
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnravelling the role of triisopropylphosphane telluride in Ag(I) complexes†
Juan Carlos
Pérez-Sánchez
,
Carmen
Ceamanos
,
Raquel P.
Herrera
 and
M. Concepción
Gimeno
*
and
M. Concepción
Gimeno
*
Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC-Universidad de Zaragoza, Pedro Cerbuna 12, 50009 Zaragoza, Spain. E-mail: gimeno@unizar.es
First published on 29th August 2023
Abstract
The coordination chemistry of chalcogenide ligands has always attracted significant interest in the field of inorganic chemistry, especially for soft metals such as those of group 11. Despite the scarcity of research on phosphane tellurides, we report on the synthesis and characterisation of five novel silver complexes containing the phosphane telluride ligand, TeP(iPr)3, along with other ancillary ligands such as mono or diphosphanes. Spectroscopic studies were performed to investigate the behaviour of these complexes, including their redox properties, as demonstrated by the 1,1′-diphenylphosphaneferrocene (dppf) silver derivatives. Additionally, these complexes showcase remarkable rapid interchange equilibrium, revealing silver species with distinctive Ag2Te2 cores and a combination of bridging and terminal TeP(iPr)3 ligands. A promising avenue for further investigation and potential applications emerges.
Introduction
Compounds containing chalcogen–phosphorus bonds, specifically those of the P–E and P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E types, where E is O, S, Se, or Te, have garnered significant attention in both organic and inorganic chemistry due to their diverse synthetic versatility and unique chemical properties.1 Organotellurium ligands, in particular, possess unique chemical properties that make them valuable building blocks for a wide range of applications, including catalysis,2 materials science,3 and electronics.4
E types, where E is O, S, Se, or Te, have garnered significant attention in both organic and inorganic chemistry due to their diverse synthetic versatility and unique chemical properties.1 Organotellurium ligands, in particular, possess unique chemical properties that make them valuable building blocks for a wide range of applications, including catalysis,2 materials science,3 and electronics.4
Among these compounds, Lawesson's reagent5 and its selenide analogue Woollins’ reagent6 are well-known examples of chalcogen-based compounds that have proven their effectiveness in transforming carbonyl groups into thio- and selenocarbonyl moieties, respectively (Fig. 1).
 | ||
| Fig. 1 Structure of Lawesson's and Woollins’ reagents. | ||
However, despite these well-known examples, phosphane tellurides have received relatively little attention in comparison.1
TePR3 compounds (where R = alkyl or aryl) are known for possessing a P–Te bond with lower bond energy compared to their chalcogen congeners.7 This lower bond energy results in these phosphanes being thermally and photochemically unstable species. In recent years, phosphane tellurides have garnered interest for their unique properties, such as their tendency to decompose in the presence of Lewis acids8 or their kinetic lability.9 These features have led to their application as Te0 carriers, enabling the formation of multiple-sized clusters10 or semiconductor materials.11 Consequently, they have emerged as a promising source of elemental telluride in various electronic applications, particularly in the production of a wide range of semiconducting metal telluride nanomaterials, ranging from nanotubes to dendritic structures.12
The versatility of phosphane tellurides is evident in their applications, highlighting Te as an elemental low-dimensional van der Waals material with exotic physical properties and significant potential in electronic devices and spintronics.13
In the context of coordination chemistry, phosphane tellurides have demonstrated soft-base behaviour, making them attractive ligands for soft metal ions due to the dipolar nature of the P+–Te− bond.14
Due to the aforementioned reasons, it is anticipated that phosphane tellurides would be superior ligands for soft metal ions in comparison to the corresponding phosphane oxide, sulphide, or selenide. This feature makes them particularly appealing for use in combination with coinage metals. In this topic, our group has described a wide range of oxide, sulphide and selenophosphane complexes.15 Notably, as far as we are aware, only a few examples of group 11 metal complexes featuring a M–TePR3 (M = Cu, Ag, or Au; R = Ph or iPr) bond have been reported (Fig. 2).16
 | ||
| Fig. 2 Some of the phosphane telluride group 11 metal complexes previously described. | ||
Therefore, we aim to report the synthesis of new silver complexes bound to a phosphane telluride, starting from some ubiquitous silver phosphane-complexes. By exploring the potential of these new complexes, we hope to contribute to the growing body of knowledge on the chemistry of telluride–phosphorus bonds and their potential applications in various fields.
