
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent progress and applications enabled via electrochemically triggered and controlled chain-growth polymerizations
Boyu
Zhao
and
Paul
Wilson
 *
*
University of Warwick, Department of Chemistry, Coventry, CV4 7AL, UK. E-mail: p.wilson.1@warwick.ac.uk
First published on 4th April 2023
Abstract
The use of external stimuli to trigger and/or control polymer synthesis has become a prominent research theme over the last 20 years. With the need for more sustainable methods of polymer synthesis and production, interest in this area has intensified in the last decade. External stimuli include, but are not limited to; heat, light, ultrasound and electricity. Relative to their chemically mediated counterparts, these methods of stimulus are non-invasive, atom economic and offer excellent spatial and temporal control over reaction progress. In particular, the use of an electric field to deliver electrons as reagents in electrosynthesis is considered to be an emerging green and sustainable solution in organic and polymer synthesis. Through modulating the applied potential/current precise control over the kinetics and thermodynamics of electron-transfer reactions can be achieved, which can enhance the efficiency and selectivity of targeted chemical transformations. In this review, we have captured the recent progress made in electrochemically triggered and controlled polymer synthesis techniques, focusing on chain-growth polymerizations including radical, ionic and ring-opening polymerizations and applications enabled by the use of an electric field to perform such reactions.
 Boyu Zhao | Boyu Zhao is a Ph.D. candidate in the Chemistry Department at the University of Warwick. He completed his undergraduate studies in the Physics Department at Southeast University (China), focusing on applied physics (graduating in 2017). He then obtained a master's degree (MSc) from the Chemistry Department at the University of Warwick in 2019. He is currently a fourth-year doctoral student in the Chemistry Department at the University of Warwick. His primary research is focused on eATRP and attempting to translate these reactions down to nanoscale reactors. |
 Paul Wilson | Paul Wilson is a Royal Society Tata University Research Fellow in the Department of Chemistry at the University of Warwick. He studied chemistry at the University of Bristol (MSci, 1st Class, 2006) before completing a PhD in organic chemistry (Warwick, 2010). He previously held a Leverhulme Trust Early Career Fellowship (Warwick, 2016–2019). His URF is focused on developing methods for nanoscale synthesis and nanofabrication, through combining the capabilities of scanning electrochemical probe microscopy techniques with electrochemically-mediated organic, macromolecular and supramolecular chemistry. More broadly, the team are developing platforms for (macro)molecular electrosynthesis/functionalization across a variety of length scales. |
Introduction
Over the last two decades, with the development of more sustainable synthetic and production procedures, (macro)molecular electrosynthesis has experienced a renaissance in academic research and industrial interest.1 This is due to the inherently ‘green’ nature of electrochemistry which increases the sustainability and reduces the carbon footprint of electrochemical processes/reactions relative to chemically driven processes. From an environmental point of view, electrochemistry is an enabling technology providing platforms for (i) sustainable generation of power/energy (solar, wind, water, etc.); (ii) valorization of waste streams; (iii) real-time monitoring of redox processes across a variety of length scales; (iv) synthesis of complex (macro)molecules and materials fabrication.2–6In the context of electrochemical synthesis and catalysis,7,8 the use of electrons as reagents for redox reactions has a number of benefits compared to stoichiometric chemical redox agents. Many of the chemical redox agents employed are toxic or form toxic by-products so replacing these with electrons minimises the need for intensive purification processes. Thus, the use of electrons either directly (at the electrode interfaces) or indirectly, via mediators, significantly improves atom economy. Moreover, fine control over the potential/current during electrolysis can provide kinetic, thermodynamic and spatial control over electron-transfer reactions which can enhance selectivity of chemical transformations.9 Indeed, the control conferred by electrolysis coupled with the relatively mild reaction conditions required, has been shown to enable selective functionalization of complex molecules that would be challenging to achieve with standard chemical redox agents.10 Electrosynthesis has also been shown to be compatible with so-called ‘green’ solvents including water (homogeneous and in dispersed phases),11,12 ionic liquids13 and deep eutectic solvents.14 The use of mediators can improve reaction and energy efficiency whilst waste streams are minimized by the ability to control the stoichiometry of desired reagents or reactive intermediates required to drive the reactions to completion. Finally, the in situ generation of reactive intermediates using electrochemistry can be coupled with real-time monitoring during electrolysis, using advanced electroanalytical methods, which plays an important role in understanding and improving the selectivity of reactions whilst also enhancing the overall safety of the reaction process as a whole.4
In chain-growth polymerizations, the generation of reactive intermediates (typically cations, anions, radicals) is crucial to initiate and control (for controlled/‘living’ polymerizations) the reactions. One strategy for improving synthesis and processes is to replace chemically and energy intensive (multiple steps) methods with milder processes that leave a reduced carbon footprint. One way to achieve this is to use external stimuli e.g. photo-, sono- and electrochemical intervention to replace chemical reagents with photons, ultrasound and electrons to mediate the generation of the required reactive intermediates and control the overall polymerization process.15
Herein, the use of electrochemistry to trigger and control chain-growth polymerizations is reviewed. Specifically, the focus will be on controlled/living polymerizations including reversible deactivation radical polymerization (RDRP), ionic polymerization and ring-opening polymerization. The electrochemical synthesis of conducting polymers proceeding via chain-growth polymerization is therefore outside the remit of this contribution and readers are directed elsewhere for information.16
Radical chain-growth polymerization
Free radical polymerization (FRP)
In recent decades, with the development of polymer synthesis technologies, a number of synthetic methods enabling precise control of molecular weight have been discovered.17 However, free radical polymerization (FRP) is still the most industrially relevant method, employed in the preparation low-density polyethylene,18 polystyrene,19 polyvinyl chloride,20 polymethyl methacrylate,21 polyacrylonitrile,22 polyvinyl acetate,23 styrene–butadiene rubber,24 nitrile rubber,25 neoprene,26etc. In conventional FRP, the reaction process can be divided into four main steps. Initiation and propagation steps result in polymer chain growth and a build-up in molecular weight whilst termination and chain transfer processes stop chain growth and in the latter case this leads to initiation of new polymer chains. The initiation step involves generation of radical intermediate and its first addition to a vinyl monomer. There are several approaches to radical generation including: thermolysis,27 photolysis,28 redox reactions,29 self-initiation of monomer30 and electrolysis.31 Typically, unstable molecules capable of undergoing homolysis to form radicals are employed as initiators. Common initiators that undergo thermal decomposition to form radicals are azo-compounds and peroxides, such as 2,2′-azobis(2-methylpropionitrile) AIBN,32 1,1′-azobis(1-cyclohexanecarbonitrile) ABCN33 and benzoyl peroxide34 that all contain relatively weak bonds that undergo homolysis when heated.The use of light to generate radicals has emerged as a powerful strategy for polymer synthesis and advanced materials fabrication.35 For conventional photo-initiated polymerizations, light sources are usually short wavelength high energy ultraviolet (UV) light (λ = 100–400 nm).36 Research tends to focus on trying to develop less energy intensive (long wavelength) reactions. Modern light sources are currently available for photo-initiated FRP, such as light-emitting diodes (LEDs).35
The most energy efficient initiation methods for generating radical intermediates are based on redox reactions.37 Compared with other methods, redox-controlled FRP is typically performed in the absence of external energy sources. For example, Fe2+ ions play an important role in the redox system in Fenton-type reactions. In this redox reaction, one electron is transferred from Fe2+ to a peroxide molecule leading to decomposition and the formation of a radical, Fe3+ and OH−.
These chemical redox reactions allude to the potential of achieving similar electron transfer processes using electrodes, negating the need for the addition of chemical oxidants or reductants. Indeed, electrochemistry has been employed in polymer synthesis and materials fabrication for decades.38 In electrochemically mediated FRP (eFRP), free radicals capable of initiating polymerization are generated at electrodes. Polymerization can then occur at the electrode surface or the radical can migrate away from electrode surface and initiate polymerization in the bulk solution. After electro-initiation, eFRP is subject to the usual elementary stages of FRP i.e. propagation for chain growth, chain transfer and termination, to obtain polymer. The setup of an eFRP reaction cell is similar to a typical electrosynthesis reaction cell (Fig. 1). The reaction cell mainly consists of 2 or 3 electrodes (working, counter, and reference electrodes) assembled in a divided or undivided cell.39 Subramanian's group used eFRP to polymerize acrylonitrile in aqueous solution, with insoluble polyacrylonitrile formed as coating on a cathode.38 Electro-initiated polymerization on electrodes is suitable for commonly used vinyl monomers including methyl methacrylate and acrolein.38
 | ||
| Fig. 1 Schematic depiction of common electrochemical cell configurations including divided (C and D) and undivided configurations (A and B) for; potentiostatic reactions (B and D) consisting of a working (red), counter (green) and reference (grey) electrode; galvanostatic reactions (A and C) consisting of a working (red), counter (green) electrodes. | ||
Generation of the reactive radical intermediates can require the application of an overpotential (η = Eapp − E1/2), where Eapp is the applied potential and E1/2 is the half-wave potential. The overpotential (η) describes the additional energy required to drive the reaction forward at an appreciable rate and can depend on the choice of electrode materials.40 Metals, as materials with high conductivity, are common choices for electrodes in electropolymerization. Commercial metals such as Pb,41 Sn,42 Pt,43 Bi,44 Fe,45 and Al46 are all suitable electrode materials.47 However, Subramanian's group first proposed to use graphite fiber as an electrode for electropolymerization.38 The working electrode (WE) usually needs better conductivity and a higher specific surface area compared to the counter electrode (CE). Reticulated vitreous carbon (RVC) with a high specific surface area and nickel foam are examples of electrode materials with good conductivity and high specific surface area. In addition to electrodes, electrochemical cells capable of mediating eFRP also require electrolytes, solvents, initiators, and monomers.
In 1949, Dineen et al. reported eFRP of acrylic acid47 highlighting the potential of eFRP. In 1960, Smith and coworkers exploited Kolbe electrolysis to generate radicals capable of initiating FRP of vinyl acetate, methyl methacrylate and vinyl chloride in aqueous electrolyte solutions.48 In the following years, MMA was used as a monomer to successfully obtain polymers in homogenous media,49 polar media,50 alcohol solutions,51 and aqueous sulfuric acid.52 In 1976, Pistoia's group successfully polymerized acrylonitrile in aqueous sulfuric acid53 whilst acrylamide was also employed as a monomer in eFRP.54 The above success proves that electro-initiated polymerization can be applied to the most commonly used polymer synthesis. The electro-initiated polymerization strategy has also been employed to prepare polymeric materials such as polymer networks,55 surface-attached responsive gel layers,56 and functionalized surfaces via electrografting.57
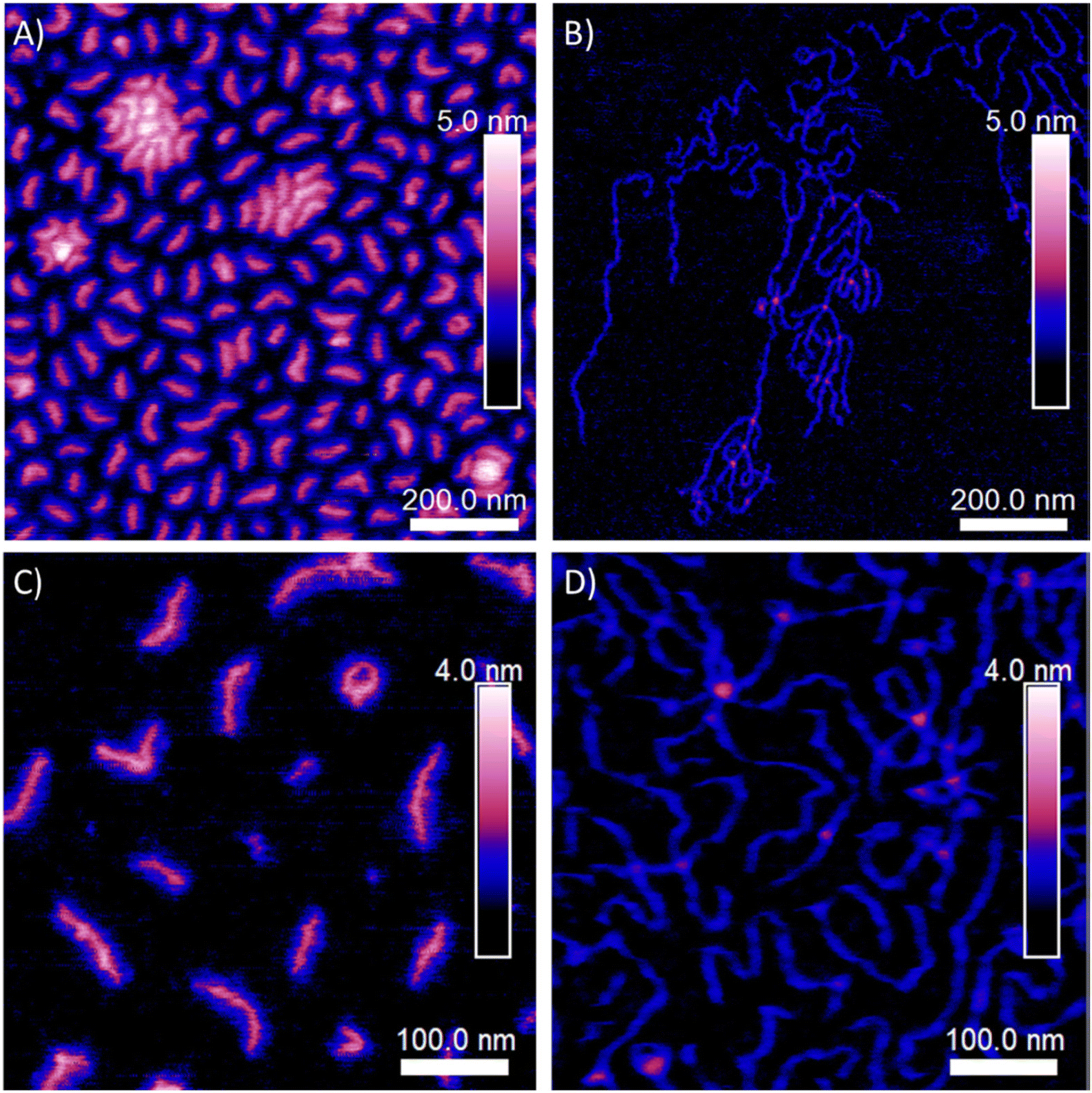
Wu's group reported eFRP of N-isopropylacrylamide (NIPAM) in the presence of a cross-linking agent (N,N′-methylenebisacrylamide) in a three-electrode system (Fig. 2).58 The generation of free radicals was electrochemically controlled leading to formation poly-NIPAM microgels. They found that the size of the microgels could be controlled by varying the applied potential. Moreover, the reaction electrodes were shown to be reusable which is attractive from a sustainability point of view.
 | ||
| Fig. 2 (A) Proposed reaction scheme for the eFRP synthesis of PNIPAM microgels. (B) DLS size distribution of PNIPAM microgels (■, □; in the presence of MBAAm in the synthesis) and PNIPAM particles (●, ○; in the absence of MBAAm in the synthesis; as a control) at t = 3 h (solid symbols) and t = 24 h (open symbols). (C) Representative TEM images of PNIPAM microgels at t = 3 h. Reproduced (adapted) from ref. 58 with permission from the Royal Society of Chemistry, copyright 2015. | ||
Most recently, Theato and co-workers reported eFRP of reactive monomers, 2,6-difluorophenyl acrylate (DFPA), pentafluorophenyl acrylate (PFPA), and glycidyl methacrylate (GMA) using a commercial and standardized Electrasyn 2.0 device.31 Using commercially available (IKA), standardized zinc (anode) and graphite (cathode) electrodes, initiation was achieved by galvanostatic cathodic reduction (−1 mA or −4 mA) of 4-fluorobenzenediazonium tetrafluoroborate to generate aryl radicals. Incorporation of the fluorine tag enabled Mn to be determined by 19F NMR spectroscopy as well as SEC. The functional groups present in each monomer were electrochemically inert under the reaction conditions and were readily used for post-polymerization modification exemplifying the potential to synthesise reactive polymer scaffolds using eFRP (Scheme 1).
 | ||
| Scheme 1 Schematic overview of eFRP. (A) Electrochemical decomposition of a fluorine-labelled aromatic diazonium salt. (B) eFRP of DFPA, PFPA, and GMA. (C) Post-polymerization modification of the reactive polymer scaffolds using a fluorine-labelled amine for the preparation of polyamides and poly(β-amino alcohols). Reproduced from ref. 31. with permission from the Royal Society of Chemistry, copyright 2021. | ||
Reversible deactivation radical polymerization (RDRP)
Over the last 30–40 years, the development and advances in our understanding of reversible deactivation radical polymerization (RDRP) techniques has had a dramatic effect of what is synthetically possible in polymer chemistry.59 Through control of a dynamic equilibrium between dormant (Pn-X) and active (Pn˙) species, RDRP enables fine control over the radical concentration during polymerization which significantly reduces termination events. Consequently, it is possible to retain and control chain-end functionality which provides access to complex polymer compositions (e.g. diblock, multiblock, gradient copolymers)60 and architectures (star, hyperbranched, graft, bioconjugates etc.).61,62 This has expanded the scope of potential applications of polymers derived from radical polymerization into areas such as healthcare,63 3D-printing,64 and nanotechnology.65 The most common RDRP methods are nitroxide mediated polymerization (NMP),66 atom transfer radical polymerization (ATRP)67 and reversible addition fragmentation chain transfer (RAFT) polymerization.68 Thus far, ATRP and RAFT have been shown to lend themselves to electrochemical intervention whereby an applied potential or current can be used to generate the active radical intermediates (Pn˙) required to trigger and/or mediated polymer synthesis.Electrochemical atom transfer radical polymerization (eATRP)
Atom transfer radical polymerization (ATRP) was independently reported by Sawamoto and Matyjaszewski,69,70 in the mid-1990s and it is one of the most extensively developed areas of polymer science.71 Compared with FRP, ATRP offers better control over molecular weight, dispersity and end-group enabling precise compositional and topological control for the design of advanced materials.72 ATRP is also a useful tool for surface-initiated polymerization73 and micropatterning via translation to probe-based methods such as scanning electrochemical cell microscopy (SECCM).74 The choice of monomers compatible with ATRP is another advantage. For example, styrenes,75 (meth)acrylates,76 (meth)acrylamides,77N-vinylpyrrolidone,78 acrylonitrile,79 vinyl acetate,80 and vinyl chloride81 are all compatible with ATRP. Considering the breadth of monomers used it is important that ATRP can be achieved in both aqueous82 and non-aqueous solution.83In Cu-mediated ATRP biochemical,84 photochemical,85 electrochemical86 and mechanochemical87 methods have all been developed, enabling external control over polymerization reactions, as the radical concentration can be accurately controlled. Each of these ATRP methods offers a more environmentally benign and industrially friendly approach to synthesis compared to conventional ATRP.88 From a mechanistic point of view, ATRP was developed from atom transfer radical addition (ATRA).67,89 In general it is important to control the equilibrium (KATRP) between dormant alkyl (R-X) or macromolecular (Pn-X) halides and the propagating radicals (R˙/Pn˙) that undergo reversible redox reactions with Cu-complexes (Scheme 2).90
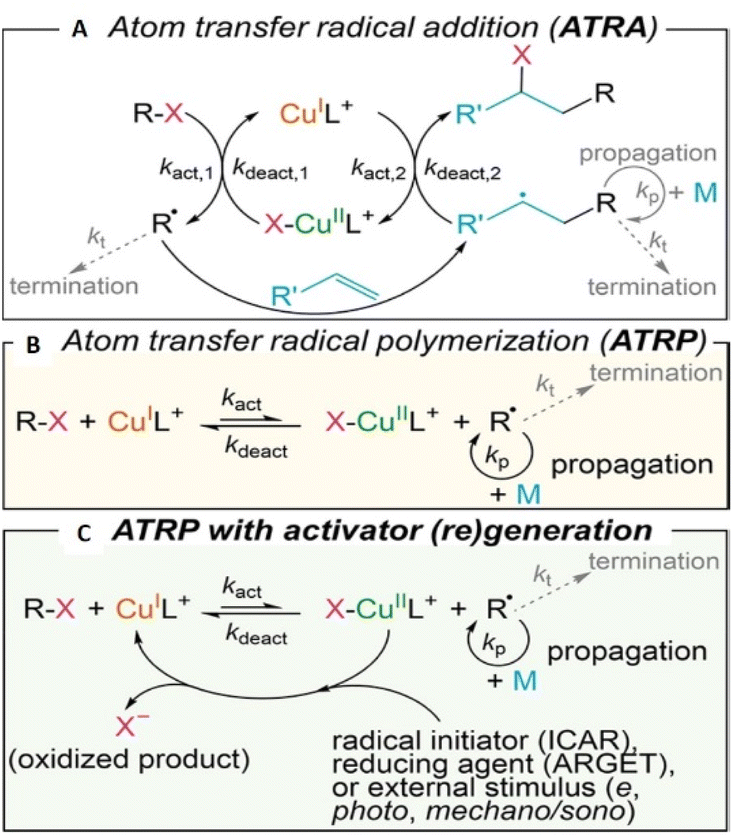 | ||
| Scheme 2 Mechanistic summary of Cu-mediated (A) ATRA, (B) ATRP, and (C) ATRP with continuous (re)generation of the CuIL+ activator promoted by external stimuli. Reprinted with permission from ref. 67. Copyright 2022 American Chemical Society. | ||
In 2017, Matyjaszewski and his team wrote an excellent and extensive review on eATRP, covering the development and applications of eATRP to date.91 Readers are direct to this for information on the traditional reaction set-up, mechanism, optimization and application of eATRP between 2011–2017, along with the advantages and limitations of the technique. For brevity, here we will briefly describe the concept of eATRP again, then focus on the progress and application of eATRP since 2017.
In eATRP, the catalyst concentration is manipulated by an electric field which can be used to move between active and dormant states. The active, yet oxidatively labile CuIL complex can be formed in situ when a potential (Eapp) or current (Iapp) is applied to promote a one electron reduction of an inactive CuIIL precursor.92 Several parameters, such as the applied current (Iapp), applied potential (Eapp), and total charge passed (Q) can be defined in eATRP to allow selection of the desired concentration of redox-active catalytic species.91 As well as the electric field, other relevant parameters that can influence the outcome of eATRP include stirring rate and diffusion,93 reaction solvent94 and supporting electrolyte.95
Electricity, as an external stimulus, drives the reaction of eATRP, and is a critical factor affecting the reaction process and outcome.96 Prior to the reaction, CV (cyclic voltammetry), is used to investigate the redox activity of the reaction solutions. The appropriate applied potential can be selected for eATRP through the obtained reduction and oxidation potentials of the copper/ligand system employed. Thus, the standard reduction potential (E1/2 = (Epa + Epc)/2) of Cu-complexes employed can be estimated from CV as the half-sum of cathodic (Epc) and anodic (Epa) peak potentials.97E1/2 values are strongly dependent on the nature of Cu-salt and ligand employed to form the Cu-complex.98 When CuBr2 forms complexes with different ligands, the corresponding values of Epc and Epa are different, so E1/2 values are also different. For example, complexes formed using ligands commonly employed in eATRP, e.g. TPMA and Me6TREN form reducing Cu-complexes as indicating by the negative E1/2 values (Fig. 3).99
 | ||
Fig. 3 Cyclic voltammetry recorded on a GC electrode at v = 0.1 V s−1, room temperature of (A) CuIIMe6Tren (8.8 mM) in H2O/OEGMA300 (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) + 0.1 M Et4NBF4 in the absence (black line) and presence (dashed red line) of 2-hydroxyethyl 2-bromoisobutyrate (HEBiB). E1/2 = −0.40 V; Ep,c = −0.51 V; Ep,a = −0.28 V.99 (B) CuIITPMA (8.8 mM) in H2O/OEGMA300 (9:1 v/v) + 0.1 M Et4NBF4 in the absence (black line) and presence (dashed red line) of 2-hydroxyethyl 2-bromoisobutyrate (HEBiB). E1/2 = −0.21 V; Ep,c = −0.31 V; Ep,a = −0.12 V. (C) CuII(NPPI)2 (8.8 mM) in H2O/OEGMA300 (9:1 v/v) + 0.1 M Et4NBF4 in the absence (black line) and presence (dashed red line) of 2-hydroxyethyl 2-bromoisobutyrate (HEBiB). E1/2 = 0.02 V; Ep,c = −0.14 V; Ep,a = 0.17 V. Reproduced from ref. 99 with permission from the Royal Society of Chemistry, copyright 2022. :1 v/v) + 0.1 M Et4NBF4 in the absence (black line) and presence (dashed red line) of 2-hydroxyethyl 2-bromoisobutyrate (HEBiB). E1/2 = −0.40 V; Ep,c = −0.51 V; Ep,a = −0.28 V.99 (B) CuIITPMA (8.8 mM) in H2O/OEGMA300 (9:1 v/v) + 0.1 M Et4NBF4 in the absence (black line) and presence (dashed red line) of 2-hydroxyethyl 2-bromoisobutyrate (HEBiB). E1/2 = −0.21 V; Ep,c = −0.31 V; Ep,a = −0.12 V. (C) CuII(NPPI)2 (8.8 mM) in H2O/OEGMA300 (9:1 v/v) + 0.1 M Et4NBF4 in the absence (black line) and presence (dashed red line) of 2-hydroxyethyl 2-bromoisobutyrate (HEBiB). E1/2 = 0.02 V; Ep,c = −0.14 V; Ep,a = 0.17 V. Reproduced from ref. 99 with permission from the Royal Society of Chemistry, copyright 2022. | ||
Applications enabled by eATRP
A direct application of eATRP is the synthesis of polymers with complex compositions and architectures, such as block copolymers,100 copolymer brushes,101 and star polymers.84 In addition, initiators can be adsorbed onto 2D and 3D surfaces to form brush-like graft (co)polymers through eATRP.102 Moreover, the procedure is tolerant to small amount of oxygen making it amenable to applications in biochemistry.103 For example, Matyjaszewski's and co-workers produced well-defined polymers and DNA–polymer bioconjugates via low-volume eATRP in the presence of oxygen.104 Kong's group found that signal amplification enabled by eATRP could be used to develop an electrochemical aptasensor for the detection of bisphenol A with sensitivity as low as 59 aM.105Complex polymer architectures from eATRP
eATRP has been successfully used for the preparation of polymers with complex architectures, benefitting from the high chain-end fidelity conferred and minimization of termination events e.g. coupling between multifunctional macromolecules.106,107 Star polymers, block copolymers, and multi-block copolymers can be synthesized with precise control over molecular weight and low dispersity values employing low catalyst loadings (as low as 10 ppm).108 eATRP is an efficient technique for synthesizing star polymers.109 A 5-arm star polymer was synthesized using α-D-glucose-Br5 containing five initiating groups in the core. In 2017, Chmielarz and co-workers used oligo(ethylene glycol) acrylate (OEGA) as monomer, and a five arm star polymer was synthesized by eATRP.110 The conversion can reach 98% and the dispersity of the POEGA was as low as Đm = 1.15. Matyjaszewski's group used n-butyl acrylate (BA) as a monomer to synthesize star polymers and bottlebrush polymers respectively.111 Bottlebrush polymers were synthesized from two multi-functional ATRP macro-initiators derived from poly(hydroxyethyl methacrylate) (PHEMA) with different chain lengths (DPn = 316, 1632). Simplified eATRP (seATRP) of BA from the multi-functional macro-initiators yielded bottlebrush polymers with Mn > 500000 g mol−1. Cleaving polymer chains from backbone revealed that the polymerization process was well controlled (Đm = 1.07–1.32). The topology of the brushes formed could be varied based on the DPn of the PHEMA backbone or the DPn of the PBA sidechains (Fig. 4). Multi-functional vitamin molecules have also been used to form the core of star polymers synthesized by eATRP. Through the precise control of the molecular weight and dispersity, these products have the potential to be used as drug delivery systems.112
 | ||
| Fig. 4 AFM images of: (A) and (C) PBiBEM316-based PBA brushes and (B) and (D) PBiBEM1632-based PBA brushes spin-casted on a flat silicon substrate. Reprinted from ref. 111. Copyright 2020, with permission from Elsevier. | ||
eATRP is a versatile, clean technique compatible with range of water-soluble, biocompatible monomers. As the main component of living organisms, water is a necessary medium for many reactions, so eATRP in aqueous solution is of broad interest. For example, HEMA and OEOA have been used to obtain polymers with predetermined molecular weight and low dispersity using low catalyst loadings and high monomer content (up to 50 vol%).108
eATRP is also an effective technique for the synthesis of copolymers, and Gennaro and co-workers reported the use of eATRP to synthesize poly(acrylonitrile) and poly(acrylonitrile-butyl acrylate) copolymers as precursors for N-doped mesoporous carbons.113 The material contained high nitrogen content, remarkable O2 reduction activity and 40% selectivity for H2O2 production.
Surface initiated eATRP
Surface-initiated (SI)-eATRP was developed to perform polymerizations from initiator-functionalized surfaces.114 Silicon wafers, which are semiconductors, are an important part of integrated circuits in electronic devices for applications in biomedical and microelectronic devices such as biosensors.115 Modification of silicon surfaces is therefore an important factor in the fabrication of new hybrid materials for microelectronics and nanoelectronics. seATRP employing a sacrificial initiator, has been used as a grafting technique to prepare modified silicon wafer surfaces. The synthesis of copolymer brushes of poly(2-hydroxyethyl acrylate) (PHEA) and hydrophobic poly(tert-butyl acrylate) (PtBA) was performed at Cu-catalyst loadings as low as 6 ppm.102 The level of control enabled by electrochemical intervention was verified by surface characterization including AFM (Fig. 5).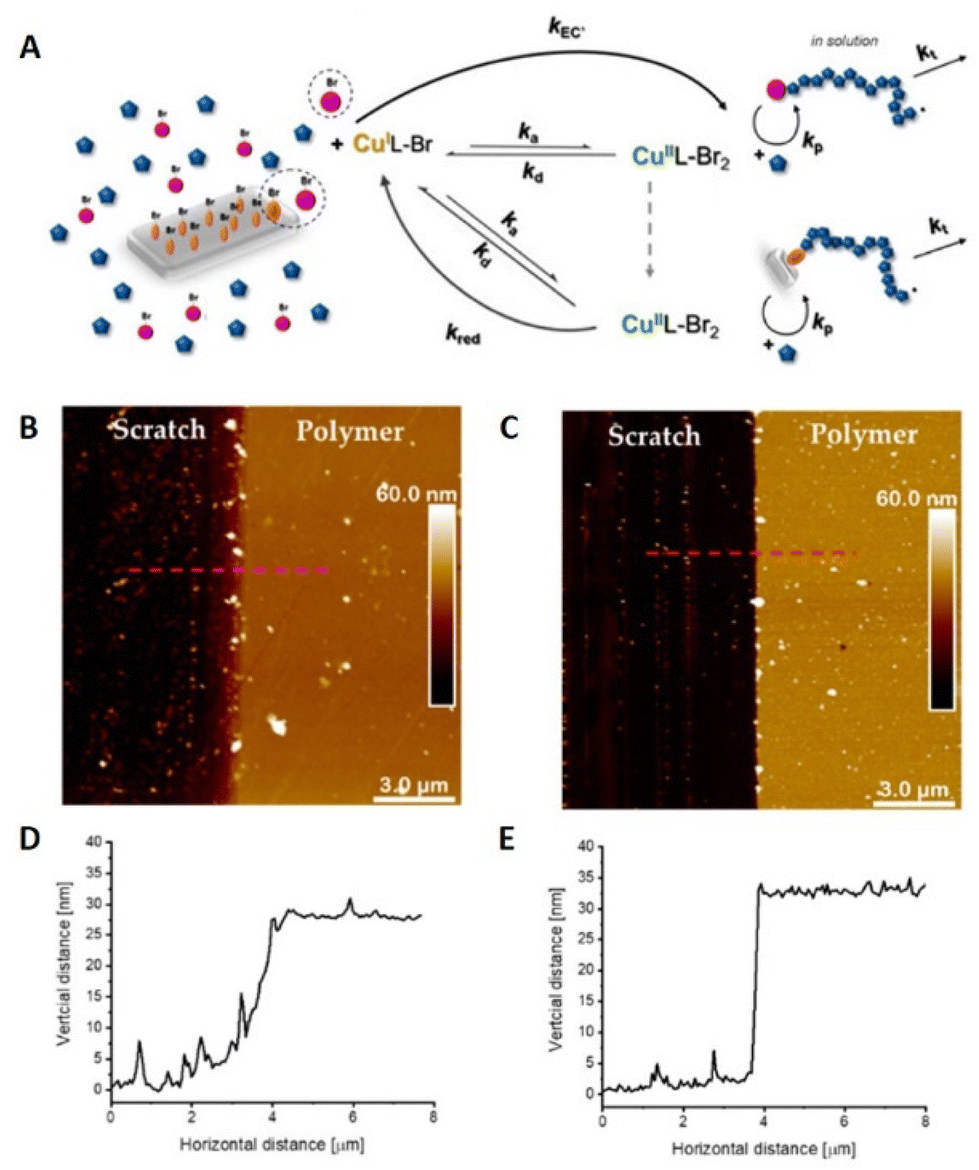 | ||
| Fig. 5 (A) Schematic representation of SI-seATRP in the presence of a sacrificial initiator. (B and D) AFM image and height profile of PHEA grafted surface ([Cu] = 26 ppm, Eapp = −190 mV, 28 ± 1 nm thickness). (C and E) AFM image and height profile of PHEA-b-tBA grafted surface ([Cu] = 6 ppm, Eapp = −120 mV, 33 ± 1 nm thickness). Reproduced from ref. 102. Copyright 2020, with permission from MPDI. | ||
Grafting polymer brushes via SI-eATRP has well-known advantages, including the use of low catalyst concentrations, no need for an inert atmosphere, and the opportunity to control polymer composition and structure.116 Downard's group reported that simultaneous Cu-catalysed electro-‘click’ and SI-eATRP reactions can be used for one-pot grafting strategy with a controlled density anchor sites.117 In 2019, Inagi's group prepared gradient polyelectrolyte brushes using a combination of bipolar electrochemistry and eATRP, and described layer-by-layer (LbL) deposition on gradient polyelectrolyte brushes to form films.118 Similarly, polyethersulfone (PES) membranes modified by SI-eATRP of glucose allylamide and cyclodextrin acrylic acid were demonstrated to have significantly improved hydrophilicity and antifouling properties compared to membranes prepared by common methods.119 In addition to preparing thin films, nanoparticle copolymer brushes can also be synthesized by SI-seATRP. Synthesis of densely grafted (co)polymer brushes, including poly(tert-butyl acrylate) (PtBA), poly(tert-butyl acrylate)-b-poly(styrene) (PtBA-b-PS), and poly(tert-butyl acrylate)-b-poly(butyl acrylate) (PtBA-b-PBA), from 15.8 nm silica nanoparticles (NPs) via SI-seATRP under constant potential electrolysis conditions has been reported.120 The polymers exhibited narrow molecular weight distribution (Đm = 1.20–1.32), and the polymer-grafted nanoparticles had excellent size uniformity (Fig. 6).
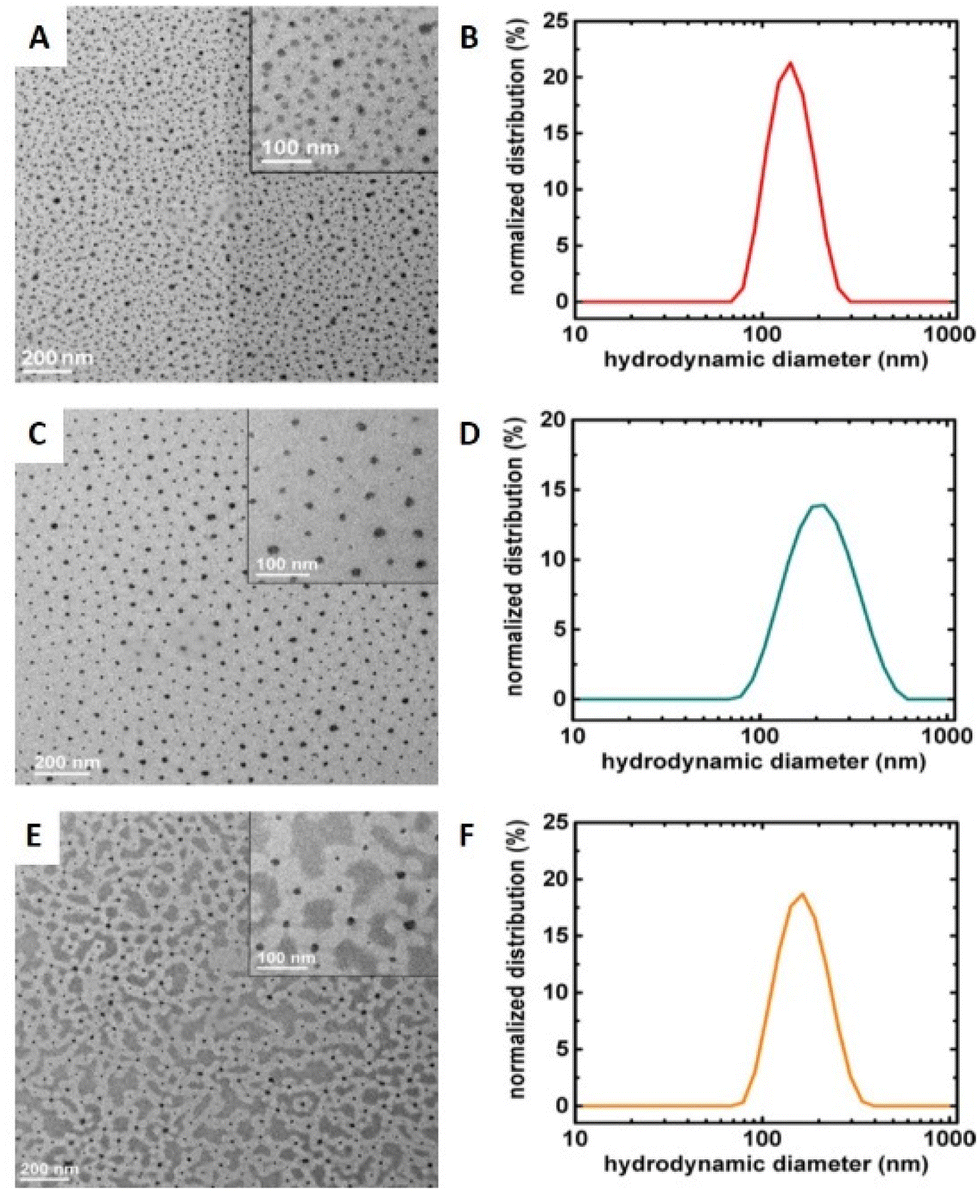 | ||
| Fig. 6 Surface-initiated seATRP employed for the formation of co-polymer brush coated Si-nanoparticles. TEM images and DLS hydrodynamic size distributions by intensity (in THF) of co-polymer brushes grafted on silica NPs via seATRP: (A and B) PtBA, (C and D) PtBA-b-PBA, (E and F) PtBA-b-PS. Reprinted with permission from ref. 120. Copyright 2017 American Chemical Society. | ||
eATRP in biochemical detection
Over the last 2 decades, SI-ATRP has emerged as a robust and sensitive method for signal amplification for the detection of clinically relevant biomolecules, including proteins, nucleic acids and antigens.121,122 Through careful selection of (co)monomers, a desirable signal tag can be controllably incorporated into the polymer brushes created to confer a signal output. For example, using conventional ATRP methodologies, ATRP-initiator-functionalized probes were used to detect and bind to specific DNA fragments immobilized on a substrate. Subsequent ATRP of HEMA resulted in changes in opacity as a result of formation of poly(HEMA) at the specific site of attachment. As a proof of concept this demonstrated that SI-ATRP could be employed to detect point mutations in DNA with a limit of detection of 1.0 nM.123The application of SI-eATRP to these signal amplification strategies has been explored to help simplify the overall process by negating the need for constant deoxygenation and/or use of chemical additives to promote regeneration of the active catalyst. In 2021, Kong's group used thiolated peptide nucleic acids (PNA) immobilized on a gold electrode substrate to specifically detect and bind lung cancer DNA fragments.124 ATRP-initiator-functionalized-graphene oxide (GO) was immobilized onto the PNA–DNA heteroduplexes using phosphate–Zr4+–carboxylate prior to eATRP of ferrocenyl methacrylate (FcMMA) as a signal tag. Using square wave voltammetry (SWV), ultrasensitive detection of lung cancer DNA with a limit of detection as low as 0.213 aM was achieved. Using a similar strategy, a signal amplification method for CYFRA 21-1 DNA, which is a crucial biomarker closely associated with non-small cell lung cancer, has been developed.125 In this work, the PNA–DNA heteroduplexes were functionalized with ATRP initiating groups via linkage to hyaluronic acid (HA) in the place of GO leading to limits of detection as low as 9.04 aM.
The recruitment of a large number of ferrocene (Fc) redox tags for signal amplification has also been employed for the detection of proteins. Prostate-specific antigen (PSA) proteins were immobilized onto a gold substrate via the N-terminal cysteine residue.126 Selective cleavage the peptides resulted in the formation carboxylate groups which were used to non-covalently attach α-bromophenylacetic acid via carboxylate–Zr4+–carboxylate chemistry. Subsequent eATRP of FcMMA and evaluation via SWV resulted in a limit of detection for PSA as low as 3.2 fM (Fig. 7).126
 | ||
| Fig. 7 Schematic representation of a cleavage-based electrochemical PSA biosensor with signal amplification enabled by eATRP. Reprinted with permission from ref. 126. Copyright 2020 ACS. | ||
In the same year, Niu's group reported an electrochemical biosensor based on the use of eATRP grafting of FcMMA as an amplification strategy for ultrasensitive determination of trypsin activity.127 The strategy employed for PSA detection was followed, using trypsin to cleave the target surface bound proteins to expose the carboxylate groups required to introduce the ATRP initiator via the Zr4+ linkage. Optimization resulted in limited of detection in the pM.
Thermally responsive protein imprinted polymers (TPIPs) have also been developed as electrochemical biosensors. Using haemoglobin (Hb) as a template the TPIPs were prepared on gold-deposited Zn-nanoflowers via immobilization of an thiol-functionalized ATRP initiator onto the deposited gold to enable eATRP of NIPAM and methacrylic acid (MAA) as comonomers in the presence of N,N-methylene bis-acrylamide (MBA).128 Upon removal of the template, the performance of the biosensor in Hb detection was evaluated using differential pulse voltammetry (DPV), indicating a detection limit of 3.1 × 10−14 mg L−1.
In addition to the detection of DNA and proteins, this methodology has also been used for the detection of glucose. A boronic acid functionalized gold surface was used to form a layer of glucose on the surface of the electrode. Reaction of the free hydroxyl groups remaining at the surface with 2-bromoisobutyryl bromide introduced initiating sites for SI-eATRP of FcMMA.129 This strategy for ultrasensitive detection of glucose presented a good linear relationship between oxidation current of ferrocene and glucose concentrations in the range from 1.0 nM to 10 μM with an extremely low limit of detection of 0.32 nM (R2 = 0.996). This strategy showed good specificity for glucose detection under physiological conditions and high reliability in human serum.
‘Plug-and-play’ simplified eATRP
The development of eATRP has been driven by thorough investigations into the reaction conditions and reaction parameters such as; exploring the effect of the ligand structure on the ensuing complexes,130 the development of oxygen tolerant eATRP,131 investigating the effect of electrode materials,107 concurrent chemical-electrochemical methods132 and the development of methods that use a commercially available, standardized reaction devices.133 The ElectraSyn 2.0 device is an integrated reaction device manufactured by IKA. It consists of a cap that can supply energy to connected electrodes whilst fitted to an appropriate reaction vial. The reaction device can support up to three electrodes: namely a working electrode (WE), counter electrode (CE) and reference electrode (RE). A wide range of standardized electrodes are commercially available and readily interchangeable in the device cap which can provide enormous synthetic scope and improve reproducibility of reactions under development.We reported the development of a simplified ‘plug-and-play’ approach to aqueous eATRP in 2021.134 Using Pt (WE), sacrificial Al (CE) and Ag/AgCl (RE) electrodes, CuI/TPMA was generated by reduction of CuII/TPMA at the WE when a suitable potential was applied (Eapp = −0.15–−0.25 V). The rate of reaction was shown to increase at more reducing Eapp values compared to E1/2. Well-controlled polymerization of PEGA480 (Đm = 1.17–1.31) was obtained and Mn = 9000–55000 g mol−1 were achieved (Fig. 8). Crucially, electrochemical control was verified by sequential steps of applied potential and no applied potential, which showed that polymerization only occurred during the steps in which potential was applied to the reaction mixture in order to generate the required CuI/TPMA. Conveniently, the reaction device can be connected to a mobile phone app and the I v t graphs associated with the polymerization can be monitored and recorded in real time allowing the total charge passed during the course of the reaction to be determined. This information can be used to design Iapp reaction profiles that enable a simplified 2-electrode (no RE required), galvanostatic eATRP which has been used for the polymerization of PEGA480 and N-hydroxyethyl acrylamide.133
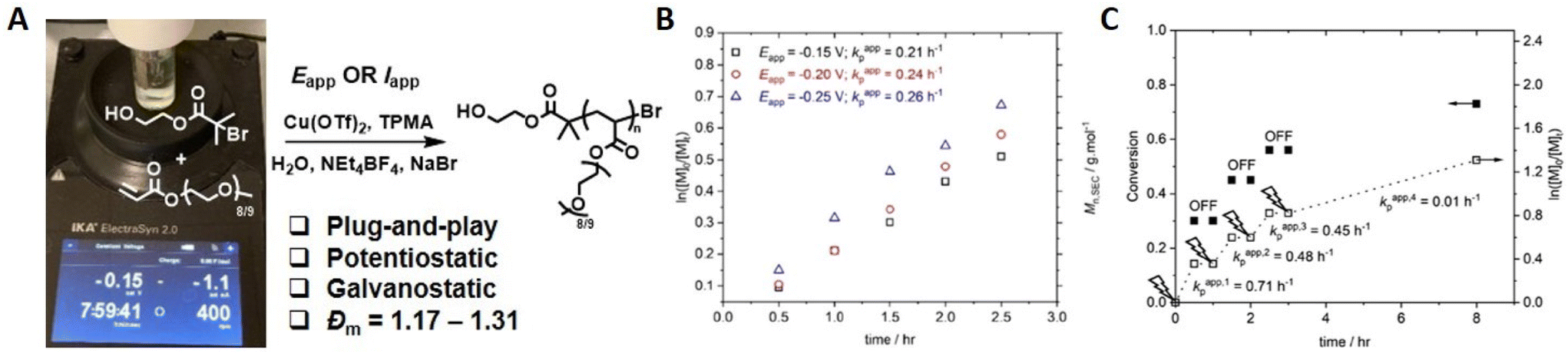 | ||
| Fig. 8 (A) Schematic representation of ‘plug-and-play’ aqueous eATRP using a commercial ElectraSyn 2.0 device. For [PEGA480]:[HEBiB]:[Cu(OTf)2]:[TPMA]:[NaBr] = [38]:[1]:[0.15]:[0.45]:[0.15]; (B) first order kinetic plots for the eATRP of PEGA480 as a function of Eapp. (C) Evolution of the Mn,SEC and Đm with conversion (Eapp = −0.20 V). Reproduced from ref. 134 with permission from the Royal Society of Chemistry, copyright 2021. | ||
The development of eATRP of acrylates and acrylamides has shown that CuI/TPMA and CuI/Me6Tren, which are considered to be more reducing complexes, can result in efficient formation of well controlled polymers in organic and aqueous media. With little attention given to less reducing Cu-complexes, particularly those containing N-alkyl pyridineimine (NAPI) ligands, we have also explored these complexes in our ‘plug-and-play’ aqueous eATRP setup. Under continuous electrolysis conditions polymerization of oligo(ethylene glycol) methyl ether methacrylate (Mn = 300 g mol−1; OEGMA300) yielded POEGMA with good control over molecular weight distribution (Đm < 1.35).99 Interestingly, these polymerizations are not under complete electrochemical control, as monomer conversion continues when electrolysis is halted during reactions in which sequentially steps of applied potential and no applied potential are performed. Alternatively, it was shown that the extent and rate of polymerization depends upon an initial period of electrolysis with seATRP using CuII(NAPI)2 following an electrochemically-triggered, rather than electrochemically mediated, ATRP mechanism (Fig. 9). The electrochemically-triggered mechanism distinguishes CuII(NAPI)2 complexes from the more active CuIIL complexes that have been previously reported. High conversions (>90%) were achieved when targeting a range of molecular weights (DPn,th = 20–200) which yielded POEGMA with good control over the molecular weight distribution (Đm < 1.35).
 | ||
| Fig. 9 For triggered seATRP of [OEGMA300]:[HEBiB]:[CuBr2]:[NPPI] = [20]:[1]:[0.5]:[1.25]; (A) conversion vs. time plot for polymerizations with different tEapp. (B) Pseudo first order kinetic plots for polymerizations for tEapp = 5 min, kappp = 0.0028 min−1; tEapp = 10 min, kappp = 0.0046 min−1; tEapp = 20 min, kappp = 0.0218 min−1; tEapp = 30 min, kappp = 0.0425 min−1. (C) SEC in THF showing the evolution of the molecular weight distribution after electrolysis (Eapp = −0.08 V, tEapp = 30 min, solid line) and at 10 minutes intervals after the potential was removed (Eapp = 0 V, dashed lines, final Mn,SEC = 9300 g mol−1, Đm = 1.33). Reproduced from ref. 99 with permission from the Royal Society of Chemistry, copyright 2022. | ||
Simplified 2-electrode (no RE required), galvanostatic eATRP is amenable to translation to flow electrolysis, taking steps towards improving the economic and environmental impacts of ATRP. To this end Kuhn and Junkers have reported the first continuous-flow self-supporting seATRP reaction using a sonicated microtubular reactor (Fig. 10).135 The use of acoustic streaming, achieved through sonication of the reactor, is beneficial for the handling of viscous mixtures which can be problematic in flow chemistry. Thus, the polymerization of acrylate monomers was fast reaching ∼80% conversion within 27 minutes at ambient temperatures. The evolution of molecular weight was found to be linear, with good agreement between Mn and Mn,th and dispersities as low as Đm = 1.24 achieved through optimization of key reaction parameters such as flow rate, residence time and Iapp. There is plenty of scope for further optimization with respect to scale and electrochemical selectivity but the work lays a good foundation for overcoming the limitations of scale associated with eATRP.
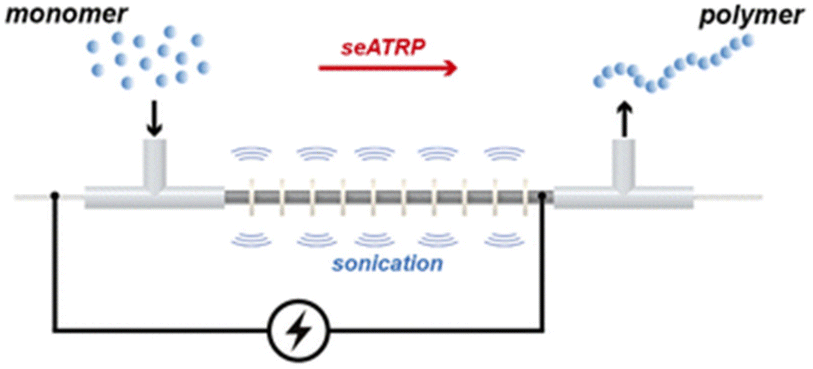 | ||
| Fig. 10 Schematic representation of continuous-flow self-supported seATRP using a sonicated microreactor. Reproduced from ref. 135 with permission from the Royal Society of Chemistry, copyright 2022. | ||
eATRP and ionic liquids
Electrolytes are essential additives in organic eATRP136 and aqueous eATRP,137 as they allow current to flow through the electrochemical cell. One drawback of eATRP is the need to efficiently remove these electrolytes before the polymers can be used in a given application. Ionic liquids (ILs) are attractive alternative solvents for eATRP as liquid salts which are capable of supporting current flow in the absence of additional electrolyte additives.138 In addition, ILs have been shown to enable convenient polymer purification through efficient separation of the polymer product from solvent, with solvent recycling also possible.139 Gennaro and his team reported eATRP of methyl acrylate (MA) in the ionic liquid 1-butyl-3-methylimidazolium trifluoromethanesulfonate ([BMIm] [OTf])140 targeting degrees of polymerization from 276 to 828, achieving well controlled polymers with Đm = 1.1 at >90% conversion in 2 h.In 2020, ionic liquid monomers were used in eATRP to prepare polymeric ionic liquids (PILs). Yue Sun's group used eATRP to prepare superoxide dismutase (SOD) imprinted PILs and reported their use as electrochemical sensors.141 In addition, Lan Xu's group polymerized 1-vinyl-3-propionate imidazolium tetrafluoroborate (VPI+ BF4−) in aqueous media, and that the PILs could be obtained targeting DP from 100 to 300 with the Đm < 1.3.142
Electrochemical reversible addition fragmentation chain transfer polymerization (eRAFT)
In 1998, Zard's group143 and Moad's group144 separately reported the mechanism of reversible addition fragmentation chain transfer polymerization (RAFT). In conventional RAFT, the initiation step is the same as that of FRP, and reaction progress depends on a constant flux of free radicals throughout the reaction.145 Propagating radical species can then react with the RAFT chain transfer agent (CTA) to establish a pre-equilibrium between active and dormant species from which the R-group of the CTA fragments to initiate a new chain. Propagating radicals created from the R-group then also reversibly react with the CTA to create a degenerate chain transfer equilibrium from controlled polymerization can readily proceed. RAFT typically provides excellent control over polymer chains with molecular weights from 1000 to 100000 g mol−1.146 RAFT can be used for the synthesis of precision polymer materials, such as multi-block copolymers,147 star polymers,148 grafting149 and (co)polymerization in supercritical fluids.150
Similarly to FRP, initiation in RAFT polymerizations can be achieved by thermolysis,151 photolysis152 and redox reactions,153 the latter of which lends itself to electrochemical intervention, providing easily tunable parameters for preparation of well-defined polymers in a spatiotemporally controlled manner under mild conditions.154 It was hypothesized that initiating radicals for eRAFT could be generated from direct electrolysis of the RAFT agent or redox active initiator or indirectly by the way of a redox mediator.
Johannsmann and co-workers reported electrochemically triggered RAFT polymerization at an electrode surface (Fig. 11).155 They reported that the addition different CTAs in acidic and basic media could influence the thickness and morphology of the PNIPAM films formed. They noted that the CTAs did not possess electrochemical activity on their own, but did change the electrochemical behavior of the initiator (APS), minimizing micro-gel formation and leading to improved film formation. In 2017, Matyjaszewski and co-workers used 4-bromobenzenediazonium tetrafluoroborate (BrPhN2+) as the redox active initiator for eRAFT (Fig. 12).156 At ambient temperatures, polymerization of BA was carried out under galvanostatic conditions. Electroreduction of BrPhN2+ resulted in formation aryl radicals that initiated polymerization of BA in solution. Under potentiostatic conditions, conversions were limited due to the formation of a passivating organic layer derived from the aryl radicals formed at the electrode surface. However, under galvanostatic conditions conversions of >80% were possible. The authors noted that the use of BrPhN2+ to create radicals was essential as direct electrolysis on the CTAs under investigation 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (DDMAT) and 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPAD) resulted in the formation of carbanions by a 2-electron transfer process. The same group performed a broader electrochemical evaluation of common dithiobenzoate, trithiocarbonates and dithiocarbamates CTAs and found that direct electrolysis of the CTA in the presence of monomer resulted in the loss of the CTA leading to uncontrolled polymerizations.154
 | ||
| Fig. 11 Schematic representation for the first report of eRAFT performed at an electrode surface. Reprinted from ref. 155. Copyright 2010, with permission from Elsevier. | ||
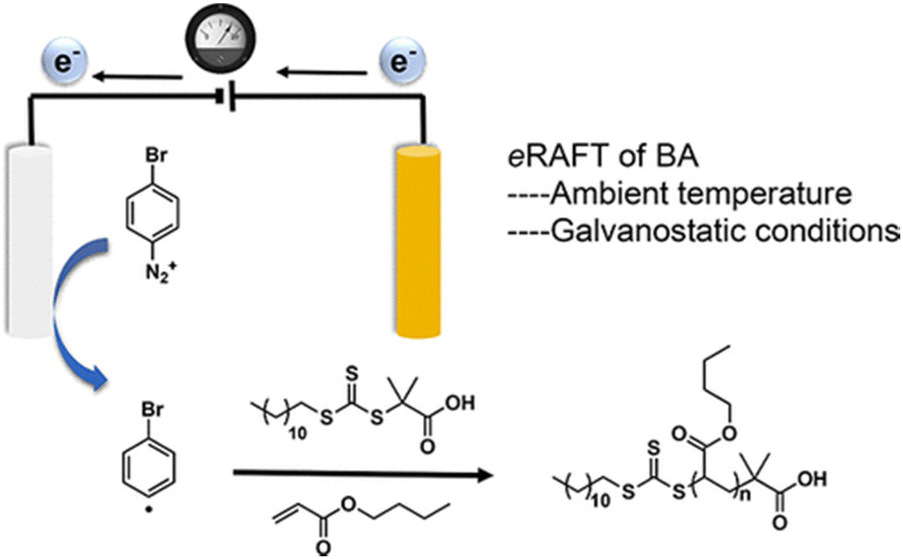 | ||
| Fig. 12 Schmatic representation of eRAFT initiated by electrochemically triggered decomposition of BrPhN2+. Reprinted with permission from ref. 156. Copyright 2017 American Chemical Society. | ||
Moad and co-workers have performed CV analysis of different CTAs to investigate the role of the R group and Z group on the redox properties.157 The major reduction peak moves to more cathodic potentials in the series dithiobenzoates > trithiocarbonates > heteroaromatic dithiocarbamates > xanthates ∼ N-alkyl-N-aryldithiocarbamates, due to the Z-group influence on thiocarbonyl bond reactivity (Fig. 13). Analysis of CVs across a range of scan rates revealed that kinetic control over the reduction mechanism is influenced by both the charge transfer rate and chemical reaction rate.
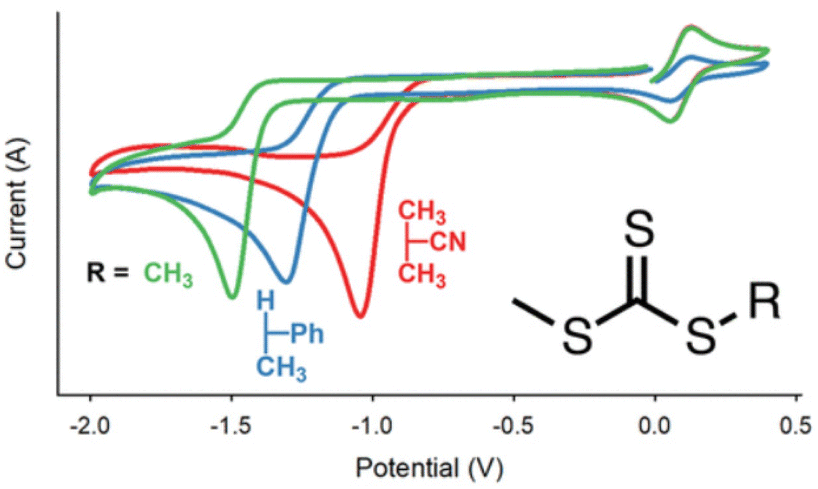 | ||
| Fig. 13 Schematic representation of the effect of the R group on the redox activity of trithiocarbamate CTAs. Reprinted with permission from ref. 157. Copyright 2019 American Chemical Society. | ||
Over-reduction of the CTA at electrode surfaces can be overcome by addition of redox mediators, such as tetraphenylporphryn (TPP), to shuttle electrons from the electrode to the CTA in the bulk solution. Mediator loadings as low as [CTA]/[TPP] = [1]:[0.01] were shown to promote controlled polymerization of acrylates. The reactions were slow, reaching <20% within 6 hours, but good agreement between Mn (2100 g mol−1) and Mn,th (2800 g mol−1) and low dispersities were achieved (Đm = 1.16) which was not the case in the absence of TPP. Anthraquinone (AQ) has also been used as redox mediator for electrochemical reduction of 4-cyano-4-(((dodecylthio)carbonothioyl)thio)pentanoic acid (CDTPA) for eRAFT of methyl methacrylate (Fig. 14).158 The polymerization of MMA (DP = 100) reached 67% conversion within 24 h at ambient temperature, with good control over the molecular weight distribution (Đm = 1.19). The control of the polymerization and retention of the RAFT end-group was verified by chain extension of the PMMA macro-CTA with styrene via thermally initiated RAFT.
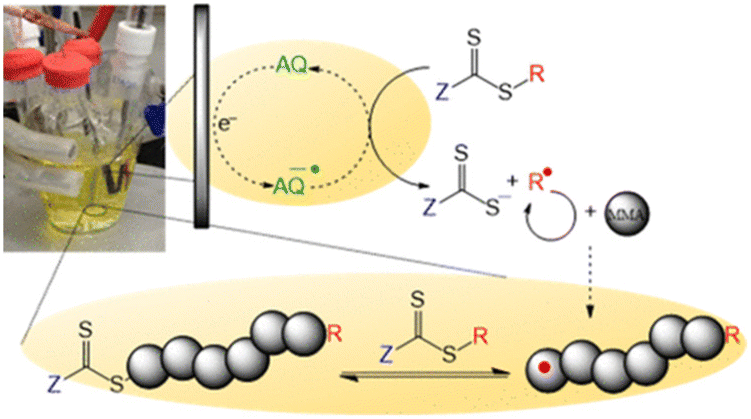 | ||
| Fig. 14 Schematic representation of eRAFT mediated by electron transfer from an anthraquinone organic electrocatalyst. Reprinted with permission from ref. 158. Copyright 2020 American Chemical Society. | ||
Applications enabled by eRAFT
RAFT is one of the most robust and versatile methods for controlling radical polymerization.159 Compared to eATRP, eRAFT is a new technology that requires further exploration and optimization. For example Matyjaszewski and co-workers have reported a dual eATRP and eRAFT system to achieve controlled polymerization of BA using EBiB as an ATRP initiator and DDMAT as a RAFT CTA.160 Addition of 1–2% DDMAT (relative to EBiB) as a CTA, improved the dispersity of PBA formed from Đm = 1.41 to Đm = 1.25 at very low catalyst loadings (10 ppm Cu). The external control of the electric field for this polymerization was exemplified by switching the potential ‘on’ and ‘off’ to observe periods of polymerization and no polymerization respectively. However, the DDMAT only served to improve the reaction control and was not directly activated by the eATRP catalyst system. Conversely, when the CTA was changed to cyano-1-methylethyldiethyldithiocarbamate (MANDC), direct activation by CuI, generated by electroreduction of a Cu-dithiocarbamate (Cu(DC)2) complex, resulted in polymerization of MMA (DP = 200; 500 ppm Cu) with good agreement between Mn (14300 g mol−1) and Mn,th (14800 g mol−1), low dispersity (Đm = 1.31) and good retention of the chain end as exemplified by chain extension with BMA using eATRP and styrene using conventional ATRP.161
eRAFT has been used to synthesize well-defined homo, gradient,157 diblock, triblock,162 and star polymers163 as well as more complex structures, including microgels155 and polymer brushes.164 In 2020, eRAFT for controlled emulsion polymerization of styrene at ambient temperature was reported. A common redox initiating system comprised of ferric sulfate hydrate (FeIII2(SO4)3·xH2O), EDTA, sodium formaladehyde sulfoxylate (SFS) as reductant, and ammonium persulfate (APS) as oxidant was adapted by replacement of SPS with an electrode to perform initial electron transfer to the Fe-EDTA complex formed in situ. Fe-EDTA retains the role of reductant shuttling electrons from the electrode to the APS which is oxidized leading to the formation of sulfate radical ion initiating radicals.165 The eRAFT emulsion polymerization reaction configuration consisted of a three-electrode bulk electrolysis cell with a glassy carbon (GC) rod counter electrode, GC rod working electrode, and an Ag/AgCl reference electrode. In a ‘surfactant-free’ system using poly(N,N-dimethyl acrylamide)-block-poly(butyl acrylate) (PDMAm-b-PBA) as the macro-RAFT agent, emulsion polymerization of styrene was shown to reach >99% conversion in less than 2 hours at room temperature yielding a latex (Zaverage = 90–120 nm; PDI = 0.2), of PDMAm-b-PBA-b-polystyrene with low dispersity (1.20 ≤ Đm ≤ 1.25). Despite the level control indicated by molecular weight data, the authors observed a viscous rather than free flowing latex, alluding to scope for further optimization.
Similar to eATRP, eRAFT has been shown to achieve signal amplification for detection of proteins and DNA. Niu's group reported a coenzyme-mediated signal cleavage-based electrochemical biosensor to interrogate trypsin activity. The strategy relies on the tethering of peptides/proteins to a gold-electrode surface via N-terminal cysteine residues prior to selective tryptic cleavage to incorporate RAFT CTAs via carboxylate–Zr4+–carboxylate interactions. Electroreduction of the NAD+ coenzyme at the gold electrode surface is then exploited to fragment the CTA and generate a surface-tethered active radical capable of recruiting FcMMA as a redox tag during the propagation process. Using electroanalytical methods including SWV and CV the signal generated by recruitment of FcMMA at the cleavage sites allows for highly selective detection of trypsin activity with the detection limit as low as 18.2 μU mL−1 (∼72.8 pg mL−1) (Fig. 15).166
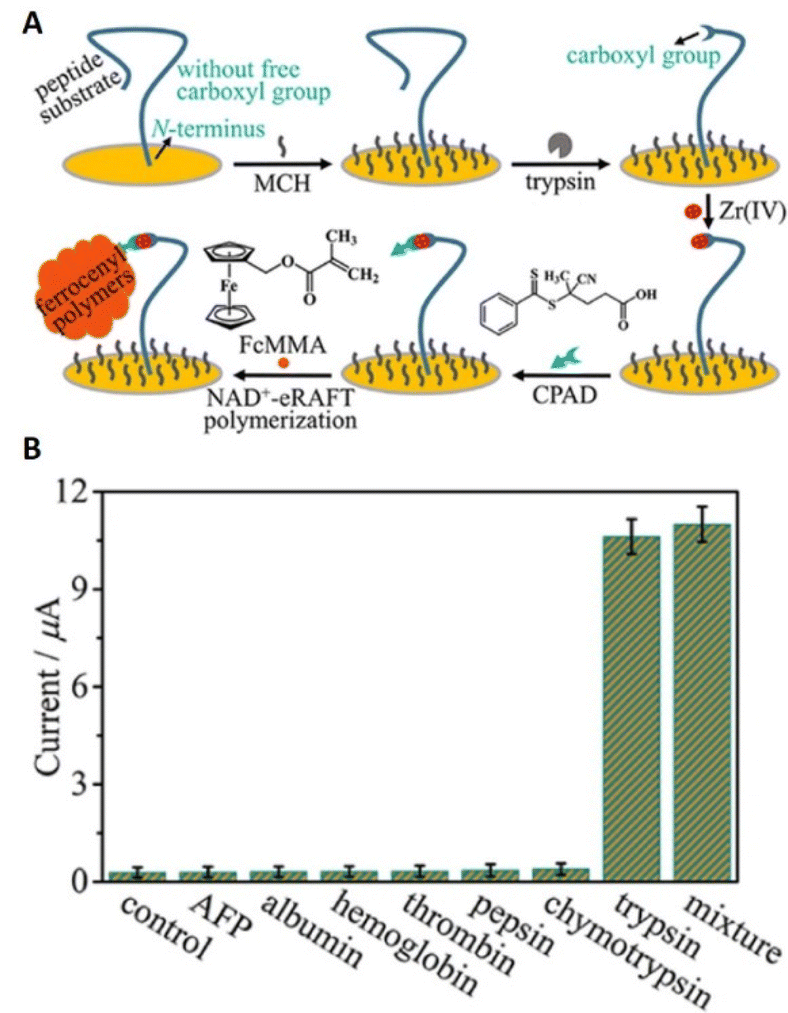 | ||
| Fig. 15 (A) Schematic representation of eRAFT signal amplification for electrochemical interrogation of trypsin activity. (B) Selectivity for trypsin exemplified by differences in currents response toward trypsin and the control proteins. Trypsin, 0.7 ng mL−1 (∼175 μU mL−1); others, 7.0 ng mL−1. Reprinted with permission from ref. 166. Copyright 2021 American Chemical Society. | ||
The same group has developed a high-sensitivity electrochemical biosensor for protein kinase activity using eRAFT polymerization.167 They anchored a carboxyl-containing CTA to the phosphorylation site, and performed eRAFT of FcMMA under constant potential conditions. Through eRAFT polymerization, polymer chains containing a large number of electroactive Fc tags were grafted from each phosphorylation site, thereby significantly amplifying the electrochemical detection signal. The results also showed that the fabricated biosensor was highly selective with a very low detection limit of 1.02 mU mL−1. The same group proposed a cleavage-based electrochemical sensor for matrix metalloproteinases (MMP).168 eRAFT polymerization was suitable for the grafting of Fc-functionalized polymers for signal output. The MMP sensor is simple to operate, low in cost, high in sensitivity and good in selectivity and it is suitable for inhibitor screening and MMP detection in complex serum samples.
In addition to being used in protein detection, eRAFT has been used for ultrasensitive detection of DNA. Niu's group described an ultrasensitive and highly selective electrochemical DNA biosensor by utilizing eRAFT as a signal amplification strategy.169 The strategy is similar to the eATRP method reported above wherein PNA probes are immobilized onto gold substrates prior to specific hybridization with targeted DNA strands to which RAFT CTAs can be linked via phosphate–Zr4+–carboxylate linkages. Subsequent RAFT polymerization of FcMMA, initiated by electrochemical decomposition of the aryl diazonium salts, generates a site specific signal with a detection limit as low at 4.1 aM.
An alternative amplification strategy based on eRAFT and in situ metallization for electrochemical detection of DNA has also been reported.170 This strategy again involves immobilization of PNA probes and hybridization with targeted DNA strands to which RAFT CTAs can be linked via phosphate–Zr4+–carboxylate linkages. Subsequently, glycosyloxyethyl methacrylate (GEMA) was assembled on the electrode surface by SI-eRAFT to form the surface bound glucose glycopolymer. Sodium periodate (NaIO4) oxidation of hydroxyl groups present in the polymer brushes generated aldehyde groups which subsequently reduced silver ions to silver nanoparticles (AgNPs) leading to in situ deposition on the electrode surface (Fig. 16). The AgNPs are used as electroactive tags, to detect the content of the target DNA through the characteristic Ag/Ag+ redox transition, monitored by DPV.
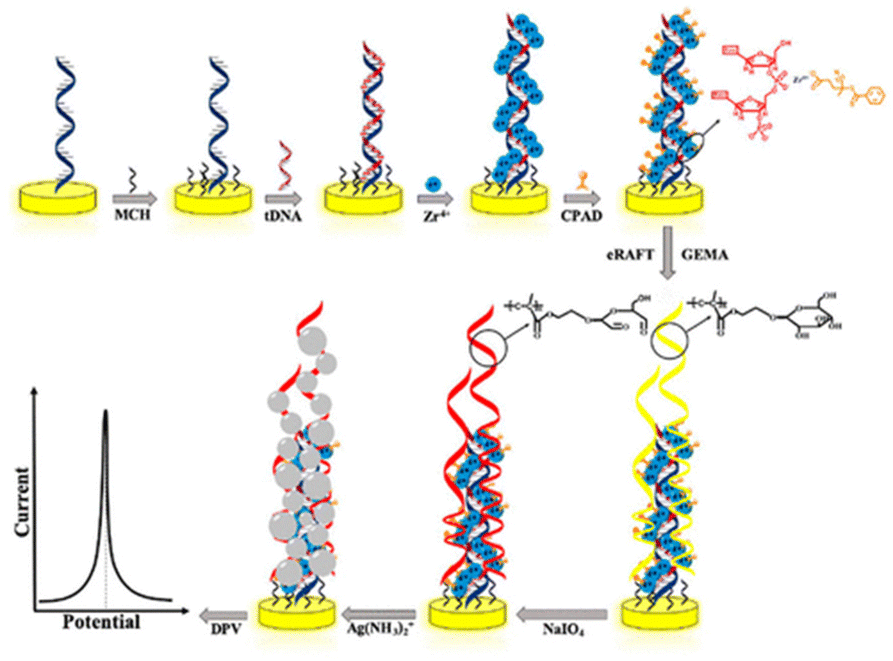 | ||
| Fig. 16 Schematic representation of the electrochemical detection of DNA via eRAFT. Reprinted with permission from ref. 170. Copyright 2019 American Chemical Society. | ||
Ionic polymerization
Electrochemical anionic polymerization
Ionic polymerization171 can be divided into anionic polymerization172 and cationic polymerization173 according to the nature of the reactive intermediate formed. Electrochemical anionic polymerizations (eAP) are chain-growth processes, with propagation occurring through a reactive carbanion chain-end.174 Under appropriate conditions, eAP shares the characteristics of conventional AP with chain termination or chain transfer reactions absent as the reactions demonstrate the characteristics of a living polymerization. At the end of the polymerization, adding quenching agents (water,175 alcohol,176 acid,177 amine,178 epoxides179) can controllably quench the living anionic chain-end to introduce a range of functionalities at the polymer chain end. The generation of a carbanion is a reductive process and in eAP three possible modes of initiation have been described, namely:(1) Direct electron transfer from cathode to monomer:180
| M + e− ⇌ M− |
(2) Electron transfer to monomer from Na metal deposited on the cathode:
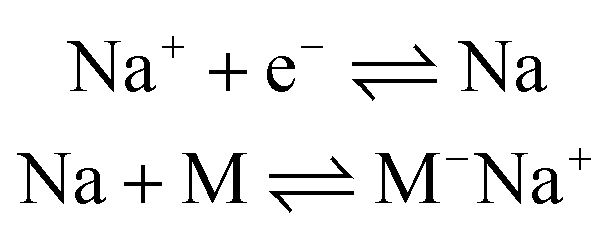
(3) Initiation by pyridyl radical anions formed by reduction of pyridine:181
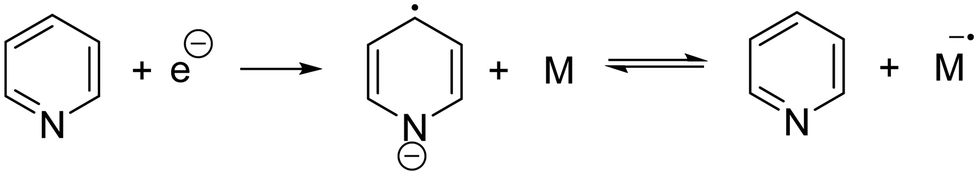
As early as 1957, Yang and coworkers reported using styrene as a monomer, which was triggered by electrons to form polystyrene at the cathode.182 In 1970, Bhadani's group proposed a mechanism of eAP in detail for the first time, and introduced the electrochemical anionic polymerization of 4-vinylpyridine in pyridine in detail.181 The reaction was carried out under galvanostatic conditions. The effects of monomer concentration, current density, polymerization rate, molecular weight, and electrochemical efficiency were analyzed with molecular weights ranging from 305000 to 400000 g mol−1. The following year, Shevchuk's group attempted to synthesize oligomeric acrylate esters by eAP.183 Their group tried to use silver as the cathode material for the first time, providing an example for future scientists to explore different electrode materials.
Electrochemical cationic polymerization
In addition to eAP, electrochemical cationic polymerization eCP has been utilized to produce polymers.184 As reported above, direct reductive electrolysis of RAFT CTAs, results in loss of the CTA and uncontrolled polymerization. However, Fors and co-workers recently demonstrated that cationic intermediates of RAFT CTAs (dithiocarbamates) can be generated via oxidative electrolysis in the presence of an organic nitroxyl radical mediator (TEMPO).184In the presence of an oxidizing current (1 mA) vinyl ether monomers including ethyl- (EVE), n-propyl- (PVE), n-butyl- (BVE), isobutyl- (IBVE) and 2-chloroethyl vinyl ether (Cl-EVE) undergo controlled polymerization to yield poly(vinyl ethers) with good agreement between Mn and Mn,th and low dispersity (Đm < 1.20). The polymerization demonstrated high temporal control, being switched ‘on’ (oxidizing current, 1 mA) and ‘off’ (reducing potential, −875 mV vs. Fc+/Fc) in the presence and absence of the oxidizing current (Fig. 17). High chain end fidelity was exemplified by chain extension of a poly(EVE) macro-CTA (5100 g mol−1; Đm = 1.18), synthesized by eCP, by addition of IBVE to the anodic chamber of the electrochemical cell and resumption of electrolysis to yield poly(EVE-b-IBVE) (8000 g mol−1; Đm = 1.20). From a mechanistic point of view, the authors suggest that in the presence of the oxidizing current TEMPO gets oxidized at the anode forming a cation that reacts with the dithiocarbamate CTA to form a second cationic intermediate (Fig. 18). Upon fragmentation TEMPO is regenerated along with a dithiocarbamate radical and oxocarbenium ion capable of undergoing cationic polymerization. When a reducing potential is applied the dithiocarbamate gets reduced to an anion which can react with the propagating cation to halt polymerization.
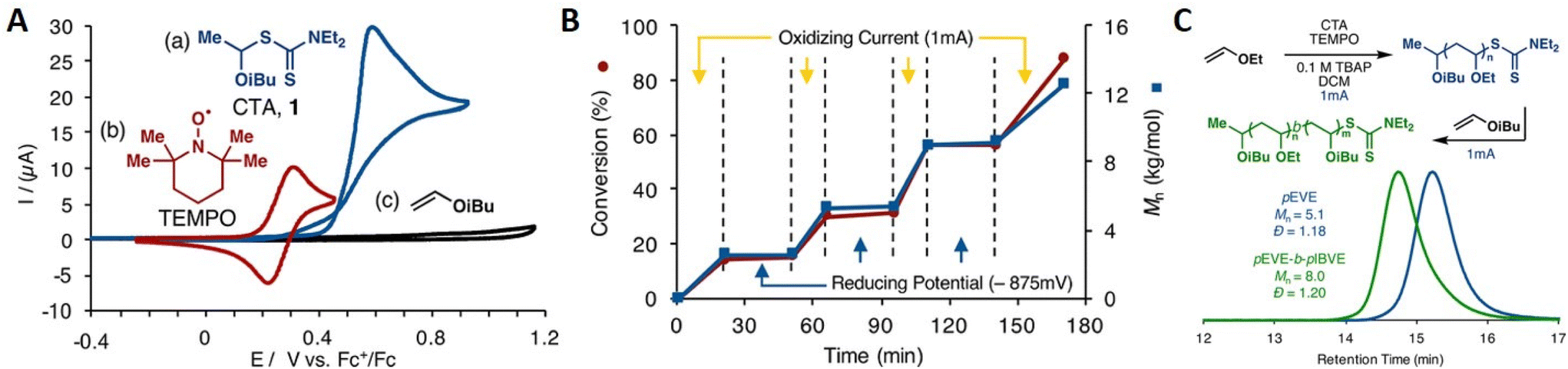 | ||
| Fig. 17 (A) Distinct redox activity of TEMPO (red, 5 × 10−4 M) and dithiocarbamate CTA (blue, 1 × 10−3 M) demonstrated by CV and IBVE (black, 1 × 10−3 M) in 0.1 M tetrabutylammonium perchlorate in dichloromethane at 20 mV s−1. (B) Temporal control of polymer chain growth in the presence and absence of Iapp. (C) SEC traces showing molecular weight distribution for the chain extension of poly(EVE) yielding poly(EVE-b-IBVE). Reprinted with permission from ref. 184. Copyright 2018 American Chemical Society. | ||
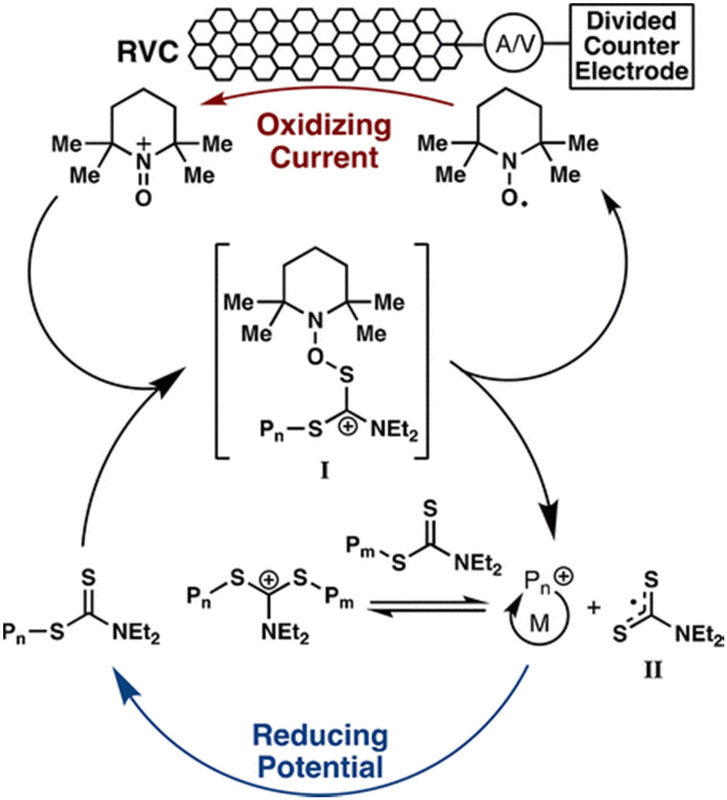 | ||
| Fig. 18 Proposed catalytic cycle of the TEMPO mediated polymerization of vinyl ethers. Reprinted with permission from ref. 184. Copyright 2018 American Chemical Society. | ||
In the same year, Yan's group proposed eCP using 2,3-dicholor-5,6-dicyano-1,4-benzoquinone (DDQ) as an electrocatalyst.185 Similar to the hypothesis of Fors, it was proposed that that DDQ could be formed in situ from its diphenolate species (DDQ2−) in the presence of the an oxidizing potential. DDQ then oxidizes the trithiocarbonate CTA, in this case, to give a carbocation intermediate for chain propagation and a trithiocarbonate radical side product capable of reversibly terminating the propagating chain in the presence of a reductive potential. Vinyl ether monomers EVE, PVE, BVE, IBVE and Cl-EVE all underwent controlled eCP yielding poly(vinyl ethers) with good agreement between Mn and Mn,th and low dispersity (1.15 ≤ Đm < 1.25). Temporal control was demonstrated by switching between oxidizing and reducing potentials and chain extensions confirmed access to block copolymers using this eCP methodology.185 The mechanism proposed above was supported by following a reaction using electron paramagnetic resonance (EPR) to monitor the formation and consumption of the semiquinone radical species of DDQ formed during the eCP mechanism.
An advantage of using thiocarbonyl-based CTA in electrochemically mediated chain-growth polymerizations is the ability to switch between radical and cationic mechanism as a function of the applied potential/current. For example, Yan, Fors and de Alaniz have reported methods that enable selective polymerization in mixtures of acrylate and vinyl ether monomers. Initially, Yan proposed an electroselective interconversion between eCP and eRAFT mechanisms using a trithiocarbonate CTA enabled by a dual electrocatalyst system.186 In the presence of both DDQ2− and nicotinamide adenine dinucleotide hydrate (NAD+) they reported an electrochemically controlled mechanistic shift that promotes selective activation (by DDQ) of eCP under oxidizing potentials (+1.20 V) for the polymerization vinyl ethers. Whilst eRAFT can be mediated (by NADH) under reducing potentials (−0.80 V) for polymerization of acrylates. From mixed monomer and electrocatalyst formulations, a variety of complex vinyl ether-co-acrylate polymer compositions are accessible by varying number and length of switches between alternating potentials. Moreover, the methodology has been translated to flow chemistry which can significantly improve the efficiency of the preparation of these complex polymers that are challenging to synthesise by traditional polymerization methods.
Alternatively, Fors and de Alaniz have developed methods capable of switching between photochemical and electrochemical activation for the polymerization of acrylates and vinyl ethers respectively. Ferrocene was employed by Fors and co-workers to mediate eCP of IBVE in the presence of an oxidizing current and a dithiocarmate CTA. To realize the dual catalytic system, Ir(ppy)3 and MA were added to the reaction mixture and shown to have little effect on the eCP yielding poly(IBVE) with good agreement between Mn and Mn,th (8800 g mol−1; Đm = 1.15). However, in the absence the electric field and under irradiation with 456 nm blue light, no photo-RAFT polymerization of MA was observed. Considering that polymerization was observed under the same conditions but in the absence of Fc and electrolyte it was hypothesized that Fc was quenching the excited state of the photocatalyst and that this could be overcome by reducing the loading of the Fc electrocatayst. With a 5-fold reduction in the concentration of Fc (1 mol% to 0.2 mol%), eCP and photo-RAFT processes were readily interchangeable and accessible by switching between periods of electrolysis and irradiation providing access to a range of (multi)block copolymers (Fig. 19).187
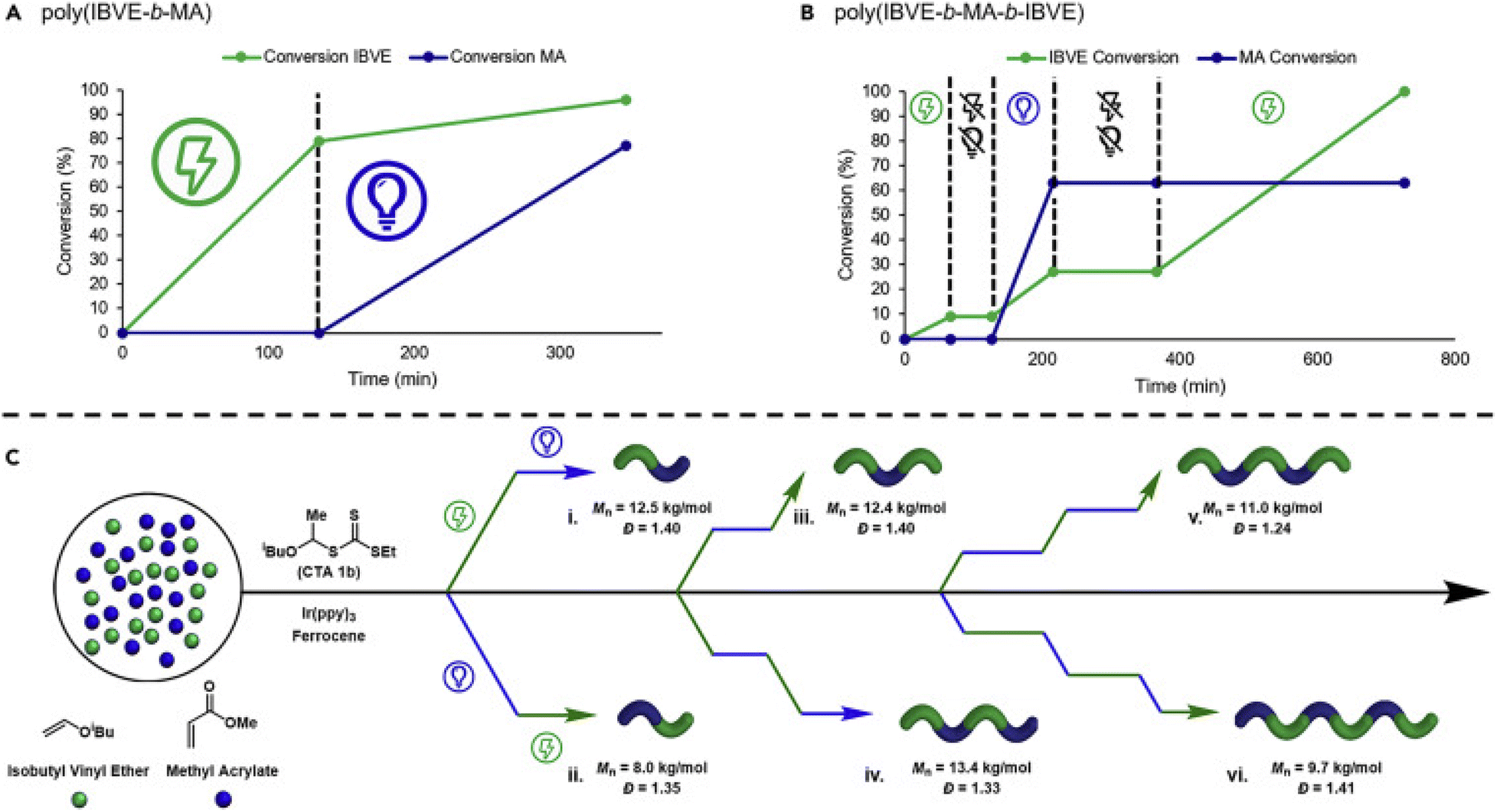 | ||
| Fig. 19 Switching between eCP and photo-RAFT through changing the external stimulus can enable efficient synthesis of polymers with complex compositions. Conversion of IBVE (green line) and MA (blue line) for dual stimuli switching by using alternating application of photochemical or electrochemical stimuli to synthesize: (A) poly(IBVE-b-MA), (B) poly(IBVE-b-MA-b-IBVE). (C) Exemplification of the complexity in composition enabled by eCP and photo-RAFT: (Ci) poly(IBVE-b-MA), (Cii) poly(MA-b-IBVE), (Ciii) poly(IBVE-b-MA-b-IBVE), (Civ) poly(IBVE-b-MA-b-IBVE-b-MA), (Cv) poly(IBVE-b-MA-b-IBVE-b-MA-b-IBVE), and (Cvi) poly(MA-b-IBVE-b-MA-b-IBVE-b-MA-b-IBVE). Reprinted from ref. 187. Copyright 2020, with permission from Elsevier. | ||
A related dual catalytic system employing a single organocatalyst (10-phenylphenothiazine, PTH) that serves as both the photo- and electrocatalyst has also been reported for the controlled copolymerization of IBVE and MA in the presence of a trithiocarbonate CTA. The electrochemical route, for cationic polymerization of IBVE, follows an eCP mechanism whereby the PTH is oxidized to a radical cation that can react with the CTA leading to formation of a cationic species, capable of undergoing propagation, and a radical intermediate of the CTA. Conversely, the photo-RAFT route, for radical polymerization of MA, was performed under irradiation with a 380 nm LED, resulting in excitation of PTH, the excited state of undergoes single electron transfer with the CTA resulting in fragmentation to form a radical species capable of undergoing propagation and a radical intermediate of the CTA.188 This dual catalytic approach enabled the synthesis of complex copolymer compositions derived from distinct mechanistic routes triggered by switching the stimulus. The control of the polymerization was exemplified by chain extension and block copolymerization resulting in di- and tri-block copolymers with dispersities of Đm = 1.42 for poly(IBVE-b-MA) and Đm = 1.29 for poly(MA-b-IBVE-b-MA) (Fig. 20).
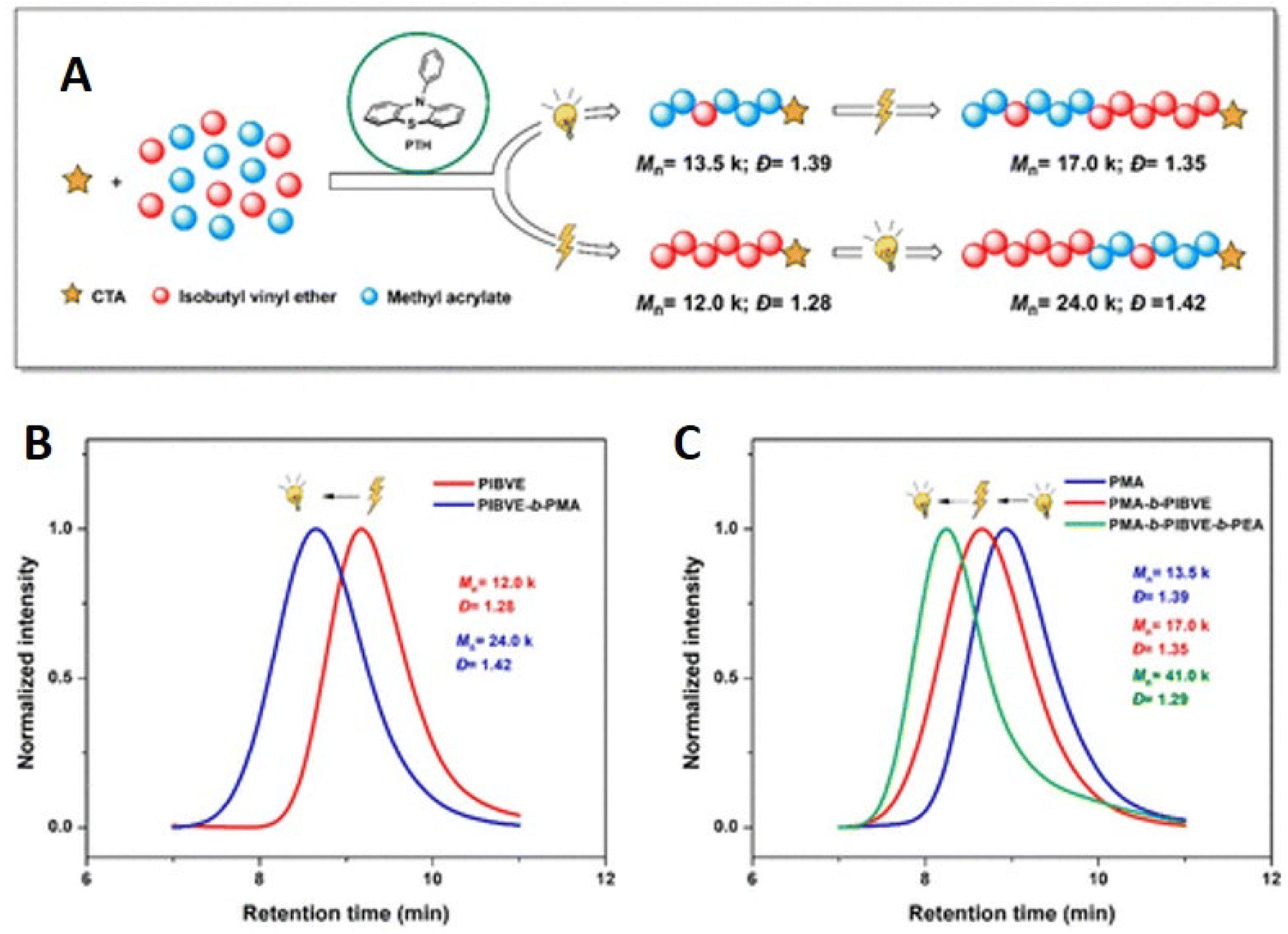 | ||
| Fig. 20 Synthesis of di- and triblock copolymers using orthogonal electro- or photochemical stimulus. (A) PTH catalyzed orthogonal stimuli controlled on-demand polymerization mechanism switching; (B) GPC traces of PIBVE homopolymer (red trace) and PIBVE-b-PMA diblock copolymer (blue trace); (C) GPC traces of PMA homopolymer (blue trace), PMA-b-PIBVE diblock copolymer (red trace), and PMA-b-PIBVE-b-PEA triblock copolymer (green trace). Reprinted with permission from ref. 188. Copyright 2021 American Chemical Society. | ||
Ring-opening polymerization
Developments in (organo)catalytic ring-opening polymerization (ROP) of cyclic esters and carbonates enables the design and synthesis if polyesters and polycarbonates respectively with excellent control over the molecular weight distribution.189 These polymer groups are of interest in the fields of polymer and materials chemistry,190 nanotechnology191 and 3D-printing,192 as alternatives to polyacylics and polyolefins, owing to their biocompatibility and improved (bio)degradation profiles.193,194 The latter can be tuned by copolymerization of different ROP monomer families using switchable catalysis which is best exemplified by the work Williams,195–201 Byers202–205 and Diaconescu,206–211 the latter two of which have focused on chemical redox switches to effect the selectivity of the ROP mechanism by manipulating the oxidation state of a catalytic transition metal centre, a strategy that naturally lends itself to electrochemical intervention.There are some significant advantages to using electrochemistry to perform redox switchable ROP. It negates the need for additional redox reagents which improves atom economy and can simplify purification processes. Redox conditions can be accurately controlled through the applied potential or current and broader redox/electrochemical windows can be achieved by careful choice of solvents and electrolytes.212,213
In 2018, Byers and his team reported the electrochemically triggered and switchable ROP of lactide and cyclohexene oxide (CHO) using Fe-alkoxide complexed with bis(imino)pyridine ligands (Fig. 21).214 It was hypothesized that the Fe2+ complex could activate ROP of lactide but be dormant for ROP of CHO, whilst oxidation to Fe3+ would halt ROP of lactide and active ROP of CHO. Homopolymerizations of each monomer demonstrated temporal control, proving this hypothesis. In reaction solutions containing both lactide and CHO (1:5), it was shown that on application of reducing potential (2.3 V vs. Li/Li+; Fe3+ → Fe2+) ROP of lactide occurred exclusively. Upon switching to an oxidizing potential (3.7 V vs. Li/Li+; Fe2+ → Fe3+) ROP of CHO commenced. Immediately after the switch a small amount of lactide polymerization continued but this became negligible as oxidation of Fe2+ to Fe3+ was completed. The order of the polymerization could also be switched i.e. CHO then lactide, with comparable control. Anchoring the Fe-catalyst to TiO2 nanoparticles which was deposited onto fluorine-doped tin oxide substrated enabled this method to be translated to the solid state with retention of the electrochemically switchable characteristics observed in solution.215
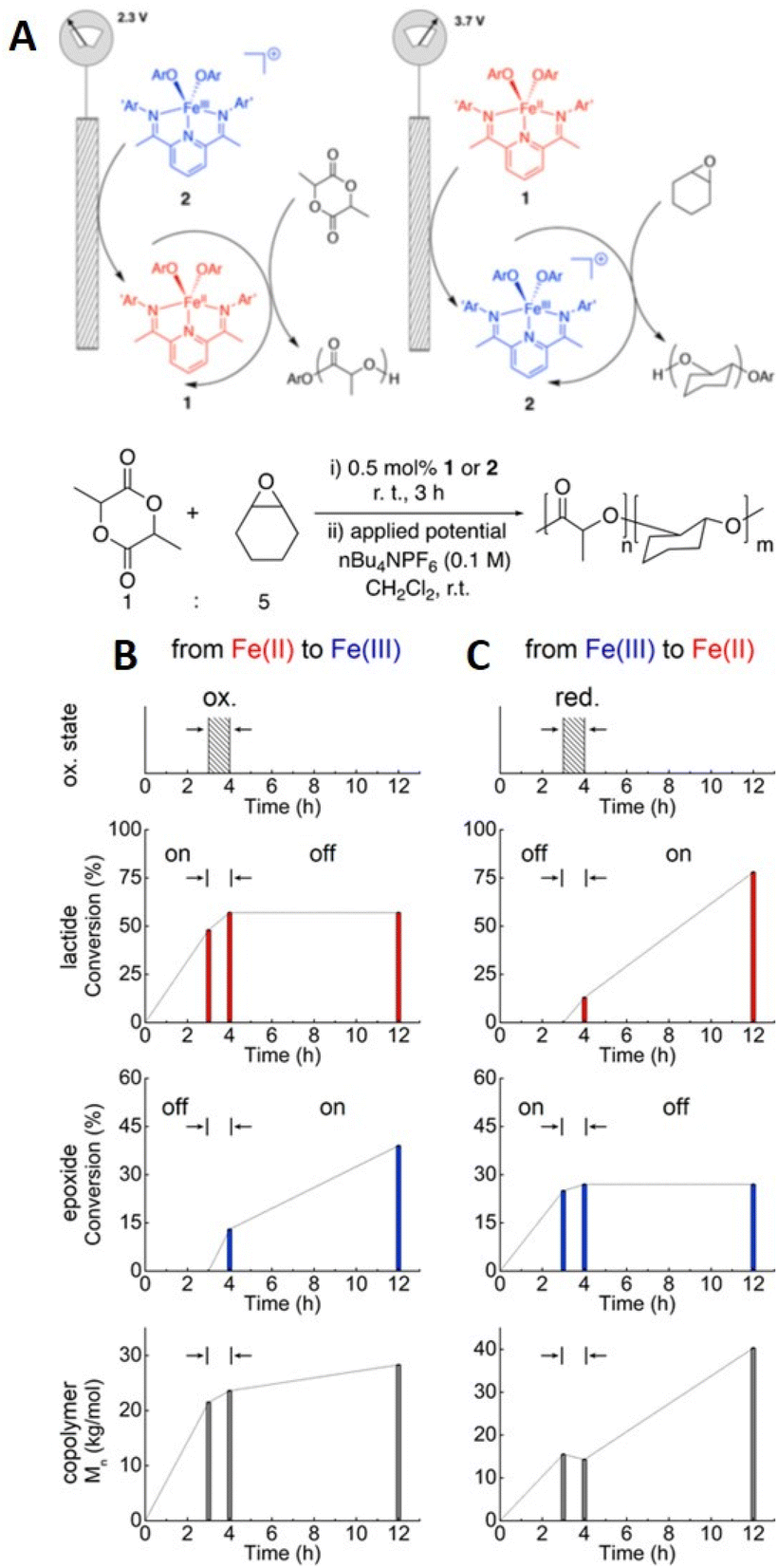 | ||
| Fig. 21 (A) Schematic representation ROP exploiting Eapp to change switch Fe-catalyst activity for lactide and epoxide polymerization. Fe-oxidation states (top plot), monomer conversions (middle two plots), and copolymer molecular weight (bottom plot) for one pot e-switchable polymerization reactions of lactide and epoxide: (B) Fe2+ to Fe3+ switch. (C) Fe3+ to Fe2+ switch. Reprinted (adapted) with permission from ref. 214. Copyright 2018 American Chemical Society. | ||
The electrochemical redox-switch strategy for ROP has also been applied to the synthesis of ABC triblock and ABAB tetrablock copolymers using (salfan)Zr-(OtBu)2 (salfan = 1,1′-di(2-tert-butyl-6-N-methylmethylenephenoxy)ferrocene). Using a glassy carbon electrode, in situ switching of the applied potential alters the Zr oxidation state and monomer selectivity. Using lactide, CHO and β-butyrolactone complex copolymers were accessible with molecular weights of 7000–26000 g mol−1 and good dispersities (Đm = 1.1–1.5) which is comparable to reaction performed using chemical redox switching.
An alternative approach to polyesters via redox-controlled ROP involves the use of redox-controlled acids.216 Fors has shown that when Fc is tethered to an acidic functional group, (electro)chemical oxidation of Fc2+ to Fc3+ could promote ROP via an activated monomer mechanism. In a model ROP of caprolactone (CL) in DCM, initiated by benzyl alcohol, ferrocenyl (phenyl)phosphonic acid was added and electrolyzed under constant anodic potential (1.4 V vs. Ag/AgCl) in a divided electrochemical cell containing a Pt anode and RVC cathode. The anodic potential was applied for 9.5 h to trigger the polymerization by oxidation of Fc2+ to Fc3+ which significantly decreases the pKa of the tethered acid group and promotes CL activation. The polymerization was allowed to proceed for a further 16 h in the absence of an electric field. A cathodic potential (0 V vs. Ag/AgCl) was then applied for 3 h to reduce Fc3+ to Fc2+, increasing the pKa of the acid group to reversibly terminated the polymerization. Switching between anodic and cathodic potentials demonstrated temporal control over the polymerization. However, the authors noted an observed tailing to low molecular weight during these experiments which they attributed to competitive oxidation of the alcohol chain end.
Electrochemical ROP (eROP) of O-carboxyanhydrides has also been reported.217 Tong and coworkers reported a Co/Zn bimetallic electrocatalyst for eROP for the stereoselective polymerization of enantiopure O-carboxyanhydrides (OCA) yielding isotactic polyesters with high molecular weight (>140 kg mol−1) and low dispersities (Đm < 1.1) (Fig. 22). Racemic OCAs are also compatible yielding syndiotactic polymers while stereoblock copolymers were realized by controlling the feed of enantiopure and racemic monomers.217 Polymerizations were performed under galvanostatic conditions in an undivided cell fitted with an Mg anode (WE) and Pt cathode (CE). The optimization conditions for the polymerization of OCAs employed BnOH as initiator, THF as solvent, tetrabutylammonium hexafluorophosphate as electrolyte. When a constant current of 4.0 mA was applied for 2 h at 0 °C, polymerization proceed smoothly with Mn increasing linearly with time yielding polymers with Đm < 1.10. The polymerization exhibited a good degree of temporal control with polymerization proceeding during electrolysis but being significantly retarded in the absence of electrolysis. The authors postulate that Co/Zn electrocatalyst system can activate polymerization via coupled sequential Co-oxidation, cathodic decarboxylation and transmetallation events resulting in formation of a Zn-alkoxide species capable of promoting polymerization. Further mechanistic investigations and exploration of the scope of the reaction are being performed by the group. Development of eROP in this context is attractive from a scale-up point of view considering that industrial production of lactide requires high temperatures and solutions provided by photoredox strategies are currently limited by scale.
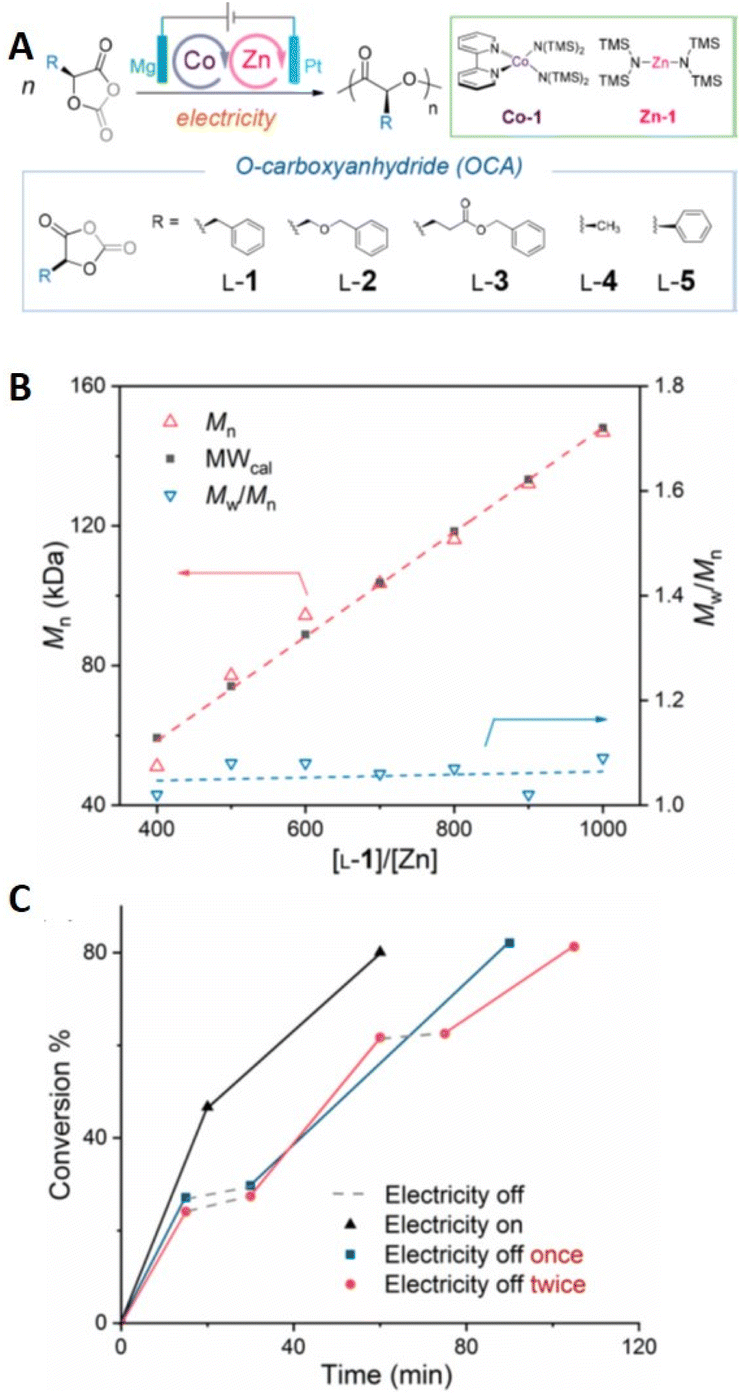 | ||
| Fig. 22 (A) Schematic representation of electrochemical ROP (eROP) of O-carboxyanhydrides using Co/Zn complexes. (B) Plots of Mn and molecular weight distribution (Mw/Mn; Đm) of poly(L-1) vs. [L-1]/[Zn-1] ratio at 0 °C. (C) Demonstration of temporal control as a function of the electric field through relationship between reaction conversion in the presence and absence of Iapp. Data relates to the polymerization of L-1 ([Co-1]/[Zn-1]/[BnOH] = 1/1/1, [L-1] = 130.2 mM, T = 0 °C using Mg(+)/Pt(−) electrodes and 4 mA current. Reprinted (adapted) with permission from ref. 217. Copyright 2020 American Chemical Society. | ||
Conclusions & outlook
The global blueprint for improved sustainability is based on the 17 Sustainable Development Goals (SDG) identified by the UN in 2015. The aim of these goals is to promote peace and prosperity for people and the planet for generations to come. In the midst of our ‘polymer age’ one of the most important challenges for polymer scientists and engineers is the development, implementation and growth of sustainable industrialisation and innovation which is summarized by SDG #9 ‘industry, innovation and infrastructure’. Over the last 2 decades, advances in polymer synthesis allow us, as researchers, to design and synthesize polymers with complex compositions and architectures to fit a range of targeted applications. The use of external stimuli to control the chemistry employed to synthesize polymers has allowed catalyst loadings to be reduced, improved atom economy and reduced the energy footprint, providing more sustainable approaches to polymer synthesis. Advances in electroanalytic, electrocatalytic and electrosynthetic methods have been at the forefront of this progress. Our understanding of the mechanistic and operational requirements for controlled electropolymerization has resulted in the emergence of viable sustainable strategies for the radical, ionic and ring opening polymerization both in solution and at 2D and 3D surfaces. Control over the polymerization in solution and at electrode surface can be intimately controlled by the electric field. This allows for precise control, where necessary, over the molecular weight distribution, chain end fidelity and therefore the polymer composition and architecture in solution, whilst the morphology of films and brushes formed can be controlled at surfaces. The complexity of the operational set-up of electrosynthetic reactions has been markedly simplified, with numerous examples of commercial and standardized hardware available for batch and flow electrosynthesis. This coupled with the development of more simplified electrochemical cell configurations holds great promise for the scale up of the electrochemical polymerization methods discussed in this review. The future of electrochemically controlled and triggered polymerization will undoubtable focus on addressing the current limitations of scale through the development of electrosynthetic and electrocatalytic polymerization in flow. Moreover, integrating automation into such processes and interfacing with artificial intelligence through machine learning algorithms will expedite progress towards realizing sustainable synthesis and production of functional and reactive polymer scaffolds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. W. thanks the Royal Society and Tata companies for the award of a University Research Fellowship (URF\R1\180274).References
- M. Yan, Y. Kawamata and P. S. Baran, Angew. Chem., Int. Ed., 2018, 57, 4149–4155 CrossRef CAS PubMed.
- F. A. Plamper, Colloid Polym. Sci, 2014, 292, 777–783 CrossRef CAS.
- E. C. R. McKenzie, S. Hosseini, A. G. C. Petro, K. K. Rudman, B. H. R. Gerroll, M. S. Mubarak, L. A. Baker and R. D. Little, Chem. Rev., 2022, 122, 3292–3335 CrossRef CAS PubMed.
- C. Sandford, M. A. Edwards, K. J. Klunder, D. P. Hickey, M. Li, K. Barman, M. S. Sigman, H. S. White and S. D. Minteer, Chem. Sci., 2019, 10, 6404–6422 RSC.
- E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302–308 CrossRef CAS PubMed.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- M. C. Leech and K. Lam, Nat. Rev. Chem., 2022, 6, 275–286 CrossRef.
- K. D. Moeller, Chem. Rev., 2018, 118, 4817–4833 CrossRef CAS PubMed.
- E. J. Horn, B. R. Rosen, Y. Chen, J. Tang, K. Chen, M. D. Eastgate and P. S. Baran, Nature, 2016, 533, 77–81 CrossRef CAS PubMed.
- F. Marken and J. D. Wadhawan, Acc. Chem. Res., 2019, 52, 3325–3338 CrossRef CAS PubMed.
- F. Harnisch and U. Schröder, ChemElectroChem, 2019, 6, 4126–4133 CrossRef CAS.
- R. Barhdadi, C. Courtinard, J. Y. Nédélec and M. Troupel, Chem. Commun., 2003, 3, 1434–1435 RSC.
- O. Hosu, M. M. Bârsan, C. Cristea, R. Săndulescu and C. M. A. Brett, Electrochim. Acta, 2017, 232, 285–295 CrossRef CAS.
- Y. N. Zhou, J. J. Li, Y. Y. Wu and Z. H. Luo, Chem. Rev., 2020, 120, 2950–3048 CrossRef CAS PubMed.
- C. Li, H. Bai and G. Shi, Chem. Soc. Rev., 2009, 38, 2397–2409 RSC.
- R. Whitfield, N. P. Truong and A. Anastasaki, Angew. Chem., Int. Ed., 2021, 60, 19383–19388 CrossRef CAS PubMed.
- C. P. Bokis, S. Ramanathan, J. Franjione, A. Buchelli, M. L. Call and A. L. Brown, Ind. Eng. Chem. Res., 2002, 41, 1017–1030 CrossRef CAS.
- M. K. Georges, R. P. N. Veregin, P. M. Kazmaier, G. K. Hamer and M. Saban, Macromolecules, 1994, 27, 7228–7229 CrossRef CAS.
- A. E. Hamielec, R. Gomez-Vaillard and F. L. Marten, J. Macromol. Sci., Chem., 2006, 17, 1005–1020 Search PubMed.
- A. Oral, M. A. Tasdelen, A. L. Demirel and Y. Yagci, Polymer, 2009, 50, 3905–3910 CrossRef CAS.
- Z. Shen, J. Zhong, S. Jiang, W. Xie, S. Zhan, K. Lin, L. Zeng, H. Hu, G. Lin, Y. Lin, S. Sun and Z. Shi, ACS Appl. Mater. Interfaces, 2022, 14, 41022–41036 CrossRef CAS PubMed.
- C. Wolf and W. Burchard, Makromol. Chem., 1976, 177, 2519–2538 CrossRef CAS.
- J. Xu, A. Zhang, T. Zhou, X. Cao and Z. Xie, Polym. Degrad. Stab., 2007, 92, 1682–1691 CrossRef CAS.
- P. Verge, S. Peeterbroeck, L. Bonnaud and P. Dubois, Compos. Sci. Technol., 2010, 70, 1453–1459 CrossRef CAS.
- J. T. Maynard and W. E. Mochel, J. Polym. Sci., 1955, 18, 227–234 CrossRef CAS.
- S. Ishii, A. Sezai, Y. Kohsaka, S. Deguchi and M. Osada, Ind. Eng. Chem. Res., 2022, 177, 111452 Search PubMed.
- M. Rahal, H. Bidotti, S. Duval, B. Graff, T. Hamieh, J. Toufaily, F. Dumur and J. Lalevée, Eur. Polym. J., 2022, 177, 111452 CrossRef CAS.
- L. Jiang, Z. Liu, J. Liu, S. He, X. Wu and W. Shao, SSRN, 2022, 4167806 Search PubMed.
- H. Riazi, A. Shamsabadi, M. C. Grady, A. M. Rappe and M. Soroush, Ind. Eng. Chem. Res., 2018, 57, 532–539 CrossRef CAS.
- E. Molle, S. Frech, T. Grüger and P. Theato, Polym. Chem., 2021, 12, 5970–5978 RSC.
- C. Chatgilialoglu and J. Lalevée, Molecules, 2012, 17, 527–555 CrossRef CAS.
- R. Techie-Menson, C. K. Rono, A. Etale, G. Mehlana, J. Darkwa and B. C. E. Makhubela, Mater. Today Commun., 2021, 28, 102721 CrossRef CAS.
- G. Moad, D. H. Solomon, S. R. Johns and R. I. Willing, Macromolecules, 1982, 15, 1188–1191 CrossRef CAS.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
- N. Awwad, A. T. Bui, E. O. Danilov and F. N. Castellano, Chem, 2020, 6, 3071–3085 CAS.
- A. S. Sarac, Prog. Polym. Sci., 1999, 24, 1149–1204 CrossRef CAS.
- R. V. Subramanian, Electric Phenomena in Polymer Science, 1979, pp. 33–58 Search PubMed.
- Y. N. Zhou, J. J. Li, Y. Y. Wu and Z. H. Luo, Chem. Rev., 2020, 120, 2950–3048 CrossRef CAS PubMed.
- D. M. Heard and A. J. J. Lennox, Angew. Chem., Int. Ed., 2020, 59, 18866–18884 CrossRef CAS PubMed.
- A. D. Mani, M. Deepa, P. Ghosal and C. Subrahmanyam, Electrochim. Acta, 2014, 139, 365–373 CrossRef CAS.
- M. Z. Ansari, S. A. Ansari and S. H. Kim, J. Energy Storage, 2022, 53, 105187 CrossRef.
- D. M. Heard and A. J. J. Lennox, Angew. Chem., Int. Ed., 2020, 59, 18866–18884 CrossRef CAS PubMed.
- A. Wang, W. Hong, L. Yang, Y. Tian, X. Qiu, G. Zou, H. Hou and X. Ji, Small, 2020, 16, 2004022 CrossRef CAS PubMed.
- Y. Fang, Z. Chen, L. Xiao, X. Ai, Y. Cao and H. Yang, Small, 2018, 14, 1703116 CrossRef PubMed.
- C. Ding, C. Li, H. Tian, Y. Tong, W. Huang and Q. Zhang, Batteries Supercaps, 2022, 5, e202200160 CAS.
- E. Dineen, T. C. Schwan and C. L. Wilson, J. Electrochem. Soc., 1949, 96, 226 CrossRef CAS.
- W. B. Smith and H. G. Gilde, J. Am. Chem. Soc., 1960, 82, 659–661 CrossRef CAS.
- B. L. Funt and K. C. Yu, J. Polym. Sci., 1962, 62, 359–367 CrossRef CAS.
- M. Albeck, M. Königsbuch and J. Relis, J. Polym. Sci., Part A-1: Polym. Chem., 1971, 9, 1375–1386 CrossRef CAS.
- M. Albeck and J. Relis, J. Polym. Sci., Part A-1: Polym. Chem., 1971, 9, 2963–2975 CrossRef CAS.
- G. Pistoia, A. Ricci and M. A. Voso, J. Appl. Polym. Sci., 1976, 20, 2441–2450 CrossRef CAS.
- G. Pistoia, B. Scrosati and M. A. Voso, Eur. Polym. J., 1976, 12, 53–57 CrossRef CAS.
- T. R. Balasubramanian and V. Mahadevan, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 2685–2693 CrossRef CAS.
- C. Decker, R. Vataj and A. Louati, Prog. Org. Coat., 2004, 50, 263–268 CrossRef CAS.
- J. Reuber, H. Reinhardt and D. Johannsmann, Langmuir, 2006, 22, 3362–3367 CrossRef CAS PubMed.
- M. Cécius, R. Jérôme and C. Jérôme, Macromol. Rapid Commun., 2007, 28, 948–954 CrossRef.
- S. Yan, Q. Wu, A. Chang, F. Lu, H. C. Xu and W. Wu, Polym. Chem., 2015, 6, 3979–3987 RSC.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- M. Ouchi and M. Sawamoto, Polym. J., 2017, 50, 83–94 CrossRef.
- T. A. Wright, R. C. Page and D. Konkolewicz, Polym. Chem., 2019, 10, 434–454 RSC.
- S. Pearson, C. St Thomas, R. Guerrero-Santos and F. D'Agosto, Polym. Chem., 2017, 8, 4916–4946 RSC.
- P. Gurnani and S. Perrier, Prog. Polym. Sci., 2020, 102, 101209 CrossRef CAS.
- A. Bagheri, C. M. Fellows, C. Boyer, A. Bagheri, C. M. Fellows and C. Boyer, Adv. Sci., 2021, 8, 2003701 CrossRef CAS PubMed.
- Y. Tokura, Y. Jiang, A. Welle, M. H. Stenzel, K. M. Krzemien, J. Michaelis, R. Berger, C. Barner-Kowollik, Y. Wu and T. Weil, Angew. Chem., Int. Ed., 2016, 55, 5692–5697 CrossRef CAS PubMed.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed.
- F. Lorandi, M. Fantin and K. Matyjaszewski, J. Am. Chem. Soc., 2022, 144, 15413–15430 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Chem. – Asian J., 2013, 8, 1634–1644 CrossRef CAS PubMed.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- K. Matyjaszewski, Macromol. Symp., 1996, 111, 47–61 CrossRef CAS.
- K. Matyjaszewski, Adv. Mater., 2018, 30, 1706441 CrossRef PubMed.
- M. Flejszar and P. Chmielarz, Materials, 2019, 12, 3030 CrossRef CAS PubMed.
- E. E. Oseland, Z. J. Ayres, A. Basile, D. M. Haddleton, P. Wilson and P. R. Unwin, Chem. Commun., 2016, 52, 9929–9932 RSC.
- L. Bai, L. Zhang, Z. Zhang, Y. Tu, N. Zhou, Z. Cheng and X. Zhu, Macromolecules, 2010, 43, 9283–9290 CrossRef CAS.
- M. C. Mackenzie, A. R. Shrivats, D. Konkolewicz, S. E. Averick, M. C. McDermott, J. O. Hollinger and K. Matyjaszewski, Biomacromolecules, 2015, 16, 236–245 CrossRef CAS PubMed.
- M. Teodorescu and K. Matyjaszewski, Macromolecules, 1999, 32, 4826–4831 CrossRef CAS.
- V. Mishra and R. Kumar, Polym. Sci., Ser. B, 2019, 61, 753–761 CrossRef.
- Z. Huang, J. Chen, L. Zhang, Z. Cheng and X. Zhu, Polymer, 2016, 8, 59 CrossRef.
- R. Nicolaÿ, Y. Kwak and K. Matyjaszewski, Chem. Commun., 2008, 5336–5338 RSC.
- Z. Huang, C. Feng, H. Guo and X. Huang, Polym. Chem., 2016, 7, 3034–3045 RSC.
- M. Fantin, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2015, 48, 6862–6875 CrossRef CAS.
- C. M. R. Abreu, L. Fu, S. Carmali, A. C. Serra, K. Matyjaszewski and J. F. J. Coelho, Polym. Chem., 2017, 8, 375–387 RSC.
- J. Pollard, O. Rifaie-Graham, S. Raccio, A. Davey, S. Balog and N. Bruns, Anal. Chem., 2020, 92, 1162–1170 CrossRef CAS PubMed.
- T. G. Ribelli, D. Konkolewicz, S. Bernhard and K. Matyjaszewski, J. Am. Chem. Soc., 2014, 136, 13303–13312 CrossRef CAS PubMed.
- M. Fantin, S. Park, Y. Wang and K. Matyjaszewski, Macromolecules, 2016, 49, 8838–8847 CrossRef CAS PubMed.
- Z. Wang, X. Pan, L. Li, M. Fantin, J. Yan, Z. Wang, Z. Wang, H. Xia and K. Matyjaszewski, Macromolecules, 2017, 50, 7940–7948 CrossRef CAS.
- S. Dworakowska, F. Lorandi, A. Gorczyński and K. Matyjaszewski, Adv. Sci., 2022, 9, 2106076 CrossRef CAS PubMed.
- T. Pintauer and K. Matyjaszewski, Chem. Soc. Rev., 2008, 37, 1087–1097 RSC.
- T. G. Ribelli, S. M. Wahidur Rahaman, J.-C. Daran, P. Krys, K. Matyjaszewski and R. Poli, Macromolecules, 2016, 49, 7749–7757 CrossRef CAS.
- P. Chmielarz, M. Fantin, S. Park, A. A. Isse, A. Gennaro, A. J. D. Magenau, A. Sobkowiak and K. Matyjaszewski, Prog. Polym. Sci., 2017, 69, 47–78 CrossRef CAS.
- F. Lorandi, M. Fantin and K. Matyjaszewski, J. Am. Chem. Soc., 2022, 144, 15413–15430 CrossRef CAS PubMed.
- Y. Wang, L. Fu and K. Matyjaszewski, ACS Macro Lett., 2018, 7, 1317–1321 CrossRef CAS PubMed.
- P. Pavan, F. Lorandi, F. de Bon, A. Gennaro and A. A. Isse, ChemElectroChem, 2021, 8, 2450–2458 CrossRef CAS.
- D. A. Corbin, B. G. McCarthy and G. M. Miyake, Polym. Chem., 2020, 11, 4978 RSC.
- F. Lorandi, M. Fantin, A. A. Isse and A. Gennaro, Curr. Opin. Electrochem., 2018, 8, 1–7 CrossRef CAS.
- E. M. Espinoza, J. A. Clark, J. Soliman, J. B. Derr, M. Morales and V. I. Vullev, J. Electrochem. Soc., 2019, 166, 3175–3187 CrossRef.
- D. Dhar, G. M. Yee and W. B. Tolman, Inorg. Chem., 2018, 57, 9794 CrossRef CAS PubMed.
- B. Zhao, F. Pashley-Johnson, B. A. Jones and P. Wilson, Chem. Sci., 2022, 13, 5741–5749 RSC.
- P. Chmielarz, Polymer, 2016, 102, 192–198 CrossRef CAS.
- B. Li, B. Yu, W. T. S. Huck, W. Liu and F. Zhou, J. Am. Chem. Soc., 2013, 135, 1708–1710 CrossRef CAS PubMed.
- M. Flejszar, P. Chmielarz, K. Wolski, G. Grześ and S. Zapotoczny, Materials, 2020, 13, 3559 CrossRef CAS PubMed.
- G. Szczepaniak, L. Fu, H. Jafari, K. Kapil and K. Matyjaszewski, Acc. Chem. Res., 2021, 54, 1779–1790 CrossRef CAS.
- Y. Sun, S. Lathwal, Y. Wang, L. Fu, M. Olszewski, M. Fantin, A. E. Enciso, G. Szczepaniak, S. Das and K. Matyjaszewski, ACS Macro Lett., 2019, 603–609 CrossRef CAS.
- M. Li, Z. Guo, X. Zheng, H. Yang, W. Feng and J. Kong, Analyst, 2019, 144, 5691–5699 RSC.
- P. Krys and K. Matyjaszewski, Eur. Polym. J., 2017, 89, 482–523 CrossRef CAS.
- F. de Bon, F. Lorandi, J. F. J. Coelho, A. C. Serra, K. Matyjaszewski and A. A. Isse, Chem. Sci., 2022, 13, 6008 RSC.
- A. Michieletto, F. Lorandi, F. de Bon, A. A. Isse and A. Gennaro, J. Polym. Sci., 2020, 58, 114–123 CrossRef CAS.
- H. Ding, S. Park, M. Zhong, X. Pan, J. Pietrasik, C. J. Bettinger and K. Matyjaszewski, Macromolecules, 2016, 49, 6752–6760 CrossRef CAS.
- P. Chmielarz, Polymer, 2017, 62, 642–649 CAS.
- I. Zaborniak, P. Chmielarz, M. R. Martinez, K. Wolski, Z. Wang and K. Matyjaszewski, Eur. Polym. J., 2020, 126, 109566 CrossRef CAS.
- P. R. Rodrigues and R. P. Vieira, Eur. Polym. J., 2019, 115, 45–58 CrossRef CAS.
- E. Trevisanello, F. de Bon, G. Daniel, F. Lorandi, C. Durante, A. A. Isse and A. Gennaro, Electrochim. Acta, 2018, 285, 344–354 CrossRef CAS.
- J. Wu, H. Song, D. Li, W. Zhao, W. Zhang, L. Kang, F. Ran and C. Zhao, Surf. Interfaces, 2017, 8, 119–126 CrossRef CAS.
- Z. Chen and V. G. Harris, J. Appl. Phys., 2012, 112, 081101 CrossRef.
- W. Yang and F. Zhou, Biosurf. Biotribol., 2017, 3, 97–114 CrossRef.
- T. Wu, E. R. Lankshear and A. J. Downard, ChemElectroChem, 2019, 6, 5149–5154 CrossRef CAS.
- N. Shida, H. Nishiyama, I. Tomita and S. Inagi, Chem. Lett., 2019, 48, 1174–1177 CrossRef CAS.
- D. Li, J. Wu, S. Yang, W. Zhang, X. Niu, Y. Chen and F. Ran, New J. Chem., 2018, 42, 2692–2701 RSC.
- P. Chmielarz, J. Yan, P. Krys, Y. Wang, Z. Wang, M. R. Bockstaller and K. Matyjaszewski, Macromolecules, 2017, 50, 4151–4159 CrossRef CAS.
- Q. Hu, Q. Wang, G. Sun, J. Kong and X. Zhang, Anal. Chem., 2017, 89, 9253–9259 CrossRef CAS PubMed.
- L. Yuan, W. Wei and S. Liu, Biosens. Bioelectron., 2012, 38, 79–85 CrossRef CAS PubMed.
- X. Lou, M. S. Lewis, C. B. Gorman and L. He, Anal. Chem., 2005, 77, 4698–4705 CrossRef CAS PubMed.
- Q. Liu, J. Liu, H. Yang, X. Wang, J. Kong and X. Zhang, Microchem. J., 2021, 160, 105766 CrossRef CAS.
- L. Zhao, H. Yang, X. Zheng, J. Li, L. Jian, W. Feng and J. Kong, Biosens. Bioelectron., 2020, 150, 111895 CrossRef CAS PubMed.
- Q. Hu, S. Gan, Y. Bao, Y. Zhang, D. Han and L. Niu, Anal. Chem., 2020, 92, 15982–15988 CrossRef CAS PubMed.
- Q. Hu, Y. Bao, S. Gan, Y. Zhang, D. Han and L. Niu, Biosens. Bioelectron., 2020, 165, 112358 CrossRef CAS PubMed.
- Y. Sun, S. Li, Y. Yang, X. Feng, W. Wang, Y. Liu, M. Zhao and Z. Zhang, J. Electroanal. Chem., 2019, 848, 113346 CrossRef CAS.
- Q. Liu, K. Ma, D. Wen, Q. Wang, H. Sun, Q. Liu and J. Kong, J. Electroanal. Chem., 2018, 823, 20–25 CrossRef CAS.
- W. Tang, Y. Kwak, W. Braunecker, N. v. Tsarevsky, M. L. Coote and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 10702–10713 CrossRef CAS PubMed.
- F. de Bon, R. G. Fonseca, F. Lorandi, A. C. Serra, A. A. Isse, K. Matyjaszewski and J. F. J. Coelho, Chem. Eng. J., 2022, 445, 136690 CrossRef CAS.
- F. de Bon, F. Lorandi, J. F. J. Coelho, A. C. Serra, K. Matyjaszewski and A. A. Isse, Chem. Sci., 2022, 13, 6008–6018 RSC.
- M. Mohammed, B. A. Jones and P. Wilson, Polym. Chem., 2022, 13, 3460–3470 RSC.
- B. Zhao, M. Mohammed, B. A. Jones and P. Wilson, Chem. Commun., 2021, 57, 3897–3900 RSC.
- S. Zhang, T. Junkers and S. Kuhn, Chem. Sci., 2022, 13, 12326–12331 RSC.
- J. Luo, C. Durante, A. Gennaro and A. A. Isse, Electrochim. Acta, 2021, 388, 138589 CrossRef CAS.
- N. Bortolamei, A. A. Isse, A. J. D. Magenau, A. Gennaro and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 50, 11391–11394 CrossRef CAS PubMed.
- E. Kim, J. Han, S. Ryu, Y. Choi and J. Yoo, Materials, 2021, 14, 4000 CrossRef CAS PubMed.
- A. Jiménez-Victoria, R. D. Peralta-Rodríguez, E. Saldívar-Guerra, G. Y. Cortez-Mazatán, L. de A. A. Soriano-Melgar and C. Guerrero-Sánchez, Polymer, 2022, 14, 3475 Search PubMed.
- F. de Bon, M. Fantin, A. A. Isse and A. Gennaro, Polym. Chem., 2018, 9, 646–655 RSC.
- Y. Yang, Y. Sun, M. Jin, R. Bai, Y. Liu, Y. Wu, W. Wang, X. Feng and S. Li, Electroanalysis, 2020, 32, 1772–1779 CrossRef CAS.
- W. Hu and L. Xu, Macromol. Chem. Phys., 2021, 222, 2000348 CrossRef CAS.
- P. Corpart, D. Charmot, S. Z. Zard, T. Biadatti and D. Michelet, inventors; Rhodia Chimie SAS, assignee. United States patent US6153705, 2000 Search PubMed.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- K. Matyjaszewski, Macromolecules, 2020, 53, 495–497 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- M. Khan, T. R. Guimarães, K. Choong, G. Moad, S. Perrier and P. B. Zetterlund, Macromolecules, 2021, 54, 736–746 CrossRef CAS.
- L. Xiang, M. Qiu, M. Shang and Y. Su, Polymer, 2021, 222, 123669 CrossRef CAS.
- J. C. Foster, S. C. Radzinski and J. B. Matson, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2865–2876 CrossRef CAS.
- J. Jennings, M. Beija, J. T. Kennon, H. Willcock, R. K. O'Reilly, S. Rimmer and S. M. Howdle, Macromolecules, 2013, 46, 6843–6851 CrossRef CAS.
- A. Postma, T. P. Davis, G. Moad and M. S. O'Shea, Macromolecules, 2005, 38, 5371–5374 CrossRef CAS.
- J. Li, M. Zhang, J. Zhu and X. Zhu, Polymer, 2019, 11, 1722 CAS.
- M. Park, K. Kim, A. K. Mohanty, H. Y. Cho, H. Lee, Y. Kang, B. Seo, W. Lee, H. B. Jeon and H.-J. Paik, Macromol. Rapid Commun., 2020, 41, 2000399 CrossRef CAS PubMed.
- F. Lorandi, M. Fantin, S. Shanmugam, Y. Wang, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2019, 52, 1479–1488 CrossRef CAS.
- J. Bünsow, M. Mänz, P. Vana and D. Johannsmann, Macromol. Chem. Phys., 2010, 211, 761–767 CrossRef.
- Y. Wang, M. Fantin, S. Park, E. Gottlieb, L. Fu and K. Matyjaszewski, Macromolecules, 2017, 50, 7872–7879 CrossRef CAS PubMed.
- L. T. Strover, A. Cantalice, J. Y. L. Lam, A. Postma, O. E. Hutt, M. D. Horne and G. Moad, ACS Macro Lett., 2019, 8, 1316–1322 CrossRef CAS PubMed.
- L. T. Strover, A. Postma, M. D. Horne and G. Moad, Macromolecules, 2020, 53, 10315–10322 CrossRef CAS.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- Y. Wang, M. Fantin and K. Matyjaszewski, Macromol. Rapid Commun., 2018, 39, 1800221 CrossRef PubMed.
- Y. Wang, M. Fantin and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 376–381 CrossRef CAS.
- A. Muslim, D. Malik and M. Hojiahmat, Chem. Pap., 2015, 69, 1512–1518 CAS.
- J. Liu, H. Duong, M. R. Whittaker, T. P. Davis and C. Boyer, Macromol. Rapid Commun., 2012, 33, 760–766 CrossRef CAS PubMed.
- M. C. R. Tria, C. D. T. Grande, R. R. Ponnapati and R. C. Advincula, Biomacromolecules, 2010, 11, 3422–3431 CrossRef CAS PubMed.
- C. Bray, G. Li, A. Postma, L. T. Strover, J. Wang, G. Moad, C. Bray, G. Li, A. Postma, L. T. Strover, J. Wang and G. Moad, Aust. J. Chem., 2020, 74, 56–64 CrossRef.
- Q. Hu, L. Su, Z. Chen, Y. Huang, D. Qin and L. Niu, Anal. Chem., 2021, 93, 9602–9608 CrossRef CAS PubMed.
- Q. Hu, J. Kong, D. Han, Y. Zhang, Y. Bao, X. Zhang and L. Niu, Anal. Chem., 2019, 91, 1936–1943 CrossRef CAS PubMed.
- Q. Hu, L. Su, Y. Mao, S. Gan, Y. Bao, D. Qin, W. Wang, Y. Zhang and L. Niu, Biosens. Bioelectron., 2021, 178, 113010 CrossRef CAS PubMed.
- H. Sun, W. Xu, B. Liu, Q. Liu, Q. Wang, L. Li, J. Kong and X. Zhang, Anal. Chem., 2019, 91, 9198–9205 CrossRef CAS PubMed.
- Q. Hu, J. Kong, D. Han, L. Niu and X. Zhang, ACS Sens., 2019, 4, 235–241 CrossRef CAS PubMed.
- M. Pitsikalis, Ionic polymerization, 2013 Search PubMed.
- N. Hadjichristidis, H. Iatrou, S. Pispas and M. Pitsikalis, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3211–3234 CrossRef CAS.
- Y. Yaǧci and I. Reetz, Prog. Polym. Sci., 1998, 23, 1485–1538 CrossRef.
- R. P. Quirk, Encycl. Polym. Sci. Technol., 2002, 5, 111–164 Search PubMed.
- U. S. Nandi, P. Ghosh and S. R. Palit, Nature, 1962, 195, 1197–1198 CrossRef CAS.
- X. W. Han, O. Daugulis and M. Brookhart, J. Am. Chem. Soc., 2020, 142, 15431–15437 CrossRef CAS PubMed.
- T. Furuncuoǧlu, I. Uǧur, I. Degirmenci and V. Aviyente, Macromolecules, 2010, 43, 1823–1835 CrossRef.
- C. H. Bamford and E. F. T. White, Trans. Faraday Soc., 1956, 52, 716–727 RSC.
- J. Zhang, W. Pointer, G. Patias, L. Al-Shok, R. A. Hand, T. Smith and D. M. Haddleton, Eur. Polym. J., 2023, 183, 111755 CrossRef CAS.
- M. Szwarc, M. Levy and R. Milkovich, J. Am. Chem. Soc., 1956, 78, 2656–2657 CrossRef CAS.
- S. N. Bhadani and G. Parravano, J. Polym. Sci., Part A-1: Polym. Chem., 1970, 8, 225–235 CrossRef CAS.
- J. Y. Yang, W. E. McEwen and J. Kleixberg, J. Am. Chem. Soc., 1957, 79, 5833–5834 CrossRef CAS.
- T. E. Lipatova, G. S. Shapoval and Y. S. Shevchuk, Polym. Sci. U.S.S.R., 1971, 13, 2747–2755 CrossRef.
- B. M. Peterson, S. Lin and B. P. Fors, J. Am. Chem. Soc., 2018, 140, 2076–2079 CrossRef CAS PubMed.
- W. Sang and Q. Yan, Angew. Chem., Int. Ed., 2018, 57, 4907–4911 CrossRef CAS PubMed.
- J. Zhu, X. Hao and Q. Yan, Sci. China: Chem., 2019, 62, 1023–1029 CrossRef CAS.
- M. J. Supej, B. M. Peterson and B. P. Fors, Chem, 2020, 6, 1794–1803 CAS.
- A. Nikolaev, Z. Lu, A. Chakraborty, L. Sepunaru and J. R. de Alaniz, J. Am. Chem. Soc., 2021, 143, 12278–12285 CrossRef CAS PubMed.
- G. Becker and F. R. Wurm, Chem. Soc. Rev., 2018, 47, 7739–7782 RSC.
- A. P. Dove, Chem. Commun., 2008, 6446–6470 RSC.
- J. Fan, Y. P. Borguet, L. Su, T. P. Nguyen, H. Wang, X. He, J. Zou and K. L. Wooley, ACS Macro Lett., 2017, 6, 1031–1035 CrossRef CAS PubMed.
- M. Zhang, A. Vora, W. Han, R. J. Wojtecki, H. Maune, A. B. A. Le, L. E. Thompson, G. M. McClelland, F. Ribet, A. C. Engler and A. Nelson, Macromolecules, 2015, 48, 6482–6488 CrossRef CAS.
- M. J. L. Tschan, R. M. Gauvin and C. M. Thomas, Chem. Soc. Rev., 2021, 50, 13587–13608 RSC.
- J. Su, G. Xu, B. Dong, R. Yang, H. Sun and Q. Wang, Polym. Chem., 2022, 13, 5897–5904 RSC.
- T. T. D. Chen, Y. Zhu and C. K. Williams, Macromolecules, 2018, 51, 5346–5351 CrossRef CAS.
- T. Stößer, D. Mulryan and C. K. Williams, Angew. Chem., Int. Ed., 2018, 57, 16893–16897 CrossRef PubMed.
- T. Stößer, G. S. Sulley, G. L. Gregory and C. K. Williams, Nat. Commun., 2019, 10, 1–9 CrossRef PubMed.
- G. S. Sulley, G. L. Gregory, T. T. D. Chen, L. Peña Carrodeguas, G. Trott, A. Santmarti, K. Y. Lee, N. J. Terrill and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 4367–4378 CrossRef CAS PubMed.
- W. T. Diment, T. Stößer, R. W. F. Kerr, A. Phanopoulos, C. B. Durr and C. K. Williams, Catal. Sci. Technol., 2021, 11, 1737–1745 RSC.
- N. Yuntawattana, G. L. Gregory, L. P. Carrodeguas and C. K. Williams, ACS Macro Lett., 2021, 10, 774–779 CrossRef CAS PubMed.
- A. C. Deacy, G. L. Gregory, G. S. Sulley, T. T. D. Chen and C. K. Williams, J. Am. Chem. Soc., 2021, 143, 10021–10040 CrossRef CAS PubMed.
- A. B. Biernesser, K. R. D. Chiaie, J. B. Curley and J. A. Byers, Angew. Chem., Int. Ed., 2016, 55, 5251–5254 CrossRef CAS PubMed.
- K. R. Delle Chiaie, L. M. Yablon, A. B. Biernesser, G. R. Michalowski, A. W. Sudyn and J. A. Byers, Polym. Chem., 2016, 7, 4675–4681 RSC.
- K. R. Delle Chiaie, A. B. Biernesser, M. A. Ortuño, B. Dereli, D. A. Iovan, M. J. T. Wilding, B. Li, C. J. Cramer and J. A. Byers, Dalton Trans., 2017, 46, 12971–12980 RSC.
- M. A. Ortuño, B. Dereli, K. R. D. Chiaie, A. B. Biernesser, M. Qi, J. A. Byers and C. J. Cramer, Inorg. Chem., 2018, 57, 2064–2071 CrossRef PubMed.
- X. Wang, A. Thevenon, J. L. Brosmer, I. Yu, S. I. Khan, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2014, 136, 11264–11267 CrossRef CAS PubMed.
- E. M. Broderick, N. Guo, T. Wu, C. S. Vogel, C. Xu, J. Sutter, J. T. Miller, K. Meyer, T. Cantat and P. L. Diaconescu, Chem. Commun., 2011, 47, 9897–9899 RSC.
- J. Wei and P. L. Diaconescu, Acc. Chem. Res., 2019, 52, 415–424 CrossRef CAS PubMed.
- A. Lai, Z. C. Hern and P. L. Diaconescu, ChemCatChem, 2019, 11, 4210–4218 CrossRef CAS.
- R. Dai and P. L. Diaconescu, Dalton Trans., 2019, 48, 2996–3002 RSC.
- S. M. Quan, X. Wang, R. Zhang and P. L. Diaconescu, Macromolecules, 2016, 49, 6768–6778 CrossRef CAS.
- V. Pande and V. Viswanathan, ACS Energy Lett., 2017, 2, 60–63 CrossRef CAS.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- M. Qi, Q. Dong, D. Wang and J. A. Byers, J. Am. Chem. Soc., 2018, 140, 5686–5690 CrossRef CAS PubMed.
- M. Qi, H. Zhang, Q. Dong, J. Li, R. A. Musgrave, Y. Zhao, N. Dulock, D. Wang and J. A. Byers, Chem. Sci., 2021, 12, 9042–9052 RSC.
- M. J. Supej, E. A. McLoughlin, J. H. Hsu and B. P. Fors, Chem. Sci., 2021, 12, 10544–10549 RSC.
- Y. Zhong, Q. Feng, X. Wang, J. Chen, W. Cai and R. Tong, ACS Macro Lett., 2020, 9, 1114–1118 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |