
Transition metal-catalyzed reactivity of carbenes with boronic acid derivatives for arylation (alkylation) and beyond
Akanksha
Babbar†
 ,
Pokhriyal
Yamini†
,
Mohammad
Saleem†
and
Dongari
Yadagiri
*
,
Pokhriyal
Yamini†
,
Mohammad
Saleem†
and
Dongari
Yadagiri
*
Laboratory of Organic Synthesis and Catalysis, Department of Chemistry, IIT Roorkee, 247667, Uttarakhand, India. E-mail: yadagiri.dongari@cy.iitr.ac.in; Web: https://yadagiridongari.wixsite.com
First published on 14th August 2023
Abstract
This comprehensive review article discussed the reactivity of carbenes with boronic acid derivatives for the one-pot synthesis of diarylmethanes, difluoromethylated arenes, aryl and alkyl boron compounds, arylacetic acid derivatives, furan derivatives, and many other compounds. We have summarized the arylation, vinylation, and alkylation of carbenes utilizing various transition metals, viz. palladium, rhodium, copper, and platinum, for the construction of carbon–carbon bonds, carbon–boron bonds, and beyond through the cross-coupling strategy. The reason for the increasing popularity of these novel methodologies is their application in the synthesis and late-stage functionalization of biologically active compounds and natural products. Notably, organoboron compounds are exemplified as versatile synthetic intermediates for constructing various bonds.
 Akanksha Babbar | Akanksha Babbar received her BSc(H) in Chemistry from Panjab University in 2020. Then, she completed her MSc (Chemistry) degree from the Institute of Technology, Ropar, in 2022. Currently, she is a PhD student working under the supervision of Prof. Dongari Yadagiri at the Indian Institute of Technology, Roorkee. Her research interests include functionalization of unactivated C–H bonds via metal catalysis. |
 Pokhriyal Yamini | Pokhriyal Yamini received her BSc from Meerut College, Chaudhary Charan Singh University, Meerut, in 2019. Then, she completed her MSc (Chemistry) degree from R.G.P.G. Degree College, Chaudhary Charan Singh University, Meerut, in 2021. Currently, she is a PhD student working under the supervision of Prof. Dongari Yadagiri at the Indian Institute of Technology, Roorkee. Her research interests include carbene insertion reactions. |
 Mohammad Saleem | Mohammad Saleem received his BSc from Anubis Degree College, Mahatma Jyotiba Phule Rohilkhand University, Bareilly, in 2019. Then, he completed his MSc (Chemistry) degree from the Indian Institute of Technology, Tirupati, in 2022. Currently, he is a PhD student working under the supervision of Prof. Dongari Yadagiri at the Indian Institute of Technology, Roorkee. His research interests include functionalization of unactivated C–H bonds. |
 Dongari Yadagiri | Dongari Yadagiri received his PhD in 2016 from the Indian Institute of Technology (IIT) Madras, India, with Prof. P. Anbarasan. He was a postdoctoral researcher (2017–2020) with Prof. V. Gevorgyan at the University of Illinois at Chicago. Later, he moved to the University of Texas at Dallas with his advisor. In 2021, he began his independent research career as Assistant Professor at IIT Roorkee, in the Department of Chemistry. His research interests are focused on organic synthesis and catalysis. |
1. Introduction
Cross-coupling reactions play a vital role in constructing bonds, such as carbon–carbon and carbon–heteroatom bonds, in synthetic organic chemistry.1 This strategy is applied in various fields, e.g., synthesizing organic frameworks and biologically active molecules in pharmaceuticals, polymer chemistry, and many other transformations.2 The C–C and C–heteroatom bond formation happens when two fragments from two electronically different coupling partners get connected through a reactive intermediate. The reaction intermediates are highly short-lived and reactive species which can be trapped during a reaction. In this context, carbenes are highly reactive intermediates in organic chemistry and can undergo many transformations for the synthesis of synthetic intermediates, and these are summarized elsewhere.3–8In this review, we have discussed the reactivity of carbenes with aryl/alkyl boronic acids and their derivatives. Diazo compounds are versatile reagents in organic chemistry for synthesizing carbenes, and these carbenes could be generated by either thermal or photochemical decomposition of diazo compounds.4 Similarly, metallocarbenes are generated from a suitable metal catalyst using diazocarbonyl compounds and related precursors.5 Thus, carbenes/metallocarbenes are reactive intermediates in organic chemistry and are widely used in the synthesis of natural products and pharmaceuticals.6 The reactivity of metallocarbenes depends on the structure of the carbenes. They can undergo various types of chemical transformations based upon the nature of the groups attached, e.g., donor carbenes (I), donor–donor carbenes (II), donor–acceptor carbenes (III), acceptor–acceptor carbenes (IV), and acceptor carbenes (V) (Fig. 1).7
 | ||
| Fig. 1 Types of metallocarbenes. | ||
1.1 Reactivity of metallocarbenes
In general, due to their electrophilic nature, metallocarbenes react with various nucleophiles and can construct various bonds, e.g., C–H, N–H, O–H, B–H, Si–H, Ge–H, Sn–H, P–H, etc, through insertion reactions. Furthermore, C–C bond formation, cascade reactions, and ylide formation followed by subsequent cyclization, annulation-type reactions, dimerization, and many other transformations (Fig. 2) take place in a single step.8 | ||
| Fig. 2 Reactivity of metallocarbenes. | ||
So far, many reviews have covered the reactivity of metallocarbenes with various nucleophiles. This review showcases the recent developments in transition metal-catalyzed reactions for the formation of carbon–carbon bonds, carbon–boron bonds, and beyond with the aid of boronic acids or esters with carbenes through the cross-coupling pathway. We have covered different carbene precursors along with boronic acids and esters for arylation, alkylation, and beyond. Organoboron compounds are useful in organic chemistry to synthesize valuable molecules.9
2. Palladium-catalyzed reactions
2.1 α-Diazocarbonyl compounds and N-tosylhydrazones as carbene precursors
The Suzuki–Miyaura cross-coupling reaction is well known for forming C–C bonds in modern organic synthesis.1,2 The widespread reactivity and popularity of this reaction are due to the utilization of various non-toxic boronic acids, mild preparation methods, and tolerance of various functional groups. Extending the scope of this reaction in 2008, the Wang group reported an agile methodology for Pd-catalyzed carbon–carbon bond formation with α-diazocarbonyl compounds and arylboronic acid for the synthesis of α-aryl substituted α,β-unsaturated carbonyl compounds (Scheme 1).10 Various α-diazocarbonyl compounds 1 and arylboronic acids 2 reacted well under the optimized conditions, giving the corresponding products 3 in good to moderate yields (Scheme 1b).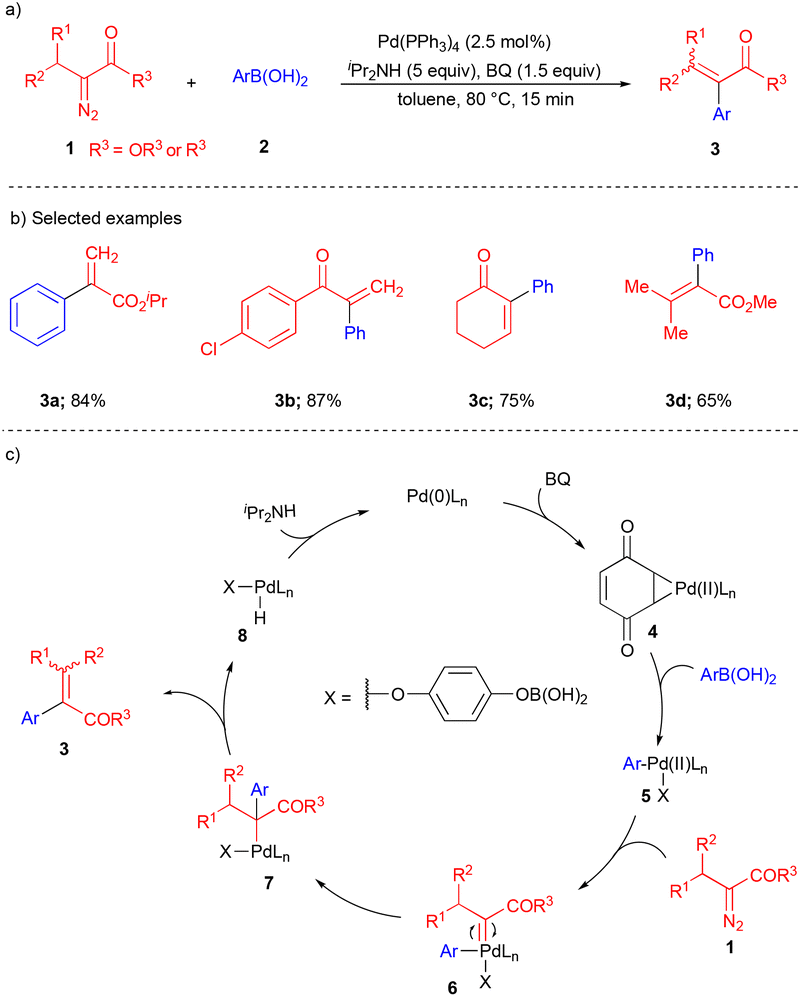 | ||
| Scheme 1 Pd-catalyzed reaction of α-diazocarbonyl compounds. | ||
First, Pd(0) undergoes oxidative addition to give species 4 (Scheme 1c) by using benzoquinone (BQ) as an oxidant. The next step is transmetalation with arylboronic acid to generate 5 (Scheme 1c). Then, the intermediate 5 reacts with the α-diazocarbonyl compound to generate Pd–carbene 6, which upon migratory insertion of the aryl group gives 7 (Scheme 1c). This is followed by β-hydride elimination, which delivers the corresponding α-aryl containing α,β-unsaturated carbonyl derivative 3 and the catalyst is ready for the next catalytic cycle in the presence of a base. Based on the control experiments, the oxidant BQ plays an important role. In the absence of BQ, product 3 was observed in trace amounts.
In 2010, the Wang group explored different carbene precursors such as N-tosylhydrazones to synthesize diaryl ethylenes (Scheme 2).11N-Tosylhydrazones are precursors for diazo compounds in the presence of base; it is well known as the Bamford–Stevens reaction.12 A variety of diaryl ethylenes 10 (Scheme 2) were synthesized using a Pd-catalyst via oxidative cross-coupling of N-tosylhydrazone derivatives 9 with aryl boronic acid 2 using a catalytic amount of CuCl along with oxygen as the oxidant to deliver the corresponding products in high yields.
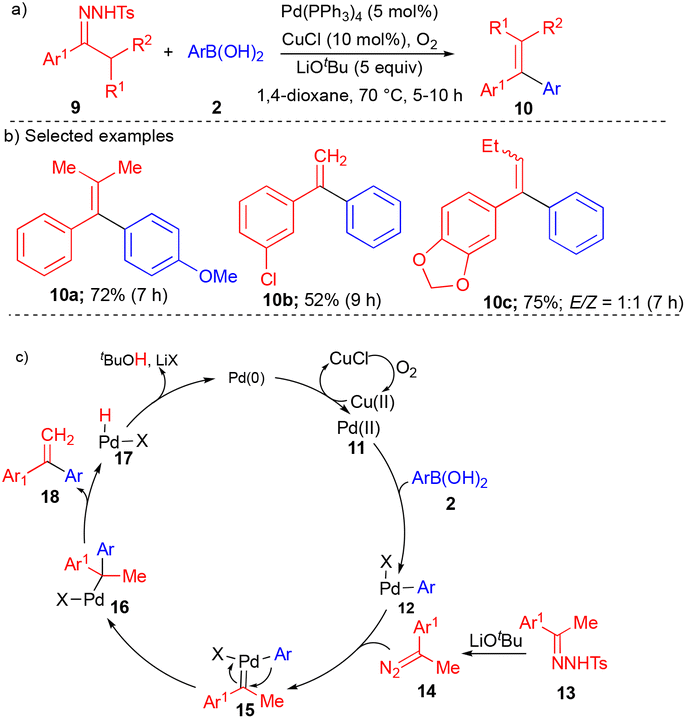 | ||
| Scheme 2 Pd-catalyzed reactions with N-tosylhydrazones. | ||
Various substituted N-tosylhydrazone derivatives 9 reacted well with arylboronic acids, giving the diaryl ethylene derivatives 10 in good to moderate yields (Scheme 2b). Based on the control experiments, the product formation could be explained by the initial formation of Pd(II) from Pd(0) in the presence of copper chloride and oxygen, followed by transmetallation with arylboronic acid to form 12. Pd–carbene complex 15 was generated from a Pd catalyst and acetophenone-derived N-tosylhydrazone 13 with the aid of lithium tert-butoxide (LiOtBu). Pd–carbene 15 underwent migratory insertion and subsequent β-hydride elimination to form diaryl ethylene derivatives 18 (Scheme 2c).
Shortly afterwards, the Yu group reported the Pd-catalyzed cross-coupling reaction between arylboronic acid 2 and α-aryl diazoacetate 19 to access (E)-α,β-diarylacrylates 20 (Scheme 3) in a highly stereoselective manner (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).13
:1).13
 | ||
| Scheme 3 Pd-catalyzed synthesis of α,β-diarylacrylates. | ||
In this transformation, 1,10-phenanthroline (phen) is essential for high stereoselectivity. Boric acid (B(OH)3) is used as an additive for the dissociation of the acetate ligand of intermediate 22 to generate 23 (Scheme 3c). Next, α-aryl diazoacetate 19 reacts with 23 to form reactive Pd–carbene 24, which undergoes migratory insertion to generate 25 and subsequent β-hydride elimination to deliver the desired α,β-diarylacrylates 20 (Scheme 3c). α,β-Diarylacrylates 20 were formed from intermediate 26, wherein two bulky aryl groups are in trans arrangement. The reaction tolerated a variety of substitutions on diazoesters 19 and arylboronic acids 2 to give trisubstituted alkenes 20 in good yields (Scheme 3b). Interestingly, α-indole containing diazoacetate also reacted very well under the optimized conditions, giving the corresponding product 20b (Scheme 3b).
Likewise, α-diazocarbonyl compounds 2-diazonaphthoquinones were also reported to react well with arylboronic acids for synthesizing 2-aryl-1-naphthols by the Kitamura group.14 2-Diazonaphthoquinone 29, derived from 1-naphthol and 2-azido-1,3-dimethylimidazolinium chloride,15 when treated with arylboronic acids 2 using a Pd(II) catalyst produced the corresponding biaryl compounds 30 (Scheme 4a) with good yields. The authors shed light on the mechanism; first, transmetalation of arylboronic acid 2 with Pd(OAc)2 in the presence of KF takes place to generate the intermediate 33 (Scheme 4c), which further reacts with 29 to form Pd–carbene 34. Consequentially, migratory insertion of the aryl group followed by protonation in the presence of acetic acid forms the desired product 2-aryl-1-naphthol 30 (Scheme 4c).
 | ||
| Scheme 4 Pd-catalyzed reaction of 2-diazonaphthoquinones. | ||
In 2014, the Wang group reported the cross-coupling between alkenyl boronic acids and cyclic α-diazocarbonyls to synthesize 1,3-dienes in an oxidative fashion.16 Since 1,3-diene moieties are found to be useful for the synthesis of various biologically important molecules and natural products, their utilization is broadly explored in synthetic organic chemistry. Here, the authors used the reaction of vinylboronic acids 37 with cyclic α-diazocarbonyls 36 to synthesize diverse substituted 1,3-diene compounds 38 bearing a ring (Scheme 5a). This reaction tolerated differently substituted vinylboronic acids with various diazocarbonyl compounds, e.g., heteroatom-containing cyclic α-diazocarbonyls such as lactone-based compounds are also suitable for this transformation (Scheme 5b). The ring size of cyclic α-diazocarbonyl compounds noticeably affected the product's yield. Cyclic α-diazocarbonyls compounds derived from the corresponding 5, 6, 7-membered ketones, and beyond gave excellent yields of the products (Scheme 5b). In the case of 5-membered cyclic α-diazocarbonyls, the yield was drastically decreased. Furthermore, the authors also explored acyclic diazocarbonyl compounds to synthesize 1,3-dienes. However, they yielded a mixture of stereoisomeric 1,3-dienes 38d (Scheme 5b).
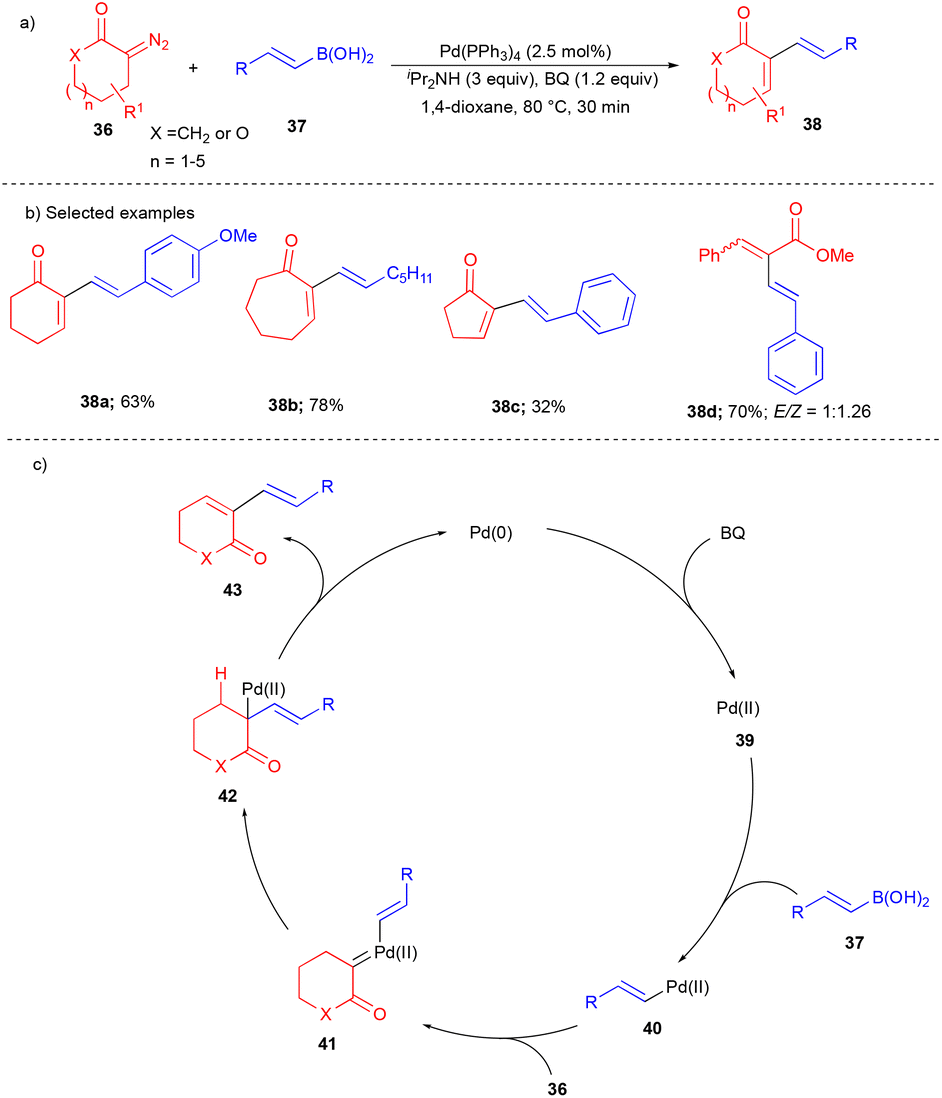 | ||
| Scheme 5 Pd-catalyzed synthesis of 1,3-dienes. | ||
The oxidation of Pd(0) by BQ generated the Pd(II) 39 species. Later, it underwent transmetalation with vinylboronic acids 37 to generate the intermediate 40. Next, cyclic α-diazocarbonyls 36 reacted with intermediate 40 to give Pd–carbene 41. Vinyl group migratory insertion gave 42, and then β-hydride elimination afforded 1,3-diene 43 (Scheme 5c).
In 2016, the Szabó group extended the reaction of allylboronic acids with α-diazoketones for the selective synthesis of linear allylic carbonyl compounds17 bearing opposite regioselectivity (Scheme 6) compared to the Cu-catalyzed cross-coupling reaction reported by the Szabó group (Scheme 32),18 which is discussed in section 4 on Cu-catalyzed transformations. Aromatic and aliphatic substituted α-diazoketones 45 reacted well with various allylboronic acid derivatives 44. Electronics on the aromatic ring of α-diazoketones 45 did not play any role in the outcome of the yields (Scheme 6b). However, alkyl substituted allylboronic acid gave the E/Z isomers 46c (Scheme 6b); further disubstituted and δ-disubstituted allylboronic acids were observed with diminished diastereoselectivity. Interestingly, unlike other Pd-catalyzed reactions, this transformation did not require any external oxidant. However, the authors used CuI as an additive to facilitate the transmetalation step and to improve the yield.
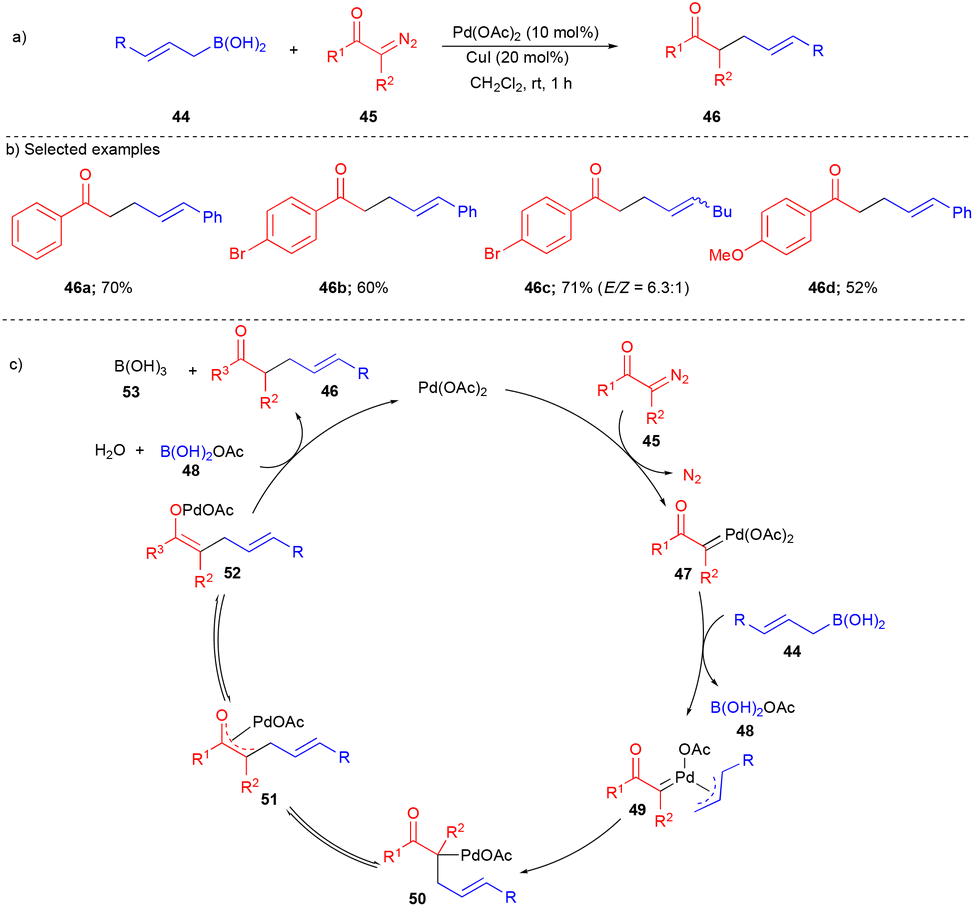 | ||
| Scheme 6 Synthesis of linear allylic carbonyl compounds. | ||
According to the proposed mechanism, the in situ generated Pd–carbene intermediate 47 undergoes transmetalation with allylboronic acid 44 to form η3-allylpalladium carbene 49. Based on the observed E/Z isomers from substituted allylboronic acid 44, species 49 undergoes η3–η1–η3 isomerization.
Next, η3-allylpalladium carbene 49 undergoes isomerization followed by migratory insertion resulting in oxa-η3-intermediate 51via50. Later, Pd-enolate 52 is generated from the oxa-η3-intermediate 51, and then the catalyst is regenerated by forming product 46 (Scheme 6c); the proposed mechanism is well supported via control experiments.
In pursuit of performing dearomative functionalization with electronically unbiased arenes as limiting reagents, Muto, Yamaguchi, and coworkers reported the dearomative C–C bond formation of bromoarene 54 with diazo compound 55 and allylboronate 56 (Scheme 7) in the presence of a Pd-catalyst.19 Here, Pd–carbene intermediate 59 formed on reaction with the diazo compound underwent migratory insertion to form a key intermediate, i.e., π-benzyl complex 60, followed by transmetalation of allylboronate 56 and reductive elimination to furnish the dearomatized compound 57. In this transformation, allylboronate 56 is not directly involved in the reaction with carbenes.
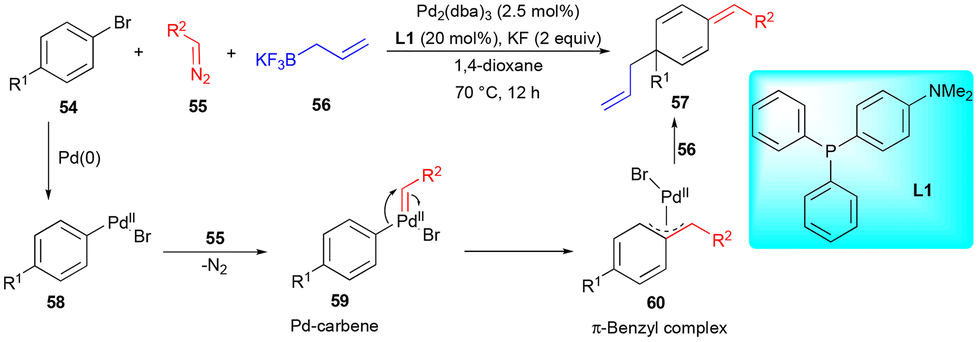 | ||
| Scheme 7 Dearomative functionalization. | ||
The Wang group continued their interest in cross-coupling reactions. In 2021, they reported that different tosylhydrazone derivatives reacted with bis(pinacolato)diborane (B2pin2) 61 in the presence of a Pd-catalyst to furnish the oxidative borylated product (Scheme 8).20 By varying the substitution on tosylhydrazones (Scheme 8a), the authors obtained different substituted borylated compounds such as tetrasubstituted alkenylboronates 63, trisubstituted alkenylboronates 65, exocyclic alkenylboronates 67 and 1,1-disubstituted alkenylboronates 69 by using the secondary substituted 62, a primary alkyl group substituted 64, cyclic substituted 66 and methyl-substituted N-tosylhydrazones 68, respectively, with B2pin261 (Scheme 8a). Electronically diverse substituted groups were tolerated well and furnished the corresponding products in moderate to good yields (Scheme 8b). Furthermore, the authors showed the utility of this transformation for the Suzuki–Miyaura cross-coupling reaction in a one-pot manner.
 | ||
| Scheme 8 Pd-catalyzed synthesis of alkenylboronates. | ||
2.2 Enynones as carbene precursors
In 2015, the Wang group explored different carbene precursors with boronic acids for synthesizing substituted furan derivatives in the presence of a Pd-catalyst.21 They utilized conjugated enynones 70 as furyl carbene precursors (Scheme 9). Under the optimized conditions, the scope of this reaction was examined between alkenylboronic acids 71 having substituted aryl, heteroaryl with different substituted enynones 70 which results in alkenyl furan derivatives and conjugated dienes 72 with high E-selectivity in good to excellent yields (Scheme 9b). The initial Pd(0) was converted into Pd(II) species 73 by oxidation with BQ. Next, transmetalation of activated Pd-catalyst 73 with boronic acid generated intermediate 74. Furyl Pd–carbene complex 76 was generated via activation of the alkyne moiety of enynone 75 by the Pd(II) intermediate 74 (Scheme 9c) and then migratory insertion of furyl Pd–carbene complex 76 followed by β-hydride elimination resulted in the formation of product 78. Pd(II) complex 79 was converted into the Pd(0) species with the aid of a base through reductive elimination. The authors reasoned the high E-selectivity of the product through selective β-hydride elimination from the intermediate 77 (Scheme 9c). | ||
| Scheme 9 Enynones as carbene precursors. | ||
Likewise, oxidative borylation of conjugated enynones with a diboron reagent was also explored by Wang's group.22 The various conjugated enynones 70 reacted with bis(pinacolato)diboron and its derivatives 81 in the presence of a Pd-catalyst (Scheme 10), 2,6-dimethylbenzoquinione (2,6-DMBQ) as an oxidant and PPh3 as a ligand to deliver furyl-substituted alkenylboronates 83. The mechanism is similar to that shown in Scheme 9. The in situ generated Pd–carbene 82 underwent boryl group migratory insertion followed by β-hydride elimination to deliver various substituted tri- and tetra-substituted alkenylboronates (Scheme 10b). Due to the steric hindrance of furyl and boryl groups, majorly the E-selectivity of the product was observed.
 | ||
| Scheme 10 Oxidative borylation of conjugated enynones with diboron compounds. | ||
2.3 Pd-catalyzed difluorocarbene transfer reactions
Fluorinated compounds play a significant role in biologically active molecules due to the intrinsic properties of fluorine.23 But the strategies to incorporate a difluoromethyl group (CF2H) as compared to a trifluoromethylated (CF3) group and a monofluorinated (F) group are relatively less explored as they suffer from functional group incompability.24 A transition metal coupled with difluoromethyl carbene uniquely installs the “CF2H” group. Nevertheless, forming a transition-metal difluorocarbene complex and transferring this “CF2” group25 are cumbersome.In 2016, the first example of Pd-catalyzed transfer of difluorocarbene was reported by the Zhang group26 for the difluoromethylation of arylboronic acids 84 using ethyl bromodifluoroacetate (BrCF2COOEt) 85 as a difluorocarbene precursor (Scheme 11). Electronically diverse substituted arylboronic acids, heteroarylboronic acids such as thiophene and carbazole, and dibenzofuran-based boronic acids were subjected to difluoromethylation and moderate to good yields of difluoromethylated compounds 86 were observed (Scheme 11b). In addition, biologically relevant molecules containing boronic acid also reacted well under the optimized conditions.
 | ||
| Scheme 11 Pd-catalyzed transfer of difluorocarbene using BrCF2COOEt. | ||
Based on the control experiments, the mechanism involves the initial formation of BrCF2COOK 87 from BrCF2COOEt/K2CO3 with the help of hydroquinone resulting in the in situ generation of difluorocarbene, and the subsequent treatment with Pd(II) delivers Pd-difluorocarbene 88 (Scheme 11c) and transmetallation with arylboronic acid produces the intermediate 89. Finally, protonolysis affords the difluoromethylated arenes 90 with the regeneration of Pd(II)(Ln) species (Scheme 11c). The authors reasoned that the role of Fe(acac)3 is ambiguous, as it could form a complex with difluorocarbene and then transfer to Pd. Also, styrene acts as a coordinative species which can improve the reactivity of the iron complex. Furthermore, the authors found that the reaction takes place via a difluorocarbene mechanism by the trapping experiment of difluorocarbene with pyridine-2-thiol.27 It further supported the difluorocarbene mechanism.
Shortly after that, the Xiao group reported the difluorocarbene transfer reaction with boronic acid to synthesize difluoromethylated arenes and olefins in the presence of a Pd-catalyst.28 Here, the authors utilized Ph3P+CF2CO2− (PDFA) 92 as the precursor for difluorocarbene.29 Pd(PPh3)4 reacted with PDFA and yielded the Pd![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CF2 trimeric complex 94 (Scheme 12a), which was unanimously confirmed by X-ray crystallography. Based on experimental evidence, the authors proposed the mechanism. Decarboxylative dissociation of PDFA 92 generated the PdCF2 complex 95 (Scheme 12b) upon coordination with Pd, followed by protonation to produce complex 97. In this transformation, 1,3-cyclopentadione 96 and Ca(OH)2 act as proton sources to form an enolate for the generation of 97, followed by transmetalation/reductive elimination to furnish the difluoromethylated arenes and olefins 100 (Scheme 12b). Electron-rich and neutral arylboronic acids gave the products in moderate yields (Scheme 12c). Interestingly, vinylboronic acid also reacted well under the optimized conditions and gave the difluoromethylated olefins with only a single stereoisomer 93e (Scheme 12c).
CF2 trimeric complex 94 (Scheme 12a), which was unanimously confirmed by X-ray crystallography. Based on experimental evidence, the authors proposed the mechanism. Decarboxylative dissociation of PDFA 92 generated the PdCF2 complex 95 (Scheme 12b) upon coordination with Pd, followed by protonation to produce complex 97. In this transformation, 1,3-cyclopentadione 96 and Ca(OH)2 act as proton sources to form an enolate for the generation of 97, followed by transmetalation/reductive elimination to furnish the difluoromethylated arenes and olefins 100 (Scheme 12b). Electron-rich and neutral arylboronic acids gave the products in moderate yields (Scheme 12c). Interestingly, vinylboronic acid also reacted well under the optimized conditions and gave the difluoromethylated olefins with only a single stereoisomer 93e (Scheme 12c).
 | ||
| Scheme 12 Difluoromethylation of arenes/olefins using PDFA. | ||
Next, the Zhang group reported difluoromethylation of arylboronic acids and their derivatives using chlorodifluoromethane (ClCF2H) 102 as a difluorocarbene precursor.30 They explored the substrate scope in the presence of a Pd-catalyst with boronic acids and boronic esters 101 and ClCF2H 102 (Scheme 13). This methodology is also applied to late-stage difluoromethylation of bioactive molecules such as δ-vitamin E 103a, ezetimibe 103b, and indometacin 103c (Scheme 13b). It was found that the difluoromethylated arene was not derived from the palladium complex 104, but it was derived from the arylboronic acid derivative itself 101a (Scheme 13c).
 | ||
| Scheme 13 ClCF2H as a difluorocarbene source. | ||
Later, in 2019, the Zhang group, in collaboration with Houk's group, reported the controllable catalytic difluoromethylation of arylboronic acids (Scheme 14) by utilizing the nucleophilicity and electrophilicity of the PdCF2 species by changing oxidation state of Pd(0) and Pd(II) respectively.31 The authors used diethyl (bromodifluoromethyl)phosphonate (BrCF2PO(OEt)2) 107 as a difluorocarbene precursor. By controlling the nucleophilic and electrophilic catalytic activity of PdCF2, four different kinds of products were synthesized with good yields (Scheme 14b). Difluoromethylated arenes 108, tetrafluoromethylated arenes 109, difluoroacetyl 110, and tetrafluoropropanoyl arenes 111 were accessed from the corresponding aryl/heteroarylboronic acid derivatives 106 and BrCF2PO(OEt)2107 (Scheme 14). For electron-withdrawing group substituted substrates, LiCl was used as an additive. Interestingly, PdIICF2 in the presence of H2O can serve as a CO precursor. For the four different types of products, reaction conditions played an important role, e.g., the higher concentration of 107 allowed the synthesis of difluoromethylated 111 and tetrafluoroethylated 109. At lower temperatures, the carbonylation products 110 and 111 were observed (Scheme 14).
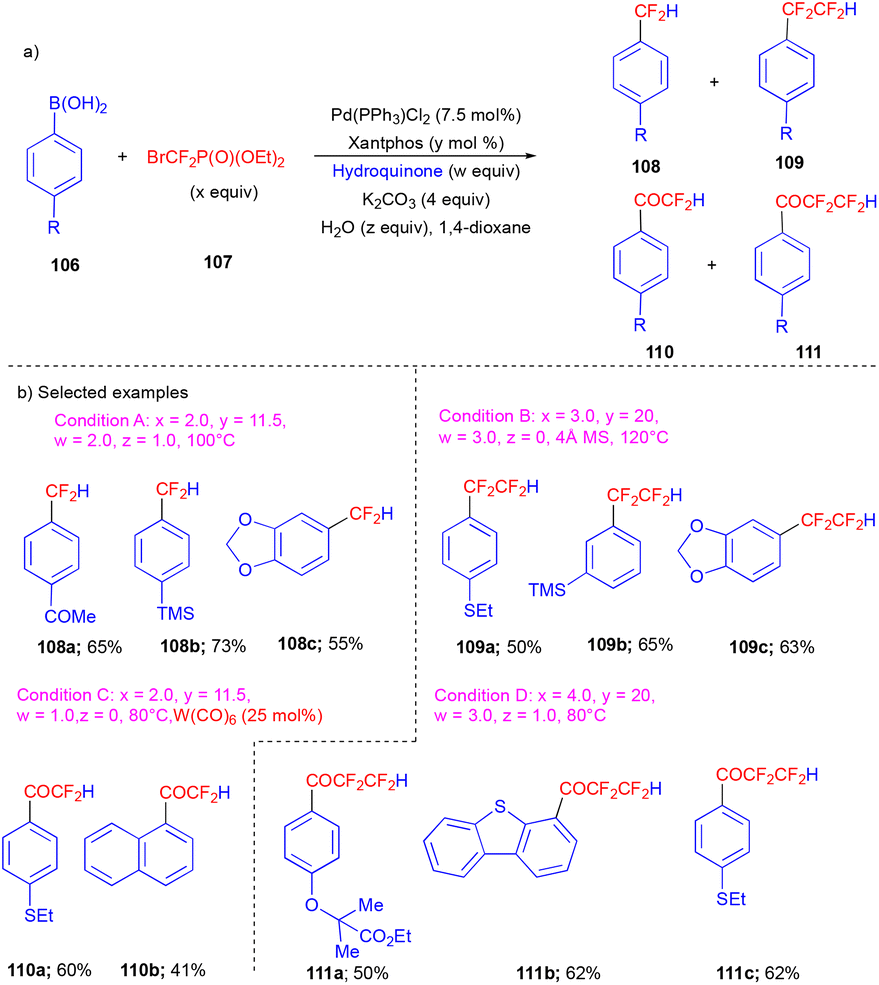 | ||
| Scheme 14 Controllable catalytic difluorocarbene transfer using BrCF2PO(OEt)2. | ||
By combining experimental details and density functional theory (DFT) calculations, the authors proposed a mechanism which is different from the Pd(II)/Pd(II) catalytic system. In contrast to the previous report, difluorocarbene is generated from 107 in the presence of a base and hydroquinone and then treated with Pd(0) to obtain the Pd(0)CF2113 species, which upon subsequent protonation gave the intermediate 114 (Scheme 15). The generated 114 then underwent transmetallation followed by reductive elimination to deliver ArCF2H 108. Next, competitive difluorocarbene insertion with the intermediate furnished 116, which underwent difluoromethyl migration to generate the CF2 elongated species 117. Then 117 reacted with arylboronic acid via transmetalation, and reductive elimination of 118 formed the tetrafluoroethylated arene 109. Theoretical calculations revealed that PdIICF2 (Scheme 15) is the electrophilic center which, on hydrolysis, generates CO. CO insertion into the σ-bond of Pd–Ar in 118 followed by reductive elimination gives 111. For synthesizing difluoromethyl ketone 110, an excess of the CO source was used to reduce the difluorocarbene concentration.
 | ||
| Scheme 15 Mechanism for controllable transfer of difluorocarbene. | ||
As discussed in previous reports, a proton source is required for the difluoromethylation of aryl/vinylboronic acids with difluorocarbene in the presence of a Pd-catalyst.26–28 In 2020, the Xiao group utilized an electrophilic carbon source for a three-component cross-coupling reaction32 between arylboronic acids, sulfonium salts, and difluorocarbene to synthesize difluoromethylated compounds with arylboronic acids via Pd-catalyzed transfer of difluorocarbene (Scheme 16). First, Ph2IOTf was employed as a phenyl cation equivalent, resulting in compound 123 with 40% 19F-NMR yield. Interestingly, when they switched to trifluoromethyl sulfonium salts 122, the product was observed in 70% 19F-NMR yield (Scheme 16a).
 | ||
| Scheme 16 Three-component coupling using PDFA. | ||
The difluoromethylated compounds were synthesized in moderate yields from arylboronic acids 2, sulfonium salts 122, and PDFA 92via a three-component approach in the presence of a Pd-catalyst (Scheme 16c). Likewise, in Scheme 12, for the initial formation of Pd0CF2124 from a Pd-catalyst and PDFA 92, the authors proposed a mechanism that involves nucleophilic reactivity of Pd0CF2124, which then reacts with sulfonium salts 122 and results in the generation of 126 (Scheme 16b). Its transmetallation with arylboronic acid 2 and finally reductive elimination furnish the product 123 (Scheme 16b).
2.4 Pd-catalyzed other transformations
In 2014, the Wang group utilized allenyl ketones as coupling partners with aryl/vinyl boronic acids to synthesize functionalized furans (Scheme 17).33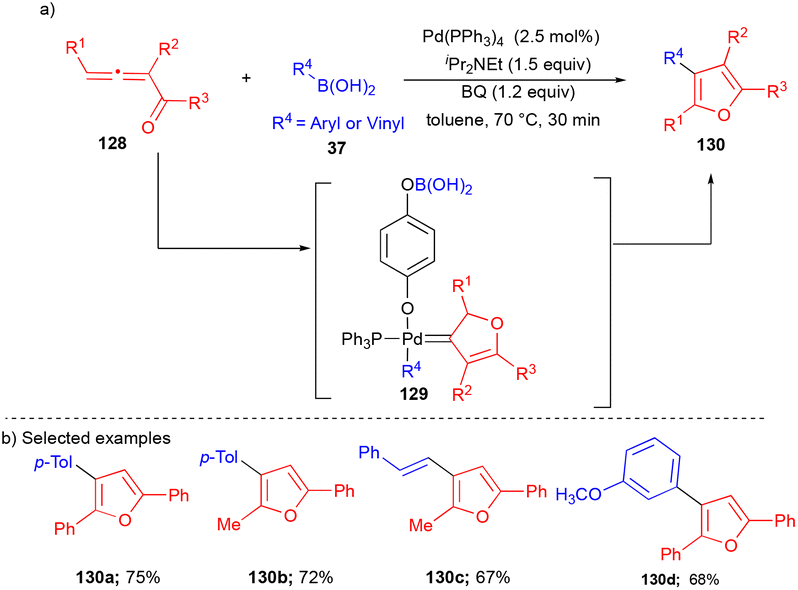 | ||
| Scheme 17 Allenyl ketones as carbene precursors. | ||
Allenyl ketones 128 as precursors of carbene, the Pd-catalyst in the presence of BQ and aryl/alkenyl boronic acid 37 generated the vinyl carbene intermediate 129, which upon subsequent migratory insertion followed by β-hydride elimination delivered the corresponding product 130. Various aryl/alkenyl boronic acids reacted well with allenyl ketone derivatives 128 to synthesize aryl, naphthyl, heteroaryl, terminal olefin, and halogen-containing furan derivatives in good yields (Scheme 17b). Interestingly, this reaction was scaled up tenfold and a comparable yield was observed, showing this transformation's efficiency.
In 2017, Wang and Lu's group reported the cross-coupling reaction between 3-diazoindolin-2-imines 131 and arylboronic acids 2 to synthesize 2-amino-3-arylindoles 135 (Scheme 18).34 This mechanism differs slightly from that of the cross-coupling reaction of Pd–carbene with arylboronic acids. First, Pd(OAc)2 reacted with 131 to form PdII–carbene 132 (Scheme 18b), which then reacted with boronate complex 133, which was formed from arylboronic acid 2 and triethylamine (Et3N). The aryl group migration from 133 to 132 generated the intermediate 134 (Scheme 18b). Finally, migration of Pd from C to N followed by protonolysis gave product 135.
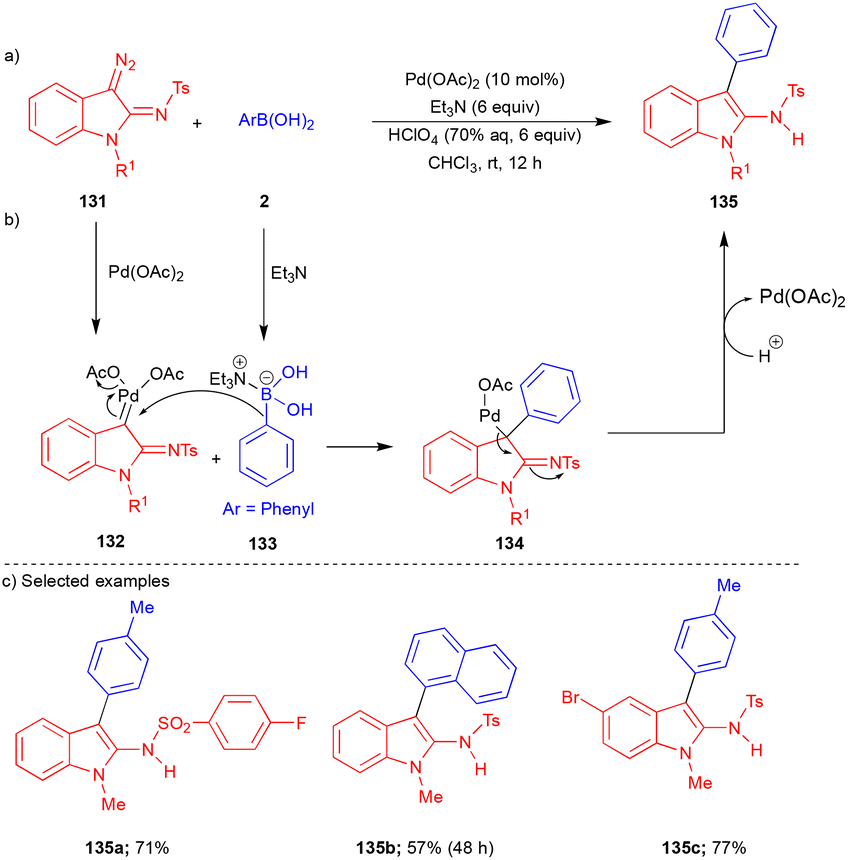 | ||
| Scheme 18 Synthesis of 2-amino-3-arylindoles. | ||
Electronically diverse 131 and arylboronic acids 2 reacted well under the optimized conditions, giving a broad spectrum of 2-amino-3-arylindoles 135 (Scheme 18c). Unfortunately, heteroaryl, alkyl and alkenylboronic acids did not react under the optimized conditions.
To find effective coupling partners, the Wang group, in 2018, reported the coupling of alkyl chromium(0) Fischer carbenes 136 with organoboronic acids 91 to yield ketones and α,β-unsaturated ketones 137 (Scheme 19).35 Based on the observed product, two pathways were proposed. Initially, the formation of 138 took place through path A, which includes alkyl carbene transfer to palladium to form intermediate 143 (Scheme 19c), and then migratory insertion followed by β-hydride elimination and subsequent hydrolysis resulted in ketone 148 (Scheme 19c).
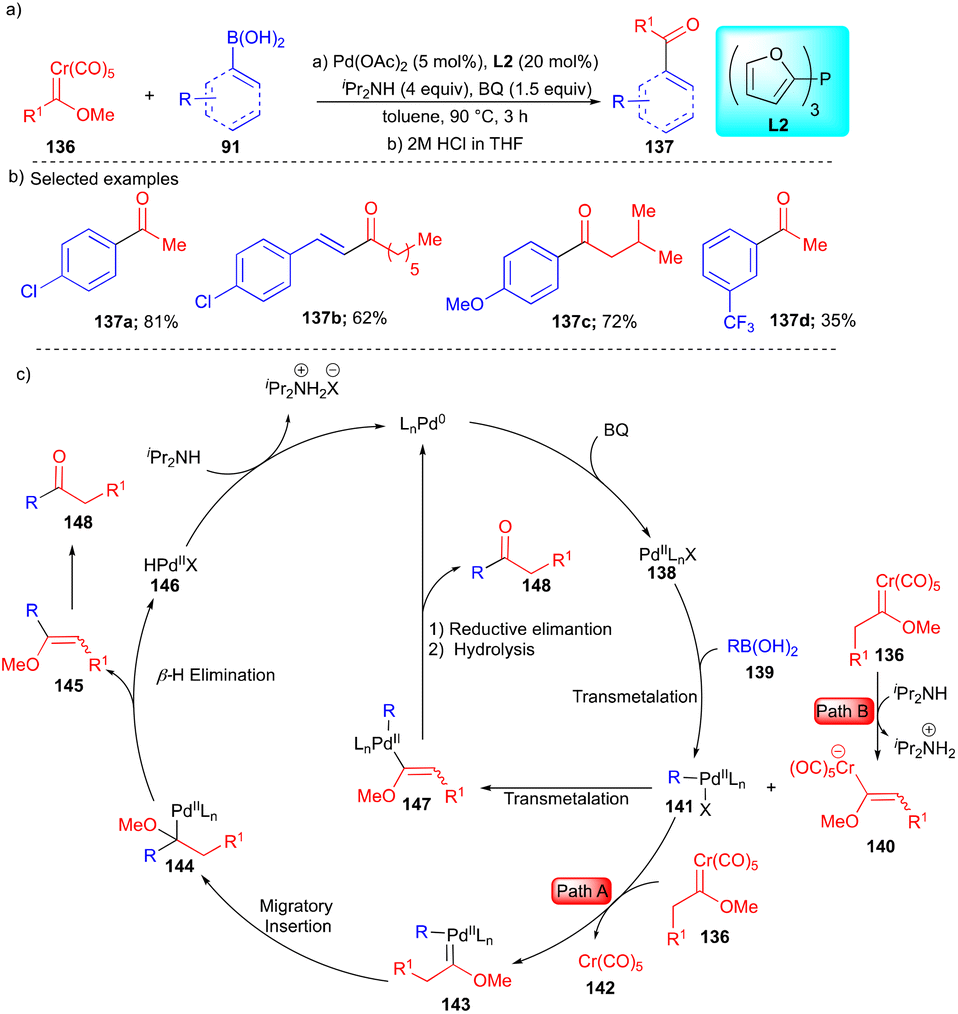 | ||
| Scheme 19 Cross-coupling reactions using alkyl chromium(0) Fischer carbene. | ||
In path B, deprotonation of alkyl chromium(0) Fischer carbenes in the presence of iPr2NH generated intermediate 140, followed by vinyl ether group transfer from 141 to intermediate 140, which resulted in palladium intermediate 147. Further reductive elimination and hydrolysis delivered ketone 148 (Scheme 19c). The authors reasoned that path B is more favorable, based on the control experiments. Both aryl and vinylboronic acids were explored along with alkyl chromium(0) Fischer carbenes and moderate to good yields of the substituted ketones 137 were observed (Scheme 19b).
In 2019, the Song group reported an elegant transformation for the Pd-catalyzed synthesis of amidinium salts and diaryl ketones using thiourea and thioamides as carbene precursors.36 Treatment of thiourea 149 with arylboronic acid 2 provided the cyclic and acyclic amidinium salts 150 (Scheme 20).
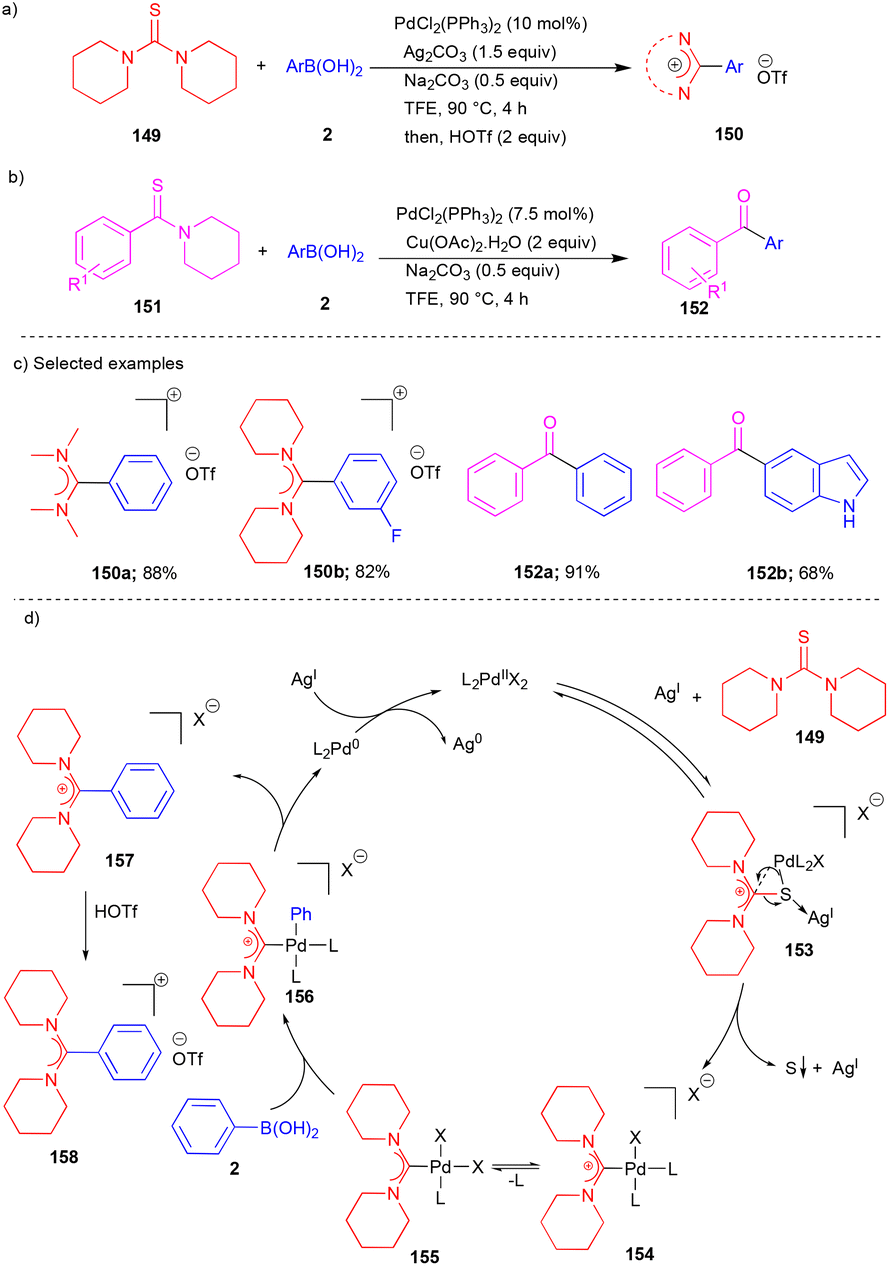 | ||
| Scheme 20 Pd-catalyzed reactions of thioureas and thioamides. | ||
Interestingly, this methodology was expanded to synthesize unsymmetrical diaryl ketones 152 from thioamides 151, which include electronically and sterically diverse substituted, heteroaryl, and biologically important diaryl ketones with good yields (Scheme 20b). In this transformation, a Ag salt plays a dual role in desulfurization, forming a Pd–aminocarbene complex and acting as an oxidant for converting Pd(0) into Pd(II). With the aid of the Ag salt, Pd(II) coordinated with thiourea 149 to form intermediate 153 (Scheme 20d). Furthermore, intermediate 153 underwent desulfurization and extrusion of sulfur with the aid of the silver salt and delivered the Pd(II) complex 154, which is in equilibrium with its neutral form 155 (Scheme 20d). Next, it underwent transmetalation/reductive elimination and gave the amidinium salt 158 in the presence of HOTf.
3. Rhodium catalyzed cross-coupling reactions
3.1 Utilizing triazoles as carbene precursors
Suitably substituted 1,2,3-triazoles undergo ring-chain isomerism via extrusion of nitrogen to form α-diazo imines, well known as Dimorth rearrangement.37 In 2012, the Fokin group reported the synthesis of enamines (Scheme 21) in the presence of the (Rh2(S-PTAD)4)-catalyst via the reaction between N-sulfonyl-1,2,3-triazoles and arylboronic acids.38 The broad array of N-sulfonyl-1,2,3-triazoles 159 when treated with sterically hindered arylboronic acids 2 containing different functional groups gave enamines 160 in good yields (Scheme 21b) with a high isomeric ratio. The authors also showed the synthesis of 3-aryl indoles 161 by integration of Rh and Cu via a semi one-pot process. Initially, Rh-catalyzed arylation of 2-bromo phenyl substituted N-sulfonyl-1,2,3-triazoles 159 with arylboronic acid 2 and then filtration followed by C–N cross-coupling resulted in 3-aryl indole derivatives 161 by using a Cu-catalyst (Scheme 21a). | ||
| Scheme 21 Arylation of Rh-azavinyl carbenes. | ||
Rhodium azavinyl carbene 162 (Scheme 21c) was generated from N-sulfonyl-1,2,3-triazoles in the presence of a Rh-catalyst.5 The lone pair of the imine nitrogen of 162 coordinates with the boron atom of the arylboronic acids 2 in a reversible manner and results in the formation of the intermediate 164. Next, the aryl group migrates from 164 to 165 in an irreversible manner shows the stereochemical outcome to yield the high isomeric product followed by protonolysis would deliver enamine 160 (Scheme 21c). CaCl2 is an additive to generate trimeric arylboroxine 163 from arylboronic acid 2.
In 2016, Miura and Murakami's group showed an interesting transformation between triazoles 166, 9-borabicyclo[3.3.1]nonane 167 (9-BBN–H), and aldehydes 172 in the presence of Rh2(OPiv)4 as a catalyst for the stereoselective synthesis of 1,3-amino alcohols 173.39 At first, in situ generated rhodium azavinyl carbene 168 from 166, which underwent C–C bond rotation and then reacted with 9-BBN–H 167 to form the intermediate 169 (Scheme 22a) via the coordination of the nitrogen lone pair of the imine with a vacant p-orbital of boron. Next, intramolecular hydride transfer formed the zwitterionic intermediate 170 followed by loss of Rh to give boron aza-enolate (E)-171. Then, it underwent an aza–aldol reaction with aldehyde 172 followed by DIBAL–H reduction to form the corresponding syn-stereochemistry of 1,3-amino alcohol 173via a six-membered transition state (TS) having a boat-like conformation (Scheme 22b).
 | ||
| Scheme 22 Rh-catalyzed synthesis of 1,3-amino alcohols. | ||
Various syn-1,3-amino alcohols were synthesized with good yields and stereoselectivity from N-sulfonyl-1,2,3-triazoles 166, 9-BBN–H 167, and aldehydes 172 (Scheme 22c). Interestingly, the authors showed a sequential addition of the starting material from terminal alkynes via Cu-catalyzed triazole synthesis40 with sulfonyl azides and then Rh-catalyzed B–H insertion followed by an aza–aldol reaction. Finally, DIBAL–H reduction gave 173 in a one-pot manner.
The Wang group explored conjugated enynones 174 as carbene precursors to synthesize furan containing triarylmethanes 175 (Scheme 23)41 in the presence of a Rh-catalyst. They synthesized electronically diverse substituted furan containing triarylmethanes 175 in good yields from boronic acids and enynones (Scheme 23b). It was observed that the alkyne moiety's aryl group on enynones played a significant role in obtaining a good yield of the corresponding products (Scheme 23b).
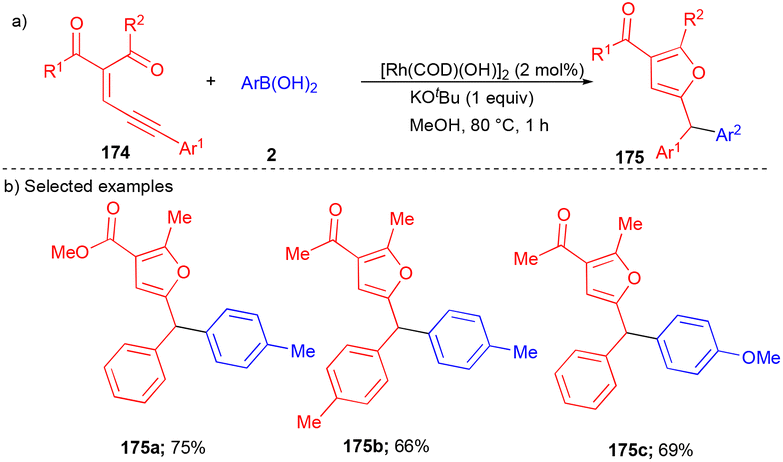 | ||
| Scheme 23 Rh-catalyzed reactions using enynones as carbene precursors. | ||
3.2 Utilizing diazo precursors
The Yu group reported the cascade reaction of arylboronic esters, aryl diazoesters, and alkyl halides to synthesize α,α-heterodiaryl carboxylic acids with the [Rh(COD)OH]2 catalyst.42 A broad spectrum of quaternary carbon-containing α,α-heterodiaryl carboxylic acids 179 (Scheme 24) were synthesized through the three-component coupling strategy in a one-pot manner from diverse substituted arylboronic esters 177, aryl diazoesters 176, and alkyl halides 178.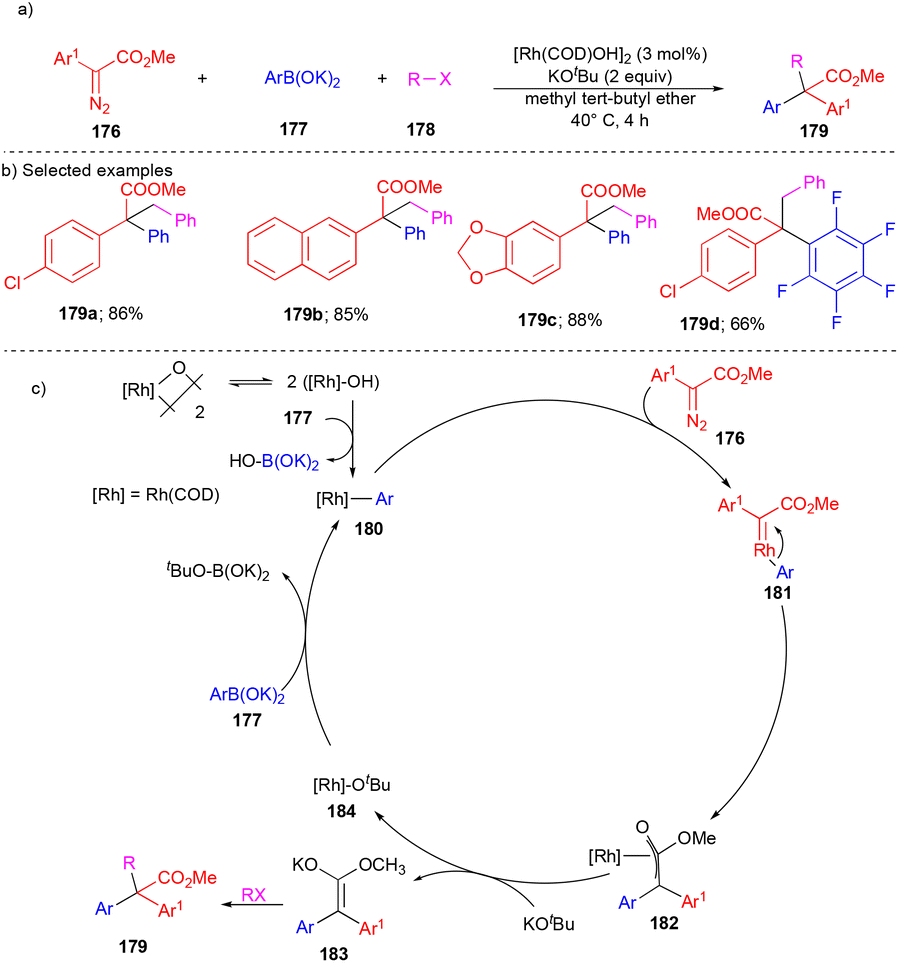 | ||
| Scheme 24 Rh-catalyzed synthesis of diaryl carboxylic acids. | ||
First, Rh(I) undergoes transmetalation with arylboronic esters 177, forms the aryl Rh(I) complex 180, and then reacts with diazoester 176, giving Rh–carbene 181. Later, it undergoes migratory insertion to form oxa-π-allylrhodium 182 (Scheme 24c). Based on the control experiments, the exchange of intermediate 182 with KOtBu gives the potassium enolate intermediate 183, which reacts with an alkyl halide to deliver the corresponding α,α-heterodiaryl carboxylic acids 179 (Scheme 24c).
Later, the same group explored N-chlorosuccinimides for the three-component reaction in the presence of the RhIII(Cp*)(OAc)2-catalyst, which resulted in the synthesis of α-aryl–α-chloro carbonyl derivatives 187 (Scheme 25).43 Arylrhodium(III) complex 191 generated from arylboronic acid 2 (Scheme 25c) when treated with 185 resulted in the formation of Rh(III)–carbene 192 and then migratory insertion formed the Rh(III)–diketonate complex 193. Next, chlorination proceeded via the coordination of N-chlorosuccinimide to 193, giving complex 194 and resulting in the product 195 through nucleophilic displacement by diketonate adduct 194 (Scheme 25c). Control experiments well supported the proposed mechanism. The substrate scope of arylboronic acids 2 and acceptor–acceptor carbene precursor 185 was well explored with N-chlorosuccinimides for chloroarylation to deliver moderate yields of 187 (Scheme 25b). Interestingly, alkene-tethered diazoester reacted chemoselectively and resulted in the corresponding product 187c in a good yield.
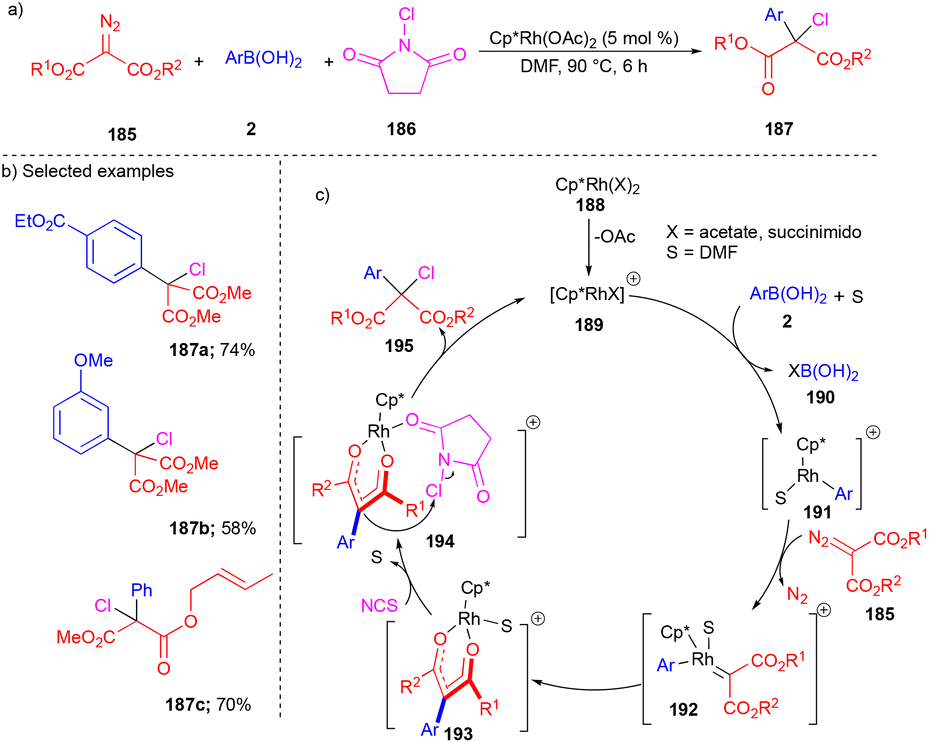 | ||
| Scheme 25 Rh-catalyzed cascade reaction. | ||
In 2015, the Anbarasan group reported the arylation of α-diazocarbonyl compounds with arylboronic acids by using RhCl[COD]2.44In situ generated arylrhodium(I) complex 198 (Scheme 26c) reacts with α-diazocarbonyl compounds 199 to generate rhodium carbene 200, which on 1,2 aryl migration gives 201, which is in equilibrium with its tautomer 202. This is followed by protodemetalation to afford the desired product 203 (Scheme 26c).
 | ||
| Scheme 26 Rh-catalyzed arylation. | ||
This methodology gave a broad spectrum of compounds under the optimized conditions from substituted α-diazocarbonyl compound 196 and various arylboronic acids 2, affording the products in moderate to good yields and with excellent chemoselectivity (Scheme 26b). Furthermore, the authors also showed the synthesis of the core structure of diclofensine, a therapeutically important molecule.
C(aryl)–C(sp3) bond formation is widely known for diazo compounds via cross-coupling reactions. In contrast, the formation of C(sp3)–C(sp3) bonds is rare. To address this problem, the Yu group reported the reaction between potassium alkyltrifluoroborate 204 and diazomalonate 205 with the aid of the [Cp*RhCl2]2 catalyst (Scheme 27).45 The formation of σ-alkylrhodium complex 207 (Scheme 27b) through transmetallation of alkyltrifluoroborate 204 in the presence of the Rh(III)-catalyst was confirmed by ESI-MS analysis. Thus, 207 reacted with diazomalonate 205 and led to the synthesis of Rh(III)–carbene 208, followed by migratory insertion to form rhodium(III)–alkyl complex 209. Furthermore, protonation delivers product 206 (Scheme 27b). On the other hand, alkyl migration followed by nitrogen elimination through the concerted mechanism could not be avoided.
 | ||
| Scheme 27 Rh-catalyzed reaction of alkyltrifluoroborate with diazomalonate. | ||
A wide variety of alkyltrifluoroborates 204 and diazomalonates 205 reacted well and gave the C(sp3)–C(sp3) bond containing compounds 206via the cross-coupling reaction (Scheme 27c). Based on the observed reactivity of alkene-tethered diazo compounds 205, it is suggested that the alkyl migratory insertion reaction is much faster than intramolecular cyclopropanation.
In 2016, the Wang group utilized the CF3-bearing N-tosylhydrazones 210 as carbene precursors (Scheme 28) for arylation with arylboronates 211 to synthesize 1,1-difluoro-2,2-diarylalkenes 212.46 Due to the broad applications of fluorinated compounds, the authors explored N-tosylhydrazones 210, which are derived from trifluoromethylketones. Moderate yields of 1,1-difluoro-2,2-diarylalkenes were obtained (Scheme 28b) from N-tosylhydrazones 210 and arylboronates 211 under the optimized conditions. The mechanism involves the initial formation of aryl rhodium species 214 through the transmetalation step. Next, the in situ generated diazo compound 216, from CF3-bearing N-tosylhydrazone 215, reacted with 214 to form the intermediate 217 and then subsequent migratory insertion followed by protonation resulted in the desired compound 212 (Scheme 28c).
 | ||
| Scheme 28 Synthesis of 1,1-difluoro-2,2-diarylalkene derivatives. | ||
Similarly, in 2017, the Wang group explored the reactivity of α-diazo phosphonates for cross-coupling reactions with arylboronic acids (Scheme 29). It was shown that the synthesis of various substituted diarylmethylphosphonates 220 was achieved in the presence of a Rh-catalyst.47 The scope was explored with substituted boronic acids 2 and α-diazo phosphonates 219. Electron-donating and sterically hindered substrates of arylboronic acids 2 resulted in high yields (Scheme 29b). In addition, substituted diazo phosphonates 219 showed good functional group tolerance. First, aryl rhodium(I) species 214 reacted with α-diazo phosphonates 221, and then the subsequent migratory insertion followed by protonation delivered the final product 224 (Scheme 29c).
 | ||
| Scheme 29 Rh-catalyzed synthesis of diarylmethylphosphonates. | ||
In 2023, Liu and Chang's group reported the Rh(III)-catalyzed [4 + 2] annulation reaction to synthesize 9,10-phenanthrenes.48 The reaction involves the initial transmetalation of [1,1′-biphenyl]-2-ylboronic acid 225 with the Rh(III)-catalyst followed by C–H activation to form the Rh–carbene intermediate 230 with β-keto ester 226. Then migratory insertion and subsequent cyclization followed by dehydration deliver the 9,10-phenanthrenes 227 (Scheme 30b).
 | ||
| Scheme 30 Rh(III)-catalyzed synthesis of 9,10-phenanthrenes. | ||
The authors explored the substrate scope with electronically biased substrates (Scheme 30c). However, in some cases, mixtures of regioisomers were observed. Substitution of boronic acid with halogen and alkyl groups at the o, m, and p positions gave the product in moderate yields (Scheme 30c).
4. Copper-catalyzed reactions
In 2011, the Cu-catalyzed three-component Mannich reaction reported by the Schaus group49 using arylboranes and carbonyl imines with diazoesters resulted in the isocyanate Mannich product, which was trapped with various nucleophiles (Scheme 31). Initially, they treated Ph–9-BBN 235 with diazoester 236 and carbonyl imines 237 in the presence of Cu(OTf)2 to obtain isocyanate, which when quenched with alcohols delivered the product 238 with good yields. The scope is very broad. Aryl and alkyl substituted diazoesters 236 gave the desired products with good yields, but the diastereoselectivity (dr) was diminished (Scheme 31b). Electron-withdrawing or donating group substituted aryl-bearing boranes 235 reacted well (Scheme 31b). Furthermore, the asymmetric Mannich reaction is also shown by the authors by using chiral auxiliary derived phenyl diazoacetate esters. The attack of in situ generated enolate 239 (Scheme 31c) on the vacant p-orbital of boron, followed by aryl group migration via extrusion of N2, results in the formation of boron enolate 241. Next, the boron enolate reacts with imine 237 to afford borylated carbamate 242 (Scheme 31c), and then elimination of B–R2O–9-BBN gives the isocyanate product 243 that could be trapped in situ by alcohol. | ||
| Scheme 31 Three-component reaction via the boron enolate Mannich reaction. | ||
In 2015, the Szabó group reported the synthesis of C(sp3)–C(sp3) containing compounds from α-diazoketones and allylboronic acid (Scheme 32) using a catalytic amount of copper(I) thiophene-2-carboxylate (CuTC) via the cross-coupling strategy.18 Highly branched selective homoallyl compounds were synthesized with excellent yields and in a highly regioselective manner from aryl and alkyl substituted α-diazoketones 45 and allylboronic acid 44 (Scheme 32b), and in the case of aliphatic α-diazocarbonyl compounds, the desired product 245 was obtained with good yields at a lower temperature. Interestingly, geraniol boronic acid also reacted well regioselectively and gave the branched selective product 245d which contains a quaternary carbon (Scheme 32b).
 | ||
| Scheme 32 Cu-catalyzed reaction of allylboronic acids with α-diazoketones. | ||
The temperature influenced the diastereoselectivity. When the authors performed the reaction at 0 °C, the dr was increased when boronic acid was derived from cinnamylboronic acid 245c (Scheme 32b). α-Diazoketones were converted into copper carbene intermediate 244via nitrogen molecule extrusion, which when reacted with allylboronic acid 44 gave the homoallylic carbonyl compound 245 (Scheme 32a). However, the mechanism is not very clear.
In 2017, the same group explored cyclic allylboronic acids in the presence of a Cu-catalyst with α-diazoketones (Scheme 33).50 Cyclic allylboronic acids 246 reacted very well with substituted α-diazoketones 247, affording the product 248 (Scheme 33a) with good yields and high diastereoselectivity. Diminished yield and diastereoselectivity were observed when the reaction was performed with disubstituted α-diazoketone 247. Interestingly, the configuration of cyclic allylboronic acids was found to be maintained. However, the yield and diastereoselectivity of cis-cyclic allylboronic acid 248b were lower than those of trans-cyclic allylboronic acid 248c. In situ generated Cu–carbene 250 (Scheme 33b) underwent syn SE2′-type transmetalation with cyclic allylboronic acids 246via intermediate 251, and then migratory insertion followed by protonation delivered the product 248.
 | ||
| Scheme 33 Reaction of cyclic allylboronic acid with α-diazoketones. | ||
5. Miscellaneous cross-coupling reactions
In 2001, Srebnik developed a novel platinum-catalyzed methodology with diazomethane 258 insertion into bis(pinacolato)diborane 61 to synthesize highly efficient sp3-carbon substituted 1,1-diboron 259 (Scheme 34).51 Later, the same group explored substituted diazocarbene precursor 263 by increasing the temperature and changing the solvent from diethyl ether to toluene to synthesize borylated compounds 264 (Scheme 34b).52 First, Pt–carbene 261 was formed from the reaction of diazomethane with 260, then migratory insertion of 261 formed 262, and finally, reductive elimination furnished the diborylated compound 259. | ||
| Scheme 34 Pt-catalyzed borylation. | ||
Later, in 2014, this concept was extended by the Kingsbury group with non-stabilized diazoalkanes 263 and B2pin2 to furnish gem-diborylalkanes 265 (Scheme 35).53 Alkyl/aryl, alkyl/alkyl substituted gem-diborylalkanes 265 were synthesized with good yields (Scheme 35b).
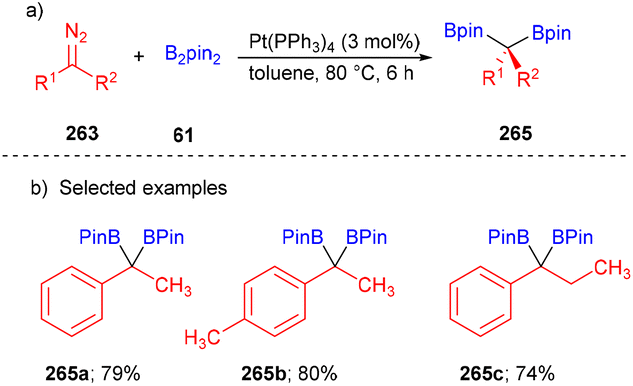 | ||
| Scheme 35 Pt-catalyzed diborylation of substituted diazoalkanes. | ||
In 2020 Huang's group reported the use of a sewage sludge (SW) surface-modified catalyst for the reaction of α-diazocarbonyl compounds with arylboronic acids as a greener approach to synthesize diarylacetate derivatives without using any strong base (Scheme 36).54 After screening various acids and additives, the SW was modified by treating with HClO4 to obtain the product in a high yield.
 | ||
| Scheme 36 Sewage sludge catalyzed cross-coupling reaction. | ||
Electronically diverse substituted α-diazo compounds 266 and arylboronic acids 2 were explored and gave moderate to good yields of diarylacetate derivatives 267 (Scheme 36b). This methodology was also applied for synthesizing biologically active molecules such as diclofensine and darifenacin. The mechanism for this reaction is quite unclear. However, the authors suggested hydrogen bonding of the OH group of arylboronic with SW (HClO4), which increased the Lewis acidity of boronic acids. Aryl group migration with extrusion of molecular nitrogen followed by protonolysis produced diarylacetate derivatives 267.
6. Summary and outlook
This review article discussed the reactivity of metallocarbenes with aryl/vinyl/allyl/alkyl boronic acid derivatives via TM-catalyzed cross-coupling to incorporate groups such as aryl, vinyl, allyl, alkyl, boron and others into various organic molecules derived from carbene precursors. Thus, carbenes are derived from α-diazocarbonyl compounds, tosylhydrazones, difluorocarbene precursors, enynones, N-sulfonyl-1,2,3-triazoles, allenyl ketones, thioureas, thioamides, and alkyl chromium(0) Fischer carbene precursors and the reactions performed in the presence of TM-catalysts such as Pd, Cu, Rh, and Pt are discussed. With the emergence of new carbene precursors, different boronic acid derivatives as coupling partners have paved the way to construct various carbon–carbon, carbon–boron, and carbon–halogen bonds. This cross-coupling strategy led to the incorporation of groups such as aryl/alkyl and others through late-stage functionalization and difluoromethyl incorporation into biologically active compounds. Furthermore, this concept is also useful for synthesizing heterocyclic compounds such as indole derivatives, furan derivatives, and boron group incorporated furans.Most of the discussed strategies showed only racemic compounds. So far, no examples of asymmetric catalysis have been shown for cross-coupling between metallocarbene and boronic acid derivatives. Also, there is ample room for discoveries in this area by implementing mild and efficient protocols, i.e., using less harsh conditions and utilizing different carbenes other than donor/acceptor carbenes with various transition metals, and gaining more mechanistic insights for new advances through this review.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the DST-SERB-CRG (CRG/2022/000442), New Delhi, India, for financial support. A. B. is thankful for the PMRF fellowship, P. Y. is thankful for the UGC fellowship, and M. S. thanks IIT Roorkee for his MHRD fellowship.References
- (a) N. Miyaura, Cross-Coupling Reactions: A Practical Guide, Springer, 2000 Search PubMed; (b) A. de Meijere and F. Diederich, Metal-Catalyzed Cross-Coupling Reactions, Wiley, 2004 CrossRef; (c) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed.
- All the references therein. (a) J. F. Hartwig, Acc. Chem. Res., 1998, 31, 852–860 CrossRef CAS; (b) J. P. Wolfe, S. Wagaw, J.-F. Marcoux and S. L. Buchwald, Acc. Chem. Res., 1998, 31, 805–818 CrossRef CAS; (c) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed; (d) E. I. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738–6764 CrossRef CAS PubMed; (e) G. Marzano, C. V. Ciasca, F. Babudri, G. Bianchi, A. Pellegrino, R. Po and G. M. Farinola, Eur. J. Org. Chem., 2014, 6583–6614 CrossRef CAS; (f) E. J. Tollefson, L. E. Hanna and E. R. Jarvo, Acc. Chem. Res., 2015, 48, 2344–2353 CrossRef CAS PubMed; (g) D. Haas, J. M. Hammann, R. Greiner and P. Knochel, ACS Catal., 2016, 6, 1540–1552 CrossRef CAS; (h) D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed; (i) A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS PubMed; (j) M. J. West, J. W. B. Fyfe, J. C. Vantourout and A. J. B. Watson, Chem. Rev., 2019, 119, 12491–12523 CrossRef CAS PubMed; (k) E. Palao, E. López, I. Torres-Moya, A. de La Hoz, Á. Díaz-Ortiz and J. Alcázar, Chem. Commun., 2020, 56, 8210–8213 RSC; (l) N. Matłok, S. Lachowicz, J. Gorzelany and M. Balawejder, Molecules, 2020, 25, 2596 CrossRef PubMed; (m) V. Lacanau, F. Bonneté, P. Wagner, M. Schmitt, D. Meyer, F. Bihel, C. Contino-Pépin and D. Bourgeois, ChemSusChem, 2020, 13, 5224–5230 CrossRef CAS PubMed.
- (a) T. Ye and M. A. McKervey, Chem. Rev., 1994, 94, 1091–1160 CrossRef CAS; (b) A. Padwa and D. J. Austin, Angew. Chem., Int. Ed. Engl., 1994, 33, 1797–1815 CrossRef; (c) A. Padwa and M. David Weingarten, Chem. Rev., 1996, 96, 223–270 CrossRef CAS PubMed; (d) M. P. Doyle, Modern catalytic methods for organic synthesis with diazo compounds: from cyclopropanes to ylides, Wiley, 1998 Search PubMed; (e) M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911–935 CrossRef CAS PubMed; (f) J. Barluenga and C. Valdes, Angew. Chem., Int. Ed., 2011, 50, 7486–7500 CrossRef CAS PubMed; (g) Z. Liu and J. Wang, J. Org. Chem., 2013, 78, 10024–10030 CrossRef CAS PubMed; (h) Y. Xia, Y. Zhang and J. Wang, ACS Catal., 2013, 3, 2586–2598 CrossRef CAS; (i) T. Wang and A. S. K. Hashmi, Chem. Rev., 2021, 121, 8948–8978 CrossRef CAS PubMed; (j) Z. Bao and J. Wang, Synlett, 2023 DOI:10.1055/a-2106-1799.
- (a) Ł. W. Ciszewski, K. Rybicka-Jasińska and D. Gryko, Org. Biomol. Chem., 2019, 17, 432–448 RSC; (b) Z. Yang, M. L. Stivanin, I. D. Jurberg and R. M. Koenigs, Chem. Soc. Rev., 2020, 49, 6833–6847 RSC; (c) J. Durka, J. Turkowska and D. Gryko, ACS Sustainable Chem. Eng., 2021, 9, 8895–8918 CrossRef CAS; (d) C. Empel, C. Pei and R. M. Koenigs, Chem. Commun., 2022, 58, 2788–2798 RSC; (e) R. D. C. Gallo, G. Cariello, T. A. C. Goulart and I. D. Jurberg, Chem. Commun., 2023, 59, 7346–7360 RSC.
- (a) B. Chattopadhyay and V. Gevorgyan, Angew. Chem., Int. Ed., 2012, 51, 862–872 CrossRef CAS PubMed; (b) H. M. L. Davies and J. S. Alford, Chem. Soc. Rev., 2014, 43, 5151–5162 RSC; (c) M. Jia and S. Ma, Angew. Chem., Int. Ed., 2016, 55, 9134–9166 CrossRef CAS PubMed; (d) P. Kumar, V. Garg, M. Kumar and A. K. Verma, Chem. Commun., 2019, 55, 12168–12171 RSC; (e) D. Yadagiri, M. Rivas and V. Gevorgyan, J. Org. Chem., 2020, 85, 11030–11046 CrossRef CAS PubMed; (f) M. Akter, K. Rupa and P. Anbarasan, Chem. Rev., 2022, 122, 13108–13205 CrossRef CAS PubMed.
- (a) H. M. L. Davies and Y. Lian, Acc. Chem. Res., 2012, 45, 923–935 CrossRef CAS PubMed; (b) K. A. Mix, M. R. Aronoff and R. T. Raines, ACS Chem. Biol., 2016, 11, 3233–3244 CrossRef CAS PubMed; (c) S. Happy, M. Junaid and D. Yadagiri, Chem. Commun., 2023, 59, 29–42 RSC.
- All the references therein. (a) J. R. Fulton, V. K. Aggarwal and J. de Vicente, Eur. J. Org. Chem., 2005, 1479–1492 CrossRef CAS; (b) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS PubMed; (c) H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857–1869 RSC; (d) J. Barluenga and C. Valdés, Angew. Chem., Int. Ed., 2011, 50, 7486–7500 CrossRef CAS PubMed; (e) D. Gillingham and N. Fei, Chem. Soc. Rev., 2013, 42, 4918–4931 RSC; (f) C. Werlé, R. Goddard, P. Philipps, C. Farès and A. Fürstner, J. Am. Chem. Soc., 2016, 138, 3797–3805 CrossRef PubMed; (g) Q. Q. Cheng, Y. Deng, M. Lankelma and M. P. Doyle, Chem. Soc. Rev., 2017, 46, 5425–5443 RSC; (h) Y. Xia and J. Wang, Chem. Soc. Rev., 2017, 46, 2306–2362 RSC; (i) Y. Xia and J. Wang, J. Am. Chem. Soc., 2020, 142, 10592–10605 CrossRef CAS PubMed; (j) D. Zhu, L. Chen, H. Fan, Q. Yao and S. Zhu, Chem. Soc. Rev., 2020, 49, 908–950 RSC.
- All the references therein. (a) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861–2904 CrossRef CAS PubMed; (b) C. Peng, G. Yan, Y. Wang, Y. Jiang, Y. Zhang and J. Wang, Synthesis, 2010, 4154–4168 CAS; (c) H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857–1869 RSC; (d) H. M. L. Davies and Y. Lian, Acc. Chem. Res., 2012, 45, 923–935 CrossRef CAS PubMed; (e) K. Wang and J. Wang, Synlett, 2019, 30, 542–551 CrossRef CAS; (f) B. D. Bergstrom, L. A. Nickerson, J. T. Shaw and L. W. Souza, Angew. Chem., Int. Ed., 2011, 133, 6864–6874 Search PubMed.
- (a) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed; (b) P. C. Trippier and C. McGuigan, Med. Chem. Commun., 2010, 1, 183–198 RSC; (c) D. G. Hall, Boronic Acids: Preparation and Applications in Organic Synthesis Medicine and Materials, Wiley-VCH, 2nd edn, 2011 CrossRef; (d) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed; (e) W. Meng, X. Feng and H. Du, Acc. Chem. Res., 2018, 51, 191–201 CrossRef CAS PubMed; (f) K. Yang and Q. Song, Acc. Chem. Res., 2021, 54, 2298–2312 CrossRef CAS PubMed.
- C. Peng, Y. Wang and J. Wang, J. Am. Chem. Soc., 2008, 130, 1566–1567 CrossRef CAS PubMed.
- X. Zhao, J. Jing, K. Lu, Y. Zhang and J. Wang, Chem. Commun., 2010, 46, 1724–1726 RSC.
- J. R. Fulton and V. K. Aggarwal, Eur. J. Org. Chem., 2005, 1479–1492 CrossRef CAS.
- Y.-T. Tsoi, Z. Zhou, A. S. C. Chan and W.-Y. Yu, Org. Lett., 2010, 12, 4506–4509 CrossRef CAS PubMed.
- M. Kitamura, R. Sakata and T. Okauchi, Tetrahedron Lett., 2011, 52, 1931–1933 CrossRef CAS.
- M. Kitamura, N. Tashiro, R. Sakata and T. Okauchi, Synlett, 2010, 2503–2505 CrossRef CAS.
- Y. Xia, Y. Xia, Z. Liu, Y. Zhang and J. Wang, J. Org. Chem., 2014, 79, 7711–7717 CrossRef CAS PubMed.
- M. C. Belhomme, D. Wang and K. J. Szabó, Org. Lett., 2016, 18, 2503–2506 CrossRef CAS PubMed.
- A. Das, D. Wang, M. C. Belhomme and K. J. Szabó, Org. Lett., 2015, 17, 4754–4757 CrossRef CAS PubMed.
- M. Komatsuda, H. Kato, K. Muto and J. Yamaguchi, ACS Catal., 2019, 9, 8991–8995 CrossRef CAS.
- Y. Ping, R. Wang, Q. Wang, T. Chang, J. Huo, M. Lei and J. Wang, J. Am. Chem. Soc., 2021, 143, 9769–9780 CrossRef CAS PubMed.
- Y. Xia, R. Ge, L. Chen, Z. Liu, Q. Xiao, Y. Zhang and J. Wang, J. Org. Chem., 2015, 80, 7856–7864 CrossRef CAS PubMed.
- Y. Ping, T. Chang, K. Wang, J. Huo and J. Wang, Chem. Commun., 2019, 55, 59–62 RSC.
- (a) J. A. Erickson and J. I. McLoughlin, J. Org. Chem., 1995, 60, 1626–1631 CrossRef CAS; (b) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed.
- T. Besset, C. Schneider and D. Cahard, Angew. Chem., Int. Ed., 2012, 51, 5048–5050 CrossRef CAS PubMed.
- W. Zhou, W.-J. Pan, J. Chen, M. Zhang, J.-H. Lin, W. Cao and J.-C. Xao, Chem. Commun., 2021, 57, 9316–9329 RSC.
- Z. Feng, Q.-Q. Min and X. Zhang, Org. Lett., 2016, 18, 44–47 CrossRef CAS PubMed.
- V. P. Mehta and M. F. Greaney, Org. Lett., 2013, 15, 5036–5039 CrossRef CAS PubMed.
- X.-Y. Deng, J.-H. Lin and J.-C. Xiao, Org. Lett., 2016, 18, 4384–4387 CrossRef CAS PubMed.
- J. Zheng, J.-H. Lin, J. Cai and J.-C. Xiao, Chem. – Eur. J., 2013, 19, 15261–15266 CrossRef CAS PubMed.
- Z. Feng, Q.-Q. Min, X.-P. Fu, L. An and X. Zhang, Nat. Chem., 2017, 9, 918–923 CrossRef CAS PubMed.
- X.-P. Fu, X.-S. Xue, X.-Y. Zhang, Y.-L. Xiao, S. Zhang, Y.-L. Guo, X. Leng, K. N. Houk and X. Zhang, Nat. Chem., 2019, 11, 948–956 CrossRef CAS PubMed.
- Z.-W. Xu, W. Zhang, J.-H. Lin, C.-M. Jin and J.-C. Xiao, Chin. J. Chem., 2020, 38, 1647–1650 CrossRef CAS.
- Y. Xia, Y. Xia, R. Ge, Z. Liu, Q. Xiao, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 3917–3921 CrossRef CAS PubMed.
- H. Ding, S. Bai, P. Lu and Y. Wang, Org. Lett., 2017, 19, 4604–4607 CrossRef CAS PubMed.
- K. Wang, J. Yang, X. Yao and J. Wang, Chem. – Asian J., 2018, 13, 3165–3168 CrossRef CAS PubMed.
- S. Mai, W. Li, X. Li, Y. Zhao and Q. Song, Nat. Commun., 2019, 10, 5709–5720 CrossRef PubMed.
- (a) O. Dimroth, Justus Liebigs Ann. Chem., 1909, 364, 183–226 CrossRef CAS; (b) T. Horneff, S. Chuprakov, N. Chernyak, V. Gevorgyan and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 14972–14974 CrossRef CAS PubMed . See also ref. 5.
- N. Selander, B. T. Worrell, S. Chuprakov, S. Velaparthi and V. V. Fokin, J. Am. Chem. Soc., 2012, 134, 14670–14673 CrossRef CAS PubMed.
- T. Miura, T. Nakamuro, S. Miyakawa and M. Murakami, Angew. Chem., Int. Ed., 2016, 55, 8732–8735 CrossRef CAS PubMed.
- J. Raushel and V. V. Fokin, Org. Lett., 2010, 12, 4952–4955 CrossRef CAS PubMed.
- Y. Xia, L. Chen, P. Qu, G. Ji, S. Feng, Q. Xiao, Y. Zhang and J. Wang, J. Org. Chem., 2016, 81, 10484–10490 CrossRef CAS PubMed.
- Y.-T. Tsoi, Z. Zhou and W.-Y. Yu, Org. Lett., 2011, 13, 5370–5373 CrossRef CAS PubMed.
- F.-N. Ng, Y.-F. Lau, Z. Zhou and W.-Y. Yu, Org. Lett., 2015, 17, 1676–1679 CrossRef CAS PubMed.
- J. Ghorai and P. Anbarasan, J. Org. Chem., 2015, 80, 3455–3461 CrossRef CAS PubMed.
- Y.-S. Lu and W.-Y. Yu, Org. Lett., 2016, 18, 1350–1353 CrossRef CAS PubMed.
- Z. Zhang, W. Yu, Q. Zhou, T. Li, Y. Zhang and J. Wang, Chin. J. Chem., 2016, 34, 473–476 CrossRef CAS.
- Y. Zhou, Y. Zhang and J. Wang, Chin. J. Chem., 2017, 35, 621–627 CrossRef CAS.
- M. Tian, L. Yang, B. Liu and J. Chang, Chin. J. Chem., 2023, 41, 1327–1332 CrossRef CAS.
- Y. Luan and S. E. Schaus, Org. Lett., 2011, 13, 2510–2513 CrossRef CAS PubMed.
- D. Wang and K. J. Szabó, Org. Lett., 2017, 19, 1622–1625 CrossRef CAS PubMed.
- H. A. Ali, I. Goldberg and M. Srebnik, Organometallics, 2001, 20, 3962–3965 CrossRef CAS.
- H. A. Ali, I. Goldberg, D. Kaufmann, C. Burmeister and M. Srebnik, Organometallics, 2002, 21, 1870–1876 CrossRef CAS.
- A. J. Wommack and J. S. Kingsbury, Tetrahedron Lett., 2014, 55, 3163–3166 CrossRef CAS.
- Z. Zhang, Y. Yu, Y. Xie, T. Hughes, J. Xu, F. Huang and H. Huang, Green Chem., 2020, 22, 4165–4173 RSC.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |