
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rod-like nanostructures through amphiphilic OPE-porphyrin self-organization†
Chiara M. A.
Gangemi‡
 ,
Maria A.
Castriciano‡
,
Ester
D'Agostino
,
Andrea
Romeo
,
Paola M.
Bonaccorsi
,
Anna
Barattucci
* and
Luigi
Monsù Scolaro
*
,
Maria A.
Castriciano‡
,
Ester
D'Agostino
,
Andrea
Romeo
,
Paola M.
Bonaccorsi
,
Anna
Barattucci
* and
Luigi
Monsù Scolaro
*
Dipartimento di Scienze Chimiche, Biologiche, Farmaceutiche ed Ambientali, Università degli Studi di Messina, V.le F. Stagno D'Alcontres 31, 98166 Messina, Italy. E-mail: lmonsu@unime.it
First published on 21st September 2023
Abstract
A new amphiphilic monosubstituted porphyrin functionalized by a β-D-glucoside terminated oligophenylenethylene (OPE) able to self-arrange into nano-aggregates in polar solvents has been synthesized and fully characterized in its monomeric and aggregated forms.
Non-covalent multichromophoric assemblies are highly investigated due to their involvement either in many fundamental processes, such as photosynthesis,1 or applications such as photodynamic therapy (PDT)2 or photovoltaics.3 In biological processes, the requirement for vectorial processes, i.e. energy and/or electron transfer, is rigidity and fixed geometry among the various components, usually accomplished through embedding the species in bilayer lipid membranes or protein scaffolds. In this respect, several chromophore-based systems have been proposed and complex architectures have been realized through self-assembling processes, exploiting a variety of specific interactions.4 Porphyrins are a large class of natural and synthetic compounds that have been widely used as versatile building blocks. They possess an extended aromatic region with peculiar electronic features, dominated by very intense absorption bands in the visible range of the spectrum (B- and Q-bands) and usually intense fluorescence emission.5,6 Self-aggregation can spontaneously occur depending on a variety of non-covalent interactions, generally dominated by π-stacking. The electronic coupling between adjacent chromophores depends on the geometrical arrangement of the interacting chromophores, classified into H-type (face-to-face) or J-type (edge-to-edge) dispositions, exhibiting bands shifted to higher and lower energies, respectively.7 Intriguing new properties could be added to supramolecular species by introducing appropriate pendant moieties at the periphery of these compounds:8–11 (i) charged or ionisable groups allow electrostatic contacts, driving the formation of even extended aggregates, such as in the case of J-aggregates of sulphonatophenyl porphyrins12–15 and (ii) carbohydrates could promote hydrogen bonding interactions and potentially induce chirality in eventual supramolecular assemblies.16 In this respect, the choice of good solvent/poor solvent mixtures allows for achieving a variety of nanostructures, depending on various parameters, including the ratio and shape of the hydrophobic vs. hydrophilic parts. Also in an aqueous environment, the medium properties in terms of pH and/or ionic strength, together with the temperature, have a deep impact on the kinetics and the final nano-architecture.17 Furthermore, since carbohydrate-recognized proteins are usually overexpressed on the membrane of certain tumor cells, the introduction of one18–23 up to four sugar moieties on a porphyrin has led to several targeted PDT applications.24–27 Another interesting class of chromophores are oligo-phenylene-ethynylenes (OPEs). These systems have been widely exploited as rigid spacers able to electronically couple acceptor and donor supramolecular porphyrinoid based dyads28–30 or to provide optimal separation in self-assembled monolayers31 or in semiconductor nanoparticles for electronic applications.32 An appropriate functionalization with carbohydrates affords systems with potential application in photodynamic therapy,33–36 such as fluorescent biomarkers37,38 or inhibitors of bacterial growth.39 To the best of our knowledge, only one example has been reported so far in the literature on the synthetic procedure coupling a mono-substituted porphyrin with a carbohydrate moiety through an OPE based bridge.40
Hence, on these premises, we designed a new amphiphilic system consisting of a free-base porphyrin macrocycle, monosubstituted at the meso-position by an oligophenylenethylene (OPE) moiety bearing a dimethylamino substituent at one of the aromatic residues and a β-D-glucoside termination.33,35 The introduction of the hydrophilic sugar termination, together with the ionisable group, was intended to study the supramolecular behaviour of the amphiphilic species for the construction of new biocompatible aggregates. The OPE residue was chosen for a dual purpose: (i) to use the conjugated fragment as a spacer between the two functional groups of the amphiphilic system33–36 and, (ii) as a chromophore, to study the mutual influence and the potential communication between the porphyrin and the OPE residues. The strategy employed for the synthesis of the bis-chromophoric system GAP (Scheme 1) involves the step-by-step modular approach to the preparation of the OPE chain. Starting from aminoaryl sugar derivative 1,35 the conjugated chain elongation to 2 was realized through Pd(0) mediated cross-coupling with an excess of 1,4-diiodobenzene and in the presence of a stoichiometric amount of Ag2O. The last reactant is reported to cause in situ direct desilylation of the terminal acetylenic residue,41 so avoiding the alternative use of basic conditions for deprotection,42 also responsible for the undesired deacetylation of the sugar residue that could cause low solubility in polar organic solvents. The reaction was complete in 1.5 hours and the iodoarene 2 was obtained in 60% yield.
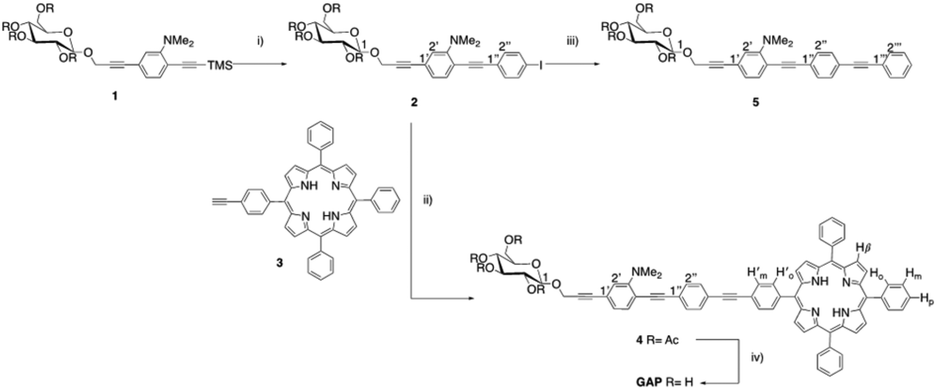 | ||
| Scheme 1 Synthetic route to GAP. (i) 1,4-Diiodobenzene, Pd(PPh3)4, Ag2O, THF/DMF (1/1), 60 °C, 1 h 30 min; (ii) Pd(PPh3)4, Et3N, DMF, 70 °C, 1 h 30 min; (iii) ethynylbenzene, Pd(PPh3)4, Et3N, DMF, 70 °C, 2 h; and (iv) NH4OHaq, MeOH/THF (1/1), RT, overnight. | ||
Copper-free Heck–Cassar–Sonogashira cross-coupling of 2 with monofunctionalized porphyrin 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 42,43 leads to chromophore 4, and then quantitatively deprotected to amphiphilic GAP using an aqueous ammonia solution in THF/MeOH. Finally, the trimeric OPE 5 was synthesized as a model compound for the spectroscopic characterization of 4 and GAP. The cross-coupling of 2 with an excess of commercial phenylacetylene afforded 5 in almost quantitative yields. Due to its amphiphilic nature the complete characterization of GAP was conducted in a CDCl3/CD3OD mixture (13:1, see ESI, Fig. S1–S11†). The absorption spectra of GAP were recorded in chloroform wherein no aggregation occurred as confirmed by the linearity of Beer's law (ε421 nm = 3.25 × 105 M−1 cm−1) (see ESI, Fig. S12†) and by the resonance light scattering (RLS) profile, which is comparable in intensity with the neat solvent (data not shown). A typical absorption spectrum (see ESI, Fig. S13†) mainly exhibits features of the porphyrin moiety in the visible range with a B-band located at 421 nm accompanied by four Q-bands at 517, 552, 591, and 647 nm. However, the presence of the OPE unit can be assessed by an additional low intensity feature in the UV region as confirmed by the spectroscopic behaviour of the trimeric OPE 5 taken as a reference (see ESI, Fig. S14†). The circular dichroism (CD) spectrum shows no induced CD signal in the porphyrin absorption region indicating no coupling with the chiral sugar residue in the side chain on the aromatic ring (see ESI, Fig. S15†). GAP is strongly emissive in solution and its calculated fluorescence quantum yield value (Φ = 0.033 in CHCl3) is comparable with that reported in the literature for similar porphyrins.44 The fluorescence emission spectrum displays the typical two-banded pattern (654 and 718 nm) and the emission decay shows a mono-exponential profile with a long-living lifetime value (8 ns) ascribable to the porphyrin in its monomeric form.5 In order to tune the extent of aggregation, which is well known to strongly depend on the nature of the media, we investigated the system on increasing the polarity of the solvent moving from chloroform to methanol (CHCl3/MeOH, 50/50 v/v and pure MeOH). In these solvents, the spectra remain substantially unchanged exhibiting only a slight hypsochromic shift of the B-band with respect to CHCl3 due to the less hydrophobic environment around the porphyrin (Δλ = −3 nm and −6 nm, respectively; see ESI, Fig. S16†).5
