
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From thermoplastic polyurethane to covalent adaptable network via reversible photo-crosslinking of a biobased chain extender synthesized from caffeic acid†
Antoine
Duval
 *ab and
Luc
Avérous
*a
*ab and
Luc
Avérous
*a
aBioTeam/ICPEES-ECPM, UMR CNRS 7515, Université de Strasbourg, 25 rue Becquerel, 67087 Strasbourg Cedex 2, France. E-mail: antoine.duval@unistra.fr; luc.averous@unistra.fr
bSoprema, 15 rue de Saint-Nazaire, 67025 Strasbourg, France
First published on 3rd May 2023
Abstract
An original segmented thermoplastic polyurethane (TPU) has been synthesized with a biobased aromatic chain extender prepared from caffeic acid (EC-diol). Some reference TPUs were also synthesized with aliphatic or aromatic chain extenders, for comparison. The TPUs were characterized by NMR, FTIR, TGA, DSC, DMA and tensile tests. The TPU based on EC-diol presents the characteristic features of thermoplastic elastomers, with phase segregation between soft and hard segments, leading to remarkable mechanical and thermal properties. Thanks to the peculiar structure of EC-diol, the corresponding TPU presents unsaturated esters as pendant chains, which are able to dimerize under UV irradiation by [2 + 2] cycloaddition, leading to crosslinking. The photodimerization was followed by UV-vis and FTIR spectroscopies. It is complete in about 2 h on thin films but is limited by the penetration depth of the UV radiation, and longer exposure is required to fully crosslink the TPU in the bulk. Photocrosslinking leads to significant changes in the physical properties of the material, with for instance an increased rigidity. When irradiated with UV light of shorter wavelength, the photodimerization is reversible, leading to decrosslinking. However, the reversibility is only partial. This study thus shows an original TPU able to reversibly crosslink upon UV irradiation at different wavelength, but also points some limitations of this dissociative covalent adaptable network for developing thick materials, because of the well-known limited penetration depth of the UV light.
Introduction
Thermoplastic polyurethanes (TPUs) are a class of highly versatile polymer materials, whose properties can easily be tuned by adjusting their macromolecular architecture, leading to a large range of industrial applications. They are conventionally synthesized in two-steps: first a prepolymer is obtained by reaction of a long polyol with an excess of diisocyanate, then a short diol is added as chain extender. The obtained TPUs possess a block copolymer structure, with soft segments (SS) constituted by a long polyol and hard segments (HS) formed by reaction between the diisocyanate and the chain extender. In HS, the high density of urethane linkages allows the formation of hydrogen bonds acting as physical crosslinks, leading to rubber-like properties. Thermodynamic incompatibility between SS and HS can further induce phase separation giving rise to a multiphase structure, where rigid HS domains can act as reinforcement of a soft matrix constituted by the SS.1–3 This specific morphology brings remarkable properties, as in the case of thermoplastic elastomers.In the frame of the recent development of the circular bioeconomy and to develop sustainable materials, a large range of renewable and performing TPU have been synthesized from different biobased building blocks,4–10 even at an industrial level.11 A very rich chemistry with new architectures has then been developed. To increase the sustainability and to decrease the environmental impact of these materials, both the beginning (biogenic carbon) and the end of life of these polymers must be simultaneously considered.12,13
Hydroxycinnamic acids, such as p-coumaric acid, ferulic acid or caffeic acid, are interesting biobased molecules with a high potential in the development of innovative and performing materials.14–18 They can be (i) extracted from lignocellulosic biomass,19 (ii) bioproduced by engineered microbes (fermentation) or (iii) obtained by more conventional (green) chemical synthesis (Fig. 1a).20 Since they contain both hydroxyl and carboxyl functional groups, they have often been used as monomers in the synthesis of homo- or co-polyesters, and integrated into a large range of macromolecular architectures.21–33 They have also been introduced as pendant side chains by post-functionalization of synthetic or natural polymers.34,35
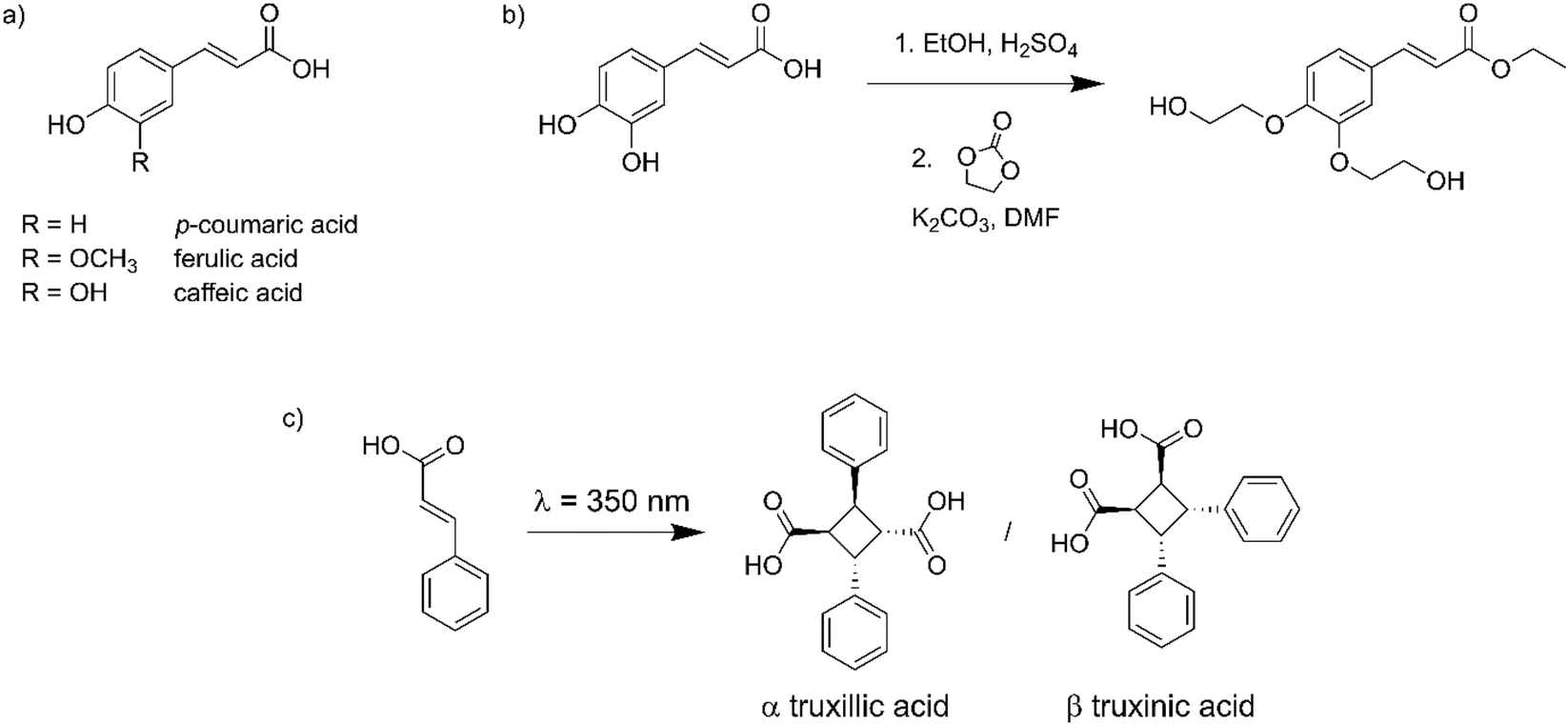 | ||
| Fig. 1 (a) Chemical structure of hydroxycinnamic acids, (b) synthesis of EC-diol from caffeic acid, (c) dimerization of cinnamic acid showing the two main stereoisomers. | ||
Cinnamic acid and its derivatives are known for more than a century for their ability to dimerize by [2 + 2] cycloaddition upon exposure to UV light. Up to 11 stereoisomers can potentially be obtained, but the most common are α-truxillic and β-truxinic acid, resulting from head-to-tail or head-to-head dimerization, respectively (Fig. 1c).36 The dimerization is reversible, since exposure to shorter wavelengths results in cleavage of the cyclobutane ring. Cinnamic acid derivatives have been used in polymer systems to prepare covalent adaptable networks (CANs),37,38 allowing the design of self-healing and memory shape materials thanks to the reversible dimerization.39–44
A single study reported the inclusion of cinnamic acid derivatives in TPUs. Wu et al.45 used a diol presenting a pendent cinnamoyl moiety, N,N-bis(2-hydroxyethyl)cinnamamide, in combination with a polycaprolactone-diol (PCL-diol), a poly(L-lactic acid)-diol and hexamethylenediisocyanate (HDI) to synthesize TPUs. The cinnamoyl photosensitive groups were included as pendant side chains in the SS. Their reversible photocycloaddition was exploited to prepare light-induced shape memory materials. A temporary shape was first fixed by photo-crosslinking under irradiation at 365 nm. Then, the initial shape could be recovered by irradiation at lower wavelength (254 nm), leading to the cleavage of the cyclobutane rings. Several studies focused on the synthesis of TPUs carrying other photoactive groups, such as coumarins46–50 or anthracenes.51,52 The materials could be crosslinked and de-crosslinked several times by irradiation at different wavelengths, although a significant loss in efficiency was often observed after several cycles.47,49–51 The reversible crosslinking was also exploited to design self-healing materials, capable of healing cracks, or welding two parts together after being cut.47–49,51
Recently, our group synthesized an original biobased diol from caffeic acid and ethylene carbonate (EC-diol, Fig. 1b).53 EC-diol is an interesting biobased building block with two highly reactive primary hydroxyl groups and an unsaturated ester. It was used in the synthesis of original polyesters by enzymatic polymerization. Given the peculiar structure of EC-diol, the aromatic groups are included into the polymer backbone, leading to enhanced thermal properties, while the photoactive unsaturated esters end up as pendant chains, resulting in high reactivity in photo-crosslinking reactions.53
The structure of EC-diol also makes it an attractive building block for the synthesis of TPUs. In this study, we report the synthesis, characterization, and reversible photo-crosslinking of a TPU based on EC-diol, which was used as chain extender. The TPU has been extensively characterized by NMR, FTIR, DSC, TGA, DMA and tensile tests. To better understand the impact of EC-diol on the structure and physical behavior of the TPU, two model TPUs were synthesized for comparison with the same SS but different chain extenders, 1,4-butanediol and 1,2-bis(hydroxyethyl)catechol, respectively. The ability of EC-diol to dimerize upon light exposure was later exploited to achieve the reversible crosslinking of the TPU by [2 + 2] photocycloaddition of the pendant unsaturated esters (Fig. 2). The kinetics of the photo-crosslinking was followed by spectroscopic techniques (UV-vis and FTIR), and changes in physical properties of the TPU were thoroughly evaluated.
