
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvaluation of bioorthogonally applicable tetrazine–Cy3 probes for fluorogenic labeling schemes†
Evelin
Albitz
 ab,
Krisztina
Németh
a,
Gergely
Knorr
*a and
Péter
Kele
*a
ab,
Krisztina
Németh
a,
Gergely
Knorr
*a and
Péter
Kele
*a
aChemical Biology Research Group, Institute of Organic Chemistry, Research Centre for Natural Sciences, Magyar tudósok krt. 2, H-1117, Budapest, Hungary. E-mail: kele.peter@ttk.hu
bHevesy György PhD School of Chemistry, Eötvös Loránd University, Pázmány Péter sétány 1/a, H-1117, Budapest, Hungary
First published on 23rd August 2023
Abstract
The fluorogenic features of three sets of tetrazine–Cy3 probes were evaluated in bioorthogonal tetrazine–cyclooctyne ligation schemes. These studies revealed that the more efficient, internal conversion-based quenching of fluorescence by the tetrazine modul is translated to improved fluorogenicity compared to the more conventional, energy transfer-enabled design. Furthermore, a comparison of directly conjugated probes and vinylene-linked tetrazine–Cy3 probes revealed that more intimate conjugation of the tetrazine and the chromophore results in more efficient IC-based quenching even in spectral ranges where tetrazine exhibits diminished modulation efficiency. The applicability of these tetrazine-quenched fluorogenic Cy3 probes was demonstrated in the fluorogenic labeling schemes of the extra- and intracellular proteins of live cells.
Introduction
Recent advances in optical microscopy techniques have highlighted the profound implications of bioorthogonally applicable fluorogenic probes particularly in the context of live cell imaging schemes. The fluorescence of such probes is generally quenched by a rationally installed bioorthogonal motif (e.g., azide1 or tetrazine2), whose transformation in a highly specific ligation scheme results in a considerable increase in emission intensity at a given wavelength.3 In combination with synthetic biology approaches, such as genetic code expansion for the installation of non-canonical amino acids (ncAAs4) bearing bioorthogonal functional groups or fusion protein technology to bind bioorthogonalized substrates of Halo-,5 CLIP-6 and SNAP-tags7 covalently, bioorthogonal labeling schemes have become a powerful method for the site-specific and selective labeling of intra- and extracellular proteins of interest (POIs). From a super-resolution microscopy perspective, the minimal linkage error in the case of labeling schemes of ncAA-tagged POIs with a small-molecular probe is notable.8,9 Amongst bioorthogonal reactions, inverse electron demand Diels–Alder (IEDDA) reactions of tetrazines and strained cycloalkynes/alkenes are extremely useful due to the fast kinetics of such transformations.10,11 Furthermore, the tetrazine moiety is also capable of quenching the emission of fluorescent cores. Consequently, various tetrazine-quenched fluorogenic probes have been developed.2,12–20 Tetrazines can exert their quenching effects through several mechanisms. These include photoinduced electron transfer (PET12) or energy transfer (FRET, TBET) processes.14–20 These concepts have been successfully applied to various fluorophores (e.g. coumarin,12 BODIPY,13,16 phenoxazine,18 xanthene,14 and cyanine21). The quenching efficiency of the tetrazine, which is reflected in the increase in fluorescence intensity upon the IEDDA reaction, however, varies greatly. In general, tetrazines are more efficient in terms of quenching in the case of fluorophores emitting in the blue–green region of the spectrum, while fluorogenicity (i.e., I/I0) drops sharply upon entering the yellow regime.22 Recently, several novel approaches have emerged to improve the fluorogenicity of probes emitting in the biologically more benign wavelength range. These approaches include multiple fluorogenicity23 or energy transfer via Dexter mechanism.24 Recently, our group reported on the development of fluorogenic dyads with improved yellow–red fluorogenicity due to an energy transfer-based relay mechanism.25 At the same time, He and co-workers presented a set of red-emitting BODIPY–tetrazine probes with high turn-on values due to a twisted intramolecular charge transfer quenching mechanism,26 while the Wu group showed examples of excellent fluorogenic probes by installing an amino-tetrazine at the meso-position of several NIR-emitting cores.27 These strategies indeed resulted in improved fluorogenic features, however, at the cost of synthetic ease.Park and co-workers described monochromophoric systems, in which the tetrazine is an integral part of the π-system of a Seoul-Fluor chromophore. In this design, the excited chromophore relaxes to a lower-lying dark state corresponding to the n–π* transition of the tetrazine via internal conversion.20 Later, a similar non-radiative, internal conversion-based relaxation pathway was proposed by us for vinyl-tetrazine-appended coumarin fluorophores and photocages.15,28 While this robust quenching mechanism efficiently modulates UV or blue-excitable cores, it has limitations in red-absorbing chromophores due to the energy levels of the corresponding excited states.27,29 Cyanines are very popular probes in the field of fluorescence imaging due to their high brightness and readily tuneable photophysical properties;30 therefore cyanine probes with improved fluorogenic behavior are always welcomed by the scientific community, especially if an appended bioorthogonal moiety facilitates the specific ligation of such probes to target structures. Not long ago, we reported on two sets of tetrazine-functionalized cyanine probes.21 We have found that in the case of probes that link the cyanine to the tetrazine through a phenylene motif (TBET design), the fluorogenic performance is lower (i.e., 4× increase) compared to probes where a vinylene linkage was applied (i.e., 14× increase). Since the vinylene linker allows direct conjugation of the tetrazine and the cyanine, the improved fluorogenic behaviour was attributed to a more efficient quenching mechanism that involves internal conversion to a dark, low-lying n–π* transition-derived excited state of the tetrazine. We wished to elaborate whether a more intimate connection between the tetrazine and the cyanine core allows improved fluorogenic behavior.