42,43 leads to chromophore 4, and then quantitatively deprotected to amphiphilic GAP using an aqueous ammonia solution in THF/MeOH. Finally, the trimeric OPE 5 was synthesized as a model compound for the spectroscopic characterization of 4 and GAP. The cross-coupling of 2 with an excess of commercial phenylacetylene afforded 5 in almost quantitative yields. Due to its amphiphilic nature the complete characterization of GAP was conducted in a CDCl3/CD3OD mixture (13:1, see ESI, Fig. S1–S11†). The absorption spectra of GAP were recorded in chloroform wherein no aggregation occurred as confirmed by the linearity of Beer's law (ε421 nm = 3.25 × 105 M−1 cm−1) (see ESI, Fig. S12†) and by the resonance light scattering (RLS) profile, which is comparable in intensity with the neat solvent (data not shown). A typical absorption spectrum (see ESI, Fig. S13†) mainly exhibits features of the porphyrin moiety in the visible range with a B-band located at 421 nm accompanied by four Q-bands at 517, 552, 591, and 647 nm. However, the presence of the OPE unit can be assessed by an additional low intensity feature in the UV region as confirmed by the spectroscopic behaviour of the trimeric OPE 5 taken as a reference (see ESI, Fig. S14†). The circular dichroism (CD) spectrum shows no induced CD signal in the porphyrin absorption region indicating no coupling with the chiral sugar residue in the side chain on the aromatic ring (see ESI, Fig. S15†). GAP is strongly emissive in solution and its calculated fluorescence quantum yield value (Φ = 0.033 in CHCl3) is comparable with that reported in the literature for similar porphyrins.44 The fluorescence emission spectrum displays the typical two-banded pattern (654 and 718 nm) and the emission decay shows a mono-exponential profile with a long-living lifetime value (8 ns) ascribable to the porphyrin in its monomeric form.5 In order to tune the extent of aggregation, which is well known to strongly depend on the nature of the media, we investigated the system on increasing the polarity of the solvent moving from chloroform to methanol (CHCl3/MeOH, 50/50 v/v and pure MeOH). In these solvents, the spectra remain substantially unchanged exhibiting only a slight hypsochromic shift of the B-band with respect to CHCl3 due to the less hydrophobic environment around the porphyrin (Δλ = −3 nm and −6 nm, respectively; see ESI, Fig. S16†).5
Evident spectroscopic changes have been observed when methanol/water at different v/v ratio values are used as solvents. More specifically, as shown in Fig. 1, the addition of increasing amounts of water to a porphyrin solution in methanol, up to a 40/60 water/methanol (v/v) ratio, induces a consistent bathochromic shift (Δλ = +16 nm) together with a concomitant broadening of the B-band. The corresponding fluorescence emission spectra display a substantial quenching with respect to the sample in pure methanol in line with the fluorescence emission intensity decays that show a bi-exponential behaviour with two lifetime values of 1.6 ns (relative amplitude 80%) and 5.6 ns (relative amplitude 20%), respectively. The RLS spectrum exhibits a peak in proximity to the main absorption band indicating the presence of aggregated species (see ESI, Fig. S17†). According to RLS theory, this spectroscopic evidence suggests that the aggregates should be constituted by at least 25 interacting porphyrin units, with a strong coupling among their electronic transition moments.45,46 This hypothesis has been definitively confirmed by dynamic light scattering (DLS) measurements which reveal the presence of well-dispersed nanometer-sized porphyrin aggregates (RH = 200 ± 50 nm; see ESI, Fig. S18†).
