
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effective synthesis, development and application of a highly fluorescent cyanine dye for antibody conjugation and microscopy imaging†
Dénes
Szepesi Kovács‡
abc,
Bence
Kontra
 def,
Balázs
Chiovini
g,
Dalma
Müller
fhi,
Estilla Zsófia
Tóth
cfj,
Péter
Ábrányi-Balogh
abc,
Lucia
Wittner
cj,
György
Várady
k,
Gábor
Turczel
l,
Ödön
Farkas
m,
Michael C.
Owen
no,
Gergely
Katona
g,
Balázs
Győrffy
chi,
György Miklós
Keserű
*abc,
Zoltán
Mucsi
*den,
Balázs J.
Rózsa
*dgp and
Ervin
Kovács‡
*eq
def,
Balázs
Chiovini
g,
Dalma
Müller
fhi,
Estilla Zsófia
Tóth
cfj,
Péter
Ábrányi-Balogh
abc,
Lucia
Wittner
cj,
György
Várady
k,
Gábor
Turczel
l,
Ödön
Farkas
m,
Michael C.
Owen
no,
Gergely
Katona
g,
Balázs
Győrffy
chi,
György Miklós
Keserű
*abc,
Zoltán
Mucsi
*den,
Balázs J.
Rózsa
*dgp and
Ervin
Kovács‡
*eq
aMedicinal Chemistry Research Group, HUN-REN Research Centre for Natural Sciences, H-1117 Budapest, Hungary
bDepartment of Organic Chemistry and Technology, Budapest University of Technology and Economics, H-1111 Budapest, Hungary
cNational Laboratory for Drug Research and Development, H-1117 Budapest, Hungary
dBrain Vision Center, H-1094 Budapest, Hungary
eFemtonics Ltd., H-1094 Budapest, Hungary
fSemmelweis University Doctoral School, H-1085 Budapest, Hungary
gFaculty of Information Technology and Bionics, Pázmány Péter Catholic University, H-1444 Budapest, Hungary
hOncology Biomarker Research Group, HUN-REN Research Centre for Natural Sciences, H-1117 Budapest, Hungary
iDepartment of Bioinformatics, Semmelweis University, H-1094, Budapest, Hungary
jIntegrative Neuroscience Research Group, HUN-REN Research Centre for Natural Sciences, H-1117 Budapest, Hungary
kMolecular Cell Biology Research Group, HUN-REN Research Centre for Natural Sciences, H-1117 Budapest, Hungary
lNMR Research Laboratory, HUN-REN Research Centre for Natural Sciences, H-1117 Budapest, Hungary
mDepartment of Organic Chemistry, Eötvös Loránd University, H-1117 Budapest, Hungary
nInstitute of Chemistry, University of Miskolc, Miskolc H-3515, Hungary
oHigher Education and Industrial Cooperation Centre, University of Miskolc, Miskolc H-3515, Hungary
pLaboratory of 3D Functional Network and Dendritic Imaging, HUN-REN Institute of Experimental Medicine, H-1083 Budapest, Hungary
qPolymer Chemistry and Physics Research Group, HUN-REN Research Centre for Natural Sciences, H-1117 Budapest, Hungary. E-mail: kovacs.ervin@ttk.hu; Tel: +3613826570
First published on 18th October 2023
Abstract
An asymmetric cyanine-type fluorescent dye was designed and synthesized via a versatile, multi-step process, aiming to conjugate with an Her2+ receptor specific antibody by an azide–alkyne click reaction. The aromaticity and the excitation and relaxation energetics of the fluorophore were characterized by computational methods. The synthesized dye exhibited excellent fluorescence properties for confocal microscopy, offering efficient applicability in in vitro imaging due to its merits such as a high molar absorption coefficient (36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 816 M−1 cm−1), excellent brightness, optimal wavelength (627 nm), larger Stokes shift (26 nm) and appropriate photostability compared to cyanines. The conjugated cyanine–trastuzumab was constructed via an effective, metal-free, strain-promoted azide–alkyne click reaction leading to a regulated number of dyes being conjugated. This novel cyanine-labelled antibody was successfully applied for in vitro confocal imaging and flow cytometry of Her2+ tumor cells.
816 M−1 cm−1), excellent brightness, optimal wavelength (627 nm), larger Stokes shift (26 nm) and appropriate photostability compared to cyanines. The conjugated cyanine–trastuzumab was constructed via an effective, metal-free, strain-promoted azide–alkyne click reaction leading to a regulated number of dyes being conjugated. This novel cyanine-labelled antibody was successfully applied for in vitro confocal imaging and flow cytometry of Her2+ tumor cells.
Introduction
Fluorescence imaging plays an increasingly important role in biochemical applications, such as fluorescence-guided surgery, photodynamic treatments, and diagnostic protocols. However, since living cells can be damaged by high energy light, there is an increasing demand for red-shifted fluorescent dyes. The use of low-energy fluorescence would make this application more suitable for cancer treatment, particularly in identifying tumors,1–3 since the ability to identify and distinguish healthy cells from tumour cells, both rapidly and reliably, is a crucial task in cancer diagnosis and drug development. The use of specific antibodies to label tumour cells exclusively and with high selectivity is an effective means of accomplishing this, and has led to recent research interest in in vitro and in vivo fluorescence methods based on antibody conjugation.4–7Cyanine fluorescent probes can be used as sensors to identify small molecules, but are equally suitable complex systems containing proteins and DNA.8–11 Cyanines, such as Cy5® and IRDye800, have been used to identify drug binding sites by linking them to small molecules like cariprazine,12 or biomolecules such as sugars, peptides, and proteins (Fig. 1A).13 Moreover, the cyanine core has been used for organelle targeting3,14–17 and DNA labelling, and as a therapeutic in emerging photodynamic applications (merocyanines or pentamethine cyanine dyes, Fig. 1B and C).9,18–21
