
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccelerated nitroxide-mediated polymerization of styrene and butyl acrylate initiated by BlocBuilder MA using flow reactors†
Ryo
Takabayashi
 a,
Stephan
Feser
b,
Hiroshi
Yonehara
c,
Ilhyong
Ryu
de and
Takahide
Fukuyama
*a
a,
Stephan
Feser
b,
Hiroshi
Yonehara
c,
Ilhyong
Ryu
de and
Takahide
Fukuyama
*a
aDepartment of Chemistry, Graduate School of Science, Osaka Metropolitan University, Sakai, Osaka 599-8531, Japan. E-mail: fukuyama@omu.ac.jp
bBYK-Chemie GmbH, Abelstrasse 45, 46483 Wesel, Germany
cBYK Japan K.K., 3-29 Ichigaya-Honmuracho, Shinjuku-ku, Tokyo 162-0845, Japan
dInstitution for Research Promotion, Osaka Metropolitan University, Sakai, Osaka 599-8531, Japan
eDepartment of Applied Chemistry, National Yang Ming Chiao Tung University (NYCU), Hsinchu 30010, Taiwan
First published on 21st September 2023
Abstract
The nitroxide-mediated radical polymerization (NMP) of styrene (St) and butyl acrylate (BA) using BlocBuilder MA under continuous flow reaction conditions showed higher conversions and smaller polydispersity index (Đ) values compared with batch reaction conditions. Adding a small amount of acetol or malononitrile accelerated the NMP reaction without sacrificing Đ values.
Flow reaction technology has attracted intense attention in terms of the excellent mass and heat diffusion, and has found numerous applications in synthetic organic chemistry.1,2 The use of flow reactors in chemical processing has many advantages over batch reactors, including the high-speed mixing of substrates and reagents, and precise control of the reaction temperature and the residence time (reaction time). Moreover, systems that utilize the properties of the flow reaction technology have been appearing, e.g., gas–liquid reactions,3 continuous production,4 parameter optimization5 and automated synthesis6 in flow reactors. Despite the initial concern regarding the clogging of microchannels by the polymers that are formed, the potential of flow polymerization is now recognized as a viable methodology for polymerization.7–10 As has been shown for many flow chemical processes, rapid and accurate heat management is a distinct advantage over other approaches in flow polymerization. This can be attributed to the high surface-to-volume ratios that can be used in this process. Indeed, polymerization reactions of alkenes are, in principle, highly exothermic due to the formation of stronger C–C σ bonds at the cost of breaking weaker C–C π bonds, which often results in uncontrolled reactions due to the inefficient removal of heat from batch reactors. The heat-management problem is much more severe in polymerization conducted in a large batch reactor for scaling up.11,12 The chemical and pharmaceutical industries widely acknowledge the advantages of the continuous production of various materials using compact flow reactors, which allows the large changes in surface-to-volume ratios that occur on upscaling to be avoided.13–16 We have long been involved in studies on using flow reactors in radical reactions versus batch processes, irrespective of whether they are thermally or photochemically induced.17,18 In joint studies with the Studer group, we previously reported that (BA–St) di-block copolymers consisting of BA and St could be synthesized in a highly controlled manner using a continuous flow reactor and a designed TEMPO-type nitroxide.19 We are now particularly interested in the industrial potential of using flow NMP reactions using an industrially available alkoxyamine, such as BlocBuilder MA (BB MA) which was developed and supplied by Arkema K.K.,20,21 especially as to whether the flow polymerization of BA–St di-block copolymer would be achieved in higher conversions.
Research over the past two decades has focused on the reversible-deactivation radical polymerization (RDRP) of alkenes for the synthesis of well-defined polymers possessing low Đ values.22 In this regard, research reports for the flow update of atom transfer radical polymerization (ATRP)23,24 and reversible addition–fragmentation chain-transfer (RAFT)25,26 are abundant, but, strangely, only a few reports regarding the use of flow for nitroxide-mediated polymerization (NMP)27 have appeared. NMP is known to control polymerization of styrene and acrylate-type monomers well and has the simple reaction mechanisms which usually do not require any purification process.28 It should be noted that there are many benefits associated with the use of flow reactors in conjunction with NMP, since typical protocols for this process involve elevated reaction temperatures higher than 100 °C.29 NMP often requires a lengthy reaction time to achieve a high conversion of monomers. Accordingly, shortening reaction time is always beneficial not to waste unreacted monomers unless it does not negatively affect polymerization. For instance, our previous work demonstrated the polymerization of BA–St di-block copolymers in flow reactors with Đ = 1.26, but the total conversion was 76% for a total 4-hour residence time.19 In this work, higher conversions with relatively low Đ values were intended. To achieve a higher conversion in a relatively short reaction time while suppressing the increase in Đ values in NMP, the addition of ascorbic acid was reported to show positive results in the polymerization of St in 2004.30 Subsequently, α-hydroxy carbonyl compounds (e.g., acetol)31,32 and active methylene compounds (e.g., malononitrile)33,34 were used in batch reactors to accelerate NMP. To our knowledge, the combination of NMP using BlocBuilder MA (BB MA) and such additives has yet to be reported, irrespective of batch or flow polymerization. In this study, we studied a synergistic effect by the use of additives and flow reactors, in the hope of strengthening the potentials of flow NMP. Herein, we report that continuous flow NMP using BB MA, coupled with effective additives such as α-hydroxy carbonyl compounds or active methylene compounds, is highly beneficial for achieving a high conversion of monomers with a relatively low Đ value in a shortened reaction time.
All NMP reactions, irrespective of whether they were used for batch or flow, were conducted in the presence of BB MA, an alkoxyamine supplied by Arkema K.K., and 2-methoxypropyl acetate (PMA) as a solvent with or without an additive, such as acetol and malononitrile, at various temperatures. All reagents were used as received without any purification. The conversions were evaluated gravimetrically. The number-average molecular mass (Mn) and polydispersity index (Đ) of the resulting polymers were determined by gel permeation chromatography (GPC) analysis.
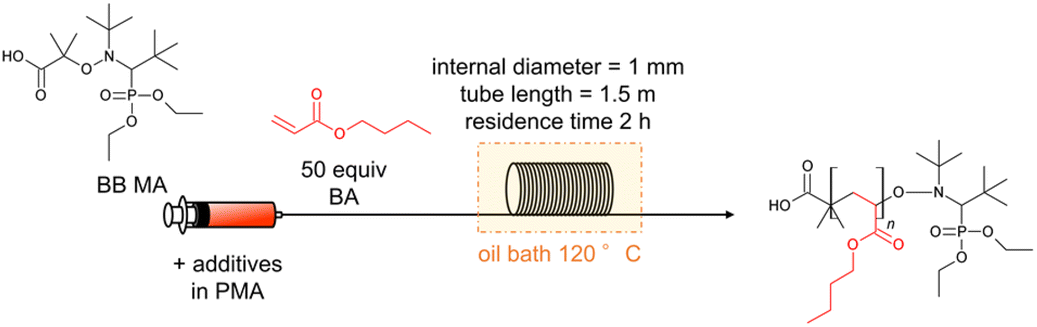
We first investigated the polymerization of St and BA using BB MA as the initiator. A 35 mL test tube was used for batch reactions, and a stainless-steel tubular reactor having a 1.5 m length and a 1 mm internal diameter (i.d.) was used for the flow reactions. Table 1 lists the conversion of the monomers, Mn, and Đ values of the obtained polymers. The polymerization of St was conducted with 0.26 M of BB MA's concentration and a BB MA/St ratio = 1/50 in PMA at 120 °C for 2 hours, and that of BA was conducted with 0.18 M of BB MA's concentration and a BB MA/BA ratio = 1/50 in PMA at 120 °C for 2 hours with a flow rate of 0.59 mL h−1. The theoretical molecular weight of these polymers was expected to be 5600 and 6800 g mol−1, respectively. The batch polymerization of St resulted in a 57% conversion with Đ = 1.14 (Table 1, entry 1), and the flow polymerization reaction resulted in a 63% conversion with Đ = 1.09 (Table 1, entry 2). The batch polymerization of BA resulted in a 41% conversion with Đ = 1.32 (Table 1, entry 3), and the flow polymerization resulted in a 56% conversion with Đ = value of 1.16 (Table 1, entry 4). While slightly higher conversion and lower Đ values were observed in the flow polymerization of St compared to the batch polymerization, the tendency to favour flow polymerization was more evident in the polymerization of BA. We speculate that the advantages owe to more efficient heat transfer in the small channels of the flow reactor, which seems more apparent in the case of polymerization of BA, more exothermic than that of St, as the Yoshida group previously observed.7 Well-controlled temperature in a flow system seems beneficial for well-controlled polymerization to prevent undesired reactions such as chain termination. Polymerization of BA in flow reactors with different lengths of flow reactors was also conducted, while the residence time was fixed to 2 hours. Polymerization in a 0.5 m flow reactor with a flow rate of 0.20 mL h−1 gave a 65% conversion with Mn = 4398 g mol−1 and Đ = 1.29 (Table 1, entry 5), and that of in a 3 m flow reactor gave a 76% conversion with Mn = 4951 g mol−1 and Đ = 1.26 with a flow rate of 1.18 mL h−1 (Table 1, entry 6). Coupled with the data using a 1.5 m flow reactor (Table 1, entry 4), we can say that flow rates affect polymerization under highly concentrated conditions. In the following experiments, the length of flow reactors was fixed to 1.5 m.

After observing favourable results for flow NMP using BB MA, we then examined the effects of α-hydroxy carbonyl compounds and active methylene compounds in accelerating the reaction. Among α-hydroxy carbonyl compounds examined for the flow NMP of BA (Table 2, entries 2–4), we found that adding 0.20 equiv. of acetol against BBMA dramatically accelerated the NMP of BA, resulting in a 92% conversion with Đ = 1.39 (Table 2, entry 4). In contrast, the addition of acetoin or α-hydroxy-γ-butyrolactone caused only a slight increase in the conversion. Among the active methylene compounds (Table 2, entries 15–18), malononitrile proved to be the most effective, giving an 89% conversion with Đ = 1.33 (Table 2, entry 18). Encouraged by the acceleration in reaction rate caused by adding acetol or malononitrile, we then investigated the amounts of additives needed for achieving optimal results. When 0.16–0.25 equiv. of acetol was used, high conversions with acceptable Đ values were observed (Table 2, entries 4–6). Increasing the amount of acetol further gave the polymers with higher Đ values despite higher conversions (Table 2, entries 7 and 8). Adding 1.0 or 2.0 equiv. of malononitrile resulted in high conversions and lower Đ values (Table 2, entries 18 and 19). Increasing the amount of additive up to 4.0 equiv., resulted in an excellent conversion of 94% but with a higher Đ value of 1.64 (Table 2, entry 20). Fig. 1 visualizes information concerning the tendency of the shift towards higher molecular weights and higher Đ values for increasing amounts of acetol or malononitrile. Polymerization of BA using 0.2 equiv. of acetol or 2.0 equiv. of malononitrile in different residence times was investigated as well (Table 2, entries 4, 9 and 10 for acetol, and entries 18, 21 and 22 for malononitrile). Both the conversions and Đ values increased as residence time was extended. The amount of monomer was also changed to 100, 200, 500 equiv. against BBMA with the addition of acetol or malononitrile without changing the residence time (Table 2, entries 11–13 for acetol, and entries 23–25 for malononitrile). For both cases, the tendency that the conversions decreased as the amount of monomer increased was observed. Indeed, when the amount of BA is 200 or 500 equiv., conversions dramatically dropped to 56%, 34% in the case of acetol, and 55% and 34% in the case of malononitrile. These results suggest that optimization of the amount of the additives and the other reaction conditions is required to reach a good balance between conversions and Đ values. We also examined the effects of acetol and malononitrile for use in batch polymerization. The batch polymerization of BA in the presence of acetol (0.2 equiv.) gave a 49% conversion with Đ = 1.38 (Table 2, entry 14), and that in the presence of malononitrile (2.0 equiv.) resulted in a 60% conversion with Đ = 1.33 (Table 2, entry 26), while in the case of no additives, only a 41% conversion with Đ = 1.32 was obtained (Table 1, entry 3). The acceleration effect by acetol or malononitrile was also observed for the flow polymerization of St. While the polymerization of St with a BB MA/St ratio = 1/50 in PMA at 120 °C for 2 hours without any additive gave a 63% conversion with Đ = 1.09, the polymerization with acetol (0.2 equiv.) showed an 87% conversion with Đ = 1.20 and that with malononitrile (2.0 equiv.) gave a 79% conversion with Đ = 1.20 respectively. The acceleration in NMP by α-hydroxy carbonyl compounds can be explained by assuming that the chain termination step is extended, as other researchers have previously reported.31,32 In principle, chain termination such as recombination and disproportionation are inevitable in radical polymerization reactions, including NMP, and it would cause the accumulation of free nitroxide in the NMP reaction system. The free nitroxide then has more chances to cap the propagating polymer chain, thus hampering the propagation of the polymer growth. α-Hydroxy carbonyl compounds capable of reacting with the free nitroxide would prevent the accumulation of it. On the other hand, in the acceleration in NMP by active methylene compounds, it is assumed that the acidic hydrogen in the active methylene compounds forms hydrogen bonds between the oxygen and the nitrogen in the nitroxide, weakening the bond between the nitroxide and the polymer terminus, leading to the smooth dissociation of the dormant species between the propagating polymer and the nitroxide.33,34
 | ||
Fig. 1 GPC charts for optimization of the amounts of acetol (top) and malononitrile (bottom) in the polymerization of BA. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Against BB MA in mol. Against BB MA in mol. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Additive | Additive's amounta | Conv. (%) | M n (g mol−1) | Đ |
| a Against BB MA in mol. b Residence time is 1 hour. c Residence time is 3 hours. d BA 100 equiv. e BA 200 equiv. f BA 500 equiv. g In batch reactors. | |||||
| 1 | No (control) | — | 56% | 4347 | 1.16 |
| 2 | Acetoin | 0.2 | 65% | 4615 | 1.22 |
| 3 | α-Hydroxy-γ-butyrolactone | 0.2 | 72% | 4808 | 1.24 |
| 4 | Acetol | 0.2 | 92% | 5453 | 1.39 |
| 5 | Acetol | 0.16 | 84% | 5184 | 1.29 |
| 6 | Acetol | 0.25 | 97% | 5441 | 1.45 |
| 7 | Acetol | 0.5 | 99% | 5095 | 1.71 |
| 8 | Acetol | 1.0 | 100% | 5518 | 1.92 |
| 9b | Acetol | 0.2 | 71% | 4353 | 1.40 |
| 10c | Acetol | 0.2 | 96% | 5117 | 1.66 |
| 11d | Acetol | 0.2 | 95% | 8029 | 1.79 |
| 12e | Acetol | 0.2 | 56% | 11989 |
1.36 |
| 13f | Acetol | 0.2 | 34% | 17520 |
1.33 |
| 14g | Acetol | 0.2 | 49% | 3291 | 1.38 |
| 15 | Acetylacetone | 2.0 | 64% | 4948 | 1.26 |
| 16 | Ethyl acetoacetate | 2.0 | 81% | 5048 | 1.30 |
| 17 | Diethyl malonate | 2.0 | 76% | 4942 | 1.25 |
| 18 | Malononitrile | 2.0 | 89% | 5360 | 1.33 |
| 19 | Malononitrile | 1.0 | 81% | 5313 | 1.22 |
| 20 | Malononitrile | 4.0 | 94% | 4681 | 1.64 |
| 21b | Malononitrile | 2.0 | 63% | 4369 | 1.30 |
| 22c | Malononitrile | 2.0 | 94% | 4984 | 1.60 |
| 23d | Malononitrile | 2.0 | 80% | 8555 | 1.44 |
| 24e | Malononitrile | 2.0 | 55% | 12675 |
1.30 |
| 25f | Malononitrile | 2.0 | 34% | 20027 |
1.32 |
| 26g | Malononitrile | 2.0 | 60% | 4120 | 1.33 |
At this point, all experiments concerning flow polymerization in the present study were carried out using a 1.5 m flow reactor whose i.d. is 1 mm, with a flow rate of 0.59 mL h−1 (0.54 g h−1). To confirm that scaling up this reaction is feasible, we then conducted the polymerization of BA using a tubular reactor having a larger diameter size (i.d. 4.25 mm) and a longer length (10 m) with a flow rate of 74.3 mL h−1 (68.5 g h−1). While the polymerization of BA conducted with MA/BA ratio = 1/50 in PMA at 120 °C for 2 hours using the smaller flow reactors (1.5 m flow reactor with i.d. 1 mm) gave a 56% conversion with Mn = 4347 g mol−1 and Đ = 1.20, the polymerization using the larger flow reactor gave a 41% conversion with Mn = 3143 g mol−1 and Đ = 1.26. Although both the conversions and the Đ values are slightly inferior to those obtained for the smaller-size reactor, it still permitted this type of BA polymer to be controlled reasonably well on scaling-up.
Considering all the clarified factors (temperature, residence time, and additive), we attempted to carry out a controlled synthesis of a BA–St di-block copolymer using a serially connected flow system (Scheme 1). The 1st BA block was polymerized at 120 °C with a residence time of 2 h with a flow rate of 0.59 mL h−1, in the presence of acetol (0.2 equiv.) or malononitrile (2.0 equiv.) as an additive. The St monomer in PMA was then fed into the flow system with a flow rate of 0.59 mL h−1 and mixed with the reaction mixture using a micromixer, MiChS α600, having a channel diameter size of 0.6 mm. While heating at 120 °C was not sufficient for the polymerization of St (Table 1, entry 2), the 2nd St block was polymerized at 140 °C for another 2 h of residence time. The total flow rate was 1.18 mL h−1. The continuous flow di-block polymerization with acetol and malononitrile resulted in a 93% conversion with Đ = 1.56 and a 95% conversion with Đ = 1.69, respectively, while flow di-block copolymerization without an additive also resulted in a total 94% conversion with Đ = 1.50. As a reference, the polymerization of a BA–St di-block copolymer without additives was conducted in batch reactors. The 1st block BA was polymerized at 120 °C for 3 hours, which gave a 68% conversion with Đ = 1.26, and then the 2nd block St was polymerized at 140 °C for 2 hours. In total, it gave an 86% conversion with Đ = 1.78. Fig. 2 visualizes the changes in GPC charts for (1) without additives in flow reactors, (2) without additives in batch reactors, (3) with malononitrile (2.0 equiv.) in flow reactors, and (4) with acetol (0.2 equiv.) in flow reactors. These results obtained in the absence of an additive in flow reactors appeared to be inconsistent with the results shown in Table 2 since the 1st BA polymerization without an additive gave only a 56% conversion with Đ = 1.16 (Table 2, entry 1). Considering the conversion for the polymerization of BA in the previous experiments (Table 2, entries 1, 4 and 18), these results led us to postulate that the high conversion of 94% would be due to the formation of less defined di-block structures in the absence of the additives. We therefore conclude that these additives contribute to the formation of well-defined di-block copolymers with BB MA in a shorter reaction time. In our previous work, polymerization of a BA–St di-block copolymer took 4 hours in total and gave a 76% conversion with Đ = 1.26.19 This flow system showed better conversions while suppressing the increase of Đ values by the addition of acetol or malononitrile. Although the alkoxyamines and the other reaction conditions are different, a faster reaction was achieved in this work.
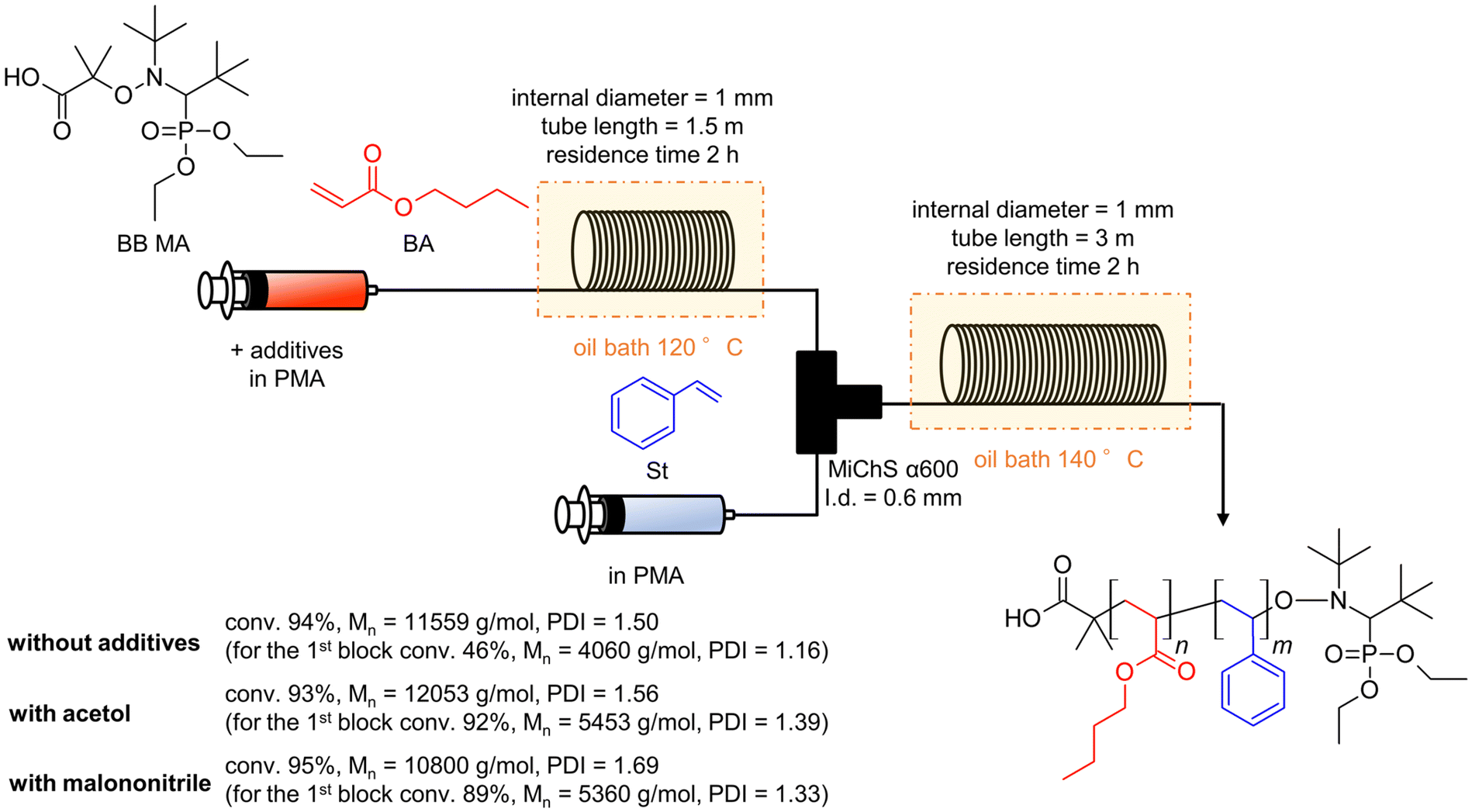 | ||
| Scheme 1 BB MA-induced continuous flow di-block copolymerization of BA and St. | ||
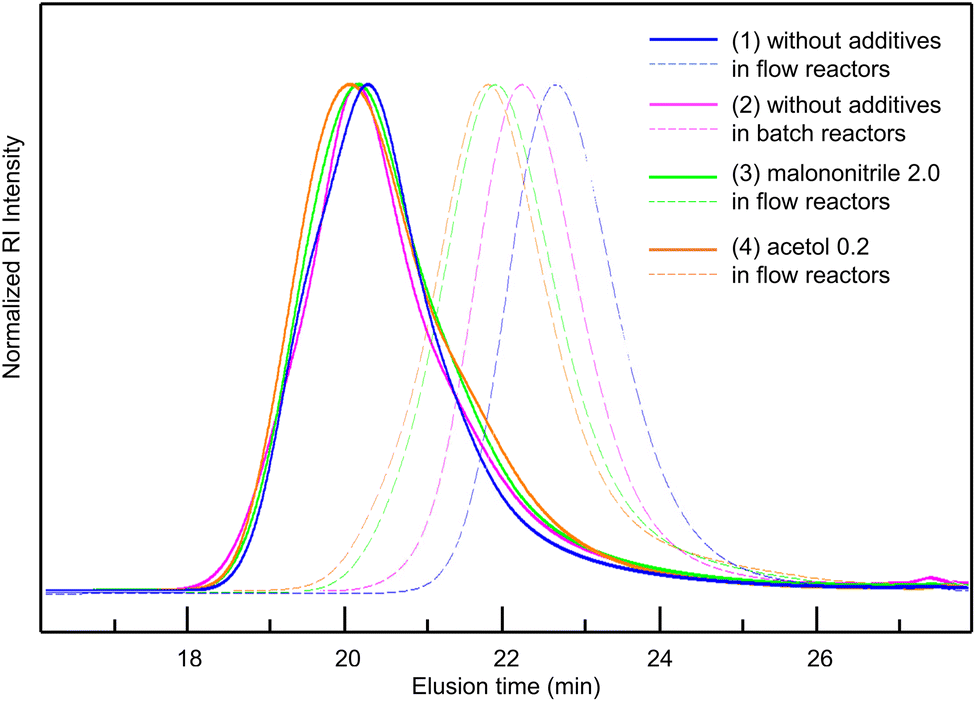 | ||
| Fig. 2 GPC charts of BA–St block copolymer (1) without additives in flow reactors (2) without additives in batch reactors (3) with malononitrile (2.0 equiv.) in flow reactors (4) with acetol (0.2 equiv.) in flow reactors. The dashed line: 1st block, the solid line: 1st + 2nd block. | ||
In conclusion, we report herein that the NMP of styrene and butyl acrylate can be achieved in a highly controlled manner by using BB MA as the initiator. Both the conversion and Đ values were improved when flow reactors were used and this difference was more apparent in the polymerization of BA. The use of an additive such as acetol or malononitrile was found to significantly accelerate the polymerization of BA, and consequently the conversion was dramatically improved without a significant increase in Đ values. The accelerating effect was much more remarkable in flow reactors than in batch reactors. BA–St di-block copolymerization was also successfully conducted with these additives using continuous flow reactors with conversions higher than 90% and a relatively low Đ such as 1.6 being achieved. The feasibility of scaling up the reaction with flow reactors was also confirmed by increasing the tubular reactor size from 1.5 m with 1 mm i.d. to 10 m with 4.35 mm i.d.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to express our appreciation to Arkema K.K. for providing us of the BlocBuilder MA. This work was supported by a JSPS grant (B). IR thanks NSTC (MOST112-2113-MA49-013) and the Center for Emergent Functional Matter Science at NYCU for additional support (111W20253).Notes and references
- J. Jiao, W. Nie, T. Yu, F. Yang, Q. Zhang, F. Aihemaiti, T. Yang, X. Liu, J. Wang and P. Li, Chem. – Eur. J., 2021, 27, 4817–4838 CrossRef CAS PubMed.
- Y. Hayashi, J. Org. Chem., 2021, 86, 1–23 CrossRef CAS PubMed.
- W. C. Fu, P. M. MacQueen and T. F. Jamison, Continuous Flow Strategies for Using Fluorinated Greenhouse Gases in Fluoroalkylations, Chem. Soc. Rev., 2021, 50(13), 7378–7394 RSC.
- R. W. Toh, M. Patrzałek, T. Nienałtowski, J. Piątkowski, A. Kajetanowicz, J. Wu and K. Grela, ACS Sustainable Chem. Eng., 2021, 9(48), 16450–16458 CrossRef CAS PubMed.
- Y. Ashikari, T. Tamaki, Y. Takahashi, Y. Yao, M. Atobe and A. Nagaki, Front. Chem. Eng., 2022, 3, 819752 CrossRef.
- C. Liu, J. Xie, W. Wu, M. Wang, W. Chen, S. B. Idres, J. Rong, L.-W. Deng, S. A. Khan and J. Wu, Nat. Chem., 2021, 13(5), 451–457 CrossRef CAS PubMed.
- T. Iwasaki and J. Yoshida, Macromolecules, 2005, 38, 1159–1163 CrossRef CAS.
- A. Nagaki and J. Yoshida, Advances in Polymer Science, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012 Search PubMed.
- N. Zaquen, M. Rubens, N. Corrigan, J. Xu, P. B. Zetterlund, C. Boyer and T. Junkers, Prog. Polym. Sci., 2020, 107, 101256 CrossRef CAS.
- M. H. Reis, F. A. Leibfarth and L. M. Pitet, ACS Macro Lett., 2020, 9, 123–133 CrossRef CAS PubMed.
- M.-C. Chevrel, S. Hoppe, D. Meimaroglou, L. Falk and A. Durand, Macromol. React. Eng., 2016, 10, 354–363 CAS.
- A. Nagaki and J. Yoshida, in Controlled polymerization and polymeric structures, Springer, International Publishing, 2013, pp. 1–50 Search PubMed.
- T. Junkers, Macromol. Chem. Phys., 2017, 218, 1600421 CrossRef.
- D. Kohlmann, M.-C. Chevrel, S. Hoppe, D. Meimaroglou, D. Chapron, P. Bourson, C. Schwede, W. Loth, A. Stammer, J. Wilson, P. Ferlin, L. Falk, S. Engell and A. Durand, Macromol. React. Eng., 2016, 10, 339–353 CrossRef CAS.
- B. J. Doyle, P. Elsner, B. Gutmann, O. Hannaerts, C. Aellig, A. Macchi and D. M. Roberge, Org. Process Res. Dev., 2020, 24, 2169–2182 CrossRef CAS.
- M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Org. Process Res. Dev., 2020, 24, 1802–1813 CrossRef CAS.
- T. Fukuyama and I. Ryu, Radical Chemistry by Using Flow Microreactor Technology, in Encyclopedia of Radicals in Chemistry, Biology, and Materials, ed. A. Studer and C. Chatgilialoglu, Wiley, Chichester, 2012, vol. 2, pp. 1243–1258 Search PubMed.
- T. Fukuyama, T. Kasakado, M. Hyodo and I. Ryu, Photochem. Photobiol. Sci., 2022, 21, 761–775 CrossRef CAS PubMed.
- I. Ryu, A. Studer, T. Fukuyama and Y. Kajihara, Synthesis, 2012, 2555–2559 CrossRef.
- J. Couturier, O. Guerret, D. Bertin, D. Gigmes, S. Marque, P. Tordo, F. Chauvin and P. Dufils, WO Pat, 2004014926, 2004 Search PubMed.
- J. Nicolas, B. Charleux, O. Guerret and S. Magnet, Macromolecules, 2004, 37, 4453–4463 CrossRef CAS.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- T. Noda, A. J. Grice, M. E. Levere and D. M. Haddleton, Eur. Polym. J., 2007, 43, 2321–2330 CrossRef CAS.
- M. Müller, M. F. Cunningham and R. A. Hutchinson, Macromol. React. Eng., 2008, 2, 31–36 CrossRef.
- C. H. Hornung, A. Postma, S. Saubern and J. Chiefari, Macromol. React. Eng., 2012, 6, 246–251 CrossRef CAS.
- S. Parkinson, N. S. Hondow, J. S. Conteh, R. A. Bourne and N. J. Warren, React. Chem. Eng., 2019, 4, 852–861 RSC.
- C. Rosenfeld, C. Serra, C. Brochon and G. Hadziioannou, Chem. Eng. Sci., 2007, 62, 5245–5250 CrossRef CAS.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- M. K. Georges, J. L. Lukkarila and A. R. Szkurhan, Macromolecules, 2004, 37, 1297–1303 CrossRef CAS.
- B. Keoshkerian, M. Georges, M. Quinlan, R. Veregin and B. Goodbrand, Macromolecules, 1998, 31, 7559–7561 CrossRef CAS.
- A. Debuigne, T. Radhakrishnan and M. K. Georges, Macromolecules, 2006, 39, 5359–5363 CrossRef CAS.
- H. Jianying, L. Jian, L. Minghua, L. Qiang, D. Lizong and Z. Yousi, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5246–5256 CrossRef.
- Z. Zhu, G. Shan and P. Pan, RSC Adv., 2016, 6, 97995–98000 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3py00709j |
| This journal is © The Royal Society of Chemistry 2023 |