DOI:
10.1039/D2QI01801B
(Research Article)
Inorg. Chem. Front., 2023,
10, 211-217
Sumanene-functionalised bis(terpyridine)–ruthenium(II) complexes showing photoinduced structural change and cation sensing†
Received
18th August 2022
, Accepted 1st November 2022
First published on 5th November 2022
Abstract
Four luminescent Ru(II) complexes (C1–C4) with sumanene-functionalised terpyridine ligands (L1 and L2) were synthesised. The presence of the sumanene moiety enabled the emergence of the ligand-centred emission and resulted in relatively easy rotation around the sumanene–terpyridine bond axis. These eventually resulted in the emergence of dual emission via the Twisted Intramolecular Charge Transfer (TICT) mechanism. This was initiated by the electron transfer from sumanene to the terpyridine–metal centre while absorbing a photon, and then the electron was converged by the electron back transfer to the phenyl moiety from the terpyridine–metal centre. Further experiments revealed the susceptible Li+ sensing ability of the complexes, which was probably attributable to the cation–π interaction on the convex side of sumanene.
Introduction
In the design strategy of sensing materials based on coordination compounds, metal fragments are used as the reporter units that change physical properties such as colour, emission intensity, and electrochemical potential by interacting with the target ions and molecules.1 In this context, Ru(II) complexes are useful scaffolds and widely applied in optical sensors due to their photosensitising ability induced by a long enough high-energy excited state to facilitate the desired electron-/energy-transfer process or photochemical reaction.2,3 In particular, [Ru(terpy)]2+ (terpy = 2,2′:6′,2′′-terpyridine) avoids diastereomeric problems in preparation, which is often observed in [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) and allows high stability.4,5 Various applications of [Ru(terpy)]2+-based systems are reported, in which donor or acceptor moieties and/or π-conjugation systems are introduced6–12 to overcome the intrinsic low emission properties of [Ru(terpy)]2+. Some selected Ru(II) complexes also show dual emissions, which occurred simultaneously from the LC and MLCT states or different MLCT states.13–18 In particular, in the case of a highly conjugated [Ru(terpy)]2+ complex, it is confirmed that the dual emission is caused by the electronic coupling of the π–π* and the MLCT states due to the structure planarisation in the excited state, which is the potential strategy for the preparation of white emitters.19,20 However, the application of the [Ru(terpy)]2+ system in sensing materials draws much less attention compared to the effort to improve its emission properties, probably because of the difficulty in combining the emission property improvement and effective target recognition.
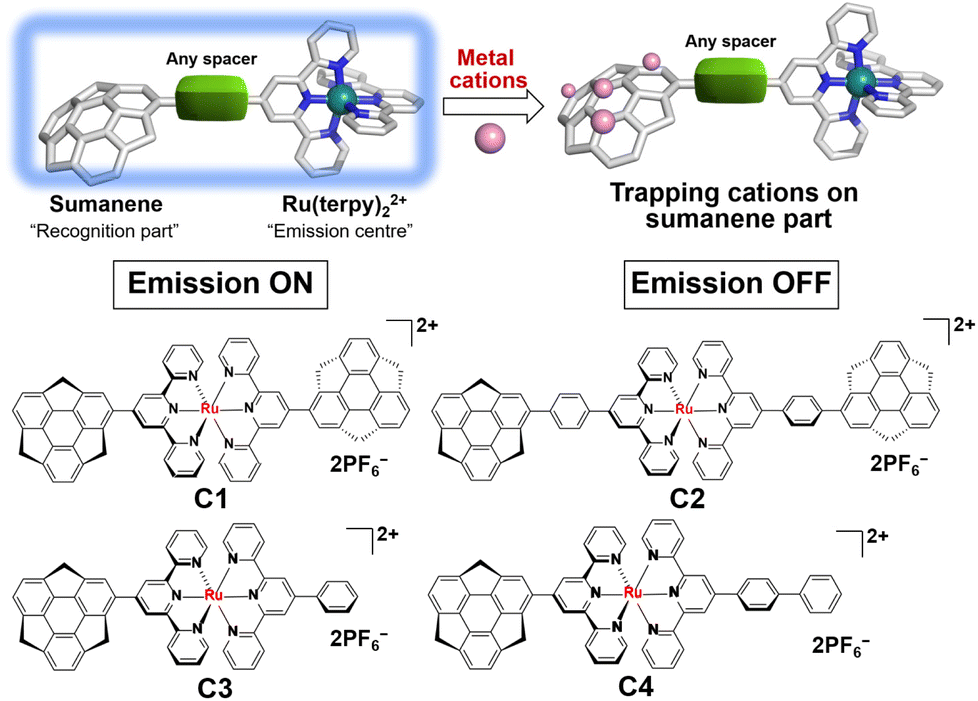
Sumanene is a fullerene fragment and therefore possesses a unique curved structure.21 This structural feature has been the source of the emergence of unique physical properties of various kinds of sumanene derivatives.22–27 In addition, it is worth noting that sumanene shows a metal cation trapping ability. In the early stage, Sastry et al. systematically explored Li+, Na+, K+ and Cu+ complexes of sumanene in terms of the formation of cation–π interactions by employing ab initio quantum mechanical tools.28 They concluded that the complex, in which the metal ion binds to the convex side of the 6-membered rim ring (Fig. 1) is the most stable. This assumption was experimentally proven by the X-ray structural characterisation of a novel complex of six potassium ions sandwiched by convex faces of two sumanene anions.29 We also recently reported the Cs+ sensing ability of neutral sumanene.30–32
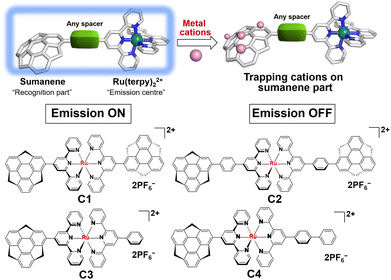 |
| | Fig. 1 A concept of sumanene–Ru(II) cation sensors and the structures of C1–C4. | |
Based on the above-reported knowledge about the high scalability of the synthesis of [Ru(terpy)]2+ complexes for various applications and the cation trapping ability of curved π-conjugated sumanene skeletons, we newly designed and synthesised a total of four homo- and heteroleptic complexes C1–C4 containing the stable [Ru(terpy)]2+ complex (Fig. 1). The experimental results showed that all the complexes were emissive at room temperature in solution upon excitation of the ligand-centred π–π* band. Significantly, C2, C3 and C4 exhibited dual emission properties caused by the molecular twisting in the excited state, which was evaluated by spectroscopic and theoretical analyses. The cation sensing abilities of C1–C4 were also investigated and it was found that all of them work as Li+ detectors, probably via the cation–π interactions.
Results and discussion
Syntheses of ligands and complexes
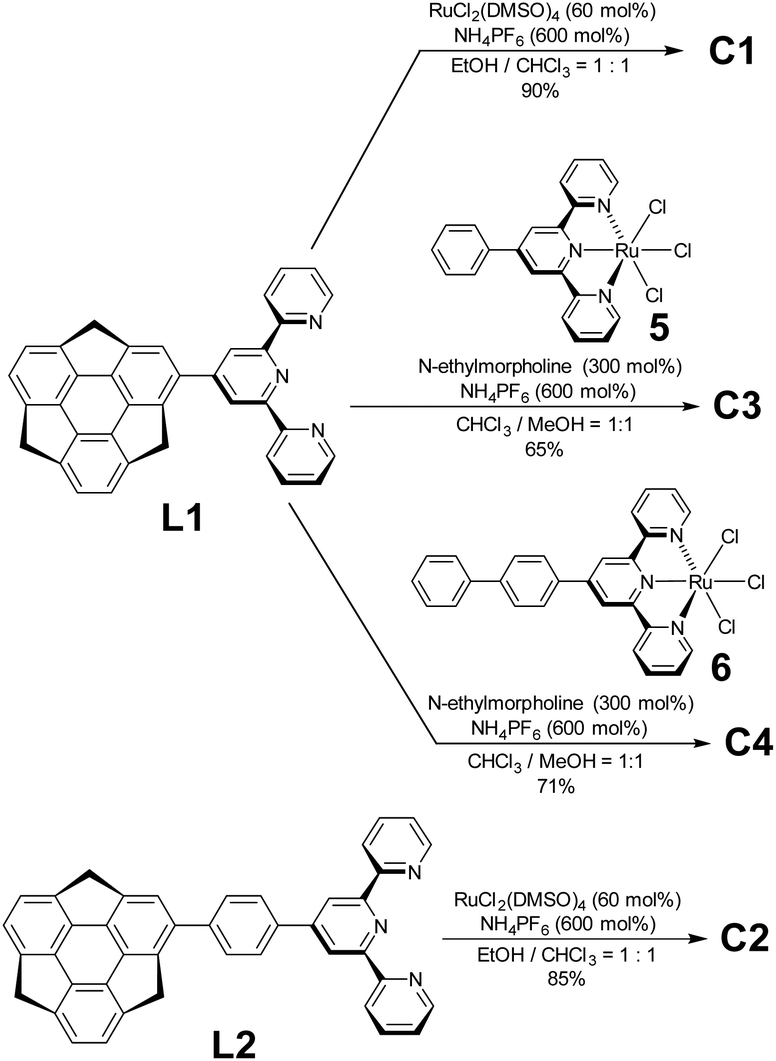
Regarding ligand preparation, 4-sumanenylterpyridine L1 was synthesised using the Kröhnke reaction from formylsumanene33 and the phenylene-tethered derivative L2 using the Pd-catalyzed Suzuki–Miyaura coupling reaction from bromosumanene,24 respectively (Scheme S1, ESI†). The complexation of L1 with 50 mol% of RuCl2(DMSO)4 was performed in an EtOH/CHCl3 mixed solution and a following counterion exchange reaction with an excess amount of NH4PF6 yielded C1 as red solids (Schemes 1 and S3, ESI†). The same method was used to obtain the red solid C2 using L2 as the ligand. Unsymmetrical C3 and C4 were prepared by the complexation of L1 with a stoichiometric amount of Ru2+ complexes 5 and 6 (Schemes S2 and S4, ESI†).34 In addition to C1–C4, the control complexes SC1–SC3 without sumanene units were also prepared (Scheme S5, ESI†). Their characterisation was performed using 1H NMR, correlation spectroscopy (COSY) NMR, and matrix-assisted laser desorption-ionisation time-of-flight mass spectrometry (MALDI-TOF-MS). The optimized structures obtained by density functional theory (DFT) calculations showed that the terpyridine moieties in the complexes were flat and that the coordination sphere did not show significant distortions (Fig. S1, ESI†). The torsion angles of the sumanene and terpy moieties in all the complexes were around 45°.
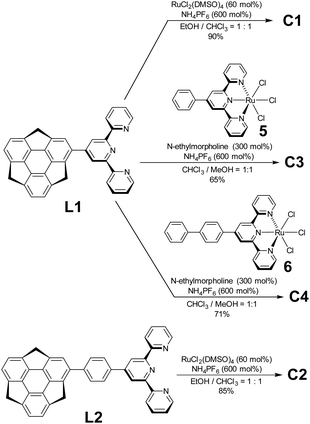 |
| | Scheme 1 Synthesis of C1, C2, C3 and C4. | |
Photophysical properties of C1–C4
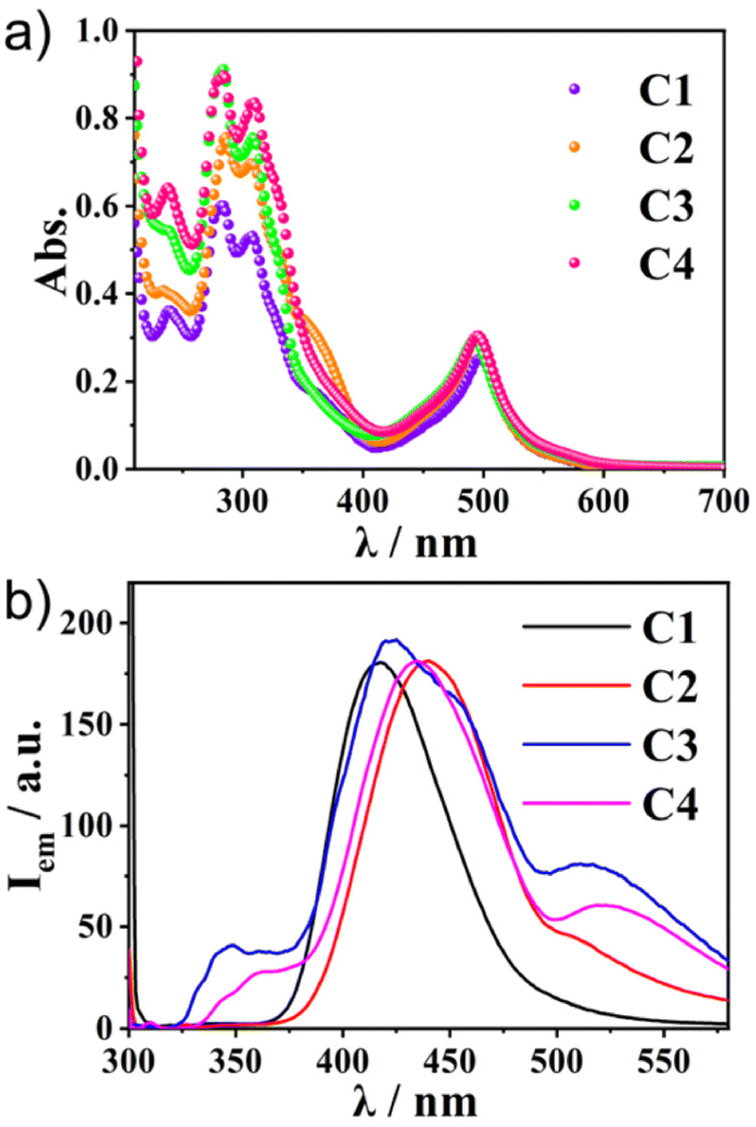
The photophysical properties of the two ligands were first investigated by UV–vis absorption and fluorescence spectroscopy. The UV–vis spectra of L1 and L2 diluted in CH3CN showed absorption peaks at 245 and 295 nm. Both L1 and L2 showed strong emissions at 420 nm (Fig. S2, ESI†). The details of the optical properties of C1–C4 were next investigated (Fig. 2a and Table 1). All the absorption spectra of the complexes exhibited similar patterns—intense bands around 280–350 nm and around 500 nm. Time-dependent (TD)-DFT calculations revealed that the former band was composed of the sumanene π–π* transition (280–350 nm) and the ligand-to-ligand charge transfer (LLCT) from sumanene to the terpyridine core (350–400 nm) (Fig. S3 and S4, ESI†). The strong absorption peak at around 500 nm was attributable to the mixture of Ru(II) to terpyridine charge transfer (MLCT) and sumanene to terpyridine charge transfer (LLCT) bands.
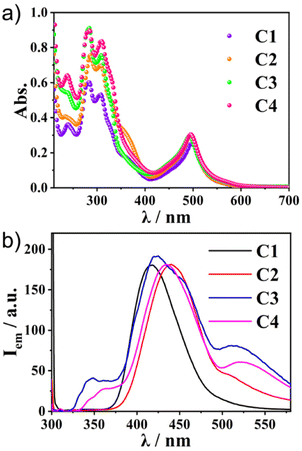 |
| | Fig. 2 (a) The absorption and (b) photoluminescence spectra (λex = 300 nm) of C1–C4 in CH3CN (1.0 × 10−5 M) at 300 K. | |
Table 1 Photophysical properties of complexes C1–C4 at 300 K
| Complex |
λ
abs
[nm] (ε × 104 [M−1 cm−1]) |
λ
em
,
[nm] (Φem × 10−2) |
|
In CH3CN (1.0 × 10−5 M).
λ
ex = 300 nm.
|
|
C1
|
283 (6.0), 308 (5.3), 494 (2.5) |
403 (0.8) |
|
C2
|
286 (7.5), 309 (6.9), 497 (3.0) |
442 (4.3) |
|
C3
|
287 (9.1), 309 (7.5), 492 (3.1) |
424 (7.9), 511 (3.2) |
|
C4
|
290 (9.1), 308 (8.3), 495 (3.1) |
434 (8.4), 534 (3.3) |
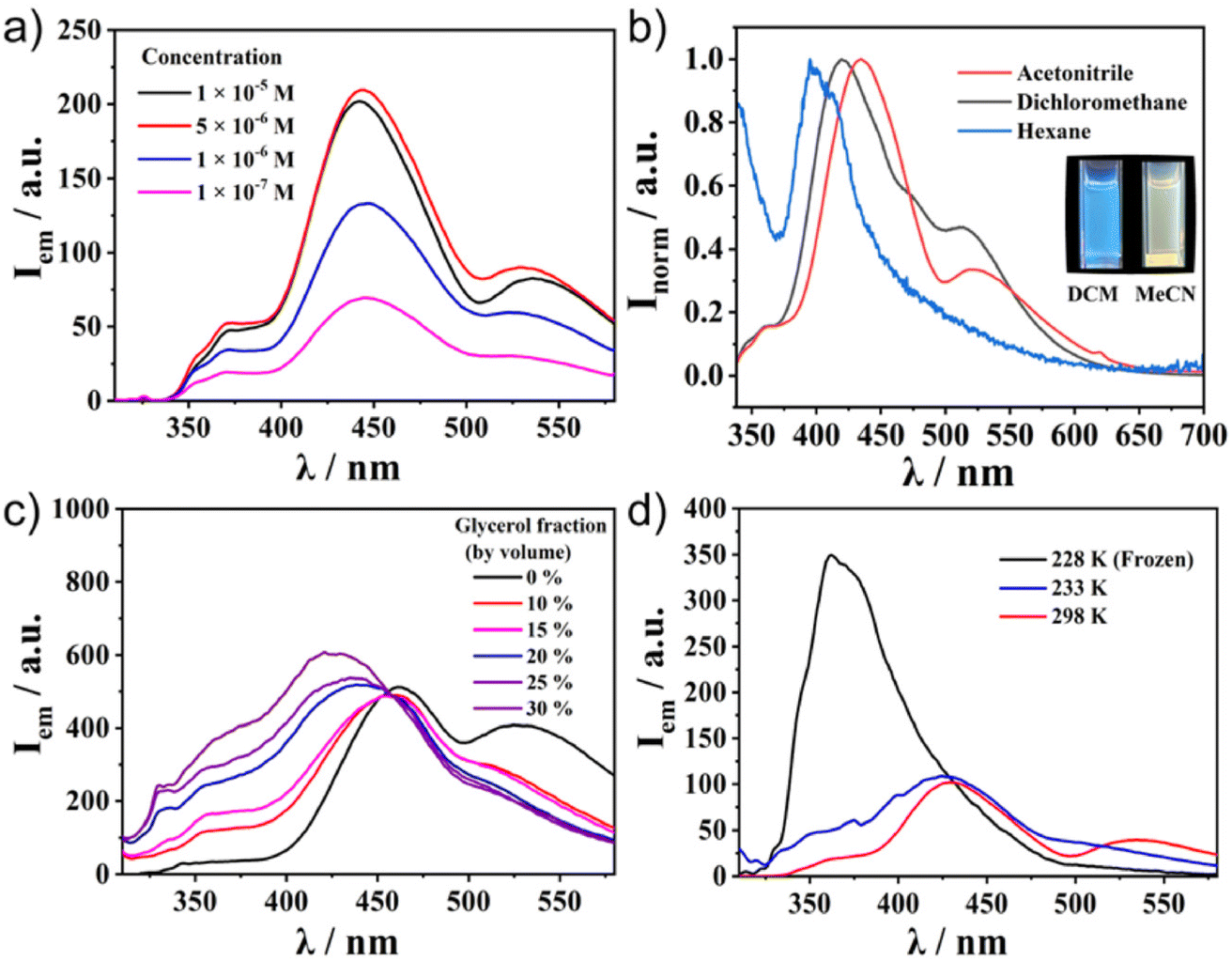
Meanwhile, the emission spectra of C1–C4 exhibited significant differences depending on the molecular structures (Fig. 2b). All the Ru(II) complexes showed only weak emission at around 650 nm upon excitation of the MLCT band (510 nm) at 298 K (Fig. S5, ESI†). Meanwhile, they exhibited one broad, strong emission band at 400–500 nm upon excitation at the π–π* band (300 nm). This was in contrast to most [Ru(terpy)2]2+ complexes, in which the internal conversion process is very fast and efficient, and therefore, the ligand centre (LC) emission is quenched effectively to give only weak emission. Indeed, the control compounds SC1–SC3 did not show any strong emission at around 500 nm (Fig. S6, ESI†), indicating that the presence of sumanene induced a different relaxation process from conventional [Ru(terpy)2]2+ complexes. Furthermore, one small shoulder peak at 500 nm in C2 and one additional broadened band at 530 nm in C3 and C4 were also observed (Fig. 2b). To reveal the source of them, we first chose C4 as a representative and investigated its concentration effect on the emission properties (Fig. 3a). As a result, it was found that C4 started to show aggregation-induced quenching at more than 5 × 10−6 M, while the emission intensity decreased at lower concentrations (<1 × 10−6 M). However, the second emission bands at more than 500 nm were retained even at 1 × 10−7 M, indicating that the source of the second band existed in the intramolecular process rather than the intermolecular one, such as the formation of some specific excimers. The same tendencies were confirmed in C2 and C3 (Fig. S7, ESI†). The emission spectra of C4 in a CH3CN/water mixture with a water fraction from 0% to 70% did not show a significant emission intensity change at 534 nm, which also supported there being no contribution of the intermolecular process to the generation of the second emission (Fig. S8, ESI†).
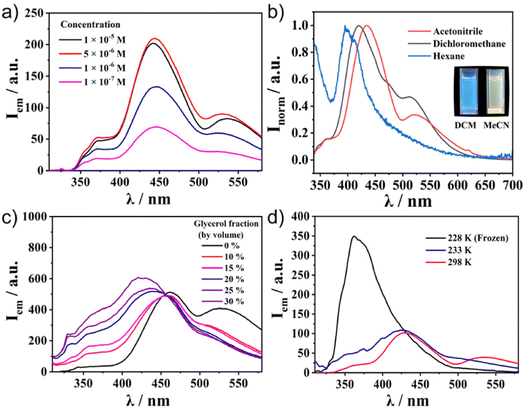 |
| | Fig. 3 Photoluminescence spectra of C4 in (a) CH3CN at different concentrations and (b) 1.0 × 10−5 M different solvents. (c) 1.0 × 10−5 M CH3CN/water/glycerol mixtures with different glycerol fractions and (d) 1.0 × 10−5 M CH3CN solution at different temperatures. Excitation wavelength λex = 300 nm. | |
These experimental results indicated the significant contribution of the intramolecular process. Considering this system's flexibility, especially at the connecting bond between sumanene and the phenyl ring, Twisted Intramolecular Charge Transfer (TICT) was the most likely reason to explain the phenomena. It is known that TICT is an electron transfer process that occurs upon photoexcitation in molecules that usually consist of donor and acceptor parts linked by a single bond. Following intramolecular twisting, the TICT state returns to the ground state through red-shifted emission. The emission properties are potentially environment-dependent. In this context, we next performed solvent, viscosity, and temperature-dependent emission property investigations (Fig. 3b–d). The emission spectral envelop of C1–C4 showed substantial solvatofluorochromism. With an increase in solvent polarity from hexane to CH3CN, the emission peak gradually shifted to lower energy (Fig. 3b† and S9, ESI†). Such a clear bathochromic shift was displayed by the emission colour change. This indicated the presence of the charge-transferred structures of C1–C4 in the excited state. Furthermore, the emission spectra of C4 in the CH3CN/water/glycerol mixture showed a significant decrease in the second emission intensity by the increment of the system viscosity with the generation of a new emission peak at 420 nm (Fig. 3c). This was because of the restriction of the molecular rotation in a viscous medium. Temperature-dependent measurements also indicated the contribution of intramolecular rotation to emergence of the second emission (Fig. 3d). When the temperature decreased to 228 K, the second emission disappeared and only one intense peak remained. These results confirmed that the key to exhibiting the dual emission was the geometrical arrangement of the fluorophores controlled by the intramolecular rotation. The lifetime measurements of C1–C4 at their different emission maxima also supported the contribution of TICT (Fig. S10–S13, ESI†). For ligand-centred emission, C1 exhibited a fluorescence lifetime longer than those of C2, C3, and C4. This is explained by C1 having a symmetric structure without the phenyl linker, making the emission somewhat transition forbidden. This corresponds to its low quantum yield (0.8%, Table 1). In the case of the second emission observed around 500 nm, the lifetimes of both C3 and C4 were longer than those with a shorter wavelength. This indicated that the second emission came through the molecular rotation process. In addition, the negative amplitudes of the fitting parameters (A′1, see ESI†) for both of the longer wavelength fluorescence lifetimes were cogent evidence for TICT (Table S2, ESI†).35,36 Another important observation is that Ru complexed without a sumanene unit, as seen in SC1–SC3, did not show dual emission (Fig. S6, ESI†). This fact indicates that the rotation of the sumanene skeleton in the excited state of the complexes having phenylene tether triggered the exhibition of the second emission.
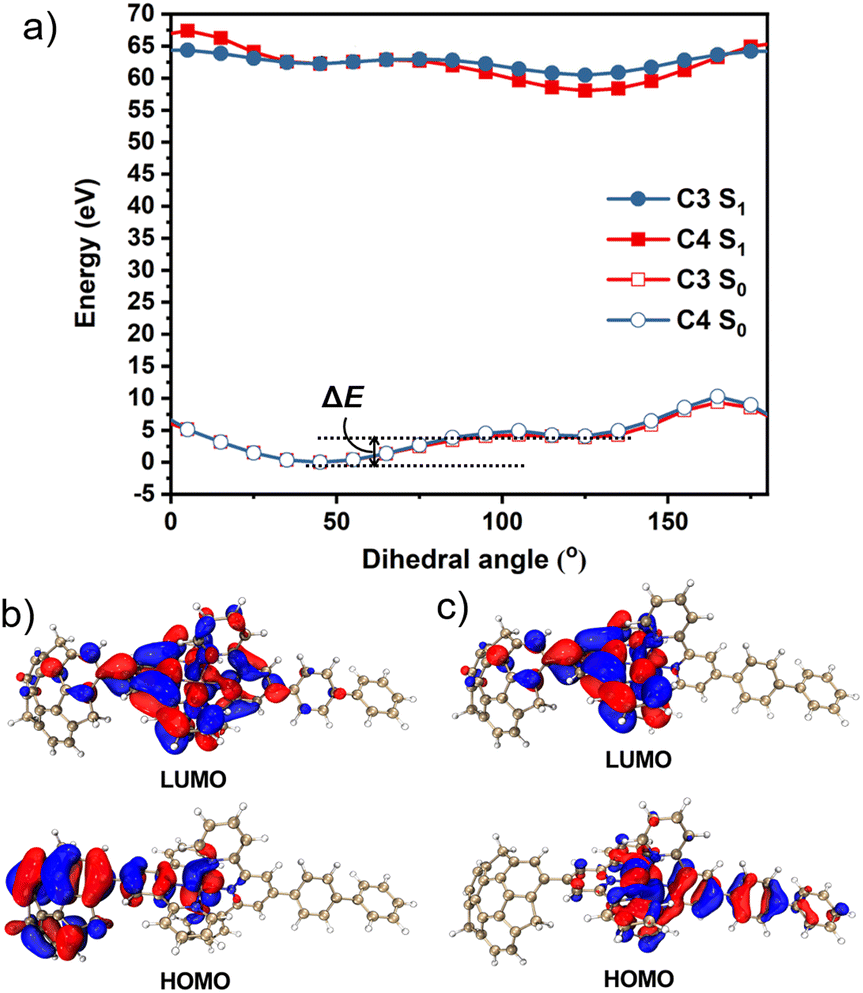
To obtain further information, we calculated the ground state (S0) and excited state (S1) potential energy surfaces of C3 and C4 in CH3CN using the DFT/TD-DFT method at the PBE0/SDD-6-31G(d) level of theory. The optimisation results suggested that the dihedral angles of sumanene–terpyridine in both C3 and C4 were 45° in the S0 state, whereas they changed to 125° in the S1 state (Fig. S1, ESI†). These results were further investigated by potential energy scans (Fig. 4a). In the calculations, the sumanene–terpyridine dihedral angles of both the S0 and S1 structures were systematically fixed between 0° and 180°, while the other parts were relaxed. The calculation results showed two minima at both the S0 and S1 states in each complex. The rotation energy barriers, ΔE, here defined as the energy difference between the two minima, were much smaller in the excited state (C3: 0.63 kcal mol−1 and C4: 0.44 kcal mol−1) than in the ground state (C3: 9.43 kcal mol−1 and C4: 7.67 kcal mol−1). The relatively low rotation energy in S1 probably allowed the photoinduced structural change in the present systems.37 MOs in the minimum energy states enabled a more detailed discussion about the mechanism (Fig. 4b, c and S14–S17, ESI†). The TD-DFT calculations revealed that the contributing orbitals for both the S1 → S0 and T1 → S0 transitions were mainly the HOMO and LUMO. The simulated emission wavelengths also confirm no contribution of the T1 → S0 transition to the second emission (Table S3, ESI†). In the case of C4, the LUMOs of both S0 and S1 were delocalised onto the terpyridine–metal centre, while the HOMOs were located on the sumanene skeleton in the S0 state and on the terpyridylphenyl unit in the S1 state. This means that an electron transfer from sumanene to the terpyridine-metal centre occurred while absorbing a photon that was followed by a molecular twisting caused during the excitation process. The same tendency was confirmed in C2 and C3 while not in C1. This is explained by the molecular symmetry which was affected by the phenyl linker. Indeed, the high molecular symmetry of C1 resulted in a symmetrical HOMO distribution in the S0 state, while the asymmetrical C3, C4, and phenyl bridged C2 did not. However, the TD-DFT simulated data showed an almost negligible effect of the symmetrical HOMO distribution of C1 on its absorption properties (Fig. S3, ESI†). Meanwhile, the absence of the phenyl linker resulted in the disappearance of the pass for the second emission, which was caused by the electron back transfer to the phenyl moiety from the terpyridine–metal centre. The very small amount of the HOMO distribution on the phenyl moiety in the S1 state of C2 was probably reflected by the weaker intensity of the second emission compared with those of C3 and C4. These results were consistent with the experimental observation, especially at low temperatures and under highly viscous conditions in which the molecular motions were significantly suppressed to restrict the molecular twisting.
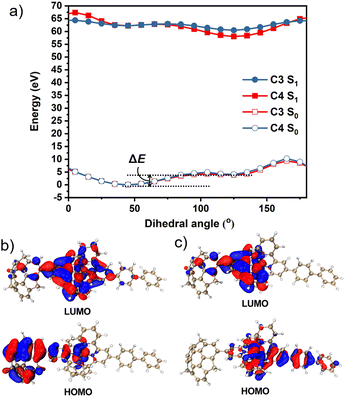 |
| | Fig. 4 (a) Potential energy scan of the S0 and S1 states of C3 and C4versus the intramolecular dihedral angle between the sumanene and the terpyridine moieties in CH3CN. The HOMO and LUMO orbitals of C4 at the (b) S0 and (c) S1 energy minimum geometry. | |
Cation sensing property
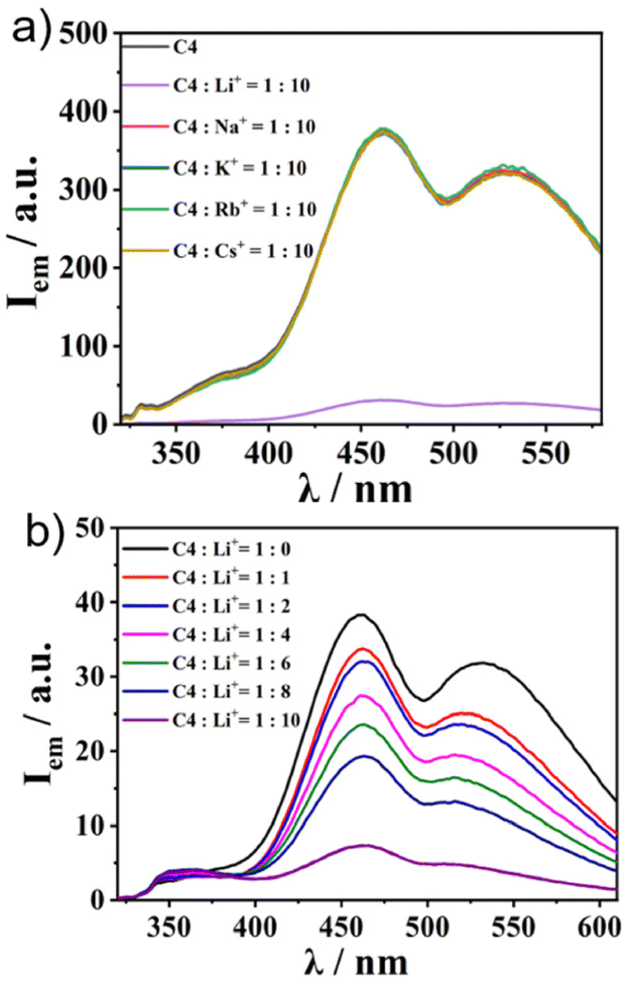
Finally, we applied the recently reported cation trapping ability of sumanene30–32 for cation sensing via emission colour or intensity change. To achieve this, we chose C4 as the representative. We checked the emission properties of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 solutions of C4 and various kinds of PF6− salts of alkali metals (in CH3CN/water = 1:1 (v/v)) (Fig. 5a). While no change in the emission intensity of C4 was observed in the cases of the addition of Na+, K+, Rb+, and Cs+, C4 exclusively exhibited a “turnoff” fluorescence behaviour upon the addition of Li+. The Li+ concentration-dependent experiment showed a gradual decrease in emission intensity when the Li+ amount was increased (Fig. 5b). The same tendency was observed in the other three complexes (Fig. S18, ESI†), while the controls SC1–SC3 did not (Fig. S19, ESI†), indicating that the Li+ sensing ability was attributable to the sumanene core. The Li+ sensing abilities of C1–C4 were evaluated by Stern–Volmer plots based on the emission titration data (Table 2 and Fig. S20, ESI†). The quenching process of C4 was not in a simple linear shape, indicating the presence of some acceleration effects for quenching under high Li+ concentration conditions. The detection limits of Li+ in CH3CN/water of C1–C4 range from 5 to 160 μM (Table 2 and Fig. S21, ESI†). In particular, C2 exhibited a lower detection limit than the reported lithium-ion sensing materials.38 Further analyses using Job's plot (Fig. S22, ESI†) revealed the 1:8 binding mode for the di-sumanenyl complexes (C1 and C2), whilst the 1:4 mode was found for the mono-sumanenyl complexes (C3 and C4), indicating that four Li+ were trapped in each sumanene core via cation–π interactions, in which electrostatics play essential roles.28
:10 solutions of C4 and various kinds of PF6− salts of alkali metals (in CH3CN/water = 1:1 (v/v)) (Fig. 5a). While no change in the emission intensity of C4 was observed in the cases of the addition of Na+, K+, Rb+, and Cs+, C4 exclusively exhibited a “turnoff” fluorescence behaviour upon the addition of Li+. The Li+ concentration-dependent experiment showed a gradual decrease in emission intensity when the Li+ amount was increased (Fig. 5b). The same tendency was observed in the other three complexes (Fig. S18, ESI†), while the controls SC1–SC3 did not (Fig. S19, ESI†), indicating that the Li+ sensing ability was attributable to the sumanene core. The Li+ sensing abilities of C1–C4 were evaluated by Stern–Volmer plots based on the emission titration data (Table 2 and Fig. S20, ESI†). The quenching process of C4 was not in a simple linear shape, indicating the presence of some acceleration effects for quenching under high Li+ concentration conditions. The detection limits of Li+ in CH3CN/water of C1–C4 range from 5 to 160 μM (Table 2 and Fig. S21, ESI†). In particular, C2 exhibited a lower detection limit than the reported lithium-ion sensing materials.38 Further analyses using Job's plot (Fig. S22, ESI†) revealed the 1:8 binding mode for the di-sumanenyl complexes (C1 and C2), whilst the 1:4 mode was found for the mono-sumanenyl complexes (C3 and C4), indicating that four Li+ were trapped in each sumanene core via cation–π interactions, in which electrostatics play essential roles.28
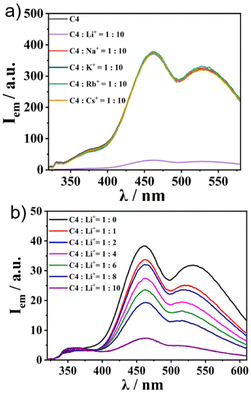 |
| | Fig. 5 The cation sensing ability of C4. (a) Emission spectra of C4 and C4:cation = 1:10 solutions (1.0 × 10−5 M in CH3CN/water = 1:1, λex = 300 nm). (b) Emission spectral change of C4 after Li+ titration (1.0 × 10−4 M in CH3CN/water = 1:1, λex = 300 nm). | |
Table 2 Li+ sensing abilities of complexes C1–C4 at 300 K
| Complex |
Stern–Volmer constant/KSV |
Detection limit [nM] |
|
C1
|
0.73 × 103 M−1 |
160 |
|
C2
|
3.03 × 103 M−1 |
5 |
|
C3
|
5.05 × 103 M−1 |
40 |
|
C4
|
5.14 × 103 M−1 |
35 |
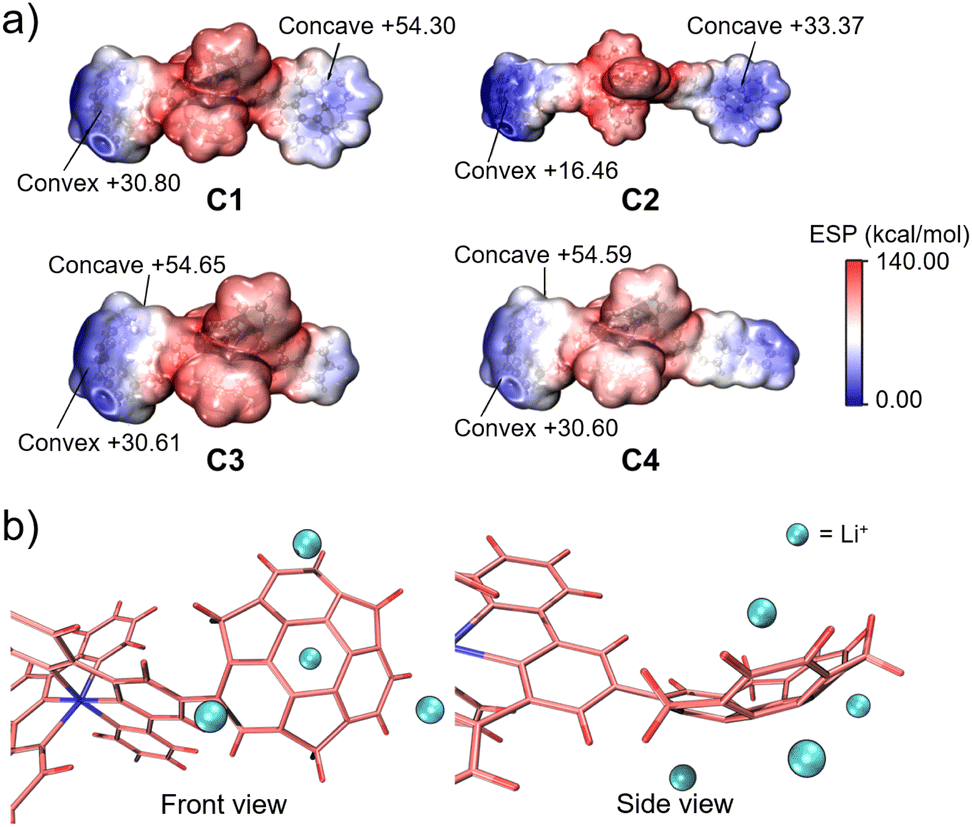
In this context, we attempted to find the trend of binding capacity by calculating the electrostatic potentials of C1–C4 using Multiwfn (Fig. 6a).39 The results showed that the most electron-negative moieties were at the convex side of sumanene in all the complexes. Thus, the structures of the complexes were hypothesised to have the metal ion bound to the convex side of the 6-membered ring of sumanene (Fig. 6b). The differences in the electron densities on the sumanene skeleton of the complexes also explained the variations in the ion trapping abilities. The comparison of electron density at the convex side of the four complexes showed that C2 possessed the highest electron negativity (Fig. 6a). This result well matched the experimental finding that C2 exhibited the lowest detection limit. However, it should be considered that the electrostatic contribution is not the sole element for controlling the stability of the cation–π interactions. The energy decomposition study reveals that sumanene polarisation due to ions is also crucial in Li+–sumanene interactions, with much larger polarisation energy than Na+ and K+.19 This might explain why only Li+ could be adsorbed in such electropositive complexes.
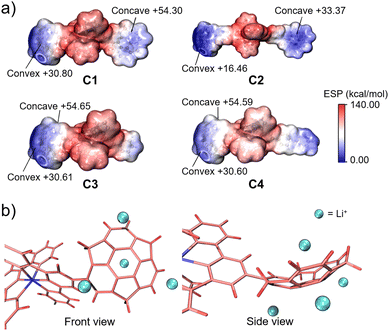 |
| | Fig. 6 (a) ESP plots of C1, C2, C3 and C4. (b) Schematic model of Li+ binding on the sumanene skeleton of C4. | |
Conclusions
We reported sumanene-ligated four Ru(II)–terpyridine complexes C1–C4. All the complexes showed higher emission ability than conventional terpyridylphenyl complexes upon excitation at the ligand-centred π–π* band. In particular, the ones containing phenylene parts exhibited the dual emission involved by the TICT, which was caused by a quick intramolecular rotation due to the low rotation barrier of sumanene and the next terpyridine in the excited state. The cation recognition experiments also revealed the susceptible Li+ trapping ability of the complexes at the sumanene cores. Our findings will contribute to further understanding the bowl-shaped effect of the curved-π based emissive materials. They will also extend the applicability of sumanene-based sensing materials.
Author contributions
Y. Y. designed the study and supervised the project. J. H. performed the syntheses of all the ligands and complexes, spectroscopic measurements and theoretical calculations. Y. T. performed the lifetime measurements of the complexes. All the authors discussed the results and co-wrote the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to express their sincere gratitude to Prof. Yi-Tsu Chan and Ms. Linting Lin (Taiwan University) for fruitful discussions about molecular synthesis. This work was supported by a Grant-in-Aid for Transformative Research Areas “Science of 2.5 Dimensional Materials” (No. JP21H05233) and JSPS KAKENHI (JP19H00912 and JP20H00400). The theoretical calculations were performed at the Research Centre for Computational Science, Okazaki, Japan (Project: 22-IMS-C068). H. J. thanks the Otsuka Toshimi Scholarship Foundation for kindly providing scholarships (19-S5 and 20-S4).
References
- M. H. Keefe, K. D. Benkstein and J. T. Hupp, Luminescent sensor molecules based on coordinated metals: A review of recent developments, Coord. Chem. Rev., 2000, 205, 201–228 CrossRef CAS.
- N. H. Damrauer, G. Cerullo, A. Yeh, T. R. Boussie, C. V. Shank and J. K. McCusker, Femtosecond Dynamics of Excited-State Evolution in [Ru(bpy)3]2+, Science, 1997, 275, 54–57 CrossRef CAS PubMed.
- P. G. Bomben, K. C. D. Robson, B. D. Koivisto and C. P. Berlinguette, Cyclometalated ruthenium chromophores for the dye-sensitised solar cell, Coord. Chem. Rev., 2012, 256, 1438–1450 CrossRef CAS.
- J.-P. Sauvage, J.-P. Collin, J.-C. Chambron, S. Guillerez and C. Coudret, Ruthenium(II) and Osmium(II) Bis(terpyridine) Complexes in Covalently-Linked Multicomponent Systems: Synthesis, Electrochemical Behavior, Absorption Spectra, and Photochemical and Photophysical Properties, Chem. Rev., 1994, 94, 993–1019 CrossRef CAS.
- H. Hofmeier and U. S. Schubert, Recent developments in the supramolecular chemistry of terpyridine-metal complexes, Chem. Soc. Rev., 2004, 33, 373–399 RSC.
- Y.-Q. Fang, N. J. Taylor, G. S. Hanan, F. Loiseau, R. Passalacqua, S. Campagna, H. Nierengarten and A. Van Dorsselaer, A Strategy for Improving the Room temperature Luminescence Properties of Ru(II) Complexes with Tridentate Ligands, J. Am. Chem. Soc., 2002, 124, 7912–7913 CrossRef CAS.
- D. Mondal, M. Bar, S. Mukherjee and S. Baitalik, Design of Ru(II) Complexes Based on Anthraimidazoledione-Functionalized Terpyridine Ligand for Improvement of Room-Temperature Luminescence Characteristics and Recognition od Selective Anions: Experimental and DFT/TD-DFT Study, Inorg. Chem., 2016, 55, 9707–9724 CrossRef CAS PubMed.
- A. Graczyk, F. A. Murphy, D. Nolan, V. Fernández-Moreira, N. J. Lundin, C. M. Fitchett and S. M. Draper, Terpyridine-fused polyaromatic hydrocarbons generated via cyclohydrogenation and used as ligand in Ru(II) complexes, Dalton Trans., 2012, 41, 7746–7754 RSC.
- D. Maity, S. Das, S. Mardanya and S. Baitalik, Synthesis, Structural Characterization, and Photophysical, Spectroelectrochemical, and Anion-Sensing Studies of Heteroleptic Ruthenium(II) Complexes Derived from 4′-Polyaromatic-Substituted Terpyridine Derivatives and 2,6-Bis(benzimidazole-2-yl)pyridine, Inorg. Chem., 2013, 52, 6820–6838 CrossRef CAS.
- S. Baitalik, X. Wang and R. H. Schmehl, A Trimetallic Mixed Ru(II)/Fe(II) Terpyridyl Complex with A Long-Lived Excited State in Solution at Room Temperature, J. Am. Chem. Soc., 2004, 126, 16304–16305 CrossRef CAS PubMed.
- P. Pal, S. Mukherjee, D. Maity and S. Baitalik, Synthesis, Structural Characterization, and Luminescence Switching of Diarylethene-Conjugated Ru(II)-Terpyridine Complexes by trans-cis Photoisomerization: Experimental and DFT/TD-DFT Investigation, Inorg. Chem., 2018, 57, 5743–5753 CrossRef CAS PubMed.
- S. H. Wadman, M. Lutz, D. M. Tooke, A. L. Spek, F. Hartl, R. W. A. Havenith, G. P. M. van Klink and G. van Koten, Cyclometalated RuII Complexes with Improved Octahedral Geometry: Synthesis and Photophysical Properties, Inorg. Chem., 2009, 48, 1887–1900 CrossRef CAS PubMed.
- G. J. Wilson, A. Launikonis, W. H. F. Sasse and A. W. H. Mau, Excited-State Processes in Ruthenium(II) Bipyridine Complexes Containing Covalently Bound Arenes, J. Phys. Chem. A, 1997, 101, 4860–4866 CrossRef CAS.
- D. S. Tyson, C. R. Luman, X. Zhou and F. N. Castellano, New Ru(II) Chromophores with Extended Excited-State Lifetimes, Inorg. Chem., 2001, 40, 4063–4071 CrossRef CAS PubMed.
- L. Song, J. Feng, X. Wang, J. Yu, Y. Hou, P. Xie, B. Zhang, J. Xiang, X. Ai and J. Zhang, Dual Emission from 3MLCT and 3ILCT Excited States in a New Ru(II) Diimine Complex, Inorg. Chem., 2003, 42, 3393–3395 CrossRef CAS PubMed.
- T. E. Keyes, C. M. O'Connor, U. O'Dwyer, C. G. Coates, P. Callaghan, J. J. McGarvey and J. G. Vos, Isotope and Temperature Dependence od Dual Emission in a Mononuclear Ruthenium(II) Polypyridyl Compound, J. Phys. Chem. A, 1999, 103, 8915–8920 CrossRef CAS.
- E. C. Glazer, D. Magde and Y. Tor, Ruthenium Complexes That Breaks the Rules: Structural Features Controlling Dual Emission, J. Am. Chem. Soc., 2007, 129, 8544–8551 CrossRef CAS.
- J. Wang, E. A. Medlycott, G. S. Hanan, F. Loiseau and S. Campagna, The multichromophore approach: A case of temperature controlled switching between single and dual emission in Ru(II) polypyridyl complexes, Inorg. Chim. Acta, 2007, 360, 876–884 CrossRef CAS.
- R. Siebert, A. Winter, B. Diezek, U. S. Schubert and J. Popp, Dual Emission from Highly Conjugated 2,2′:6′,2′′-Terpyridine Complexes−A Potential Route to White Emitters, Macromol. Rapid Commun., 2010, 31, 883–888 CrossRef CAS PubMed.
- R. Siebert, A. Winter, U. S. Schubert, B. Dietzek and J. Popp, The molecular mechanism of dual emission in terpyridine transition metal complexes−ultrafast investigations of photo-induced dynamics, Phys. Chem. Chem. Phys., 2011, 13, 1606–1617 RSC.
- H. Sakurai, T. Daiko and T. Hirao, A Synthesis of Sumanene, a Fullerene Fragment, Science, 2003, 301, 1878 CrossRef CAS.
- H. Sakurai, T. Daiko, H. Sakane, T. Amaya and T. Hirao, Structural Elucidation of Sumanene and Generation of Its Benzylic Anions, J. Am. Chem. Soc., 2005, 127, 11580–11581 CrossRef CAS PubMed.
- T. Amaya, H. Sakane, T. Muneishi and T. Hirao, Bowl-to-bowl inversion of sumanene derivatives, Chem. Commun., 2008, 765–767 RSC.
- T. Amaya, S. Seki, T. Moriuchi, K. Nakamoto, T. Nakata, H. Sakane, A. Saeki, S. Tagawa and T. Hirao, Anisotropic Electron Transport Properties in Sumanene Crystal, J. Am. Chem. Soc., 2009, 131, 408–409 CrossRef CAS PubMed.
- M. Li, J.-Y. Wu, K. Sambe, Y. Yakiyama, T. Akutagawa, T. Kajitani, T. Fukushima, K. Matsuda and H. Sakurai, Dielectric Response of 1,1-Difluorosumanene Caused by an In-Plane Motion, Mater. Chem. Front., 2022, 6, 1752–1758 RSC.
- H. Kojima, M. Nakagawa, R. Abe, F. Fujiwara, Y. Yakiyama, H. Sakurai and M. Nakamura, Thermoelectric and Thermal Transport Properties in Sumanene Crystals, Chem. Lett., 2018, 47, 524–527 CrossRef CAS.
- Y. Shoji, T. Kajitani, F. Ishiwari, Q. Ding, H. Sato, H. Anetai, T. Akutagawa, H. Sakurai and T. Fukushima, Hexathioalkyl sumanenes: an electron-donating buckybowl as a building block for supramolecular materials, Chem. Sci., 2017, 8, 8405–8410 RSC.
- D. Vijay, H. Sakurai, V. Subramanian and G. N. Sastry, Where to bind in buckybowls? The dilemma of a metal ion, Phys. Chem. Chem. Phys., 2012, 14, 3057–3065 RSC.
- S. N. Spisak, Z. Wei, N. J. O'Neil, A. Y. Rogachev, T. Amaya, T. Hirao and M. A. Petrukhina, Convex and Concave Encapsulation of Multiple Potassium Ions by Sumanenyl Anions, J. Am. Chem. Soc., 2015, 137, 9768–9771 CrossRef CAS PubMed.
- A. Kasprzak, A. Kowalczyk, A. Jagielska, B. Wagner, A. M. Nowicka and H. Sakurai, Tris(ferrocenylmethidene)sumanene: synthesis, photophysical properties and applications for efficient caesium cation recognition in water, Dalton Trans., 2020, 49, 9965–9971 RSC.
- A. Kasprzak and H. Sakurai, Disaggregation of a sumanene-containing fluorescent probe towards highly sensitive and specific detection of caesium cations, Chem. Commun., 2021, 57, 343–346 RSC.
- A. Kasprzak and H. Sakurai, Site-selective cation-π interaction as a way of selective recognition of the caesium cation using sumanene-functionalised ferrocenes, Dalton Trans., 2019, 48, 17147–17152 RSC.
- B. B. Shrestha, S. Karanjit, G. Panda, S. Higashibayashi and H. Sakurai, Synthesis of Substituted Sumanenes by Aromatic Electrophilic Substitution Reactions, Chem. Lett., 2013, 42, 386–388 CrossRef CAS.
- Z. Zhang, H. Wang, X. Wang, Y. Li, B. Song, O. Bolarinwa, R. A. Reese, T. Zhang, X. Q. Wang, J. Cai, B. Xu, M. Wang, C. Liu, H. B. Yang and X. Li, Supersnowflakes: Stepwise Self-Assembly and Synamic Exchange of Rhombus Star-Shaped Supramolecules, J. Am. Chem. Soc., 2017, 139, 8174–8185 CrossRef CAS PubMed.
- T. Yoshihara, S. I. Druzhinin and K. A. Zachariasse, Fast Intermolecular Charge Transfer with a Planar Rigidized Electron Donor/Acceptor Molecule, J. Am. Chem. Soc., 2004, 126, 8535–8539 CrossRef CAS.
- A. N. Cammidge, V. H. M. Goddard, H. Goppe, N. L. Harrison, D. L. Hughes, C. J. Schubert, B. M. Sutton, G. L. Watts and A. J. Whitehead, Org. Lett., 2006, 8, 4071–4074 CrossRef CAS.
- Z. Piontkowski and D. W. McCamant, Excited-State Planarization in Donor-Bridge Dye Sentisitizers: Phenylene versus Thiophene Bridges, J. Am. Chem. Soc., 2018, 140, 11046–11057 CrossRef CAS.
- E. Villemin and O. Raccurt, Optical lithium sensors, Coord. Chem. Rev., 2021, 435, 213801 CrossRef CAS.
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data and tables, theoretical calculation details, and copies of the NMR spectra of the compounds. See DOI: https://doi.org/10.1039/d2qi01801b |
|
| This journal is © the Partner Organisations 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab,
Youhei
Takeda
*ab,
Youhei
Takeda