
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards panchromatic Fe(II) NHC sensitizers via HOMO inversion†
Anil Reddy
Marri
 ab,
Bogdan
Marekha
cd,
Thomas
Penfold
e,
Stefan
Haacke
*c and
Philippe C.
Gros
*a
ab,
Bogdan
Marekha
cd,
Thomas
Penfold
e,
Stefan
Haacke
*c and
Philippe C.
Gros
*a
aUniversité de Lorraine, CNRS, L2CM, F-54000 Nancy, France. E-mail: philippe.gros@univ-lorraine.fr
bDepartment of Chemistry, North Carolina State University, 851 Main Campus Drive, Raleigh, North Carolina 27695-8204, USA
cUniversité de Strasbourg, CNRS, IPCMS, F-67000 Strasbourg, France
dMax-Planck-Institute for Medical Research, D-69120 Heidelberg, Germany
eChemistry-School of Nat. & Env. Sci., Newcastle University, Newcastle Upon-Tyne, UK
First published on 14th November 2022
Abstract
A combined experimental and theoretical study of iron(II) complexes with pyridyl N-heterocyclic carbene ligands is presented, with a new focus on the effect of extending the ligands with thiophenes of variable number. In agreement with recent theoretical predictions by the Jakubikova group, these ligands induce a substantial mixture of the occupied t2g and π HOMO orbitals, as manifested by a near 80 nm red-shift of the 1MLCT transition and a 2–3 fold increase of the molar extinction coefficient. The thiophene moieties permit delocalisation of the MLCT state on the ligands, which results in excited state lifetimes in the 13–18 ps range, almost twice as much as for the reference compound lacking the thienyl substituents. Relaxation into 3MC states remains the main 3MLCT quenching mechanism, and the effect of the number of thiophenes in lengthening the 3MLCT lifetime is qualitatively consistent with a reduction in the 3MLCT–3MC energy gap.
Introduction
The replacement of noble metals by earth-abundant substitutes is at the forefront of current research1–4 aimed at developing sustainable photoactive materials required for solar energy conversion or photocatalysis. Iron is by far the most abundant metal in Earth's crust and appears a promising candidate to replace the ruthenium gold standard that is in the same column of the periodic table.Ruthenium polypyridine complexes have been applied extensively for sensitization in Dye-sensitized solar cells (DSSCs).5–10 Their photophysical and redox properties facilitate efficient light harvesting through long-lived Metal-to-Ligand Charge Transfer (MLCT) states which are responsible for electron transfer into semiconductors and efficient ground state regeneration after excitation.11–13
In contrast Fe(II)-polypyridines have short-lived MLCT states due to an ultrafast intersystem crossing populating their low-lying metal centred (MC) states.14–17 To date increasing the ligand field strength has been the strategy of choice to increase the energy of the unreactive low-lying ligand field states. Ligands based on σ-donating NHC (N-heterocyclic carbenes). The MLCT lifetime has reached tens of picoseconds for complexes with tridentate C^N^C18–20 and bidentate C^N ligands,21–24 and nanoseconds for compounds bearing tridentate C^C^C ligands,25 making it possible for iron to be used in photochemical applications such as iron-sensitized DSSCs.26–31 However, apart from the recently published Fe(II) complex with phenN,N′^C ligands,32 this improvement in lifetime was obtained at the sacrifice of achieving the panchromatic absorption required for solar energy conversion applications. Recently, “HOMO inversion” has emerged as a promising concept in the field.33–35 This approach aims to adjust the filled orbital energetic levels in order to mix metal t2g and ligand molecular orbitals thus switching the HOMO localization from the metal to the ligand (Fig. 1). This approach was demonstrated experimentally by the Herbert group35 to design new panchromatic iron(II) complexes using ligands combining amido donors with benzannulated phenanthridine and quinoline. The covalent amido–Fe bond deeply strengthened the ligand field subsequently producing long lived excited states with up to 2.5 ns lifetime.
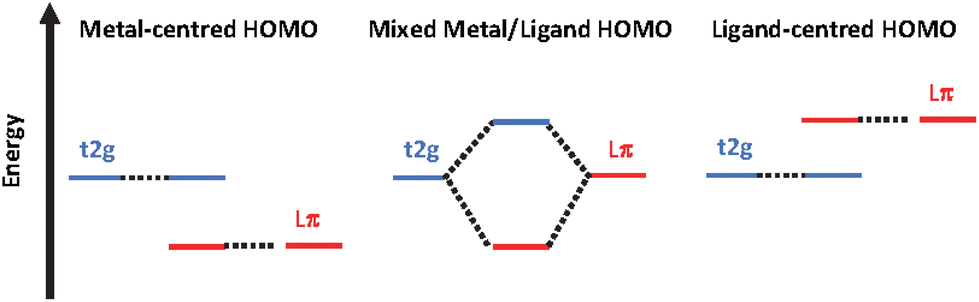 | ||
| Fig. 1 Energetic diagrams illustrating the HOMO inversion concept. | ||
The group of E. Jakubikova33 has reported a computational work on a series of iron(II) polypyridine complexes and predicted that the ligand π-delocalization should promote notable changes in absorption features and suggested that the same effect could be expected with pyridylNHC Fe(II) complexes. To our knowledge besides this exclusively computational work, the effect of π-delocalization and the impact of HOMO inversion on the photophysical and electronic properties of pyridylNHC Fe(II) complexes has not been investigated yet.
Herein we report the synthesis of complexes CTh, CTh2 and CTh3 designed with thiophene and fused polythiophenes conjugated with the central pyridine ring of the pyridylNHC ligand (Fig. 2) and the investigation of their photophysical properties by means of ultra-fast transient absorption. Complex C0 is the proto-typical [Fe(bmip)2]2+, where bmip = 2,6-bis(3-methyl-imidazole-1-ylidine)-pyridine, published by the group of Wärnmark.18,36 We demonstrate that, as compared to C0, extending the delocalization promotes an important red-shift of the pyridine-MLCT absorption band and an impressive increase of the molar extinction coefficients. The excited state lifetimes were also increased substantially by comparison with the unsubstituted complexes while they were slightly impacted by the conjugation extent.
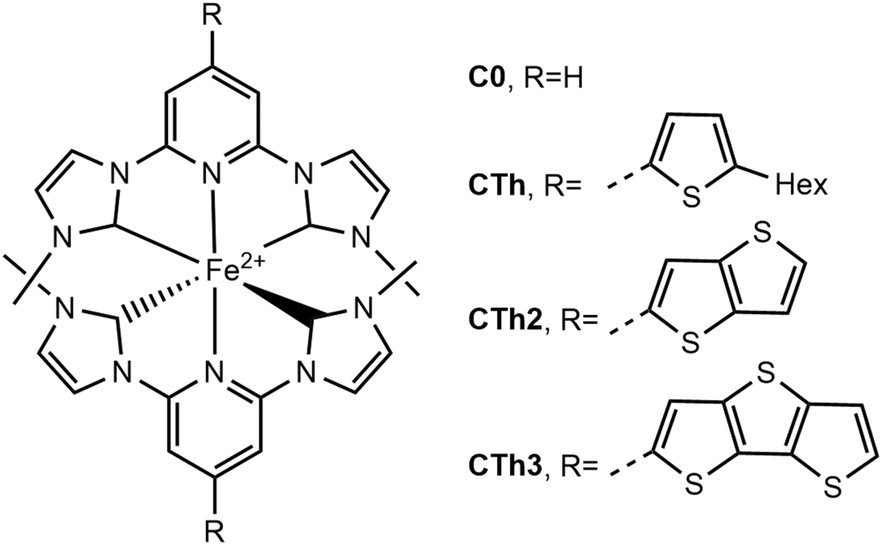 | ||
| Fig. 2 Complexes studied in this work. The counterion is PF6− throughout. | ||
Results and discussion
Synthesis of ligands and complexes
The imidazolium salts L1, L2 and L3, that are the required precursors of the pyridyl-NHC ligands to be coordinated to iron, were first synthesized (Scheme 1, see ESI† for experimental details). L1 was obtained following our reported procedure.27L2 and L3 were obtained in good yields respectively by quaternization of pyridylimidazoles 3 and 4 resulting from a Suzuki coupling of the bromo derivative 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27 with the appropriate preformed thienyl boron pinacol esters that were obtained by a lithiation–borylation sequence on the parent thienyl compounds (see ESI†).37,38 The target complexes were finally obtained in good to moderate yield applying an in situ coordination protocol. FeCl2 was first reacted with the appropriate ligand precursor, then t-BuOK was added to generate the carbene moiety. The coordination yield was found dependent on the number of thiophenes attached to the central pyridine. This agrees with the weakening of the pyridine nitrogen coordination ability by conjugation with the extended π-system.
27 with the appropriate preformed thienyl boron pinacol esters that were obtained by a lithiation–borylation sequence on the parent thienyl compounds (see ESI†).37,38 The target complexes were finally obtained in good to moderate yield applying an in situ coordination protocol. FeCl2 was first reacted with the appropriate ligand precursor, then t-BuOK was added to generate the carbene moiety. The coordination yield was found dependent on the number of thiophenes attached to the central pyridine. This agrees with the weakening of the pyridine nitrogen coordination ability by conjugation with the extended π-system.
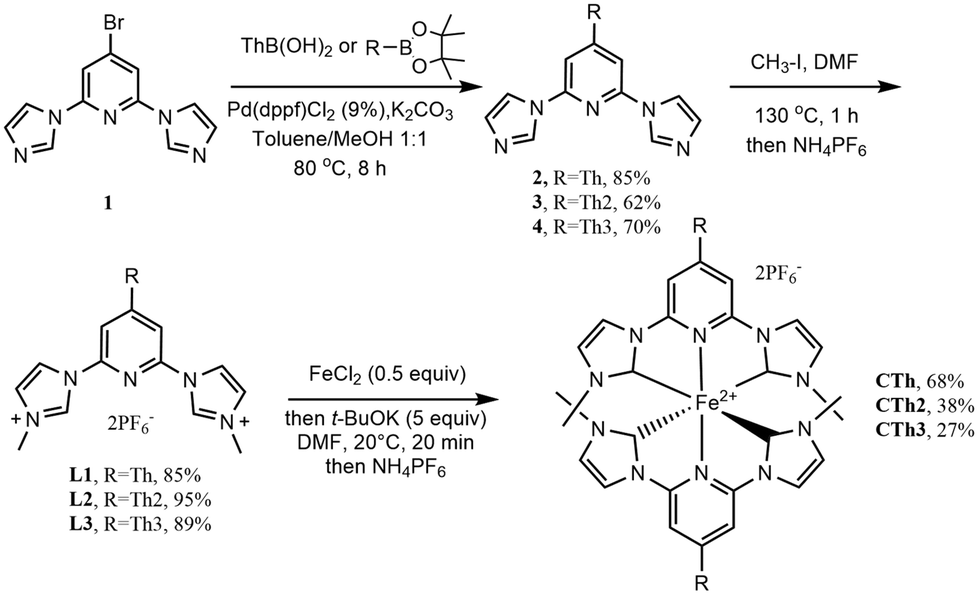 | ||
| Scheme 1 Synthesis of ligand precursors and thienyl substituted complexes. | ||
Electronic properties of complexes
The UV-Vis absorption spectra of the three complexes in deaerated acetonitrile solution are shown in Fig. 3 and the data concerning the lowest energy bands are collected in Table 1. The complexes display three distinct absorption bands. The band at higher energy (250–350 nm) is assigned to ligand-centered π–π* transitions, while the two other bands at lower energy, correspond to Fe-NHC transitions (340–420 nm) and Fe-pyridine MLCT transitions (410–650 nm). In CTh3, the very broad π–π* absorption band of the dithienothiophene moiety masks the Fe-NHC MLCT band.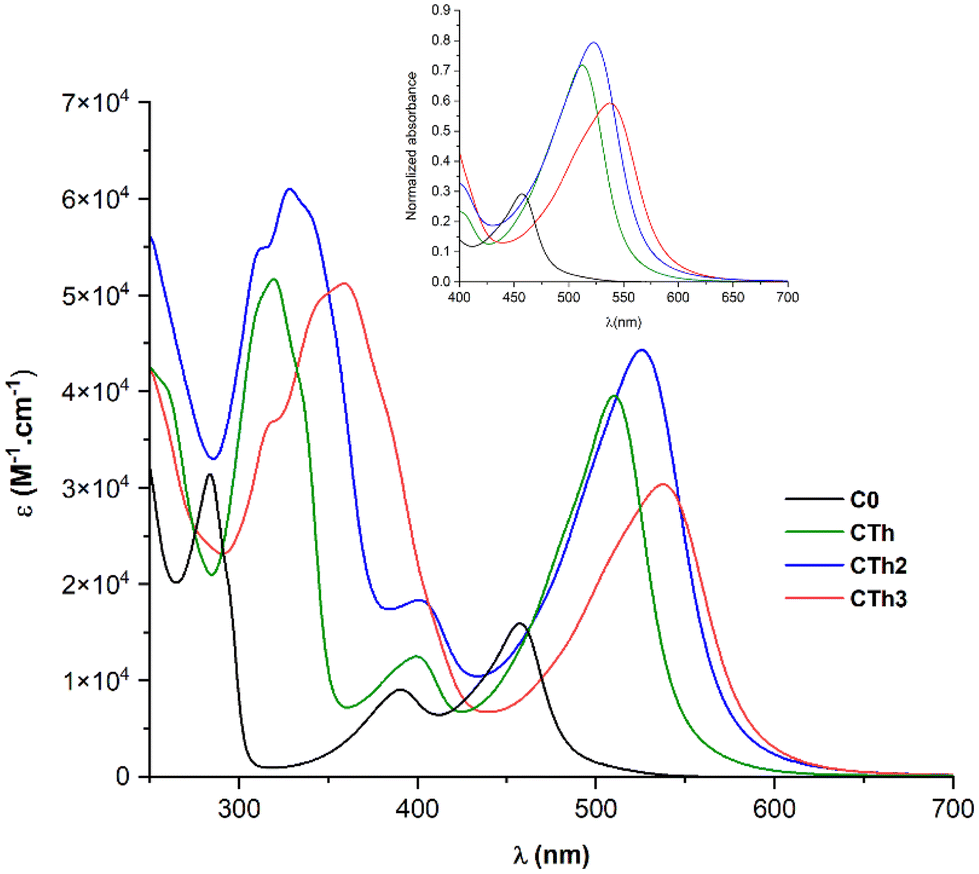 | ||
| Fig. 3 UV-Vis spectra of complexes in acetonitrile. Inset: normalized absorbance for the lowest energy band. | ||
| Complex | λ max (nm) (ε(M−1 cm−1)) | E ox (FeII/FeIII) (V SCE−1) | E Red (V SCE−1) | ΔE |
|---|---|---|---|---|
|
C018,22 |
284 (31433) |
0.80 (rev) | −1.95 (irr) | 2.75 |
| 390 (9077) | ||||
| 457 (15955) |
||||
| CTh | 319 (51619) |
0.68 (rev) | −1.69 (qr) | 2.37 |
| 399 (12519) |
||||
| 516 (39330) |
||||
| CTh2 | 328 (61027) |
0.71 (rev) | −1.56 (qr) | 2.27 |
| 401 (18354) |
||||
| 526 (44300) |
||||
| CTh3 | 359 (51200) |
0.73 (rev) | −1.52 (irr) | 2.25 |
| 535 (30240) |
As a general trend, the substitution of complex C0 with thiophenes promoted a notable bathochromic effect on both the π–π* and Fe-pyridine MLCT bands and the amplitude of the red-shift was directly related to the number of thiophene units (Δλ = 78 nm going from C0 to CTh3). In contrast, the thiophenes had no impact on the Fe-carbene MLCT band the transition energy of which remained constant.
Focusing on the lowest-energy portion of the spectrum, the MLCT Fe-pyridine band, CTh, CTh2 and CTh3 present a broad and intense absorption, ranging from 450 to 620 nm. Among all, CTh3 showed the broadest and most red shifted absorption (535 nm, ε = 30240 M−1 cm−1) and CTh2 the highest extinction coefficient (526 nm, ε = 44300 M−1 cm−1).
The cyclic voltammetry data (Table 1) globally confirm the UV-Vis absorption spectroscopy results. The electrochemical gap markedly decreased when comparing CTh to C0 in agreement with the 59 nm redshift of the MLCT band upon addition of one thiophene unit on the central pyridine. Also corroborating the absorption spectra, a decrease of the gap was observed albeit in a lesser extent going from CTh to CTh3.
Table 2 gathers the computed energy and oscillator strengths for the lowest singlet excited states of C0, CTh, CTh2 and CTh3 that are associated with the lowest band in the absorption spectrum. Consistent with the experimental observations, a red shift of the lowest transitions of CTh, CTh2 and CTh3 compared to C0 is observed and this increases with the delocalization of the MLCT state. The HOMO–LUMO gap is slightly larger than experimentally measured, but exhibits the same trend as the cyclic voltammetry data in Table 1.
| Complex | λ (nm) (f) | ε HOMO/eV | ε LUMO/eV | ΔE/eV |
|---|---|---|---|---|
| C0 | 518 (0.00163) | −5.33 | −2.02 | 3.31 |
| 518 (0.00162) | ||||
| CTh | 615 (0.00340) | −5.29 | −2.41 | 2.88 |
| 614 (0.00370) | ||||
| CTh2 | 628 (0.00290) | −5.32 | −2.56 | 2.76 |
| 627 (0.00260) | ||||
| CTh3 | 637 (0.00430) | −5.33 | −2.62 | 2.71 |
| 637 (0.00340) |
The HOMO and LUMO have been plotted for all the complexes (Fig. 4 and ESI†). Fig. 4 clearly evidenced a significant enhancement of the mixing between the ligand and metal orbitals in the HOMO as the thiophene units are included. The Fe 3d orbitals contribution to the HOMO decreased with the increase of the number of thienyl units in the substituent.
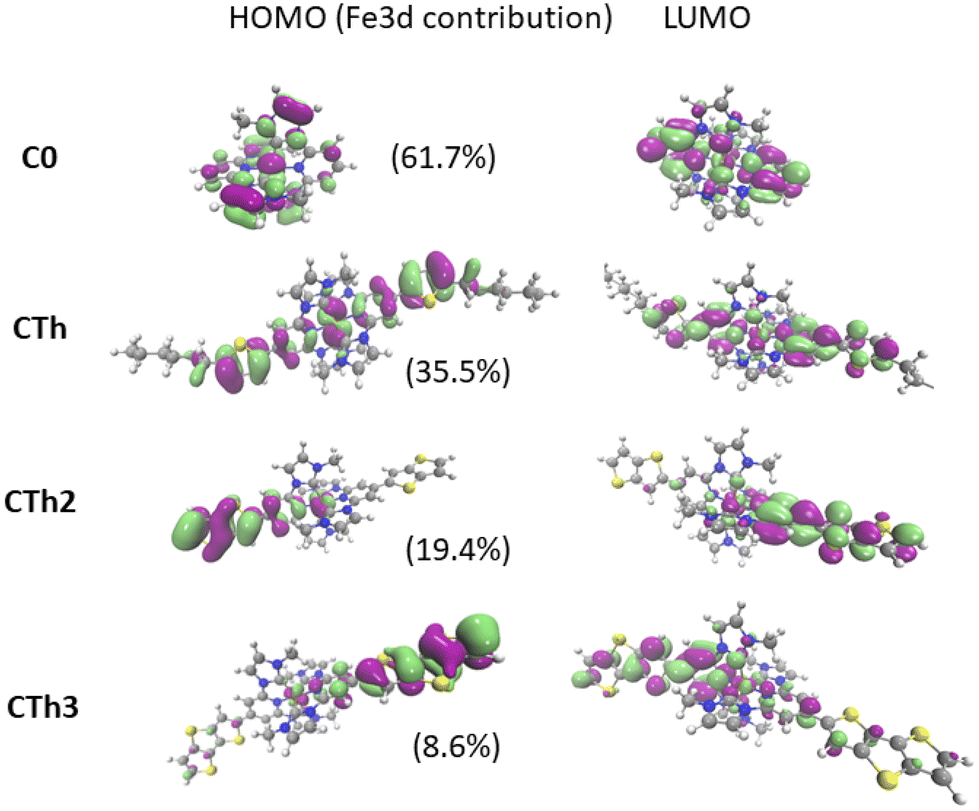 | ||
| Fig. 4 HOMO–LUMO plots for all the complexes including the contribution of Fe 3d orbitals in HOMO. | ||
Excited state dynamics and relaxation
Temporal evolution of the fs-TAS signal of the CTh series of molecules bears a number of common features and therefore will be illustrated on the example of CTh3 (Fig. 5, see the ESI† for data on the other molecules). In particular, upon optical excitation of the Fe-pyridine MLCT transition by the pump pulse the differential transient spectra in the 360–680 nm spectral range reveal two negative and two positive features. The former ones (<390 nm, 500–580 nm) come from the depletion of the population of the initial state (ground state bleaching, GSB) and peak around the absorption maxima of the Fe-pyridine and Fe-NHC MLCT transitions (Fig. 5B). In turn, the positive features originate from the excited state absorption (ESA) of the states populated by the pump pulse. Notably, the high-energy ESA (400–470 nm) shows twice the intensity of the low energy ESA feature (>580 nm).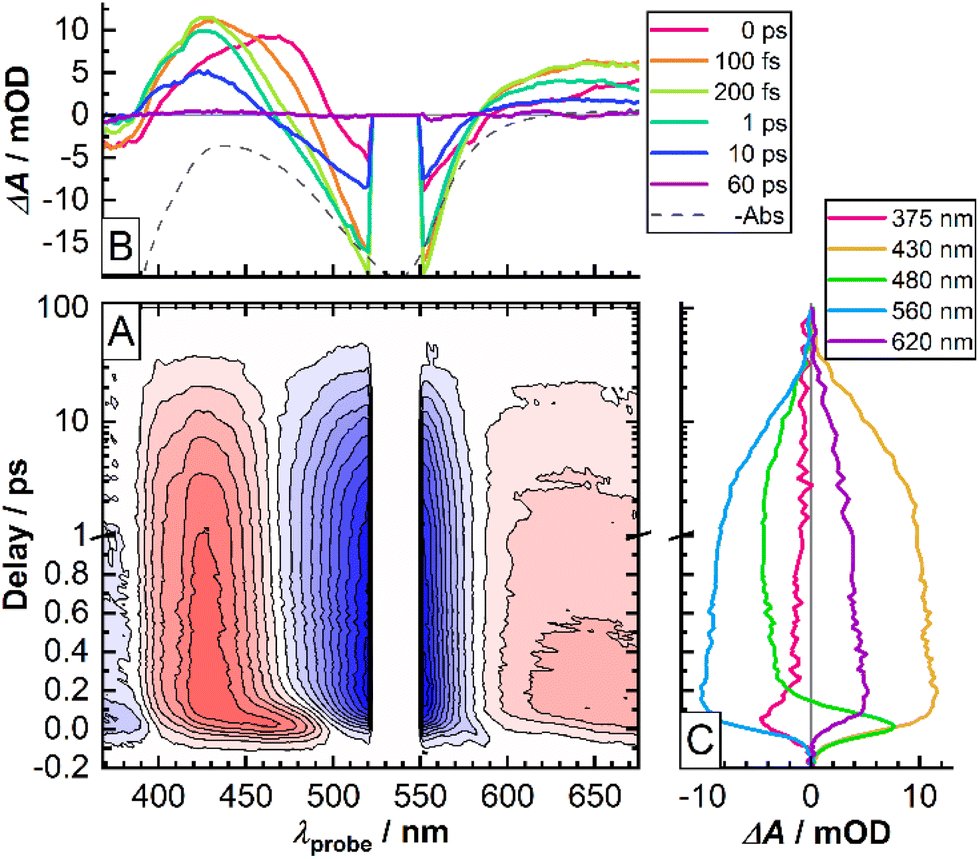 | ||
| Fig. 5 Fs-TAS data for CTh3. (A) Two-dimensional color-plot as a function of pump–probe delay and probe wavelength. Positive signals (induced absorption) are in shades of red, whereas negative ones (bleach) are in shades of blue. Contour lines are drawn at 10% intervals of the absolute maximum value. The data around 535 ± 10 nm were omitted due to large noise of scattered pump light overlaying the TAS signal. The time axis is logarithmic above 1 ps delay. (B) Transient spectra at selected delays. Inverse-signed steady-state UV-vis absorption spectrum is shown for comparison (dashed line). (C) Kinetic traces at selected probe wavelengths. | ||
The initially observed spectral features exhibit an ultrafast evolution on the sub-200 fs time scale. Namely, the high-energy ESA undergoes a pronounced blue-shift, which is most evident by the zero-crossing point moving from 500 nm at 0 ps to ∼475 nm at 200 fs (Fig. 5B and 480 nm trace in Fig. 5C). The low-energy ESA shows similar development. Also, a somewhat delayed rise of the low-energy GSB band suggests that it overlaps with the ultrafast evolving ESA. Further temporal evolution shows little spectral variation and rather reflects intensity decay on the ∼15 ps time scale. The ESA and GSB features decay concomitantly thereby evidencing complete relaxation of the excited state population back to the ground state.
To provide a quantitative framework to the observed fs-TAS results and given the concerted nature of temporal evolution of different transient spectral features, we performed a global analysis39 using GloTarAn software40 assuming a sequential decay model convoluted with a Gaussian instrument response function (IRF). The data could be satisfactorily described by a sum of 4 exponential functions (see the ESI† for the discussion on the relevance of the 4th exponential function). The corresponding decay time constants are reported in Table 3 and the evolution associated difference spectra (EADS) for CTh3 are shown in Fig. 6.
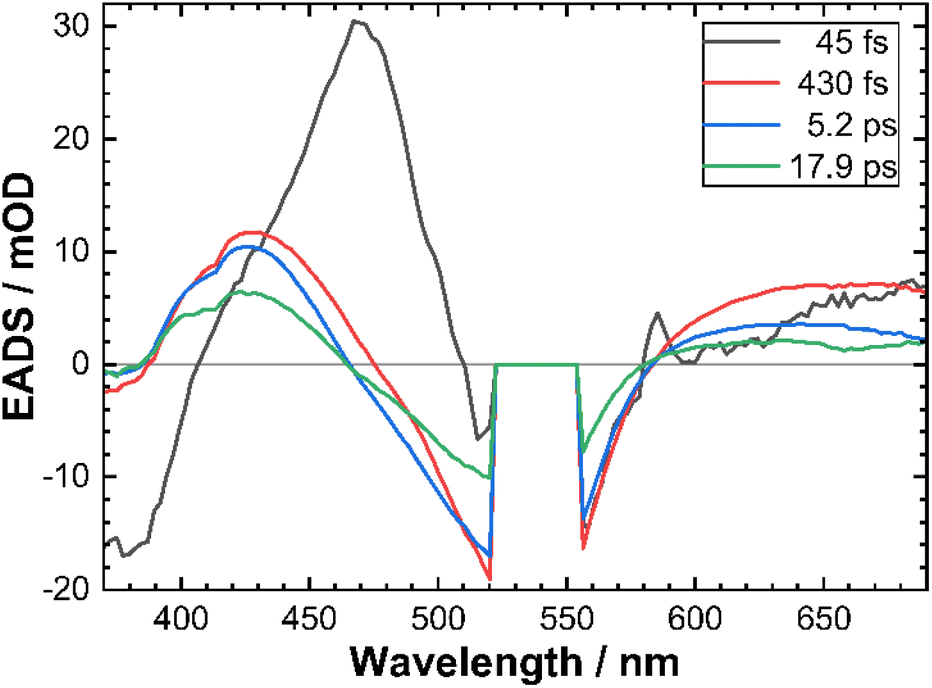 | ||
| Fig. 6 Evolution associated difference spectra (EADS) obtained from global analysis of CTh3 fs-TAS data. The corresponding decay time constants are indicated in the legend. | ||
In accordance with the qualitative analysis of the experimental data, the first sub-100 fs component revealed by the global analysis accounts for the initial blue-shift of both high and low-energy ESA bands. The corresponding spectral profile of EADS also suggests that the high-energy part of the ESA feature is very intense as it largely compensates the GSB signal around the pump wavelength. The other sub-ps component bears some residual features of this transient blue-shift of the high energy ESA and corresponds to a significant part of the intensity decay of the low energy ESA on a sub-picosecond time scale (Fig. 5B). The two slower components mostly reveal the complete intensity decay back to the ground state with very little changes in band shape.
The physical picture of the photodynamics of these molecules upon MLCT optical excitation that emerges from these results resembles previous results from us19,20,41–43 and others18,44 on similar tridentate complexes with 4 NHC ligands. Namely, the initially created 1MLCT state is converted through an ultrafast (<100 fs) spin-crossover to a hot non-emissive (no noticeable stimulated emission observed) 3MLCT state which then undergoes a sub-ps vibrational relaxation. This thermalized triplet state then returns to the ground state on the ps time-scale. In particular the last, τ4 ∼ 18 ps, step corresponds to the return to the ground state, involving ultrafast relaxation through lower energy 3MC states, in accordance with the relaxation cascade observed for the parent pyridylNHC Fe(II) complexes.18,19,41–43 In particular the reference complex C0 was studied in detail,18,36 demonstrating a 9.0 ps lifetime of the 3MLCT state (corresponding to τ4).
The τ3 = 5.2 ps is a relevant time constant since it is needed for an improved quality of the fits, in particular in the low energy bleach part (see Fig. S18†). However, unlike the 0.43 ps component, its EADS does not show a significant difference in spectral shape with the 17.9 ps EADS, unlike similar time components we found in previous work with diazinyl-NHC ligands.20 This suggests two alternatives for its assignment. It could be due to a residual slower thermalization process which involves vibrational modes that are less strongly coupled to the optical transitions and which therefore leads to little spectral variation. Alternatively, τ3 and τ4 may correspond to two different 3MLCT–3MC relaxation pathways in the multi-dimensional excited state landscape. As a side remark, a parallel population of both 3MLCT and 3MC on the sub-picosecond time scale36,43,45 cannot be excluded, since population of the latter states go commonly un-noticed in UV-VIS transient absorption experiments.42
Concerning the influence of the number of fused thiophene rings in the CTh series of molecules on the excited state dynamics one can see from the results of the global analysis (Table 3) as well from the bleach recovery kinetics (Fig. 7) that increasing the number of fused thiophene rings and thereby promoting the electron delocalization in the MLCT state leads to a moderate slow-down of all the steps of the excited state relaxation. Nonetheless, this delocalization effect is apparently not enough to alter the overall relaxation cascade mechanism observed for related Fe-NHC complexes.
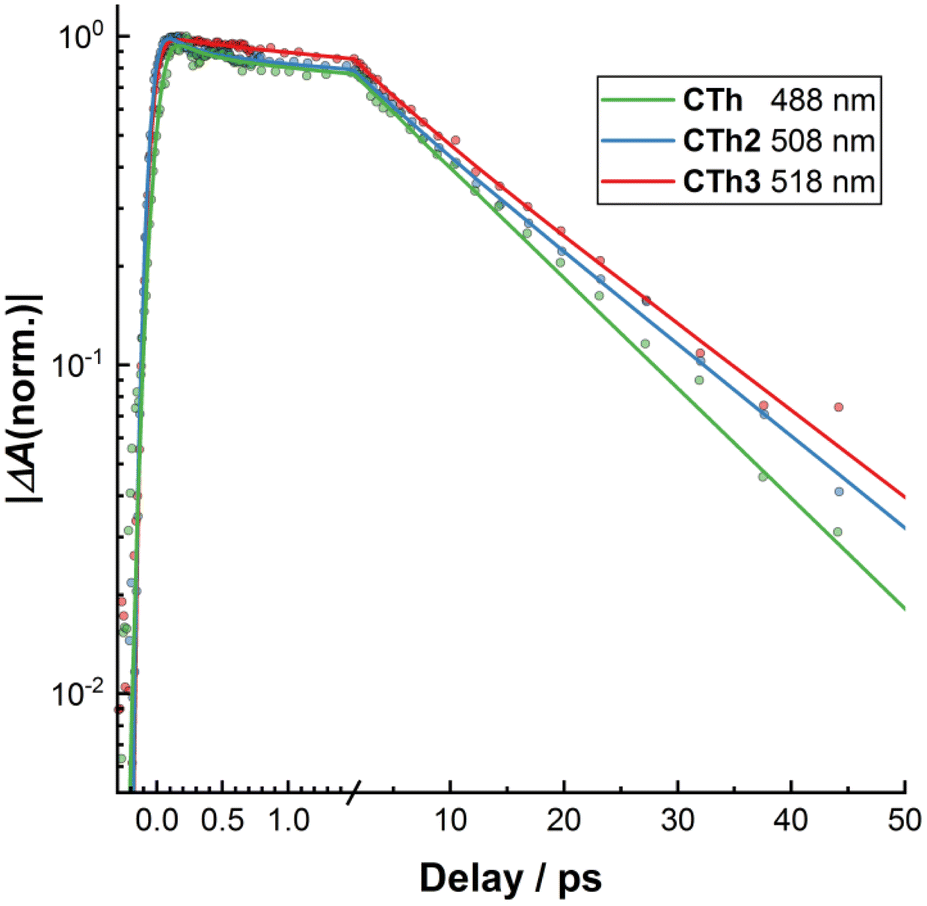 | ||
| Fig. 7 Semi-log plot of normalized kinetics of the bleach recovery of CTh, CTh2 and CTh3. Dots are measured datapoints whereas lines are results of the global fit (see text). | ||
A Jablonski diagram showing the computed energetics of the key states for the series of complexes is shown in Fig. 8. While the MLCT states show a distinct red shift, the 3MC states, which mix very little with the fused thiophene units (see spin density in Fig. S20†) remain largely unchanged among the CTh, CTh2 and CTh3 series compared to C0. The geometry of all the important structures are provided in the ESI.†
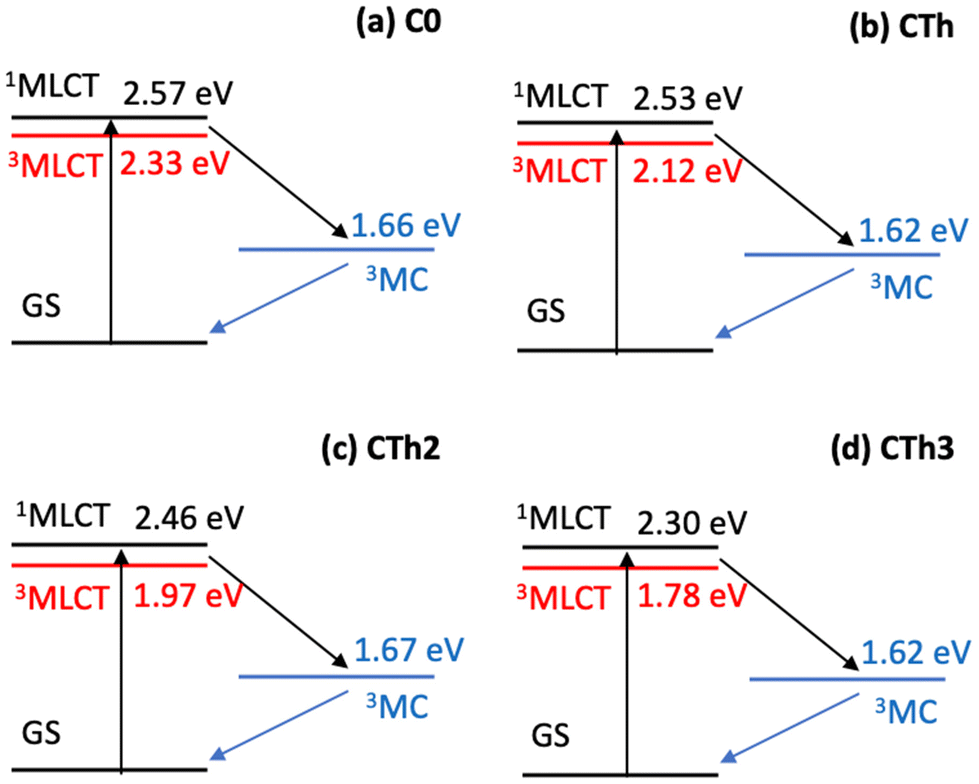 | ||
| Fig. 8 Computed Jablonski diagrams of the important excited states in (a) C0, (b) CTh, (c) CTh2 and (d) CTh3. Using TDDFT, the energies of the 1,3MLCT states were computed at the 3MLCT optimized geometry and the energies of the 3MC was computed at the 3MC optimized geometry (1MLCT and 3MLCT states have similar geometries). Both geometries were computed using UKS and the relevant geometries are in the ESI.† | ||
Given the significant structural reorganization between the 3MLCT and 3MC states, the decreasing energy gap (ΔE) between the two states as the number of thiophene units is increased may introduce a small barrier to the transformation between the two states, responsible for the increase in τ3 and τ4 observed experimentally. This counterintuitive effect is highlighted in Fig. 9. Indeed, and as in Marcus theory for electron transfer,46 the energy barrier between the states decreases, and reaches zero only when ΔE is equal to the structural reorganization energy. A similar behavior was recently also put forward in a study on solvent effects of Fe complexes.47
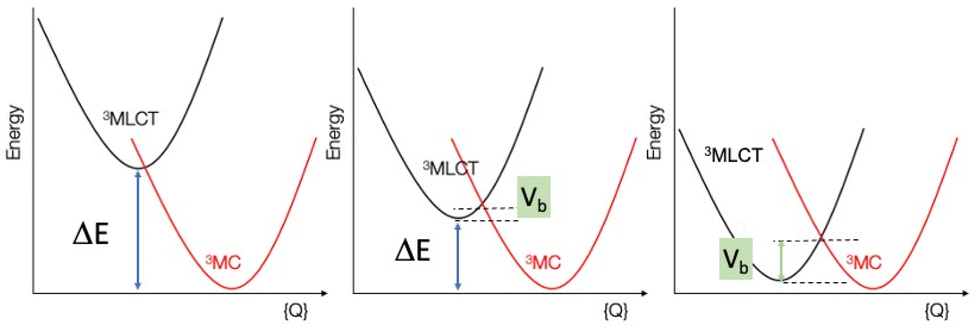 | ||
| Fig. 9 Schematic representation of the 3MLCT and the 3MC states approximated as harmonic potentials along an effective structural coordinate Q. When the energy gap ΔE decreases (left to right), the barrier Vb between 3MLCT and 3MC increases, leading to a longer 3MLCT lifetime. | ||
Conclusions
In conclusion, we present here a combined experimental and theoretical study of iron(II) complexes with pyridyl NHC ligands conjugated with thiophene and fused polythiophenes. As predicted theoretically,33 these ligands induce a substantial mixture of the occupied t2g and π HOMO orbitals, as manifested by a near 80 nm red-shift of the 1MLCT transition and a 2–3-fold increase of the extinction coefficient. The thiophene moieties allow for a delocalisation of the MLCT state on the ligands, which results in excited state lifetimes in the 13–18 ps range, almost twice as much as for the reference compound lacking the thiophene side groups. Relaxation into 3MC states remains the main 3MLCT quenching mechanism, and the effect of the number of thiophenes in lengthening the 3MLCT lifetime is qualitatively consistent with a stabilisation of the 3MLCT energy due to increased charge delocalisation. As a result, the 3MLCT–3MC energy gap is reduced (Fig. 8), which leads to an increased barrier for the excited state structural relaxation involved in the 3MLCT–3MC transition (Fig. 9). Works are in progress to further amplify the mixing between metal and ligand orbitals via a wider π-extended delocalization in various parts of the ligands.Experimental
General
Solvents and commercially available reagents were used as received. Thin layer chromatography (TLC) was performed by using silica gel 60 F-254 (Merck) plates and visualized under UV light. Chromatographic purification was performed by using silica gel 60 (0.063–0.2 mm per 70–230 mesh). 1H (400 MHz) and 13C NMR (100 MHz) spectra were taken on a DRX400 Bruker spectrometer at ambient temperature. The chemical shifts (δ), were calibrated by using either tetramethylsilane (TMS) or signals from the residual protons of the deuterated solvents and are reported in parts per million (ppm) from low to high field, NMR signal assignments for L2, L3, CTh, CTh2 and CTh3 are detailed in the ESI.†High-resolution mass spectrometry (HRMS) data was obtained by using Bruker micrOTOF-Q spectrometer. UV-vis spectra were recorded in a 1 cm path length quartz cell on a LAMBDA 1050 (PerkinElmer), spectrophotometer. Cyclic voltammetry was performed on a Radiometer PST006 potentiostat using a conventional three-electrode cell. The saturated calomel electrode (SCE) was separated from the test compartment using a bridge tube. The solutions of studied complexes (0.2 mM) were purged with argon before each measurement. The test solution was acetonitrile containing 0.1 M Bu4NPF6 as supporting electrolyte. The working electrode was a vitreous carbon rod (1 cm2) wire and the counter-electrode was a 1 cm2 Pt disc. After the measurement, ferrocene was added as the internal reference for calibration. All potentials were quoted versus SCE. In these conditions the redox potential of the couple Fc+/Fc was found at 0.39 V. In all the experiments the scan rate was 100 mV s−1.
Synthesis of complexes
27 (88 mg, 0.126 mmol) in 2 mL of anhydrous DMF was added anhydrous FeCl2 (8 mg, 0.063 mmol). Then t-BuOK (0.071 g, 0.630 mmol) was added to the above mixture and stirred at room temperature for 20 min. A saturated aqueous solution of NH4PF6 was added (10 ml) and the precipitate was collected by filtration. The crude product was further purified by silica gel column chromatography eluting with an acetone/H2O/KNO3(sat) = 10/3/0.5 mixture. The reddish fraction was collected and after evaporation of acetone, the left solution was treated with a saturated solution of NH4PF6. The desired complex precipitate was then filtered, washed with distilled water followed by ether, and dried under vacuum. CTh was obtained as a reddish solid (50 mg, 68% yield). 1H NMR (400 MHz, CD3CN, δ ppm): 8.12 (d, J = 2.19 Hz, 2H), 7.95 (s, 2H), 7.84 (d, J = 3.72 Hz, 1H), 7.06 (s, J = 3.72 Hz, 1H), 7.04 (d, J = 2.19 Hz, 2H), 2.99 (t, J = 7.51 Hz, 2H), 2.62 (s, 6H), 1.79 (q, 2H), 148–1.35 (m, 6H), 0.95 (t, J = 7.09 Hz, 3H). 13C NMR (100 MHz, CD3CN, δ ppm): 201.1, 154.5, 151.4, 144.5, 138.0, 128.7, 127.2, 127.0, 116.1, 101.6, 35.3, 32.0, 31.8, 30.5, 29.0, 22.9, 14.0. ESI-HRMS calcd for C46H54FeN10S2m/z = 433.1656. Found: 433.1653 [M − 2PF6].
Computational details
All complexes were optimized in the ground state and 3MC and 3MLCT states using DFT within the approximation of the B3LYP* exchange and correlation functional48 and a def2-SVP basis set49 as implemented within the ORCA quantum chemistry package.50 While this is only valid for the lowest state of each multiplicity, both the 3MC and 3MLCT are the lowest triplets at their respective optimized geometries. Numerical frequencies calculations were carried out on the geometries optimized to ensure no negative frequencies. TDDFT calculations, used to calculate the excited state energies of each complex used the Tamm–Dancoff approximation51 and were also performed using B3LYP*. All simulations were performed using a polarisable continuum model with the parameters of acetonitrile.Fs-TAS measurements
The home-built transient absorption spectrometer has been described in detail elsewhere52,53 and only substantial parameters will be given here. The pump pulses were tuned to be in resonance with the absorption maxima of the studied compounds (510 nm for CTh, 525 nm for CTh2 and 535 nm for CTh3). The probe light covering 360–700 nm range was generated by focusing 40 fs 800 nm pulses of the primary laser source in a 2 mm thick CaF2 plate mounted on a motorized linear stage to reduce thermal effects. Sample solutions (1 mM in dry acetonitrile) were circulated in a flow cell with 200 μm optical pathlength (Starna). The probe beam was focused to ca. 60 μm Gaussian spot diameter (1/e2). The pump spot size was ca. 150 μm in diameter (1/e2) and typical maximal energy densities were 0.25 mJ cm−2 (in the center of the Gaussian beam profile) to keep the fraction of excited molecules below 5%. Solvent subtraction and correction for wavelength-dependent time-zero were performed using previously established protocols52,53 using the neat solvent data acquired under identical conditions.Author contributions
A. R. M. performed the synthesis and ground state characterization of ligands and complexes. B. M. performed transient absorption spectroscopy experiments and analysed the results with S. H., T. P. performed DFT computations and analysed the results. P. C. G. conceived the project and wrote the manuscript with B. M., T. P. and S. H.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Strasbourg team is grateful to Dr O. Crégut for expert help in laser maintenance and development of data acquisition software, and for the funding provided by the ITI 2021–28 program of the University of Strasbourg, CNRS and Inserm, supported by IdEx Unistra (ANR 10 IDEX 0002), by SFRI STRAT'US project (ANR 20 SFRI 0012) and EUR QMAT ANR-17-EURE-0024 under the framework of the French Investments for the Future Program. The L2CM thanks the French Agence Nationale de Recherche (ANR-16-CE07-0013-02), Lorraine Université d'Excellence (IMPACT N4S) and the European Regional Development Funds (Programme opérationnel FEDER-FSE Lorraine et Massif des Vosges 2014-2020/“Fire Light” project: “Photo-bio-active molecules and nanoparticles” for financial support and grants to A. R. M. The L2CM also warmly thanks S. Parant for the electrochemical experiments and F. Dupire from the mass spectrometry MassLor platform of Lorraine. TJP acknowledges the EPSRC through grant EP/V010573/1 for support.References
- T. Duchanois, L. Liu, M. Pastore, A. Monari, C. Cebrián, Y. Trolez, M. Darari, K. Magra, A. Francés-Monerris, E. Domenichini, M. Beley, X. Assfeld, S. Haacke and P. Gros, NHC-Based Iron Sensitizers for DSSCs, Inorganics, 2018, 6, 63 CrossRef.
- O. S. Wenger, Photoactive Complexes with Earth-Abundant Metals, J. Am. Chem. Soc., 2018, 140, 13522–13533 CrossRef CAS PubMed.
- S. Kaufhold and K. Wärnmark, Design and Synthesis of Photoactive Iron N-Heterocyclic Carbene Complexes, Catalysts, 2020, 10, 132 CrossRef CAS.
- P. Dierks, Y. Vukadinovic and M. Bauer, Photoactive iron complexes: more sustainable, but still a challenge, Inorg. Chem. Front., 2022, 9, 206–220 RSC.
- B. O'Regan and M. Grätzel, A Low-Cost, High-Efficiency Solar Cell Based on Dye-Sensitized Colloidal TiO2 Films, Nature, 1991, 353, 737–740 CrossRef.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Mueller, P. Liska, N. Vlachopoulos and M. Graetzel, Conversion of light to electricity by cis-X2bis(2,2′-bipyridyl-4,4′-dicarboxylate)ruthenium(II) charge-transfer sensitizers (X=Cl-, Br-, I-, CN-, and SCN-) on nanocrystalline titanium dioxide electrodes, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
- Md. K. Nazeeruddin, S. M. Zakeeruddin, J.-J. Lagref, P. Liska, P. Comte, C. Barolo, G. Viscardi, K. Schenk and M. Graetzel, Stepwise assembly of amphiphilic ruthenium sensitizers and their applications in dye-sensitized solar cell, Coord. Chem. Rev., 2004, 248, 1317–1328 CrossRef CAS.
- K. C. D. Robson, P. G. Bomben and C. P. Berlinguette, Cycloruthenated sensitizers: improving the dye-sensitized solar cell with classical inorganic chemistry principles, Dalton Trans., 2012, 41, 7814–7829 RSC.
- M. G. Lobello, K.-L. Wu, M. A. Reddy, G. Marotta, M. Grätzel, M. K. Nazeeruddin, Y. Chi, M. Chandrasekharam, G. Vitillaro and F. De Angelis, Engineering of Ru(II) dyes for interfacial and light-harvesting optimization, Dalton Trans., 2014, 43, 2726–2732 RSC.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, Md. K. Nazeeruddin and M. Grätzel, Dye-sensitized solar cells with 13% efficiency achieved through the molecular engineering of porphyrin sensitizers, Nat. Chem., 2014, 6, 242–247 CrossRef CAS.
- S. Ardo and G. J. Meyer, Photodriven heterogeneous charge transfer with transition-metal compounds anchored to TiO2 semiconductor surfaces, Chem. Soc. Rev., 2009, 38, 115–164 RSC.
- A. Listorti, C. Creager, P. Sommeling, J. Kroon, E. Palomares, A. Fornelli, B. Breen, P. R. F. Barnes, J. R. Durrant, C. Law and B. O'Regan, The mechanism behind the beneficial effect of light soaking on injection efficiency and photocurrent in dye sensitized solar cells, Energy Environ. Sci., 2011, 4, 3494–3501 RSC.
- A. Listorti, B. O'Regan and J. R. Durrant, Electron Transfer Dynamics in Dye-Sensitized Solar Cells, Chem. Mater., 2011, 23, 3381–3399 CrossRef CAS.
- J. E. Monat and J. K. McCusker, Femtosecond Excited-State Dynamics of an Iron(II) Polypyridyl Solar Cell Sensitizer Model, J. Am. Chem. Soc., 2000, 122, 4092–4097 CrossRef CAS.
- G. Auböck and M. Chergui, Sub-50 fs photoinduced spin crossover in [Fe(bpy)3]2+, Nat. Chem., 2015, 7, 629–633 CrossRef.
- A. L. Smeigh, M. Creelman, R. A. Mathies and J. K. McCusker, Femtosecond Time-Resolved Optical and Raman Spectroscopy of Photoinduced Spin Crossover: Temporal Resolution of Low-to-High Spin Optical Switching, J. Am. Chem. Soc., 2008, 130, 14105–14107 CrossRef CAS.
- M. Cammarata, R. Bertoni, M. Lorenc, H. Cailleau, S. Di Matteo, C. Mauriac, S. F. Matar, H. Lemke, M. Chollet, S. Ravy, C. Laulhé, J.-F. Létard and E. Collet, Sequential Activation of Molecular Breathing and Bending during Spin-Crossover Photoswitching Revealed by Femtosecond Optical and X-Ray Absorption Spectroscopy, Phys. Rev. Lett., 2014, 113, 227402 CrossRef PubMed.
- Y. Liu, T. Harlang, S. E. Canton, P. Chábera, K. Suárez-Alcántara, A. Fleckhaus, D. A. Vithanage, E. Göransson, A. Corani, R. Lomoth, V. Sundström and K. Wärnmark, Towards longer-lived metal-to-ligand charge transfer states of iron(II) complexes: an N-heterocyclic carbene approach, Chem. Commun., 2013, 49, 6412 RSC.
- L. Liu, T. Duchanois, T. Etienne, A. Monari, M. Beley, X. Assfeld, S. Haacke and P. C. Gros, A new record excited state 3MLCT lifetime for metalorganic iron(II) complexes, Phys. Chem. Chem. Phys., 2016, 18, 12550–12556 RSC.
- M. Darari, E. Domenichini, A. Francés-Monerris, C. Cebrián, K. Magra, M. Beley, M. Pastore, A. Monari, X. Assfeld, S. Haacke and P. C. Gros, Iron(II) complexes with diazinyl-NHC ligands: impact of π-deficiency of the azine core on photophysical properties, Dalton Trans., 2019, 48, 10915–10926 RSC.
- A. Francés-Monerris, K. Magra, M. Darari, C. Cebrián, M. Beley, E. Domenichini, S. Haacke, M. Pastore, X. Assfeld, P. C. Gros and A. Monari, Synthesis and Computational Study of a Pyridylcarbene Fe(II) Complex: Unexpected Effects of fac/mer Isomerism in Metal-to-Ligand Triplet Potential Energy Surfaces, Inorg. Chem., 2018, 57, 10431–10441 CrossRef.
- K. Magra, E. Domenichini, A. Francés-Monerris, C. Cebrián, M. Beley, M. Darari, M. Pastore, A. Monari, X. Assfeld, S. Haacke and P. C. Gros, Impact of the fac/mer Isomerism on the Excited-State Dynamics of Pyridyl-carbene Fe(II) Complexes, Inorg. Chem., 2019, 58, 5069–5081 CrossRef CAS PubMed.
- K. Magra, M. Darari, E. Domenichini, A. Francés-Monerris, C. Cebrián, M. Beley, M. Pastore, A. Monari, X. Assfeld, S. Haacke and P. C. Gros, Photophysical Investigation of Iron(II) Complexes Bearing Bidentate Annulated Isomeric Pyridine-NHC Ligands, J. Phys. Chem. C, 2020, 124, 18379–18389 CrossRef CAS.
- K. Magra, A. Francés-Monerris, C. Cebrián, A. Monari, S. Haacke and P. C. Gros, Bidentate Pyridyl–NHC Ligands: Synthesis, Ground and Excited State Properties of Their Iron(II) Complexes and the Role of the fac/mer Isomerism, Eur. J. Inorg. Chem., 2022, 7, e202100818 Search PubMed.
- M. Bauer, J. Steube, A. Päpcke, O. Bokareva, T. Reuter, S. Demeshko, R. Schoch, S. Hohloch, F. Meyer, K. Heinze, O. Kühn and S. Lochbrunner, Janus-type dual emission of a Cyclometalated Iron(III) complex, Research Square, 2020 Search PubMed , in review.
- E. Marchini, M. Darari, L. Lazzarin, R. Boaretto, R. Argazzi, C. A. Bignozzi, P. C. Gros and S. Caramori, Recombination and regeneration dynamics in FeNHC (II)-sensitized solar cells, Chem. Commun., 2020, 56, 543–546 RSC.
- A. Reddy Marri, E. Marchini, V. D. Cabanes, R. Argazzi, M. Pastore, S. Caramori and P. C. Gros, Record power conversion efficiencies for iron(II)-NHC-sensitized DSSCs from rational molecular engineering and electrolyte optimization, J. Mater. Chem. A, 2021, 9, 3540–3554 RSC.
- M. Becker, C. E. Housecroft and E. C. Constable, Electrolyte Tuning in Iron(II)-Based Dye-Sensitized Solar Cells: Different Ionic Liquids and I2 Concentrations, Materials, 2021, 14, 3053 CrossRef CAS PubMed.
- M. Becker, V. Wyss, C. E. Housecroft and E. C. Constable, The influence of alkyl chains on the performance of DSCs employing iron(II) N-heterocyclic carbene sensitizers, Dalton Trans., 2021, 50, 16961–16969 RSC.
- L. Lindh, O. Gordivska, S. Persson, H. Michaels, H. Fan, P. Chábera, N. W. Rosemann, A. K. Gupta, I. Benesperi, J. Uhlig, O. Prakash, E. Sheibani, K. S. Kjaer, G. Boschloo, A. Yartsev, M. Freitag, R. Lomoth, P. Persson and K. Wärnmark, Dye-sensitized solar cells based on Fe N-heterocyclic carbene photosensitizers with improved rod-like push-pull functionality, Chem. Sci., 2021, 12, 16035–16053 RSC.
- A. R. Marri, E. Marchini, V. D. Cabanes, R. Argazzi, M. Pastore, S. Caramori, C. A. Bignozzi and P. C. Gros, A Series of Iron(II)–NHC Sensitizers with Remarkable Power Conversion Efficiency in Photoelectrochemical Cells, Chem. – Eur. J., 2021, 27, 16260–16269 CrossRef CAS PubMed.
- W. Leis, M. A. Argüello Cordero, S. Lochbrunner, H. Schubert and A. Berkefeld, A Photoreactive Iron(II) Complex Luminophore, J. Am. Chem. Soc., 2022, 144, 1169–1173 CrossRef CAS.
- S. Mukherjee, D. E. Torres and E. Jakubikova, HOMO inversion as a strategy for improving the light-absorption properties of Fe(II) chromophores, Chem. Sci., 2017, 8, 8115–8126 RSC.
- J. D. Braun, I. B. Lozada, C. Kolodziej, C. Burda, K. M. E. Newman, J. van Lierop, R. L. Davis and D. E. Herbert, Iron(II) coordination complexes with panchromatic absorption and nanosecond charge-transfer excited state lifetimes, Nat. Chem., 2019, 11, 1144–1150 CrossRef CAS PubMed.
- J. D. Braun, I. B. Lozada and D. E. Herbert, In Pursuit of Panchromatic Absorption in Metal Coordination Complexes: Experimental Delineation of the HOMO Inversion Model Using Pseudo-Octahedral Complexes of Diarylamido Ligands, Inorg. Chem., 2020, 59, 17746–17757 CrossRef CAS PubMed.
- K. Kunnus, M. Vacher, T. C. B. Harlang, K. S. Kjær, K. Haldrup, E. Biasin, T. B. van Driel, M. Pápai, P. Chabera, Y. Liu, H. Tatsuno, C. Timm, E. Källman, M. Delcey, R. W. Hartsock, M. E. Reinhard, S. Koroidov, M. G. Laursen, F. B. Hansen, P. Vester, M. Christensen, L. Sandberg, Z. Németh, D. S. Szemes, É. Bajnóczi, R. Alonso-Mori, J. M. Glownia, S. Nelson, M. Sikorski, D. Sokaras, H. T. Lemke, S. E. Canton, K. B. Møller, M. M. Nielsen, G. Vankó, K. Wärnmark, V. Sundström, P. Persson, M. Lundberg, J. Uhlig and K. J. Gaffney, Vibrational wavepacket dynamics in Fe carbene photosensitizer determined with femtosecond X-ray emission and scattering, Nat. Commun., 2020, 11, 634 CrossRef CAS PubMed.
- S. Alesi, F. Di Maria, M. Melucci, D. J. Macquarrie, R. Luque and G. Barbarella, Microwave-assisted synthesis of oligothiophene semiconductors in aqueous media using silica and chitosan supported Pd catalysts, Green Chem., 2008, 10, 517–523 RSC.
- J. K. Lee, H. S. Kim and S. J. Yun, Synthesis of organic dye containing an alkylenesulfanyl-bridged bithienyl π-linker and its use in dye-sensitized solar cells, J. Photochem. Photobiol., A, 2014, 275, 47–53 CrossRef CAS.
- I. H. M. van Stokkum, D. S. Larsen and R. van Grondelle, Global and target analysis of time-resolved spectra, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 82–104 CrossRef CAS.
- J. J. Snellenburg, S. P. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, Glotaran: A Java-Based Graphical User Interface for the R Package TIMP, J. Stat. Softw., 2012, 49(3), 1–22 Search PubMed.
- T. Duchanois, T. Etienne, C. Cebrián, L. Liu, A. Monari, M. Beley, X. Assfeld, S. Haacke and P. C. Gros, An Iron–Based Photosensitizer with Extended Excited–State Lifetime: Photophysical and Photovoltaic Properties, Eur. J. Inorg. Chem., 2015, 2015, 2469–2477 CrossRef CAS.
- C. Cebrián, M. Pastore, A. Monari, X. Assfeld, P. C. Gros and S. Haacke, Ultrafast Spectroscopy of Fe(II) Complexes Designed for Solar–Energy Conversion: Current Status and Open Questions, ChemPhysChem, 2022, 23(7), e202100659 Search PubMed.
- F. Hainer, N. Alagna, A. Reddy Marri, T. J. Penfold, P. C. Gros, S. Haacke and T. Buckup, Vibrational Coherence Spectroscopy Identifies Ultrafast Branching in an Iron(II) Sensitizer, J. Phys. Chem. Lett., 2021, 12, 8560–8565 CrossRef CAS PubMed.
- P. Zimmer, L. Burkhardt, A. Friedrich, J. Steube, A. Neuba, R. Schepper, P. Müller, U. Flörke, M. Huber, S. Lochbrunner and M. Bauer, The Connection between NHC Ligand Count and Photophysical Properties in Fe(II) Photosensitizers: An Experimental Study, Inorg. Chem., 2018, 57, 360–373 CrossRef CAS PubMed.
- H. Tatsuno, K. S. Kjær, K. Kunnus, T. C. B. Harlang, C. Timm, M. Guo, P. Chàbera, L. A. Fredin, R. W. Hartsock, M. E. Reinhard, S. Koroidov, L. Li, A. A. Cordones, O. Gordivska, O. Prakash, Y. Liu, M. G. Laursen, E. Biasin, F. B. Hansen, P. Vester, M. Christensen, K. Haldrup, Z. Németh, D. Sárosiné Szemes, É. Bajnóczi, G. Vankó, T. B. Van Driel, R. Alonso-Mori, J. M. Glownia, S. Nelson, M. Sikorski, H. T. Lemke, D. Sokaras, S. E. Canton, A. O. Dohn, K. B. Møller, M. M. Nielsen, K. J. Gaffney, K. Wärnmark, V. Sundström, P. Persson and J. Uhlig, Hot Branching Dynamics in a Light–Harvesting Iron Carbene Complex Revealed by Ultrafast X–ray Emission Spectroscopy, Angew. Chem., Int. Ed., 2020, 59, 364–372 CrossRef CAS.
- R. A. Marcus, On the Theory of Oxidation–Reduction Reactions Involving Electron Transfer. I, J. Chem. Phys., 1956, 24, 966–978 CrossRef CAS.
- K. S. Kjær, K. Kunnus, T. C. B. Harlang, T. B. Van Driel, K. Ledbetter, R. W. Hartsock, M. E. Reinhard, S. Koroidov, L. Li, M. G. Laursen, E. Biasin, F. B. Hansen, P. Vester, M. Christensen, K. Haldrup, M. M. Nielsen, P. Chabera, Y. Liu, H. Tatsuno, C. Timm, J. Uhlig, V. Sundstöm, Z. Németh, D. S. Szemes, É. Bajnóczi, G. Vankó, R. Alonso-Mori, J. M. Glownia, S. Nelson, M. Sikorski, D. Sokaras, H. T. Lemke, S. E. Canton, K. Wärnmark, P. Persson, A. A. Cordones and K. J. Gaffney, Solvent control of charge transfer excited state relaxation pathways in [Fe(2,2′-bipyridine)(CN)4, Phys. Chem. Chem. Phys., 2018, 20, 4238–4249 RSC.
- M. Reiher, O. Salomon and B. A. Hess, Reparameterization of hybrid functionals based on energy differences of states of different multiplicity, Theor. Chem. Acc., 2001, 107, 48–55 Search PubMed.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- F. Neese, Software update: The ORCA program system—Version 5.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- S. Hirata and M. Head-Gordon, Time-dependent density functional theory within the Tamm–Dancoff approximation, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- T. Roland, J. Léonard, G. Hernandez Ramirez, S. Méry, O. Yurchenko, S. Ludwigs and S. Haacke, Sub-100 fs charge transfer in a novel donor–acceptor–donor triad organized in a smectic film, Phys. Chem. Chem. Phys., 2012, 14, 273–279 RSC.
- L. Liu, P. Eisenbrandt, T. Roland, M. Polkehn, P.-O. Schwartz, K. Bruchlos, B. Omiecienski, S. Ludwigs, N. Leclerc, E. Zaborova, J. Léonard, S. Méry, I. Burghardt and S. Haacke, Controlling charge separation and recombination by chemical design in donor–acceptor dyads, Phys. Chem. Chem. Phys., 2016, 18, 18536–18548 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2qi01903e |
| This journal is © the Partner Organisations 2023 |