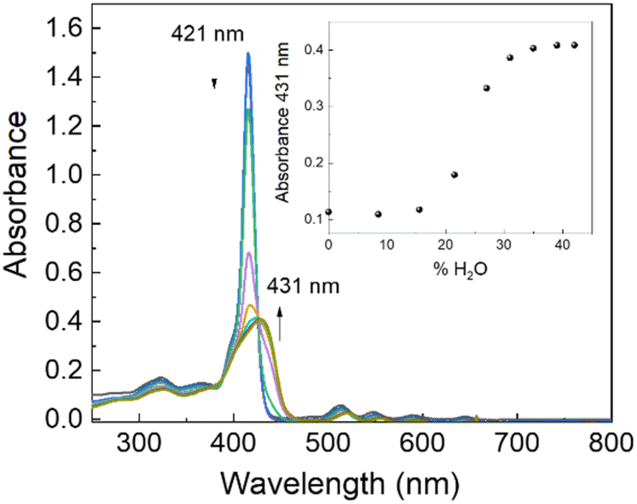 | ||
| Fig. 1 UV/Vis spectral change of GAP in methanol on increasing the water percentage (v/v). GAP in methanol. [GAP] = 7 μM, T = 298 K. | ||
Depolarized RLS measurements can give useful insights into the geometrical disposition of the chromophores in large aggregates. The value of the depolarization ratio ρv(90) calculated for the investigated samples is 0.125, from which a slip angle φ = 40 or 50° between adjacent porphyrin planes can be calculated, assuming a parallel arrangement of the transition moments of the exciton-coupled chromophores (Fig. 2a).45 This angle is in line with the observed J-type shift of the B-band to a lower energy with respect to the isolated monomer. Considering also that these aggregates are CD silent (data not shown), a tilt angle close to 90° could be envisaged between the transition moments (Fig. 2b).47 The presence of supramolecular aggregates in solution is ascribable to the ability of both porphyrins and OPEs to establish π-stacking interactions together with the presence of free glucosidic units, which allow, in principle, the formation of intermolecular hydrogen bonds that stabilize the porphyrin network.