Results and discussion
Synthesis and characterisation
Coordination chemistry between phosphane tellurides and silver has not been studied further since the pioneering work reported by W.-W. du Mont and co-workers.16b In this way, we have tested the reactions of TeP(iPr)3 against Ag(I) derivatives bearing mono or diphosphanes as ancillary ligands, such as [Ag(OTf)(PPh3)], AgOTf and [Ag(OTf)(dppf)] (Scheme 1).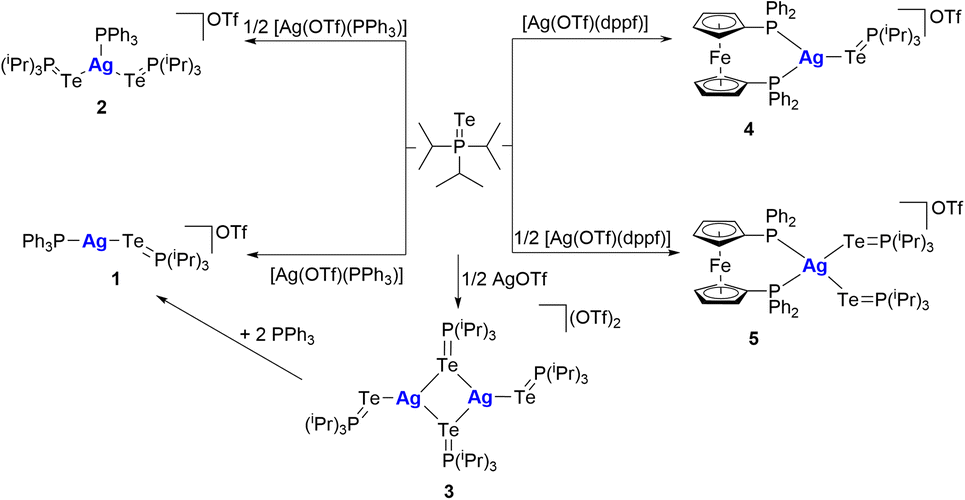 | ||
| Scheme 1 Synthesis of silver complexes 1–5. | ||
The reaction of [Ag(OTf)(PPh3)] with TeP(iPr)3 in stoichiometries of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2 yields complexes 1 and 2, respectively, as pale-yellow solids that are stable in air (Scheme 1). However, in solution, they rapidly decompose by loss of tellurium, resulting in the formation of a tellurium mirror that can be visually observed within one hour. Despite this decomposition, the complexes can be detected and characterised by NMR spectroscopy before degradation occurs. The 1H NMR spectra of both complexes show the expected resonances associated with phenyl protons, a multiplet that corresponds to the CH group of triisopropylphosphane, and a doublet of doublets assigned to the protons of the CH3 group of the phosphane. The integrals of each signal are consistent with the stoichiometry proposed for the complex formation. The 31P{1H} NMR spectra of the complexes show two resonances (Fig. S2†). For complex 1, the signal associated with TeP(iPr)3 appears at δP = 48.3 ppm and exhibits satellites due to the 1J(31P, 125Te) coupling, which presents a value around 15% lower than that of free TeP(iPr)3 (1J(31P, 125Te) = 1669.8 Hz, CD2Cl2, 298 K).17 Another signal appears at δP = 9.9 ppm with the same intensity as the previous one but with a wide singlet aspect due to phosphorus coordinated to the Ag atom as a result of the 107Ag, and 109Ag coupling not resolved at room temperature.
:1 and 1:2 yields complexes 1 and 2, respectively, as pale-yellow solids that are stable in air (Scheme 1). However, in solution, they rapidly decompose by loss of tellurium, resulting in the formation of a tellurium mirror that can be visually observed within one hour. Despite this decomposition, the complexes can be detected and characterised by NMR spectroscopy before degradation occurs. The 1H NMR spectra of both complexes show the expected resonances associated with phenyl protons, a multiplet that corresponds to the CH group of triisopropylphosphane, and a doublet of doublets assigned to the protons of the CH3 group of the phosphane. The integrals of each signal are consistent with the stoichiometry proposed for the complex formation. The 31P{1H} NMR spectra of the complexes show two resonances (Fig. S2†). For complex 1, the signal associated with TeP(iPr)3 appears at δP = 48.3 ppm and exhibits satellites due to the 1J(31P, 125Te) coupling, which presents a value around 15% lower than that of free TeP(iPr)3 (1J(31P, 125Te) = 1669.8 Hz, CD2Cl2, 298 K).17 Another signal appears at δP = 9.9 ppm with the same intensity as the previous one but with a wide singlet aspect due to phosphorus coordinated to the Ag atom as a result of the 107Ag, and 109Ag coupling not resolved at room temperature.