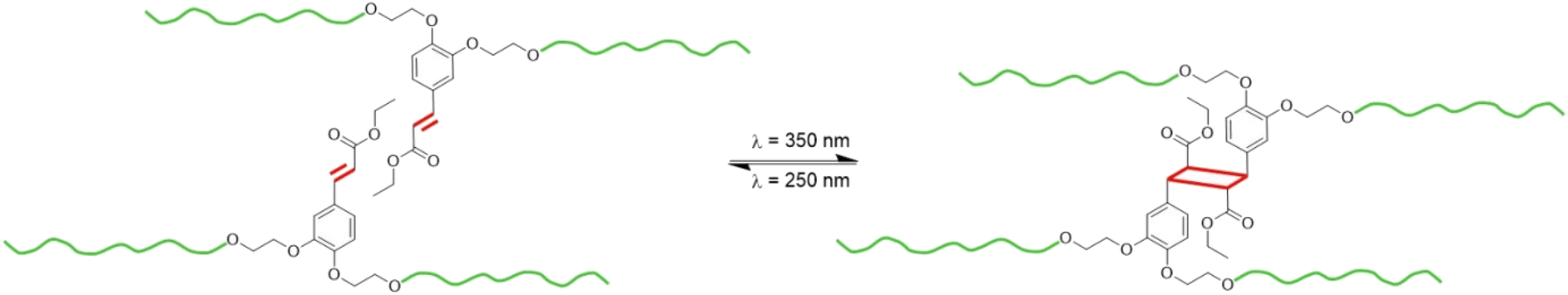 | ||
| Fig. 2 Reversible crosslinking by [2 + 2] photocycloaddition of TPU prepared from EC-diol. | ||
Experimental part
Materials
Pyrocatechol (PC, 99+%) and 1,4-butanediol (1,4-BDO; 99+%) were purchased from Acros Organics. Caffeic acid (3-(3,4-dihydroxyphenyl)acrylic acid, 95%) was purchased from Fluorochem, and ethylene carbonate (EC, 99%) from Alfa Aesar. 4,4′-Methylene bis(phenyl isocyanate) (MDI, Suprasec® 1306) was obtained from Huntsmann (USA). Polytetrahydrofuran (Mn = 1000 g mol−1, PTHF 1000) was obtained from BASF. It was dried under vacuum at 40 °C prior to use.Synthesis of pyrocatechol-diol (PC-diol, 1,2-bis(hydroxyethyl)catechol)
Pyrocatechol (5.51 g, 50 mmol), K2CO3 (1.38 g, 10 mmol) and EC (9.25 g, 105 mmol) were introduced in a three-necked round bottom flask. The mixture was stirred under an argon flux for 1 h at 150 °C, after which it was cooled down to room temperature in a water bath. The crude was then dissolved in 100 mL chloroform and extracted twice with 100 mL brine. The organic layer was then dried over MgSO4, and evaporated to dryness in a rotary evaporator, yielding 6.66 g of a white powder (67% yield). The product was further purified by recrystallization from ethyl acetate.δ H (400 MHz, DMSO) 7.02–6.93 (2H, m), 6.91–6.82 (2H, m), 4.80 (2H, t, J 2.9), 3.97 (4H, t, J 5.2), 3.70 (4H, q, J 5.2).
Synthesis of ethyl caffeate-diol (EC-diol, ethyl 3,4-bis(hydroxyethyl)caffeate)
EC-diol was synthesized in two steps from caffeic acid, as previously reported.53 First, ethyl caffeate was prepared quantitatively by Fisher esterification of caffeic acid in ethanol. Then ethyl caffeate (3.50 g, 16.8 mmol), K2CO3 (0.47 g, 3.4 mmol) and EC (3.11 g, 35.3 mmol) were introduced in a three-necked round bottom flask. Anhydrous DMF (100 mL) was added, and the mixture was stirred under an argon flux for 3 h at 130 °C, before to be cooled down to room temperature in a water bath. 400 mL ethyl acetate were then added, and the organic phase was washed with brine (250 mL), saturated NaHCO3 solution (250 mL) and brine (250 mL). The organic layer was then dried over MgSO4 and evaporated to dryness in a rotary evaporator, yielding a brown solid. The product was further purified by column chromatography (gradient of ethyl acetate and methanol), yielding 2.30 g of a pale beige powder (47% yield).δ H (400 MHz, DMSO) 7.56 (1H, d, J 16.0), 7.37 (1H, d, J 2.0), 7.22 (1H, dd, J 8.5, 2.0), 7.00 (1H, d, J 8.4), 6.53 (1H, d, J 16.0), 4.84 (2H, dt, J 9.2, 5.5), 4.17 (2H, q, J 7.1), 4.04 (4H, dt, J 7.7, 5.1), 3.72 (4H, q, J 5.2), 1.25 (3H, t, J 7.1).
TPUs synthesis
TPUs were synthesized in two steps, as depicted on Scheme 1. First, a prepolymer was synthesized from PTHF 1000 and MDI, with an NCO/OH index of 2.0. PTHF 1000 was introduced in a three-necked round bottom flask, which was placed in an oil bath regulated at 80 °C, and extensively flushed with argon. MDI was then added via syringe. The mixture was stirred with a mechanical stirrer and allowed to react for 1 h at 80 °C. The chain extender (1,4-BDO, PC-diol or EC-diol) was dissolved in about 5 mL of dry THF and added to the prepolymer via syringe. The amount of chain extender was adjusted to obtain an NCO/OH index of 1.0. The mixture was stirred with high agitation (500 rpm), before to be poured on a Teflon plate when the viscosity started to increase. The final TPUs were obtained after curing in an oven at 80 °C overnight. They were then compression molded into films of 1 mm thickness using a hot press (LabTech Engineering Company Ltd) and molds of 10 × 10 cm2. The polymers were first heated at 150 °C without pressure for 10 min, then pressed at 160 bar for 20 min, and finally cooled down to room temperature with a water circulating system within the plates.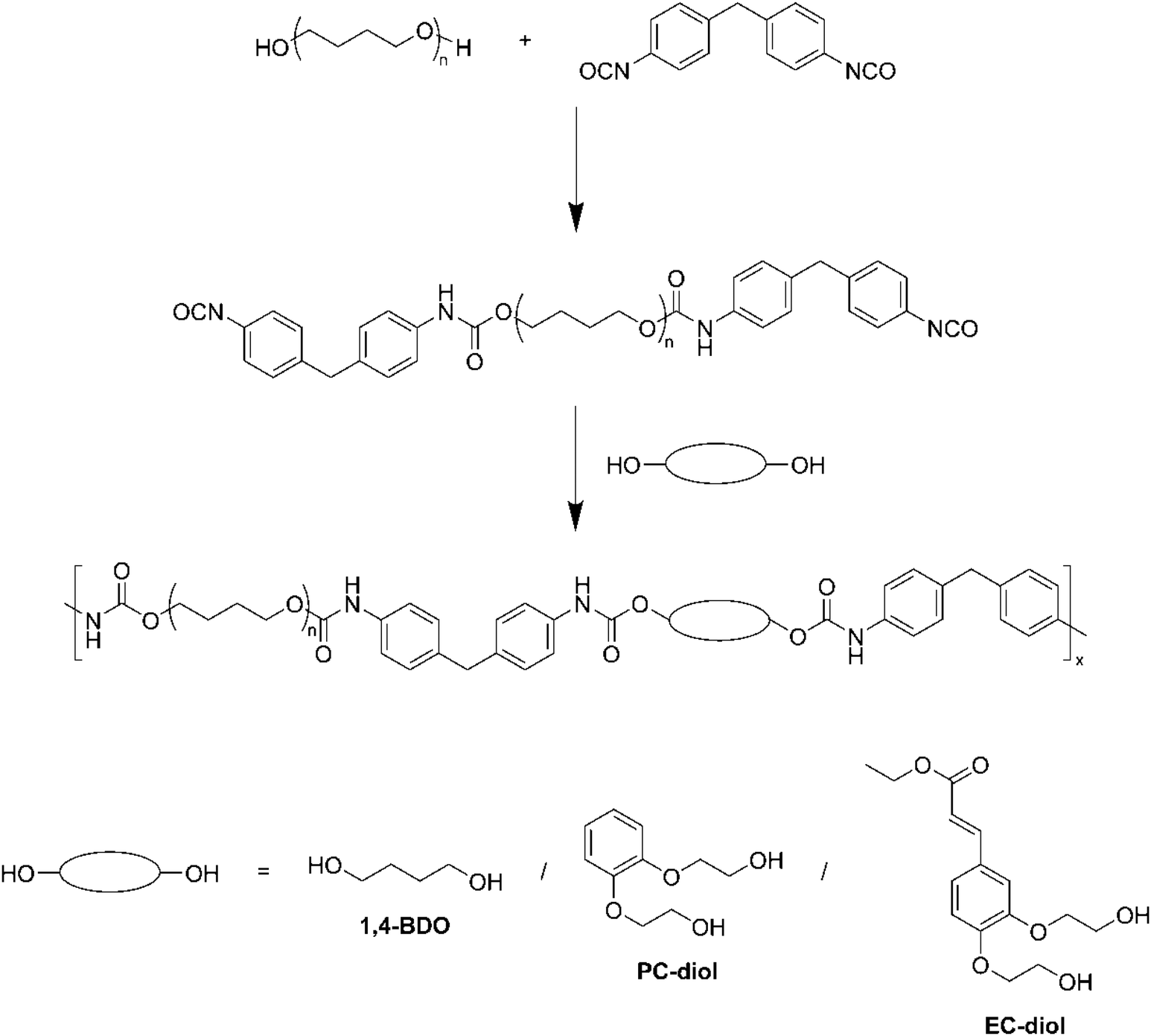 | ||
| Scheme 1 TPUs two-step synthesis. | ||
The TPUs have been synthesized with the same molar ratio between PTHF, MDI and chain extender. Since the chain extenders have different molar masses, the HS contents of the TPUs, calculated on a weight basis according to eqn (1), are different. HS contents are 37.5, 41.5 and 44.7 wt% for TPU·BDO, TPU·PC and TPU·EC, respectively.
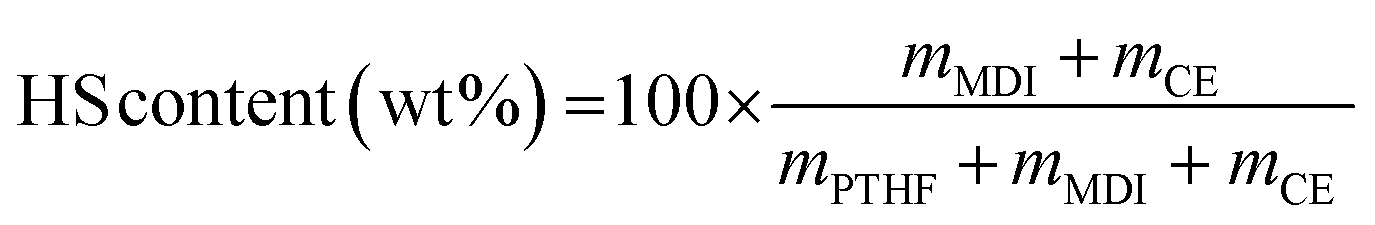 | (1) |
Photo-crosslinking of TPU
TPU samples were irradiated in a photoreactor (LZC-4Xb, Luzchem Research Inc) equipped with 14 UV lamps (8 W) with emission centered at 355 nm. For the photo-crosslinking study by UV-Vis spectroscopy, 10 mg TPU was dissolved in 1 mL of chloroform/TFA mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v). 20 μL of the solution were then spread on a microscope quartz slide, and the solvent was allowed to evaporate under a fume hood. The quartz slide was then irradiated, and UV-Vis spectra were recorded at regular intervals on a Shimadzu UV-2600 spectrometer in the 200–500 nm range. To study the cyclo-reversion reaction, the photoreactor was equipped with 14 UV lamps (8 W) with emission centered at 254 nm. The quartz slide was irradiated and UV-Vis spectra were recorded at regular intervals.
:1 v/v). 20 μL of the solution were then spread on a microscope quartz slide, and the solvent was allowed to evaporate under a fume hood. The quartz slide was then irradiated, and UV-Vis spectra were recorded at regular intervals on a Shimadzu UV-2600 spectrometer in the 200–500 nm range. To study the cyclo-reversion reaction, the photoreactor was equipped with 14 UV lamps (8 W) with emission centered at 254 nm. The quartz slide was irradiated and UV-Vis spectra were recorded at regular intervals.
For the photo-crosslinking study by FTIR, TPU films of 1.0, 0.4 or 0.2 mm thickness prepared by compression molding were placed on a rotating plate in the photoreactor. FTIR spectra were recorded at regular intervals on both sides, the irradiated one and the non-irradiated one. Finally, for photo-crosslinking prior to DSC, DMA and tensile tests, corresponding samples were cut from films of 1 mm thickness and irradiated in the photoreactor for 48 h, with turning them upside down after 24 h, to ensure complete photo-crosslinking in the bulk.
Memory shape experiments
A strip of TPU (approximately 45 × 10×0.2 mm3) was used for memory shape experiments. Two marks were drawn on the sample to define the initial length (l0). The sample was then elongated to l1, fixed in position on a quartz plate by two clamps and irradiated at 365 nm for 6 h. One clip was removed, and the length of the sample was measured (l2). Finally, the sample was irradiated at 254 nm for 1 h, and the final length (l3) was measured. Scheme and pictures of the setup are available in the ESI (Fig. S19†).The strain fixity (Rf) and strain recovery (Rr) were calculated using eqn (2) and (3), respectively:
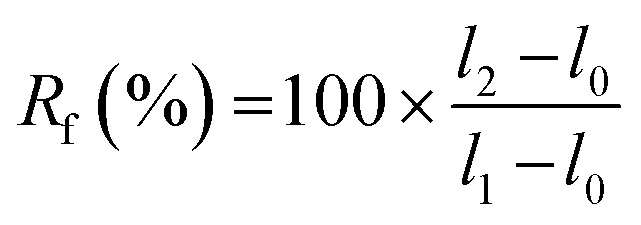 | (2) |
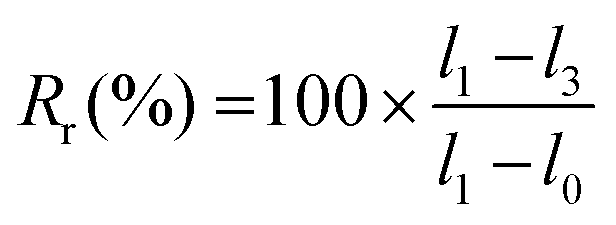 | (3) |
Polymer characterizations
1H NMR spectra were recorded on a Bruker 400 MHz spectrometer. About 20 mg samples were dissolved in 600 μL of CDCl3/trifluoroacetic acid mixture (5:1 v/v) and 16 scans were collected at 25 °C.
Fourier Transform Infrared (FTIR) spectroscopy was performed on a ThermoScientific Nicolet iS10 spectrometer equipped with a diamond ATR probe at room temperature. 32 scans were collected at a resolution of 4 cm−1.
Thermogravimetric analysis (TGA) was performed on a Q5000 apparatus (TA instruments). Samples (1 to 3 mg) were heated from 30 to 700 °C under nitrogen, at a heating rate of 20 °C min−1. The temperatures where 95% of initial mass remains are reported as T95%, whereas the temperatures at the peak of derivative weight loss (DTG curves) are reported as Tdeg.
Differential Scanning Calorimetry (DSC) was conducted on a DSC25 apparatus (TA instruments). The samples were first cooled to −80 °C and maintained at temperature for 3 min, followed by heating to 250 °C at a rate of 10 °C min−1.
Dynamic mechanical analysis (DMA) was carried out in torsion mode on a Discovery HR-3 apparatus (TA instruments), using rectangular samples (about 25 mm × 10 mm × 1 mm). Measurements were performed from −70 to 250 °C at a heating rate of 3 °C min−1 and a frequency of 1 Hz with 0.05% strain.
Uniaxial tensile tests were performed on a Zwick Roll dynamometer equipped with a 5 kN load cell. Dumbbell-shaped samples with an effective length of 20 mm, a width of 4 mm and a thickness of about 1 mm were used. The tensile measurements were performed using a preload of 0.1 N and pulling speed of 100 mm min−1 until sample failure. Elongation at break (εbreak), tensile strength (σmax) and Young's modulus (E) are reported as average of 5 to 6 experiments with the corresponding standard deviations. The plotted stress – strain curves were selected to be representative of the average values.
Results and discussion
Synthesis of TPUs
The TPUs were synthesized in two steps, as depicted on Scheme 1. Prepolymers with isocyanate chain ends were first prepared from PTHF and excess MDI, before adding the selected chain extender to obtain the final TPUs. EC-diol was used to prepare a TPU able to crosslink by photocycloaddition of the pendant unsaturated ester (Fig. 2). To evaluate its influence on the TPU structure and properties, two reference TPUs were prepared for comparison, with selected aliphatic or aromatic chain extenders. A first model TPU was prepared with 1,4-BDO as chain extender, since this system is extensively described in the literature. A second model TPU was synthesized with the aromatic PC-diol, which shares the same hydroxyethyl catechol structure as EC-diol but without unsaturated ester pendant chain.1H NMR confirmed the expected structure of the TPUs (ESI, Fig. S5 and S6†). Signals from the unsaturated side chains of EC-diol are present (Fig. 3), indicating that they were unaffected by the polymerization reaction. The TPUs could not be analyzed by size exclusion chromatography (SEC) because they were insoluble in common SEC solvents, such as THF or chloroform, as often with high molar masses TPUs.
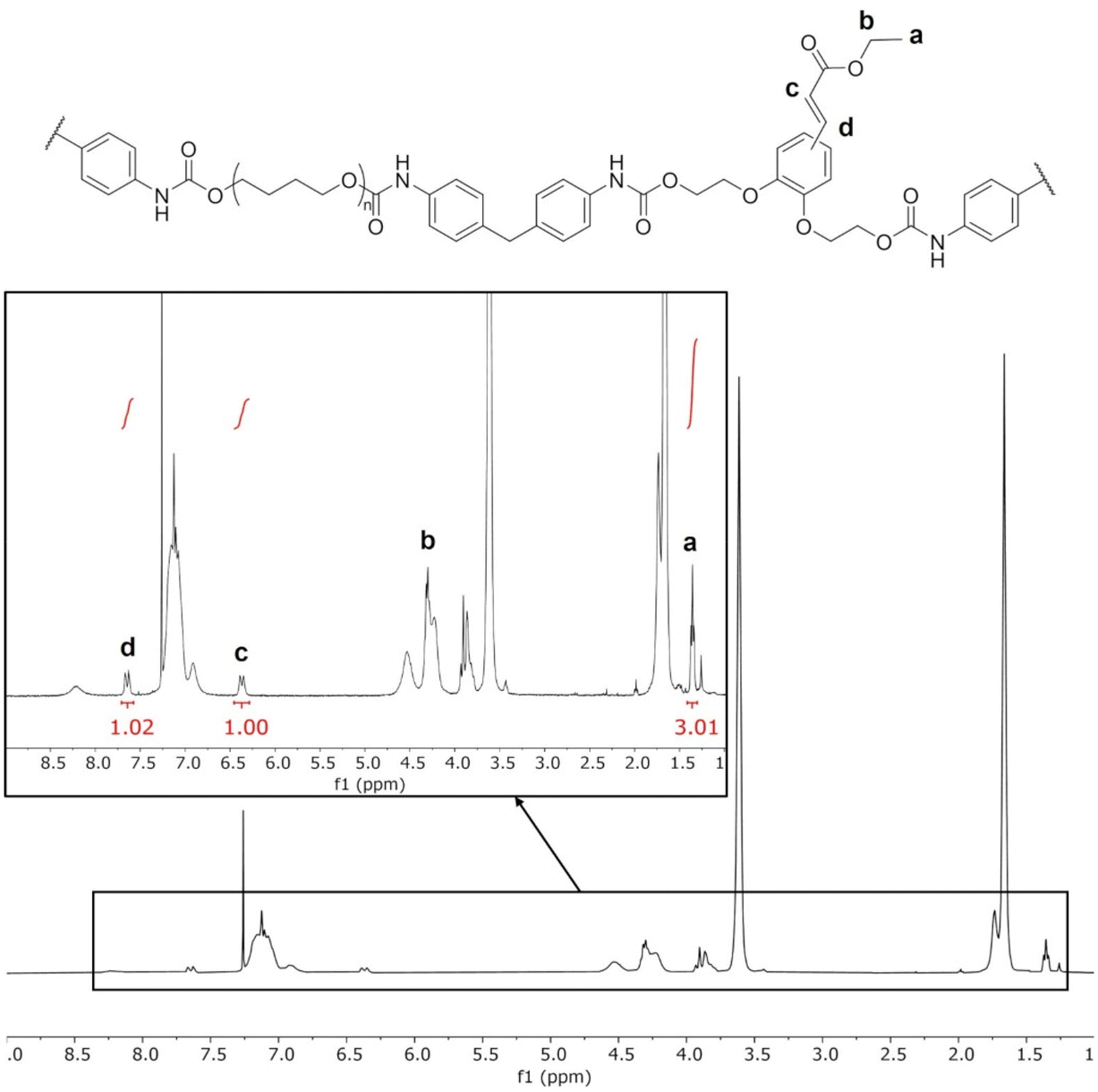 | ||
| Fig. 3 1H NMR of TPU·EC. Assignation of the unsaturated ester side chains are indicated on the inset. | ||
FTIR spectroscopy was used to monitor the level of hydrogen bonding of the carbonyl groups, and gain insights into the organization of the TPUs depending on the chain extender (Fig. 4). For TPU·BDO, 2 distinct peaks are located at 1700 and 1730 cm−1. They correspond to non-hydrogen bonded C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and strongly hydrogen bonded CO, respectively.54,55 The first ones correspond to CO in urethane linkages connecting SS, whereas the latter can be assigned to CO in urethane linkages within HS. The low wavenumber indicates strong hydrogen bonding and is characteristic of crystalline HS.56,57
O and strongly hydrogen bonded CO, respectively.54,55 The first ones correspond to CO in urethane linkages connecting SS, whereas the latter can be assigned to CO in urethane linkages within HS. The low wavenumber indicates strong hydrogen bonding and is characteristic of crystalline HS.56,57
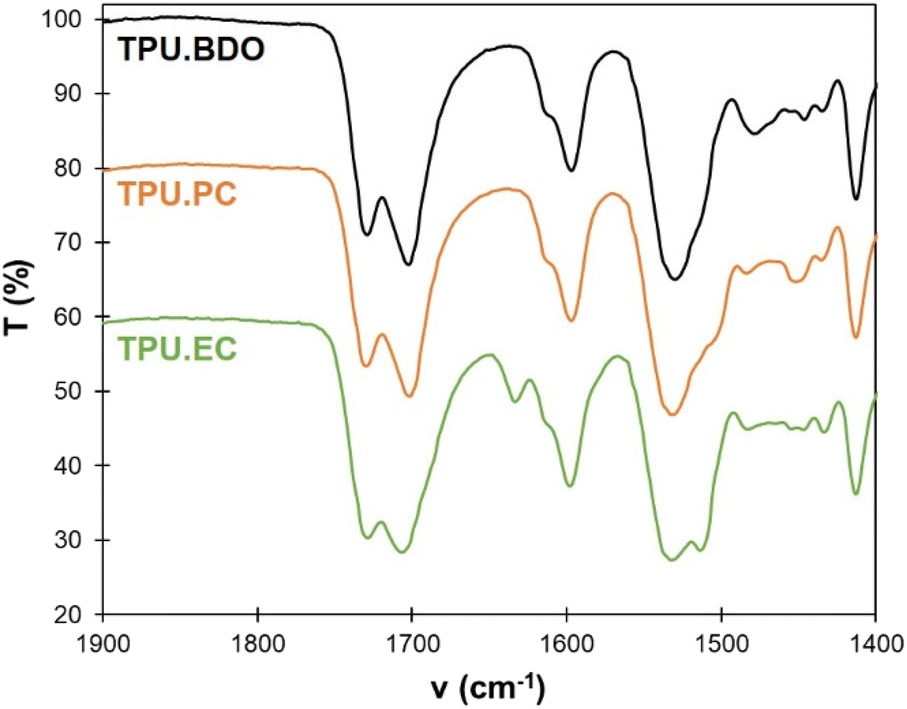 | ||
| Fig. 4 Detail of the FTIR spectra (1400–1900 cm−1) of the different TPUs. | ||
The FTIR spectrum of TPU·PC is similar to that of TPU·BDO. It thus indicates that strong hydrogen bonding occurs in the HS, which should be well-separated from the SS and probably present a crystalline structure. The spectrum of TPU·EC is different from the others. Two peaks are also observed for the CO stretch, but the second one appears at a slightly higher wavenumber, about 1705 cm−1, revealing a lower level of hydrogen bonding within HS. The ratio between both peaks is also different. However, the ester group of EC-diol also contributes to the CO stretch band, thus complicating its analysis. In addition, TPU·EC presents a peak at 1634 cm−1, which correspond to the conjugated CC bond of the unsaturated ester. It thus confirms that the CC bonds were not affected by the polymerization, and that TPU·EC presents unsaturated esters as pendant side chains.
Thermal properties of TPUs
Thermal properties of the TPUs were evaluated by DSC and TGA. Results of TGA are provided in the ESI (Fig. S7†) and in Table 1. All TPUs show a good thermal stability, with T95% ranging from 266 to 301 °C. The degradation follows a two-step process, with a first step corresponding to the HS degradation, which is dependent on the nature of the chain extender, and a second one corresponding to the PTHF segments degradation, which is common to all TPUs, with a main degradation temperature (Tdeg) above 400 °C.DSC analysis was then performed from −80 to 250 °C, since TGA showed the absence of thermal degradation within this temperature range. Results are presented on Fig. 5 and Table 1. TPU·BDO presents the typical behavior of segmented TPU with well-separated crystalline HS. It presents a Tg at low temperature that corresponds to the Tg of the SS, and an endotherm at higher temperature corresponding to the melting of the crystalline HS formed between BDO and MDI. The multiple melting endotherm can be caused by various phenomena, such as polymorphism or microphase mixing, which have been abundantly discussed in the literature.58–68 TPU·PC presents a similar behavior, but the melting endotherm ends at lower temperature compared to TPU·BDO. It means that HS formed between PC-diol and MDI can crystallize, confirming the FTIR results, but melt at lower temperature than BDO-MDI HS. TPU·EC presents a significantly different behavior. Its Tg is about 10 °C higher than that of TPU·BDO. It reveals a lower degree of phase segregation, with the presence of HS dissolved in the soft phase, leading to an increase of its Tg. In addition, no crystallinity is detected. It is likely that the presence of the pendent ester groups prevents the HS formed between EC-diol and MDI to crystallize. Similarly, the inclusion of EC-diol into polyesters led to a progressive loss of crystallinity.53
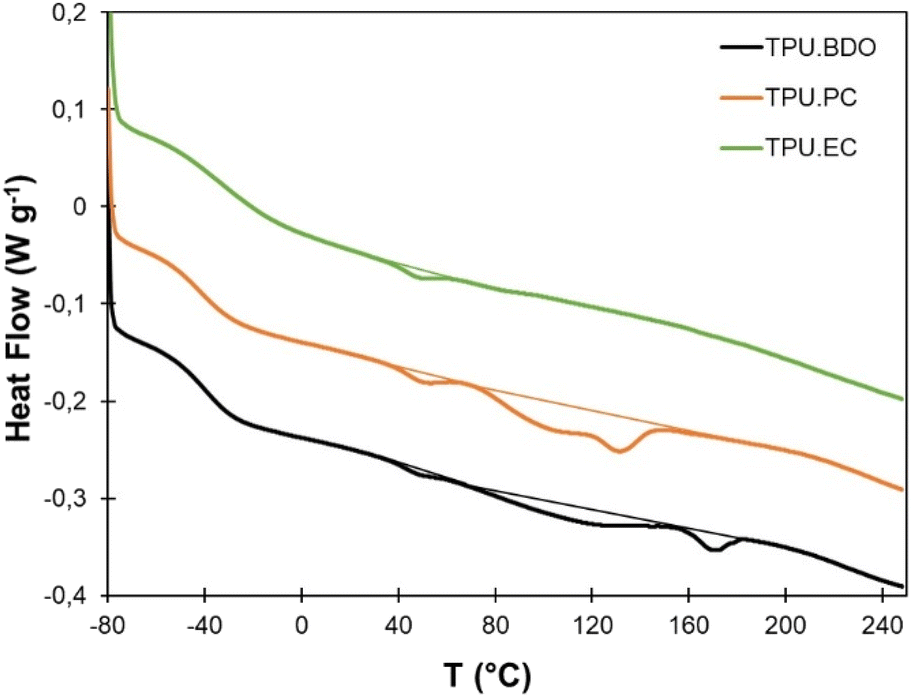 | ||
| Fig. 5 DSC of the different TPUs (10 °C min−1 temperature ramp). | ||
All TPUs present a small endotherm close to 50 °C, which can be assigned to the melting of crystalline PTHF soft segments.69,70 Pure PTHF has a melting temperature between 15 and 20 °C (Fig. S8†).
Mechanical and thermomechanical properties of TPUs
DMA results of the TPUs are shown on Fig. 6. TPU·BDO has a relaxation temperature Tα at −25 °C, which can be associated to the Tg of the SS observed by DSC. At high temperature, it presents a rubbery plateau typical of thermoplastic elastomers where the well-separated HS act as physical crosslinks. Above the melting temperature of the crystalline HS (T > 170 °C) the material starts to flow, and the storage modulus drops abruptly. TPO·PC shows the same kind of behavior, with a low Tα, but the plateau modulus is higher and extends over a narrower temperature range. Indeed, as shown by DSC, the crystalline HS melt at lower temperature than the BDO-MDI HS, leading to an earlier flow, starting around 100 °C and with an abrupt drop of the storage modulus above 120 °C. The higher plateau modulus can be explained by the slightly higher HS content, or by the increased aromatic content brought by the replacement of aliphatic BDO by aromatic PC-diol. TPU·EC has a higher Tα than the other TPUs, confirming a lower degree of phase segregation. However, it still shows a rubbery plateau extending over a wide temperature range, indicating that the HS act as physical crosslinks. The plateau modulus is lower than for the other two TPUs, probably because of the absence of crystalline structures that can play the role of reinforcements. TPU·EC starts to flow above 170 °C, similarly to TPU·BDO. However, in this case it is not related to the melting of crystalline HS nor to any thermal event detected by DSC. It likely corresponds to the relaxation of the HS.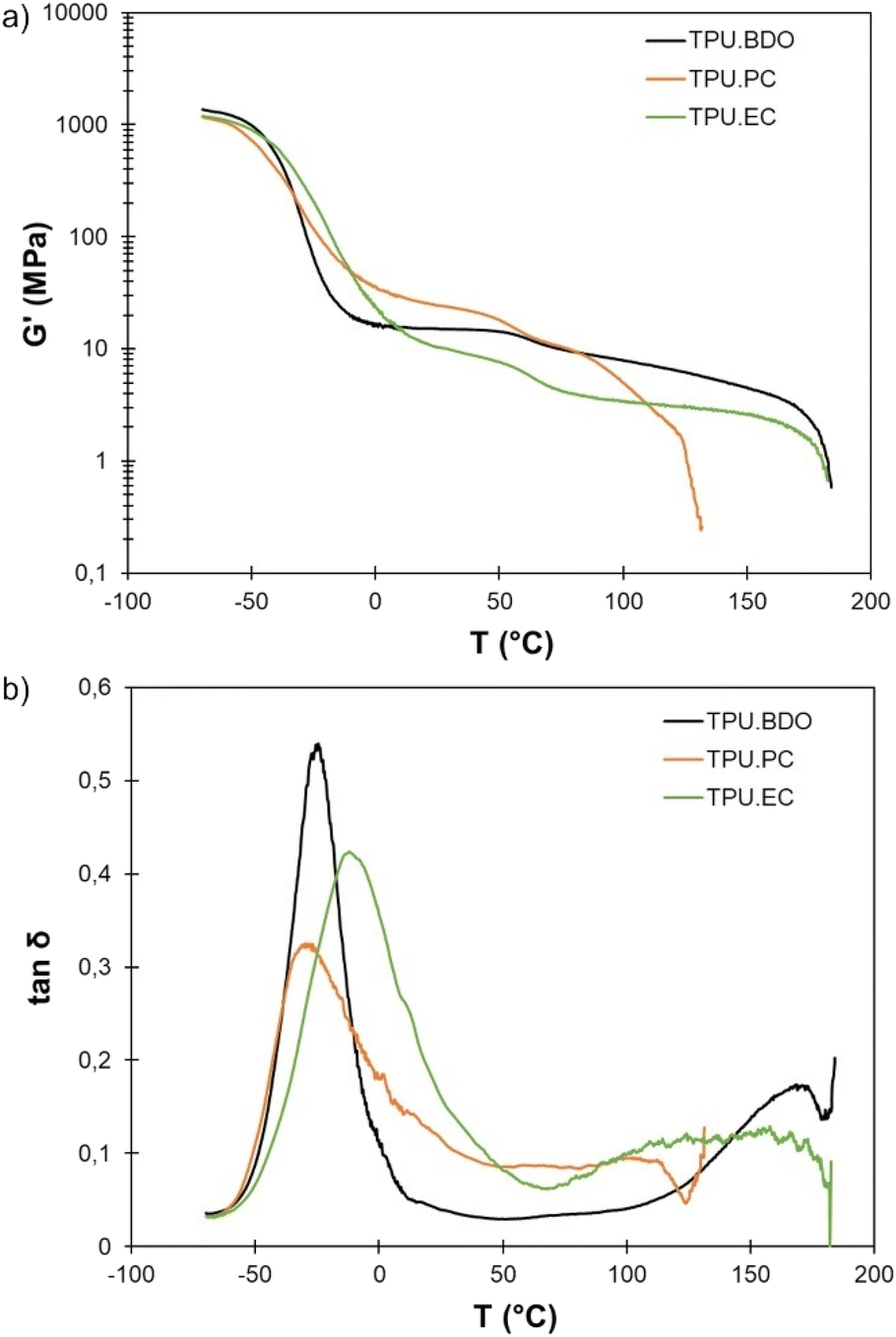 | ||
| Fig. 6 DMA of the different TPUs: (a) storage modulus G′ and (b) loss factor tanδ depending on temperature. | ||
All the TPUs also show a decrease in the storage modulus around 50 °C, which corresponds well to the thermal event observed by DSC at this temperature. This decrease in modulus is compatible with the potential melting of PTHF soft segments.
The results of uniaxial tensile tests of the TPUs are presented in Table 2. Fig. 7 shows representative stress–strain curves. The TPUs present typical behavior of thermoplastic elastomers. TPU·PC presents the highest Young's modulus, confirming the DMA results. It shows much less strain hardening than the other two TPUs, resulting in the lowest tensile strength. TPU·EC presents remarkable mechanical properties, with a tensile strength close to 30 MPa and an elongation at break higher than 1200%, close to the properties of TPU·BDO.
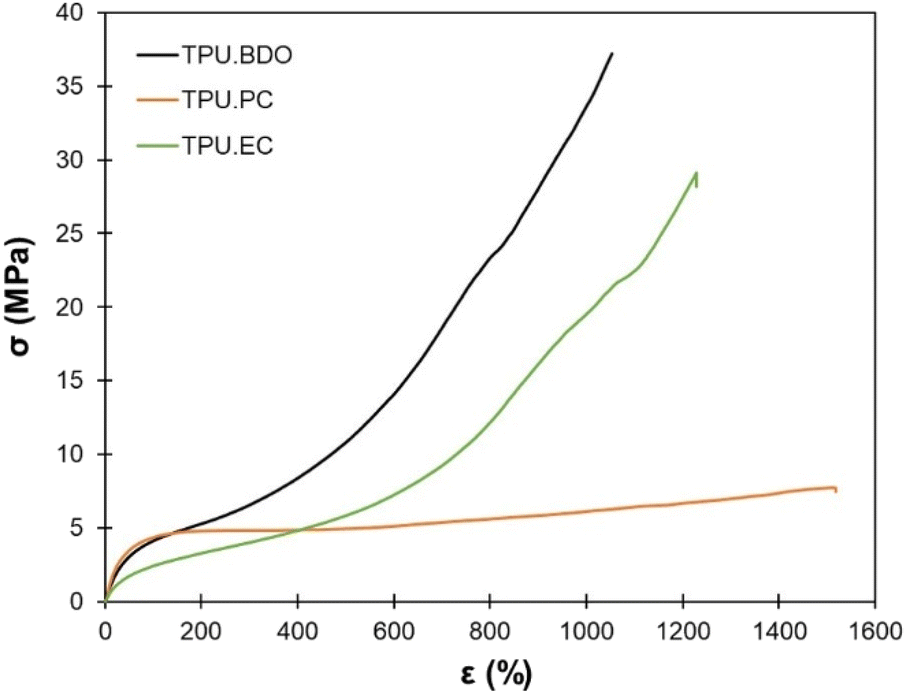 | ||
| Fig. 7 Representative stress–strain curves of the TPUs by uniaxial tensile tests. | ||
| TPU | E (MPa) | σ (MPa) | ε (%) | T α (°C) |
|---|---|---|---|---|
| TPU·BDO | 10.5 ± 0.4 | 36.7 ± 5.0 | 997 ± 100 | −25 |
| TPU·PC | 14.7 ± 1.0 | 7.6 ± 0.5 | 1530 ± 66 | −30 |
| TPU·EC | 6.5 ± 0.4 | 29.8 ± 1.5 | 1220 ± 22 | −12 |
UV-induced photo-crosslinking of TPU based on EC-diol
Cinnamic acid and its derivative are well-known for their ability to dimerize by [2 + 2] photocycloaddition triggered by UV light. The photodimerization of EC-diol was verified experimentally (more details are available in the ESI). Irradiation of a water suspension was found to be an efficient and green method to produce the dimer of EC-diol quantitatively. As confirmed earlier by 1H NMR and FTIR, TPU·EC presents the photoactive unsaturated esters as pendant side chains, making it suitable for UV-induced crosslinking (Fig. 2).The kinetics of the photo-crosslinking reaction of TPU·EC was followed by UV-vis analysis after different irradiation times of a thin film casted on a quartz plate (Fig. 8). Conjugation of the unsaturated ester with the aromatic ring on EC-diol leads to a strong absorption at 320 nm. The [2 + 2] cycloaddition causes a loss of conjugation leading to the progressive disappearance of this absorption band. Within 2 h, the absorption completely vanishes, indicating full crosslinking. Similar results were previously recorded on polyesters based on EC-diol.53
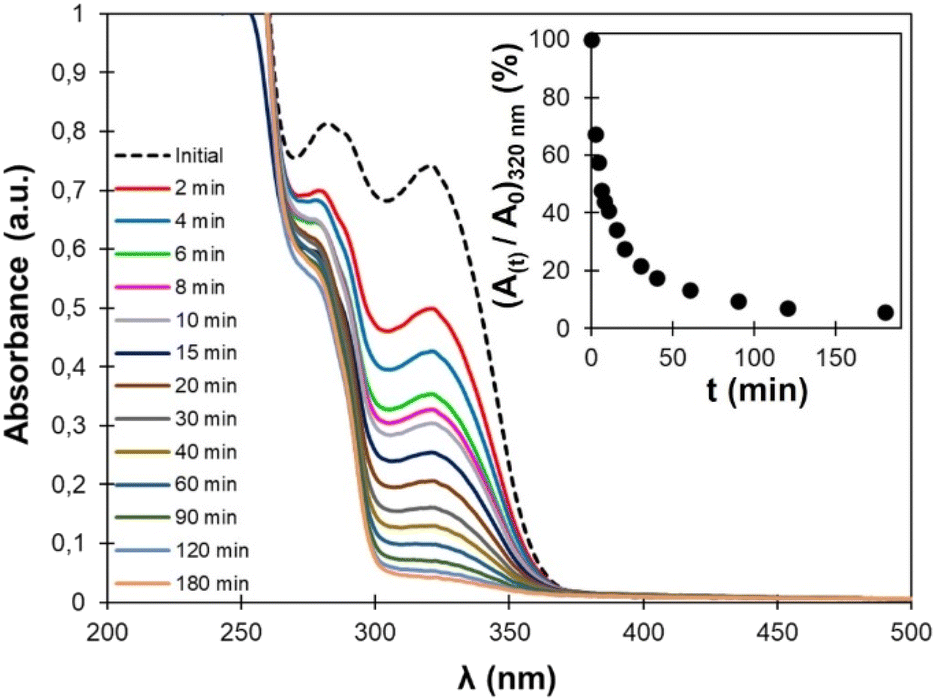 | ||
| Fig. 8 UV-Vis spectra of TPU·EC after different irradiation times at 355 nm. The inset shows the evolution of the absorbance at 320 nm (normalized by the absorbance before irradiation) with the irradiation time. | ||
FTIR was also used to follow the photo-crosslinking. As shown on Fig. 4, TPU·EC has a distinct peak at 1634 cm−1 related to the CC bond of EC-diol. Upon photoirradiation, the double bonds are consumed by the [2 + 2] cycloaddition, leading to a progressive decrease of this band. Films of 1 mm thickness were irradiated at 355 nm and analyzed by ATR-FTIR at regular intervals. Spectra are available in the ESI (Fig. S11†). The penetration depth of the ATR-FTIR analysis is estimated between 5 and 10 μm, which is much lower than the films thickness. The ATR-FTIR measurement is thus mainly performed on the surface and can be used to compare the crosslinking reaction on both sides of the film. As seen on Fig. 9, the intensity of the CC band on the irradiated side quickly decreases with irradiation time, to reach a plateau after about 1 h, in good agreement with the UV-Vis data. However, the intensity of the CC band remains constant on the non-irradiated side, meaning that photo-crosslinking only takes place on the irradiated surface, but not in bulk. The same experiment was then performed on thinner films, to evaluate the influence of the film thickness. The results are detailed in the ESI (Fig. S12–S15†). They show that crosslinking also takes place on the non-irradiated side for thin films, but only upon long time exposure. For a 0.2 mm thick film, the level of crosslinking on the non-irradiated side is roughly the same after 16 h of irradiation than after only 10 min on the irradiated side. These results illustrate the strong limitation of photo-polymerization for achieving polymerization in the bulk.41
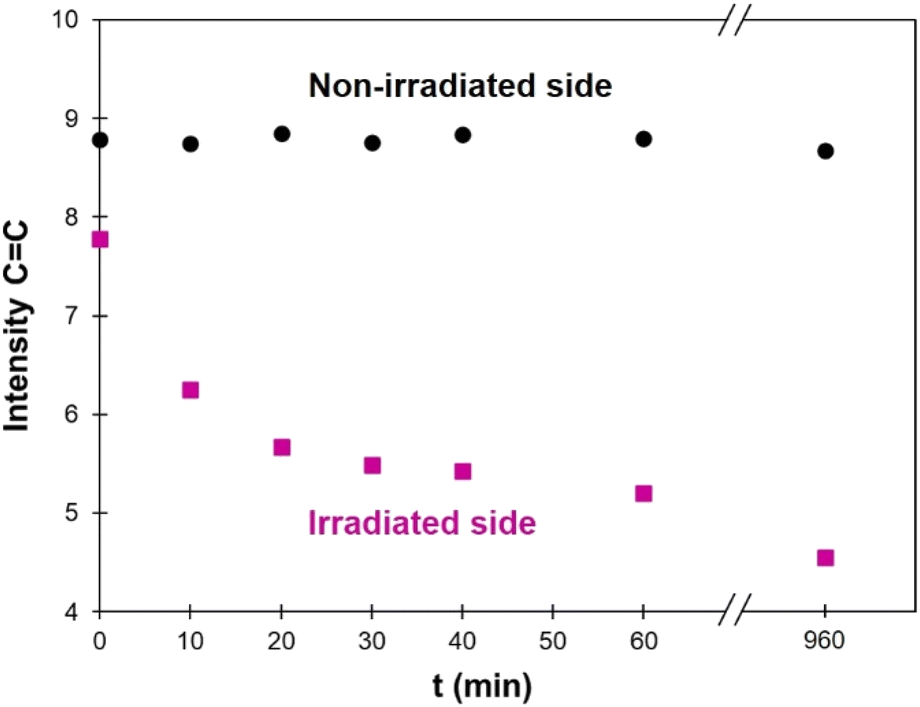 | ||
| Fig. 9 Intensity of the CC band measured by ATR-FTIR (at 1634 cm−1) on a 1 mm thick film, depending on the irradiation time (λ = 355 nm). The corresponding FTIR spectra are available in the ESI (Fig. S10†). | ||
Influence of photo-crosslinking on the properties of TPU·EC
To ensure bulk crosslinking prior to the analysis of the thermal, thermomechanical and mechanical properties, a 1 mm thick film of TPU·EC was exposed to UV light for 24 h on both sides. The thermal stability, evaluated by TGA, is not affected by the crosslinking (Fig. S16†).DMA clearly reveals the effect of UV irradiation. Tα is unaffected (Fig. 10b). This transition corresponds to the relaxation of the SS whereas the UV-induced crosslinking reaction takes place within the HS. However, the plateau modulus increases and extends over a much wider temperature range because of the crosslinking (Fig. 10a). The material did not flow even at 250 °C thanks to the presence of covalent crosslinks.
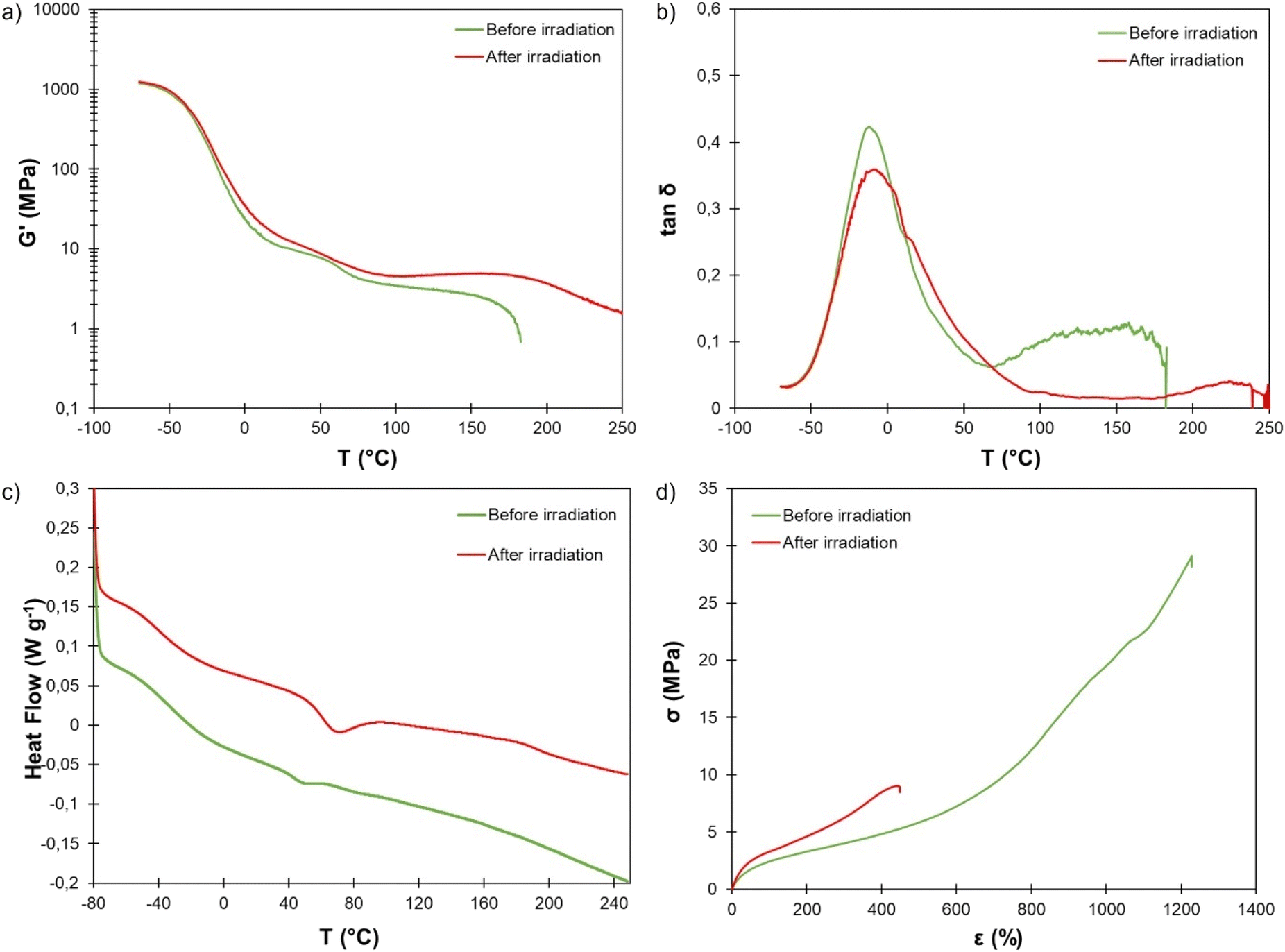 | ||
| Fig. 10 (a) Storage modulus and (b) tanδ measured by DMA, (c) DSC thermograms and (d) stress–strain curves of TPU·EC before and after photoirradiation at 355 nm. | ||
DSC further reveals the influence of the photo-crosslinking on the thermal behavior (Fig. 10c). The Tg is almost unchanged (−36 °C, against −32 °C before irradiation), confirming DMA results. Surprisingly, the endotherm visible around 50 °C, corresponding to the melting of PTHF crystalline segments,69,70 appears larger and shifted to higher temperature, around 70 °C, after the irradiation. Analysis by modulated DSC clearly shows that the endotherm is a non-reversing thermal event (Fig. S17†). It is no longer observed when a second heating run is performed (Fig. S18†). It is unclear whether this results from the photocrosslinking or only from the storage of the material at room temperature, which may have led to further crystallization of the PTHF SS. This phenomenon would require deeper investigations. An additional thermal event is also visible at higher temperature on the thermogram of the TPU after irradiation, around 190 °C. Modulated DSC shows that is a non-reversing endothermic event (Fig. S17†). It may correspond to thermally induced rupture of the cyclobutane rings, which has already been reported to occur at high temperature.71–75 As it would lead to decrosslinking, it could explain the decrease of the plateau modulus observed by DMA above 200 °C (Fig. 10a).
The mechanical properties of the irradiated TPU were finally evaluated (Fig. 10d). UV-induced crosslinking resulted in a net increase of the Young's modulus, as expected (8.2 ± 0.5 MPa, against 6.5 ± 0.4 MPa before irradiation). At a given deformation, the corresponding stress is also significantly increased by crosslinking. However, the elongation at break is reduced by more than half after crosslinking (454 ± 22 against 1220 ± 22%), leading to an important decrease of the tensile strength (8.6 ± 0.5 against 29.8 ± 1.5 MPa).
UV-induced reversible decrosslinking and memory shape properties of TPU·EC
Photo-crosslinked TPU·EC was then exposed to high-energy UVC light (λ = 254 nm). Upon exposure, the cyclobutane rings are broken, leading to decrosslinking of the material (Fig. 2). The reaction reforms the unsaturated esters, leading to the recovery of the absorbance at 320 nm on the UV-vis spectra (Fig. 11). However, the recovery of the absorbance is incomplete, only about 30% of the initial value, indicating that the decrosslinking by rupture of the cyclobutane rings is only partial.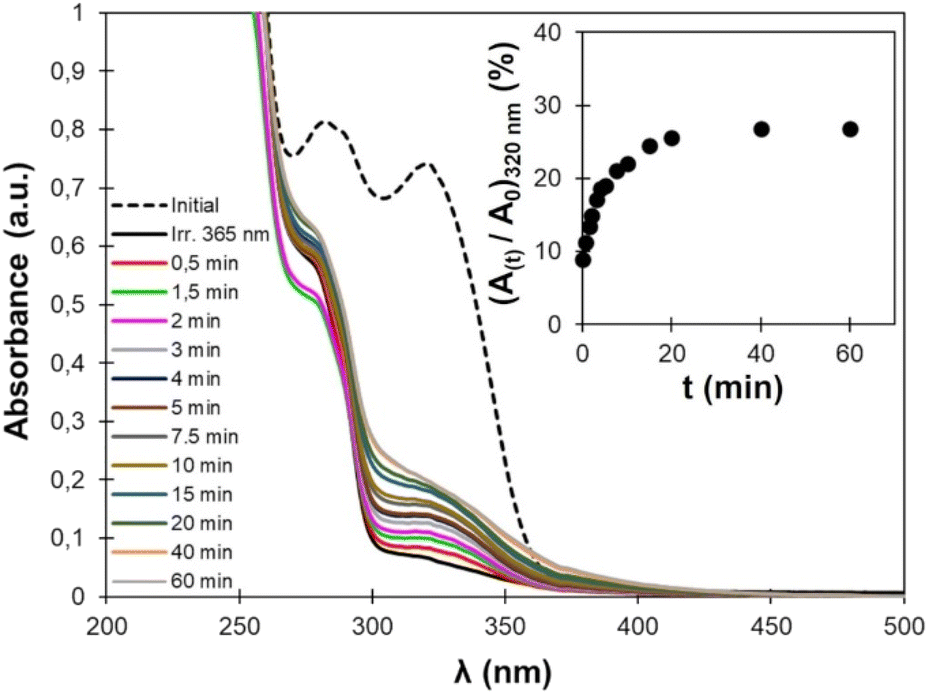 | ||
| Fig. 11 UV-Vis spectra of TPU·EC after different irradiation times at 254 nm. The inset shows the evolution of the absorbance at 320 nm (normalized by the absorbance before irradiation) with the irradiation time. | ||
Several phenomena can explain this behavior. First, the penetration depth of radiation inside the material depends on the radiation wavelength. The low wavelength UVC light (λ = 254 nm) has a low penetration depth, meaning that the cycloreversion is mainly limited to the surface since the radiation cannot reach the core of the material. Complete decrosslinking induced by UVC light has already been reported on PU crosslinked with coumarins, but only on thin films (less than 5 μm thickness).48,50 On other coumarin-based PU, only 30 to 60% of the coumarins were recovered after rupture of the dimers by UVC light.47,49 Here, the phenomenon is further accentuated by the high absorbance of the material below 250 nm, due to its high content in aromatic rings coming from EC-diol and MDI. In addition, the dimerized and cleaved forms are in equilibrium under irradiation at 254 nm, because both cycloaddition and cycloreversion are excited at this wavelength.76 Finally, the cycloreversion can proceed in a non-symmetric fashion, leading to cleaved products which are chemically different from the starting compounds.77,78 This would be the case here if the cinnamic moieties dimerize in a head-to-head (truxinates) rather than in head-to-tail configuration (truxillates).36
Several crosslinking – decrosslinking cycles were then performed by successively exposing the material to UV light of 355 nm and 254 nm wavelengths. The changes in absorbance at 320 nm were monitored by UV-vis spectroscopy and are reported in Fig. 12. As discussed above, the decrosslinking is only partial. The recovery is the same on the second cycle than on the first one. However, the decrease of absorbance is less important on the second and third cycles of irradiation at 355 nm. Such decrease in efficiency of the cycloaddition/cycloreversion cycles is caused by the phenomena described above, and has commonly been observed on e.g., coumarin-based photo-responsive TPUs.47,49
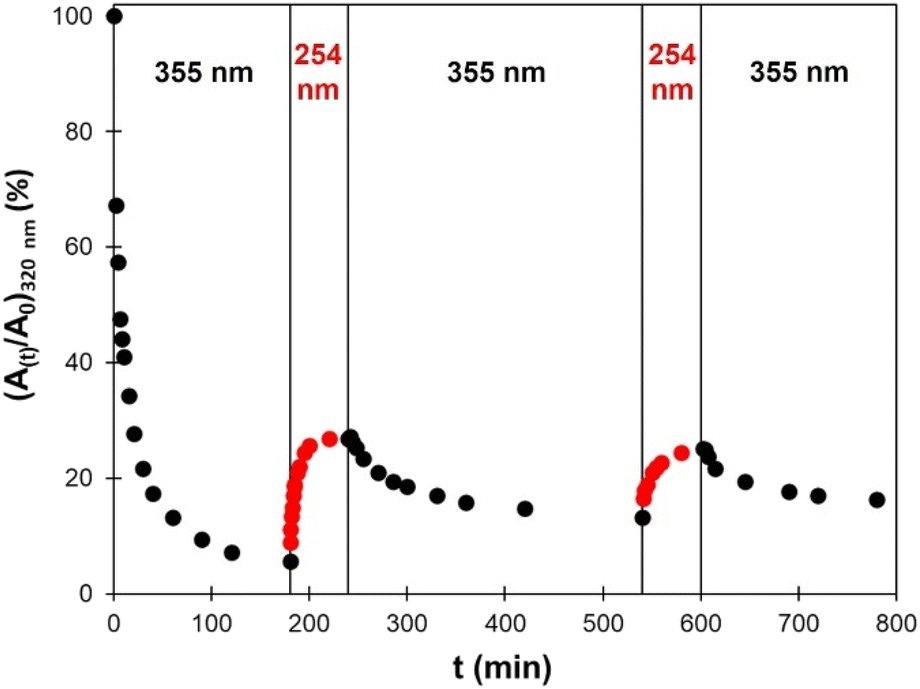 | ||
| Fig. 12 Evolution of the absorption at 320 nm measured by UV-vis spectroscopy during successive irradiations by UV light of 355 nm or 254 nm wavelengths. | ||
The UV-induced reversible crosslinking was also used to obtain memory shape properties. The memory shape properties of TPU·EC were characterized using a setup described in the ESI (Fig. S19†). A temporary shape was fixed by irradiating the material at 355 nm. The material showed a relatively modest strain fixity of 36% at 20% deformation, in the same range as some early developed systems,79 but far from highly sophisticated systems which can show strain fixity close to 100%. The modest strain fixity probably originates from the low proportion of photoactive monomer in the designed TPU (17 wt%). After irradiation at 254 nm, the material gets back to its initial shape, with a strain recovery of 93%.
Conclusion
An original TPU was successfully synthesized from a biobased aromatic chain extender prepared from caffeic acid (EC-diol), and carefully characterized by NMR, FTIR, TGA, DSC, DMA and tensile tests. TPU·EC was compared to two model TPUs prepared with aliphatic (1,4-BDO) and aromatic (PC-diol) chain extenders, to better evaluate the influence of EC-diol on the structure and properties of the TPU.TPU·EC presents all the characteristic features of thermoplastic elastomers, with phase segregation between SS and HS, leading to remarkable mechanical and thermal properties. In addition, thanks to the peculiar structure of EC-diol, TPU·EC possess unsaturated esters as pendant chains, which are able to dimerize upon UV irradiation at λ = 355 nm by [2 + 2] cycloaddition, leading to crosslinking. The photo-crosslinking reaction was monitored on thin TPU films by UV-vis spectroscopy, which reveals a successful cycloaddition that is complete in about 2 h. FTIR spectroscopy was further used to evaluate the photocrosslinking on samples of various thickness, by comparing the progress of the cycloaddition reaction on both sides of films during irradiation. It reveals that the reaction is mostly limited to the surface of the material, because of the low penetration depth of UV light. Only thin samples can be fully crosslinked, since long exposure is required to achieve crosslinking for thick materials. This mostly limits the applicability of the developed systems to thin films or coatings. The irradiation results in important modifications of the macroscopic properties of the material: its stiffness and thermomechanical stability increase because of covalent crosslinking.
Irradiation at shorter wavelength (λ = 254 nm) leads to decrosslinking by cleavage of the cyclobutane rings. The reversion has been monitored by UV-vis spectroscopy, but appears incomplete, because of (i) the well-known low penetration depth of short wavelength UV light, and (ii) the potential equilibrium between dimerized and cleaved forms. The efficiency of the cycloaddition/cycloreversion cycles also decreases after each cycle, which constitutes the main limitation for the use of this system as covalent adaptable networks (CANs). However, DSC and DMA of the photo-crosslinked TPU shows that a thermally induced cycloreversion may take place above 200 °C, which could bring another option to decrosslink the material on-demand and improve the potential of this crosslinking strategy for the design of CANs. This potential thermal pathway will be the focus of further investigations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Alfred Bazin is kindly acknowledged for his help with EC-diol purification and fruitful discussions on the project.References
- P. Król, Prog. Mater. Sci., 2007, 52, 915–1015 CrossRef
.
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed
- I. Yilgör, E. Yilgör and G. L. Wilkes, Polymer, 2015, 58, A1–A36 CrossRef
- C. Bueno-Ferrer, E. Hablot, F. Perrin-Sarazin, M. C. Garrigós, A. Jiménez and L. Averous, Macromol. Mater. Eng., 2012, 297, 777–784 CrossRef CAS
- C. Bueno-Ferrer, E. Hablot, M. del C. Garrigós, S. Bocchini, L. Averous and A. Jiménez, Polym. Degrad. Stab., 2012, 97, 1964–1969 CrossRef CAS
- M. Charlon, B. Heinrich, Y. Matter, E. Couzigné, B. Donnio and L. Avérous, Eur. Polym. J., 2014, 61, 197–205 CrossRef CAS
- L. Ugarte, B. Fernández-d'Arlas, A. Valea, M. L. González, M. a. Corcuera and A. Eceiza, Polym. Eng. Sci., 2014, 54, 2282–2291 CrossRef CAS
- T. Calvo-Correas, M. D. Martin, A. Retegi, N. Gabilondo, M. A. Corcuera and A. Eceiza, ACS Sustainable Chem. Eng., 2016, 4, 5684–5692 CrossRef CAS
- S. Wendels, B. Heinrich, B. Donnio and L. Avérous, Eur. Polym. J., 2021, 153, 110531 CrossRef CAS
- A. Duval, A. Sarbu, F. Dalmas, D. Albertini and L. Avérous, Macromolecules, 2022, 55, 5371–5381 CrossRef CAS
- H. Sardon, D. Mecerreyes, A. Basterretxea, L. Avérous and C. Jehanno, ACS Sustainable Chem. Eng., 2021, 9, 10664–10677 CrossRef CAS
- L. Ugarte, T. Calvo-Correas, I. Gonzalez-Gurrutxaga, C. Peña-Rodriguez, O. Etxeberria, M. A. Corcuera and A. Eceiza, Proceedings, 2018, 2, 1490 Search PubMed
- T. Calvo-Correas, M. Benitez, I. Larraza, L. Ugarte, C. Peña-Rodríguez and A. Eceiza, Polym. Degrad. Stab., 2022, 198, 109880 CrossRef CAS
- R. Ménard, S. Caillol and F. Allais, Ind. Crops Prod., 2017, 95, 83–95 CrossRef
- R. Ménard, S. Caillol and F. Allais, ACS Sustainable Chem. Eng., 2017, 5, 1446–1456 CrossRef
- B. M. Upton and A. M. Kasko, ACS Sustainable Chem. Eng., 2018, 6, 3659–3668 CrossRef CAS
- B. M. Upton and A. M. Kasko, Biomacromolecules, 2019, 20, 758–766 CrossRef CAS PubMed
- A. C. Fonseca, M. S. Lima, A. F. Sousa, A. J. Silvestre, J. F. J. Coelho and A. C. Serra, Polym. Chem., 2019, 10, 1696–1723 RSC
- S. D. Karlen, P. Fasahati, M. Mazaheri, J. Serate, R. A. Smith, S. Sirobhushanam, M. Chen, V. I. Tymokhin, C. L. Cass, S. Liu, D. Padmakshan, D. Xie, Y. Zhang, M. A. McGee, J. D. Russell, J. J. Coon, H. F. Kaeppler, N. de Leon, C. T. Maravelias, T. M. Runge, S. M. Kaeppler, J. C. Sedbrook and J. Ralph, ChemSusChem, 2020, 13, 2012–2024 CrossRef CAS PubMed
- A. L. Flourat, J. Combes, C. Bailly-Maitre-Grand, K. Magnien, A. Haudrechy, J.-H. Renault and F. Allais, ChemSusChem, 2021, 14, 118–129 CrossRef CAS PubMed
- A. Bazin, L. Avérous and E. Pollet, Polymers, 2021, 13, 3693 CrossRef CAS PubMed
- M. Matsusaki, A. Kishida, N. Stainton, C. W. G. Ansell and M. Akashi, J. Appl. Polym. Sci., 2001, 82, 2357–2364 CrossRef CAS
- T. Kaneko, M. Matsusaki, T. T. Hang and M. Akashi, Macromol. Rapid Commun., 2004, 25, 673–677 CrossRef CAS
- T. Kaneko, T. H. Thi, D. J. Shi and M. Akashi, Nat. Mater., 2006, 5, 966–970 CrossRef CAS PubMed
- J. Du, Y. Fang and Y. Zheng, Polymer, 2007, 48, 5541–5547 CrossRef CAS
- T. H. Thi, M. Matsusaki, D. Shi, T. Kaneko and M. Akashi, J. Biomater. Sci., Polym. Ed., 2008, 19, 75–85 CrossRef CAS PubMed
- T. H. Thi, M. Matsusaki and M. Akashi, Biomacromolecules, 2009, 10, 766–772 CrossRef CAS PubMed
- W. Dong, H. Li, M. Chen, Z. Ni, J. Zhao, H. Yang and P. Gijsman, J. Polym. Res., 2011, 18, 1239–1247 CrossRef CAS
- W. Dong, J. Ren, L. Lin, D. Shi, Z. Ni and M. Chen, Polym. Degrad. Stab., 2012, 97, 578–583 CrossRef CAS
- M. A. Ouimet, N. D. Stebbins and K. E. Uhrich, Macromol. Rapid Commun., 2013, 34, 1231–1236 CrossRef CAS PubMed
- M. A. Ouimet, J. Griffin, A. L. Carbone-Howell, W.-H. Wu, N. D. Stebbins, R. Di and K. E. Uhrich, Biomacromolecules, 2013, 14, 854–861 CrossRef CAS PubMed
- H. T. H. Nguyen, M. H. Reis, P. Qi and S. A. Miller, Green Chem., 2015, 17, 4512–4517 RSC
- H. T. H. Nguyen, G. N. Short, P. Qi and S. A. Miller, Green Chem., 2017, 19, 1877–1888 RSC
- D.-S. Lee, J.-Y. Woo, C.-B. Ahn and J.-Y. Je, Food Chem., 2014, 148, 97–104 CrossRef CAS PubMed
- C. Yang, Y. Zhou, Y. Zheng, C. Li, S. Sheng, J. Wang and F. Wu, Int. J. Biol. Macromol., 2016, 87, 577–585 CrossRef CAS PubMed
-
D. M. Bassani, in CRC Handbook of Organic Photochemistry and Photobiology, ed. W. M. Horspool and F. Lenci, Taylor&Francis, Boca Raton, FL, 2003 Search PubMed
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC
- J. M. Rochette and V. S. Ashby, Macromolecules, 2013, 46, 2134–2140 CrossRef CAS
- G. Kaur, P. Johnston and K. Saito, Polym. Chem., 2014, 5, 2171–2186 RSC
- S. Chatani, C. J. Kloxin and C. N. Bowman, Polym. Chem., 2014, 5, 2187–2201 RSC
- H. Xie, K.-K. Yang and Y.-Z. Wang, Prog. Polym. Sci., 2019, 95, 32–64 CrossRef CAS
- R. H. Aguirresarobe, S. Nevejans, B. Reck, L. Irusta, H. Sardon, J. M. Asua and N. Ballard, Prog. Polym. Sci., 2021, 114, 101362 CrossRef CAS
- J. Dahlke, S. Zechel, M. D. Hager and U. S. Schubert, Adv. Mater. Interfaces, 2018, 5, 1800051 CrossRef
- L. Wu, C. Jin and X. Sun, Biomacromolecules, 2011, 12, 235–241 CrossRef CAS PubMed
- C. Salgado, M. P. Arrieta, V. Sessini, L. Peponi, D. López and M. Fernández-García, Polym. Degrad. Stab., 2020, 178, 109204 CrossRef CAS
- R. H. Aguirresarobe, L. Martin, N. Aramburu, L. Irusta and M. J. Fernandez-Berridi, Prog. Org. Coat., 2016, 99, 314–321 CrossRef CAS
- J. Ling, M. Z. Rong and M. Q. Zhang, J. Mater. Chem., 2011, 21, 18373–18380 RSC
- J. Ling, M. Z. Rong and M. Q. Zhang, Polymer, 2012, 53, 2691–2698 CrossRef CAS
- R. Seoane Rivero, P. Bilbao Solaguren, K. Gondra Zubieta, A. Gonzalez-Jimenez, J. L. Valentin and A. Marcos-Fernandez, Eur. Polym. J., 2016, 76, 245–255 CrossRef CAS
- Y. Fang, X. Du, Z. Du, H. Wang and X. Cheng, J. Mater. Chem. A, 2017, 5, 8010–8017 RSC
- J. Van Damme, O. van den Berg, L. Vlaminck, J. Brancart, G. Van Assche and F. Du Prez, Eur. Polym. J., 2018, 105, 412–420 CrossRef CAS
- A. Bazin, A. Duval, L. Avérous and E. Pollet, Macromolecules, 2022, 55, 4256–4267 CrossRef CAS
- M. M. Coleman, K. H. Lee, D. J. Skrovanek and P. C. Painter, Macromolecules, 1986, 19, 2149–2157 CrossRef CAS
- H. S. Lee, Y. K. Wang and S. L. Hsu, Macromolecules, 1987, 20, 2089–2095 CrossRef CAS
- I. Yilgor, E. Yilgor, I. G. Guler, T. C. Ward and G. L. Wilkes, Polymer, 2006, 47, 4105–4114 CrossRef CAS
- J. Mattia and P. Painter, Macromolecules, 2007, 40, 1546–1554 CrossRef CAS
- J. Blackwell, M. R. Nagarajan and T. B. Hoitink, Polymer, 1982, 23, 950–956 CrossRef CAS
- J. Blackwell and C. D. Lee, J. Polym. Sci., Polym. Phys. Ed., 1984, 22, 759–772 CrossRef CAS
- R. M. Briber and E. L. Thomas, J. Macromol. Sci., Part B: Phys., 1983, 22, 509–528 CrossRef
- R. M. Briber and E. L. Thomas, J. Polym. Sci., Polym. Phys. Ed., 1985, 23, 1915–1932 CrossRef CAS
- G. Pompe, A. Pohlers, P. Pötschke and J. Pionteck, Polymer, 1998, 39, 5147–5153 CrossRef CAS
- J. T. Koberstein, A. F. Galambos and L. M. Leung, Macromolecules, 1992, 25, 6195–6204 CrossRef CAS
- J. T. Koberstein and A. F. Galambos, Macromolecules, 1992, 25, 5618–5624 CrossRef CAS
- J. Balko, B. Fernández-d'Arlas, E. Pöselt, R. Dabbous, A. J. Müller and T. Thurn-Albrecht, Macromolecules, 2017, 50, 7672–7680 CrossRef CAS
- A. Saiani, W. A. Daunch, H. Verbeke, J.-W. Leenslag and J. S. Higgins, Macromolecules, 2001, 34, 9059–9068 CrossRef CAS
- A. Saiani, C. Rochas, G. Eeckhaut, W. A. Daunch, J.-W. Leenslag and J. S. Higgins, Macromolecules, 2004, 37, 1411–1421 CrossRef CAS
- A. Saiani, A. Novak, L. Rodier, G. Eeckhaut, J.-W. Leenslag and J. S. Higgins, Macromolecules, 2007, 40, 7252–7262 CrossRef CAS
- Y. Camberlin and J. P. Pascault, J. Polym. Sci., Polym. Chem. Ed., 1983, 21, 415–423 CrossRef CAS
- L. Zhang, L. Chen and S. J. Rowan, Macromol. Chem. Phys., 2017, 218, 1600320 CrossRef
- K. Novak, V. Enkelmann, G. Wegner and K. B. Wagener, Angew. Chem., Int. Ed. Engl., 1993, 32, 1614–1616 CrossRef
- A. Chanthapally, G. K. Kole, K. Qian, G. K. Tan, S. Gao and J. J. Vittal, Chem. – Eur. J., 2012, 18, 7869–7877 CrossRef CAS PubMed
- J. W. Chung, Y. You, H. S. Huh, B.-K. An, S.-J. Yoon, S. H. Kim, S. W. Lee and S. Y. Park, J. Am. Chem. Soc., 2009, 131, 8163–8172 CrossRef CAS PubMed
- I.-H. Park, A. Chanthapally, Z. Zhang, S. S. Lee, M. J. Zaworotko and J. J. Vittal, Angew. Chem., Int. Ed., 2014, 53, 414–419 CrossRef CAS PubMed
- H. Amjaour, Z. Wang, M. Mabin, J. Puttkammer, S. Busch and Q. R. Chu, Chem. Commun., 2018, 55, 214–217 RSC
- H. Frisch, D. E. Marschner, A. S. Goldmann and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2018, 57, 2036–2045 CrossRef CAS PubMed
- N. Yonezawa, T. Yoshida and M. Hasegawa, J. Chem. Soc., Perkin Trans. 1, 1983, 1083–1086 RSC
- N. Yonezawa, T. Yamashita, T. Kanoe, K. Saigo and M. Hasegawa, Ind. Eng. Chem. Prod. Res. Dev., 1985, 24, 593–598 CrossRef CAS
- A. Lendlein, H. Jiang, O. Jünger and R. Langer, Nature, 2005, 434, 879–882 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00162h |
| This journal is © The Royal Society of Chemistry 2023 |