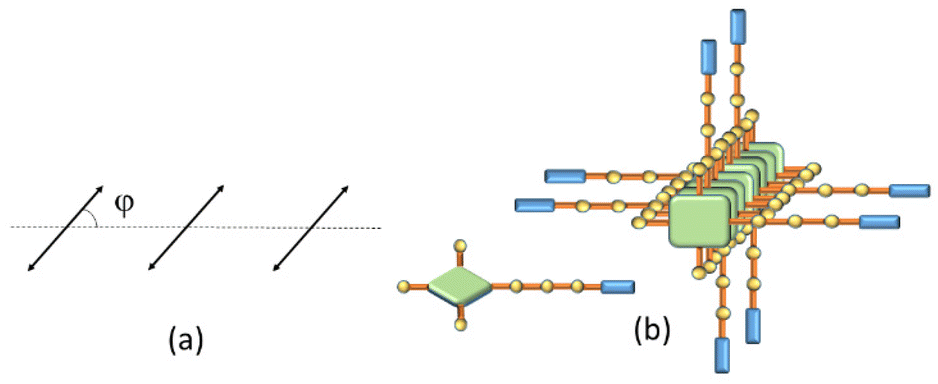 | ||
| Fig. 2 Model for (a) the geometrical arrangement showing the slip angle φ between the transition moments and the line connecting the centroids of the porphyrin rings, and (b) sketch of the porphyrin stacks. | ||
To clarify the nature of these aggregates, HCl (up to 0.1 M) was added to the 40/60 water/methanol solution. The unchanged spectroscopic features suggest that the hydrophobic porphyrin core is located within, which conversely exposes the hydrophilic glycosidic part towards the aqueous environment. Fluorescence emission experiments, performed by adding an iodide anion as a quencher, confirm this hypothesis.48 Indeed, the addition of the quencher induces only a slight bathochromic shift of the UV/Vis profile without affecting the fluorescence emission in terms of intensity and lifetimes (see ESI, Fig. S19†). Finally, a 40/60 water/methanol solution of GAP was dropped onto the silicon surface. After evaporation of the solvent, AFM microscopy (Fig. 3) shows well-defined rod-like structures with an average diameter of about 50 nm, a length of 300 nm and a thickness of about 20 nm (see ESI, Fig. S20†), in agreement with the size measured in solution. Considering that the calculated length of a single GAP unit from molecular models is around 3.9 nm, all the previous experimental evidence suggest that these nanoaggregates cannot be formed by a single stacked arrangement as shown in the model of Fig. 2b. Assuming a typical inter-porphyrin distance of 0.33–0.35 nm, a complete turn of the sugar pendant arms occurs with a pitch of ca. 1 nm. Therefore, hydrogen-bonding interactions among these moieties could be operative to bridge or interdigitate more porphyrin stacks to justify the observed diameter of the final structures.
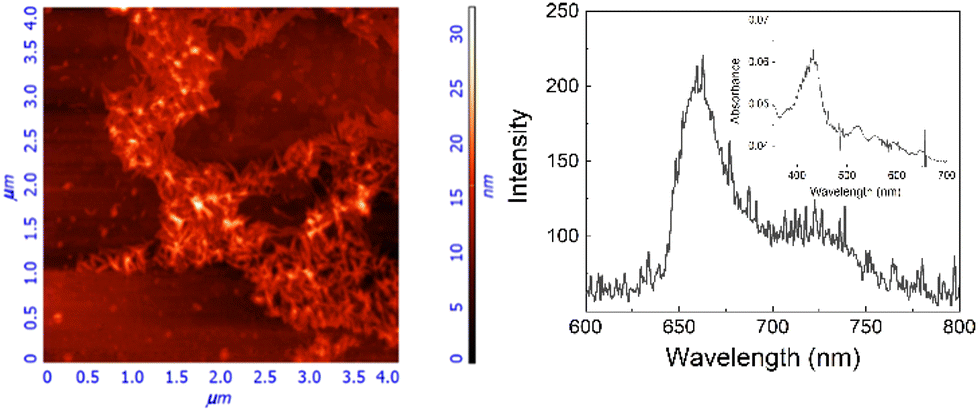 | ||
| Fig. 3 AFM image of the silicon surface (left) and fluorescence emission and absorption spectra (right and inset) of GAP aggregates. 1 μM GAP in 40/60 v/v water/methanol added dropwise and evaporated at room temperature on the silicon surface (left) and glass surface (right), λex 430nm. | ||
In conclusion, we have designed, synthesized, and fully characterized a new amphiphilic monosubstituted porphyrin functionalized by an oligophenylenethylene (OPE) with a β-D-glucoside termination. The introduction of an OPE and a hydrophilic sugar moiety at the periphery allowed the induction of the supramolecular organization of the amphiphilic species on increasing the polarity of the medium, thus affording new promising biocompatible nano-aggregates, potentially suitable for photodynamic therapy, bioimaging or drug delivery.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. Salvatore Patanè for AFM microscopy. The work was financially supported by the European Union-FSE-REACT-EU, PON Research and Innovation 2014–2020 DM.1062/2021 and Next Generation EU, PNRR Samothrace Project (ECS00000022).References
- S. Y. Cao, A. Roslawska, B. Doppagne, M. Romeo, M. Feron, F. Cherioux, H. Bulou, F. Scheurer and G. Schull, Nat. Chem., 2021, 13, 766–770 CrossRef CAS PubMed.