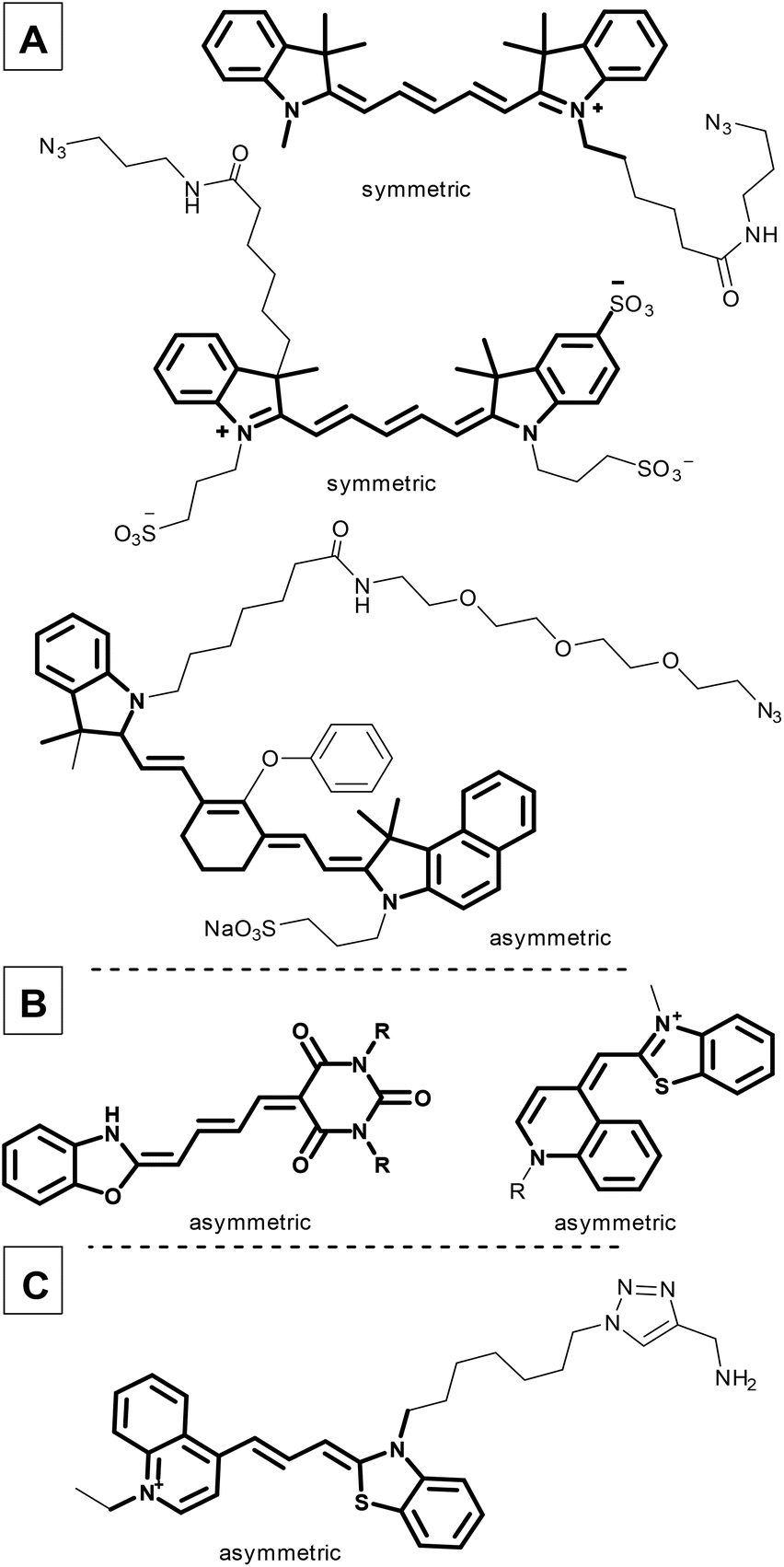 | ||
| Fig. 1 Commercially available symmetric and asymmetric cyanine dyes (A); cyanines applied in photodynamic therapy (B); a recently developed DNA probe with a cyanine moiety (C). | ||
Dyes with a polymethine linker between two nitrogen atoms with a delocalized charge are called cyanines.22 Usually both nitrogen atoms are part of heteroaromatic units, like indoles and benzothiazoles (Fig. 1). The elongated cyanine dye core (C5 or C7, depending on the number of conjugated ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH– units between the two nitrogen atoms)13 helps cyanines to become effective red-shifted fluorescent dyes. Moreover, this core has beneficial tunable photophysical properties, i.e. an emission wavelength up to the near infrared region (NIR, 510–800 nm) and an excitation wavelength between 492 nm and 780 nm. Cyanine dyes also exhibit extremely large molar absorption coefficients, reaching 100000 M−1 cm−1;13,23 however, their quantum yield is often low due to the twisted intramolecular charge transfer (TICT). The main drawback of red-shifted dyes is their limited photostability as their fluorescence fades shortly after irradiation.24
CH– units between the two nitrogen atoms)13 helps cyanines to become effective red-shifted fluorescent dyes. Moreover, this core has beneficial tunable photophysical properties, i.e. an emission wavelength up to the near infrared region (NIR, 510–800 nm) and an excitation wavelength between 492 nm and 780 nm. Cyanine dyes also exhibit extremely large molar absorption coefficients, reaching 100000 M−1 cm−1;13,23 however, their quantum yield is often low due to the twisted intramolecular charge transfer (TICT). The main drawback of red-shifted dyes is their limited photostability as their fluorescence fades shortly after irradiation.24
Two types of cyanine dyes were designed previously with symmetric (Fig. 1A) and asymmetric structures (Fig. 1B). Symmetric cyanines have two quasi-equivalent heteroaryl groups at the two ends in contrast to asymmetric cyanines, which contain different aromatic heterocycles at their termini. In general, symmetric cyanine dyes induce Stokes shifts that are too small (10–15 nm), due to the equivalent electron distributions, and are prone to self-absorption. For example, AlexaFluor25 and its competitors generally absorb light at around 650 nm and emit at 665 nm.
Furthermore, it is suggested that these dyes be used with 594 or 633 nm lasers. This range is far from the optimal excitation wavelength, which results in a weaker photon emission intensity.37 Unfortunately, there are fewer dyes that are commercially available with a λemmax near 650 nm. Finally, due to the highly hydrophobic main core, several sulfonyl groups must be added to facilitate solubility in aqueous solutions.38
In contrast, asymmetric cyanines exhibit a larger Stokes shift with increased fluorescence intensity and brightness because of the unequal electron distribution, resulting in an altered excited state structure.26–28 Analogous cyanine structures have already been used in haematology.29
In spite of the fact that the relevance of cyanine conjugated antibodies is obviously high, recently, only a few examples have been published (Table S1†).30–35
In this regard, and as a continuation of our interest in the design and synthesis of novel asymmetric dyes36–42 and antibody modification, we herein describe the development of a novel fluorescent cyanine dye.37,43,44 The strengths of our novel asymmetric cyanine are as follows: first, we increased solubility by introducing a positively charged ionic core; second, we improved its photophysical properties, in particular increasing the Stokes shift by 25 nm, which improved image quality and reduced self-absorption, achieving an excitation maximum at the desirable 633 nm wavelength, and an improving photostability; and third, we added an azide-containing linker that enables its application in azide–alkyne click reactions, which results in an optimal fluorophore–antibody ratio. Thereafter we successfully cross-linked the candidate dye to the therapeutic antibody trastuzumab. Finally, in vitro confocal microscopy was used to demonstrate the ability of the novel cyanine-labelled antibody to specifically recognize Her2+ cancer cells without labelling the Her2− cancer cell line (Scheme 1).
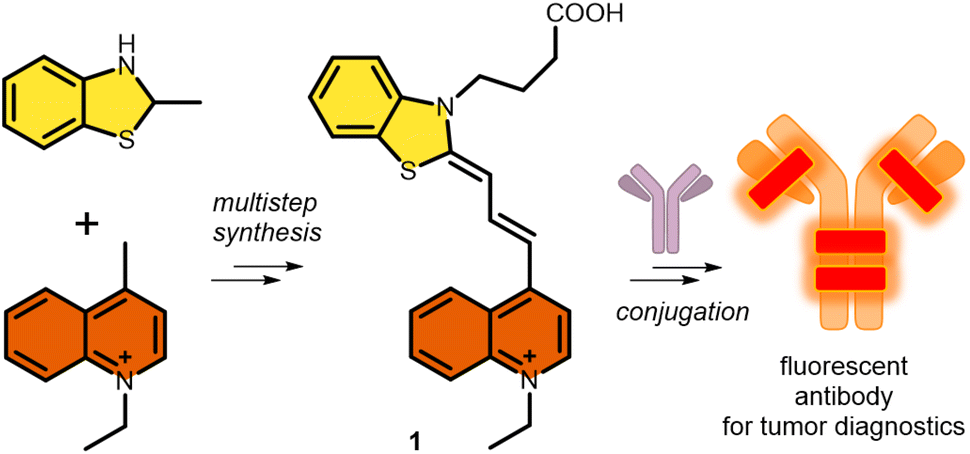 | ||
| Scheme 1 Synthetic strategy and proposed application of the cyanine (1) synthesized in this work. | ||
Results and discussion
Synthesis of a new cyanine fluorophore
The new fluorescent dye containing the ((benzo[d]thiazol-2(3H)-ylidene)prop-1-en-1-yl)quinolin-1-ium moiety was synthesized using a 5-step method. First, the N-alkylation of 2-methyl benzothioimidazole (2) was carried out using ethyl 4-bromobutyrate (3) in the presence of potassium iodide. This derivative 4 was reacted with N,N′-diphenylformimide (5), and the intermediate was subsequently reacted with N-methyl quinolone (6). Finally, deprotection using LiOH resulted in the free acid 1 (Scheme 2).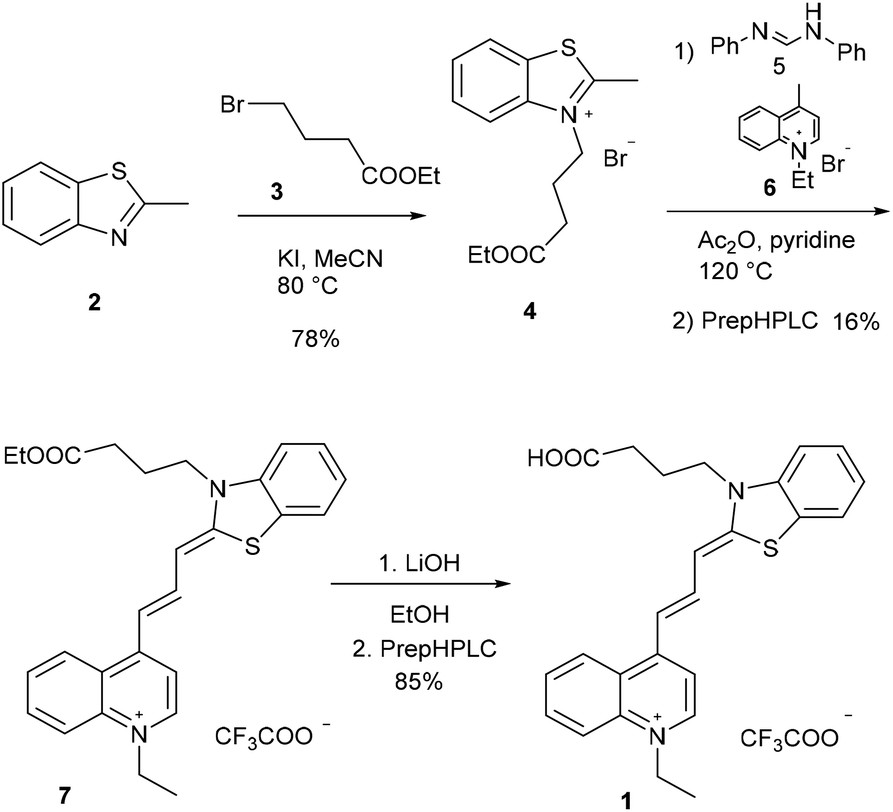 | ||
| Scheme 2 Synthetic route for cyanine 1. Compounds 1 and 7 were purified by preparative HPLC (prepHPLC) resulting in the formation of the products as TFA salts. | ||
The click reaction is applied widely in biomolecule labelling; therefore we aimed to synthesize a clickable derivative from the acid 1. At first, the activated45 NHS ester of 1 was prepared with N,N,N′,N′-tetramethyl-O-(N-succinimidyl)uronium tetrafluoroborate (TSTU), followed by the smooth acylation of 3-azidopropane-1-amine (8), resulting in 9 (Scheme 3).
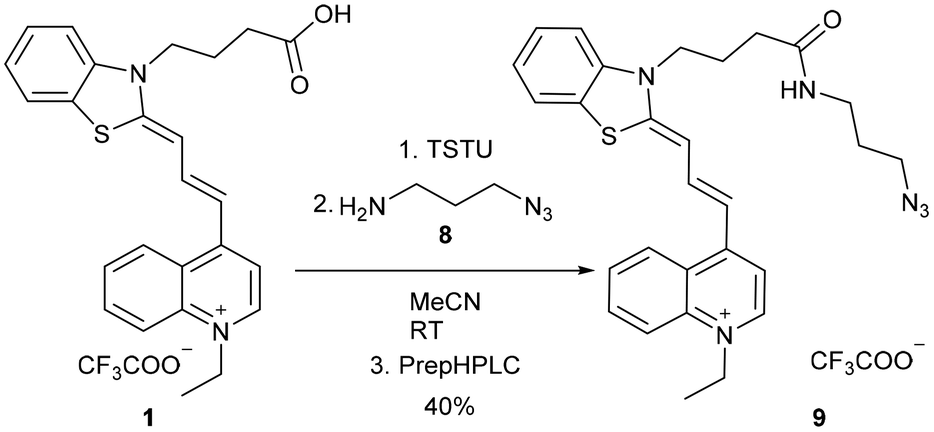 | ||
| Scheme 3 Transformation of acid 1 using a simple and fast method to NHS active ester followed by the introduction of the azide functional group. Compound 9 was purified by preparative HPLC (prepHPLC) resulting in the formation of the desired product as a TFA salt. TSTU: N,N,N′,N′-tetramethyl-O-(N-succinimidyl)uronium tetrafluoroborate. | ||
Photophysical characterization of the new fluorophore
The spectroscopic properties of the novel cyanine with the azide function (9) were investigated in detail (Fig. S15†). The wavelength of its excitation maximum (λmaxexc) is 627 nm and its molar absorption coefficient is 36816 M−1 cm−1. Furthermore, its quantum yield (0.3%) and brightness (110 M−1 cm−1) were also determined. Beneficially, the Stokes shift (26 nm) is larger than those of the most widely used commercially available cyanines. Its brightness is moderate, but this is a common phenomenon in the case of dyes that emit red light. Moreover, these spectroscopic properties generally improve9 when the rotation of the groups is restricted, which occurs when these compounds are embedded in the plasma membrane or intercalated on an antibody or between DNA strands.
Finally, to demonstrate the suitability of cyanine 9 for use in biological applications, we first investigated its photostability in HEPES buffer by continuous excitation using a 620 nm LED light source (6.1 W and 2.6 W). The original fluorescence intensity decreased to 50% after 10 minutes of irradiation at 2.6 W, and after 3 minutes of irradiation at 6.1 W, as shown in Fig. S20.† Thereafter, the bleaching rates are acceptable considering that imaging processes usually require fixed excitation of two minutes or less. Also, this emission decreasing rate is similar to that of the widely used Cy5® dye of the solvent on the photophysical properties of 9 was investigated. The absorbance in apolar solvents (such as THF, EtOAc, and DCM) has a bathochromic shift from 627 to 667 nm (Fig. S21A†).
This red-shifting can also be observed in the excitation and emission spectra. The emission intensity maximum is higher in apolar solvents. This indicates that if the dye approaches the apolar regions of the cells, it might have an increased emission wavelength (Fig. S21B†).
The effect of changes in pH in the range of 4.1 to 11.2 was not significant. This can be explained by the presence of basic nitrogen atoms with different pKa values. The varying protonation states affect the push and pull effect of the electrons within the structure, which in turn affects the spectroscopic properties of the compounds46 (Fig. S22†).
Theoretical explanation of the photochemical properties
The photochemistry of the simplified structure of 1, which only includes a methyl group on the nitrogen of the benzothiazole moiety in the cyanine dye (Fig. 2, 1* in ESI†), was studied by quantum chemical computations [B3LYP/6-31G(d,p)//PCM(w)]. The ground state A(S0) exhibits an extended geometry in a 2-D plane, capped by the two aromatic rings from both sides. In this work, we examined how the aromatic rings and alkene groups are influenced upon excitation. In order to observe these details, we applied the concepts of aromaticity and olefinicity, developed previously.47,48 Aromaticity (%) measures the degree of aromatization in the compound studied on a linear percentage scale, making benzene the perfect (100%) aromatic and cyclobutadiene the perfect antiaromatic (−100%) reference compounds. Analogously, olefinicity also measures the degree of conjugation of the alkene bond on a linear scale, defining ethylene as the non-conjugated (0%) and the allyl anion as the fully conjugated reference compounds. Both the cationic quinoline and cationic benzothiazole represent the standard aromaticities (%). These aromaticity degrees were studied and supported by nucleus independent chemical shift (NICS) values,49 as shown in Fig. 2. The calculated aromaticity (%) and NICS values correlate well. The structure includes two double bonds; however, their positions alter due to the resonance structures. The olefinicity values (%) were also calculated for all four potential double bonds. All of these values (∼100%) indicate a quite strong conjugation and also show near equivalent contributions from the resonance forms (Fig. 2).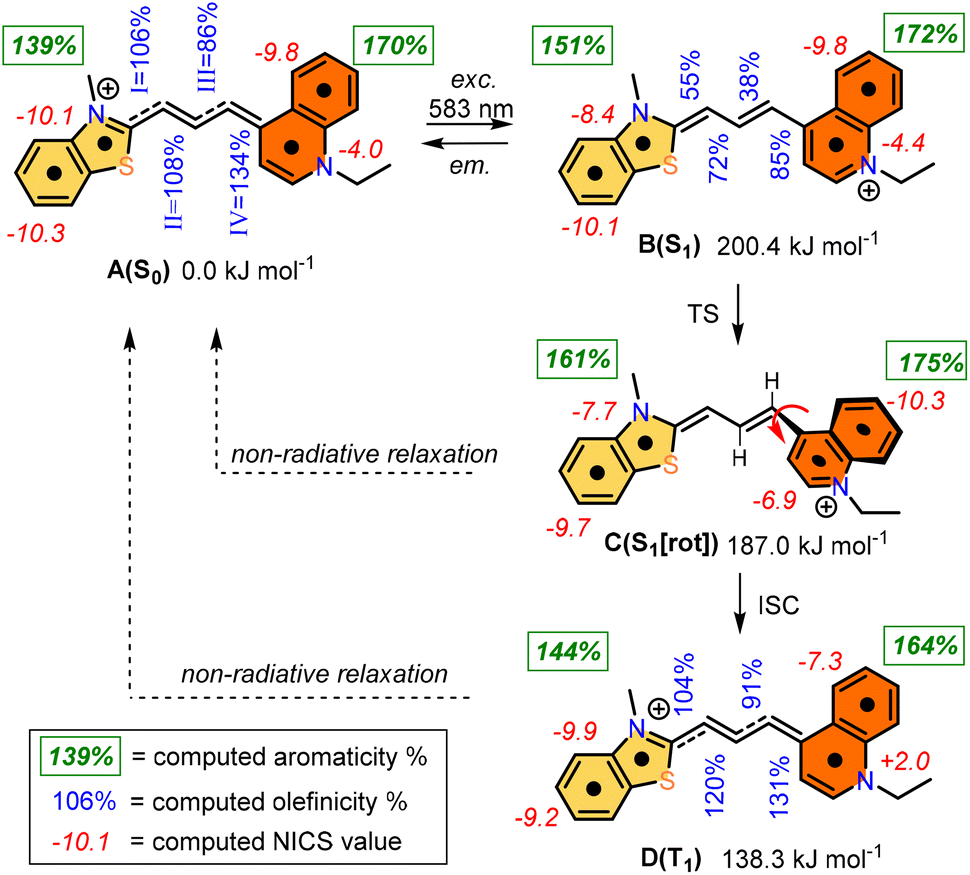 | ||
| Fig. 2 Computed photochemical processes of the cyanine dye from the A(S0) state to the B(S1), C(S1), and then D(T1) states. Green numbers refer to the aromaticity (%) for the bicyclic aromatic rings, blue numbers refer to the olefinicity values of the four potential olefinic bonds (I, II, III, and IV), while the red numbers refer to the NICS values (in ppm). | ||
The whole photochemical process was calculated, including the ground (S0), excited (S1) and triplet states (T1), using the dihedral rotation around the olefinic bond IV as the reaction coordinate. Upon photoexcitation at 583 nm, the molecule in the A(S0) state undergoes an electronic transition to B(S1) (+200.4 kJ mol−1; red curve, Fig. 3), where the aromaticity of the benzothiazole rings increased from 139% to 151%, while the corresponding values of quinoline remained constant (see Fig. 2).
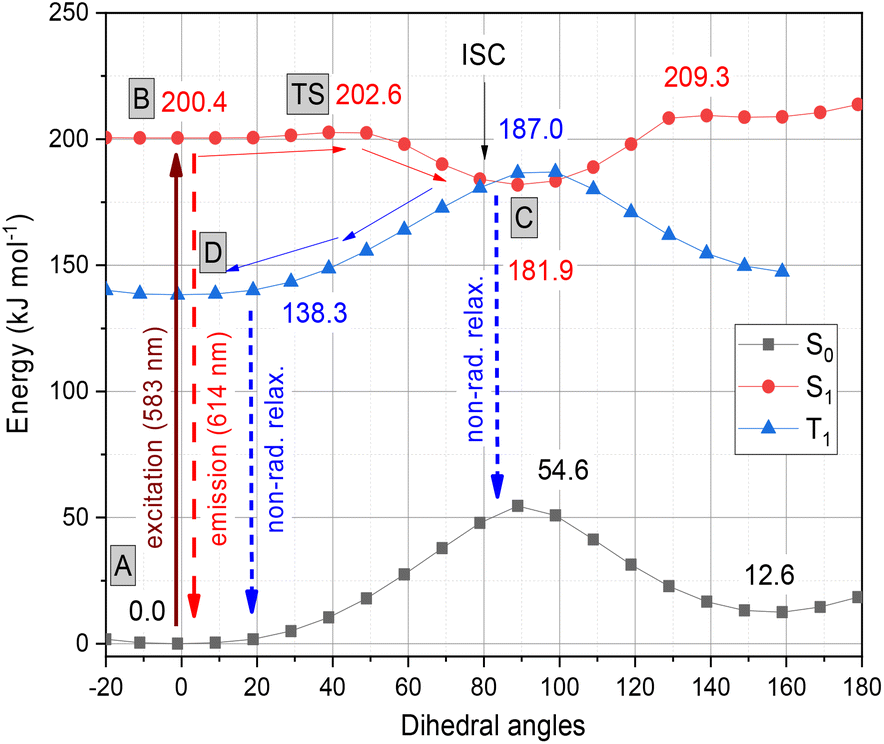 | ||
| Fig. 3 Graphical representation of the profiles calculated for the ground (S0, black), excited (S1, red) and triplet states (T1, blue) along the dihedral rotation of the double bond IV. | ||
The high transition moment from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) (f = 1.689; ε = 65550 mol, as shown in Fig. 4) indicates a strong absorption band, and the calculated fluorescence parameters are also significant (f = 1.621; ε = 62300 mol), in contrast to the low intensity observed in experiments.
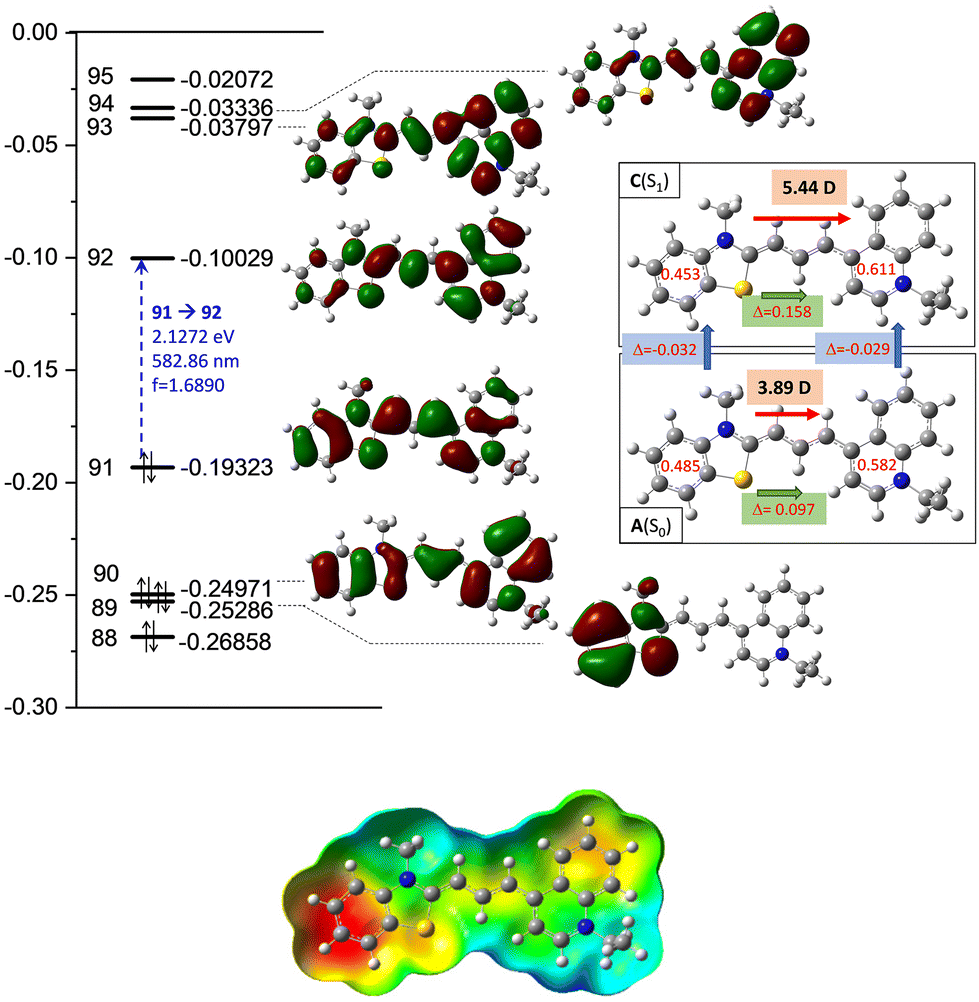 | ||
| Fig. 4 Top: detailed data of the computed molecular orbitals (MOs) and the corresponding orbital energies in Hartree of the cyanine dye. The two–two red numbers in the box attached to the ground [A(S0)] and excited state [C(S1)] structures illustrate the group charges of the aromatic rings. The green arrows show the differences in the charges of the two sites. The red arrows illustrate the dipole moment of the two states. Bottom: electrostatic potential mapped on the total density surface (isoval = 0.0004). | ||
Meanwhile, the olefinicity values in the B(S1) state decreased, showing a decrease in conjugation (Fig. 2). In the S1 state, the dihedral angle of the olefin bond IV (Fig. 2) rotated by ca. 90°, which led to the energy drop (−18.5 kJ mol−1) in C(S1) via a low-lying transition state (TS; +2.4 kJ mol−1) increasing the stability of the structure. Here, the increased stability can be attributed to the increased aromaticities of the two aromatic rings. The uncorrected energy (ΔE, kJ mol−1) curve of the olefin dihedral angle IV in the triplet state (T1; blue curve, Fig. 3) shows a maximum around 90° (187.0 kJ mol−1) and a minimum at 0° as D(T1). The excited singlet and triplet curves intersect at ∼80°, possibility enabling an inter-system crossing (ISC) from C(S1) to D(T1), resulting in non-radiative relaxation. This high quenching probability may manifest as the significant decrease in the fluorescence intensity observed in our experiments herein. Fig. 4 summarizes the molecular background of the excitation process. The molecular orbitals (MOs), computed at the same level of theory, and the corresponding orbital energies in Hartrees of the cyanine dye show that the transition is purely limited to the HOMO→LUMO transition. The group charges (sum of the atomic charges of a functional group) of the two aromatic rings attached to the ground [A(S0)] and excited state [C(S1)] structures (two–two red numbers in the box in Fig. 4) illustrate that the benzoxazole ring transfers electron density toward the quinoline ring by a value of 0.032. The green arrows show the differences in the charges of the two sites, which shifted at the excited states. The blue arrows show the evolution of the group charge distributions upon electronic excitation by photons. The red arrows illustrate the dipole moment of the two states. The electrostatic potential surface in Fig. 4 (bottom) also confirms that the quinoline group possesses the larger positive value, in contrast to that of the benzothiazole ring. This asymmetric distribution of the positive charge is responsible for the increased fluorescence intensity.
Antibody conjugation and microscopy imaging
After obtaining all these promising synthetic and spectroscopic results, with the biorthogonal azide 9, we attempted the conjugation of this derivative by an azide–alkyne click reaction using the alkyne-substituted trastuzumab antibody 11, named FCY. Given that antibodies can target specific cancer cells and deliver their fluorescent4 or cytotoxic payloads50 with high specificity, we have conjugated 9 to the human IgG trastuzumab, which has four cyclooctinyl harbors (10, Scheme 4) via a procedure described elsewhere.51 This results in the potential diagnostic tracer FCY (11) for imaging Her2+ tumor cells.52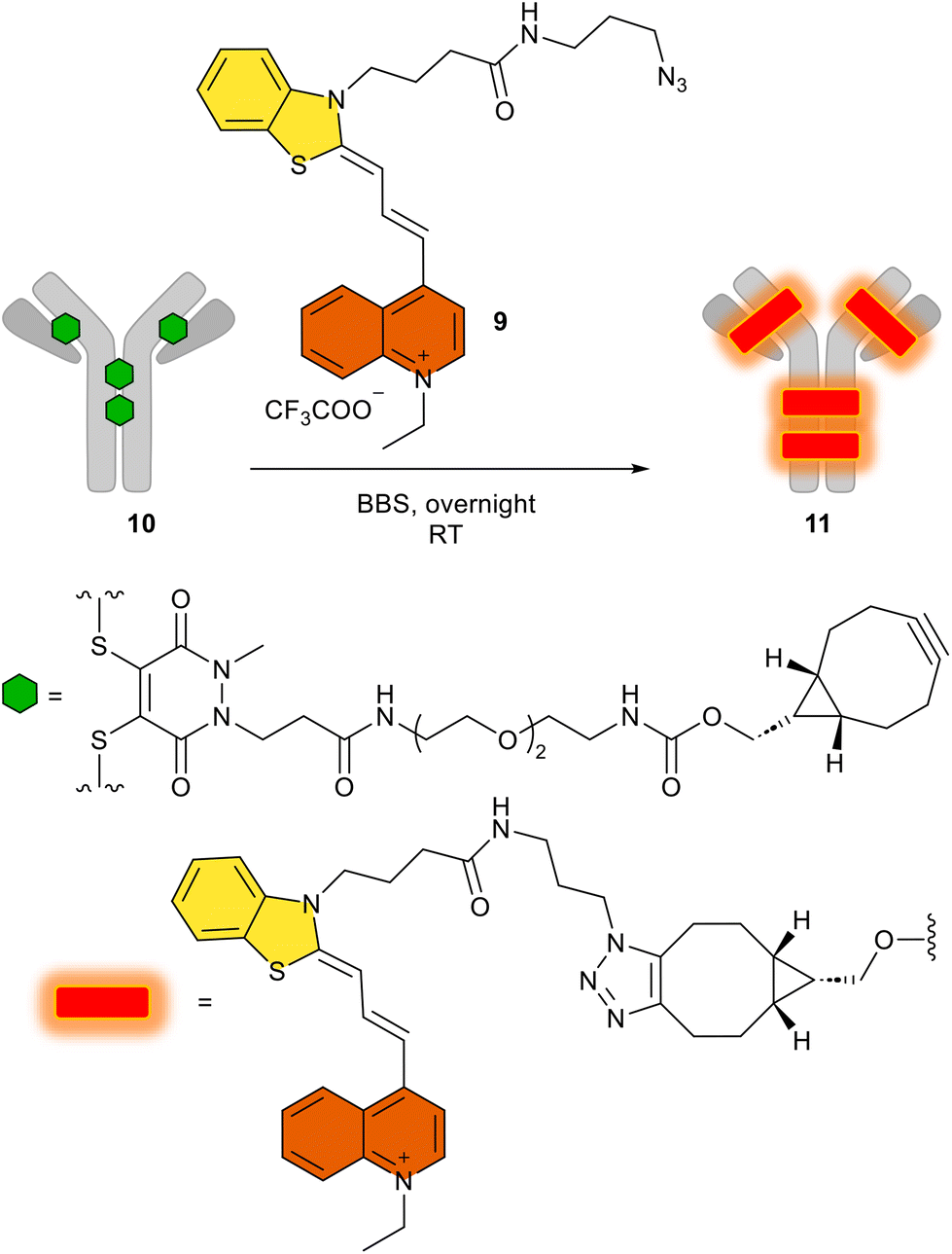 | ||
| Scheme 4 The use of 9 in the click reaction to produce the antibody–fluorophore conjugate FCY (11). BBS: borate buffered saline (pH 8.2). | ||
We then added the azido dye 9 in a copper-free click reaction generally used in biorthogonal chemistry, and the appropriate antibody conjugate FCY (11) was prepared by following a method described in detail.51 After the click reaction, excess dye was removed by buffer-exchange and the fluorophore-to-antibody ratio was determined spectroscopically using the absorbance. Using the Lambert–Beer equation, the ideal fluorophore-to-antibody ratio (FAR = 4) was confirmed (Table S2†). The homogeneity of the conjugate was shown to be 95%, as determined by SDS-PAGE, even after the click reaction (Fig. S23† IV). While keeping the SDS gel under UV light (366 nm), the fluorescent spot of the antibody–fluorophore conjugate could be seen with the naked eye (Fig. S23† V).
With the antibody–fluorophore conjugate FCY (11) in hand, we first examined the selectivity of the conjugates using flow cytometry on living cells.
We treated both NCI-N87 cells overexpressing the Her2 receptor in the membrane and Her2-negative MCF7 cells with the conjugate FCY (11). The unchanged selectivity observed by FACS indicated that the conjugates have the potential to be useful in imaging processes on living cells (Fig. S24†). Second, cell sections from the same but fixed cell lines were treated with the antibody–fluorophore conjugate 11 (Fig. 5). Confocal microscopy showed no membrane labelling for Her2-negative cells (Fig. 5G), while in the case of the Her2+ cell line, the membrane labelling was significant (red, Fig. 5C), comparable to the DAPI signal (blue, Fig. 5A). This also confirms the receptor selectivity of the modified antibody. The GFP expressed by the cells indicates the size of the whole cell and the green-labelled cells clearly separate from the red labelled cells. This phenomenon further demonstrates that the fluorescent antibody labelling can only be detected on the cell's surface (Fig. 5B).
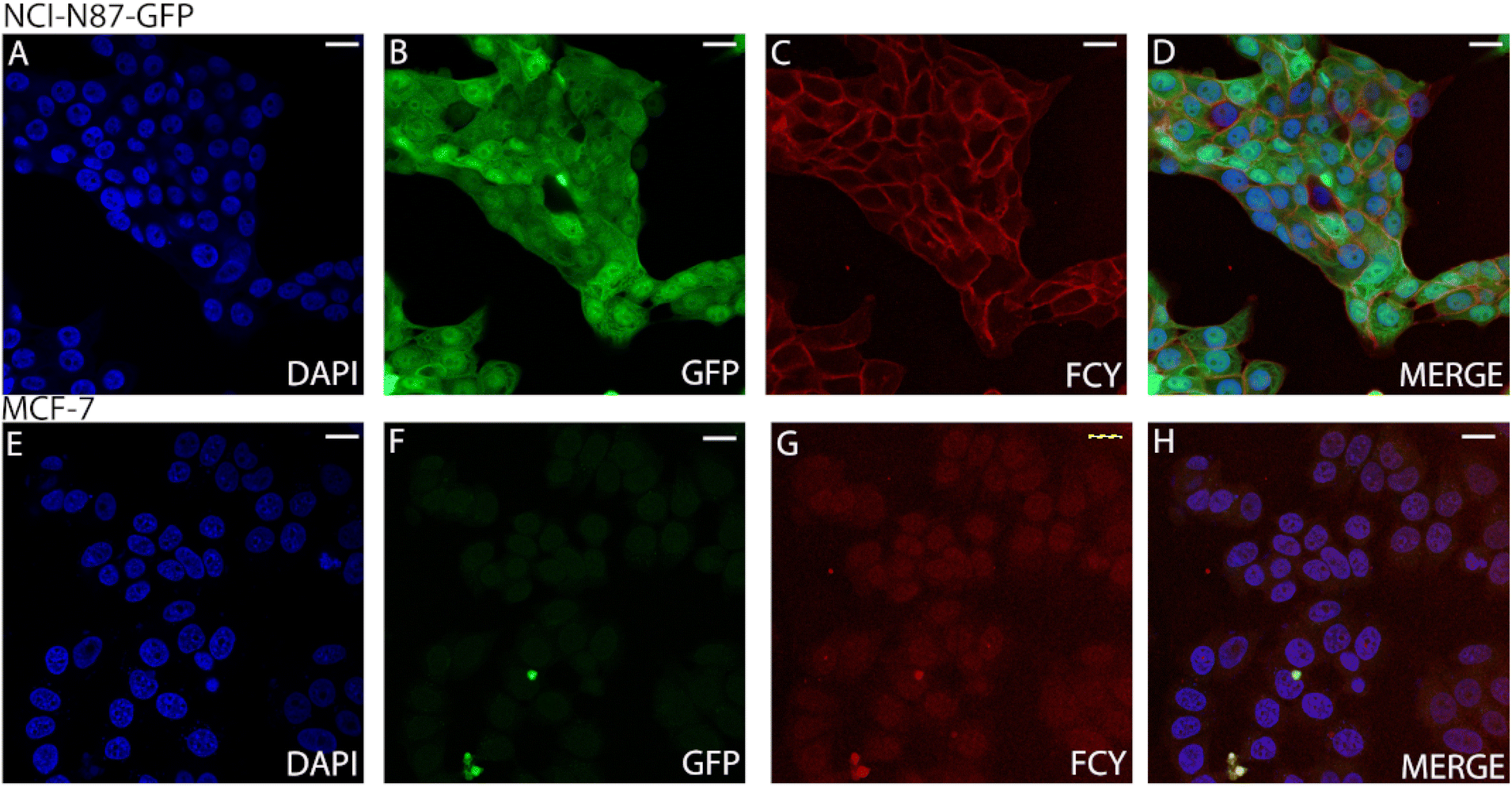 | ||
| Fig. 5 Confocal microscopy images (scale bars: 10 μm) of NCI-N87 (Her2+) (A–D) and MCF-7 (Her2−) (E–H) cells treated with the fluorophore–antibody conjugate FCY (11). (A) DAPI cell nucleus labelling of NCI-N87 cells. (B) GFP expressed by the NCI-N87 cells. (C) 11 conjugate FCY labelling of NCI-N87 cells. (D) Merged image of DAPI, GFP and 11 conjugate labelling of NCI-N87 cells. (E) DAPI cell nucleus labelling of MCF-7 cells. (F) GFP channel image of MCF-7 cells. (G) 11 conjugate (FCY) labelling of MCF-7 cells. (H) Merged image of DAPI and 11 conjugate labelling of MCF-7 cells. Excitation: 405 nm; emission: 456 nm (A, D, E and H). Excitation: 488 nm; emission 539 nm (B, D, F and H). Excitation: 633 nm; emission: 646 nm (C, D, G and H). | ||
Experimental
For experimental procedures and compound characterization (Fig. S1–S24†), computational methods and theoretical data (Tables S2–S7), see the ESI.†Conclusions
In conclusion, we have synthesized an asymmetric cyanine fluorophore with versatile conjugation points to attach to the selected antibody. Theoretical and spectroscopic studies confirmed its improved fluorescence properties compared to Cy5®, especially in terms of its optimal absorption wavelength and increased fluorescence intensity as well as larger Stokes shift. Furthermore, the acidic compound 1 can be easily modified with several reactive groups for any further goals and thus can be used in any type of biorthogonal click reaction or amide formation reaction. The dye exhibited a precise emission wavelength (λmaxem = 627 nm) with the widely used 633 nm laser line, excellent brightness, good applicability in confocal and other fluorescence microscopy techniques and appropriate photostability for imaging in aqueous systems. We explored and predicted the spectrophysical characteristics together with the potential energy surface of the ground, excited and triplet states. We concluded that the most plausible quenching mechanism of this cyanine derivative is the twisted intramolecular charge transfer (TICT). This revealed that the unrestricted rotation around the double bond in the linker chain localizes the electrons, which ‘turns off’ the dye and results in fluorescence quenching. The developed cyanine dye was subsequently equipped with an azide functional group and linked to the cyclooctyne-derived trastuzumab, prepared parallelly. Finally, this fluorophore–antibody conjugate was proven to be efficient in the selective labelling of Her2+ cells instead of Her2-negative cells in deep red, as shown by the confocal microscopy images of trastuzumab-targeted tumour cells.Author contributions
The synthesis of fluorescent dyes was carried out by EK and BK. The spectroscopic characterization was made by DSzK, GT and EK. The conjugation was prepared by DSzK. Theoretical calculations and data curation were accomplished by ZM and ÖF. Biological experiments were designed by BGy and carried out by DM and BC, and confocal microscopy was performed by EZsT and LW. Acquisition of reagents and resources for biological measurements was carried out by BGy, BR and KG. Flow cytometry was performed by GyV. Validation was accomplished by PAB, EK, ZM and GyMK. GyMK and RJB were responsible for resources; funding acquisition was accomplished by GyMK, GK, RJB and ZM and project administration was implemented by EK. EK, ZM and GyMK supervised the project. The visualization was carried out by EK, DSzK and MZ. The manuscript was written through contributions of EK, DSzK, MCO, PAB and MZ. All authors have given approval to the final version of the manuscript. DSzK and EK contributed equally.Conflicts of interest
GK and RJB are founders of Femtonics and members of its scientific advisory board. The other authors declare that no conflicts of interest exist.Acknowledgements
This research was supported by the 2018-1.3.1-VKE-2018-00032, KFI-18-2018-00097, TKP2021-EGA-42, TKP2021-NVA-15, 2020-1.1.5-GYORSÍTÓSÁV-2021-00004, 2020-2.1.1-ED-2021-00190, 2020-2.1.1-ED-2022-00208 and TKP2021-NVA-14 grants of the National Office of Science, Innovation and Technology (NKFIH). We are grateful for the support from the János Bolyai Research Scholarship (BO/799/21/7, ÚNKP-22-ME3). The authors acknowledge the supportive work of Krisztina Németh. On behalf of the Development and mechanistic study of DNA dyes (PI: Dr Ervin Kovács, ELKH Research Centre for Natural Sciences) project we are grateful for the possibility to use ELKH Cloud,53 which helped us achieve the results published in this paper.References
- Y. Li, Y. Zhou, X. Yue and Z. Dai, Adv. Healthcare Mater., 2020, 9, 1–24 Search PubMed.
- A. P. Gorka, R. R. Nani and M. J. Schnermann, Org. Biomol. Chem., 2015, 13, 7584–7598 RSC.
- L. Feng, W. Chen, X. Ma, S. H. Liu and J. Yin, Org. Biomol. Chem., 2020, 18, 9385–9397 RSC.
- J. M. Warram, E. de Boer, A. G. Sorace, T. K. Chung, H. Kim, R. G. Pleijhuis, G. M. van Dam and E. L. Rosenthal, Cancer Metastasis Rev., 2014, 33, 809–822 CrossRef CAS PubMed.
- R. Pal, H. Kang, H. S. Choi and A. T. N. Kumar, Clin. Cancer Res., 2019, 25, 6653–6661 CrossRef PubMed.
- C. P. Toseland, J. Chem. Biol., 2013, 6, 85–95 CrossRef PubMed.
- R. A. Hansen, A. Märcher and K. V. Gothelf, Bioconjugate Chem., 2022, 33, 1811–1817 CrossRef CAS PubMed.
- M. S. Chan, D. Xu, L. Guo, D. Y. Tam, L. S. Liu, Y. Chen, M. S. Wong and P. K. Lo, Org. Biomol. Chem., 2015, 13, 7307–7312 RSC.
- Y. V. Suseela, N. Narayanaswamy, S. Pratihar and T. Govindaraju, Chem. Soc. Rev., 2018, 47, 1098–1131 RSC.
- H.-Y. Peng, G. Zhang, Y.-J. Xu, R. Sun and J.-F. Ge, Org. Biomol. Chem., 2022, 20, 5558–5565 RSC.
- P. R. Bohländer and H.-A. Wagenknecht, Org. Biomol. Chem., 2013, 11, 7458–7462 RSC.
- S. Prokop, P. Ábrányi-Balogh, B. Barti, M. Vámosi, M. Zöldi, L. Barna, G. M. Urbán, A. D. Tóth, B. Dudok, A. Egyed, H. Deng, G. M. Leggio, L. Hunyady, M. van der Stelt, G. M. Keserű and I. Katona, Nat. Commun., 2021, 12, 6505 CrossRef PubMed.
- K. Ilina and M. Henary, Chem. – Eur. J., 2021, 27, 4230–4248 CrossRef CAS PubMed.
- J. Lin, K. Yang and E. J. New, Org. Biomol. Chem., 2021, 19, 9339–9357 RSC.
- C. S. Abeywickrama, H. J. Baumann, N. Alexander, L. P. Shriver, M. Konopka and Y. Pang, Org. Biomol. Chem., 2018, 16, 3382–3388 RSC.
- A. R. Nödling, E. M. Mills, X. Li, D. Cardella, E. J. Sayers, S.-H. Wu, A. T. Jones, L. Y. P. Luk and Y.-H. Tsai, Chem. Commun., 2020, 56, 4672–4675 RSC.
- Z. Yang, L. Li, J. Ling, T. Liu, X. Huang, Y. Ying, Y. Zhao, Y. Zhao, K. Lei, L. Chen and Z. Chen, Chem. Sci., 2020, 11, 8506–8516 RSC.
- N. Lange, W. Szlasa, J. Saczko and A. Chwiłkowska, Pharmaceutics, 2021, 13, 1–17 CrossRef PubMed.
- J. Atchison, S. Kamila, H. Nesbitt, K. A. Logan, D. M. Nicholas, C. Fowley, J. Davis, B. Callan, A. P. McHale and J. F. Callan, Chem. Commun., 2017, 53, 2009–2012 RSC.