For complex 2 the same resonances are observed at different chemical shift and also with the appropriate integral ratio, but the most significant change is seen in the 31P{1H} NMR spectrum (Fig. S6†). The resonance associated with phosphane telluride appears at δP = 43.8 ppm, centred between the satellites of 31P–125Te, with a coupling constant around 100 Hz higher than that exhibited by complex 1. The magnitude of the 1J(31P, 125Te) in complex 2 is comparable to that reported by du Mont and coworkers,16b for a silver tricoordinated phosphane telluride complex (Fig. 2e), which shows a 1J(31P, 125Te) = 1498.6 Hz while complex 2 displays a 1J(31P, 125Te) = 1481.6 Hz. Otherwise, complex 1 exhibits a 1J(31P, 125Te) = 1384.2 Hz, which is lower than the values previously reported for this type of complexes, ranging from 1418 to 1500 Hz. However, this value is more closely aligned to the hexameric silver complexes (Fig. 2b) reported by Chivers and coworkers.16a
Additionally, the signal associated with PPh3 bound to Ag appears as a broad singlet with an intensity half that of the other signal and at δP = 6.8 ppm, which implies a ΔδP = –3.1 ppm with respect to complex 1. This change could be attributed to a variation in the coordination environment of Ag(I) from a linear geometry to a trigonal plane geometry.18
Remarkably, the mass spectra (ESI-QTOF) of both complexes provide valuable insight into their chemical properties. For complex 1, the molecular ion is located at an m/z ratio of 659.0397. Conversely, the molecular ion of complex 2 cannot be observed due to fragmentation processes. Despite this, both spectra exhibit two prominent ions. The first is located at an m/z of 684.9936 and has a relative intensity of 100%, corresponding to the fragment [Ag{TeP(iPr)3}2]+. Additionally, both spectra show an ion at m/z 557.0874 with a relative intensity of 40%, corresponding to the species [Ag{P(iPr)3}{TeP(iPr)3}]+ resulting from a tellurium atom loss. Additionally, elemental analytical data corroborates the proposed stoichiometry for both complexes.
In addition, the reaction between Ag(OTf) and TeP(iPr)3 in a 1:2 ratio produces the complex [Ag2(TeP(iPr)3)4][OTf]2 (3) (Scheme 1). This complex is a symmetrical dimer with an inversion axis where two telluride atoms serve as a bridge between two silver atoms.16b The 1H NMR spectrum of this complex shows a doublet of septuplets at 2.44 ppm, which corresponds to the protons of the CH group of triisopropylphosphane, and a doublet of doublets at 1.34 ppm, corresponding to the protons of the methyl group.
The 31P{1H} NMR spectrum reveals a singlet signal at 47.2 ppm with a 1J(31P–125Te) = 1386.7 Hz. Notably, this coupling constant is approximately 50 Hz lower than the value measured for the dimeric complex described previously by du Mont and collaborators (Fig. 2f).16b At room temperature, this resonance appears as a narrow singlet despite the presence of two inequivalent phosphorus atoms in the molecule. This observation could indicate the existence of a dynamic process that enables the exchange of the terminal Te atoms with the bridging Te atoms, leading to the equivalence of all phosphorus atoms in the complex (Fig. 3).
 | ||
| Fig. 3 Proposed mechanism of interconversion between terminal and bridging phosphane telluride ligands (top) and variable temperature 31P{1H} NMR spectra (162 MHz, CD2Cl2) of complex 3 (bottom). The (*) denotes free TeP(iPr)3. | ||
To gain insight into the telluride bridge opening/closing process, a series of variable temperature 31P{1H} NMR experiments were conducted. The obtained NMR spectra revealed a broadening of the signal as the temperature was lowered to −80 °C (coalescence temperature). With further decrease in temperature to −90 °C, the broad signal began to split into two inequivalent signals. Finally, at −95 °C, the two signals were resolved into two distinct peaks. This result is in line with the observed kinetic lability of the Ag–Te coordination, wherein the rapid breaking of Ag–Te bond appears to be impeded only at very low temperatures.
The ESI-QTOF mass spectrum of 3 exhibits a molecular ion that corresponds to the [Ag{Te(PiPr3)}2]+ species (Fig. S25†). This observation suggests that the Ag–Te bonds, where Te acts as a bridging ligand, are more labile compared to the bonds involving terminal Te atoms. To demonstrate the reactivity of complex 3, it was mixed with two equivalents of triphenylphosphane. This reaction proceeded rapidly, leading to the dimer cleavage and the formation of two equivalents of complex 2 (Scheme 1). The 1H and 31P{1H} NMR spectra of complex 4 after addition of PPh3 was found to be identical to that of complex 2, which is consistent with the NMR observations and provides further evidence for the dynamic behaviour of the complex. The crystal structure of 3 was established by X-ray diffraction and contains an unprecedented structural framework. Complex 3 crystallises with two independent molecules in the asymmetric unit (Fig. 4), one of them is a dimer containing two tricoordinate silver atoms and four TeP(iPr)3 ligands. Two of the ligands act as bridging ligands, linking two silver atoms, whereas the other two are bonded to the silver centres in a terminal monodentate fashion (Fig. 4a).
 | ||
| Fig. 4 (a and b) Crystal structure of both of the asymmetric molecules of complex 3.25 Ellipsoids are shown at 50% probability level. Hydrogen atoms, the triflate anions (for 3a and 3b) and triisopropyl groups (for 3b) are omitted for clarity. | ||
The Ag–Te distances in the present complex are 2.7081(3) Å and 2.7136(3) Å for the terminal ligands, and in the range 2.7945(3) to 2.8244(3) Å for the bridging ligands. The latter are comparable to those found in the [Ag2{N(SO2Me)2}{TeP(iPr)3}4][N(SO2Me)2] complex (2.8117(6) and 2.8024(6) Å), for which bridging telluride phosphanes are present.16a For the first molecule (Fig. 4a), the P–Te bond distances are 2.4164(6) Å and 2.4038(7) Å for the bridging and terminal tellurophosphanes, respectively. For the second molecule (Fig. 4b), the P–Te bond distances fall within the range of 2.4090(6) Å to 2.4246(6) Å. These bond distances agree with previously reported values for phosphane telluride silver complexes16b (Fig. 2d–f), where both terminal and bridging ligands displayed P–Te bond distances ranging from 2.40 to 2.43 Å. Such distances are also typical for organophosphorus(V) tellurides with formal double bonds, PVTe.
Additionally, a short Ag⋯Ag distance of 3.0388(3) Å is observed, which can be interpreted as a weak bonding interaction within the range for reported argentophilic interactions.19 The second molecule is an oligomer which features two Ag2Te2 cores bridged by two phosphane telluride units (Fig. 4b).
In addition, the terminal phosphane telluride units make weak Te⋯Te contacts with a distance of 3.802 Å, which is below the sum of van der Waals radii for Te⋯Te (4.12 Å).20 This intermolecular interaction leads to the formation of a supramolecular structure within one of the asymmetric molecules in the unit cell of complex 3. These secondary bonding interactions originally coined by Alcock21 have been previously identified as a chalcogen bond (ChB) and are more pronounced for heavier chalcogen atoms as tellurium.22 It has been defined by IUPAC as net attractive interaction between an electrophilic region associated with a chalcogen atom in a molecular entity and a nucleophilic region in another.23
Considering the importance of these interactions, we conducted a diffusion-ordered NMR (DOSY) experiment to study the behaviour of complex 3 in solution, particularly because the asymmetric unit of its single crystal structure contains two different molecular species. The DOSY spectrum of complex 3 at room temperature (see ESI, Fig. S13 and S14†), shows that all proton resonances display the same diffusion coefficient in CD2Cl2 (1.2 × 10−9 m2 s−1), showing that in solution complex 3 behaves as a single entity.
In pursuit of a deeper understanding of the reactivity and coordination behaviour of this phosphane telluride, we decided to expand our scope of complexes, by incorporating 1,1′-bis(diphenylphosphane)ferrocene (dppf) as ligand, having in mind the interesting redox properties that the ferrocene moiety could provide.24 We synthesised two analogues of complexes 1 and 2. Complexes 4 and 5 were prepared by reacting complex [Ag(OTf)(dppf)] with one or two equivalents of TeP(iPr)3, respectively (Scheme 1).
The 1H NMR spectra of complexes 4 and 5 exhibit resonances characteristic of the cyclopentadienyl rings of ferrocene and those assigned to phosphane telluride moiety, whose integrations are consistent with the proposed stoichiometries. The 31P{1H} NMR spectrum of complex 4 displays two signals (Fig. S16†), with one at 44.9 ppm attributed to the TePR3 unit and another wide signal at −0.4 ppm corresponding to the phosphorus atoms of the dppf ligand attached to the Ag atom. Meanwhile, the 31P{1H} NMR spectrum of complex 5 exhibits a similar pattern to that of complex 4, with some differences (Fig. S20†). Firstly, there is around an 18% decrease in the coupling constant (at room temperature) 31P–125Te compared to complex 4. Secondly, the signal of the phosphorus atoms attached to Ag appears at 2 ppm at higher field. We speculate that these differences may originate from a change in the geometric environment of the Ag atom, as was previously observed for complexes 1 and 2.
Positive-ion ESI-QTOF mass spectra reveals a molecular cation peak at m/z 951.0526 (57%) for complex 4, while complex 5 does not show the cation molecular peak but does the same fragment at m/z 951.0514 (54%). The radical cation [Ag(dppf)Te]˙+ is the most intense peak (100%) observed in both spectra with m/z 790.9189 (Fig. S26 and S27†). Elemental analytical data corroborates the proposed stoichiometry for complexes 4 and 5.
The structure of complex 4 was also determined by X-ray diffraction (Fig. 5). It is a tricoordinated mononuclear complex with a terminal TeP(iPr)3 ligand bonded to the Ag(dppf) fragment. The distance Ag–Te 2.7109(5) Å is similar to the Ag–Te bond distance found in the terminal phosphane telluride ligands of complex 3. Furthermore, the P–Te bond distance was determined to be 2.4034(6) Å, which is nearly identical to the P–Te bond distance found for the terminal tellurophosphanes in complex 3. Both complexes exhibit P–Te bond distances which are slightly longer (ca. 4 pm) than the bond distance of the free ligand (2.363(1) Å),17 which could be attributed to coordination to the silver atom, as previously observed for other silver complexes.16
 | ||
| Fig. 5 Crystal structure of complex 4.25 Ellipsoids are shown at 50% probability level. Hydrogen atoms and the triflate anion are omitted for clarity. | ||
The angle P–Ag–P of 110.53(2)° represents the main distortion from the ideal trigonal planar geometry.
In certain cases, a correlation has been reported between the values of d(P–Te) and 1J(31P, 125Te). Typically, the usual P–Te bond distances for phosphorus(V) tellurides are associated with large 1J(31P, 125Te) (>1000 Hz). In our case, both complexes 3 and 4 display similar P–Te bond distances, but their coupling constants differ by approximately 65 Hz. However, with these data, we have not found any significant correlation between d(P–Te) and 1J(31P, 125Te), other than both falling within the range for a phosphorus(V) telluride ligand.
Electrochemical studies
Organophosphorus(V) telluride species are known to exhibit rich redox behaviour26a and act as reactive sources of tellurium. Complex 4 was chosen as a simple model to investigate the possible redox transfer processes between the ferrocene moiety and phosphane telluride (Fig. 6).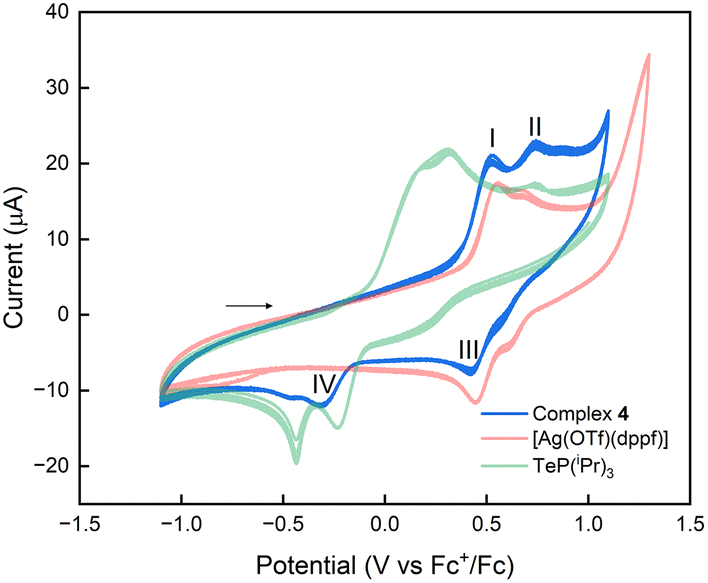 | ||
| Fig. 6 Superimposition of cyclic voltammograms of 0.5 mM solutions of 4 (blue), [Ag(OTf)(dppf)] (red) and TeP(iPr)3 (green) in CH2Cl2/0.1 M [Bu4N][PF6] at 250 mV s−1, in the electrochemical window of −600 mV to 1600 mV, referenced against Fc+/Fc couple. | ||
Upon closer examination of the voltammogram, the first redox wave exhibited quasi-reversible behaviour, which can be attributed to the (dppf)Ag moiety. This redox process involves the FeII → FeIII (peak I) and FeIII → FeII (peak III) events, resulting in E1/2 = 0.490 V. Notably, this potential is shifted by approximately −30 mV compared to the half-wave potential of the [Ag(OTf)(dppf)] complex. The oxidation peak II at Ep,a = 0.768 V can be attributed to an irreversible oxidative process involving TeP(iPr)3 fragment. It is well established that these species can participate in oxidative coupling processes, leading to the formation of dicationic species such as [(iPr3)P–Te3–P(iPr)3]2+.26c Interestingly, this process occurs at a potential +45 mV compared to the free TeP(iPr)3, indicating that the coordination to the (dppf)Ag moiety stabilises the phosphane, causing oxidation to occur at higher potentials.
Finally, the reduction peak IV at Ep,c = –0.337 V can be assigned to the reduction of tellurium species previously formed to another oxidation state, such as elemental tellurium or tritelluride anions Te32−.26b The results of the electrochemical studies suggest that complex 4 could potentially participate in redox transfer processes, further studies will be needed to gain a comprehensive knowledge into the electronic transfer processes in organophosphorus(V) telluride species.
Conclusions
In summary, our study provides insights into the coordination chemistry of the overlooked TeP(iPr)3 ligand, which has proven to be a versatile and valuable building block for the synthesis of silver complexes. While the chalcogenide analogues of TeP(iPr)3 have been extensively studied in combination with coinage metals, this phosphane has been relatively underexplored. We have reported a novel crystal structure featuring two distinct types of molecules, each containing Ag2Te2 moieties. Additionally, low-temperature NMR experiments have highlighted the dynamic equilibrium of the dimeric complex. Furthermore, by using the dppf ligand, we have synthesised new complexes that exhibit significantly higher stability in solution, enabling a comprehensive study of their redox properties using the ferrocene moiety. Overall, the use of TeP(iPr)3 in obtaining molecular complexes remains a compelling area of research, as a deeper understanding of its potential for tellurium transfer and its versatility in coordination chemistry is still required.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Agencia Estatal de Investigación (PID2019-104379RB-C21, PID2022-136861NB-I00 and PID2020-117455GB-I00/AEI/10.13039/501100011033) and Gobierno de Aragón (Research Group E07_23R) for financial support of our research. J. C. Pérez-Sánchez also thanks the Spanish Ministerio de Universidades for a predoctoral grant (FPU21/01888).Notes and references
- A. Nordheider, J. D. Woollins and T. Chivers, Organophosphorus–tellurium chemistry: from fundamentals to applications, Chem. Rev., 2015, 115, 10378–10406 CrossRef CAS PubMed.
- (a) G. Kedarnath and V. K. Jain, Pyridyl and pyrimidyl chalcogen (Se and Te) compounds: a family of multi utility molecules, Coord. Chem. Rev., 2013, 257, 1409–1435 CrossRef CAS; (b) P. Oswal, A. Arora, S. Gairola, A. Datta and A. Kumar, Organosulfur, organoselenium, and organotellurium ligands in the development of palladium, nickel, and copper-based catalytic systems for Heck coupling, New J. Chem., 2021, 45, 21449–21487 RSC; (c) A. Arora, P. Oswal, G. K. Rao, S. Kumar, A. K. Singh and A. Kumar, Catalytically active nanosized Pd9Te4 (telluropalladinite) and PdTe (kotulskite) alloys: first precursor-architecture controlled synthesis using palladium complexes of organotellurium compounds as single source precursors, RSC Adv., 2021, 11, 7214–7224 RSC; (d) A. Arora, P. Oswal, G. K. Rao, S. Kumar, A. K. Singh and A. Kumar, Tellurium-ligated Pd(II) complex of bulky organotellurium ligand as a catalyst of Suzuki coupling: first report on in situ generation of bimetallic alloy ‘telluropalladinite’ (Pd9Te4) nanoparticles and role in highly efficient catalysis, Catal. Lett., 2022, 152, 1999–2011 CrossRef CAS.
- (a) V. K. Jain and G. Kedarnath, Applications of metal selenium/tellurium compounds in materials science, Phys. Sci. Rev., 2019, 4, 20170127 Search PubMed; (b) J. Su, K. Liu, F. Wang, B. Jin, Y. Guo, G. Liu, H. Li and T. Zhai, Van der Waals 2D transition metal tellurides, Adv. Mater. Interfaces, 2019, 6, 1900741 CrossRef CAS; (c) S. Sen, S. Bera and N. Pradhan, Maneuvering tellurium chemistry to design metal–telluride heterostructures for diverse applications, Chem. Mater., 2022, 34, 9329–9343 CrossRef CAS.
- (a) Photonic Applications of Tellurite Glasses, ed. V. Rivera and D. Manzani, Springer, Cham, Switzerland, 2017 Search PubMed; (b) R. S. Chauhan, A. Kumar and P. Prabhu, Synthesis of palladium tellurolate complexes derived from hemi-labile tellurolate ligands and studies their reactivity as gas sensing materials, Inorg. Chim. Acta, 2019, 487, 395–397 CrossRef CAS; (c) M. J. Watts, P. Hatton, R. Smith, T. Fiducia, A. Abbas, R. Greenhalgh, J. M. Walls and P. Goddard, Chlorine passivation of grain boundaries in cadmium telluride solar cells, Phys. Rev. Mater., 2021, 5, 035403 CrossRef CAS.
- J. Perregaard, S. Scheibye, H. J. Meyer, I. Thomsen and S.-O. Lawesson, Studies on organophosphorus compounds XVIII. Oxidation of tertiary alicyclic amines with elemental sulfur in hexamethylphosphoric triamide (HMPA). Oxidative rearrangements of hexahydroazepines and octahydroazocines to bis(3-pyrrolyl)polysulfides, Bull. Soc. Chim. Belg., 1977, 86, 679–691 CrossRef CAS.
- M. J. Pilkington, A. M. Z. Slawin, D. J. Williams, P. T. Wood and J. D. Woollins, The preparation and characterization of binary phosphorus-selenium rings, Heteroat. Chem., 1990, 1, 351–355 CrossRef CAS.
- N. Sandblom, T. Ziegler and T. Chivers, A density functional study of the bonding in tertiary phosphine chalcogenides and related molecules, Can. J. Chem., 1996, 74, 2363–2371 CrossRef CAS.
- A. M. Brodie, G. A. Rodley and C. J. Wilkins, Cobalt(II) complexes with trimethylphosphine selenide, J. Chem. Soc. A, 1969, 2927–2929 RSC.
- W.-W. du Mont, Die Reaktion von Tri-t-butylphosphan und Tri-t-butylarsan mit Selen und Tellur: Bildung inerter und labiler Chalkogenophosphorane und -arsorane, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1985, 40b, 1453–1456 CrossRef CAS.
- J. Wachter, Metal telluride clusters – From small molecules to polyhedral structures, Eur. J. Inorg. Chem., 2004, 1367–1378 CrossRef CAS.
- M.-Y. Su, X.-Y. Li and J.-T. Zhang, Telluride semiconductor nanocrystals: progress on their liquid-phase synthesis and applications, Rare Met., 2022, 41, 2527–2551 CrossRef CAS.
- D. Jamwal and S. K. Mehta, Metal telluride nanomaterials: facile synthesis, properties and applications for third generation devices, ChemistrySelect, 2019, 4, 1943–1963 CrossRef CAS.
- G. Qiu, A. Charnas, C. Niu, Y. Wang, W. Wu and P. D. Ye, The resurrection of tellurium as an elemental two-dimensional semiconductor, npj 2D Mater. Appl., 2022, 6, 17 CrossRef CAS.
- T. Chivers and R. S. Laitinen, Tellurium: a maverick among the chalcogens, Chem. Soc. Rev., 2015, 44, 1725–1739 RSC.
- See for example: (a) M. C. Gimeno, P. G. Jones, A. Laguna and C. Sarroca, 1,1′-Bis(diphenylthiophosphoryl)ferrocene as a trans-chelating ligand in gold(I) and silver(I) complexes, J. Chem. Soc., Dalton Trans., 1995, 3563–3564 RSC; (b) M. C. Gimeno, P. G. Jones, A. Laguna and C. Sarroca, Coordination chemistry of 1,1′-bis(diphenylthiophosphoryl)ferrocene (dptpf) towards silver(I). Crystal structure of the polymeric complex [Ag2(μ-dptpf){(SPPh2)2CH2}2]n[ClO4]2n, J. Chem. Soc., Dalton Trans., 1998, 1277–1280 RSC; (c) S. Canales, O. Crespo, M. C. Gimeno, P. G. Jones and A. Laguna, Coordination chemistry of 1,1′-bis(diphenylselenophosphoryl)ferrocene (dpspf) towards Group 11 elements. Crystal structures of [Ag(dpspf){(SPPh2)2CH2}]OTf and [Au(dpspf)][AuCl2], J. Organomet. Chem., 2000, 613, 50–55 CrossRef CAS; (d) M. C. Gimeno, P. G. Jones, A. Laguna, C. Sarroca and M. D. Villacampa, 1,1′-Bis(diphenyloxophosphoryl)ferrocene (dpopf) as ligand in silver(I) complexes. Crystal structure of [Ag(dpopf)(PPh3)2]ClO4, Inorg. Chim. Acta, 2001, 316, 89–93 CrossRef CAS; (e) S. Canales, M. D. Villacampa, A. Laguna and M. C. Gimeno, Coordination studies of the ferrocenyl phosphine selenide ligand FcCON(CH2CH2PPh2Se)2, J. Organomet. Chem., 2014, 760, 84–88 CrossRef CAS.
- (a) M. C. Copsey, A. Panneerselvam, M. Afzaal, T. Chivers and P. O'Brien, Syntheses, X-ray structures and AACVD studies of group 11 ditelluroimidodiphosphinate complexes, Dalton Trans., 2007, 1528–1538 RSC; (b) C. Daniliuc, C. Druckenbrodt, C. G. Hrib, F. Ruthe, A. Blaschette, P. G. Jones and W.-W. du Mont, The first trialkylphosphane telluride complexes of Ag(I): molecular, ionic and supramolecular structural alternatives, Chem. Commun., 2007, 2060–2062 RSC; (c) D. J. Eisler, S. D. Robertson and T. Chivers, Gold complexes of ditelluridoimidodiphosphinate ligands – Reversible oxidation of Au(I) to Au(III) via insertion of gold into a phosphorus–tellurium bond, Can. J. Chem., 2009, 87, 39–46 CrossRef CAS; (d) A. Nordheider, A. M. Z. Slawin, J. D. Woollins and T. Chivers, A Silver(I) Iodide Complex of a Tellurophosphorane, Z. Anorg. Allg. Chem., 2015, 641, 405–407 CrossRef CAS.
- N. Kuhn, G. Henkel, H. Schumann and R. Fröhlich, Die Bindungsverhältnisse in Phosphantelluriden. Eine empirische NMR-Studie und die Kristallstruktur von (iso-C3H7)3PTe, Z. Naturforsch., B: J. Chem. Sci., 1990, 45, 1010–1018 CrossRef CAS.
- M. Altaf and H. Stoeckli-Evans, Silver(I) tertiary phosphine complexes: Influence of the anion on the structural and spectroscopic properties, Polyhedron, 2010, 19, 701–708 CrossRef.
- H. Schmidbaur and A. Schier, Argentophilic Interactions, Angew. Chem., Int. Ed., 2015, 54, 746–784 CrossRef CAS PubMed.
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, Consistent van der Waals radii for the whole main group, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed.
- N. W. Alcock, Secondary bonding to nonmetallic elements, Adv. Inorg. Chem. Radiochem., 1972, 15, 1–58 CrossRef CAS.
- (a) P. Scilabra, G. Terraneo and G. Resnati, The chalcogen bond in crystalline solids: a world parallel to halogen bond, Acc. Chem. Res., 2019, 52, 1313–1324 CrossRef CAS PubMed; (b) L. Vogel, P. Wonner and S. M. Huber, Chalcogen bonding: an overview, Angew. Chem., Int. Ed., 2019, 58, 1880–1891 CrossRef CAS PubMed; (c) Y. V. Torubaev, A. V. Rozhkov, I. V. Skabitsky, R. M. Gomila, A. Frontera and V. Y. Kukushkin, Heterovalent chalcogen bonding: supramolecular assembly driven by the occurrence of a tellurium(II)⋯Ch(I) (Ch = S, Se, Te) linkage, Inorg. Chem. Front., 2022, 9, 5635–5644 RSC.
- C. B. Aakeroy, D. L. Bryce, G. R. Desiraju, A. Frontera, A. C. Legon, F. Nicotra, K. Rissanen, S. Scheiner, G. Terraneo, P. Metrangolo and G. Resnati, Definition of the chalcogen bond (IUPAC Recommendations 2019), Pure Appl. Chem., 2019, 91, 1889–1892 CrossRef CAS.
- (a) G. Pilloni, B. Longato and B. Corain, Heteropolymetallic complexes of 1,1′-bis(diphenylphosphino)ferrocene (dppf): VII. Redox behaviour of dppf, J. Organomet. Chem., 1991, 420, 57–65 CrossRef CAS; (b) L. Fabbrizzi, The ferrocenium/ferrocene couple: a versatile redox switch, ChemTexts, 2020, 6, 22 CrossRef CAS.
- CCDC 2271147 (3) and 2271148 (4) contains the supplementary crystallographic data for this paper. Crystallographic details and relevant structural parameters are given in the ESI.†.
- (a) A. Cisar and J. D. Corbett, Synthesis and crystal structure of a salt containing the tritelluride(2−) anion, Inorg. Chem., 1977, 16, 632–635 CrossRef CAS; (b) N. Kuhn and H. Schumann, Phosphorus tellurium compounds. II. Tellurophosphonium cations, Phosphorus Sulfur Relat. Elem., 1986, 26, 199–201 CrossRef CAS; (c) N. Kuhn, H. Schumann and R. Boese, Oxidative coupling of tellurophosphoranes, a route to phosphane stabilised tritellurium dications, J. Chem. Soc., Chem. Commun., 1987, 1257–1258 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2271147 (3) and 2271148 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi01485a |
| This journal is © the Partner Organisations 2023 |