- L. X. Sun, J. Wang, B. C. Yang, X. X. Wang, G. X. Yang, X. Q. Wang, Y. Y. Jiang, T. Y. Wang and J. Z. Jiang, RSC Adv., 2021, 11, 10061–10074 RSC.
- G. Charalambidis, K. Karikis, E. Georgilis, B. L. M'Sabah, Y. Pellegrin, A. Planchat, B. Lucas, A. Mitraki, J. Boucle, F. Odobel and A. G. Coutsolelos, Sustainable Energy Fuels, 2017, 1, 387–395 RSC.
- J.-H. Fuhrhop and J. Köning, Membranes and Molecular Assemblies: The Synkinetic Approach, The Royal Society of Chemistry, Cambridge, 1994, 10.1039/9781847551368-fp001.
- N. C. Maiti, M. Ravikanth, S. Mazumdar and N. Periasamy, J. Phys. Chem. B, 1995, 99, 17192–17197 CrossRef CAS.
- W. I. White, The Porphyrins, Academic Press, New York, 1978 Search PubMed.
- E. G. McRae and M. Kasha, Physical Processes in Radiation Biology, Academic Press, New York, 1964 Search PubMed.
- C. M. A. Gangemi, R. Randazzo, M. E. Fragalà, G. A. Tomaselli, F. P. Ballistreri, A. Pappalardo, R. M. Toscano, G. Trusso Sfrazzetto, R. Purrello and A. D'Urso, New J. Chem., 2015, 39, 6722–6725 RSC.
- M. A. Castriciano, R. Zagami, M. Trapani, A. Romeo, S. Patane and L. M. Scolaro, Chirality, 2015, 27, 900–906 CrossRef CAS PubMed.
- M. A. Castriciano, A. Romeo, N. Angelini, N. Micali, A. Longo, A. Mazzaglia and L. M. Scolaro, Macromolecules, 2006, 39, 5489–5496 CrossRef CAS.
- L. M. Scolaro, C. Donato, M. Castriciano, A. Romeo and R. Romeo, Inorg. Chim. Acta, 2000, 300, 978–986 CrossRef.
- A. Romeo, M. A. Castriciano, I. Occhiuto, R. Zagami, R. F. Pasternack and L. M. Scolaro, J. Am. Chem. Soc., 2014, 136, 40–43 CrossRef CAS PubMed.
- R. Zagami, A. Romeo, M. A. Castriciano and L. M. Scolaro, J. Mol. Liq., 2021, 332, 115801 CrossRef CAS.
- R. Zagami, A. Romeo, M. A. Castriciano and L. MonsùScolaro, Chem. – Eur. J., 2017, 23, 70–74 CrossRef CAS PubMed.
- R. Zagami, M. A. Castriciano, A. Romeo, M. Trapani, R. Pedicini and L. M. Scolaro, Dyes Pigm., 2017, 142, 255–261 CrossRef CAS.
- M. Stefanelli, F. Mandoj, G. Magna, R. Lettieri, M. Venanzi, R. Paolesse and D. Monti, Molecules, 2020, 25, 4544 CrossRef CAS PubMed.
- R. Zagami, M. A. Castriciano, A. Romeo and L. M. Scolaro, J. Porphyrins Phthalocyanines, 2023, 27, 463–470 CrossRef CAS.
- J. P. C. Tome, E. M. P. Silva, A. Pereira, C. M. A. Alonso, M. A. F. Faustino, M. Neves, A. C. Tome, J. A. S. Cavaleiro, S. A. P. Tavares, R. R. Duarte, M. F. Caeiro and M. L. Valdeira, Bioorg. Med. Chem., 2007, 15, 4705–4713 CrossRef CAS PubMed.
- J. P. C. Tome, M. Neves, A. C. Tome, J. A. S. Cavaleiro, A. F. Mendonca, L. S. N. Pegado, R. Duarte and M. L. Valdeira, Bioorg. Med. Chem., 2005, 13, 3878–3888 CrossRef CAS PubMed.
- F. Giuntini, F. Bryden, R. Daly, E. M. Scanlan and R. W. Boyle, Org. Biomol. Chem., 2014, 12, 1203–1206 RSC.
- F. Figueira, L. M. O. Lourenco, M. Neves, J. A. S. Cavaleiro and J. P. C. Tome, J. Porphyrins Phthalocyanines, 2020, 24, 330–339 CrossRef CAS.
- A. Fadlan, H. Tanimoto, T. Ito, Y. Aritomi, M. Ueno, M. Tokuda, S. Hirohara, M. Obata, T. Morimoto and K. Kakiuchi, Bioorg. Med. Chem., 2018, 26, 1848–1858 CrossRef CAS PubMed.
- M. C. Bennion, M. A. Burch, D. G. Dennis, M. E. Lech, K. Neuhaus, N. L. Fendler, M. R. Parris, J. E. Cuadra, C. F. Dixon, G. T. Mukosera, D. N. Blauch, L. Hartmann, N. L. Snyder and J. V. Ruppel, Eur. J. Org. Chem., 2019, 6496–6503 CrossRef CAS PubMed.
- X. Zheng and R. K. Pandey, Anticancer Agents Med. Chem., 2008, 8, 241–268 CrossRef CAS PubMed.
- S. Singh, A. Aggarwal, N. Bhupathiraju, G. Arianna, K. Tiwari and C. M. Drain, Chem. Rev., 2015, 115, 10261–10306 CrossRef CAS PubMed.
- M. Rosa, N. Jedryka, S. Skorupska, I. Grabowska-Jadach and M. Malinowski, Int. J. Mol. Sci., 2022, 23, 11321 CrossRef CAS PubMed.
- M. Lupu, P. Maillard, J. Mispelter, F. Poyer and C. D. Thomas, Photochem. Photobiol. Sci., 2018, 17, 1599–1611 CrossRef CAS PubMed.
- A. Lembo, P. Tagliatesta, D. M. Guldi, M. Wielopolski and M. Nuccetelli, J. Phys. Chem. A, 2009, 113, 1779–1793 CrossRef CAS PubMed.
- E. Goransson, J. Boixel, J. Fortage, D. Jacquemin, H. C. Becker, E. Blart, L. Hammarstrom and F. Odobel, Inorg. Chem., 2012, 51, 11500–11512 CrossRef PubMed.
- M. P. Eng, J. Martensson and B. Albinsson, Chem. – Eur. J., 2008, 14, 2819–2826 CrossRef CAS PubMed.
- S. Watcharinyanon, D. Nilsson, E. Moons, A. Shaporenko, M. Zharnikov, B. Albinsson, J. Martensson and L. S. O. Johansson, Phys. Chem. Chem. Phys., 2008, 10, 5264–5275 RSC.
- J. Rochford and E. Galoppini, Langmuir, 2008, 24, 5366–5374 CrossRef CAS PubMed.
- A. Lara-Pardo, A. Mancuso, S. Simon-Fuente, P. M. Bonaccorsi, C. M. A. Gangemi, M. A. Moline, F. Puntoriero, M. Ribagorda, A. Barattucci and F. Sanz-Rodriguez, Org. Biomol. Chem., 2023, 21, 386–396 RSC.