- L. N. Schulte, B. Heinrich, H. Janga, B. T. Schmeck and O. Vázquez, Angew. Chem., Int. Ed., 2018, 57, 11564–11568 CrossRef CAS PubMed.
- X. Peng, T. Wu, J. Fan, J. Wang, S. Zhang, F. Song and S. Sun, Angew. Chem., Int. Ed., 2011, 50, 4180–4183 CrossRef CAS PubMed.
- L. A. Ernst, R. K. Gupta, R. B. Mujumdar and A. S. Waggoner, Cytometry, 1989, 10, 3–10 CrossRef CAS PubMed.
- S. Luo, E. Zhang, Y. Su, T. Cheng and C. Shi, Biomaterials, 2011, 32, 7127–7138 CrossRef CAS PubMed.
- A. Bera, D. Bagchi and S. K. Pal, J. Phys. Chem. A, 2019, 123, 7550–7557 CrossRef CAS PubMed.
- C. Gebhardt, M. Lehmann, M. M. Reif, M. Zacharias and T. Cordes, ChemPhysChem, 2020, 22, 1566–1583 CrossRef PubMed.
- H. Kim, H. S. Choi, S.-K. Kim, B. Il Lee and Y. Choi, Theranostics, 2017, 7, 952–961 CrossRef CAS PubMed.
- M. M. Deken, D. L. Bos, W. S. F. J. Tummers, T. L. March, C. J. H. van de Velde, M. Rijpkema and A. L. Vahrmeijer, EJNMMI Res., 2019, 9, 98 CrossRef CAS PubMed.
- E. R. Thapaliya, S. M. Usama, N. L. Patel, Y. Feng, J. D. Kalen, B. St. Croix and M. J. Schnermann, Bioconjugate Chem., 2022, 33, 718–725 CrossRef CAS PubMed.
- B. Xu, B. Zhang and Y. Kuang, US Pat., US20090023129A1, 2009 Search PubMed.
- E. A. te Velde, T. Veerman, V. Subramaniam and T. Ruers, Eur. J. Surg. Oncol., 2010, 36, 6–15 CrossRef CAS PubMed.
- D. Kobzev, C. Prasad, D. Walunj, H. Gotman, O. Semenova, A. Bazylevich, L. Patsenker and G. Gellerman, Eur. J. Med. Chem., 2023, 252, 115298 CrossRef CAS PubMed.
- K. Jeong, S. H. Kong, S. W. Bae, C. R. Park, F. Berlth, J. H. Shin, Y. S. Lee, H. Youn, E. Koo, Y. S. Suh, D. J. Park, H. J. Lee and H. K. Yang, J. Gastric Cancer, 2021, 21, 191–202 CrossRef PubMed.
- L. Sampath, S. Kwon, S. Ke, W. Wang, R. Schiff, M. E. Mawad and E. M. Sevick-Muraca, J. Nucl. Med., 2007, 48, 1501–1510 CrossRef CAS PubMed.
- D. Kobzev, C. Prasad, D. Walunj, H. Gotman, O. Semenova, A. Bazylevich, L. Patsenker and G. Gellerman, Eur. J. Med. Chem., 2023, 252, 115298 CrossRef CAS PubMed.
- K. Sano, M. Mitsunaga, T. Nakajima, P. L. Choyke and H. Kobayashi, Breast Cancer Res., 2012, 14, R61 CrossRef CAS PubMed.
- D. Sóvári, A. Kormos, O. Demeter, A. Dancsó, G. M. Keserű, M. Milen and P. Ábrányi-Balogh, RSC Adv., 2018, 8, 38598–38605 RSC.
- D. Szepesi Kovács, I. Hajdu, G. Mészáros, L. Wittner, D. Meszéna, E. Z. Tóth, Z. Hegedűs, I. Ranđelović, J. Tóvári, T. Szabó, B. Szilágyi, M. Milen, G. M. Keserű and P. Ábrányi-Balogh, RSC Adv., 2021, 11, 12802–12807 RSC.
- A. Jancsó, E. Kovács, L. Cseri, B. J. Rózsa, G. Galbács, I. G. Csizmadia and Z. Mucsi, Spectrochim. Acta, Part A, 2019, 218, 161–170 CrossRef PubMed.
- E. Kovács, L. Cseri, A. Jancsó, F. Terényi, A. Fülöp, B. Rózsa, G. Galbács and Z. Mucsi, Eur. J. Org. Chem., 2021, 2021, 5649–5660 CrossRef.
- A. Csomos, B. Kontra, A. Jancsó, G. Galbács, R. Deme, Z. Kele, B. J. Rózsa, E. Kovács and Z. Mucsi, Eur. J. Org. Chem., 2021, 2021, 5248–5261 CrossRef CAS.
- B. Chiovini, D. Pálfi, M. Majoros, G. Juhász, G. Szalay, G. Katona, M. Szőri, O. Frigyesi, C. Lukácsné Haveland, G. Szabó, F. Erdélyi, Z. Máté, Z. Szadai, M. Madarász, M. Dékány, I. G. Csizmadia, E. Kovács, B. Rózsa and Z. Mucsi, ACS Omega, 2021, 6, 15029–15045 CrossRef CAS PubMed.
- A. Csomos, E. Kovács, M. Madarász, F. Z. Fedor, A. Fülöp, G. Katona, B. Rózsa and Z. Mucsi, Sens. Actuators, B, 2024, 398, 134753 CrossRef CAS.
- L. Petri, P. A. Szijj, Á. Kelemen, T. Imre, Á. Gömöry, M. T. W. Lee, K. Hegedűs, P. Ábrányi-Balogh, V. Chudasama and G. M. Keserű, RSC Adv., 2020, 10, 14928–14936 RSC.
- D. Szepesi Kovács, B. Chiovini, D. Müller, E. Z. Tóth, A. Fülöp, P. Ábrányi-Balogh, L. Wittner, G. Várady, Ö. Farkas, G. Turczel, G. Katona, B. Győrffy, G. M. Keserű, Z. Mucsi, B. J. Rózsa and E. Kovács, ACS Omega, 2023, 8, 22836–22843 CrossRef PubMed.
- E. Kovács, B. Rózsa, A. Csomos, I. G. Csizmadia and Z. Mucsi, Molecules, 2018, 23, 2859 CrossRef PubMed.
- M. Nagy, D. Rácz, Z. L. Nagy, P. P. Fehér, S. L. Kovács, C. Bankó, Z. Bacsó, A. Kiss, M. Zsuga and S. Kéki, Sci. Rep., 2019, 9, 8250 CrossRef PubMed.
- Z. Mucsi, B. Viskolcz and I. G. Csizmadia, J. Phys. Chem. A, 2007, 111, 1123–1132 CrossRef CAS PubMed.
- Z. Mucsi, G. A. Chass, B. Viskolcz and I. G. Csizmadia, J. Phys. Chem. A, 2009, 113, 7953–7962 CrossRef CAS PubMed.
- P. V. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- P. Khongorzul, C. J. Ling, F. U. Khan, A. U. Ihsan and J. Zhang, Mol. Cancer Res., 2020, 18, 3–19 CrossRef CAS PubMed.
- C. Bahou, D. A. Richards, A. Maruani, E. A. Love, F. Javaid, S. Caddick, J. R. Baker and V. Chudasama, Org. Biomol. Chem., 2018, 16, 1359–1366 RSC.
- T. A. D. Smith, Br. J. Radiol., 2010, 83, 638–644 CrossRef CAS PubMed.
- M. Héder, E. Rigó, D. Medgyesi, R. Lovas, S. Tenczer, F. Török, A. Farkas, M. Emődi, J. Kadlecsik, G. Mező, Á. Pintér and P. Kacsuk, InfTars, 2022, 22, 128 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ob01471a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |