
Thermoresponsive “irreversible” property change of POSS-crosslinked PNIPAAm hydrogels†
Shohei
Ida
 *a,
Tenki
Hikida
a,
Atsumi
Kawai
a,
Tomonari
Matsuda
a,
Souma
Suzuki
a,
Hiroaki
Imoto
b,
Kensuke
Naka
b and
Shokyoku
Kanaoka
*a
*a,
Tenki
Hikida
a,
Atsumi
Kawai
a,
Tomonari
Matsuda
a,
Souma
Suzuki
a,
Hiroaki
Imoto
b,
Kensuke
Naka
b and
Shokyoku
Kanaoka
*a
aDepartment of Materials Science, Faculty of Engineering, The University of Shiga Prefecture, 2500 Hassaka, Hikone, Shiga 522-8533, Japan. E-mail: ida.s@mat.usp.ac.jp; kanaoka.s@mat.usp.ac.jp
bFaculty of Molecular Chemistry and Engineering, Graduate School of Science and Technology, Kyoto Institute of Technology, Goshokaido-cho, Matsugasaki, Sakyo-ku, Kyoto 606-8585, Japan
First published on 14th April 2023
Abstract
Control over the reversibility of property changes of a stimuli-responsive polymer hydrogel such as transient fixation and on-demand recovery is a key challenge in realizing a sophisticated function as observed in natural soft tissues. Inspired by the denaturation process of proteins, we focused on incorporation of a highly hydrophobic component into a thermoresponsive hydrogel to induce internal structural fixation by hydrophobic aggregation upon thermoresponsive macroscopic shrinking of a gel network. A polyhedral oligomeric silsesquioxane (POSS) compound with 8 ammonium cation groups was employed as a crosslinker for gel synthesis through end-crosslinking of telechelic poly(N-isopropylacrylamide) (PNIPAAm) with an activated ester terminus, which was prepared by reversible addition–fragmentation chain-transfer (RAFT) polymerization. POSS-crosslinked PNIPAAm gels were obtained with good reproducibility under the reaction conditions in the presence of a large amount of a base. The product gels exhibited reversible swelling/deswelling in water against temperature change; however, their appearance did not return to the original transparency once the gel was heated in water. This behavior contrasts sharply with the reversible change in appearance with a normal PNIPAAm gel. The “irreversible” change in turbidity was likely to result from the aggregation of the POSS moieties induced by macroscopic volume shrinking of the POSS-containing PNIPAAm gels. This internal structural change also led to an appreciable change in mechanical properties: an increase in Young's modulus and elongation. In addition, the turbid POSS-crosslinked gels after a heating/cooling cycle returned to almost transparent ones by immersion in a highly polar solvent such as dimethylsulfoxide.
Introduction
Thermoresponsive hydrogels, which change their properties in response to a temperature change, have been attracting much attention as materials with autonomously dynamic functions for potential application in diverse fields including sensing materials, actuators, carriers in a drug delivery system, and soft robotics.1–8 Since the response behavior of a thermoresponsive hydrogel is based on a balance between hydrophilic and hydrophobic sites in the structure (i.e., the former governs hydration contributing to swelling and the latter induces aggregation of the network),9 a design achieving an appropriate balance of hydrophilicity/hydrophobicity in the structure contributes to the control of the responsive behavior of thermoresponsive hydrogels.10,11In general, the responsive behavior of thermoresponsive hydrogels is reversible. A change in properties induced by a temperature change is temporary, and the properties return to the original state when an external temperature is set back to the initial condition. On the other hand, proteins, an important component of natural hydrogels, undergo an irreversible change in a higher-order structure upon heating due to dissociation and reformation of non-covalent intramolecular interactions such as hydrophobic interactions.12–14 Such denaturation alters the properties of protein hydrogels drastically and irreversibly as observed in the coagulation of a boiled egg, which does not return to the initial sol state (a raw egg) after cooling. This denaturation behavior would be an attractive feature to furnish an artificial hydrogel material because it contributes, for example, to the fixation of gel properties in a desired state and suppression of the deterioration of mechanical properties derived from swelling, which often accompanies conventional thermoresponsive hydrogels.
A key to realize a denaturation-like process with an artificial hydrogel to incorporate a component capable of strong aggregation into a thermoresponsive polymer network: structural fixation by strong aggregation moieties is possible when a matrix gel shrinks in response to a temperature change (Fig. 1a). Such structural fixation is a key mechanism for a shape-memory hydrogel,15–17 which is able to temporarily fix a macroscopic shape and recover to the original shape by forming and deconstructing physical crosslinking (hydrophobic interactions, hydrogen bonds, etc.) in the network upon external stimuli such as temperature. The control of properties by reorganization of physical interactions was also observed on annealing a crystalline polymer material.18,19 Thus, the presence of an aggregation component inducing physical crosslinking in a thermoresponsive hydrogel network would be crucial to an “irreversible” property change derived from fixation of the internal structure upon thermal stimuli.
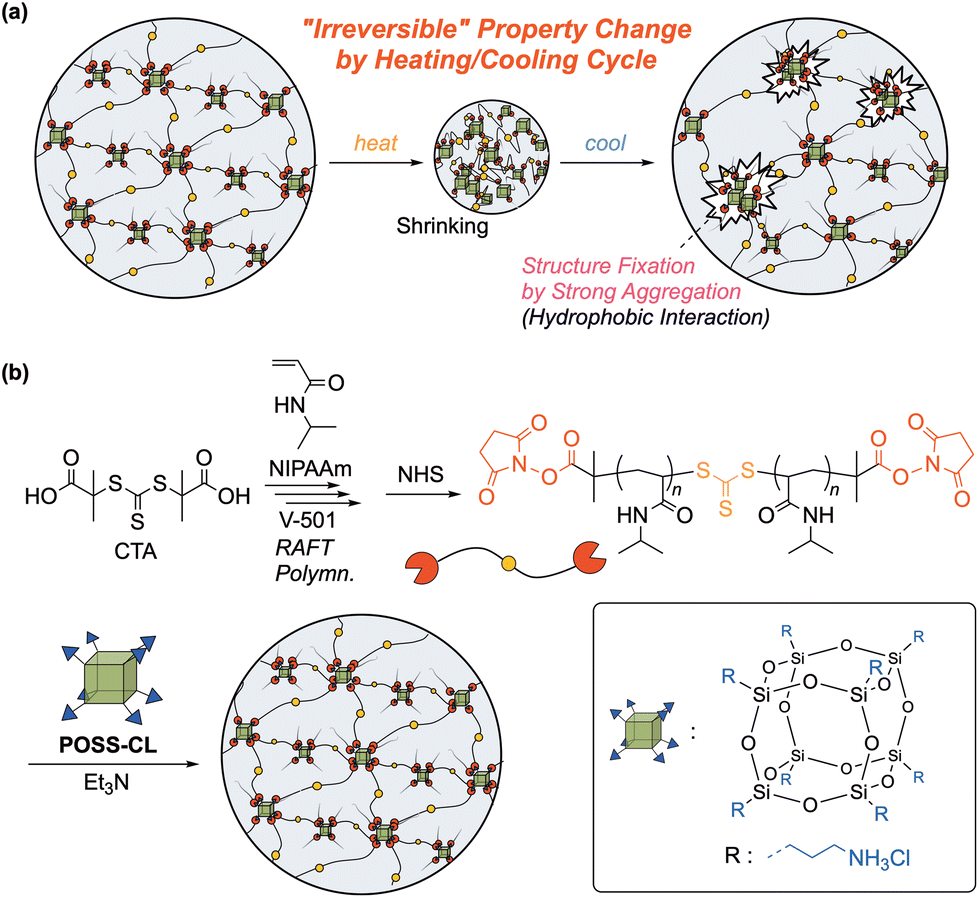 | ||
| Fig. 1 (a) Thermoresponsive irreversible property change induced by a strong aggregation effect in a hydrogel network, and (b) synthetic scheme of the end-crosslinked PNIPAAm gel with a POSS crosslinker as an aggregation component. | ||
In this study, we focused on a polyhedral oligomeric silsesquioxane (POSS) compound (Fig. 1b), rigid and highly hydrophobic, as an aggregation component. Typical POSS compounds consist of (RSiO1.5)8 with a siloxane nanosized cage and 8 functional groups (R) bonded to the corner Si atoms.20,21 Since POSS compounds have an inorganic core surrounded by organic substituents, they exhibit good dispersibility at the molecular level to a variety of matrixes, and POSS derivatives can also be multifunctional, serving as a crosslinker. The hybridization of POSS is reported to improve the mechanical properties of a matrix polymer.21–24 Incorporation of POSS into a hydrogel is also intriguing, and, for example, improvement of the mechanical properties and rapid swelling/shrinking behavior of POSS-hybridized gels were reported.25–30 In addition, Naka et al. recently found a significantly reduced dissolution rate of POSS-linked poly(azomethine) film in a solvent after casting, although this polymer was well-dissolved in the same solvent during polymerization.31 This unique behavior may be attributed to an enhanced entanglement based on the bulky structure of the POSS moieties in the main chain. Such a strongly cohesive and hydrophobic POSS structure is a promising candidate as an aggregation component for structural fixation in an “irreversible” thermoresponsive gel (Fig. 1a). Thus, we examined the synthesis of a new class of POSS-containing poly(N-isopropylacrylamide) (PNIPAAm) gels, typical thermoresponsive gels,32–34 using a POSS-based crosslinker (Fig. 1b). In order to control the aggregation and entanglement behavior of POSS moieties, the control of the distance between neighboring POSS units in a network would be crucial. Therefore, we utilized an end-crosslinking system for gel synthesis, in which a uniform telechelic polymer precursor is treated with a multifunctional POSS crosslinker to produce a gel with a uniform distance between crosslinking points.35–37
In this study, we examined gel synthesis utilizing a POSS derivative with 8 amino groups at the corners as a crosslinker for the reaction with telechelic PNIPAAm having an activated ester terminus (Fig. 1b). The product gel irreversibly changed the appearance and mechanical properties in heating/cooling cycles. We investigated the effect of the molecular weight of the telechelic polymer precursors on the responsive behavior to discuss the mechanism of this unique behavior. We also examined the possibility of restoring the altered properties to the initial state by re-dispersing aggregated POSS moieties.
