
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGuest exchange in a biomimetic ZnII cavity-complex: kinetic control by a catalytic water, through pore selection, 2nd sphere assistance, and induced-fit processes†
N.
Nyssen
ab,
N.
Giraud
 a,
J.
Wouters
c,
I.
Jabin
b,
L.
Leherte
*c and
O.
Reinaud
*a
a,
J.
Wouters
c,
I.
Jabin
b,
L.
Leherte
*c and
O.
Reinaud
*a
aLaboratoire de Chimie et de Biochimie Pharmacologiques et Toxicologiques, CNRS UMR 8601 Université Paris Cité, 45 Rue des Saints Pères, 75006 Paris, France. E-mail: Olivia.Reinaud@parisdescartes.fr
bLaboratoire de Chimie Organique, Université Libre de Bruxelles (ULB), Avenue F. D. Roosevelt 50, CP160/06, B-1050 Brussels, Belgium
cLaboratory of Structural Biological Chemistry, Unit of Theoretical and Structural Physical Chemistry, Department of Chemistry, NAmur Research Institute for LIfe Sciences (NARILIS), Namur Institute of Structured Matter (NISM), NAmur MEdicine & Drug Innovation Center (NAMEDIC), University of Namur, Rue de Bruxelles 61, B-5000 Namur, Belgium
First published on 9th August 2023
Abstract
The kinetics of ligand exchange at a ZnII center confined in a biomimetic environment is studied. The metal ion is bound to a TMPA unit covalently capping the calix[6]arene core. As such, the system is rigidified in a cone conformation open to the solvent at the large rim. At the small rim, the methyl groups of the anisole units lead to steric crowding, thus defining a small pore leading to the metal center. The exchange of an organic guest ligand (MeCN) for another (EtCN) is studied by 1H NMR spectroscopy. It is shown that it is very slow under dry conditions (several hours at mM concentrations), but much faster in the presence of appreciable amounts of water. Kinetic analyses indicate that the exchange process involves the transient formation of an aqua complex that was independently synthesized and well characterized. Computer studies suggest that transient water coordination first occurs through the small rim pore, in exo-position. The water molecule then flips in endo-position, as the MeCN guest decoordinates and exits from the cavity through the large rim. This allows another organic guest ligand to get in, with release of the catalytic water to the solvent. Steered molecular dynamics simulations also show the assistance of the second coordination sphere of the metal ion for the exo/endo flipping process of the water ligand through H-bonding to the oxygenated small rim of the calixarene macrocycle. Importantly, it appeared that only water can play such a catalytic role, due to size selection by the pores defined by the calixarene small rim. From a biological point of view, it further substantiates the importance of water molecules and micro-environment on metal ion lability.
Introduction
ZnII coordination in biology is important, displaying structural (e.g. Zn fingers) or functional roles (Zn enzymes).1–3 In these biological structures, ZnII is coordinated to 3 or 4 amino-acid residues. In enzymes, an extra site is generally occupied by a water molecule that is the reactant (in hydrolases) or that is displaced by the reactant (e.g. a phosphate derivative as in alkaline phosphatases4 or an alcohol in alcohol dehydrogenase5). For all these enzymes, ligand exchange at the metal center is a key step in the catalysis. Ligand exchange is also a key factor that determines the stability of a structural site (inertness), or the Zn homeostasis. These exchange processes are difficult to study within the biological systems. Therefore, small model complexes are currently elaborated and studied to get specific insights into mechanisms at the molecular level.Poly-aza tripods are classically used to mimic the poly-histidine rich biological sites found in metallo-enzymes. One of them, namely TMPA (Tris(2-PyridylMethyl)Amine), has been long studied for Fe and Cu biomimetic complexes. Less has been reported with Zn. While few examples of TMPA Zn complexes exist in literature, the main area of focus has been the utilization of ZnII complexes in the cleavage of phosphodiester bonds. In those catalytic transformations, the kinetically active form was determined to be the corresponding Zn-aqua complex.6 The introduction of hydrogen bond donors has been shown to decrease the pKa of the bound water molecule and enhance the activity of such complexes, while also favoring the binding of phosphates ions.7,8 Indeed, it has long been recognized that the microenvironment around metal ions is a major factor that controls the reactivity/stability of Zn centers. One way to explore the supramolecular aspects relative to this microenvironment is to construct a well-defined and tunable microstructure around the metal ion labile site and evaluate the impact on its properties.9 Such a strategy allows to control the second coordination sphere of the metal ion, but also cavity effects related to ligand exchange. Being open to the solvent, the cavity acts not only as a receptor pocket, but also as a pore that will allow or not exogenous molecules to reach the metal center, thereby controlling its reactivity.
Ligands based on the calix[6]arene macrocycle connected to a tripodal nitrogenous core at the small rim are good mimics of the histidine-rich protein cavity.10,11 One of them, namely calix[6]TMPA (L, Scheme 1), presents a structure that is highly congested although open to the solvent. The calix[6]arene macrocycle is tris-methylated on alternate position of the phenol units, and covalently capped by a TMPA unit through short methylene linkers to the three other phenol units. As such, access to the metal ion bound in the TMPA cap through the small rim pores is highly restricted by steric congestion between the methoxy substituents and the cap methylene linkers.
 | ||
| Scheme 1 Guest exchange G/G′ equilibrium inside the cavity of [Zn(G)L]2+. | ||
In a recent study,12 we reported the synthesis of the corresponding ZnII acetonitrilo complex that is 5-coordinate with a guest acetonitrile molecule bound in the calixarene cavity. A thermodynamic study of ligand exchanges monitored by NMR spectroscopy showed that the macrocyclic structure is rigidified in a cone conformation. However, it keeps some flexibility as the three anisole “walls” can rotate along the calix methylene groups for a few degrees, as a function of the size of the included guest ligand. Along our studies, we noticed some surprisingly slow processes: first, coordination of the ZnII dication required heating for hours; second, the exchange of the MeCN guest for EtCN took several hours to reach equilibrium under some experimental conditions. These observations stand in strong contrast with the behavior of simple TMPA metal complexes, devoted of cavity, for which metalation and ligand exchange proceed almost instantaneously. Wanting to know more about these phenomena, we undertook a detailed study relative to the kinetics of exchange between two representative organic guest ligands (G and G′, Scheme 1), namely MeCN and EtCN, which is the subject of the present article.
Results
Characterization of the aqua complex
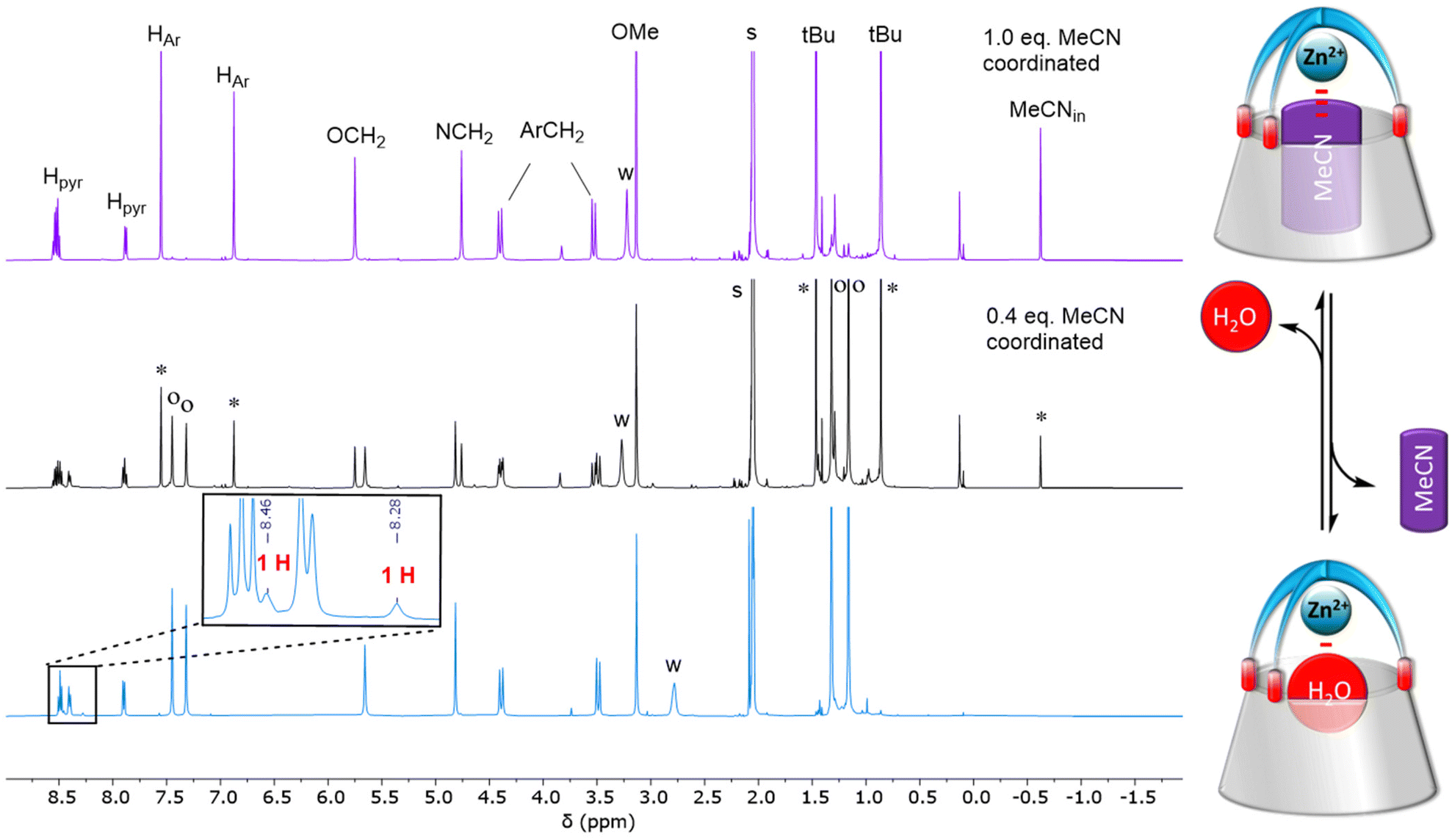
Complex [Zn(MeCN)L](ClO4)2 was synthesized as previously reported, by reacting the calixarene ligand L with a stoichiometric amount of the ZnII perchlorate salt in MeCN. Noticeably, the complexation process required heating the solution for several hours. Indeed, protonation of L occurs immediately in solution in a first step and complexation necessitates the displacement of the proton by the metal ion in a second slow, but thermodynamically favored, step.12 The dicationic complex was isolated as the acetonitrile complex and characterized by 1H NMR spectroscopy. The overall signature attested to a C3v symmetry with the ZnII center bound to the TMPA cap and one molecule of acetonitrile sitting inside the calixarene cavity as attested by its high field δ shift (−0.62 ppm). We noticed however that, when dissolved in wet acetone, a second species was present. The latter did not correspond to the protonated ligand and vanished upon addition of small amounts of MeCN. Suspecting that it could correspond to the aqua complex, we repeated the complexation process with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of ligand L and ZnII salt in a non(/poorly)-coordinating solvent, namely acetone. The complexation also required heating the solution for several hours. The isolated complex was characterized by various spectroscopies. The IR spectrum confirmed the presence of two perchlorates as counterions (see the ESI†). The 1H NMR spectrum (Fig. 1, bottom-blue) is typical of a C3v symmetrical structure with a cone conformation for the calix[6]arene macrocycle. As for the nitrile complex, the pyridyl protons are low field shifted compared to the free ligand, thus attesting to the coordination of the Lewis acidic metal ion ZnII. The two sets of aromatic HAr and tBu protons display closer δ shifts for the aqua complex (Δδ ∼ 0.2 ppm) than for the MeCN complex (∼0.7 ppm). This suggests an average straighter conformation for the calixarene host, consistently with the smaller size of the guest water molecule. The coordinated water molecule could be identified under relatively dry conditions, through two signals at 8.28 and 8.46 ppm, each one integrating for one proton. These two signals vanished upon D2O addition. Saturation transfer experiments with free water and NOESY measurements further confirmed their attribution (see the ESI†). These low-field shifted δ values indicate that the water molecule is bound to the Lewis acidic ZnII center and sits at the level of the small rim of the calixarene structure, away from the anisotropic cone of the aromatic walls. It is noteworthy that each proton of the coordinated water molecule is differentiated from the other. This reflexes strong electrostatic interaction with the oxygen atoms belonging to the calixarene small rim that slows down the rotation of the water molecule inside the structure. The differentiation of the two protons can be due to a stronger interaction of one of the water protons with a calixarene oxygen atom. Addition of an aliquot of MeCN to this aqua complex led to the appearance of a high-field peak at −0.62 ppm corresponding to the MeCN ligand included in the cavity (Fig. 1, top-purple), thus attesting to the formation of the MeCN complex. The exchange was slow at the 1H NMR shift time scale, but fast at the experimental time scale. A careful titration of the aqua complex by acetonitrile (see the ESI†) confirmed that one water molecule is coordinated to the ZnII ion, and no extra water guest is present, either in the cavity or in exo-position.
:1 mixture of ligand L and ZnII salt in a non(/poorly)-coordinating solvent, namely acetone. The complexation also required heating the solution for several hours. The isolated complex was characterized by various spectroscopies. The IR spectrum confirmed the presence of two perchlorates as counterions (see the ESI†). The 1H NMR spectrum (Fig. 1, bottom-blue) is typical of a C3v symmetrical structure with a cone conformation for the calix[6]arene macrocycle. As for the nitrile complex, the pyridyl protons are low field shifted compared to the free ligand, thus attesting to the coordination of the Lewis acidic metal ion ZnII. The two sets of aromatic HAr and tBu protons display closer δ shifts for the aqua complex (Δδ ∼ 0.2 ppm) than for the MeCN complex (∼0.7 ppm). This suggests an average straighter conformation for the calixarene host, consistently with the smaller size of the guest water molecule. The coordinated water molecule could be identified under relatively dry conditions, through two signals at 8.28 and 8.46 ppm, each one integrating for one proton. These two signals vanished upon D2O addition. Saturation transfer experiments with free water and NOESY measurements further confirmed their attribution (see the ESI†). These low-field shifted δ values indicate that the water molecule is bound to the Lewis acidic ZnII center and sits at the level of the small rim of the calixarene structure, away from the anisotropic cone of the aromatic walls. It is noteworthy that each proton of the coordinated water molecule is differentiated from the other. This reflexes strong electrostatic interaction with the oxygen atoms belonging to the calixarene small rim that slows down the rotation of the water molecule inside the structure. The differentiation of the two protons can be due to a stronger interaction of one of the water protons with a calixarene oxygen atom. Addition of an aliquot of MeCN to this aqua complex led to the appearance of a high-field peak at −0.62 ppm corresponding to the MeCN ligand included in the cavity (Fig. 1, top-purple), thus attesting to the formation of the MeCN complex. The exchange was slow at the 1H NMR shift time scale, but fast at the experimental time scale. A careful titration of the aqua complex by acetonitrile (see the ESI†) confirmed that one water molecule is coordinated to the ZnII ion, and no extra water guest is present, either in the cavity or in exo-position.
 | ||
| Fig. 1
1H NMR spectra (300 K, 500 MHz) of [Zn(H2O)L](ClO4)2 dissolved in acetone-d6 before (bottom-blue) and after the addition of MeCN (ca. 0.5 and 1 equiv. middle-black and top-purple, respectively). Inset: Zoom on the low-field region to show the peaks associated to the water ligand. The spectra where a nitrile was added were recorded immediately after the addition. o: aqua complex, *MeCN complex. | ||
Kinetic study of the acetonitrile/propionitrile exchange
The exchange between two nitriles was followed by 1H NMR spectroscopy by recording multiple spectra over the course of several hours of a solution of one nitrile complex in presence of a known amount of the other nitrile in deuterated acetone. A representative example is shown in Fig. 2. The amount of each complex was calculated by integrating the two signatures in slow exchange on the NMR shift time scale (ESI†). However, we noticed that the rate of exchange varied not only with the amount of MeCN or EtCN, but also with the amount of water: under “dry” conditions, it was slow, and under “wet” conditions it was much faster. In light of the ability of water to be complexed to the metal center, we hypothesized that the exchange mechanism involves its transient coordination (pre-equilibrium displayed in Scheme 2). Hence, starting from the MeCN complex dissolved in acetone-d6, we followed by 1H NMR spectroscopy, as a function of time, the exchange process of the MeCN guest for EtCN, varying three factors: [MeCN], [EtCN], and [H2O]. Table 1 summarizes the conditions of 5 different experiments A–E (the detailed quantifications are reported in the ESI†). Qualitatively, these results show that the rate of exchange increases when the concentration of MeCN decreases (experiments A–C) and when the concentration of EtCN and water increases (experiments C–E). | ||
| Fig. 2 1H NMR spectra (300 K, 500 MHz, acetone-d6) recorded during the titration of G = MeCN by G′ = EtCN. Initial conditions in (a) [Zn(MeCN)L]2+ = 3.7 mM, [free MeCN] = 92 mM, [EtCN] = 0 mM, [H2O] = 20 mM. Percentage of EtCN complex in solution: (a) 0%, (b) 17%, (c) 38%, (d) 41%, (e) 59%, (f) 71%. This experiment is reported as entry A in Table 1. Full conversion corresponds to the amount of EtCN complex formed at equilibrium under these experimental (71%). The “t (50% conversion)” is then the time at which we have 35.5% of EtCN complex in solution. *: free EtCN. | ||
 | ||
| Scheme 2 Proposed pathway for the guest exchange involving the transient formation of the aqua complex. | ||
| Experiment # | [Zn(MeCN)L]2+ (mM) | [EtCN] (mM) | [MeCN] (mM) | [Water] (mM) | t (50% conversion) (min) |
|---|---|---|---|---|---|
| a 86% conversion was already achieved at the first measurement after 5 min. b The first measurement after 5 min already showed full conversion. Full conversion refers to the quantity of [Zn(EtCN)L]2+ at equilibrium. | |||||
| A | 3.7 | 10 | 96 | 20 | 23 |
| B | 2.0 | 10 | 36 | 16 | 19 |
| C | 3.1 | 9 | 15 | 15 | 7 |
| D | 2.2 | 66 | 14 | 18 | <5a |
| E | 2.8 | 70 | 14 | 129 | ≪5b |
Consistently, the more the pre-equilibrium is displaced to the right, towards the formation of the aqua complex, the faster the formation of the EtCN complex (Scheme 2). The rate of exchange of the acetonitrile guest for the propionitrile guest was expressed in eqn (1) according to the hypothesis made in Scheme 2. Importantly, in none of the experiments reported in Table 1, the aqua complex was detected in the 1H NMR spectra. This is obviously related to the large relative affinities of nitrile guests that occupies the calixarene cavity compared to water that is too small and lies above. Indeed, from titration experiments, we found that KMeCN/H2O = 200 ± 12 (see the ESI†), and knowing that KEtCN/MeCN = 30 ± 1,12 we can deduce that KEtCN/H2O = 6000 ± 540. The kinetic data measured in experiments A, B, C (Table 1 and Fig. 3) were then analyzed according to eqn (1)–(4) obtained by applying the steady state approximation for aqua intermediate concentration:
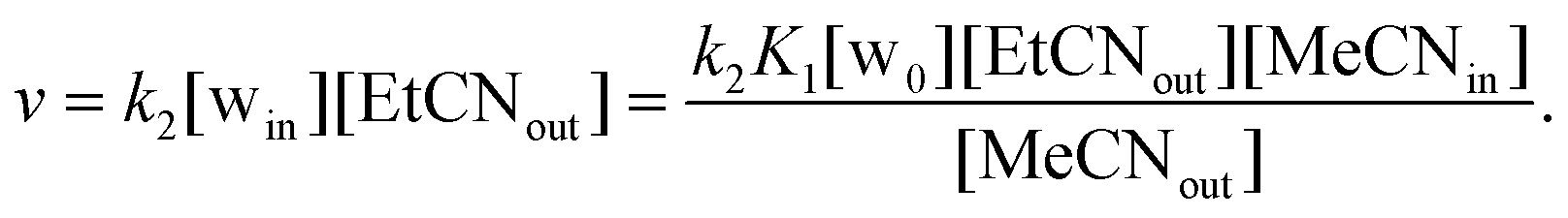 | (1) |
 | ||
| Fig. 3 Kinetic fit of the 1H NMR analyses. For the expression of F(x), see (eqn (4)). A, B, C refers to experiments of Table 1. | ||
Setting that x = [MeCNin] and c = [total calixarene], we obtain:
 | (2) |
 | (3) |
 | (4) |
Fig. 3 shows full agreement with the working hypothesis as the data recorded for the 3 different experiments show a linear relationship in agreement with eqn (4). The exchange kinetic constant is thus: k2 = 0.090 ± 0.004 mM−2 min−1. From this value, we can deduce that k−2 = 0.015 ± 0.002 × 10−3 mM−2 min−1.
NOESY measurement
As the MeCN/H2O exchange process is much faster than that of MeCN/EtCN, we have chosen to monitor it by NMR using EXchange SpectroscopY (EXSY). For this purpose, a solution containing a mixture of the MeCN and aqua complexes was prepared, and we have recorded a 1H–1H 2D NMR NOESY spectrum on this sample. On the resulting data, we clearly observe several sets of correlations for the complex, revealing a slow exchange on the NMR shifts time scale between the MeCN and the aqua form. Fig. 4 shows the region of the resulting 2D spectrum where this correlation pattern is observed. We have thus recorded a series of EXSY spectra, varying the mixing time to monitor this slow exchange process (a representative 2D map acquired at 600 MHz is described in the ESI†). The volume of the resolved correlation patterns was measured for each mixing time value. The resulting build-up curves that were obtained for the selected diagonal and cross peaks arising from the exchange process between the aqua and MeCN complexes can be modelled using modified Bloch equations accounting for proton longitudinal relaxation and the formal equilibrium:
 | ||
| Fig. 4 Selected region of a 2D 1H–1H NOESY spectrum (500 MHz) of a solution containing a mixture of the aqua and MeCN Zn complex in acetone-d6. This spectrum was recorded with a mixing time of 1.5 s. The sample was prepared with 0.9 eq. of MeCN and 200 eq. of H2O. Correlations reflecting the exchange process between MeCN and H2O in the cavity of the Zn complex are highlighted with a dotted circle. Other regions of interest of this 2D map are shown together with the 1D 1H NMR spectrum in ESI.† | ||
The analysis of the NOESY correlations involving (i) an aromatic proton from the calixarene ligand, and (ii) the methyl protons from MeCN, both undergoing a chemical exchange between the aqua and MeCN complexes, yielded k1 values from 2.2 to 11 min−1 mM−2 and k−1 = 0.02 ± 0.02 min−1 mM−2. This result yields a calculated value for KMeCN/H2O = k1/k−1 = 110 to 500, in good agreement with the value of 200 measured by titration (see above and Scheme 2). These kinetic data, when compared to those measured for the EtCN/H2O exchange (k2 = 0.090 mM−2 min−1 and k−2 = 0.015 × 10−3 mM−2 min−1), indicate that both, entrance and exit of the nitrilo guest are three orders of magnitude faster for MeCN than for EtCN. Hence, the rates are obviously related to the size of the guest. This corresponds to the energy cost to open the small rim door constituted by the 3 tBu substituents of the most rigid aromatic units, namely those connected to the TMPA cap. Such a kinetic differentiation is reminiscent of a previous study reported for a CuI complex obtained with a calix[6]arene functionalized by three α-methylpyridyl groups. Indeed, it was shown that part of the activation energy for the exit of the acetonitrile guest stems from the tBu door into which the guest “bump” during the dissociative process.13
In order to clarify the role of the water molecule and the reason for which the aqua-complex is involved as a necessary intermediate for the exchange between two nitrile guests, we have carried out molecular computing studies. The idea was to start with the acetonitrile complex and identify the pathway through which the water molecule becomes in interaction with the ZnII center, whereas the MeCN guest leaves the cavity.
Steered molecular dynamics simulation of the guest-water exchange
To favor the guest-water exchange in the calixarene structure, Molecular Dynamics (MD) and Steered MD (SMD) simulations of a calixarene–MeCN complex interacting with a water molecule were carried out in vacuum at 300 K using the program GROMACS2020,14 as detailed in the ESI.† Water and the small molecules were modeled using the TIP3P and CHARMM27 force fields, respectively. The simulations were carried out with four atom position restraints placed on the N atoms of the calixarene cap, while restraining the four Zn–Ncap distances to avoid exaggerated displacement of the Zn2+ ion during the approach and the insertion of water inside the calixarene cavity. The system was first optimized using a steepest descent algorithm (Fig. 5, structure A). Then, the MD total equilibration stage duration was set to 2100 ps. At the end of this stage, the water molecule more closely interacts with ZnII (Fig. 5, structure B). Afterwards, the SMD stage was carried out for a period of 60 ps. A pulling force was applied to the center of mass of MeCN to progressively evacuate MeCN from the calixarene cavity. As illustrated in the ESI,† the time-dependence of the water interaction energy with its neighborhood allows to model the substitution of MeCN as a stepwise process. | ||
| Fig. 5 Stepwise substitution of MeCN guest by a water molecule following an associative pathway, as obtained from the 2160 ps MD and SMD trajectories in vacuum at 300 K. Initial optimized structure – A, snapshot at 1701 ps of the MD trajectory (coordination of H2O – B), at t = 29.8 of the SMD trajectory (insertion stage – I1), 29.9 ps (insertion stage – I2), 30.0 ps (insertion stage – I3), and 60.0 ps (final structure – I4). Semi-empirical MP7 optimized structure (C). Dotted lines refer to selected interatomic distances (in Å). Green dotted lines depict hydrogen bonds. The folded tBu moiety is displayed using ball and stick representations in I4 and C. Inset from top to bottom (obtained from the 60 ps long SMD trajectory): variation of the NMeCN–ZnII–Owater angle and Hwater⋯OMe distances; tBu–calixarene short-range interaction energy profile. | ||
To support the observation, the 60 ps long SMD simulation was continued by a 4000 ps conventional MD run using initial random atom velocities. Unfolding–refolding events were observed with exchange between the tBu moieties until t = 1576.3 ps (see the ESI†). Then, tBu exchange stops and the last folded conformation remains present until the end of the simulation. In this structure, a strong H-bond connects regularly one of the hydrogen atoms of the water guest to an anisole unit during 4% of the time, a percentage equally shared by both H atoms of the water molecule.
The computer simulations were also conducted with acetone as a solvent instead of vacuum and similar results were obtained (see the ESI†). The water molecule first gets close to the calixarene structure and deforms it while it coordinates ZnII. In the solvated case however, H2O is first hydrogen bonded to two acetone molecules. Then, similarly to the vacuum case, it forms a hydrogen bond with a methoxy group of the calixarene. The simulations were also performed with EtCN as an initial guest instead of MeCN, and same results were obtained for the substitution process. Finally, when the water molecule initially out of the cavity is replaced by MeCN, close interaction with the Zn center in exo-position was observed but at a distance that is much too long to correspond to a coordination bond. Consistently, the interaction energy of the exo-MeCN molecule is not significantly reduced compared to other positions. It remains very mobile and, after a while, tends to be trapped at the level of the cap of the calixarene. Similar results were obtained with MeOH replacing H2O (see the ESI†). Coordination of the MeCN and MeOH molecules by ZnII are then triggered by removing the endo-ligand MeCN during the whole MD simulations. It likely favors the strong deformation of the calixarene structure, but it does not initiate the insertion of the molecule that stays outside the calixarene cavity. Nevertheless, the insertion of MeOH can be artificially produced by the application of a steering force to pull the ligand inside the calixarene through the small rim. Such a SMD simulation was carried out using the same methodology as used for pulling the endo-ligand outside the cavity. Interestingly, MeCN does not enter the calixarene using that approach. Hence, water is the only ligand to spontaneously enter the calixarene cavity through the small rim: it is small enough to flip from the exo-position to the endo-position through the small pore existing at the calixarene small rim, whereas MeCN and MeOH appear to be either too large or not energetically stabilized at the level of the small pore. This suggests that the only (or most probable) pathway for an organic ligand to coordinate the metal center in endo-position, inside the cavity, is to go through the large rim of the calix-funnel. This inclusion process is, likely for a steric reason, impossible when the cavity is already occupied by an organic guest, whereas a water guest can get in from the small rim.
In summary, the simulations show that the preliminary step for a ligand to enter the cavity through the calixarene small rim is to be immobilized strongly enough, through a coordination bond to the metal center to the calixarene structure. Without such binding, the ligand remains free to move around the calixarene structure. Then, if the exo-ligand is small enough, it can flip into the cavity when the guest ligand leaves, thereby avoiding the formation of an “empty cavity” intermediate during guest exchange. Hence, water seems very unique as it can bind to ZnII in exo-position and flip into the cavity in endo-position, thus allowing the guest–Zn bond cleavage and the exit of the guest, and conversely. Quite interestingly, and subtly, the key flipping process involves transient assistance of an oxygen atom present at the small rim of the calixarene acting as H-bond acceptor in the second coordination sphere of the metal ion.
Discussion
It may appear surprising that a simple dissociative mechanism for guest-exchange is not at work in this system. Indeed, direct displacement of the guest would leave a 4-coordinate intermediate, which should not be so unstable. One explanation could rely on the rigidity of the system. Indeed, the capping TMPA unit of ligand L constrains the calixarene macrocycle in a cone conformation with aromatic units lying alternatively in in/out position relative to the cavity. This alternate cone conformation is the opposite to the one adopted by the more flexible ligands: three imidazole arms17 or a TREN (tris(2-aminoethyl)amine) unit.18 The relatively straight conformation adopted by calixarene L is explained by the fact that the aromatic units connected to the cap are obliged to sit in a position that projects their oxygen atom toward the outside (Fig. 6). Consequently, the three other aromatic units, bearing methoxy groups, are constrained to orient their oxygen atom toward the inside. This creates a high steric hindrance that defines the pore for exo-binding and allows 2nd sphere assistance for water exo-binding and endo-flipping. Importantly also, the rigidified calix core of ZnIIL restricts the induced-fit process observed with the other systems that can adopt a flattened conformation, thus shrinking the cavity space in the absence of guest. As a result, the energy cost for leaving a fully empty cavity is probably much higher with L; it explains the fact that the water ligand flips into the cavity whereas the guest leaves. The resulting new 5-coordinate intermediate then undergoes partial insertion of one of its tBu substituent present at the large rim in order to optimize the cavity filling. This leads to a fully stable aqua complex that could be independently synthetized and fully characterized.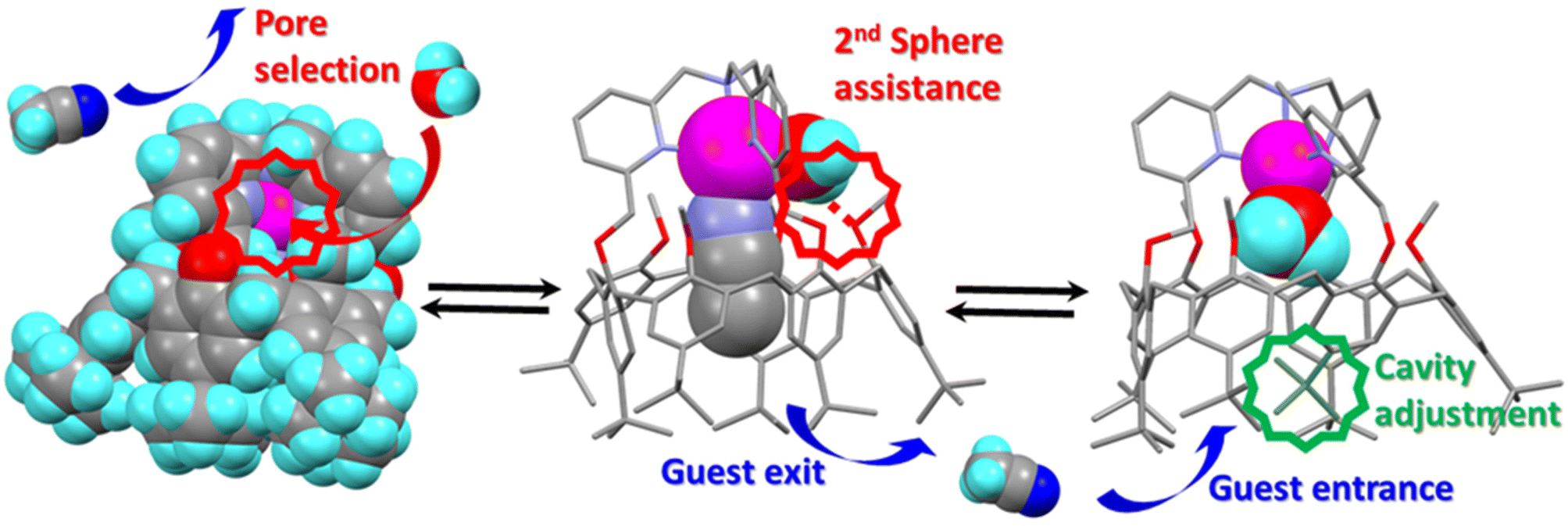 | ||
| Fig. 6 Key steps for the guest exchange catalyzed by a water molecule evidencing: (left) pore-size selection for the exo-access to the metal center; (center) H-bonding assistance for the water ligand flipping from an exo to an endo position; (right) tBu partial insertion for filling the cavity space. | ||
Finally, it is worth noting that the tBu group that is partially included in the cavity space of the aqua complex constitutes a gate that needs to be open for guest leaving and guest entering. These in/out phenomena may explain the impressive difference of rate exchange for the MeCN and EtCN guests in both directions. The corresponding difference in activation energies (ca. 3 orders of magnitude ∼ 17 kJ mol−1) may correspond to the interaction energy difference between the tBu gate and the ethyl vs. methyl group of the guest during the leaving and entering processes. Interestingly, such an inertness under dry conditions was previously observed19 with the Cu complexes of the very same ligand L and it was shown that water guests regulate its redox activity.20 In the present study, we evidence the role of water for ligand exchange, a role that was not elucidated before.
Conclusion
In conclusion, we have described a very special system where the ZnII cation is embedded in a nitrogenous cap connected to a cavity open to the solvent, for which guest ligand exchange is under the control of a single water molecule. The exchange process involves a first associative step where a water molecule binds the Zn center in exo-position, thus leading to an octahedral intermediate. The second step is the intramolecular substitution of the endo-bound guest (G) by the exo-bound water to give rise to a 5-coordinate aqua Zn complex with concomitant expulsion of the initial guest (G). In virtue of the micro-reversibility principle, the substitution of the endo-bound aqua ligand by a new organic guest (G′) follows the same pathway, with entrance of the new guest (G′) at the large rim, and expulsion of the water molecule by the side, at the small rim. Hence, the water molecule acts as a catalyst and is very unique in its catalytic role: it first binds to the metal center in an exo-position, and flips into the pore at the calixarene small rim with the assistance of an oxygen atom of the host structure acting as H-bond acceptor. Other studied molecules are too large or do not, under the selected calculation conditions, spontaneously initiate the insertion step through coordination to ZnII, which is required before insertion under the current calculation conditions. As a spectacular result, G exchanges for G′ is fast only in the presence of water, and water plays the role of a very unique catalyst that controls the kinetics of guest exchange at the metal center thanks to a pore selection and 2nd coordination sphere assistance.All in all, this study represents the second case ever reported in the literature of well-characterized molecular metal complex for which ligand exchange is under the unique control of a single water molecule.17 Here, the control relies on pore selection, 2nd sphere assistance through transient H-bonding, and host–guest induced fit. From a biological point of view, it further substantiates the importance of water molecules on metal ion lability, a property that is key for the stability of structural Zn sites in proteins, for Zn-enzyme activity, as well as for homeostasis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
NN thanks the “Ministère de l'Enseignement Supérieur et de la recherche” and “Université Paris Cité” and the CCCI funding from “Université libre de Bruxelles”. LL and JW used resources of the ‘Plateforme Technologique de Calcul Intensif (PTCI)’ (https://www.ptci.unamur.be) located at the University of Namur, Belgium, which is supported by the FNRS-FRFC under the conventions no. 2.5020.11. The PTCI is member of the ‘Consortium des Équipements de Calcul Intensif (CÉCI)’ (https://www.ceci-hpc.be), funded by the ‘Fonds de la Recherche Scientifique de Belgique (F.R.S.-FNRS)’.Notes and references
- W. N. Lipscomb and N. Sträter, Recent Advances in Zinc Enzymology, Chem. Rev., 1996, 96, 2375–2434 CrossRef CAS PubMed.
- W. Maret and Y. Li, Coordination Dynamics of Zinc in Proteins, Chem. Rev., 2009, 109, 4682–4707 CrossRef CAS PubMed.
- A. Krężel and W. Maret, The biological inorganic chemistry of zinc ions, Arch. Biochem. Biophys., 2016, 611, 3–19 CrossRef PubMed.
- B. Stec, K. M. Holtz and E. R. Kantrowitz, A revised mechanism for the alkaline phosphatase reaction involving three metal ions, J. Mol. Biol., 2000, 299, 1303–1311 CrossRef CAS PubMed.
- B. V. Plapp, B. R. Savarimuthu, D. J. Ferraro, J. K. Rubach, E. N. Brown and S. Ramaswamy, Horse Liver Alcohol Dehydrogenase: Zinc Coordination and Catalysis, Biochemistry, 2017, 56, 3632–3646 CrossRef CAS PubMed.
- G. Feng, J. C. Mareque-Rivas, R. T. M. de Rosales and N. H. Williams, A Highly Reactive Mononuclear Zn(II) Complex for Phosphodiester Cleavage, J. Am. Chem. Soc., 2005, 127, 13470–13471 CrossRef CAS PubMed.
- J. C. Mareque-Rivas, R. Prabaharan and R. T. M. de Rosales, Relative importance of hydrogen bonding and coordinating groups in modulating the zinc–water acidity, Chem. Commun., 2004, 76–77 RSC.
- J. C. Mareque-Rivas, R. T. M. de Rosales and S. Parsons, The affinity of phosphates to zinc(II) complexes can be increased with hydrogen bond donors, Chem. Commun., 2004, 610–611 RSC.
- R. Gramage-Doria, D. Armspach and D. Matt, Metallated cavitands (calixarenes, resorcinarenes, cyclodextrins) with internal coordination sites, Coord. Chem. Rev., 2013, 257, 776–816 CrossRef CAS.
- J.-N. Rebilly, B. Colasson, O. Bistri, D. Over and O. Reinaud, Biomimetic cavity-based metal complexes, Chem. Soc. Rev., 2015, 44, 467–489 RSC.
- D. Coquière, S. Le Gac, U. Darbost, O. Sénèque, I. Jabin and O. Reinaud, Biomimetic and self-assembled calix[6]arene-based receptors for neutral molecules, Org. Biomol. Chem., 2009, 7, 2485–2500 RSC.
- P. Aoun, N. Nyssen, S. Richard, F. Zhurkin, I. Jabin, B. Colasson and O. Reinaud, Selective Metal-ion Complexation of a Biomimetic Calix[6]arene Funnel Cavity Functionalized with Phenol or Quinone, Chem. – Eur. J., 2023, 29, e202202934 CrossRef CAS PubMed.
- Y. Rondelez, A. Duprat and O. Reinaud, Calix[6]arene-Based Cuprous “Funnel Complexes”: A Mimic for the Substrate Access Channel to Metalloenzyme Active Sites, J. Am. Chem. Soc., 2002, 124, 1334–1340 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- J. J. P. Stewart, Stewart Computational Chemistry, Colorado Springs, CO, USA, 2016, https://OpenMOPAC.net, last accessed 20 Dec. 2022 Search PubMed.
- Y. Rondelez, O. Sénèque, M.-N. Rager, A. F. Duprat and O. Reinaud, Biomimetic Copper(I)–CO Complexes: A Structural and Dynamic Study of a Calix[6]arene-Based Supramolecular System, Chem. - Eur. J., 2000, 6, 4218–4226 CrossRef CAS PubMed.
- E. Brunetti, L. Marcelis, F. E. Zhurkin, M. Luhmer, I. Jabin, O. Reinaud and K. Bartik, A Water Molecule Triggers Guest Exchange at a Mono-Zinc Centre Confined in a Biomimetic Calixarene Pocket: a Model for Understanding Ligand Stability in Zn Proteins, Chem. – Eur. J., 2021, 27, 13730–13738 CrossRef CAS PubMed.
- U. Darbost, X. Zeng, M.-N. Rager, M. Giorgi, I. Jabin and O. Reinaud, X-ray and Solution Structures of the First Zn Funnel Complex Based on a Calix[6]aza-cryptand, Eur. J. Inorg. Chem., 2004, 4371–4374 CrossRef CAS.
- N. Le Poul, B. Douziech, J. Zeitouny, G. Thiabaud, H. Colas, F. Conan, N. Cosquer, I. Jabin, C. Lagrost, P. Hapiot, O. Reinaud and Y. Le Mest, Mimicking the Protein Access Channel to a Metal Center: Effect of a Funnel Complex on Dissociative versus Associative Copper Redox Chemistry, J. Am. Chem. Soc., 2009, 131, 17800–17807 CrossRef PubMed.
- N. Le Poul, B. Colasson, G. Thiabaud, D. J. Dit Fouque, C. Iacobucci, A. Memboeuf, B. Douziech, J. Řezáč, T. Prangé, A. de la Lande, O. Reinaud and Y. Le Mest, Gating the electron transfer at a monocopper centre through the supramolecular coordination of water molecules within a protein chamber mimic, Chem. Sci., 2018, 9, 8282–8290 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and procedure and characterization of [Zn(H2O)L](ClO4)2, thermodynamic and kinetic measurements of guest exchanges through NMR spectroscopy, computational details. See DOI: https://doi.org/10.1039/d3qi01271a |
| This journal is © the Partner Organisations 2023 |