Results and discussion
Synthesis of fluorogenic probes
Along these considerations, we designed and synthesized three sets of Cy3 probes representing directly conjugated (1), phenylene-linked (2) and vinylene-linked (3) tetrazine–Cy3 scaffolds (Fig. 1). Each set included a presumably membrane permeable positively charged derivative and a non-membrane permeable congener with zero net charge. In order to access these probes, 4-aminobenzonitrile and 4-iodoaniline were first converted into 4-cyanophenylhydrazine (S1) and 4-iodophenylhydrazine (S5) in a diazotization-reduction sequence. Then, the indoles (S2, S3 and S6) were prepared from the hydrazines in the presence of 3-methylbutan-2-one by a Fischer reaction. Intermediate 4 was obtained in moderate yields from indole S2. Treatment of the indoles (4, S3, S6 or 2,3,3-trimethyl-3H-indole) with methyl iodide produced indolium precursors 5, S4, S7 and S8. Indolium salts S4 and S8 were converted into hemicyanines upon treatment with N,N′-diphenylformimidamide in a mixture of acetic acid and acetic anhydride. The subsequent reaction of the respective hemicyanine with indolium 5 or S7 provided probes 1a–b and cyanines 6a–b. Intermediates 6a–b were subjected to Suzuki- and Heck-type cross-coupling reactions to furnish probes 2a–b and 3a–b, respectively (Scheme 1).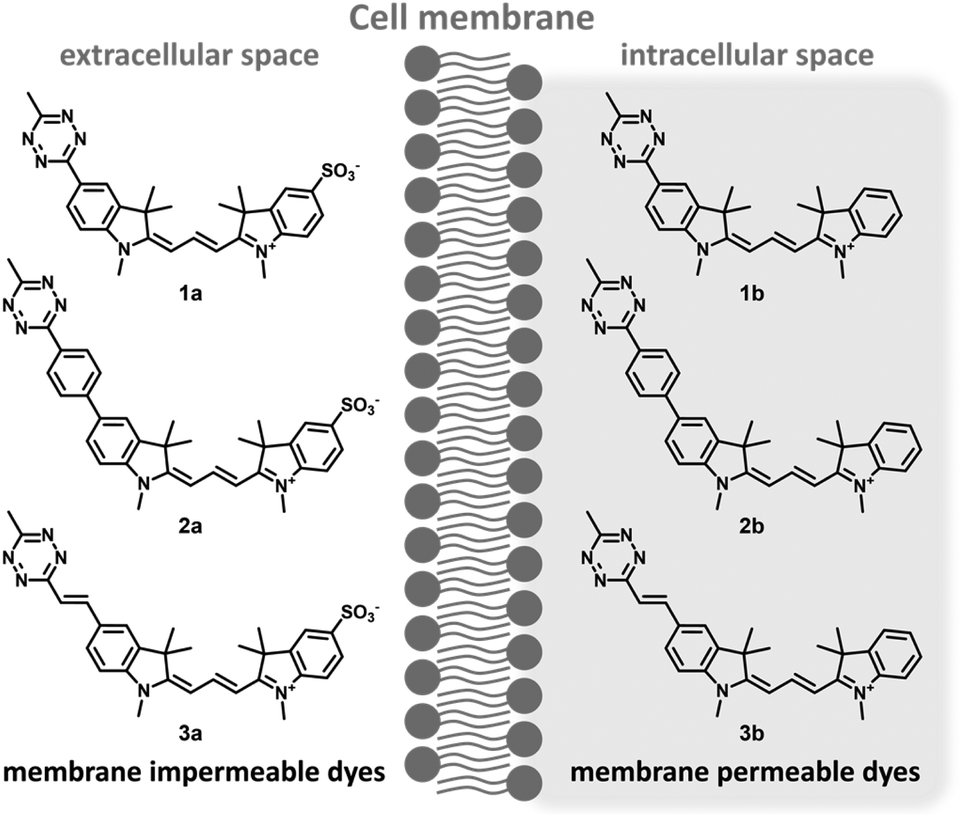 | ||
| Fig. 1 Structures of bioorthogonally applicable fluorogenic cyanines 1–3. | ||
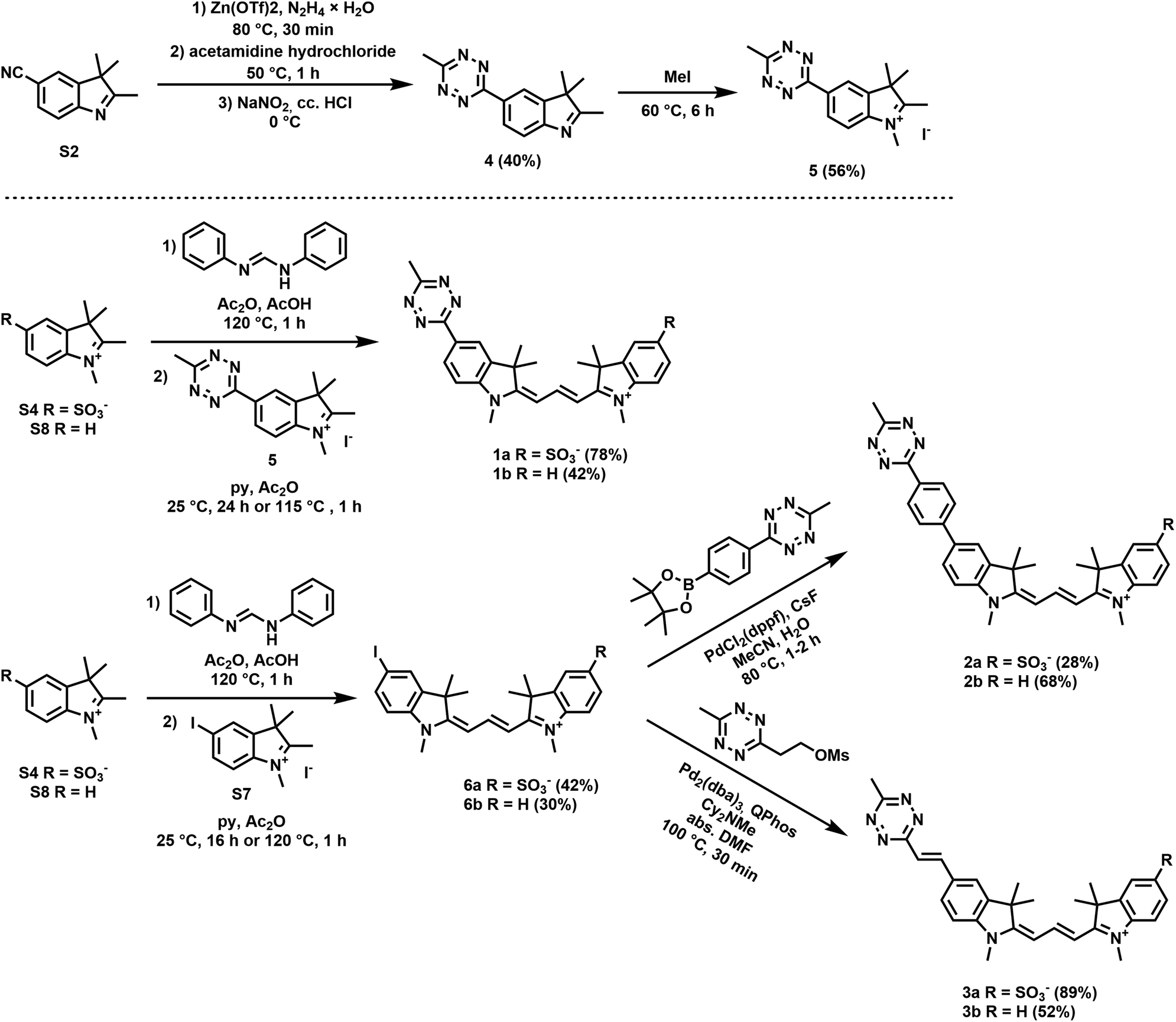 | ||
| Scheme 1 Synthesis of fluorogenic cyanines 1–3. | ||
Fluorescence and fluorogenicity studies
Next, we studied the main spectral features and fluorogenic behavior of our probes in an IEDDA reaction with (1R,8S,9S)-bicyclo[6.1.0]non-4-yn-9-ylmethanol, BCN (Table 1). As expected, the vinylene-linked derivatives (3) exhibited red-shifted absorption and emission spectra compared to 1 and 2, due to the extension of the π system. In each case, the reaction with BCN resulted in a slight blue shift of the absorption and emission maxima with the exception of 3b, where a small red shift was observed for λem. A small drop in the molar absorptivities was also detected upon reaction with BCN. The clicked products were found to be more fluorescent than their respective tetrazine congeners and the fluorescence quantum yields outweighed the decrease in the molar absorptivities, leading to brighter clicked products in each case. Very small effects on the spectral properties could be attributed to the presence of the sulfonate substituent. A comparison of the fluorescence enhancement values of the different designs revealed considerable differences of the turn-on performances. In line with our hypothesis, the highest fluorogenicity (∼20×) was observed for intimately conjugated tetrazine–cyanines, while vinylene conjugation or phenylene linkage gave lower enhancement values.| λ abs (nm) | λ em (nm) | ε × 104 (M−1 cm−1) | Φ (%) | B × 104 (ε × ϕ) | I BCN/ITetd | B BCN/BTet | |
|---|---|---|---|---|---|---|---|
| a In sodium PBS (pH = 7.4, containing 0.1% SDS). b Determined at λabs,max. c Relative to rhodamine B (ϕ = 0.65). d Determined at λem,max of the BCN conjugate. | |||||||
| 1a | 565 | 578 | 11.5 | 1.63 | 18.8 | 21.0 | 12.4 |
| 1a·BCN | 558 | 571 | 10.6 | 21.92 | 232.3 | ||
| 1b | 561 | 575 | 8.2 | 1.13 | 9.2 | 19.3 | 12.8 |
| 1b·BCN | 555 | 570 | 7.8 | 15.18 | 118.2 | ||
| 2a | 568 | 586 | 14.0 | 3.19 | 44.8 | 4.3 | 4.0 |
| 2a·BCN | 567 | 586 | 13.2 | 13.56 | 179.3 | ||
| 2b | 567 | 587 | 13.2 | 2.94 | 38.9 | 5.8 | 5.3 |
| 2b·BCN | 565 | 585 | 12.8 | 16.22 | 207.7 | ||
| 3a | 578 | 593 | 16.3 | 0.97 | 15.8 | 11.9 | 9.0 |
| 3a·BCN | 575 | 596 | 14.0 | 10.19 | 142.6 | ||
| 3b | 577 | 584 | 11.5 | 2.91 | 33.5 | 6.1 | 4.4 |
| 3b·BCN | 572 | 592 | 10.7 | 13.80 | 148.0 | ||
To gain further insight into the quenching mechanism, we conducted TD-DFT calculations at the B3LYP/6-31+G(d) level for molecules 1a, 1b, 3a, and 3b. In all cases, we found that the main excitation pathway is an S0 → S2 transition (see the ESI; Fig. S10–S13†). The S1 (LUMO) orbital was found to be localized mostly on the tetrazine and to a lesser extent on the cyanine scaffold. At the same time, the S2 (LUMO+1) molecular orbital was delocalized all over the molecule. These two excited state orbitals, however, were very close to each other; in fact, they were separated only by 0.05–0.16 eV, which is within the error limit of the calculation method.31 This implies that although the S2 state is formed predominantly corresponding to a π–π* transition of the cyanine unit as suggested by the oscillator strengths, the transition between the S1 and S2 excited states is reversible. These findings not only explain why conjugated Cy3–tetrazines are more fluorogenic than TBET-governed probes (e.g., 2a and 2b) but also justify the lower fluorogenicity of 1 and 3 compared to, e.g., related coumarin or Seoul-Fluor probes.
Confocal and STED microscopy
The notable increase in brightness in the case of directly conjugated tetrazine–Cy3 probes 1a–b upon IEDDA reactions suggests that these are promising candidates for in vivo labeling schemes. For comparison, probes 3a–b were also included in these studies. Before applying the probes to live cells, we evaluated the effects of the probes on cellular viability. Treatment of HEK293T cells with probes 1a–b or 3a–b even for prolonged incubation times (i.e., 90 minutes) either did not have any noticeable effects on viability or affected the cells only negligibly (e.g., the changes in viabilities remained within the error range) in the applied 0.3–3 μM concentration range (see the ESI; Fig. S9† for more details).For extracellular protein labeling schemes, we modified the GFP-fused insulin receptor (IR) protein site specifically with a bioorthogonalized lysine (ε-amino(BCN)-Lys) at the extracellular domain using genetic code expansion. We then treated IRK676BCN-Lys–GFP-expressing live HEK293T cells with probes 1a and 3a. After a 90-minute incubation time, the cells were washed, fixed, and subjected to confocal microscopy (Fig. 2). To our delight, fluorogenic cyanines 1a and 3a were indeed membrane impermeable and suitable for selective extracellular labeling and showed excellent co-localization with the reporter protein. We also tested the applicability of the non-sulfonated cyanines (1b and 3b) in the labeling schemes of intracellular proteins. To this end, we labeled a similarly tailored, genetically bioorthogonalized cytoskeletal protein, vimentin–GFP, modified with BCN-Lys. Live COS-7 cells expressing a VimN116BCN-Lys–GFP protein construct were treated with 1b and 3b for 90 minutes, washed, fixed and then imaged (Fig. 2). The low background and the excellent co-localization of the fluorescent signals with that of the reporter protein confirmed the membrane permeability of the probes and their suitability for specific fluorogenic labeling schemes.
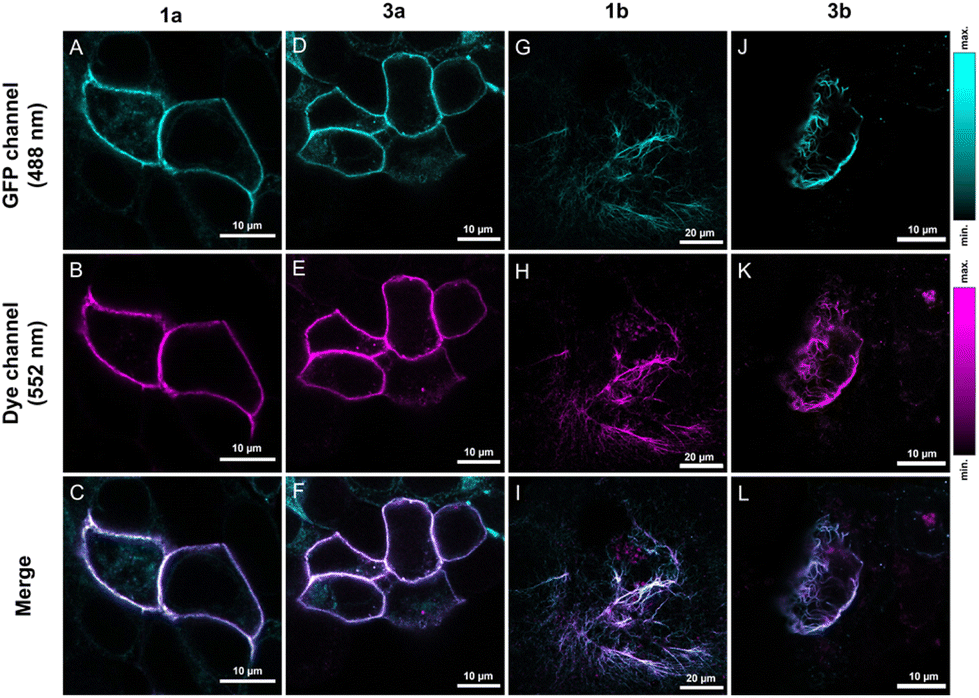 | ||
| Fig. 2 Confocal microscopy images of IRK676BCN-Lys–GFP expressing HEK293T cells (A–F) and VimN116BCN-Lys–GFP expressing COS-7 cells (G–L) treated with BCN–Lys and probes 1a (A–C), 1b (G–I), 3a (D–F) and 3b (J–L). Spectral detection: GFP: λexc: 488/λem: 500–540 nm; dye: λexc: 552/λem: 565–800 nm. Overlay pictures demonstrate the merged fluorescent signals of the reporter and dye. | ||
We also wished to demonstrate the applicability of the dyes in super-resolution (STED) microscopy. The well-known spectral properties of the Cy3 dyes25 suggest that these probes are suitable for STED microscopy equipped with a 660 nm depletion laser. We first assessed the photostability of probes 1 and 3 by multiple frame acquisition. Neither 1b nor 3b showed significant decrease in intensity over the 160-cycle period, while a slight decrease was observed for probes 1a and 3a compared to their initial values (see the ESI; Fig. S7†) suggesting the suitability of probes 1a–b and 3a–b in STED imaging. Indeed, a more detailed structure of the vimentin network was revealed by applying membrane-permeable 1b and 3b, and a more detailed image of the membrane lineage between two neighboring cells was acquired when using non-permeable 1a and 3a. Overall, the resolution characterised by the full width at half maximum (FWHM) values increased by a factor of 2–3 upon STED imaging with probes 1a–b and 3a–b (Fig. 3 and 4).
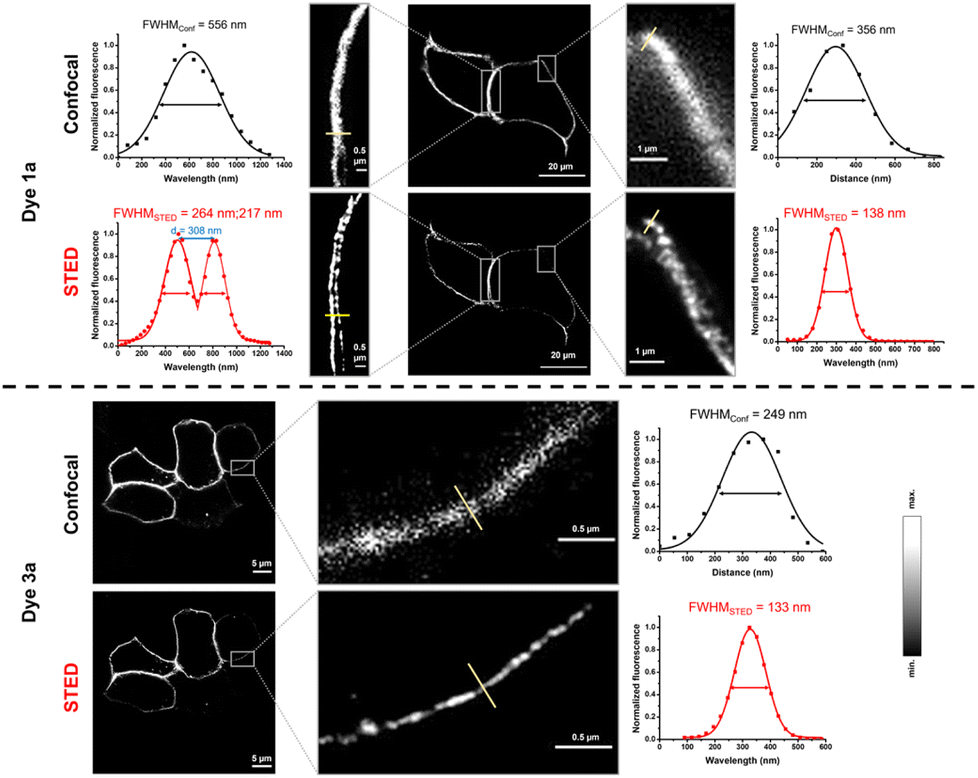 | ||
| Fig. 3 Confocal and STED images of IRK676BCN-Lys–GFP expressing HEK293T cells treated with BCN–Lys and dye 1a and 3a. λexc: 552/λem: 565–800 nm; λSTED: 660 nm (CW). FWHM: full width at half maxima. | ||
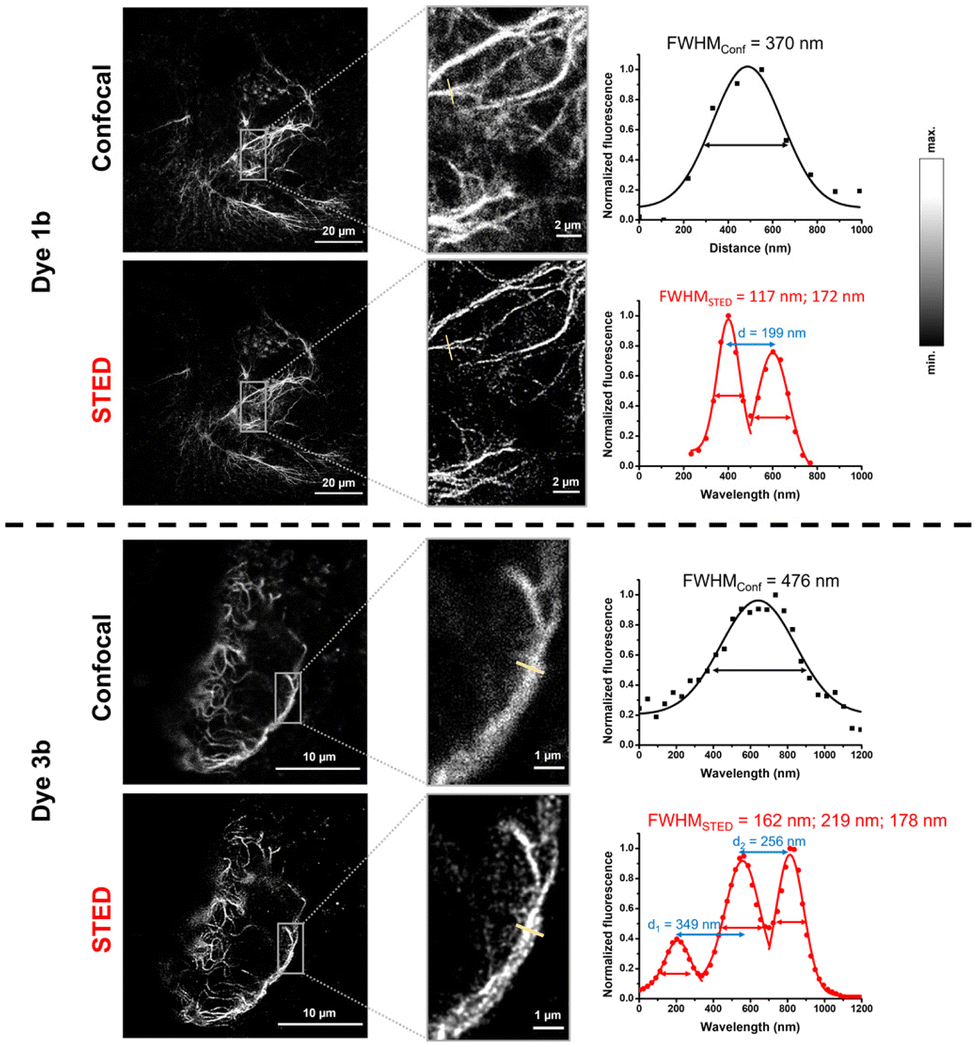 | ||
| Fig. 4 Confocal and STED images of VimN116BCN-Lys–GFP expressing COS7 cells treated with BCN-Lys and dye 1b and 3b. λexc: 552/λem: 565–800 nm; λSTED: 660 nm (CW). FWHM: full width at half maxima. | ||
Conclusions
In conclusion, we synthesized three sets of tetrazine-functionalized Cy3 derivatives following different design strategies in order to gain experimental insights into the modulation effects of the bioorthogonal unit on the fluorescence of yellow-emitting indocyanines. Our study involved directly conjugated tetrazine–cyanines (direct conjugation, 1a–b), phenylene-linked units (TBET design, 2a–b) or vinylene-conjugated systems (π-extended direct conjugation, 3a–b). Each of these sets included a membrane-permeable derivative and a non-permeable derivative. We assessed the fluorogenic characteristics of 1–3 upon reacting these probes with a strained cyclooctyne, BCN, in an IEDDA reaction. A comparison of the fluorogenicities revealed that probes 1a–b outperformed probes 2a–b and 3a–b, indicating that the more intimate conjugation of the tetrazine results in more efficient quenching of fluorescence and further confirming that internal conversion-based modulation of fluorescence is more efficient than the through-bond energy transfer-based modulation mechanism. The directly conjugated probes 1 and 3 were further evaluated in the labeling schemes of genetically bioorthogonalized extra- and intracellular proteins of live cells. To our delight, all these probes were found to be suitable for the specific labeling of target structures. Furthermore, all four probes tested were found to be applicable for STED imaging, resulting in subdiffraction imaging of extracellular insulin receptors or the intracellular vimentin network. These results indicate that fluorogenic probes quenched via internal conversion can even be effective in spectral ranges where the tetrazine exhibits diminished modulation efficiency.Experimental
Organic synthesis
The exact masses were determined with an Agilent 6230 time-of-flight mass spectrometer.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 to 0:100). 1H NMR (500 MHz, CD3OD) δ 8.77 (d, J = 1.2 Hz, 1H), 8.75 (dd, J = 8.4, 1.6 Hz, 1H), 8.70 (t, J = 13.5 Hz, 1H), 8.05 (d, J = 1.4 Hz, 1H), 8.01 (dd, J = 8.3, 1.6 Hz, 2H), 7.65 (d, J = 8.4 Hz, 1H), 7.51 (d, J = 8.3 Hz, 1H), 6.62 (d, J = 13.6 Hz, 1H), 6.58 (d, J = 13.3 Hz, 1H), 3.82 (s, 3H), 3.82 (s, 3H), 3.13 (s, 3H), 1.94 (s, 6H), 1.89 (s, 6H). 13C NMR (126 MHz, CD3OD) δ 178.3, 176.9, 168.6, 165.0, 153.0, 145.2, 144.6, 143.1, 142.3, 130.6, 130.2, 130.2, 128.3, 122.6, 121.4, 112.8, 112.3, 105.5, 104.6, 51.0, 50.5, 32.2, 31.9, 28.3, 28.0, 21.1. HRMS: m/z calcd for [C28H31N6O3S]+: 531.2172; found: 531.2177 [M + H]+.
:5 to 0:100) to yield 13.2 mg (42%) of purple crystals. 1H NMR (500 MHz, CD3OD) δ 8.76–8.73 (m, 2H), 8.68 (t, J = 13.5 Hz, 1H), 7.66 (d, J = 7.4 Hz, 1H), 7.61 (d, J = 8.9 Hz, 1H), 7.56 (d, J = 7.9 Hz, 1H), 7.51 (d, J = 7.8 Hz, 1H), 7.45 (t, J = 7.4 Hz, 1H), 6.63 (d, J = 13.7 Hz, 1H), 6.52 (d, J = 13.2 Hz, 1H), 3.83 (s, 3H), 3.72 (s, 3H), 3.12 (s, 3H), 1.94 (s, 6H), 1.87 (s, 6H). 13C NMR (126 MHz, CD3OD) δ 178.2, 175.9, 168.6, 165.0, 152.5, 143.9, 142.9, 142.5, 130.2, 130.2, 130.1, 127.6, 123.5, 122.6, 112.9, 112.5, 105.5, 103.8, 73.7, 71.5, 62.2, 51.2, 32.2, 28.4, 28.0, 21.0. HRMS: m/z calcd for [C28H31N6]+: 451.2610; found: 451.2612 [M + H]+.
:5 to 0:100). 1H NMR (500 MHz, DMSO-d6) δ 8.31 (t, J = 13.5 Hz, 1H), 8.15 (s, 1H), 8.03 (d, J = 1.4 Hz, 1H), 7.78 (dd, J = 8.3, 1.5 Hz, 1H), 7.65 (d, J = 7.4 Hz, 1H), 7.50 (d, J = 7.8 Hz, 1H), 7.46 (t, J = 7.6 Hz, 1H), 7.32 (t, J = 7.3 Hz, 1H), 7.27 (d, J = 8.4 Hz, 1H), 6.49 (d, J = 13.6 Hz, 1H), 6.40 (d, J = 13.4 Hz, 1H), 3.67 (s, 3H), 3.59 (s, 3H), 1.68 (s, 6H), 1.67 (s, 6H). 13C NMR (126 MHz, DMSO-d6) δ 174.9, 173.2, 163.0, 149.5, 142.8, 142.6, 142.6, 140.7, 137.0, 131.0, 128.6, 125.5, 122.4, 113.4, 111.7, 103.4, 102.4, 89.1, 49.0, 48.7, 31.5, 31.3, 27.2, 27.1. HRMS: m/z calcd for [C25H28IN2]+: 483.1297; found: 483.1288 [M]+.
:5 to 0:100). 1H NMR (500 MHz, DMSO-d6) δ 8.58 (d, J = 8.4 Hz, 2H), 8.38 (t, J = 13.5 Hz, 1H), 8.14 (s, 1H), 8.08 (d, J = 8.4 Hz, 2H), 7.92 (d, J = 8.2 Hz, 1H), 7.82 (s, 1H), 7.70 (d, J = 8.2 Hz, 1H), 7.59 (d, J = 8.4 Hz, 1H), 7.41 (d, J = 8.2 Hz, 1H), 6.48 (d, J = 13.5 Hz, 1H), 3.70 (s, J = 14.8 Hz, 3H), 3.67 (s, 3H), 3.02 (s, 3H), 1.78 (s, 6H), 1.72 (s, 6H). 13C NMR (126 MHz, DMSO-d6) δ 174.9, 174.2, 171.1, 167.0, 163.1, 149.4, 146.0, 144.8, 143.2, 142.9, 142.5, 141.5, 139.9, 135.8, 130.7, 127.9, 127.5, 127.3, 126.1, 121.0, 119.8, 111.9, 110.6, 103.3, 103.1, 48.9, 31.6, 27.3, 27.2, 20.8. HRMS: m/z calcd for [C34H35N6O3S]+: 607.2485; found: 607.2498 [M + H]+.
:5 to 0:100) to yield 32 mg (68%) of purple crystals. 1H NMR (500 MHz, DMSO-d6) δ 8.58 (d, J = 8.5 Hz, 2H), 8.37 (t, J = 13.5 Hz, 1H), 8.14 (d, J = 1.6 Hz, 1H), 8.08 (d, J = 8.5 Hz, 2H), 7.92 (dd, J = 8.4, 1.7 Hz, 1H), 7.65 (d, J = 7.3 Hz, 1H), 7.59 (d, J = 8.4 Hz, 1H), 7.52–7.43 (m, J = 8.1 Hz, 2H), 7.32 (td, J = 7.4, 1.5 Hz, 1H), 6.48 (apparent t containing: 6.50 (d, J = 13.7 Hz, 1H), 6.47 (d, J = 13.6 Hz, 1H)), 3.69 (s, 3H), 3.68 (s, 3H), 3.02 (s, 3H), 1.78 (s, 6H), 1.71 (s, 6H). 13C NMR (126 MHz, DMSO-d6) δ 174.7, 174.1, 167.0, 163.0, 149.4, 143.2, 143.0, 142.6, 141.4, 140.6, 135.7, 130.7, 128.5, 127.9, 127.5, 127.3, 125.3, 122.4, 121.0, 111.8, 111.6, 103.2, 102.9, 49.0, 48.8, 31.5, 27.3, 27.2, 20.8. HRMS: m/z calcd for [C34H35N6]+: 527.2923; found: 527.2922 [M + H]+.
:5 to 0:100). 1H NMR (500 MHz, DMSO-d6) δ 8.37 (t, J = 13.5 Hz, 1H), 8.31–8.27 (m, 2H), 7.91 (dd, J = 8.2, 0.8 Hz, 1H), 7.83 (d, J = 1.0 Hz, 1H), 7.72 (d, J = 16.2 Hz, 1H), 7.70 (dd, J = 8.1, 1.3 Hz, 1H), 7.51 (d, J = 8.3 Hz, 1H), 7.43 (d, J = 8.3 Hz, 1H), 6.51 (d, J = 13.6 Hz, 1H), 6.46 (d, J = 13.3 Hz, 1H), 3.69 (s, 3H), 3.66 (s, 3H), 2.96 (s, 3H), 1.76 (s, 6H), 1.72 (s, 6H). 13C NMR (126 MHz, DMSO-d6) δ 175.4, 173.8, 170.8, 165.9, 164.3, 149.5, 146.2, 144.2, 142.4, 141.3, 140.1, 139.2, 132.0, 130.2, 126.1, 121.4, 120.1, 119.8, 111.5, 110.8, 103.9, 103.2, 49.1, 48.5, 31.7, 27.3, 27.1, 20.8. HRMS: m/z calcd for [C30H33N6O3S]+: 557.2329; found: 557.2335 [M + H]+.
:5 to 0:100) to yield 13.4 mg (52%) of purple crystals. 1H NMR (500 MHz, CD3OD) δ 8.54 (t, J = 13.5 Hz, 1H), 8.31 (d, J = 16.2 Hz, 1H), 8.00 (s, 1H), 7.79 (d, J = 7.9 Hz, 1H), 7.60–7.52 (m containing: 7.57 (d, J = 16.1 Hz, 1H), 7.54 (d, J = 7.0 Hz, 1H)), 7.44 (d, J = 7.5 Hz, 1H), 7.38 (d, J = 3.6 Hz, 1H), 7.36 (d, J = 4.1 Hz, 1H), 7.32 (t, J = 7.4 Hz, 1H), 6.47 (d, J = 13.6 Hz, 1H), 6.39 (d, J = 13.3 Hz, 1H), 3.69 (s, 3H), 3.65 (s, 3H), 2.96 (s, 3H), 1.80 (s, 6H), 1.75 (s, 6H). 13C NMR (126 MHz, CD3OD) δ 177.5, 176.0, 167.7, 166.1, 159.7, 152.2, 145.7, 144.0, 142.9, 142.3, 140.9, 134.2, 131.1, 130.0, 127.2, 123.5, 122.5, 121.5, 112.7, 112.4, 104.9, 103.9, 50.9, 50.2, 32.0, 28.3, 28.0, 21.1. HRMS: m/z calcd for [C30H33N6]+: 477.2767; found: 477.2762 [M + H]+.
:5 to 0:100). 1H NMR (500 MHz, CD3OD) δ 8.77 (d, J = 1.2 Hz, 1H), 8.75 (dd, J = 8.4, 1.6 Hz, 1H), 8.70 (t, J = 13.5 Hz, 1H), 8.05 (d, J = 1.4 Hz, 1H), 8.01 (dd, J = 8.3, 1.6 Hz, 2H), 7.65 (d, J = 8.4 Hz, 1H), 7.51 (d, J = 8.3 Hz, 1H), 6.62 (d, J = 13.6 Hz, 1H), 6.58 (d, J = 13.3 Hz, 1H), 3.82 (s, 3H), 3.82 (s, 3H), 3.13 (s, 3H), 1.94 (s, 6H), 1.89 (s, 6H). 13C NMR (126 MHz, CD3OD) δ 178.3, 176.9, 168.6, 165.0, 153.0, 145.2, 144.6, 143.1, 142.3, 130.6, 130.2, 130.2, 128.3, 122.6, 121.4, 112.8, 112.3, 105.5, 104.6, 51.0, 50.5, 32.2, 31.9, 28.3, 28.0, 21.1. HRMS: m/z calcd for [C28H31N6O3S]+: 531.2172; found: 531.2177 [M + H]+.
:5 to 0:100) to yield 13.2 mg (42%) of purple crystals. 1H NMR (500 MHz, CD3OD) δ 8.76–8.73 (m, 2H), 8.68 (t, J = 13.5 Hz, 1H), 7.66 (d, J = 7.4 Hz, 1H), 7.61 (d, J = 8.9 Hz, 1H), 7.56 (d, J = 7.9 Hz, 1H), 7.51 (d, J = 7.8 Hz, 1H), 7.45 (t, J = 7.4 Hz, 1H), 6.63 (d, J = 13.7 Hz, 1H), 6.52 (d, J = 13.2 Hz, 1H), 3.83 (s, 3H), 3.72 (s, 3H), 3.12 (s, 3H), 1.94 (s, 6H), 1.87 (s, 6H). 13C NMR (126 MHz, CD3OD) δ 178.2, 175.9, 168.6, 165.0, 152.5, 143.9, 142.9, 142.5, 130.2, 130.2, 130.1, 127.6, 123.5, 122.6, 112.9, 112.5, 105.5, 103.8, 73.7, 71.5, 62.2, 51.2, 32.2, 28.4, 28.0, 21.0. HRMS: m/z calcd for [C28H31N6]+: 451.2610; found: 451.2612 [M + H]+.
:5 to 0:100). 1H NMR (500 MHz, DMSO-d6) δ 8.31 (t, J = 13.5 Hz, 1H), 8.15 (s, 1H), 8.03 (d, J = 1.4 Hz, 1H), 7.78 (dd, J = 8.3, 1.5 Hz, 1H), 7.65 (d, J = 7.4 Hz, 1H), 7.50 (d, J = 7.8 Hz, 1H), 7.46 (t, J = 7.6 Hz, 1H), 7.32 (t, J = 7.3 Hz, 1H), 7.27 (d, J = 8.4 Hz, 1H), 6.49 (d, J = 13.6 Hz, 1H), 6.40 (d, J = 13.4 Hz, 1H), 3.67 (s, 3H), 3.59 (s, 3H), 1.68 (s, 6H), 1.67 (s, 6H). 13C NMR (126 MHz, DMSO-d6) δ 174.9, 173.2, 163.0, 149.5, 142.8, 142.6, 142.6, 140.7, 137.0, 131.0, 128.6, 125.5, 122.4, 113.4, 111.7, 103.4, 102.4, 89.1, 49.0, 48.7, 31.5, 31.3, 27.2, 27.1. HRMS: m/z calcd for [C25H28IN2]+: 483.1297; found: 483.1288 [M]+.
:5 to 0:100). 1H NMR (500 MHz, DMSO-d6) δ 8.58 (d, J = 8.4 Hz, 2H), 8.38 (t, J = 13.5 Hz, 1H), 8.14 (s, 1H), 8.08 (d, J = 8.4 Hz, 2H), 7.92 (d, J = 8.2 Hz, 1H), 7.82 (s, 1H), 7.70 (d, J = 8.2 Hz, 1H), 7.59 (d, J = 8.4 Hz, 1H), 7.41 (d, J = 8.2 Hz, 1H), 6.48 (d, J = 13.5 Hz, 1H), 3.70 (s, J = 14.8 Hz, 3H), 3.67 (s, 3H), 3.02 (s, 3H), 1.78 (s, 6H), 1.72 (s, 6H). 13C NMR (126 MHz, DMSO-d6) δ 174.9, 174.2, 171.1, 167.0, 163.1, 149.4, 146.0, 144.8, 143.2, 142.9, 142.5, 141.5, 139.9, 135.8, 130.7, 127.9, 127.5, 127.3, 126.1, 121.0, 119.8, 111.9, 110.6, 103.3, 103.1, 48.9, 31.6, 27.3, 27.2, 20.8. HRMS: m/z calcd for [C34H35N6O3S]+: 607.2485; found: 607.2498 [M + H]+.
:5 to 0:100) to yield 32 mg (68%) of purple crystals. 1H NMR (500 MHz, DMSO-d6) δ 8.58 (d, J = 8.5 Hz, 2H), 8.37 (t, J = 13.5 Hz, 1H), 8.14 (d, J = 1.6 Hz, 1H), 8.08 (d, J = 8.5 Hz, 2H), 7.92 (dd, J = 8.4, 1.7 Hz, 1H), 7.65 (d, J = 7.3 Hz, 1H), 7.59 (d, J = 8.4 Hz, 1H), 7.52–7.43 (m, J = 8.1 Hz, 2H), 7.32 (td, J = 7.4, 1.5 Hz, 1H), 6.48 (apparent t containing: 6.50 (d, J = 13.7 Hz, 1H), 6.47 (d, J = 13.6 Hz, 1H)), 3.69 (s, 3H), 3.68 (s, 3H), 3.02 (s, 3H), 1.78 (s, 6H), 1.71 (s, 6H). 13C NMR (126 MHz, DMSO-d6) δ 174.7, 174.1, 167.0, 163.0, 149.4, 143.2, 143.0, 142.6, 141.4, 140.6, 135.7, 130.7, 128.5, 127.9, 127.5, 127.3, 125.3, 122.4, 121.0, 111.8, 111.6, 103.2, 102.9, 49.0, 48.8, 31.5, 27.3, 27.2, 20.8. HRMS: m/z calcd for [C34H35N6]+: 527.2923; found: 527.2922 [M + H]+.
:5 to 0:100). 1H NMR (500 MHz, DMSO-d6) δ 8.37 (t, J = 13.5 Hz, 1H), 8.31–8.27 (m, 2H), 7.91 (dd, J = 8.2, 0.8 Hz, 1H), 7.83 (d, J = 1.0 Hz, 1H), 7.72 (d, J = 16.2 Hz, 1H), 7.70 (dd, J = 8.1, 1.3 Hz, 1H), 7.51 (d, J = 8.3 Hz, 1H), 7.43 (d, J = 8.3 Hz, 1H), 6.51 (d, J = 13.6 Hz, 1H), 6.46 (d, J = 13.3 Hz, 1H), 3.69 (s, 3H), 3.66 (s, 3H), 2.96 (s, 3H), 1.76 (s, 6H), 1.72 (s, 6H). 13C NMR (126 MHz, DMSO-d6) δ 175.4, 173.8, 170.8, 165.9, 164.3, 149.5, 146.2, 144.2, 142.4, 141.3, 140.1, 139.2, 132.0, 130.2, 126.1, 121.4, 120.1, 119.8, 111.5, 110.8, 103.9, 103.2, 49.1, 48.5, 31.7, 27.3, 27.1, 20.8. HRMS: m/z calcd for [C30H33N6O3S]+: 557.2329; found: 557.2335 [M + H]+.
:5 to 0:100) to yield 13.4 mg (52%) of purple crystals. 1H NMR (500 MHz, CD3OD) δ 8.54 (t, J = 13.5 Hz, 1H), 8.31 (d, J = 16.2 Hz, 1H), 8.00 (s, 1H), 7.79 (d, J = 7.9 Hz, 1H), 7.60–7.52 (m containing: 7.57 (d, J = 16.1 Hz, 1H), 7.54 (d, J = 7.0 Hz, 1H)), 7.44 (d, J = 7.5 Hz, 1H), 7.38 (d, J = 3.6 Hz, 1H), 7.36 (d, J = 4.1 Hz, 1H), 7.32 (t, J = 7.4 Hz, 1H), 6.47 (d, J = 13.6 Hz, 1H), 6.39 (d, J = 13.3 Hz, 1H), 3.69 (s, 3H), 3.65 (s, 3H), 2.96 (s, 3H), 1.80 (s, 6H), 1.75 (s, 6H). 13C NMR (126 MHz, CD3OD) δ 177.5, 176.0, 167.7, 166.1, 159.7, 152.2, 145.7, 144.0, 142.9, 142.3, 140.9, 134.2, 131.1, 130.0, 127.2, 123.5, 122.5, 121.5, 112.7, 112.4, 104.9, 103.9, 50.9, 50.2, 32.0, 28.3, 28.0, 21.1. HRMS: m/z calcd for [C30H33N6]+: 477.2767; found: 477.2762 [M + H]+.
Spectroscopic characterization
Photophysical measurements were performed on a JASCO FP 8300 spectrofluorometer and a JASCO v750 spectrophotometer. A stock solution was prepared in DMSO from the solid dyes (1 mM). All experiments were conducted in all-sodium PBS (pH = 7.4, containing 0.1% SDS), in order to prevent aggregation. All of the dyes (1 mM in DMSO) were reacted with (1R,8S,9S)-bicyclo[6.1.0]non-4-yn-9-ylmethanol (BCN) in DMSO at room temperature. The completion of the reaction was verified by HPLC-MS. Excitation and emission spectra were recorded using 1 μM concentration of the compounds while absorbance spectra were recorded using 5 μM concentration (DMSO content was kept below 1% in each case). The excitation and emission wavelengths are given for each spectrum. Quantum yields were determined using rhodamine B as a standard. For excitation, emission and absorbance spectra, see Fig. S1–S6 in the ESI.†Protein labeling
000 cell per well) or COS-7 (15000 cell per well) cells were transferred into μ-Slide 8-well plates (Ibidi 80827) and were incubated for 40 h at 37 °C in a 5% CO2 atmosphere. In the case of HEK293T cells, Ibidi plates were pre-treated with 0.01 mg mL−1 poly-L-lysine (Sigma P5899) for 4 hours at room temperature and washed afterwards. The bioorthogonally reactive chemical reporter BCN was administered in the HaloTag substrate or in non-canonical amino acid (ncAA). Therefore, cells were transfected with 0.25 μg of Lamin-HaloTag32 plasmid using the JetPrime (Polyplus 114-07) transfection agent for four hours according to the manufacturer's protocol. A similar procedure was carried out for ncAA installation. Cells were incubated with 0.25 μg of IRK676TAG–GFP [obtained from EMBL under a Material Transfer Agreement]33 or 0.25 μg of vimentinN116TAG–GFP34 in combination with 0.25 μg of the tRNAPyl/NES–PylRSAF plasmid (obtained from EMBL under a Material Transfer Agreement)35 in the presence of 250 μM BCNendo–lysine (Sichem SC-8014) during transfection. Subsequently, the supernatant was replaced with ncAA-free medium overnight. One day after transfection, cells were labeled with the fluorescent dyes 1a, 1b, 3a and 3b at a concentration of 1–3 μM in complete DMEM (Gibco 21063-029) for 90 min at 37 °C in the dark. In the case of the HaloTag fusion protein, the Halo–BCN substrate (3 μM; 60 min)32 was added to the cells before the fluorescent labeling step. Afterwards, a two-hour washing step – with complete DMEM – was performed followed by fixation (4% PFA for 10 min at 25 °C) and quick washes – twice – with PBS prior to imaging.
Confocal and STED imaging and analysis
Confocal and STED images were acquired on a Leica TCS SP8 STED 3× microscope using the 488 nm laser for excitation of the reporter fluorescent protein, 552 nm laser for the probes and 660 nm STED (1.5 W, continuous wave) laser for depletion (at 1.17–2.48 × 109 W mm−2 nominal intensity with an output laser power of 1.2 W/32 nm × 32 nm pixel size). The images were captured using a Leica HC PL APO 100×/1.40 oil immersion objective using the Leica HyD detector. Spectral detection parameters were set as follows: excitation: 488 nm/em. range: 500–550 nm for GFP and excitation: 552 nm/em. range: 565–650 nm for compounds 1a, 1b, 3a, and 3b.Using the Huygens Professional software (SVI), we performed deconvolution for image restoration on the recorded STED images. The deconvolution process was based on the theoretical point spread function (PSF). Images were analysed using Leica Application Suite X and Fiji for ImageJ software (NIH). We selected the results of some representative line analysis for demonstration. A non-linear Gaussian curve was fitted to the normalized fluorescence intensity values using OriginPro 9 software. For characterizing the resolution efficiency, the full width at half maximum values (FWHM) were provided.
Author contributions
EA conducted the synthesis and purification of all starting components and conducted all the characterization studies. EA, GK and PK came up with the experimental design. KN designed the cellular experiments. EA and KN carried out the cellular and labeling studies. GK performed the theoretical calculations. EA, KN, GK and PK wrote the manuscript and created the figures. All authors discussed and provided comments on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present work was supported by the National Research, Development and Innovation Office of Hungary (NKFIH-K-143581 and VEKOP-2.3.3-15-2016-00011). We are also grateful for the generous support from the Eötvös Loránd Research Network (FIKU). We thank Dr György Török for his valuable help in image processing.References
- P. Shieh, V. T. Dien, B. J. Beahm, J. M. Castellano, T. Wyss-Coray and C. R. Bertozzi, J. Am. Chem. Soc., 2015, 137, 7145–7151 CrossRef CAS PubMed.
- S. K. Choi, J. Kim and E. Kim, Molecules, 2021, 26, 1868 CrossRef CAS PubMed.
- E. Kozma and P. Kele, Org. Biomol. Chem., 2019, 17, 215–233 RSC.
- T. Plass, S. Milles, C. Koehler, J. Szymański, R. Mueller, M. Wießler, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2012, 51, 4166–4170 CrossRef CAS PubMed.
- G. V. Los, L. P. Encell, M. G. Mcdougall, D. D. Hartzell, N. Karassina, D. Simpson, J. Mendez, K. Zimmerman, P. Otto, G. Vidugiris and J. Zhu, ACS Chem. Biol., 2008, 3, 373–382 CrossRef CAS PubMed.
- A. Gautier, A. Juillerat, C. Heinis, I. R. Corrêa, M. Kindermann, F. Beaufils and K. Johnsson, Chem. Biol., 2008, 15, 128–136 CrossRef CAS PubMed.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86–89 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS PubMed.
- A. I. König, R. Sorkin, A. Alon, D. Nachmias, K. Dhara, G. Brand, O. Yifrach, E. Arbely, Y. Roichman and N. Elia, Nanoscale, 2020, 12, 3236–3248 RSC.
- A. C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131–5142 RSC.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- L. G. Meimetis, J. C. T. Carlson, R. J. Giedt, R. H. Kohler and R. Weissleder, Angew. Chem., Int. Ed., 2014, 53, 7531–7534 CrossRef CAS PubMed.
- J. C. T. Carlson, L. G. Meimetis, S. A. Hilderbrand and R. Weissleder, Angew. Chem., Int. Ed., 2013, 52, 6917–6920, DOI:10.1002/anie.201301100.
- A. Wieczorek, P. Werther, J. Euchner and R. Wombacher, Chem. Sci., 2017, 8, 1506–1510 RSC.
- S. T. Shen, W. Zhang, P. Yadav, X. W. Sun and X. Liu, Mater. Chem. Front., 2023, 7, 1082–1092 RSC.
- N. K. Devaraj, S. Hilderbrand, R. Upadhyay, R. Mazitschek and R. Weissleder, Angew. Chem., Int. Ed., 2010, 49, 2869–2872 CrossRef CAS PubMed.
- J. Yang, J. Šečkute, C. M. Cole and N. K. Devaraj, Angew. Chem., Int. Ed., 2012, 51, 7476–7479 CrossRef CAS PubMed.
- G. Knorr, E. Kozma, A. Herner, E. A. Lemke and P. Kele, Chem. – Eur. J., 2016, 22, 8972–8979 CrossRef CAS PubMed.
- H. Wu, J. Yang, J. Šečkutė and N. K. Devaraj, Angew. Chem., 2014, 126, 5915–5919 CrossRef.
- Y. Lee, W. Cho, J. Sung, E. Kim and S. B. Park, J. Am. Chem. Soc., 2018, 140, 974–983 CrossRef CAS PubMed.
- G. Knorr, E. Kozma, J. M. Schaart, K. Németh, G. Török and P. Kele, Bioconjugate Chem., 2018, 29, 1312–1318 CrossRef CAS PubMed.
- E. Németh, G. Knorr, K. Németh and P. Kele, Biomolecules, 2020, 10, 397, DOI:10.3390/biom10030397.
- E. Kozma, G. Estrada Girona, G. Paci, E. A. Lemke and P. Kele, Chem. Commun., 2017, 53, 6696–6699 RSC.
- P. Werther, K. Yserentant, F. Braun, K. Grußmayer, V. Navikas, M. Yu, Z. Zhang, M. J. Ziegler, C. Mayer, A. J. Gralak, M. Busch, W. Chi, F. Rominger, A. Radenovic, X. Liu, E. A. Lemke, T. Buckup, D. P. Herten and R. Wombacher, ACS Cent. Sci., 2021, 7, 1561–1571 CrossRef CAS PubMed.
- E. Albitz, D. Kern, A. Kormos, M. Bojtár, G. Török, A. Biró, Á. Szatmári, K. Németh and P. Kele, Angew. Chem., Int. Ed., 2022, 61, e202111855 CrossRef CAS PubMed.
- L. Chen, F. Li, Y. Li, J. Yang, Y. Li and B. He, Chem. Commun., 2022, 58, 298–301 RSC.
- W. Mao, W. Chi, X. He, C. Wang, X. Wang, H. Yang, X. Liu and H. Wu, Angew. Chem., Int. Ed., 2022, 61, e202117386 CrossRef CAS PubMed.
- M. Bojtár, K. Németh, F. Domahidy, G. Knorr, A. Verkman, M. Kállay and P. Kele, J. Am. Chem. Soc., 2020, 142, 15164–15171 CrossRef PubMed.
- L. Chen, F. Li, M. Nandi, L. Huang, Z. Chen, J. Wei, W. Chi, X. Liu and J. Yang, Dyes Pigm., 2020, 177, 108313 CrossRef CAS.
- K. Klehs, C. Spahn, U. Endesfelder, S. F. Lee, A. Fürstenberg and M. Heilemann, ChemPhysChem, 2014, 15, 637–641 CrossRef CAS PubMed.
- M. I. Sorour, K. A. Kistler, A. H. Marcus and S. Matsika, J. Phys. Chem. A, 2021, 125, 7852–7866 CrossRef CAS PubMed.
- Á. Szatmári, G. B. Cserép, T. Á. Molnár, B. Söveges, A. Biró, G. Várady, E. Szabó, K. Németh and P. Kele, Molecules, 2021, 26, 4988 CrossRef PubMed.
- I. Nikic, J. H. Kang, G. E. Girona, I. V. Aramburu and E. A. Lemke, Nat. Protoc., 2015, 10, 780–791 CrossRef CAS PubMed.
- A. Egyed, A. Kormos, B. Söveges, K. Németh and P. Kele, Bioorg. Med. Chem., 2020, 28, 115218 CrossRef CAS PubMed.
- I. Nikić, G. E. Girona, J. H. Kang, G. Paci, S. Mikhaleva, C. Koehler, N. V. Shymanska, C. Ventura Santos, D. Spitz and E. A. Lemke, Angew. Chem., Int. Ed., 2016, 55, 16172–16176 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthetic procedures, spectroscopic characterization, physical data determination, details of the imaging experiments, further images and viability assessment. See DOI: https://doi.org/10.1039/d3ob01204b |
| This journal is © The Royal Society of Chemistry 2023 |