Experimental
Materials
NIPAAm (FUJIFILM Wako Pure Chemical, >98.0%) was purified by recrystallization from toluene/n-hexane. S,S′-Bis(α,α′-dimethylacetic acid) trithiocarbonate, a chain transfer agent (CTA) for RAFT polymerization, was prepared as reported in the literature.38 A POSS crosslinker carrying 8 ammonium chloride groups (POSS-CL; Hybrid Plastics Inc.), 4,4′-azobis(4-cyanovaleric acid) (V-501; FUJIFILM Wako Pure Chemical, >98.0%), N-hydroxysuccinimide (NHS; FUJIFILM Wako Pure Chemical, >98.0%), N,N′-dicyclohexylcarbodiimide (DCC; Tokyo Chemical Industry, >98%), 4-dimethylaminopyridine (DMAP; FUJIFILM Wako Pure Chemical, >98.0%), triethylamine (TEA; FUJIFILM Wako Pure Chemical, >99.0%), N,N′-methylenebisacrylamide (BIS; FUJIFILM Wako Pure Chemical, for electrophoresis, >99.0%), n-hexylamine (Tokyo Chemical Industry, >99%), 2-hydroxyethyl acrylate (Tokyo Chemical Industry, >95%), tris(2-carboxyethyl)phosphine hydrochloride (TCEP; Tokyo Chemical Industry, >98%), 1,2,3,4-tetrahydronaphthalene (tetralin; Sigma-Aldrich, 99.0%), 1,4-dioxane (FUJIFILM Wako Pure Chemical, for organic synthesis, >99.5%), dichloromethane (DCM, FUJIFILM Wako Pure Chemical, >99.5%), N,N-dimethylformamide (DMF; FUJIFILM Wako Pure Chemical, for organic synthesis, 99.5%), tetrahydrofuran (THF; FUJIFILM Wako Pure Chemical, for organic synthesis, 99.5%), dimethylsulfoxide (DMSO; FUJIFILM Wako Pure Chemical, for organic synthesis, 99.0%), chloroform (FUJIFILM Wako Pure Chemical, for organic synthesis, 99.0%), methanol (FUJIFILM Wako Pure Chemical, for organic synthesis, 99.8%), diethyl ether (FUJIFILM Wako Pure Chemical, >99.0%), 1 mol L−1 hydrochloric acid (FUJIFILM Wako Pure Chemical, for volumetric analysis), CDCl3 (Cambridge Isotope Laboratories, 99.5%), and CD3OD (Cambridge Isotope Laboratories, 99.8%) were used as received.Measurement and characterization
The number-average molecular weight (Mn), the weight-average molecular weight (Mw), and the polydispersity index (Mw/Mn) of polymers were determined by size-exclusion chromatography (SEC; a Shimadzu LC-10A system consisting of an LC-10AD precision pump and an RID-10A refractive index detector, or a Shimadzu Prominence system consisting of an LC-20AD precision pump and an RID-20A refractive index detector) in DMF containing 10 mM LiBr as an eluent at 40 °C (flow rate: 1.0 mL min−1) using three polystyrene gel columns (Shodex KF-805L). The columns were calibrated against standard poly(methyl methacrylate) samples (Agilent, Mn = 1.86 × 103–1.68 × 106).1H nuclear magnetic resonance (NMR) spectra were recorded on a JEOL JNM-LA400 spectrometer operating at 399.65 MHz, or a JEOL JNM-ECS400 operating at 399.90 MHz. The degree of polymerization (DPn) and Mn,NMR were calculated from the integral values of peaks derived from CTA and monomeric units. 29Si NMR spectra (80 MHz) were recorded on a Bruker AVANCE III 400 NMR spectrometer in CDCl3.
The swelling degree of the gels was determined by measuring the weight of the gel samples. The weight, w, of the samples after immersion in various solvents at the predetermined temperature was measured, and the swelling degree was calculated using w/w0; w0 is the weight of the as-prepared gel.
A uniaxial tensile test was conducted with a Shimadzu EZ-SX using rectangular specimens with the dimensions of ca. 2 × 10 × 20 mm. The cross-head speed was 5.0 mm min−1. The Young's modulus was calculated from the initial slope of the stress–strain curve. In this study, 5–8 specimens were prepared and tested for each condition, and the representative results are shown.
The turbidity measurement of the gels was conducted with a Jasco V-750 UV/vis spectrophotometer. Rectangular specimens (thickness: 3.0–3.7 mm) were attached to an interior side in a measurement cell, and the transmittance of 500 nm light, Texp, was measured. The value was normalized to those of the gel samples with 3.0 mm thickness using the following equation: Tn = Td/3exp, where Tn is the normalized transmittance and d (mm) is the thickness of the sample.
Synthesis of telechelic PNIPAAm
A typical example is given below. NIPAAm (11.3 g, 100 mmol), CTA (282 mg, 1.00 mmol), V-501 (28.1 mg, 0.100 mmol), tetralin (2.0 mL), and 1,4-dioxane (48 mL) were added into a 100 mL round-bottomed flask equipped with a three-way stopcock and bubbled with nitrogen for 10 minutes. The flask was placed in an oil bath kept at 60 °C for 25 h. The reaction was terminated by cooling the reaction mixture to −60 °C, and the reaction mixture was poured into diethyl ether to obtain the purified PNIPAAm carrying carboxy groups at both ends (9.93 g; DPn = 102 and Mn = 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800, both of which were calculated by 1H NMR analysis).
800, both of which were calculated by 1H NMR analysis).
The obtained PNIPAAm (9.88 g, 0.836 mmol), NHS (481 mg; 4.18 mmol), DCC (863 mg; 4.18 mmol), and DMAP (10.2 mg; 0.0836 mmol) were dissolved in 41.8 mL of DCM, and the reaction mixture was stirred at room temperature under nitrogen for 24 h. The solution was filtered, and the filtrate was poured into diethyl ether to obtain the purified telechelic PNIPAAm (9.09 g).
Gel synthesis
A typical example is given below. A telechelic PNIPAAm (DPn = 103 and Mn = 12100, 294 mg; 0.0243 mmol) was dissolved in a mixture of TEA (0.17 mL; 1.2 mmol) and DMF (0.33 mL). To this solution, 0.50 mL solution of POSS-CL in DMF/H2O (3/1, v/v) containing 7.1 mg (0.012 mmol) of POSS-CL was added, and the reaction mixture was held at room temperature for 48 h. The gelation time was determined by a tilting method checking the loss of fluidity of the reaction solution at the predetermined intervals. The obtained gel (as-prepared state) was washed by immersion in a high amount of DMF, 0.10 mol L−1 HClaq, and water in this order for several days in each step. The gel specimens after washing (the initial stage before a heating/cooling cycle) were heated at 40 °C in water for 24 h and then cooled to room temperature in water for 24 h to obtain gel samples after the heating/cooling cycle. The samples before and after the heating/cooling cycle were employed for tensile and turbidity measurements. For the turbidity measurement, rectangular specimens after a heating/cooling cycle in water were immersed in various solvents (DMSO, DMF, methanol, and THF) for 48 h to substitute the internal solvent. In the case of solvents immiscible with water (DCM and CHCl3), the gel specimens were immersed in these solvents for 48 h after immersion in THF for 24 h.
Decomposition of gel
A gel sample (ca. 1.0 mL) was immersed in THF for 24 h to substitute the internal solvent with THF. Then, the gel was immersed in 10 mL of THF containing n-hexylamine (0.017 mL; 0.13 mmol), HEA 0.013 mL (0.13 mmol) and TCEP (1.5 mg, 0.005 mmol), and the mixture was stirred at room temperature under nitrogen for 24 h. The obtained solution was poured into an excess of diethyl ether to obtain the decomposition product.Results and discussion
Synthesis of telechelic polymer precursors
Telechelic polymer precursors for gel synthesis were prepared by RAFT polymerization and the subsequent end-functionalization as reported in our previous report.35 First, NIPAAm was polymerized using S,S′-bis(α,α′-dimethylacetic acid) trithiocarbonate, a bifunctional CTA with two carboxy groups in the molecule, in conjunction with V-501 as a radical source. A variety of feed ratios of NIPAAm and CTA were examined. Under all conditions, well-controlled polymers with narrow molecular weight distributions were obtained (Mw/Mn ∼1.2; Fig. S1 and S2 and Table S1 in the ESI†).The obtained PNIPAAms were treated with NHS in DCM to convert the terminal carboxy groups into activated esters, which exhibit high reactivity to a primary amine under mild conditions.39 The almost quantitative esterification was confirmed from the 1H NMR spectra of the products, specifically the integral ratio of the signals derived from methylene protons in activated ester groups (Fig. S3 in the ESI†). The SEC curves remained unimodal after the reaction, indicating that no side reaction such as polymer coupling or main chain scission occurred (Fig. S4 in the ESI†). Thus, the telechelic polymer precursors, having an activated ester terminus with different molecular weights, were successfully prepared (P103, P152, and P197; Table 1).
| Codeb | DPn,NMR | M n,NMR | M w/Mn,SEC |
|---|---|---|---|
| a Synthetic conditions of RAFT polymerization: [NIPAAm] = 2000 mM, [CTA]/[V-501] = 10 in 1,4-dioxane at 60 °C for 25 h. Reaction conditions of end-functionalization: [PNIPAAm] = 20 mM, [NHS] = 100 mM, [DCC] = 100 mM, [DMAP] = 2.0 mM in DCM at room temperature for 24 h. b “P” and the subscript number stand for “telechelic polymer” and DPn,NMR of the polymers. | |||
| P103 | 103 | 12100 |
1.23 |
| P152 | 152 | 17700 |
1.18 |
| P197 | 197 | 22800 |
1.24 |
Gel synthesis
The reactions of the product telechelic PNIPAAms with a functionalized POSS, as a crosslinker, were examined in a mixed solvent of DMF and H2O (H2O: 12.5 vol%) at room temperature (Fig. 1b). The crosslinker POSS employed was POSS-CL, a POSS derivative containing 8 ammonium cation groups (–NH3Cl) in the molecule. No gelation proceeded even at a higher polymer concentration, whereas efficient gelation was observed in the presence of TEA (Fig. 2a). In addition, the gelation became faster with increased concentration of TEA (Fig. 2b). These results suggested that TEA converted the ammonium cations of POSS-CL to neutral amino groups (–NH2) with high reactivity to an activated ester, promoting the crosslinking reaction. The neutralization of the ammonium cations should also be quick since water, a cosolvent in the reaction, would induce the hydrolysis of the activated esters at the polymer ends. Thus, a large excess of TEA was required for effective gelation.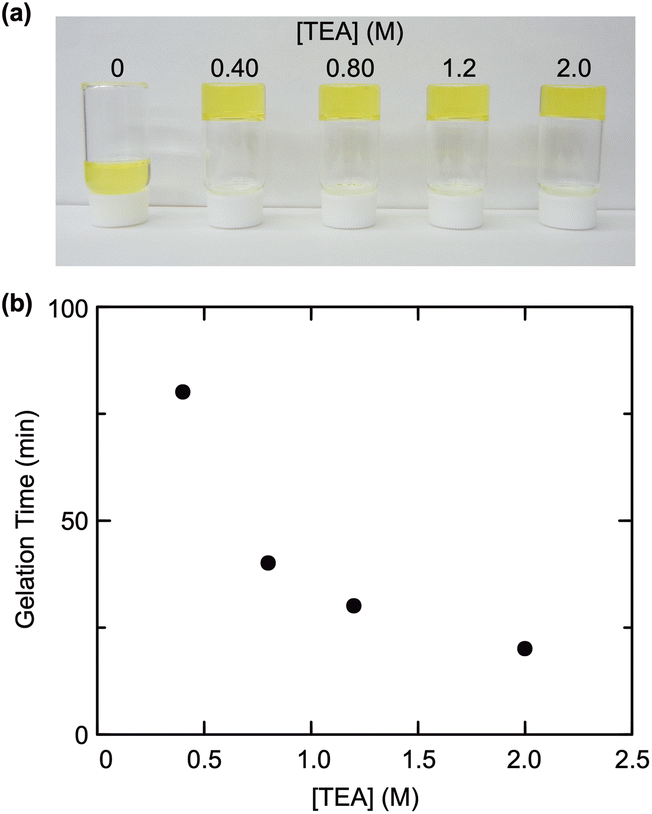 | ||
| Fig. 2 Effect of feed TEA concentration on (a) gelation behavior and (b) gelation time. Reaction conditions: [P103] = 25 mM, [POSS-CL] = 6.25 mM, [TEA] = 0–2.0 M in DMF/H2O (H2O: 12.5 vol%) at room temperature. | ||
The addition order of the reagents was found to be also important in the examined gelation reaction. Adding a DMF/H2O solution of POSS-CL to a DMF solution of PNIPAAm and TEA was essential to efficiently obtain a gel with good reproducibility. Although the neutralization of POSS-CL in advance appears reasonable, the prior mixing of TEA with a DMF/H2O solution of POSS-CL produced a phase-separated solution (Fig. S5 in the ESI†), leading to poor neutralization of POSS-CL. The decreased solubility of a neutralized POSS-CL may also affect the gelation reaction, since precipitation occurred when the ratio of DMF against water increased in the reaction solution. Thus, a gel can be obtained only when the reaction of the telechelic PNIPAAm with the crosslinker POSS can start as soon as POSS-CL is neutralized in the same reaction mixture, which was achieved by the addition of POSS-CL to the telechelic PNIPAAm solution containing TEA.
The product gels were immersed in a large amount of DMF to wash out the byproducts such as NHS produced by the amidation reaction, and then immersed in HClaq since the basic condition derived from TEA and the unreacted amino group would be harmful to base-labile trithiocarbonate groups in the network. Finally, the gels were immersed in water to substitute the internal solvent, and the obtained hydrogels were employed for the various measurements described below.
Thermoresponsive swelling behavior in water
The product POSS-crosslinked PNIPAAm gels exhibited thermoresponsive shrinking in water irrespective of the TEA concentration in the preparation (Fig. S6 in the ESI†). The gel prepared at [TEA] = 400 mM showed a higher swelling degree at a low temperature than the gels obtained at higher TEA concentrations, indicative of a low crosslinking efficiency at [TEA] = 400 mM. This swelling behavior is consistent with the relationship between TEA concentration and gelation time (Fig. 2). The longer reaction time at a low TEA concentration probably induces the hydrolysis of the activated ester end groups of the PNIPAAm prepolymer, which hinders the crosslinking reaction. The shrinking temperatures of POSS-crosslinked gels were at around 25–30 °C regardless of the TEA feed concentration in the preparation. The response temperature was slightly lower than the phase transition temperature (33 °C) of a typical PNIPAAm gel prepared by radical polymerization with BIS,32 because of the effect of the hydrophobic POSS crosslinker. The gel turned opaque upon heating, as observed with a conventional PNIPAAm gel; however, surprisingly, the gel did not return to the transparent state after cooling (Fig. 3). This lasting turbidity after heating was probably attributed to the fixation of the aggregated POSS moieties in the network induced by macroscopic shrinking of the gel upon heating. | ||
| Fig. 3 Appearance change of (a) POSS-crosslinked PNIPAAm gel and (b) BIS-crosslinked PNIPAAm gel against a heating/cooling cycle in H2O. Preparation conditions: (POSS-crosslinked gel) [P103] = 25 mM, [POSS-CL] = 6.25 mM, [TEA] = 1.2 M in DMF/H2O (H2O: 12.5 vol%) at room temperature, and (BIS-crosslinked gel) [NIPAAm] = 2500 mM, [BIS] = 25 mM, [V-70] = 5.0 mM in DMF at room temperature. | ||
Despite the continuing turbidity, the swelling degree returned to the original value after heating/cooling cycles (Fig. 4). The reaction of a longer prepolymer with the constant monomer unit concentration ([NIPAAm unit] = 2500 mM) yielded a gel (G152 and G197: the subscript number in a gel code corresponds to the DPn of the prepolymer) exhibiting a higher swelling degree as well as irreversible turbidity against a heating/cooling cycle (Fig. 4 and Fig. S7 in the ESI†). However, the turbidity of this gel was paler than that of the gel from the shorter prepolymer (G103). These results indicated that the longer prepolymer chains suppressed the aggregation of POSS moieties in the shrinking of the gel upon heating. The swelling degree of G197 at low temperatures increased from the initial value after undergoing a heating/cooling cycle. In addition, the volume in the heating state increased in all the gels examined. The increase in volume was likely attributed to the chain cleavage at the labile trithiocarbonate groups located at the center of each network chain during re-swelling after the formation of POSS aggregates at the first heating. In particular, the effect of the chain cleavage was more evident in the gel prepared from longer prepolymers (G197) because of the relatively low concentration of the network chains. Thus, an appropriate balance of POSS aggregation and chain cleavage results in a reversible volume change despite the continuing turbidity.
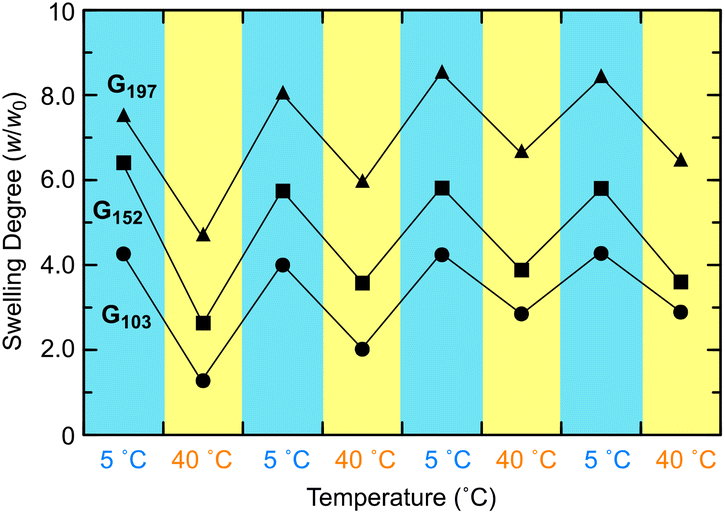 | ||
| Fig. 4 Swelling behaviors in water of POSS-crosslinked PNIPAAm gels prepared from prepolymers with different molecular weights against heating/cooling cycles. The samples are denoted as Gx using x as DPn of the prepolymers. Preparation conditions: [NIPAAm unit] = 2500 mM, [PNIPAAm] = 4 × [POSS-CL] with an excess of TEA (G103: 1.2 M, G152: 1.8 M, G197: 2.4 M) in DMF/H2O (H2O: 12.5 vol%) at room temperature. | ||
Mechanical property change by the heating/cooling cycle
The appearance change through heating/cooling cycles suggests a change in the internal structure of the gel, which would affect its mechanical properties. Thus, we investigated the effects of temperature and heating and cooling processes on the mechanical properties of the POSS-crosslinked gels. Three gel samples, prepared from prepolymers with different molecular weights, were employed for the uniaxial tensile test (Fig. 5 and Table 2). For all the POSS-crosslinked gels, the Young's modulus and elongation increased after heat treatment in contrast to a conventional PNIPAAm gel prepared by free radical polymerization of NIPAAm in the presence of a divinyl crosslinker, BIS, which shows a slight change in mechanical properties. In particular, the prepolymer with the lower molecular weight caused a significant increase in Young's modulus, as observed with G103 (Fig. 5a), while a prepolymer with a higher molecular weight resulted in an increased elongation (G197: Fig. 5c). This tendency may be due to the differences in the physical crosslinking effect based on the aggregation of POSS moieties. With a gel from a prepolymer with a low molecular weight, the relatively short distance between neighboring POSS moieties in the network and a less steric effect around POSS moieties are likely to result in a strong hydrophobic interaction between POSS units that would cause a significant increase in Young's modulus. Such physical crosslinking caused by the POSS aggregation would be hampered by longer network chains, derived from longer prepolymers. The weakened physical crosslinking is likely to give an appropriate strength, which, in general, contributes to an energy dissipation upon deformation,40 possibly resulting in a noticeable increase in elongation. Moreover, the mechanical properties of G197 after the 2nd heating/cooling cycle remained almost unchanged from those after the 1st heating/cooling cycle (Fig. S8 in the ESI†), indicating that the gel reached an equilibrium aggregation state after the 1st heating/cooling cycle.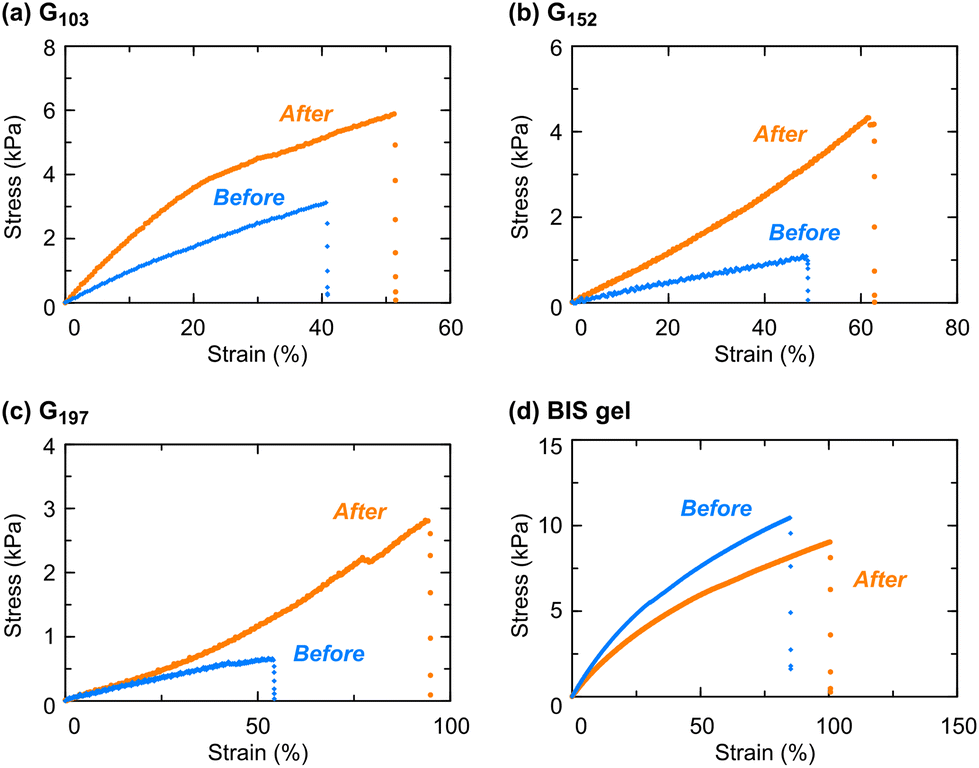 | ||
| Fig. 5 Representative stress–strain curves obtained by uniaxial tensile tests of (a)–(c) POSS-crosslinked PNIPAAm gels prepared from prepolymers with different molecular weights and (d) BIS-crosslinked PNIPAAm before and after the heating/cooling cycle. Preparation conditions of the BIS-crosslinked gel: [NIPAAm] = 2500 mM, [BIS] = 25 mM, [V-70] = 5.0 mM in DMF at room temperature. | ||
| Young's modulus (kPa) | Breaking strain (%) | |||
|---|---|---|---|---|
| Before | After | Before | After | |
| G103 | 10.2 ± 0.6 | 19.3 ± 1.3 | 40 ± 8 | 53 ± 7 |
| G152 | 2.73 ± 0.20 | 5.38 ± 0.72 | 46 ± 3 | 61 ± 13 |
| G197 | 1.58 ± 0.10 | 2.43 ± 0.46 | 50 ± 5 | 88 ± 13 |
| BIS gel | 23.8 ± 1.9 | 20.3 ± 1.0 | 81 ± 4 | 108 ± 10 |
Evaluation of POSS aggregation
In order to understand the effect of a heating/cooling cycle on the internal structure of the POSS-crosslinked PNIPAAm gels, we decomposed the POSS gels and analyzed the degradation products. The trithiocarbonate groups in the middle of the network chains derived from CTA in the prepolymers were cleaved by the aminolysis reaction41 in the presence of TCEP, as a reducing agent, and HEA, as a capping agent, which underwent the Michael addition reaction with a thiol group, preventing the dimerization of thiol groups (Fig. 6a). The degradation of the POSS-crosslinked PNIPAAm gels before applying a heating/cooling cycle yielded soluble polymers, regardless of the molecular weight of a prepolymer or the length of a network chain. The SEC curves of the decomposition products consist of three major peaks (Fig. 6b–d; black solid lines). The main peak, several times higher in molecular weight than the prepolymer (black dotted lines in Fig. 6b–d), was attributable to a POSS-centered branched polymer formed by the crosslinking reaction. The peak in the lowest molecular weight region with about half the molecular weight of the prepolymer was assigned to a cleaved polymer derived from a dangling chain with an unreacted end-group. The proportion of this unreacted polymer was small in all cases, indicating that the crosslinking reaction proceeded in high efficiency. A relatively larger amount of dangling chains was observed in the degradation product of a gel prepared from a higher molecular weight prepolymer, indicative of less efficient crosslinking due to the steric hindrance. | ||
| Fig. 6 (a) Degradation of POSS-crosslinked hydrogels by the aminolysis reaction and (b)–(d) SEC curves of the degradation product from the gels with various molecular weights (red line: degradation products from gels after a heating/cooling cycle, black solid line: degradation products from gels before a heating/cooling cycle, and black dotted line: prepolymers). | ||
The molecular weight of the decomposition product after the heating/cooling process was appreciably higher (red lines in Fig. 6b–d), compared to that before applying the heating/cooling cycle. In particular, a larger amount of a high molecular weight product was obtained from the gel prepared from the shorter prepolymer. Furthermore, the amount of the high molecular weight product remained almost unchanged even after a longer or repeated degradation reaction. The high molecular weight compound is likely a highly branched polymer containing multiple POSS linkers derived from the suppression of the cleavage reaction due to the POSS aggregation in the gel network. The low content of high molecular weight compounds in the degradation product from G197 after a heating/cooling cycle also supported that the degree of aggregation became small when the longer prepolymer was employed for the gel synthesis, as discussed in the mechanical behavior section.
The degradation product was also analyzed by 29Si NMR spectroscopy to obtain the information on the state of POSS moieties in the heating/cooling cycle. The product from G103 without a heating/cooling process exhibited no peak in the spectrum (Fig. S9 in the ESI†), probably due to the small fraction of the POSS moiety in a long PNIPAAm polymer chain. In order to increase the relative volume fraction of the POSS moiety in a network, a shorter prepolymer (DPn = 25) was used for the crosslinking reaction with POSS-CL (synthetic condition of POSS-crosslinked gel: [prepolymer] = 100 mM, [POSS-CL] = 25 mM). The 29Si NMR spectrum of the degradation product before applying a heating/cooling cycle exhibited a peak at around −66.8 ppm assignable to a silicon atom in the corner of a POSS cage (Fig. S10 in the ESI†). The same peak but no other peak was observed in the spectrum of the degraded polymer from the gel after a heating/cooling process (Fig. S10 in the ESI†). These results suggested that the state of chemical bonding of a silicon atom remained unchanged even after a heating/cooling cycle: side reactions such as a cleavage in the POSS cage or a bond exchange between POSS moieties were negligible. Thus, the continuing turbidity in the appearance and the change in mechanical properties were likely to result from the physical aggregation of POSS without any chemical side reactions.
“Renaturation” by solvent immersion
Since the “irreversible” property change of the POSS-crosslinked PNIPAAm gel is likely to originate from the POSS aggregation, an immersion into good solvents would be effective for the re-dispersion of the aggregated POSS moieties and the return of gel properties to the original state. Thus, we immersed a POSS-crosslinked PNIPAAm gel (G103) after a heating/cooling cycle in a variety of solvents (Fig. 7). In the case of chloroform and DCM, which were immiscible with water, the solvent of the samples was exchanged with THF before immersion. As a result, only the gel immersed in DMSO became translucent, whereas those immersed in other solvents were opaque or cloudy, as also demonstrated by the transmittance measurement of a gel film (Table S2 in the ESI†). Interestingly, a good solvent for PNIPAAm is not necessarily effective for regaining transparency: DMF and methanol had a moderate effect on the recovery of transparency, while chloroform and DCM augmented the turbidity. These results were in good accordance with the solubility of POSS-CL in various solvents in the presence of the base: POSS-CL was soluble in DMSO and methanol (Fig. S11 in the ESI†). The affinity of the solvents for POSS moieties was likely to play an important role in the recovery of the transparency. A highly polar solvent was shown to be efficient for re-dispersion of POSS moieties in the network. Importantly, the gel that became transparent again after the immersion in DMSO was very similar in Young's modulus and elongation to the gel in the initial state (Fig. S12 in the ESI;† the tensile test was conducted after the solvent substitution from DMSO to water). Thus, solvent immersion realizes the return of the properties or “renaturation” of POSS-crosslinked PNIPAAm gels. | ||
| Fig. 7 Appearances of POSS-crosslinked PNIPAAm gels immersed in various solvents after applying a heating/cooling cycle in water. | ||
Conclusions
In this study, we successfully synthesized POSS-crosslinked PNIPAAm hydrogels by end-crosslinking of telechelic prepolymers with activated ester ends using the multifunctional crosslinker POSS-CL having ammonium cation sites at the corners of the cage structure. The product gels were shown to undergo an “irreversible” change in the appearance and the mechanical properties. The presence of a large amount of base (Et3N) was crucial to obtain a gel with good reproducibility, since this condition enabled an effective conversion of the ammonium cations in POSS-CL into neutral amino groups with high reactivity, followed by an immediate reaction with PNIPAAm prepolymers. The product gels exhibited reversible swelling/deswelling against a temperature change in water, as is the case with a typical PNIPAAm gel; however, the appearance did not return to the original transparency after heating. This “irreversible” change in the turbidity was likely to result from the aggregation of POSS moieties induced by macroscopic volume shrinking of the product PNIPAAm gels. The internal structural change also caused an increase in Young's modulus and elongation, which was significant particularly with the gel prepared from a shorter prepolymer. Despite the strong interaction between POSS moieties, the turbidity of POSS-crosslinked gels after a heating/cooling cycle can be reversed. The transparency was regained to a large extent by immersion of the gel in DMSO. Such transient fixation of hydrogel properties is regarded analogous to the denaturation/renaturation process of natural hydrogels, and it would contribute to broadening the scope of multi-step stimuli-responsive materials.Author contributions
S.I. conceived the idea, designed the experiments, and wrote the manuscript. T.H., A.K., T.M. and S.S. designed and performed most of the experiments. H.I. and K.N. contributed to the preparation of POSS compounds and 29Si NMR analysis. S.K. supervised the project, and edited and revised the manuscript. All the authors discussed the results and contributed to the data interpretation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the Japan Society for the Promotion of Science through a Grant-in-aid for Young Scientists (B) (No. 16K17962) and a Grant-in-aid for Scientific Research (C) (No. 19K05602), for which the authors are grateful.References
- R. Pelton, Adv. Colloid Interface Sci., 2000, 85, 1–33 CrossRef CAS PubMed.
- A. Kikuchi and T. Okano, Adv. Drug Delivery Rev., 2002, 54, 53–77 CrossRef CAS PubMed.
- C. d. l. H. Alarcón, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2009, 5, 2513–2533 RSC.
- X. Lin, X. Wang, L. Zeng, Z. L. Wu, H. Guo and D. Hourdet, Chem. Mater., 2021, 33, 7633–7656 CrossRef CAS.
- K. Matsumoto, N. Sakikawa and T. Miyata, Nat. Commun., 2018, 9, 2315 CrossRef PubMed.
- X.-H. Li, C. Liu, S.-P. Feng and N. X. Fang, Joule, 2019, 3, 290–302 CrossRef CAS.
- T. Nonoyama, Y. W. Lee, K. Ota, K. Fujioka, W. Hong and J. P. Gong, Adv. Mater., 2020, 32, 1905878 CrossRef CAS PubMed.
- Q. Zhang, C. Weber, U. S. Schubert and R. Hoogenboom, Mater. Horiz., 2017, 4, 109–116 RSC.
- S. Ida, T. Kawahara, Y. Fujita, S. Tanimoto and Y. Hirokawa, Macromol. Symp., 2015, 350, 14–21 CrossRef CAS.
- S. Ida, D. Nishisako, A. Fujiseki and S. Kanaoka, Soft Matter, 2021, 17, 6063–6072 RSC.
- C. Branden and J. Tooze, Introduction to Protein Structure, Garland Publishing, New York, US, 2nd edn, 1999 Search PubMed.
- B. Alberts, A. Johnson, J. Lewis, D. Morgan, M. Raff, K. Roberts and P. Walter, Molecular Biology of The Cell, W. W. Norton & Company, New York, US, 6th edn, 2015 Search PubMed.
- T. Z. Yuan, C. F. G. Ormonde, S. T. Kudlacek, S. Kunche, J. N. Smith, W. A. Brown, K. M. Pugliese, T. J. Olsen, M. Iftikhar, C. L. Raston and G. A. Weiss, ChemBioChem, 2015, 16, 393–396 CrossRef CAS PubMed.
- Y. Osada and A. Matsuda, Nature, 1995, 376, 219–219 CrossRef CAS PubMed.
- J. Shang, X. Le, J. Zhang, T. Chen and P. Theato, Polym. Chem., 2019, 10, 1036–1055 RSC.
- C. Löwenberg, M. Balk, C. Wischke, M. Behl and A. Lendlein, Acc. Chem. Res., 2017, 50, 723–732 CrossRef PubMed.
- C. N. Rochette, S. Rosenfeldt, K. Henzler, F. Polzer, M. Ballauff, Q. Tong, S. Mecking, M. Drechsler, T. Narayanan and L. Harnau, Macromolecules, 2011, 44, 4845–4851 CrossRef CAS.
- Q. Zhang, Z. Mo, S. Liu and H. Zhang, Macromolecules, 2000, 33, 5999–6005 CrossRef CAS.
- R. H. Baney, M. Itoh, A. Sakakibara and T. Suzuki, Chem. Rev., 1995, 95, 1409–1430 CrossRef CAS.
- D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081–2173 CrossRef CAS PubMed.
- K. Tanaka, S. Adachi and Y. Chujo, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5690–5697 CrossRef CAS.
- G. Z. Li, L. Wang, H. Toghiani, T. L. Daulton, K. Koyama and C. U. Pittman, Macromolecules, 2001, 34, 8686–8693 CrossRef CAS.
- S. Ponnupandian, P. Mondal, T. Becker, R. Hoogenboom, A. B. Lowe and N. K. Singha, Polym. Chem., 2021, 12, 876–884 RSC.
- K. Zeng, L. Wang and S. Zheng, J. Phys. Chem. B, 2009, 113, 11831–11840 CrossRef CAS PubMed.
- Y. Chen, Y. Xiong, C. Peng, W. Liu, Y. Peng and W. Xu, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1494–1504 CrossRef CAS.
- D. Prządka, E. Andrzejewska and A. Marcinkowska, Eur. Polym. J., 2015, 72, 34–49 CrossRef.
- U. Haldar, M. Nandi, B. Maiti and P. De, Polym. Chem., 2015, 6, 5077–5085 RSC.
- W. Pu, F. Jiang, P. Chen and B. Wei, Soft Matter, 2017, 13, 5645–5648 RSC.
- A. Romo-Uribe and L. Albanil, ACS Appl. Mater. Interfaces, 2019, 11, 24447–24458 CrossRef CAS PubMed.
- S. Fujii, S. Minami, K. Urayama, Y. Suenaga, H. Naito, O. Miyashita, H. Imoto and K. Naka, ACS Macro Lett., 2018, 7, 641–645 CrossRef CAS PubMed.
- Y. Hirokawa and T. Tanaka, J. Chem. Phys., 1984, 81, 6379–6380 CrossRef.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- A. Halperin, M. Kröger and F. M. Winnik, Angew. Chem., Int. Ed., 2015, 54, 15342–15367 CrossRef CAS PubMed.
- S. Ida, R. Kimura, S. Tanimoto and Y. Hirokawa, Polym. J., 2017, 49, 237–243 CrossRef CAS.
- S. Ida, M. Yamawaki, T. Maruta and Y. Hirokawa, Trans. Mater. Res. Soc. Jpn., 2018, 43, 71–74 CrossRef CAS.
- S. Ida, Polym. J., 2019, 51, 803–812 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6677–6687 CrossRef CAS.
- X. Zhao, Soft Matter, 2014, 10, 672–687 RSC.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional 1H NMR spectra and SEC curves of prepolymers, appearances of samples, swelling behavior of gels, results of uniaxial tensile tests, 29Si NMR spectra of decomposition products, and transmittance of gel films. See DOI: https://doi.org/10.1039/d3py00097d |
| This journal is © The Royal Society of Chemistry 2023 |