
Amino-bridged attapulgite@perovskite nanocomposites: the role of bridge linkage in their optical properties and stabilities†
Lei
Qiu
abc,
Mengyu
Guan
a,
Wei
Wang
a,
Maxim S.
Molokeev
de,
Sergey P.
Polyutov
 d,
Zhigao
Dai
*ac and
Guogang
Li
*ab
d,
Zhigao
Dai
*ac and
Guogang
Li
*ab
aFaculty of Materials Science and Chemistry, China University of Geosciences, 388 Lumo Road, Wuhan 430074, P. R. China. E-mail: ggli@cug.edu.cn
bZhejiang Institute, China University of Geosciences, Hangzhou 311305, P. R. China
cShenzhen Institute, China University of Geosciences, Shenzhen, 518052, P. R. China. E-mail: daizhigao@cug.edu.cn
dInternational Research Center of Spectroscopy and Quantum Chemistry—IRC SQC, Siberian Federal University, Krasnoyarsk, 660041, Russia
eLaboratory of Crystal Physics, Kirensky Institute of Physics, Federal Research Center KSC SB RAS, Krasnoyarsk 660036, Russia
First published on 10th July 2023
Abstract
Perovskite-based nanocomposites have attracted significant interest due to their potential in interfacial engineering, optical modification, and stability enhancement. However, current understanding of the construction models between perovskites and guest materials is limited, and the design concept of perovskite-based nanocomposites remains unclear. Herein, we thoroughly investigated the effects of amino bridge linkages in attapulgite@perovskite nanocomposites on the crystallization kinetics, optical properties, and stabilities of perovskites. We monitored the difference in structural, compositional, and morphological characteristics. The attapulgite@perovskite nanocomposites with amino bridge linkages exhibited a smaller average size distribution of 23.8 ± 5.8 nm and demonstrated enhanced thermal stability (81% relative PL intensity after one heating–cooling cycle) and photo-stability (84% relative PL intensity after 30 h of ultraviolet light irradiation). These improvements can be attributed to effective passivation by the amino bridge linkages. Our work aims to deepen the understanding of perovskite-based nanocomposite construction and inspire new approaches for modifying their optical properties and enhancing their stability.
Introduction
Perovskites have garnered recent attention due to their remarkable optoelectronic properties, including high light-absorption coefficients,1 color purity,2 photo-luminescence quantum yields (PLQYs),3 radiative recombination4,5 and compositional direct-bandgap color tunability.6 These properties position perovskites as potential candidates for a wide range of applications, such as light-emitting diodes (LEDs),7–9 solar cells,10–12 lasers13–15 and photodetectors.16–18 However, the instability issues of perovskites, including their high sensitivity to heat and photodegradation, have hindered their practical applications. Therefore, it is crucial to explore effective approaches to enhance their stability.Various strategies, such as matrix encapsulation,19–21 surface passivation22–26 and compositional engineering,27–29 have been developed to enhance the stability of perovskites. Constructing perovskite-based nanocomposites is a popular strategy to stabilize perovskites. This approach offers several advantages, including the introduction of a more stable guest material as a matrix protector layer to prevent the direct exposure of perovskites to the environment, ensure minimal fluorescence quenching for high-quality luminescence, and provide interfacial engineering to explore unique properties for a broader range of applications.30–32 For instance, Hao et al. used rich hydroxyl groups on the surface of halloysite nanotubes to generate surface adsorption and covalent grafting to perovskite nanocrystals (NCs). This resulted in the formation of a halloysite@perovskite nanocomposite with significantly improved thermal stability and photostability.33 Li et al. designed type II heterojunction nanocomposites, specifically ZnSe@CsSnCl3 with enhanced photocatalytic activity for CO2 reduction. This improvement was attributed to efficient charge separation and reduction in the free energy required for CO2 conversion.34 Fan et al. synthesized CsPbBr3@Cs4PbBr6@silica molecular sieve nanocomposites using an advanced solid-state synthesis method. These nanocomposites exhibited remarkable stabilities against water, heat, and blue light irradiation.35 However, a lack of research on coupling models in perovskite-based nanocomposites has had a significant impact on their interfacial properties. To comprehend the features and performances of perovskite-based nanocomposites, it is crucial to study the integration of two different parts.
Attapulgite (ATP) is a promising material for constructing nanocomposites due to its large specific surface area,36,37 ease of surface functionalization,38,39 and good biocompatibility.40 Herein, we utilized ATP as a template for loading perovskite NCs and constructing highly stable perovskite-based nanocomposites. To enhance the surface properties of ATP, surface functionalization was carried out, resulting in the construction of the corresponding nanocomposite.
We conducted a systematic investigation of the correlation between the bridge linkage and the performance of the nanocomposite using zeta potential, XPS, and TEM analyses. Based on the experimental results and our hypothesis, we proposed a bridge linkage passivation mechanism that stabilizes CsPbBr3 NCs. The as-prepared nanocomposite demonstrated great potential for use in solid-state lighting applications. Our study contributes to a deeper understanding of the role of bridge linkage in perovskite-based nanocomposites and inspired us to design high-performance perovskite-based nanocomposites.
Experimental
Chemicals
Cesium carbonate (Cs2CO3, 99.5%, Acros Organics), lead(II) bromide (PbBr2, 99.999%, Aladdin), oleic acid (OA, technical grade 90%, Aldrich), oleylamine (OAm, 80–90%, Aladdin), 1-octadecene (ODE, >90%, Aladdin), toluene (98%, Aladdin), hexane (analytical reagent 97%, Sinopharm Chemical Reagent Co., Ltd, Shanghai), attapulgite (purchased from Lincang county, Yunnan province, China), (3-Aminopropyl) trimethoxysilane (APTMS, 98%, Aladdin) and aminoethylamino propyltrimethoxy silane (98%, Aladdin) were obtained for this study. All chemicals were used without further purification.Surface functionalization of ATP
The pristine ATP NRs were firstly calcined at 105 °C to remove residual organic pollutants. Taking ATP-AC6 as an example, 0.6 g of ATP and 0.5 mL of APTMS were added to 10 mL of toluene under violent stirring. The reaction temperature was set to 45 °C and the reaction time was 2 hours. The obtained crude solution was centrifuged at 6000 rpm for 5 min and the precipitate was washed with deionized water three times. As for ATP-AC8, APTMS was replaced by aminoethylamino propyltrimethoxy silane. The dosage of APTMS was set to 0.1, 0.2, 0.5, 1.0 and 2.0 mL to study the optimal volume.Synthesis of Cs-oleate
In a typical experiment, Cs2CO3 (0.8140 g), OA (5.00 mL), and ODE (50.00 mL) were loaded into a 100 mL three-necked flask and stirred under vacuum for 1 hour at 120 °C. Then, the solution was heated at 150 °C under N2 until it became clear and transparent, implying the complete reaction of Cs2CO3 and OA. The Cs-oleate solution was stored under N2 and needed to be preheated to 100 °C before further use because it precipitates at room temperature.Synthesis of ATP-AC6@CsPbBr3 nanocomposites
Typically, 0.3450 g of PbBr2 and 0.1 g of ATP-AC6 were dispersed in 25 mL of ODE in a three-necked flask. The mixture was dried and stirred at 120 °C for 2 hours under vacuum to guarantee a complete dissolution of PbBr2. Then, 2.50 mL of OA and 2.5 mL of OAm were added and the temperature was raised to 170 °C in an N2 atmosphere. When the temperature reached 170 °C, Cs-oleate (2 mL) was rapidly injected into the flask. The flask was immediately cooled down by immersing it in an ice bath after the mixture had reacted for five seconds, and then a crude colloidal solution was obtained. The above solution was centrifuged at 9000 rpm for 10 min to obtain the resulting ATP-AC6@CsPbBr3 precipitates. As for ATP@CsPbBr3 and ATP-AC8@CsPbBr3 nanocomposites, ATP-AC6 was substituted for ATP and ATP-AC8, respectively.Fabrication of LED devices
The white LED device was constructed by using the as-prepared green ATP-AC6@CsPbBr3 nanocomposite, the commercial red phosphor K2SiF6:Mn4+ and a 460 nm InGaN chip. Typically, the ATP-AC6@CsPbBr3 nanocomposite and K2SiF6:Mn4+ were evenly blended with silicone resins A and B (A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :B = 1:1) in an agate mortar. Then, the resulting mixture was coated on a 460 nm InGaN chip. The packaged devices were cured in an oven at 120 °C for 12 hours to form the resulting white LED device.
:B = 1:1) in an agate mortar. Then, the resulting mixture was coated on a 460 nm InGaN chip. The packaged devices were cured in an oven at 120 °C for 12 hours to form the resulting white LED device.
Characterization studies
The crystal structure and phase purity were characterized by Powder X-ray diffraction (PXRD), which was performed on a D8 Focus diffractometer at a scanning rate of 1° min−1 in the 2θ range from 10° to 60° with Ni-filtered Cu Kα (λ = 1.540598 Å) radiation. Fourier transform infrared (FTIR) spectra were recorded using a Fourier transform infrared spectrometer (Thermo Fisher Scientific, IS50). The morphologies were inspected using a field emission scanning electron microscope (FE-SEM, S-4800, Hitachi) and a transmission electron microscope (FEI Tecnai G2 F20). The photoluminescence spectra were recorded using a fluorescence spectrometer (Fluoromax-4P, Horiba Jobin Yvon, New Jersey, USA) equipped with a 150 W xenon lamp as an excitation source, and both excitation and emission slit widths were set up to be 1.0 nm with the width of the monochromator slits adjusted to 0.50 nm. Zeta potential quantities were measured using a Zetasizer Nano ZS system (Malvern, UK) equipped with a standard 633 nm laser. The spectra from X-ray photoelectron spectroscopy (XPS) were recorded on a Thermo ESCALAB 250XI. The thermal stability of luminescence properties was measured using a Fluoromax-4P spectrometer connected to heating equipment (TAP-02), which involved using a combined setup consisting of a xenon lamp, a Hamamatsu MPCD-7000 multichannel photodetector and a computer-controlled heater. The photoluminescence quantum yield (PLQY) was measured using an absolute PL quantum yield measurement system C9920-02 (Hamamatsu Photonics K.K., Japan). The photoluminescence decay curves were obtained from a Lecroy Waverunner 6100 Digital Oscilloscope (1 GHz) using a tunable laser (pulse width = 4 ns, gate = 50 ns) as an excitation source (Continuum Sunlite OPO).Results and discussion
ATP nanorods (NRs) are promising candidates for coupling with perovskite NCs due to their large specific surface area and excellent dispersity. SEM images reveal solid rod structures with average vertical and lateral lengths of 1330.0 and 37.8 nm, respectively (Fig. S1†). To investigate the impact of bridge linkages on the interfacial features and optical performances of perovskite-based nanocomposites, we designed a series of comparative experiments that distinguished between the presence and absence of amino bridge linkages. The pristine ATP NRs were initially calcined at 105 °C to remove residual organic pollutants, thereby eliminating steric hindrance and enhancing the exposure of Si–OH bonds for surface functionalization. For the convenience of discussion, we designated ATP NRs with/without grafted amino bridge linkages as ATP-ACX/ATP, where CX represents the carbon chain length of amino-containing silane coupling agents. A schematic diagram of the construction of ATP@CsPbBr3 and ATP-ACX@CsPbBr3 nanocomposites is depicted in Fig. 1. The entire synthetic process remains unchanged to investigate the correlation between optical performances of nanocomposites and amino bridge linkages.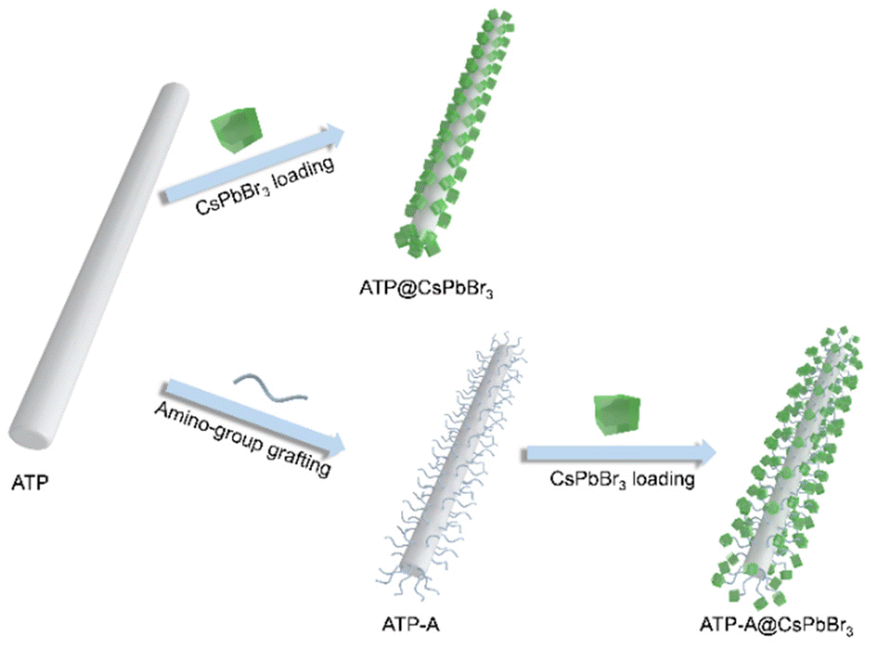 | ||
| Fig. 1 Schematic diagram of the construction of ATP@CsPbBr3 and ATP-A@CsPbBr3 nanocomposites. | ||
Fig. 2a illustrates the nature of three favored surface linkages of pristine ATP NRs obtained after surface modification processes. The surface of calcined ATP retains –OH groups, while the surface functionalized with silane coupling agents retains –NH2 groups. Amino-containing silane coupling agents used in this study include aminopropyltriethoxysilane (APTMS) and aminoethylamino propyltrimethoxy silane, which vary in their carbon chain lengths and the activity of their amino groups. Fourier transform infrared (FTIR) spectroscopy tests are conducted to examine the surface characteristics of functionalized ATP. The absorption bands between 3500 and 3750 cm−1 in ATP are ascribed to the stretching vibration absorptions of the –OH groups (Fig. 2b). In addition, the absence of –CH2 absorption bands implies the effective removal of residual organic pollutants. Upon surface modification with amino-containing silane coupling agents, the relative absorption intensities of –OH groups decrease, and absorption bands appear at 2930 and 1620 cm−1, corresponding to the stretching vibration absorptions of –CH2 and –NH2 groups, respectively. The newly appeared absorption bands originate from the amino-containing silane coupling agents, indicating the occurrence of a hydrolysis reaction between the silane and surface –OH groups and the successful grafting of amino groups onto the surface of ATP. It should be noted that APTMS with its shorter carbon chain length exhibits higher reactivity for silane grafting, although the absorption signals of –OH and –NH2 groups are weaker. This can be attributed to the high sensitivity of APTMS to water molecules. The zeta potential measurement records the surface potentials of ATP, ATP-AC6 and ATP-AC8. The results reveal that the potential value at pH = 7 changes from −10.97 to 9.27 and 4.31 mV after silane grafting (Fig. 2c). This result is consistent with previous reports, which show that the potential value of the –OH groups is negative, while that of the –NH2 group is positive.41,42 Powder X-ray diffraction (PXRD) patterns show no significant difference after silane grafting, confirming the preservation of ATP phase structure (Fig. S2†). Collectively, these results indicate the successful grafting of silane agents onto the surface of ATP and the engineering of interfacial surface characteristics.
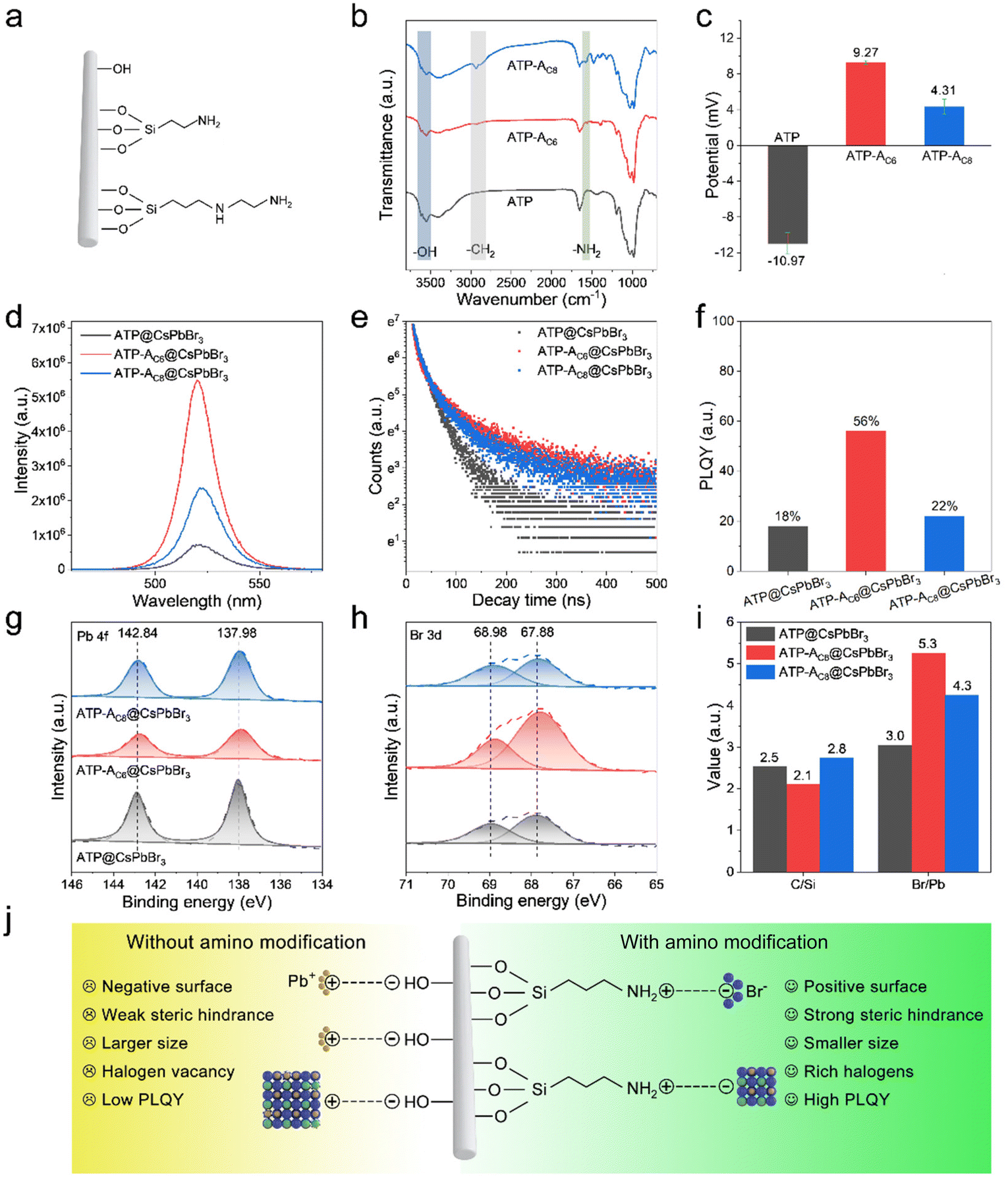 | ||
| Fig. 2 (a) Representation of three types of functional groups present on the surface of ATP. (b) FTIR spectra and (c) zeta potentials of ATP, ATP-AC6 and ATP-AC8. (d) PL spectra, (e) decay curves and (f) PLQY of ATP@CsPbBr3, ATP-AC6@CsPbBr3 and ATP-AC8@CsPbBr3. High-resolution XPS spectra of (g) Pb 4f, (h) Br 3d and (i) C/Si (Br/Pb) ratios of ATP@CsPbBr3, ATP-AC6@CsPbBr3 and ATP-AC8@CsPbBr3. (j) Schematic diagram of the growth mechanism of CsPbBr3 NCs on the surface of ATP without or with amino modification. | ||
The successful functionalization endows ATPs with different surface characteristics, and the compositional, optical and morphological properties of their corresponding nanocomposites are investigated. XRD patterns depict that the characteristic and dominant diffraction peaks of the as-prepared ATP@CsPbBr3, ATP-AC6@CsPbBr3 and ATP-AC8@CsPbBr3 nanocomposites locked at 8.50°, 19.85° and 20.84° are ascribed to the (110), (040) and (121) planes of the orthorhombic phase of ATP (PDF 31-0783), and the diffraction peaks locked at 21.57°, 30.67° and 37.8° originate from the (110), (200) and (211) planes of the cubic phase of CsPbBr3 (PDF 54-0752) (Fig. S3†). The coexistence of XRD patterns for both ATP and CsPbBr3 is powerful confirmation illustrating the successful construction of nanocomposites. Their corresponding PL spectra are varied from PL intensities and PL peak positions, where they exhibit higher PL signals and slightly blue-shifted PL peaks (Fig. 2d). The optical differences suggest that the surface characteristics of ATP have an impact on the growth of CsPbBr3 NCs. Importantly, the ATP-AC6@CsPbBr3 nanocomposite exhibits higher PL intensity than the ATP-AC8@CsPbBr3 nanocomposite, which is ascribed to the stronger interaction between ATP-AC6 and surface Br atoms of CsPbBr3 NCs for suppressed surface defects. In addition, PL decay curves of the above samples demonstrate that nanocomposites with amino bridge linkages have larger proportions of radiation recombination and longer average lifetimes (Fig. 2e). A summary of the corresponding PL decay characteristics is displayed in Table S1.† Photoluminescence quantum yield (PLQY) measurements further agree with PL decay results, which show a maximum value of 56% for the ATP-AC6@CsPbBr3 nanocomposite and a minimum value of 18% for the ATP@CsPbBr3 nanocomposite (Fig. 2f).
To explore the working mechanism of amino bridge linkages with respect to the optical performances of the above nanocomposites, the growth crystallization kinetics of CsPbBr3 NCs and interfacial properties of nanocomposites are investigated. X-ray photoelectron spectroscopy (XPS) is employed to study the coordination environment and surface properties. High resolution spectra corresponding to Pb 4f and Br 3d are displayed in Fig. 2g and h, which suggest that the binding energies of amino-functionalized nanocomposites undergo an obvious negative shift compared to those without amino bridge linkages. The shift of Br 3d orbitals to lower binding energy is mainly attributed to the effective surface passivation of amino-functionalized ATP. Self-assembled polyacrylic acid-grafted graphene oxide NRs have been reported to be utilized as surface ligands to stabilize perovskite NCs and achieve improved stability toward protic solvents and heat.43 For a comparison with a typical oleylamine ligand, amino-functionalized ATP possesses a shorter chain length, which has reduced steric hindrance and stronger binding to couple with CsPbBr3 NCs. In addition, passivation of amino-functionalized ATP has the advantage of eliminating surface halogen defects, and thus reduces the possibility of exposing inner lead atoms. The less chance of lead exposure on the surface weakens binding with surface ligands. To further prove the passivation effect of amino bridge linkages, elemental compositions and proportions were studied. The ratios of elemental proportions of C/Si in ATP@CsPbBr3, ATP-AC6@CsPbBr3 and ATP-AC8@CsPbBr3 nanocomposites are 2.5, 2.1 and 2.8, respectively (Fig. 2i). The amino-containing silane coupling agents grafted on ATP have shorter carbon chain lengths than oleylamine, so the value of the C/Si ratio would be reduced for amino-functionalized nanocomposites. However, an unusually high value is noticed for the ATP-AC8@CsPbBr3 nanocomposite, which mainly results from insufficient surface ligand passivation for the ATP@CsPbBr3 nanocomposite. The ratios of elemental proportions of Br/Pb in ATP@CsPbBr3, ATP-AC6@CsPbBr3 and ATP-AC8@CsPbBr3 nanocomposites are 3.0, 5.3 and 4.3, respectively. The higher values of the Br/Pb ratios for amino-functionalized nanocomposites imply the effective elimination of surface halogen defects.
Combining our design anticipations and experimental results, we proposed an amino-functionalized ATP passivated mechanism to control the crystallization kinetics of CsPbBr3 NCs (Fig. 2j). For nanocomposites without amino bridge linkages, the surface of the ATP features negatively charged –OH groups. Positively charged lead ions are gathered on the surface of the ATP due to electrostatic adsorption, and serve as the crystal nucleus for the growth of CsPbBr3 NCs. The ripening of CsPbBr3 NCs is rapid because of the direct contact with ATP nanorods with weak steric hindrance. For nanocomposites with amino bridge linkages, the surface of the ATP features positively charged –NH2 groups. Negatively charged bromide ions are coupled with the –NH2 groups via hydrogen bonds, and CsPbBr3 NCs are grown rich with halide. The richness of halide on the surface has been proved advantageous to eliminate surface defects and promote radiative recombination.
The morphological and compositional properties are further studied to verify the hypothesis of the ripening mechanism of CsPbBr3 NCs. For the ATP@CsPbBr3 nanocomposite, ATP NRs are severely aggregated because of the weak steric hindrance of –OH groups, where CsPbBr3 NCs are directly attached on the surface (Fig. 3a). It could be observed that CsPbBr3 NCs possess an inconsistent size distribution. Their average size is found to be 30.5 nm with an error of ±8.5 nm (Fig. 3d). As for ATP-AC6@CsPbBr3 and ATP-AC8@CsPbBr3 nanocomposites, ATP NRs exhibit better dispersibility due to the strong steric hindrance of amino-containing silica coupling agents (Fig. 3b and c). The size distributions of CsPbBr3 NCs are well matched with Gaussian fitting, demonstrating an average size of 23.8 ± 5.8 and 24.3 ± 5.5 nm, respectively (Fig. 3e and f). The smaller size distributions of CsPbBr3 NCs in amino-functionalized nanocomposites may be the root for the effective passivation of amino-containing silica coupling agents. However, pure CsPbBr3 NCs possess an average size of 12.4 ± 5.5 nm (Fig. S4†). The overripening of CsPbBr3 NCs in nanocomposites is mainly triggered by the incorporation of ATP, which works as the crystal nucleus for the heterogeneous growth of CsPbBr3 NCs.44,45 The high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images and the corresponding energy-dispersive X-ray spectroscopy (EDS) elemental mapping images of ATP-AC6@CsPbBr3 nanocomposite reveal the distributions of Si, Mg, Cs, Pb and Br elements, suggesting the uniform decoration of CsPbBr3 NCs on ATP NRs (Fig. 3g). The EDS spectrum further proves the appearance of the corresponding elements at different energy levels (Fig. 3h). These results directly confirm the successful combination of ATP NRs and CsPbBr3 NCs.
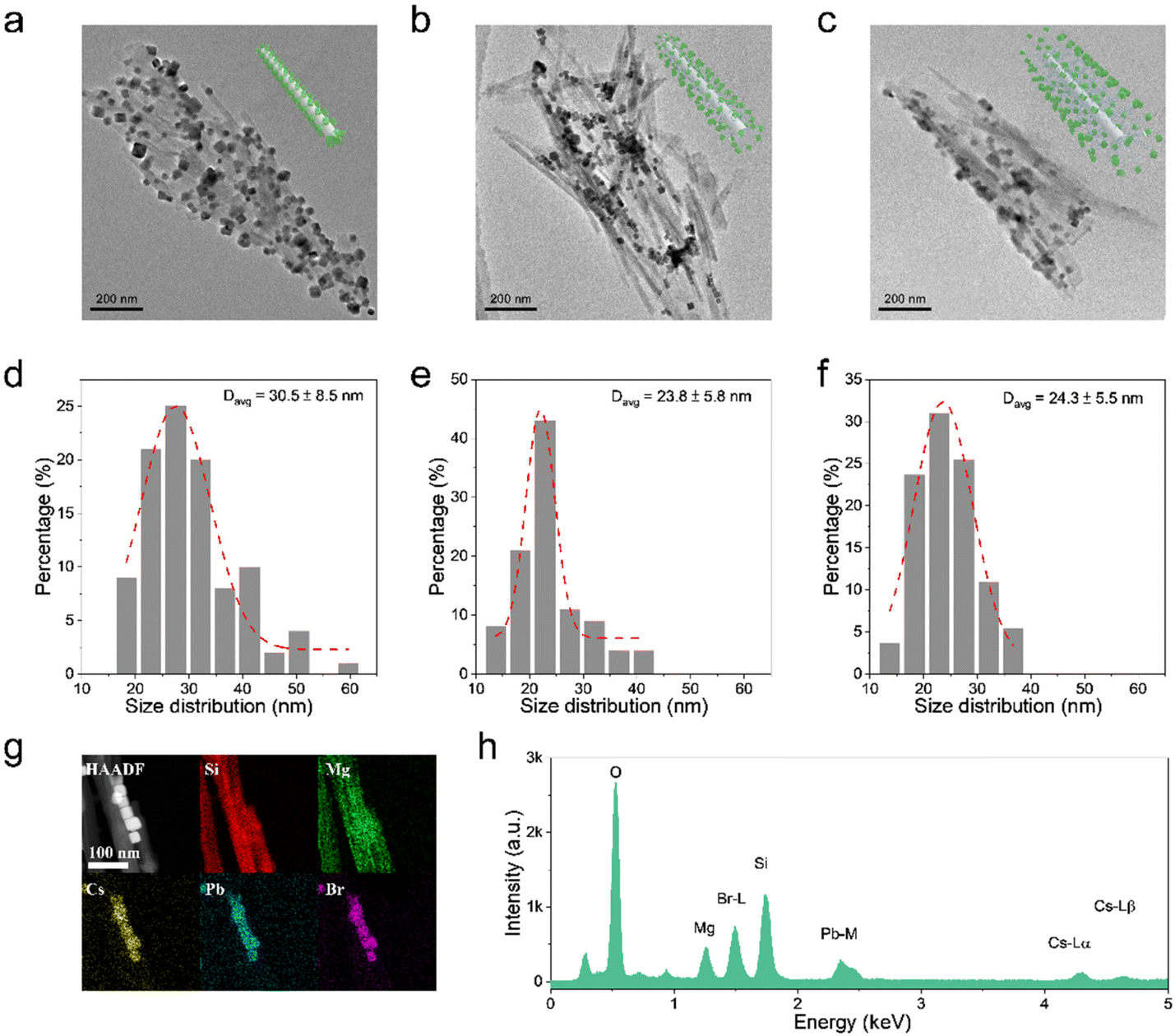 | ||
| Fig. 3 TEM images of (a) ATP@CsPbBr3, (b) ATP-AC6@CsPbBr3 and (c) ATP-AC8@CsPbBr3 nanocomposites. Inset: Schematic diagram illustrating the structures with or without amino bridge linkages. Scale bars: 200 nm. Size distributions of (d) ATP@CsPbBr3, (e) ATP-AC6@CsPbBr3 and (f) ATP-AC8@CsPbBr3 nanocomposites. (g) EDS mapping of the ATP-AC6@CsPbBr3 nanocomposite, along with corresponding STEM-HAADF image and elemental maps of Si, Mg, Cs, Pb, and Br. Scale bars: 100 nm. (h) Elemental spectrum of the ATP-AC6@CsPbBr3 nanocomposite. | ||
The working mechanism of the amino bridge linkages to perovskite-based nanocomposites has been systematically investigated, and the dosage of APTMS is further optimized. The dosage of APTMS is varied from 0.1 to 2.0 mL to explore the surface differences of ATP-AC6. FTIR spectra reveal that the intensities of the νSi–O–Si band at (980, 1032) cm−1, νN–H band at 1560 cm−1 and νC–H band at (2877, 2930) cm−1 are increased until the APTMS dosage reaches 0.5 mL, and then drop at higher APTMS dosages (Fig. 4a). The intensity variation of the vibration band demonstrates that the grafting of APTMS on the ATP surface does not increase when accompanied by increasing APTMS volume. Their corresponding zeta potential values are further recorded, which show a transfer from negative to positive and reach a maximum of 4.75 mV at 0.5 mL (Fig. 4b). The trend of the potential values is in accordance with FTIR results, demonstrating that the surface property of ATP is closely associated with the dosage of APTMS. In order to quantitatively analyze the proportion of APTMS, thermogravimetric (TG) analysis is carried out to calculate the weight ratio of APTMS. As shown in Fig. 4c, the residual weight of pristine ATP remains at 84.8% after calcination at 800 °C, while it remains at 75.8%, 71.9%, 68.0%, 69.2 and 75.1% with APTMS dosages of 0.1, 0.2, 0.5, 1.0 and 2.0 mL, respectively. The maximum grafting weight ratio of APTMS to ATP is calculated to be 24.7%. The PL spectra of their corresponding nanocomposites are depicted in Fig. 4d, implying positive correlation between the PL intensity and APTMS grafting volume. The red shift of the PL peak at low APTMS volume is attributed to insufficient passivation and overgrowth of CsPbBr3 NCs. XRD patterns demonstrate that the ATP-AC6@CsPbBr3 nanocomposite with APTMS dosage of 0.5 mL possesses a higher crystallinity (Fig. 4e). The minimum full width at half maximum (FWHM) value of 0.37135° at 2θ = 21.57° further verifies this result (Fig. S5†). In addition, the XRD diffraction peak at 2θ = 37.8° is ascribed to the (211) plane of the cubic CsPbBr3 phase, which matches well with the standard card of PDF 54-0572, as shown in Fig. 4e. The optical differences of nanocomposites suggest that the higher the grafting ratio of APTMS on the surface of ATP, the better the crystallinity and brighter the luminescence of CsPbBr3 NCs. It is well known that the greater the exposure of –OH groups on the surface of ATP the easier it is to trigger a hydrolysis reaction with APTMS.46 With increasing APTMS dosage, the number of –OH group active sites on the surface of the ATP decreases sharply. However, excess APTMS in solution has more chance of self-aggregation, which is a great barrier to effective coupling with –OH groups (Fig. 4f). This is the reason for the low grafting content on the surface of ATP at high APTMS concentrations.
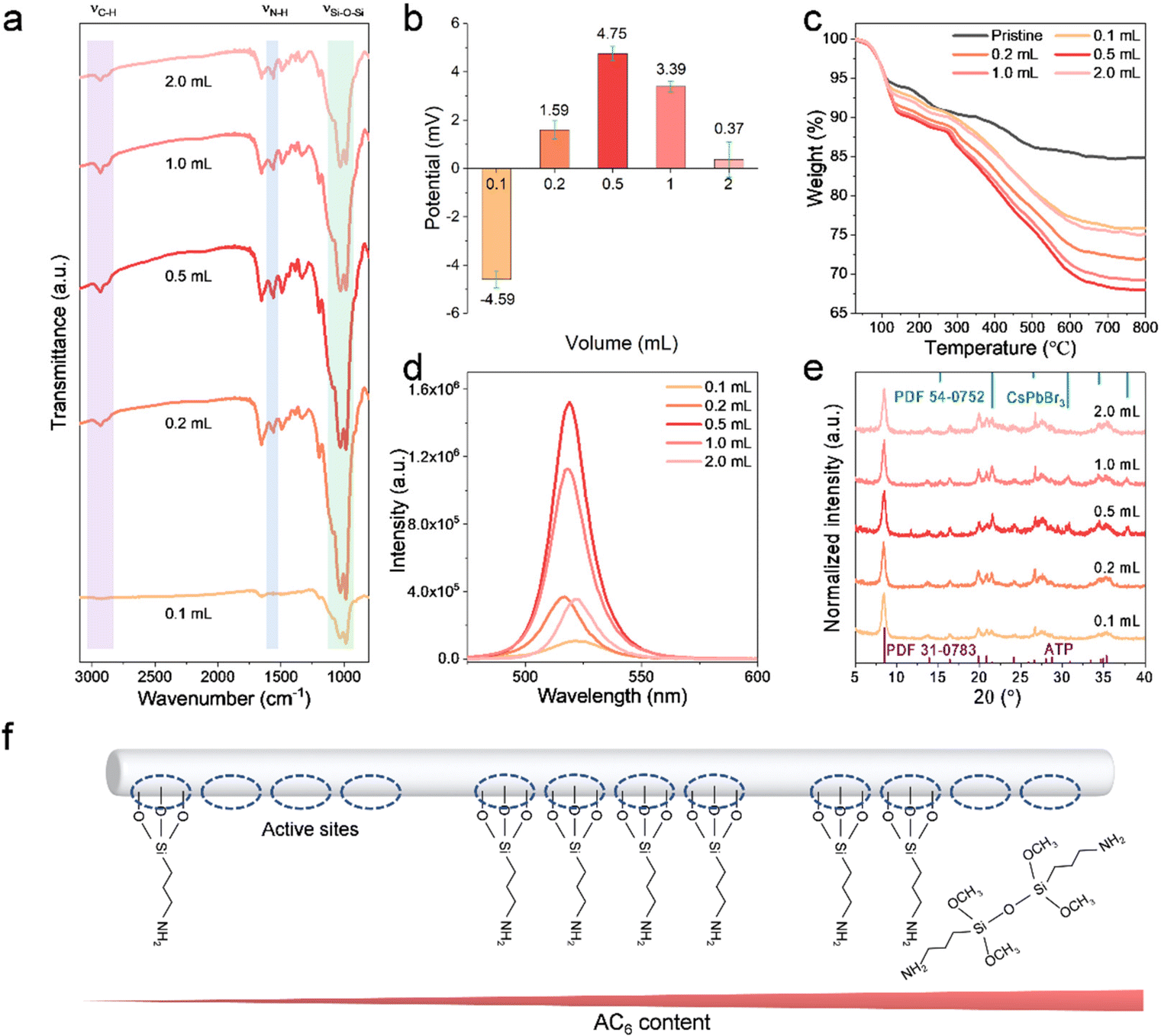 | ||
| Fig. 4 (a) FTIR spectra, (b) zeta potentials and (c) TG curves of ATP with varying APTMS modification contents. (d) PL spectra and (e) the corresponding XRD patterns of the ATP-AC6@CsPbBr3 nanocomposite with different APTMS modification contents. (f) Schematic diagram of the relationship between reactive sites and APTMS content. | ||
The excellent optical properties of the ATP-AC6@CsPbBr3 composite have been studied, and its stabilities are further investigated to evaluate its potential for practical utilization. Thermal stability involving a heating–cooling cycle is carried out using a controlled temperature system ranging from 20 to 120 °C. Fig. 5a and b displays pseudo-color images of the temperature-dependent PL for ATP@CsPbBr3 and ATP-AC6@CsPbBr3 nanocomposites. It is observed that the ATP-AC6@CsPbBr3 nanocomposite possesses a more stable fluorescence intensity at high temperature. Their corresponding PL spectra during the heating–cooling process are shown in Fig. S6.† Normalized PL intensities are subsequently provided to directly compare the relative PL values at different temperatures (Fig. 5c). These two samples both exhibit a decrease of PL intensity during the heating process, while the PL intensity is recovered during the cooling process. Differently, the fluorescence recovers to its initial intensity of 54% for ATP@CsPbBr3 and 81% for ATP-AC6@CsPbBr3. XRD patterns demonstrate that diffraction peaks belonging to CsPbBr3 NCs vanished for ATP@CsPbBr3, while they are transformed to a monoclinic phase (PDF 18-0364) for ATP-AC6@CsPbBr3 (Fig. 5d). The PL quenching with thermal excitation in perovskites is mainly attributed to lattice vibrations, which accelerate the rate of nonradiative recombination.47,48 The thermal test indicates that CsPbBr3 NCs combined with the ATP-AC6 template undergo a weaker lattice vibration during the heating process, and a quicker lattice vibration recovery to the initial state is triggered during the cooling process. This result proves the comprehensive protection of ATP-AC6 for the suppressed lattice vibrations of CsPbBr3 NCs.
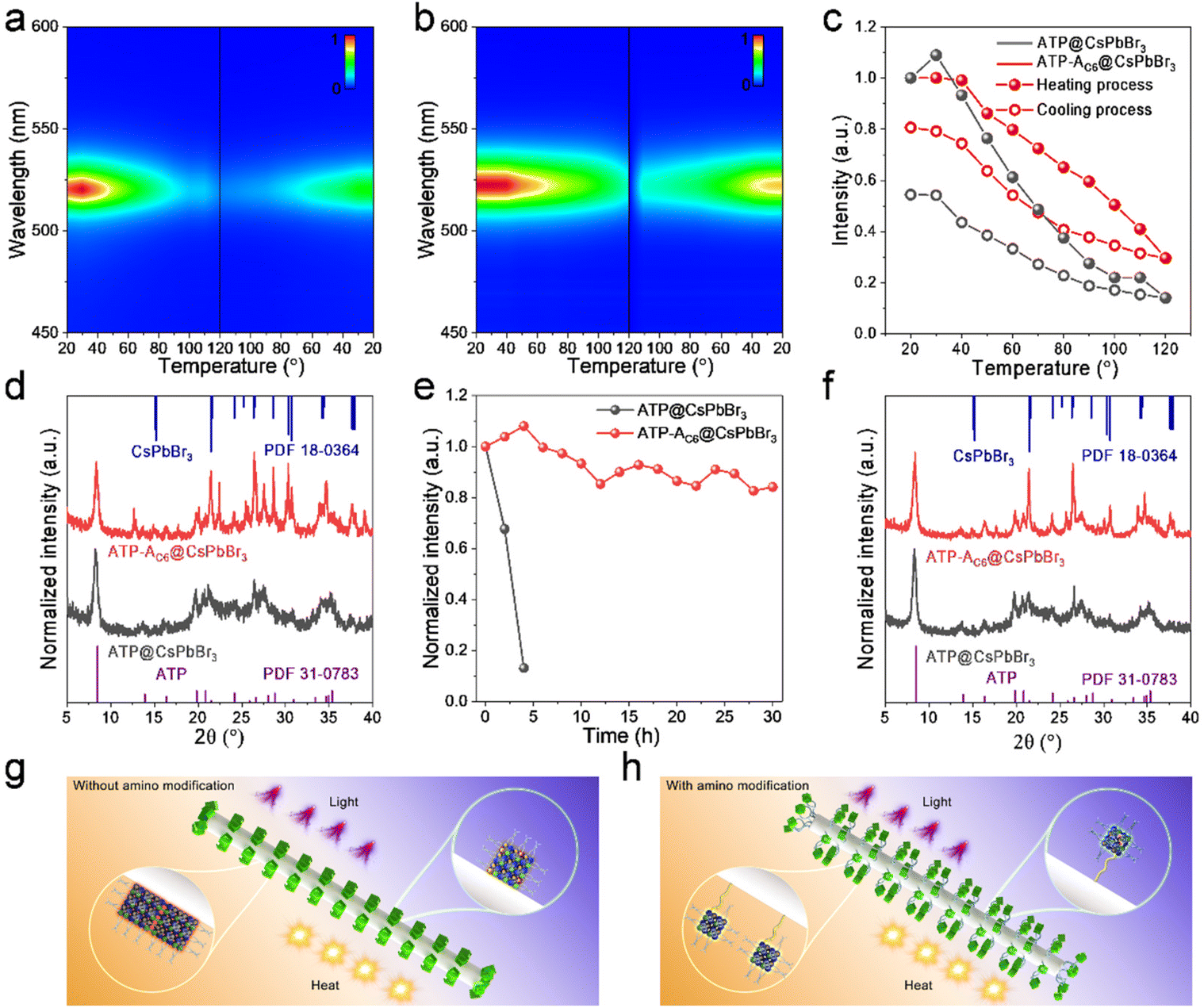 | ||
| Fig. 5 Pseudo-color images of the temperature-dependent PL for (a) ATP@CsPbBr3 and (b) ATP-AC6@CsPbBr3 nanocomposites, during a heating and cooling process. The corresponding (c) normalized PL intensities and (d) XRD patterns after the heating/cooling cycle. (e) Normalized PL intensities and (f) XRD patterns of ATP@CsPbBr3 and ATP-AC6@CsPbBr3 nanocomposites after continuous irradiation with UV light. Schematic diagram illustrating the degradation mechanism of CsPbBr3 NCs (g) without or (h) with amino bridge linkages. | ||
Furthermore, their photo-oxidation stabilities are also tested to evaluate their long-term performance against ultraviolet (UV) light. The as-prepared ATP@CsPbBr3 and ATP-AC6@CsPbBr3 nanocomposites are continuously radiated under a 365 nm UV lamp with light intensity of 9 mW cm−2 at a distance of 0.1 m. Normalized PL intensities reveal that the relative fluorescence intensity of ATP@CsPbBr3 drops to 13% after 4 hours of UV light exposure, while that of ATP-AC6@CsPbBr3 remains at 84% after 30 hours of UV light exposure (Fig. 5e). Interestingly, the PL intensity of the ATP-AC6@CsPbBr3 nanocomposite is enhanced at early irradiation times, which is mainly triggered by a photo-activated process.49,50 Their corresponding PL spectra during continuous UV irradiation are depicted in Fig. S7.† XRD patterns suggest the absence of the perovskite phase in ATP@CsPbBr3, and the coexistence of cubic (PDF 54-0752) and monoclinic (PDF 18-0364) phases of CsPbBr3 NCs in the ATP-AC6@CsPbBr3 (Fig. 5f). Taking the above results into consideration, we proposed an amino bridge linkage passivation mechanism to stabilize CsPbBr3 NCs. As for nanocomposites without amino bridge linkages, ATP and CsPbBr3 NCs are in direct contact. Owing to the heat accumulation of external environments, adjacent CsPbBr3 NCs would spontaneously merge, triggering an increase in grain size and decrease in PL properties. The integration of CsPbBr3 NCs is not reversible even when the heat source is removed. In addition, free electrons caused by UV light are easy to transfer to the ATP, which generate more halogen defects and lead to collapse of the crystal structure. As for nanocomposites with amino bridge linkages, ATP and CsPbBr3 NCs are connected via APTMS. Their good dispersion endows CsPbBr3 NCs with stable size distribution when exposed to heat, and any fluorescence decay induced by thermal lattice vibrations would be recovered. Besides, the comprehensive passivation of amino bridge linkages confines free electrons to CsPbBr3 NCs and suppresses the formation of surface defects when exposed to UV light.
Finally, the potential application of the green ATP-AC6@CsPbBr3 nanocomposite in a white LED device is evaluated by mixing it with the commercial red phosphor K2SiF6:Mn4+ on a blue InGaN chip. Fig. 6a presents the electroluminescence (EL) spectra of the white LED device under a current of 100 mA, and a photograph of the operating device is given in the inset. The device displays a luminous efficiency of 39 lm W−1, and the corresponding value of the color rendering index (CRI) is 78. As shown in Fig. 6b, the Commission Internationale de L'Eclairage (CIE) color coordinate is (0.37, 0.36) with a white light correlated color temperature (CCT) of 3981 K, which is close to the standard white emission coordinate (0.33, 0.33). As a comparison, the core parameters of the white LED device fabricated from pure CsPbBr3 NCs are also provided; this LED exhibited a luminous efficiency of 34 lm W−1, and a CRI of 54, while the CIE color coordinate is (0.30, 0.34), and the CCT is 6967 K. The detailed performance parameters of LED devices are shown in Table S2.†
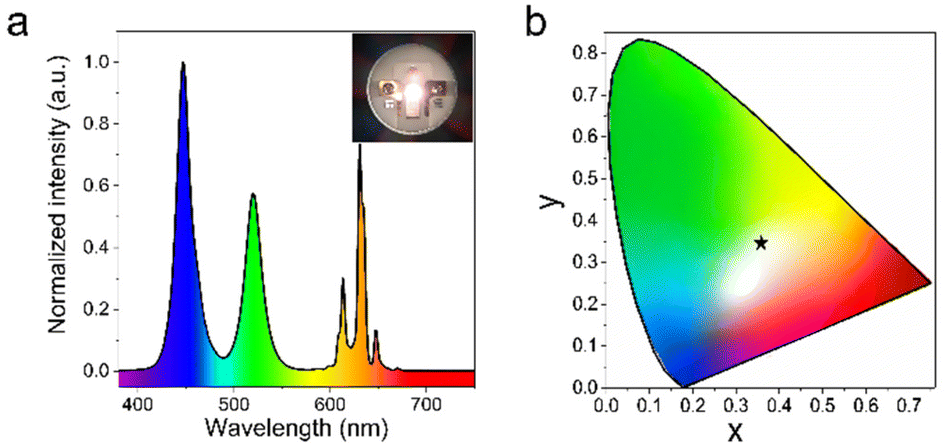 | ||
| Fig. 6 (a) Emission spectra of a white LED fabricated by depositing green ATP-AC6@CsPbBr3 nanocomposite and the commercial red phosphor K2SiF6:Mn4+ on a blue InGaN chip under a current of 100 mA. The inset shows a photograph of the operating white LED. (b) CIE color diagram of the white LED device. | ||
Conclusions
In summary, we investigated the influence of amino bridge linkages on crystallization control, optical property improvement, and stability enhancement in ATP@CsPbBr3 nanocomposites. Our results demonstrate that amino bridged linkages not only regulate the crystallization kinetics of perovskites but also act as surface ligands for passivating perovskites. The strong coupling between amino bridge linkages and perovskites results in minimized surface defects and excellent optical properties. Furthermore, we discovered that amino bridge linkages with shorter carbon chain lengths exhibit a more pronounced coupling effect, leading to enhanced and brighter luminescence and improved stability. The ATP-AC6@CsPbBr3 nanocomposite demonstrated significant potential as a light source for applications in the lighting field. This study holds crucial implications for comprehending the relationship between bridge linkages and the construction mechanism of perovskite-based nanocomposites, offering valuable insights for the design of high-performance materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant no. 52072349 and 52172162). Z. D. acknowledges support from the Fundamental Research Funds for the Central Universities, China University of Geosciences (Wuhan) (no. 162301202610), the Natural Science Foundation of Guangdong Province (2022A1515012145), the Shenzhen Science and Technology Program (JCYJ20220530162403007), and the Key Research and Development Plan of Hubei Province. G. L. acknowledges support from the National Natural Science Foundation of Zhejiang Province (LR22E020004). M. S. Molokeev and S. P. Polyutov acknowledge support from the Ministry of Science and High Education of the Russian Federation (Project no. FSRZ-2023-0006).References
- H. Zai, C. Zhu, H. Xie, Y. Zhao, C. Shi, Z. Chen, X. Ke, M. Sui, C. Chen, J. Hu, Q. Zhang, Y. Gao, H. Zhou, Y. Li and Q. Chen, Congeneric incorporation of CsPbBr3 nanocrystals in a hybrid perovskite heterojunction for photovoltaic efficiency enhancement, ACS Energy Lett., 2017, 3, 30–38 CrossRef.
- F. Zhang, H. Zhong, C. Chen, X.-g. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, Brightly luminescent and color-tunable colloidal CH3NH3PbX3 (X = Br, I, Cl) quantum dots: potential alternatives for display technology, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS PubMed.
- F. Liu, Y. Zhang, C. Ding, S. Kobayashi, T. Izuishi, N. Nakazawa, T. Toyoda, T. Ohta, S. Hayase, T. Minemoto, K. Yoshino, S. Dai and Q. Shen, Highly luminescent phase-stable CsPbI3 perovskite quantum dots achieving near 100% absolute photoluminescence quantum yield, ACS Nano, 2017, 11, 10373–10383 CrossRef CAS PubMed.
- X. Li, Y. Wu, S. Zhang, B. Cai, Y. Gu, J. Song and H. Zeng, CsPbX3 quantum dots for lighting and displays: room-temperature synthesis, photoluminescence superiorities, underlying origins and white light-emitting diodes, Adv. Funct. Mater., 2016, 26, 2435–2445 CrossRef CAS.
- M. Lorenzon, L. Sortino, Q. Akkerman, S. Accornero, J. Pedrini, M. Prato, V. Pinchetti, F. Meinardi, L. Manna and S. Brovelli, Role of nonradiative defects and environmental oxygen on exciton recombination processes in CsPbBr3 perovskite nanocrystals, Nano Lett., 2017, 17, 3844–3853 CrossRef CAS PubMed.
- X. Zhang, C. Sun, Y. Zhang, H. Wu, C. Ji, Y. Chuai, P. Wang, S. Wen, C. Zhang and W. W. Yu, Bright perovskite nanocrystal films for efficient light-emitting devices, J. Phys. Chem. Lett., 2016, 7, 4602–4610 CrossRef CAS PubMed.
- T. Chiba, K. Hoshi, Y. J. Pu, Y. Takeda, Y. Hayashi, S. Ohisa, S. Kawata and J. Kido, High-efficiency perovskite quantum-dot light-emitting devices by effective washing process and interfacial energy level alignment, ACS Appl. Mater. Interfaces, 2017, 9, 18054–18060 CrossRef CAS PubMed.
- J. Li, L. Xu, T. Wang, J. Song, J. Chen, J. Xue, Y. Dong, B. Cai, Q. Shan, B. Han and H. Zeng, 50-fold EQE improvement up to 6.27% of solution-processed all-inorganic perovskite CsPbBr3 QLEDs via surface ligand density control, Adv. Mater., 2017, 29, 1603885 CrossRef PubMed.
- H. He, S. Mei, Z. Wen, D. Yang, B. Yang, W. Zhang, F. Xie, G. Xing and R. Guo, Recent advances in blue perovskite quantum dots for light-emitting diodes, Small, 2022, 18, e2103527 CrossRef PubMed.
- J. Zhang, Z. Jin, L. Liang, H. Wang, D. Bai, H. Bian, K. Wang, Q. Wang, N. Yuan, J. Ding and S. F. Liu, Iodine-optimized interface for inorganic CsPbI2Br perovskite solar cell to attain high stabilized efficiency exceeding 14%, Adv. Sci., 2018, 5, 1801123 CrossRef PubMed.
- L. Liu, A. Najar, K. Wang, M. Du and S. F. Liu, Perovskite quantum dots in solar cells, Adv. Sci., 2022, 9, e2104577 CrossRef PubMed.
- Y. Wang, C. Duan, X. Zhang, J. Sun, X. Ling, J. Shi, L. Hu, Z. Zhou, X. Wu, W. Han, X. Liu, C. Cazorla, D. Chu, S. Huang, T. Wu, J. Yuan and W. Ma, Electroluminescent solar cells based on CsPbI3 Perovskite Quantum Dots, Adv. Funct. Mater., 2021, 32, 2108615 CrossRef.
- X. Li, Y. Wang, H. Sun and H. Zeng, Amino-mediated anchoring perovskite quantum dots for stable and low-threshold random lasing, Adv. Mater., 2017, 29, 1701185 CrossRef PubMed.
- W. Zhan, L. Meng, C. Shao, X.-g. Wu, K. Shi and H. Zhong, In situ patterning perovskite quantum dots by direct laser writing fabrication, ACS Photonics, 2021, 8, 765–770 CrossRef CAS.
- H. Yu, X. Cheng, Y. Wang, Y. Liu, K. Rong, Z. Li, Y. Wan, W. Gong, K. Watanabe, T. Taniguchi, S. Wang, J. Chen, Y. Ye and L. Dai, Waterproof perovskite-hexagonal boron nitride hybrid nanolasers with low lasing thresholds and high operating temperature, ACS Photonics, 2018, 5, 4520–4528 CrossRef CAS.
- X. Ren, Z. Li, Z. Huang, D. Sang, H. Qiao, X. Qi, J. Li, J. Zhong and H. Zhang, Environmentally Robust Black Phosphorus Nanosheets in Solution: Application for Self-Powered Photodetector, Adv. Funct. Mater., 2017, 27, 1606834 CrossRef.
- C. Xie, C. Mak, X. Tao and F. Yan, Photodetectors Based on Two-Dimensional Layered Materials Beyond Graphene, Adv. Funct. Mater., 2017, 27, 1603886 CrossRef.
- Z. Liu, Z. Zhang, X. Zhang, X. Li, Z. Liu, G. Liao, Y. Shen and M. Wang, Achieving high responsivity and detectivity in a quantum-dot-in-perovskite photodetector, Nano Lett., 2023, 23, 1181–1188 CrossRef CAS PubMed.
- Y. Wei, X. Deng, Z. Xie, X. Cai, S. Liang, P. a. Ma, Z. Hou, Z. Cheng and J. Lin, Enhancing the stability of perovskite quantum dots by encapsulation in crosslinked polystyrene beads via a swelling-shrinking strategy toward superior water resistance, Adv. Funct. Mater., 2017, 27, 1703535 CrossRef.
- H. Zhang, X. Wang, Q. Liao, Z. Xu, H. Li, L. Zheng and H. Fu, Embedding perovskite nanocrystals into a polymer matrix for tunable luminescence probes in cell imaging, Adv. Funct. Mater., 2017, 27, 1604382 CrossRef.
- H. Hu, L. Wu, Y. Tan, Q. Zhong, M. Chen, Y. Qiu, D. Yang, B. Sun, Q. Zhang and Y. Yin, Interfacial synthesis of highly stable CsPbX3/oxide janus nanoparticles, J. Am. Chem. Soc., 2018, 140, 406–412 CrossRef CAS PubMed.
- W. Yin, M. Li, W. Dong, Z. Luo, Y. Li, J. Qian, J. Zhang, W. Zhang, Y. Zhang, S. V. Kershaw, X. Zhang, W. Zheng and A. L. Rogach, Multidentate ligand polyethylenimine enables bright color-saturated blue light-emitting diodes based on CsPbBr3 nanoplatelets, ACS Energy Lett., 2021, 6, 477–484 CrossRef CAS.
- D. Yang, X. Li, W. Zhou, S. Zhang, C. Meng, Y. Wu, Y. Wang and H. Zeng, CsPbBr3 quantum dots 2.0: benzenesulfonic acid equivalent ligand awakens complete purification, Adv. Mater., 2019, 31, 1900767 CrossRef PubMed.
- J. Pan, Y. Shang, J. Yin, M. De Bastiani, W. Peng, I. Dursun, L. Sinatra, A. M. El-Zohry, M. N. Hedhili, A. H. Emwas, O. F. Mohammed, Z. Ning and O. M. Bakr, Bidentate ligand-passivated CsPbI3 perovskite nanocrystals for stable near-unity photoluminescence quantum yield and efficient red light-emitting diodes, J. Am. Chem. Soc., 2018, 140, 562–565 CrossRef CAS PubMed.
- J. Chen, D. Jia, J. Qiu, R. Zhuang, Y. Hua and X. Zhang, Multidentate passivation crosslinking perovskite quantum dots for efficient solar cells, Nano Energy, 2022, 96, 107321 Search PubMed.
- Q. Zhuang, C. Zhang, C. Gong, H. Li, H. Li, Z. Zhang, H. Yang, J. Chen and Z. Zang, Tailoring multifunctional anion modifiers to modulate interfacial chemical interactions for efficient and stable perovskite solar cells, Nano Energy, 2022, 102, 107747 CrossRef CAS.
- G. Shao, S. Liu, L. Ding, Z. Zhang, W. Xiang and X. Liang, KxCs1−xPbBr3 NCs glasses possessing super optical properties and stability for white light emitting diodes, Chem. Eng. J., 2019, 375, 122031 CrossRef CAS.
- W. J. Mir, Y. Mahor, A. Lohar, M. Jagadeeswararao, S. Das, S. Mahamuni and A. Nag, Postsynthesis doping of Mn and Yb into CsPbX3 (X = Cl, Br, or I) perovskite nanocrystals for downconversion emission, Chem. Mater., 2018, 30, 8170–8178 CrossRef CAS.
- X. Tang, W. Chen, Z. Liu, J. Du, Z. Yao, Y. Huang, C. Chen, Z. Yang, T. Shi, W. Hu, Z. Zang, Y. Chen and Y. Leng, Ultrathin, core-shell structured SiO2 coated Mn2+-doped perovskite quantum dots for bright white light-emitting diodes, Small, 2019, 15, e1900484 CrossRef PubMed.
- Y. Bai, M. Hao, S. Ding, P. Chen and L. Wang, Surface chemistry engineering of perovskite quantum dots: strategies, applications, and perspectives, Adv. Mater., 2022, 34, e2105958 CrossRef PubMed.
- C. Gong, C. Zhang, Q. Zhuang, H. Li, H. Yang, J. Chen and Z. Zang, Stabilizing buried interface via synergistic effect of fluorine and sulfonyl functional groups toward efficient and stable perovskite solar cells, Nano-Micro Lett., 2022, 15, 17 CrossRef PubMed.
- C. Zhang, Q. Zhuang, H. Li, C. Gong, H. Wang, R. Li, H. Li, Z. Zhang, H. Yang, J. Chen and Z. Zang, Modulating chemical interaction to realize bottom–up defect passivation for efficient and stable perovskite solar cells, Sol. RRL, 2022, 6, 2200512 CrossRef CAS.
- J. Hao, X. Qu, L. Qiu, G. Li, Y. Wei, G. Xing, H. Wang, C. Yan, H. S. Jang, Z. Cheng and J. Lin, One–step loading on natural mineral halloysite nanotube: an effective way to enhance the stability of perovskite CsPbX3 (X = Cl, Br, I) quantum dots, Adv. Opt. Mater., 2018, 7, 1801323 CrossRef.
- N. Li, X. Chen, J. Wang, X. Liang, L. Ma, X. Jing, D. L. Chen and Z. Li, ZnSe nanorods-CsSnCl3 perovskite heterojunction composite for photocatalytic CO2 reduction, ACS Nano, 2022, 16, 3332–3340 CrossRef CAS PubMed.
- C. Fan, H. Liu, J. Zhou, X. Dai, H. He and Z. Ye, Ultrastable and highly efficient CsPbBr3 composites achieved by dual–matrix encapsulation for display devices, InfoMat, 2023, e12417 CrossRef.
- J. Liu, J. Zhong, Z. Chen, J. Mao, J. Liu, Z. Zhang, X. Li and S. Ren, Preparation, characterization, application and structure evolution of attapulgite: from nanorods to nanosheets, Appl. Surf. Sci., 2021, 565, 150398 CrossRef CAS.
- T. Zhang, W. Wang, Y. Zhao, H. Bai, T. Wen, S. Kang, G. Song, S. Song and S. Komarneni, Removal of heavy metals and dyes by clay-based adsorbents: From natural clays to 1D and 2D nano-composites, Chem. Eng. J., 2021, 420, 127574 CrossRef CAS.
- M. Fu, X. Li, R. Jiang and Z. Zhang, One-dimensional magnetic nanocomposites with attapulgites as templates: growth, formation mechanism and magnetic alignment, Appl. Surf. Sci., 2018, 441, 239–250 CrossRef CAS.
- X. Wang, B. Mu, X. An and A. Wang, Insights into the relationship between the color and photocatalytic property of attapulgite/CdS nanocomposites, Appl. Surf. Sci., 2018, 439, 202–212 CrossRef CAS.
- P. Wu, D. Deng, J. Gao and C. Cai, Tubelike gold sphere-attapulgite nanocomposites with a high photothermal conversion ability in the near-infrared region for enhanced cancer photothermal therapy, ACS Appl. Mater. Interfaces, 2016, 8, 10243–10252 CrossRef CAS PubMed.
- H. Xu, J. Wang, X. Yang and L. Ning, Magnetically recyclable graphene oxide demulsifier adapting wide pH conditions on detachment of oil in the crude oil-in-water emulsion, ACS Appl. Mater. Interfaces, 2021, 13, 6748–6757 CrossRef CAS PubMed.
- Y. Zhang, F. Gao, B. Wanjala, Z. Li, G. Cernigliaro and Z. Gu, High efficiency reductive degradation of a wide range of azo dyes by SiO2-Co core-shell nanoparticles, Appl. Catal., B, 2016, 199, 504–513 CrossRef CAS.
- A. Pan, M. J. Jurow, F. Qiu, J. Yang, B. Ren, J. J. Urban, L. He and Y. Liu, Nanorod suprastructures from a ternary graphene oxide-polymer-CsPbX3 perovskite nanocrystal composite that display high environmental stability, Nano Lett., 2017, 17, 6759–6765 CrossRef CAS.
- B. Wang, J. Peng, X. Yang, W. Cai, H. Xiao, S. Zhao, Q. Lin and Z. Zang, Template assembled large-size CsPbBr3 nanocomposite films toward flexible, stable, and high-performance X-ray scintillators, Laser Photonics Rev., 2022, 16, 2100736 CrossRef CAS.
- J. Wang, M. Zhang, Y. Liu, Y. a. Huang, Y. Zhang, J. Jiang, H. Li, J. Chen and Z. Lin, Ultrastable highly-emissive amphiphilic perovskite nanocrystal composites via the synergy of polymer-grafted silica nanoreactor and surface ligand engineering for white light-emitting diode, Nano Energy, 2022, 98, 107321 CrossRef CAS.
- M. J. Palimi, M. Rostami, M. Mahdavian and B. Ramezanzadeh, Surface modification of Fe2O3 nanoparticles with 3-aminopropyltrimethoxysilane (APTMS): An attempt to investigate surface treatment on surface chemistry and mechanical properties of polyurethane/Fe2O3 nanocomposites, Appl. Surf. Sci., 2014, 320, 60–72 CrossRef CAS.
- X. Hu, Y. Wu, Y. Wang, L. Xu, S. Zhang, J. Wang, K. Wu, Y. Liu, Y. Li and X. Li, Surface anchoring–induced robust luminescence thermal quenching suppression in shell–free perovskite nanocrystals, Adv. Opt. Mater., 2022, 10, 2201205 CrossRef CAS.
- D. Li and G. Chen, Near-infrared photoluminescence from Ytterbium- and Erbium-codoped CsPbCl3 perovskite quantum dots with negative thermal quenching, J. Phys. Chem. Lett., 2023, 14, 2837–2844 CrossRef CAS PubMed.
- H. Li, Y. Gao, X. Ying, Y. Feng, M. Zhu, D. Zhang, G. Lu, R. Tao, Q. Cai, H. He, X. Dai, Z. Ye and J. Huang, In situ reacted multiple-anchoring ligands to produce highly photo-thermal resistant CsPbI3 quantum dots for display backlights, Chem. Eng. J., 2023, 454, 140038 CrossRef CAS.
- P. Ma, Y. Hou, Z. Chen, J. Su, L. Li, N. Liu, Z. Zhang, X. Jiang, F. Long, Y. Ma and Y. Gao, Enhanced stability of CsPbBr3 quantum dots by anchoring on the hierarchical three-dimensional layered double hydroxide, Chem. Eng. J., 2021, 425, 130471 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3qi00863k |
| This journal is © the Partner Organisations 2023 |