- C. M. A. Gangemi, A. Barattucci and P. M. Bonaccorsi, Molecules, 2021, 26, 3088 CrossRef CAS PubMed.
- E. Deni, A. Zamarron, P. Bonaccorsi, M. C. Carreno, A. Juarranz, F. Puntoriero, M. T. Sciortino, M. Ribagorda and A. Barattucci, Eur. J. Med. Chem., 2016, 111, 58–71 CrossRef CAS PubMed.
- A. Barattucci, E. Deni, P. Bonaccorsi, M. G. Ceraolo, T. Papalia, A. Santoro, M. T. Sciortino and F. Puntoriero, J. Org. Chem., 2014, 79, 5113–5120 CrossRef CAS PubMed.
- A. Mancuso, A. Barattucci, P. Bonaccorsi, A. Giannetto, G. La Ganga, M. Musarra-Pizzo, T. M. G. Salerno, A. Santoro, M. T. Sciortino, F. Puntoriero and M. L. Di Pietro, Chem. – Eur. J., 2018, 24, 16972–16976 CrossRef CAS PubMed.
- E. Arias, M. T. Mendez, E. Arias, I. Moggio, A. Ledezma, J. Romero, G. Margheri and E. Giorgetti, Sensors, 2017, 17, 1025 CrossRef PubMed.
- F. Pertici, N. Varga, A. van Duijn, M. Rey-Carrizo, A. Bernardi and R. J. Pieters, Beilstein J. Org. Chem., 2013, 9, 215–222 CrossRef CAS PubMed.
- B. Godlewski, D. Baran, M. de Robichon, A. Ferry, S. Ostrowski and M. Malinowski, Org. Chem. Front., 2022, 9, 2396–2404 RSC.
- A. Mori, T. Kondo, T. Kato and Y. Nishihara, Chem. Lett., 2001, 286–287, DOI:10.1246/cl.2001.286.
- Y.-L. Zhao, L. Liu, W. Zhang, C.-H. Sue, Q. Li, O. Š. Miljanić, O. M. Yaghi and J. F. Stoddart, Chem. – Eur. J., 2009, 15, 13356–13380 CrossRef CAS PubMed.
- A. R. McDonald, N. Franssen, G. P. M. van Klink and G. van Koten, J. Organomet. Chem., 2009, 694, 2153–2162 CrossRef CAS.
- M. Taniguchi, J. S. Lindsey, D. F. Bocian and D. Holten, J. Photochem. Photobiol., C, 2021, 46, 100401 CrossRef CAS.
- J. Parkash, J. H. Robblee, J. Agnew, E. Gibbs, P. Collings, R. F. Pasternack and J. C. de Paula, Biophys. J., 1998, 74, 2089–2099 CrossRef CAS PubMed.
- R. F. Pasternack and P. J. Collings, Science, 1995, 269, 935–939 CrossRef CAS PubMed.
- A. Rodger and J. J. Chubb, Encyclopedia of Analytical Chemistry, 2023, pp. 1–42, DOI:10.1002/9780470027318.a5402.pub3.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum Publishers, New York, 1999 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S8: 1H and 13C NMR spectra; Fig. S9–S11: ESI mass spectra; Fig. S12: Absorption spectra of GAP in chloroform and Beer's plot; Fig. S13: Absorption and emission spectra of GAP in chloroform; Fig. S14: Excitation and emission spectra of OPE in chloroform; Fig. S15: CD spectrum of GAP in chloroform; Fig. S16: Absorption spectra of GAP in chloroform, chloroform/methanol 50/50 v/v, and methanol; Fig. S17: RLS spectra of GAP in methanol and water/methanol 40/60 v/v. Fig. S18: DLS distribution of GAP in water/methanol 40/60 v/v; Fig. S19: Absorption spectrum of GAP in water/methanol 40/60 v/v and in the presence of KI; and Fig. S20: AFM image and relative profile of GAP aggregates on a silicon surface. See DOI: https://doi.org/10.1039/d3ob01385e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |