheme 5 Proposed reaction mechanism. | ||
Conclusions
To summarize, we have outlined a copper-catalyzed robust and scalable reaction of α,β-alkynic N-tosyl hydrazones and diaryliodonium triflates to construct a structurally and electronically diverse collection of N-aryl pyrazole derivatives. The developed method offers a broad scope with appreciable functional group tolerance and enables the synthesis of the final products in good yields. Notably, the reaction utilizes a copper-catalyzed tandem cyclization/tosyl-deprotection/arylation sequence, where copper plays a distinct role. The detailed mechanistic investigations comprising carefully crafted control experiments provided insight into the reaction mechanism. At present, we are engaged in unravelling further applications of copper-catalyzed tandem annulations involving DAIRs towards the construction of densely functionalized molecular entities.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SM acknowledges CSIR [02(0426)/21/EMR-II] and SERB [CRG/2022/000470] for funding and DST-FIST [SR/FST/CS-II/2019/119(C)] for the HRMS facility at IIT Jodhpur.References
- (a) S. Fustero, M. Sánchez-Roselló, P. Barrio and A. Simón-Fuentes, Chem. Rev., 2011, 111, 6984–7034 CrossRef CAS PubMed; (b) J. V. Faria, P. F. Vegi, A. G. C. Miguita, M. S. dos Santos, N. Boechat and A. M. R. Bernardino, Bioorg. Med. Chem., 2017, 25, 5891–5903 CrossRef CAS PubMed.
- (a) Z. Xu, C. Gao, Q.-C. Ren, X.-F. Song, L.-S. Feng and Z.-S. Lv, Eur. J. Med. Chem., 2017, 139, 429–440 CrossRef CAS PubMed; (b) D. J. P. Pinto, J. M. Smallheer, D. L. Cheney, R. M. Knabb and R. R. Wexler, J. Med. Chem., 2010, 53, 6243–6274 CrossRef CAS PubMed; (c) T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Carter, P. W. Collins, S. Docter, M. J. Graneto, L. F. Lee, J. W. Malecha, J. M. Miyashiro, R. S. Rogers, D. J. Rogier, S. S. Yu, G. D. Anderson, E. G. Burton, J. N. Cogburn, S. A. Gregory, C. M. Koboldt, W. E. Perkins, K. Seibert, A. W. Veenhuizen, Y. Y. Zhang and P. C. Isakson, J. Med. Chem., 1997, 40, 1347–1365 CrossRef CAS PubMed; (d) G. Li, Y. Cheng, C. Han, C. Song, N. Huang and Y. Du, RSC Med. Chem., 2022, 13, 1300–1321 RSC.
- B. S. Gerstenberger, M. R. Rauckhorst and J. T. Starr, Org. Lett., 2009, 11, 2097–2100 CrossRef CAS PubMed.
- (a) M. Tang, W. Zhang and Y. Kong, Org. Biomol. Chem., 2013, 11, 6250–6254 RSC; (b) M. C. Pérez-Aguilar and C. Valdés, Angew. Chem., Int. Ed., 2015, 54, 13729–13733 CrossRef PubMed; (c) M. C. Pérez-Aguilar and C. Valdés, Angew. Chem., Int. Ed., 2013, 52, 7219–7223 CrossRef PubMed; (d) Y. Kong, M. Tang and Y. Wang, Org. Lett., 2014, 16, 576–579 CrossRef CAS PubMed.
- (a) Y. Xia and J. Wang, Chem. Soc. Rev., 2017, 46, 2306–2362 RSC; (b) M. L. Hossain and J. Wang, Chem. Rec., 2018, 18, 1548–1559 CrossRef CAS PubMed.
- (a) M. Zora, A. Kivrak and C. Yazici, J. Org. Chem., 2011, 76, 6726–6742 CrossRef CAS PubMed; (b) M. Zora and A. Kivrak, J. Org. Chem., 2011, 76, 9379–9390 CrossRef CAS PubMed; (c) J. Qian, Y. Liu, J. Zhu, B. Jiang and Z. Xu, Org. Lett., 2011, 13, 4220–4223 CrossRef CAS PubMed; (d) X. Yu, Y.-Z. Shang, Y.-F. Cheng, J. Tian, Y. Niu and W.-C. Gao, Org. Biomol. Chem., 2020, 18, 1806–1811 RSC; (e) H.-F. Yao, F.-H. Li, J. Li, S.-Y. Wang and S.-J. Ji, Org. Biomol. Chem., 2020, 18, 1987–1993 RSC; (f) G. Ji, X. Wang, S. Zhang, Y. Xu, Y. Ye, M. Li, Y. Zhang and J. Wang, Chem. Commun., 2014, 50, 4361–4363 RSC; (g) Q. Wang, L. He, K. K. Li and G. C. Tsui, Org. Lett., 2017, 19, 658–661 CrossRef CAS PubMed.
- (a) R. Zhu, L. Xing, X. Wang, C. Cheng, D. Su and Y. Hu, Adv. Synth. Catal., 2008, 350, 1253–1257 CrossRef CAS; (b) M. Taillefer, N. Xia and A. Ouali, Angew. Chem., Int. Ed., 2007, 46, 934–936 CrossRef CAS PubMed; (c) P. Suresh and K. Pitchumani, J. Org. Chem., 2008, 73, 9121–9124 CrossRef CAS PubMed; (d) Z. Gonda and Z. Novák, Chem. – Eur. J., 2015, 21, 16801–16806 CrossRef CAS PubMed.
- (a) M. McLaughlin, K. Marcantonio, C.-y. Chen and I. W. Davies, J. Org. Chem., 2008, 73, 4309–4312 CrossRef CAS PubMed; (b) C. Despotopoulou, L. Klier and P. Knochel, Org. Lett., 2009, 11, 3326–3329 CrossRef CAS PubMed.
- J. Liu, E. Xu, J. Jiang, Z. Huang, L. Zheng and Z.-Q. Liu, Chem. Commun., 2020, 56, 2202–2205 RSC.
- (a) K. Aradi, B. L. Tóth, G. L. Tolnai and Z. Novák, Synlett, 2016, 1456–1485 CAS; (b) E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052–9070 CrossRef CAS PubMed.
- (a) F. Tinnis, E. Stridfeldt, H. Lundberg, H. Adolfsson and B. Olofsson, Org. Lett., 2015, 17, 2688–2691 CrossRef CAS PubMed; (b) J. Malmgren, S. Santoro, N. Jalalian, F. Himo and B. Olofsson, Chem. – Eur. J., 2013, 19, 10334–10342 CrossRef CAS PubMed; (c) D. I. Bugaenko, M. A. Yurovskaya and A. V. Karchava, Org. Lett., 2018, 20, 6389–6393 CrossRef CAS PubMed; (d) D. I. Bugaenko, A. A. Volkov, V. V. Andreychev and A. V. Karchava, Org. Lett., 2023, 25, 272–276 CrossRef CAS PubMed; (e) R. J. Phipps, N. P. Grimster and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 8172–8174 CrossRef CAS PubMed; (f) R. J. Phipps and M. J. Gaunt, Science, 2009, 323, 1593–1597 CrossRef CAS PubMed; (g) Y. Wang, C. Chen, S. Zhang, Z. Lou, X. Su, L. Wen and M. Li, Org. Lett., 2013, 15, 4794–4797 CrossRef CAS PubMed; (h) Y. Wang, C. Chen, J. Peng and M. Li, Angew. Chem., Int. Ed., 2013, 52, 5323–5327 CrossRef CAS PubMed.
- (a) R. K. Samanta, P. Meher and S. Murarka, J. Org. Chem., 2022, 87, 10947–10957 CrossRef CAS PubMed; (b) S. K. Parida, S. Jaiswal, P. Singh and S. Murarka, Org. Lett., 2021, 23, 6401–6406 CrossRef CAS PubMed; (c) P. Meher, R. K. Samanta, S. Manna and S. Murarka, Chem. Commun., 2023, 59, 6092–6095 RSC; (d) S. Jaiswal, S. K. Parida, S. Murarka and P. Singh, Bioorg. Med. Chem., 2022, 68, 116874 CrossRef CAS PubMed; (e) R. Budhwan, S. Yadav and S. Murarka, Org. Biomol. Chem., 2019, 17, 6326–6341 RSC; (f) R. Budhwan, G. Garg, I. N. N. Namboothiri and S. Murarka, Targets Heterocycl. Syst., 2019, 23, 27–52 CAS.
- (a) X. Zhang, J. Kang, P. Niu, J. Wu, W. Yu and J. Chang, J. Org. Chem., 2014, 79, 10170–10178 CrossRef CAS PubMed; (b) S. M. Landge, A. Schmidt, V. Outerbridge and B. Török, Synlett, 2007, 1600–1604 CAS.
- (a) R. J. Phipps, L. McMurray, S. Ritter, H. A. Duong and M. J. Gaunt, J. Am. Chem. Soc., 2012, 134, 10773–10776 CrossRef CAS PubMed; (b) N. R. Deprez and M. S. Sanford, Inorg. Chem., 2007, 46, 1924–1935 CrossRef CAS PubMed.
- (a) J. Li, H. Wang, Y. Hou, W. Yu, S. Xu and Y. Zhang, Eur. J. Org. Chem., 2016, 2388–2392 CrossRef CAS; (b) S.-K. Kang, T. Yamaguchi, J.-S. Kim, S.-C. Choi and C. Ahn, Synth. Commun., 1997, 27, 1105–1110 CrossRef CAS.
- J. Xu, P. Zhang, Y. Gao, Y. Chen, G. Tang and Y. Zhao, J. Org. Chem., 2013, 78, 8176–8183 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2261058. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ob00899a |
| This journal is © The Royal Society of Chemistry 2023 |